")
Back to Journals » International Journal of Nanomedicine » Volume 20
A Bi2O3-TiO2 Heterojunction for Triple-Modality Cancer Theranostics
Authors Zheng Z, Williams GR, Guo H, Zheng Y, Xiu M, Zhang Y, Zhang H, Wang K, Xia J, Wang Y , Zhu LM
Received 12 December 2024
Accepted for publication 15 April 2025
Published 29 April 2025 Volume 2025:20 Pages 5593—5610
DOI https://doi.org/10.2147/IJN.S511891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jie Huang
Zhiyu Zheng,1 Gareth R Williams,2 Honghua Guo,3 Yilu Zheng,1 Mengting Xiu,1 Yanyan Zhang,1 Huan Zhang,4 Kai Wang,5 Jindong Xia,3 Yu Wang,4,5 Li-Min Zhu1
1College of Biological Science and Medical Engineering, Shanghai Engineering Research Center of Nano-Biomaterials and Regenerative Medicine, Donghua University, Shanghai, People’s Republic of China; 2UCL School of Pharmacy, University College London, London, UK; 3Department of Radiology, Songjiang Hospital Affiliated to Shanghai Jiaotong University of Medicine, Shanghai, People’s Republic of China; 4International Union Laboratory on Acupuncture Based Target Discovery, International Joint Laboratory on Acupuncture Neuro-Immunology, Shanghai Research Institute of Acupuncture and Meridian, Yue Yang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 5Experiment Center for Science and Technology, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China
Correspondence: Yu Wang, Email [email protected] Li-Min Zhu, Email [email protected]
Purpose: Owing to the limitations of single-mode cancer treatments, combination therapies have attracted much attention. However, constructing a platform for combination therapies in a simple and effective way and improving the overall treatment effect remains a challenge. Our aim was to combine sonodynamic therapy, radiotherapy and chemotherapy together and improve therapeutic outcomes within one nanoplatform.
Methods: In this work, we sought to exploit the properties of nanoscale heterojunctions to this end. A multifunctional Bi2O3-TiO2@polydopamine-doxorubicin (BTPD) nanoparticle platform was constructed as an anti-cancer theranostic. Under ultrasound irradiation, the Bi2O3-TiO2 core can generate singlet oxygen to damage tumor cells. Meanwhile, the high-Z Bi2O3 can attenuate the energy of X-rays and scatter secondary electrons to enhance radiation damage in the tumor. A thin coating of polydopamine (PDA) increases the biocompatibility but also gives the particles the ability for photoacoustic imaging. Doxorubicin, a DNA repair inhibitor which can hinder tumor recovery from radiation damage, was loaded onto the PDA.
Results: A comprehensive series of in vitro and in vivo assays demonstrated that the nanoparticles were effectively taken up into cancer cells, where they could induce ROS production and cause cell death. In vivo, this led to a marked reduction in tumor volume in a murine 4T1 cancer model.
Conclusion: The formulations developed here have significant potential for future investigation and exploration in the treatment of cancer.
Keywords: sonodynamic therapy, radiosensitization, DNA repair inhibition, combination therapy
Introduction
Cancer remains one of the primary diseases threatening human health.1 Due to the difficulty in achieving a full and comprehensive treatment and the high potential for metastasis,2,3 it is particularly important to continuously improve existing treatment methods and develop targeted therapies. A number of cancer treatments are currently used in the clinic, of which chemotherapy (CT) and radiotherapy (RT) are the most commonly used.4,5 However, there are challenges in RT: healthy tissues are generally more sensitive to radiation, and different types of tumors have varying absorbance of X-rays, which may result in severe side-effects and dissatisfactory therapeutic efficacy.6–8 To solve these problems, several strategies have been developed by researchers.
X-rays can directly damage DNA by causing DNA double-strand breaks (DSBs), DNA single-strand breaks (SSBs) or ionizing water molecule to generate reactive oxygen species (ROS).5,9 While all three of these processes can be effective, cells have a complex set of countermeasures available to enable them to recover from radiation injuries. Disrupting these could be a potent way to increase radiation damage and thus improve the efficacy of RT.10,11 One method to do this is by combining RT with CT, with a particular eye on hindering cancer cells’ ability to repair radiation damage. Topoisomerase is a key enzyme in the process of DNA duplication and repair, which makes it a suitable target in the DNA damage repair (DDR) pathway.12 Many drugs have been discovered that act as topoisomerase inhibitors, such as camptothecin,13 etoposide and doxorubicin.12 These could hence be applied in concert with RT to improve therapeutic efficacy.
Another strategy to enhance RT is to elevate the absorption of X-ray energy through high-Z elements. High-Z elements can attenuate X-rays effectively through the Compton and photoelectric effects, resulting in more photoelectron and Auger electron generation.14,15 These scattered secondary electrons can directly damage DNA and produce ROS by water ionization.16 Bismuth has the highest Z of all the non-radioactive elements, and many studies have proved that bismuth-based materials have excellent radiosensitizing ability.17–19 For instance, Liu et al prepared a multifunctional ultrathin BiO2-x nanosheet which can effectively aid the absorption of X-ray energy in the tumor.20 The presence of surface defects also promoted the separation of electrons (e−) and holes (h+), which endowed it with the ability to catalyze the decomposition of H2O2 to ROS.
Sonodynamic therapy (SDT) is a non-invasive cancer treatment based on the absorption of ultrasound and subsequent generation of ROS. At present, the most frequently used sonosensitizers are a series of small molecules such as dihydrochlorin e6,21 hematoporphyrin, and its derivatives.22 Unfortunately, these small molecule sensitizers have a number of shortcomings. Many can also be used as photosensitizers, which means they can produce ROS under light exposure. This phenomenon may lead to side effects such as skin sensitization.23 Moreover, toxicity can also be caused by heavy-atom effects. Overall, these issues compromise the overall therapeutic efficacy of SDT. To overcome this challenge, some scholars have turned their attention to inorganic sonosensitive materials, with TiO2 being one of the most widely studied. However, the rapid recombination of e− and h+ in TiO2 reduces the production of ROS such that the sonodynamic therapeutic efficacy is limited.24
A heterojunction strategy can be a solution to these problems. When two semiconductors or a semiconductor and a metal are in contact with one other physically, their energy bands undergo various forms of curvature due to differences in the Fermi energy levels. The former is termed an S-S junction, while the latter is an S-M junction. This phenomenon can hinder the recombination of electrons and holes when semiconductors are excited, resulting in the generation of increased amounts of ROS. Most studies employing S-M junctions to enhance antitumor performance have utilized noble metals (eg, gold, platinum).25,26 Although these materials exhibit excellent performance, their high cost hinders practical applications. In contrast, the strategy of S-S junctions holds greater advantages in this regard.
Zhou et al designed a two-dimensional heterojunction composed of TiO2 nanosheets and oxygen-defective MnO2 dots and found that this can effectively promote e−/h+ separation and increase the production of singlet oxygen and hydroxyl radicals.24 Among the various semiconductor options, bismuth oxide not only exhibits the previously mentioned radiosensitizing effects but is also easy to prepare. Previous studies have shown that nanoscale bismuth oxide-contained materials demonstrate high biocompatibility and low toxicity.27,28 These advantages make bismuth oxide the preferred choice for this study.
There are to date few reports on the combination of SDT and RT, despite the great potential for synergistic therapy which could result from this.29,30 Herein, a Bi2O3/TiO2 nanoscale heterojunction was constructed through hydrothermal methods and calcination. Following a surface modification with polydopamine, the chemotherapeutic agent doxorubicin (DOX) could be loaded onto the platform through electrostatic interactions and π-π stacking to give Bi2O3-TiO2@PDA-DOX (BTPD) nanoparticles (NPs). As demonstrated in Figure 1, BTPD NPs accumulate at the tumor site passively through the enhanced permeability and retention effect, and the mildly acidic tumor microenvironment will accelerate degradation of the polydopamine layer. This triggers DOX release and inhibits topoisomerase II, preventing the tumor cells from repairing DNA damage caused by RT. Simultaneously, the presence of bismuth allows the nanoplatform to generate highly toxic secondary electrons and further damage DNA. With an effective separation of e− and h+ after ultrasound irradiation, BTPD is able to produce high levels of singlet oxygen, resulting in enhanced SDT. In addition, polydopamine can emit an ultrasound signal when being irradiated by near-infrared light. This endows the nanoplatform with photoacoustic imaging (PAI) ability. Therefore, this work demonstrates a multifunctional drug-loaded nanoscale heterojunction which integrates RT, enhanced SDT, CT and PAI functionality.
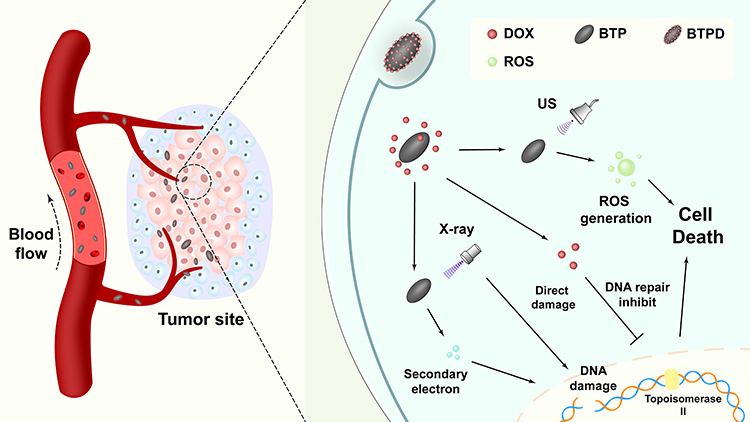 |
Figure 1 The strategy underlying this work, in which BTPD can achieve anti-cancer therapy through RT/enhanced SDT/CT. |
Materials and Methods
Materials
Tetrabutyl titanate (TBT), glacial acetic acid, nitric acid (65%), absolute ethanol, dimethyl sulfoxide (DMSO) and urea were purchased from Sinopharm Chemical Reagent Co., Ltd. Bismuth nitrate was sourced from Shanghai Yien Chemical Technology Co., Ltd. Dopamine hydrochloride was purchased from Aladdin Scientific Corp. Doxorubicin hydrochloride was obtained from Meryer (Shanghai) Biochemical Technology Co., Ltd. Tris-HCl, 1.3-diphenylisobenzofuran (DPBF) was sourced from Shanghai Macklin Biochemical Technology Co., Ltd. A calcein AM/PI kit was purchased from Bestbio Biotechnology Co., Ltd. 4′,6-diamidino-2-phenylindole (DAPI) dye solution, CCK-8 kit, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) kit, phosphate buffer (PBS, pH=7.4), penicillin–streptomycin solution and trypsin-EDTA solution were obtained from Labgic Technology Co., Ltd. Dulbecco’s Modified Eagle Medium (DMEM) and Roswell Park Memorial Institute 1640 medium (RPMI 1640) were provided by Cytiva. Fetal bovine serum (FBS) was purchased from Sigma-Aldrich. A dihydroethidium kit was obtained from Beyotime Biotech Inc. 4T1 cells, and L929 cells were purchased from iCell Bioscience Inc, a Ki67 staining kit from Cell Signaling Technology, and a γ-H2AX staining kit (ab243906) from Abcam. All chemicals were used without additional purification.
Preparation Methods
The preparation of BTPD is illustrated in Scheme 1.
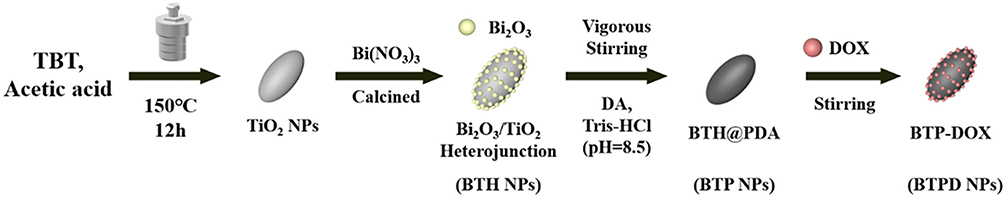 |
Scheme 1 Preparation of TiO2 NCs, BTH NPs, BTP and BTPD. |
Preparation of TiO2 nanocrystals (TiO2 NCs): the method to prepare TiO2 NCs was based on a previous study.31 Two milliliters of TBT was added to 30 mL of glacial acetic acid, and 1 mL of deionized (DI) water rapidly added dropwise to the solution. After stirring for 10 min at room temperature, the solution was transferred to a 50 mL Teflon-lined autoclave and heated at 150 °C for 12 h. The prepared TiO2 NCs were collected through centrifugation (13,000 rpm, 10 min) after the autoclave had cooled to ambient temperature. The solid product was washed with DI water and absolute ethanol three times. The TiO2 NCs were freeze-dried for subsequent use.
Preparation of Bi2O3-TiO2 heterojunction (BTH): following a previous study,32,100 mg of TiO2 NCs was dispersed in 50 mL of DI water which contained 10 μL of HNO3. About 73.0 mg of Bi(NO3)3 and 1.2 g of urea were added to the dispersion. The mixture was heated at 80 °C for 6 h under continuous stirring. The precipitate was collected through centrifugation (13,000 rpm, 10 min) and calcined at 500 °C for 3 h in air to form BTH.
Preparation of Bi2O3-TiO2@PDA (BTP): Polydopamine (PDA) can be easily formed through the self-polymerization of dopamine (DA) under alkaline conditions. Twenty milligrams of BTH nanoparticles (NPs) was dispersed in 30 mL of Tris-HCl buffer (15 mm, pH = 8.5). Twenty milliliters of DA solution of different concentrations (0.1 mg/mL, 0.2 mg/mL and 0.4 mg/mL) was added to the dispersion at a speed of 10 mL/h under vigorous stirring in dark environment. After the addition had finished, the dispersion was stirred for a further 4 h.
Characterization
Transmission electron microscopy (TEM, JEM-2100, JEOL) was used to observe the morphology and measure the size of the TiO2 NCs, BTH NPs and BTP NPs. High-resolution TEM (JEM-2100F, JEOL) was used to observe the lattice fringe and elemental distribution of BTH NPs. The composition of TiO2 NCs and BTH NPs were assessed through X-ray diffraction (XRD, Bruker D8, Bruker) and X-ray photoelectron spectroscopy (XPS, Escalab 250Xi, Thermo Fisher Scientific). Zeta potential measurements (ZS90, Malvern) and Fourier transformed infrared spectroscopy (FT-IR, Nicolet Nexus 670, NICOLET) were used to confirm the successful coating of PDA and loading of DOX.
Drug Loading
Different mass ratios (1:1, 1.5:1, 2:1) of DOX and BTP NPs were dispersed in 5 mL of DI water. These three different mass ratios correspond to 2 mg DOX+2 mg BTP NPs, 3 mg DOX+2 mg BTP NPs, and 4 mg DOX+2 mg BTP NPs, respectively. The dispersion was then stirred vigorously in the dark for 24 h. The drug loaded nanomaterials were harvested through centrifugation (13,000 rpm, 12 min), and the solid product was lyophilized for further use. To quantify the drug loading, a standard curve of DOX was constructed in water over the concentration range 5 to 120 μg/mL. The absorbance of DOX at 481 nm was measured with a UV-Vis spectrophotometer (UV-1800, Shanghai Jinghua Instrument Co., Ltd).
Measurement of Drug Loading Rate and Encapsulation Efficiency
Drug loading rate and encapsulation efficiency were determined by measuring the UV absorbance of DOX in the supernatant and calculated according to the following formulae:
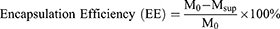
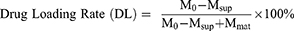
Drug Release
Drug release was measured through the dialysis method. To mimic healthy tissue and the mildly acidic tumor microenvironment, two different pH conditions were explored. One milligram of BTPD was dispersed in 2 mL of PBS (pH=7.4 or pH=6.5) and loaded in a dialysis bag (MWCO = 7000 Da). Then, the dialysis bag containing BTPD NPs was soaked in 18 mL PBS at the same pH and shaken continuously at 37 °C. At predetermined time points, 1 mL of liquid was extracted from outside the dialysis bag, and its absorbance at 481 nm was measured to calculate the amount of DOX released. An equivalent amount of fresh pre-heated PBS was added into the container to maintain a constant volume.
ROS Generation Performance
The ROS generating performance of the TiO2 NCs and BTH NPs was determined by first preparing a dispersion at 50 μg/mL in DI water. Next, 2.7 mg of DPBF was dissolved in 1 mL of DMSO to form a solution with a concentration of 10 mM. Three milliliters of TiO2 NCs or BTH NPs suspension was thoroughly mixed with 30 μL of DBPF solution in a brown sample bottle, followed by an ultrasonic treatment (1.0 MHz, 0–5 min, 40% duty cycle, 1.5 W/cm2). The absorbance at 417 nm was recorded to evaluate ROS generation.
Electron spin resonance (ESR) spectroscopy (EMXplus-6/1, Bruker) was utilized to identify the type of ROS generated under US irradiation. 2,2,6,6-Tetramethylpiperidine (TEMP) and 5.5-dimethyl-1-pyrroline-N-oxide (DMPO) were chosen as the spin traps for singlet oxygen and hydroxy radicals, respectively. The ESR signal was recorded before and after US irradiation (1.0 MHz, 40% duty cycle, 1.5 W/cm2, 5 min).
Cellular Uptake
When DOX is excited by a laser with a wavelength of 490 nm, it can emit fluorescence at 550 nm, and thus can be used to track uptake. To observe the uptake behavior of 4T1 cells, the cells were seeded in glass bottomed petri dishes at density of 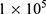 cells (2 mL) per dish. After a 24 h incubation, the old culture medium was removed. BTPD NPs were dispersed in RPMI 1640 to prepare a suspension with a DOX concentration of 5 μg/mL, and 2 mL of this BTPD suspension was added to the cell-seeded dish and co-incubated for a pre-determined time (0, 4, 12, 24 h). Subsequently, the suspension was removed, and the cells were washed three times with PBS (pH=7.4). The cells were fixed with 4% paraformaldehyde for 15 min and then washed three times. After that, the cells were stained with DAPI solution (10 μg/mL) in the dark for 5 min and washed another three times with PBS. Confocal laser scanning microscopy (CLSM; LSM700 microscope, Zeiss) was used to observe the fluorescence of DOX.
cells (2 mL) per dish. After a 24 h incubation, the old culture medium was removed. BTPD NPs were dispersed in RPMI 1640 to prepare a suspension with a DOX concentration of 5 μg/mL, and 2 mL of this BTPD suspension was added to the cell-seeded dish and co-incubated for a pre-determined time (0, 4, 12, 24 h). Subsequently, the suspension was removed, and the cells were washed three times with PBS (pH=7.4). The cells were fixed with 4% paraformaldehyde for 15 min and then washed three times. After that, the cells were stained with DAPI solution (10 μg/mL) in the dark for 5 min and washed another three times with PBS. Confocal laser scanning microscopy (CLSM; LSM700 microscope, Zeiss) was used to observe the fluorescence of DOX.
Quantification of uptake was done through flow cytometry. The same treatment as above was performed on 4T1 cells in 6-well transparent culture plates, and the cells harvested at different times (0, 2, 4, 8, 12, 24 h). The intracellular fluorescence intensity of the DOX was measured with the aid of a flow cytometer (Accuri C6, BD).
Biocompatibility and In Vitro Cytotoxicity
L929 cells and 4T1 cells were chosen to evaluate the potential cytotoxicity of the nanomaterials to healthy and tumor cells. 4T1 cells were cultured in RPMI 1640 and L929 cells in DMEM. In both cases, the media was supplemented with penicillin–streptomycin solution (100 U/mL penicillin and 100 μg/mL streptomycin) and 10% v/v FBS to give complete media.
For assays, the cells were firstly seeded in 96-well transparent cell culture plates at a density of 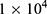 cells per well (200 μL). After incubation (37 °C, 5% CO2) for 24 h, the original culture medium was replaced with a pre-prepared RPMI 1640 or DMEM dispersion of BTP at a range of concentrations (50, 100, 150, 200, 300 μg/mL). After another 24 h incubation, the cell viability was then evaluated through the CCK-8 method.
cells per well (200 μL). After incubation (37 °C, 5% CO2) for 24 h, the original culture medium was replaced with a pre-prepared RPMI 1640 or DMEM dispersion of BTP at a range of concentrations (50, 100, 150, 200, 300 μg/mL). After another 24 h incubation, the cell viability was then evaluated through the CCK-8 method.
The cytotoxicity of different groups of materials and different treatments on 4T1 cells was also determined using the CCK-8 method. 4T1 cells were seeded into 96-well transparent cell culture plates at 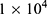 cells per well (200 μL) and incubated for 24 h in complete RPMI 1640. BTPD was dispersed in RPMI to prepare suspensions at different concentrations (to give a DOX concentration of 0.5, 1, 2, 4, 8 μg/mL). The concentration of drug-free materials was 3.5, 7, 14, 28, 56 μg/mL. After 24 h, the original media was removed and replaced with 200 μL of the BTPD suspension.
cells per well (200 μL) and incubated for 24 h in complete RPMI 1640. BTPD was dispersed in RPMI to prepare suspensions at different concentrations (to give a DOX concentration of 0.5, 1, 2, 4, 8 μg/mL). The concentration of drug-free materials was 3.5, 7, 14, 28, 56 μg/mL. After 24 h, the original media was removed and replaced with 200 μL of the BTPD suspension.
Several treatments were explored, with groups comprising: individual material treatment, ultrasound treatment, radiation treatment, and a combined treatment. For the individual material treatment group (BTPD group), cell viability was investigated with a CCK-8 kit directly after co-incubation for 24 h. For the ultrasound treatment groups (BTP+US and BTPD+US), the cells were irradiated by ultrasound (1.0 MHz, 2 W/cm2, 40% duty cycle, 3 min) after co-incubation with the relevant materials for 16 h. Cell viability was investigated using a CCK-8 kit after another 8 h incubation period. For the radiation treatment groups (BTP+X-ray and BTPD+X-ray), the cells received X-ray irradiation at a dosage of 4 Gy (tube voltage 120 kV, 385 mAs) after 16 h of incubation with the materials. After incubation for another 8 h, cell viability was measured by the CCK-8 method. For the combined treatment group (BTP+US+X-ray, BTPD+US+X-ray), after co-incubation with the test material for 16 h the cells were treated with US irradiation (1.0 MHz, 2 W/cm2, 40% duty cycle, 3 min) first and then X-ray irradiation at a dosage of 4 Gy (tube voltage 120 kV, 385 mAs). After another 8 h incubation, a CCK-8 kit was used to determine the cell viability.
Live/Dead Staining
4T1 cells were seeded in a 6-well transparent cell culture plate at 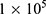 cells per well (2 mL), followed by a 24 h incubation. A series of test groups (PBS, BTP, DOX, BTPD, PBS+US+X-ray, BTP+US+X-ray, DOX+US+X-ray and BTPD+US+X-ray) were established. After 24 h, the old culture medium was replaced by 1 mL of fresh medium containing the test material (at a concentration to give a final DOX concentration of 8 μg/mL) and the cells were incubated for 16 h. Then, the US/X-ray co-treated groups were irradiated by US (1.0 MHz, 2 W/cm2, 40% duty cycle, 3 min) and X-rays (4 Gy) sequentially. All groups were incubated at 37 °C for another 8 h.
cells per well (2 mL), followed by a 24 h incubation. A series of test groups (PBS, BTP, DOX, BTPD, PBS+US+X-ray, BTP+US+X-ray, DOX+US+X-ray and BTPD+US+X-ray) were established. After 24 h, the old culture medium was replaced by 1 mL of fresh medium containing the test material (at a concentration to give a final DOX concentration of 8 μg/mL) and the cells were incubated for 16 h. Then, the US/X-ray co-treated groups were irradiated by US (1.0 MHz, 2 W/cm2, 40% duty cycle, 3 min) and X-rays (4 Gy) sequentially. All groups were incubated at 37 °C for another 8 h.
After the incubation period was complete, the culture medium was removed, and the cells were washed with PBS (pH = 7.4) 3 times. A calcein-AM/PI kit was used to differentiate living and dead cells. After diluting the stock solutions of calcein-AM and PI 1000-fold with culture medium, 1 mL of calcein-AM working solution was added to each well and the plate incubated for 10 minutes in the dark. The calcein-AM working solution was removed, and the cells were washed twice with PBS (pH = 7.4). Next, 1 mL of PI working solution was added to each well, and the plate was incubated for another 5 minutes in the dark. The PI working solution was removed, the cells washed twice, and fluorescence observed using an inverted fluorescence microscope (DMi8, Leica).
In Vitro ROS Staining
4T1 cells were seeded at density of 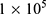 cells per well (2 mL) in a 6-well transparent cell culture plate and incubated in a 37 °C incubator for 24 h. The medium was then aspirated, followed by the addition of 1 mL of an NP suspension. The treatments were PBS, BTP, DOX, BTPD, PBS+US, BTP+US, DOX+US and BTPD+US. The concentration of BTP NPs and BTPD NPs were set as 56 μg/mL and 64 μg/mL, respectively, containing the same amount of BTP. The BTPD suspension contained the same amount of DOX as the free DOX group (8 μg/mL). After incubation for 16 hours, the culture medium was removed, and the cells were washed twice with PBS. Subsequently, 1 mL of DCFH-DA working solution (prepared with serum-free medium) was added to each well. After incubation for 10 minutes, the US treatment group was irradiated (1.0 MHz, 2 W/cm2, 40% duty cycle, 3 min). An inverted fluorescence microscope (DMi8, Leica) was used to observe the green fluorescence of 2′,7′-dichlorofluorescein (DCF) after another 20 min incubation in the dark.
cells per well (2 mL) in a 6-well transparent cell culture plate and incubated in a 37 °C incubator for 24 h. The medium was then aspirated, followed by the addition of 1 mL of an NP suspension. The treatments were PBS, BTP, DOX, BTPD, PBS+US, BTP+US, DOX+US and BTPD+US. The concentration of BTP NPs and BTPD NPs were set as 56 μg/mL and 64 μg/mL, respectively, containing the same amount of BTP. The BTPD suspension contained the same amount of DOX as the free DOX group (8 μg/mL). After incubation for 16 hours, the culture medium was removed, and the cells were washed twice with PBS. Subsequently, 1 mL of DCFH-DA working solution (prepared with serum-free medium) was added to each well. After incubation for 10 minutes, the US treatment group was irradiated (1.0 MHz, 2 W/cm2, 40% duty cycle, 3 min). An inverted fluorescence microscope (DMi8, Leica) was used to observe the green fluorescence of 2′,7′-dichlorofluorescein (DCF) after another 20 min incubation in the dark.
Establishment of Animal Model
All animal experiments were performed in accordance with the guidelines outlined in the National Research Council’s Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Donghua University (SYXK(SH)2020–0018). 4-5-week-old female Balb/c mice (obtained from Shanghai Jiesijie Experimental Animal Co., Ltd.) were used to establish a tumor-bearing mouse model. 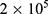 4T1 cells in 200 μL PBS (pH=7.4) were subcutaneously injected under the armpit of the left forelimb of each mouse. The tumor volume was estimated by the following formula:
4T1 cells in 200 μL PBS (pH=7.4) were subcutaneously injected under the armpit of the left forelimb of each mouse. The tumor volume was estimated by the following formula:
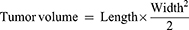
Biodistribution and Blood Compatibility
In the biodistribution study, the particles were loaded with indocyanine green (ICG) instead of DOX. To prepare the ICG loaded material, 2 mg of ICG was dissolved in 5 mL of DI water, and then 2 mg of BTP was added to the solution. After being mixed with ultrasonic assistance, the mixture was vigorously stirred at room temperature in the dark for 24 h. ICG-labelled BTP was then collected through centrifugation (13,000 rpm, 10 min).
ICG-labelled BTP was then dispersed in saline to prepare a suspension at a concentration of 800 μg/mL. 100 μL of ICG-labelled BTP dispersion was injected into a tumor-bearing mouse (n=1) through its tail vein. The near-infrared (NIR) signal at 785 nm was collected with an in vivo imaging system (IVIS Lumina Series III, PerkinElmer) at pre-determined time points (2, 4, 8, 12, 24 h).
After determining when the near-infrared signal of the tumor site reached its peak (tpeak), another tumor-bearing mouse received the same treatment and was euthanatized at t = tpeak to capture ex vivo NIR images of the major organs (heart, liver spleen lung and kidney) and tumor.
Hemolysis experiments were used to evaluate the blood compatibility of BTPD NPs. BTPD NPs were dispersed in 1 mL of saline to prepare dispersions with different concentrations (50, 100, 200, 300, 400 μg/mL). Subsequently, 2 mL of blood from healthy Balb/c mice was taken, and red blood cells were extracted by centrifugation (3000 rpm, 3 min). About 20 μL of the red blood cell suspension was added to each NPs dispersion. Positive control and negative control groups were established to give 100% hemolysis and 0% hemolysis, respectively. The positive control comprised 1 mL of deionized water with 20 μL red blood cells, and the negative control 1 mL of saline with 20 μL red blood cells. The suspensions containing red blood cells were mixed with a pipette and then placed in a refrigerator at 4 °C for 1 h. Next, the samples were centrifuged (3000 rpm, 3 min) to separate the red blood cells out. 200 μL of the supernatant from each group was transferred moved to a 96-well transparent cell culture plate and absorbance at 570 nm measured on a plate reader (Multiskan FC, Thermo Fisher Scientific). The hemolysis rates of each group were then calculated. All the experiments were performed in triplicate.
In Vivo Antitumor Efficacy
When the tumor volume reached 100 mm3, tumor-bearing mice were randomly divided into 8 groups (n=6 in each). These groups were then subjected to 8 different treatments: control: saline, group I: saline+US+X-ray, group II: free DOX, group III: free DOX+US+X-ray, group IV: BTP, group V: BTP+US, group VI: BTPD, group VII: BTPD+US+X-ray. The treatment procedure is illustrated in Scheme 2. With reference to the dosages utilized in previous studies,33 a BTP or BTPD saline suspension was intravenously injected into each mouse of groups IV–VII at a dosage of 5 mg/kg every 2 days for 14 days. Each mouse in the US groups received ultrasound irradiation (2 W/cm2, 5 min) at the tumor site 12 hours after each injection. Similarly, all mice in the X-ray group received a dose of 4 Gy of X-ray irradiation 12 hours after administration every 4 days for 14 days (ie at days 0, 4, 8 and 12). The body weight of each mouse and the tumor volume were measured every 2 days. After 14 days’ treatment, all mice were euthanized and the major organs (heart, liver, spleen, lung, kidney) and tumors were harvested. The major organs and tumor tissue were then fixed in polyformaldehyde. After dehydration and paraffin embedding, the tissues were sliced for hematoxylin and eosin (H&E), Ki67 and γ-H2AX staining.
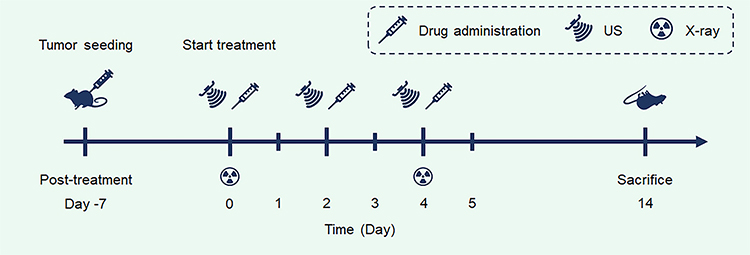 |
Scheme 2 The timeline of in vivo anti-tumor experiments. |
In Vivo ROS Generation
Another set of Balb/c tumor-bearing mice was divided into 8 groups (n=1), with the groups the same as above. Twelve hours after NP administration, ultrasound irradiation was performed on the tumor site (2 W/cm2, 5 min). The mice were sacrificed immediately, and the tumor tissue harvested, frozen in liquid nitrogen, and stored at −80 °C. The frozen sectioning technique was used to slice the samples and dihydroethidium (DHE) staining performed to observe the generation of ROS at the tumor site.
In Vivo Photoacoustic Imaging (PAI)
BTPD NPs were intravenously injected into tumor-bearing mice through the tail vein, at a dosage of 5 mg/kg. At various time points (0, 3, 6, 12, 24 h), the tumor region was subjected to PAI using a VEVO LAZR-X system (VisualSonics).
Statistical Analysis
No fewer than three parallel experiments were conducted in each experiment. Quantitative data are presented as mean ± standard deviation (S.D.), and analyzed by a two tailed Student’s t-test. P < 0.05 was taken as a statistically significant difference. Data are marked with (*) for P <0.05, (**) for P < 0.01, and (***) for P <0.001.
Results and Discussion
Characterization
The TiO2 NCs synthesized hydrothermally had uniform ellipsoid shapes with a size of approximately 77  8 nm (Figure 2a). After the precipitation of Bi2O3, the size underwent a slight increase to 108
8 nm (Figure 2a). After the precipitation of Bi2O3, the size underwent a slight increase to 108  11 nm (Figure 2b). In the deposition of the PDA coating, as demonstrated in Figures S1 and 2c, varying concentrations of DA solutions were found to yield distinct outcomes. There was no distinguishable PDA layer observed on the surface of BTH when a DA solution concentration of 0.1 mg/mL was employed. Furthermore, the formulation utilizing a 0.4 mg/mL DA solution caused particle adhesion. In contrast, the addition of 0.2 mg/mL DA solution led to a distinct translucent PDA layer on the surface of BTH NPs and another increase in size. The thickness of the PDA coating was ~9 nm, as was measured by ImageJ. The increase in particle size with successive modifications can be seen in Figure 2d.
11 nm (Figure 2b). In the deposition of the PDA coating, as demonstrated in Figures S1 and 2c, varying concentrations of DA solutions were found to yield distinct outcomes. There was no distinguishable PDA layer observed on the surface of BTH when a DA solution concentration of 0.1 mg/mL was employed. Furthermore, the formulation utilizing a 0.4 mg/mL DA solution caused particle adhesion. In contrast, the addition of 0.2 mg/mL DA solution led to a distinct translucent PDA layer on the surface of BTH NPs and another increase in size. The thickness of the PDA coating was ~9 nm, as was measured by ImageJ. The increase in particle size with successive modifications can be seen in Figure 2d.
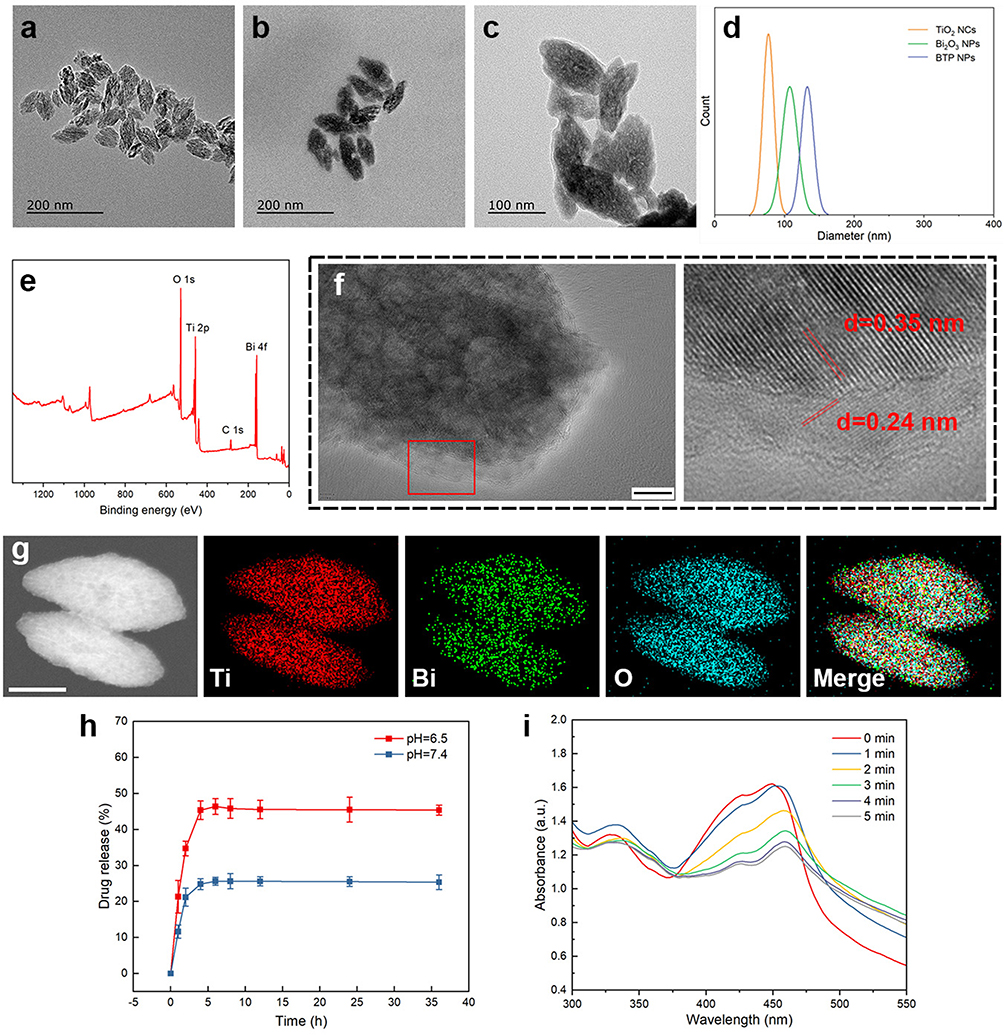 |
Figure 2 TEM images of TiO2 NCs (a), BTH NPs (b) and BTP NPs (c); size distribution (d); XPS spectrum of BTH NPs (e); HRTEM image and lattice fringes observed in the marked area (f), scale bar=10 nm; and elemental mapping of the BTH NPs (g), scale bar=50 nm; drug-release from BTPD at different pH (h), n=3; UV-Vis spectra of DPBF treated by BTH NPs (i). |
Figure S2 illustrates the XRD patterns of the TiO2 NCs and BTH NPs. Through comparison with the PDF standard substance cards (Bi2O3: PDF#76-2478, TiO2: PDF#21-1272), it can be seen that BTH was composed of anatase TiO2 and Bi2O3. The Bragg reflections at 25.3°, 37.8° and 48.0° correspond to the (101), (004) and (200) crystal planes of TiO2 and those at 28.3°, 47.1° and 55.9° are the (111), (220) and (311) planes of Bi2O3. XPS spectra are displayed in Figure 2e. The expected elements of Ti, Bi and O are all clearly present in the survey spectrum. Considering the standard binding energies detailed in the NIST XPS database, the high-resolution spectra of Ti and Bi (Figures S3 and S4) revealed the presence of Ti4+ and Bi3+ ions.34,35 Zeta potential data (Figure S5) reveal a distinct reduction from 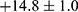 mV to
mV to 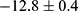 mV upon the deposition of the PDA coating on BTH, owing to the presence of myriad hydroxyl groups in PDA. These findings are all consistent with the formation of the desired system.
mV upon the deposition of the PDA coating on BTH, owing to the presence of myriad hydroxyl groups in PDA. These findings are all consistent with the formation of the desired system.
To further confirm the formation of a heterojunction, the lattice fringes of BTH NPs were imaged through HRTEM. As shown in Figure 2f, two different lattice fringes were observed on the surface of BTH. Of these, that at 0.35 nm arises from the (101) crystal plane of TiO2 and that at 0.24 nm from the (330) crystal plane of Bi2O3. The elemental mapping results in Figure 2g show a homogeneous distribution of Ti, Bi and O throughout the particles. The existence of bismuth inside the BTH NPs was attributed to the penetration of some bismuth source solution into the interior through pores or defects in the TiO2 NCs during the deposition.
FT-IR spectroscopy was obtained to verify the successful coating of PDA and loading of DOX. As shown in Figure S6, the peaks at 1622 cm −1 and 1066 cm −1 in the spectrum of BTP arise from the stretching of C=C in the aromatic ring, bending of N-H and stretching of C-N bonds in PDA.36,37 The peak at 2987 cm-1 is the C-H stretch.38 The characteristic peaks of DOX can also be seen in the BTPD spectrum (see highlighted areas in Figure S6), while they did not exist in the BTP spectrum, indicating that DOX was successfully loaded onto the surface of the BTP NPs. The change of zeta potential (Figure S5) also supported the loading of DOX: DOX has a positive charge, and the zeta potential of the BTPD NPs showed a slight increase compared with the BTP NPs. This phenomenon suggests the occurrence of electrostatic adsorption.39 Additionally, since both DOX and PDA contain aromatic ring structures, π-π stacking interactions may also contribute to the drug-loading process.40,41
Measurement of Drug Loading Rate and Encapsulation Efficiency
The drug loading rate and encapsulation efficiency were measured by UV-Vis spectroscopy based on a calibration curve (Figure S7). The DL and EE of DOX in BTPDs prepared with different mass ratios are displayed in Table 1. The highest DL and EE were calculated to be 12.6 1.5% and 9.1
1.5% and 9.1 1.2%, respectively, at a ratio of 1.5:1. This ratio was thus used for the preparation of BTPD NPs for future work.
1.2%, respectively, at a ratio of 1.5:1. This ratio was thus used for the preparation of BTPD NPs for future work.
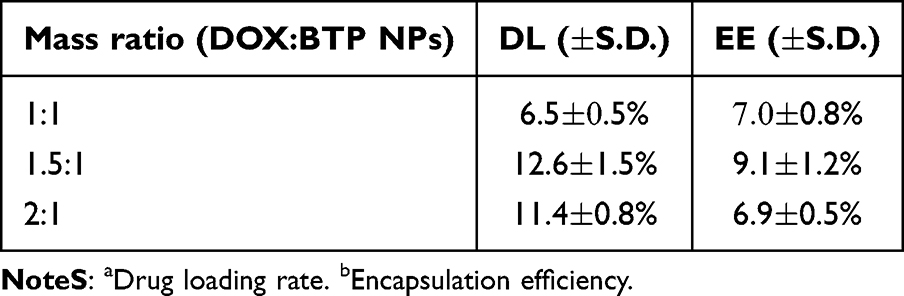 |
Table 1 DLa and EEb Results for BTPD Prepared at Different DOX:BTP NP Mass Ratios |
Drug-Release Behavior
As illustrated in Figure 2h, there was a burst release of DOX in the first 4 h, after which a plateau is observed up to 36 h. The same trend is seen at both neutral and mildly acidic conditions, except that in the latter the maximum release was 46.4%, two-fold higher than in neutral conditions. The reason for the difference may be the breaking of electrostatic interactions between DOX and PDA, which arises due to the protonation of the amine group of DOX.40,42 Since drug release is accelerated in conditions representative of the tumor microenvironment, the NPs potentially have the ability to target delivery to cancer cells and avoid off-target effects.
ROS Generation
Both the TiO2 NCs and BTH NPs have the ability to generate singlet oxygen under US irradiation. Notably, the absorbance of DPBF at 417 nm showed a greater decline in intensity with time with the BTH group than with the TiO2 group (Figures 2i and S8), indicating that more ROS was generated by BTH.
The difference in ROS generation performance between the TiO2 NCs and BTH NPs can be attributed to differences in e−/h+ pair separation. As illustrated in Figure 3a, when TiO2 is excited by US, e− move to the conduction band and h+ are left in the valence band. However, these e− and h+ can easily recombine, therefore limiting the catalytic efficiency. When Bi2O3 and TiO2 are in contact in the form of a heterojunction the excited e− move to the surface of TiO2, while the h+ are transferred to the surface of Bi2O3. This makes it much harder for the two to recombine, resulting in greater efficiency in generating ROS.43
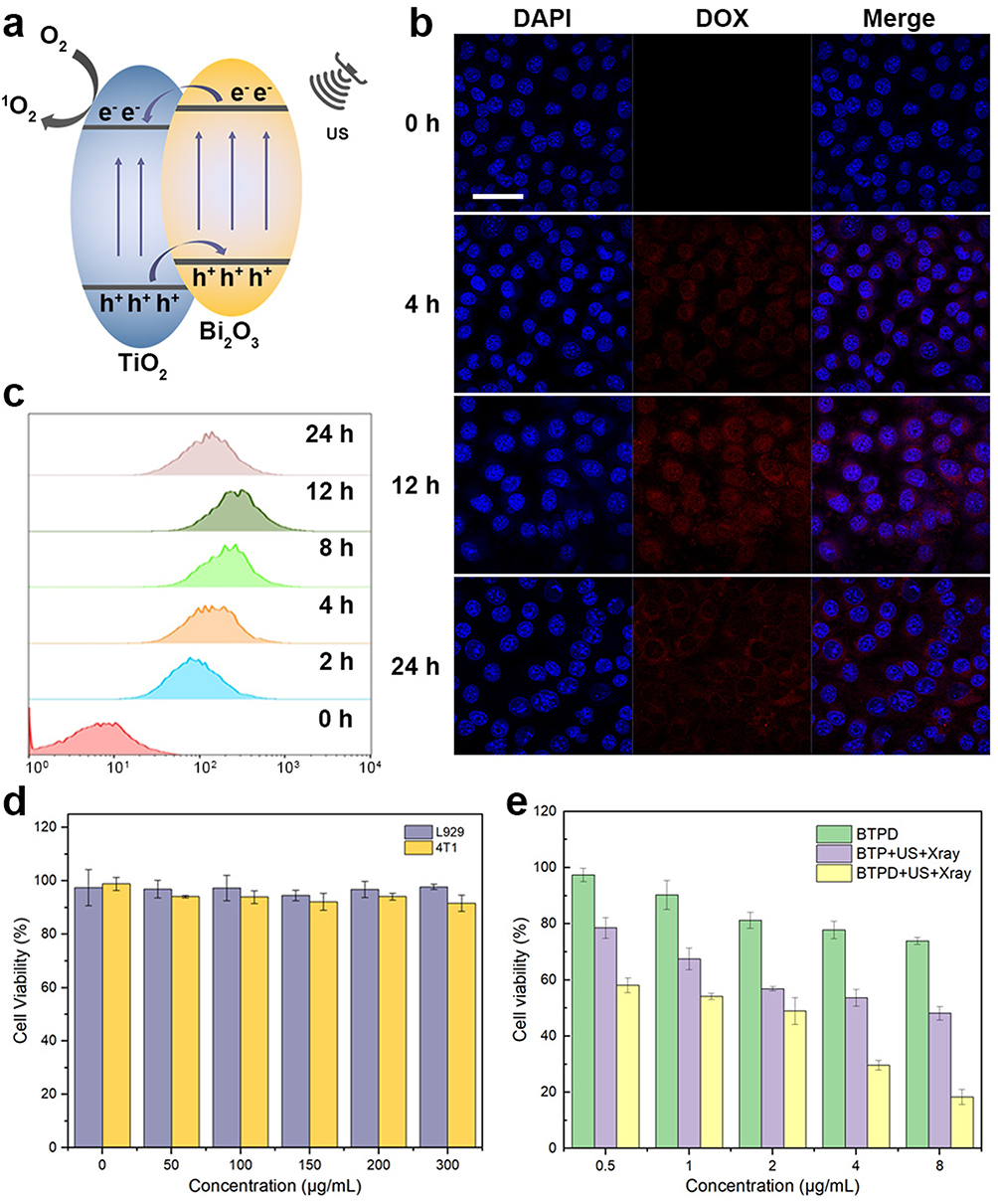 |
Figure 3 The mechanism of catalytic production of 1O2 (a); cellular uptake of BTPD NPs by 4T1 cells (b), scale bar=500 μm; quantification of cellular uptake (c); biocompatibility of BTP NPs (d); cytotoxicity of different treatments (e). |
ESR spectra allow the identification of the type of ROS generated. As illustrated in Figure S9, after ultrasound irradiation, BTH clearly exhibited a characteristic signal of singlet oxygen, whereas no such signal was observed in the absence of ultrasound irradiation. This result was consistent with the decrease in DPBF absorbance. Hydroxyl radicals, another common ROS, were also investigated using ESR. The data (Figure S10) showed no significant signals corresponding to hydroxyl radicals in the ESR spectra, regardless of ultrasound irradiation, indicating that the primary therapeutic ROS was not hydroxyl radicals.
Cellular Uptake
The cellular uptake behavior of BTPD was investigated by CLSM (Figure 3b). The uptake of DOX can be seen by dint of its red fluorescence. Stronger red fluorescence was observed in 4T1 cells after 4 and 12 h than at 0 h, showing that the uptake of BTPD was time dependent. The brightest fluorescence was seen at 12 h, followed by a decline at 24 h. Flow cytometry quantification was undertaken to verify these findings (Figure 3c). The results are in full agreement with the CLSM imaging. The amount of BTPD that was internalized by 4T1 cells increased until it reached a peak value at 12 h and then started to fall. This confirms that 12 h is a suitable time point for further experiments.
Biocompatibility and In Vitro Cytotoxicity
Two cell lines, L929 and 4T1 cells, were used to represent healthy and tumor cells for biocompatibility analysis. The results (Figure 3d) showed that the BTP NPs did not exhibit any notable toxicity (>90% viability) towards either L929 or 4T1 cells even at concentrations up to 300 μg/mL, suggesting that the BTP NPs are highly cytocompatible.
To evaluate the cytotoxicity of different treatments towards 4T1 cells, a series of CCK-8 tests were performed. The results are given in Figures 3e and S11. Overall, the combined treatment group had a greater impact on 4T1 cell viability than the individual material treatment groups, ultrasound treatment group, and radiation treatment group. The BTPD NPs, providing CT, could only reduce the cell viability of 4T1 cells by a relatively small amount. The combination of SDT and RT induced by BTP NPs with US and X-ray treatment led to greater suppression of cell viability. In comparison, the triple CT/SDT/RT obtained after treatment with BTPD+US+X-rays gave the strongest inhibitory effect. Specifically, the viability of the 4T1 cells decreased to 18.4  2.7% after incubation with BTPD NPs at a DOX concentration of 8 μg/mL and application of US and X-rays. At all concentrations, the BTPD+US+X-ray treatment gave a significantly reduced viability. Live/dead staining (Figure 4a) also confirmed that triple therapy caused the most severe cell death (denoted by red cells).
2.7% after incubation with BTPD NPs at a DOX concentration of 8 μg/mL and application of US and X-rays. At all concentrations, the BTPD+US+X-ray treatment gave a significantly reduced viability. Live/dead staining (Figure 4a) also confirmed that triple therapy caused the most severe cell death (denoted by red cells).
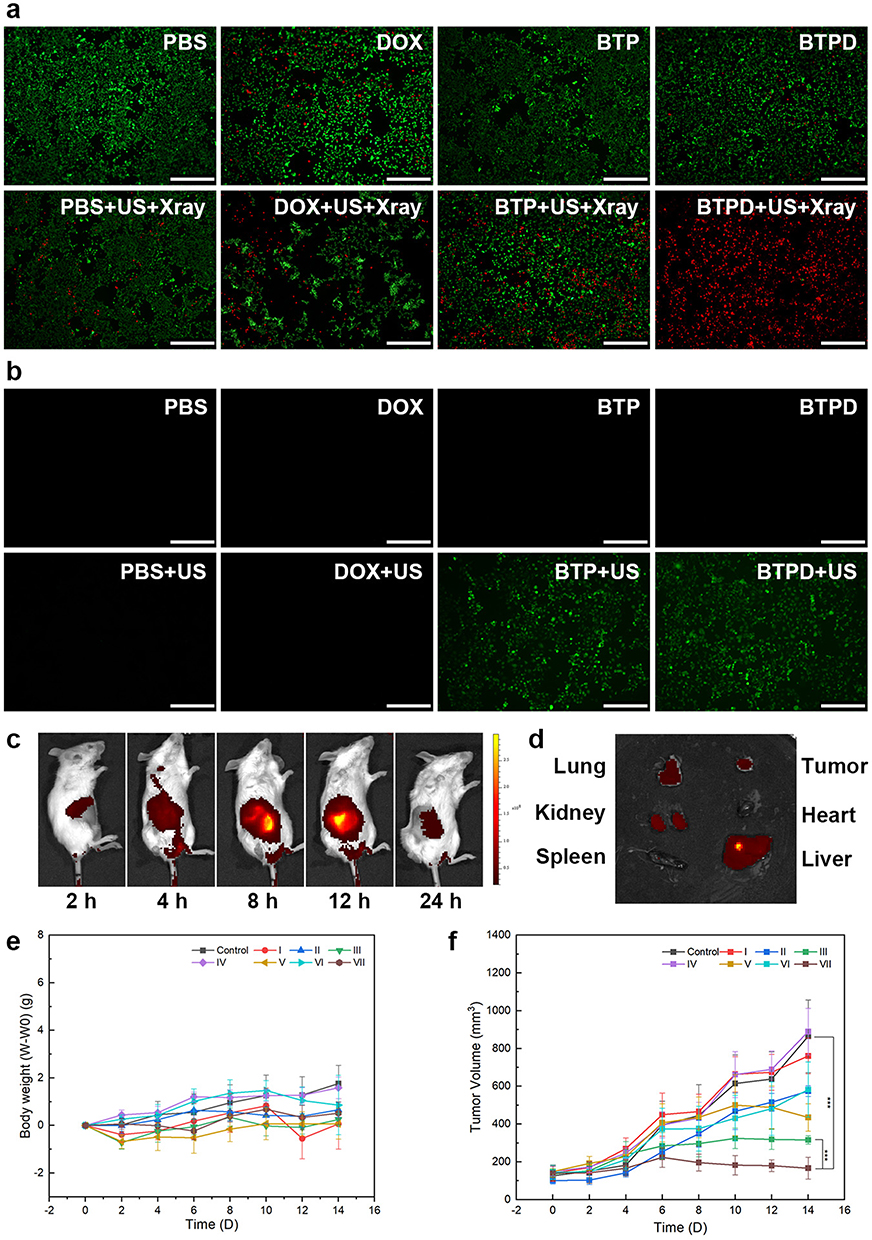 |
Figure 4 Live/dead staining images (a) and in vitro ROS staining results (b), scale bars=500 μm; biodistribution of BTPD NPs (c) and ex vivo organ images taken after 12 h (d); change of body weight with time (e); tumor volume changes with time (f). Control group: saline; group I saline+US+X-ray, group II: free DOX, group III: free DOX+US+X-ray, group IV: BTP, group (V) BTP+US+X-ray, group VI: BTPD, group VII: BTPD+US+X-ray. (***) for P < 0.001. |
In Vitro ROS Staining
To illustrate the extent of in vitro ROS generation visually, DCFH-DA was used as a fluorescent probe. DCFH-DA is a small molecule which can penetrate the cell membrane. When it reaches the cytoplasm, it can be hydrolyzed, being converted to DCFH which can no longer pass through the cell membrane. Non-fluorescent DCFH can be oxidized by intracellular ROS to DCF which emits green fluorescence. As the ROS staining results (Figure 4b) show, none of DOX, BTP NPs or BTPD NPs could generate any form of intracellular ROS without ultrasound. After being irradiated by ultrasound, no green fluorescence was observed in the PBS and DOX groups, indicating that ultrasound irradiation itself did not trigger ROS generation, and DOX is not a factor affecting intracellular ROS production. In comparison, the BTP+US group and BTPD+US group exhibited distinct green fluorescence, consistent with ROS production. This indicated that the source of ROS is BTH excited by ultrasound.
Biodistribution and Blood Compatibility
After being intravenously injected, the ICG-labelled BTP was found to accumulate at the tumor site (see Figure 4c), and over 12 h the intensity of the NIR signal continuously rose. After this, the intensity started to fall. The NIR images of isolated organs (Figure 4d) after 12 h also confirmed the enrichment of ICG-labelled BTP at the tumor site. The signals in the liver and kidneys indicated that BTPD NPs were also processed to the liver and kidney and thus can be cleared from the body. There is also a notable intensity in the lungs, the reason for which is not clear.
Blood compatibility of BTPD NPs was evaluated with a hemolysis experiment. Corresponding hemolysis rates are shown in Figure S12. Even at a concentration of 400 μg/mL the hemolysis rate was below 5%, indicating that the drug-loaded nanoplatform had good blood compatibility and should be safe for intravenous administration.
In Vivo Antitumor Efficacy
The antitumor efficacy was investigated in 4T1 tumor-bearing mice. The mice were divided into eight groups (n=6) as follows: control: saline, group I: saline+US+X-ray, group II: free DOX, group III: free DOX+US+X-ray, group IV: BTP, group V: BTP+US, group VI: BTPD, group VII: BTPD+US+X-ray. Tumor volume and the changes in body weight (Figure 4e) of each mouse were recorded every two days. There was no significant fluctuation in weight within the 14 days of treatment, indicating that the various treatments did not adversely affect the health of the mice.
The changes in the tumor volume observed during the treatment period (Figure 4f) and digital photos of the tumors after sacrifice (Figure S13) revealed that groups I and VI exhibited no obvious antitumor efficacy. The tumor sizes in groups II, V and VI were smaller, while there was a significant reduction in size in groups III and VII compared to the control group. This was because both the presence of BTP and US irradiation were indispensable for SDT, and a low dose X-ray treatment could not effectively damage the tumor tissue. In comparison, group VII demonstrated the greatest extent of tumor suppression, indicating that the combination of radiosensitization/SDT/CT was a successful strategy.
H&E staining was performed on the major organs and tumor tissues from each group (see Figure S14). The tissue morphology of the major organs did not show significant changes after the different treatments, indicating that the BTPD NPs have good biosafety. In the tumor tissues (Figure 5a), it was evident that groups I and IV exhibited dense structural organization and a significant number of nuclei, with obvious differences from the untreated control group. This indicates that the tumor tissues have not been significantly damaged. In contrast, the number of nuclei in group VII was markedly reduced, suggesting that the combined treatment of radiosensitization/SDT/CT had effectively suppressed tumor growth.
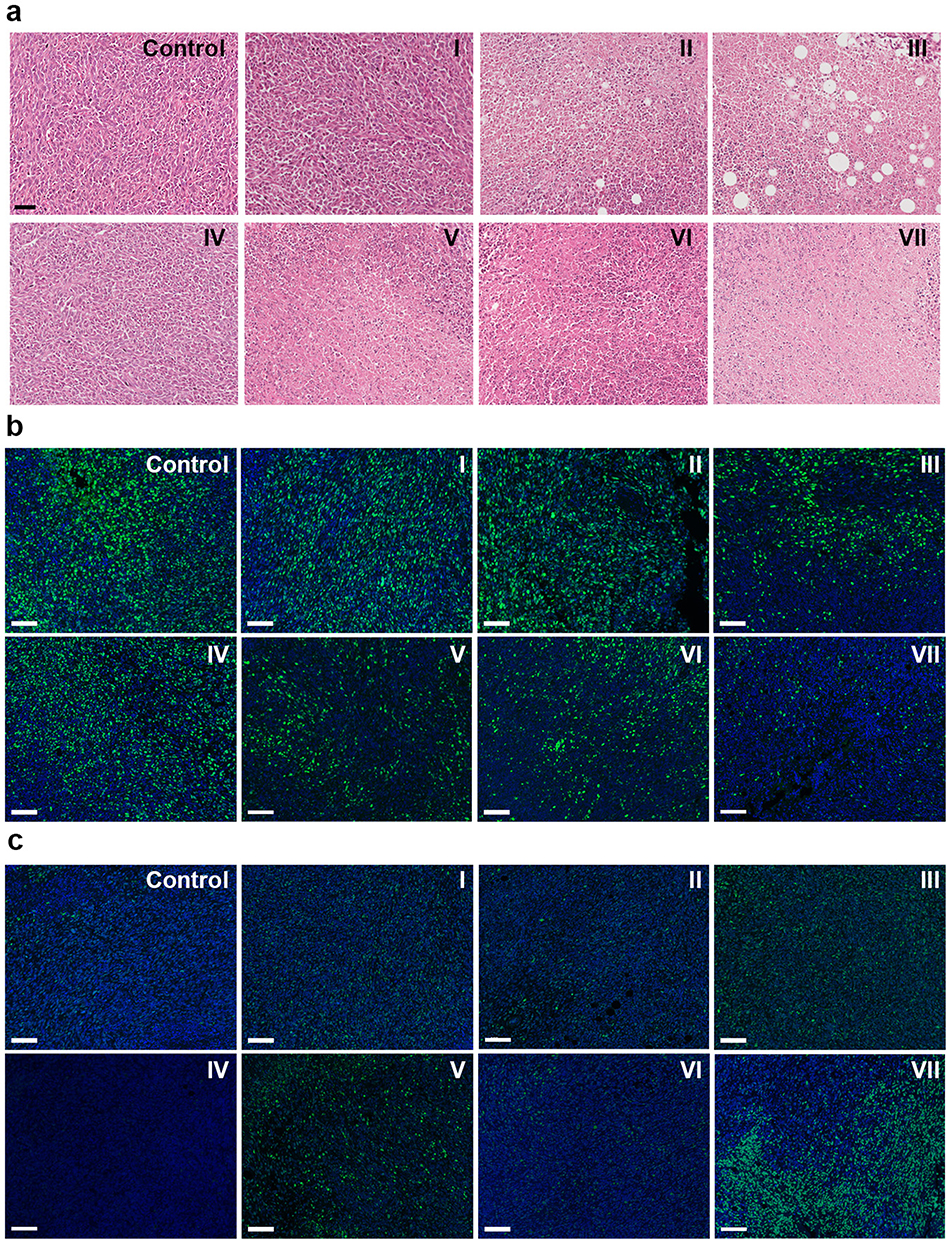 |
Figure 5 Images from H&E staining of the tumor tissue (a); scale bar=50 μm. Ki67 (b) and γ-H2AX (c) staining of tumor sections isolated after different treatments; scale bar=100 μm. Control group: saline; group I saline+US+X-ray, group II: free DOX, group III: free DOX+US+X-ray, group IV: BTP, group (V) BTP+US+X-ray, group VI: BTPD, group VII: BTPD+US+X-ray. |
To evaluate the cellular proliferation activity and DNA damage present in the tumors, Ki67 and γ-H2AX immunofluorescence staining were performed. Ki67 is a protein specifically expressed during the cell proliferation phase. Proliferative activity of the tumor can be estimated by detecting the expression level of Ki67 (green fluorescence). The results (Figure 5b) showed that the control group and group I exhibited the strongest green fluorescence, indicating that ultrasound and low-dose X-ray irradiation could not significantly affect the proliferation of tumor tissue. The green fluorescence of group II and group III showed a slight decrease compared to the control, which could be attributed to the toxicity of free DOX. In addition, the fluorescence of group III was weaker because DOX could prevent DNA repair and accumulated radiation damage. The fluorescence of group IV was similar to that of the control group, indicating that the BTP NPs could not directly affect the ability of tumor cells to proliferate. In contrast, the fluorescence intensity of group V was notably reduced, suggesting that SDT mediated by BTP NPs and radiosensitization effectively damaged tumors and inhibited cellular proliferation. The fluorescence of group VI was similar to that of group II. This is because the main way for BTPD NPs to kill tumor cells without the influence of ultrasound and X-rays is release of the chemotherapeutic agent DOX. The green fluorescence of group VII was the weakest amongst all the groups, which was a result of combined SDT, radiosensitization and DDR inhibition. Thus, under the combined treatment, the proliferation activity of tumor tissue was greatly inhibited.
H2AX is a type of histone. When DNA sustains damage, the serine at position 139 is rapidly phosphorylated, leading to the formation of γ-H2AX. In γ-H2AX staining (Figure 5c), the intensity of green fluorescence reflects the level of γ-H2AX in the tumor tissue. Hence, the stronger the green fluorescence, the more severe the radiation damage. Group IV exhibited the same performance as the control group, suggesting that BTP NPs did not cause observable DNA damage. In groups I, II, V, and VI, the tumor tissue showed weak green fluorescence, indicating that low-dose X-ray, DOX, and SDT/radiosensitization therapy had modest effects on the DNA of tumor cells. In comparison, the green fluorescence of group III was stronger, which was attributed to reduction of the activity of topoisomerase II, thereby inhibiting the ability of tumor cells to repair DNA. The strongest green fluorescence could be observed in group VII, indicating the greatest extent of radiation damage. This was the result of the combination of SDT/radiosensitization and DNA repair hindrance induced by DOX. The former elevated radiation injuries by depositing the energy of the X-rays in the tumor, and via resultant scattered highly toxic secondary electrons. At this time, tumor cells will try to activate the DNA repair process to aid in their recovery, but the presence of DOX can inhibit this and leaves more DNA single or double strand breaks, thus resulting in the accumulation of radiation injuries at the tumor site.
In Vivo ROS Generation
To evaluate ROS generation at the tumor site, DHE was used as a fluorescent probe. When DHE enters a cell, it will be easily oxidized by intracellular ROS and combined with DNA or RNA to produce red fluorescence. DHE staining of tumor sections is presented in Figure S15. The control group and all treatment groups without ultrasound irradiation (group II, IV and VI) did not exhibit significant red fluorescence, indicating that none of the free DOX nor the BTP and BTPD NPs alone can catalyze the production of ROS, which aligns with the results of in vitro cell experiments. Among all treatment groups subjected to ultrasound and X-ray irradiation, groups V and VII present strong red fluorescence, while groups I and III show no change over the control. This suggests that the primary source of ROS at the tumor site is the BTP component and its ability to drive ultrasound-mediated SDT.
PAI
The PDA coating on BTPD can absorb NIR irradiation and, as a result, emit ultrasonic signals. This property allows it to be used as a PAI agent. Following the intravenous injection of BTPD, photoacoustic signals from the tumor site were collected at predetermined time points (0, 3, 6, 12, 24 h). The photoacoustic signal at the tumor site (Figure S16) gradually increased and permeated from the periphery to the deep tumor tissue over 12 h. This was attributed to the accumulation of BTPD NPs in the tumor, which was also identified in biodistribution studies. Subsequently, the signal began to decay, indicating that BTPD NPs were metabolized and removed from the tumor. PAI is a non-invasive and powerful imaging technique, and thus this property means that the BTPD NPs could be used to monitor the tumor in real time during clinical treatments.
Compared with other studies exploring radiosensitization and SDT induced by titanium-based materials,33,44,45 the BTPD NPs result in similar tumor suppression levels at a lower dosage of X-rays and with a lower concentration of the TiO2 component. In terms of dosage, some studies based on titanium-based materials have used administration doses of 10 mg/kg or higher in mouse experiments.46,47 The fact that our study achieves comparable therapeutic effects at a lower dose demonstrates the superior efficiency of our system. Moreover, the PAI properties make it possible for the BTPD NPs to be used as a theranostic platform. This functionality is also uncommon in other multimodal therapeutic platforms. It is worth noting that some studies have reported the use of bismuth-based materials as contrast agents for CT imaging.48,49 However, the BTPD prepared in this study did not exhibit this capability, which can be attributed to the bismuth content at therapeutic concentrations being insufficient to generate distinguishable CT imaging contrast.
There remain a number of challenges which may still impede the clinical translation of BTPD. While the nanoplatform demonstrates preferential accumulation at tumor sites, its pH-dependent drug release mechanism raises concerns, as premature drug release in other mildly acidic physiological environments could diminish therapeutic efficacy at the target site. Furthermore, the potential long-term toxicity of this platform and its therapeutic efficacy against other tumor types require further investigation. Comprehensive validation through long-term follow-up studies and the collection of data across diverse cancer types will be essential.
Conclusions
In this work, a triple-modality anti-cancer therapeutic nanoplatform is reported. We first synthesized TiO2 nanocrystals (NCs) with uniform ellipsoidal shape through a hydrothermal method. Bi2O3 was then precipitated on the surface of the TiO2 NCs to form a nanoscale heterojunction. This was expected to enhance the efficiency of ROS production by promoting the separation of electrons and holes. Subsequently, a thin layer of PDA was coated on the system, endowing the nanoplatform with good biocompatibility and PAI ability. Doxorubicin, a DNA repair inhibitor, was loaded onto the PDA through electrostatic interactions and π-π stacking. The resultant BTPD NPs induced enhanced SDT, radiosensitization and DNA repair inhibition after treatment with ultrasound irradiation and low-dose X-rays. The antitumor efficacy was demonstrated through a wide range of experiments, both in vitro and in vivo. Overall, the results show that the BTPD system developed in this work could effectively suppress the growth of a tumor, suggesting that it has the potential to be a promising theranostic for cancer.
Acknowledgments
This investigation was supported by the grants 22520710400, 21WZ2501300 and 20DZ2254900 awarded by the Science and Technology Commission of Shanghai Municipality.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. Ca a Cancer J Clinicians. 2022;72(1):7–33. doi:10.3322/caac.21708
2. Zhang W, Wang F, Hu C, Zhou Y, Gao H, Hu J. The progress and perspective of nanoparticle-enabled tumor metastasis treatment. Acta Pharmaceutica Sinica B. 2020;10(11):2037–2053. doi:10.1016/j.apsb.2020.07.013
3. Kong X, Cheng R, Wang J, Fang Y, Hwang KC. Nanomedicines inhibiting tumor metastasis and recurrence and their clinical applications. Nano Today. 2021;36:101004.
4. Pei P, Wang Y, Shen W, et al. Oxygen-driven cuproptosis synergizes with radiotherapy to potentiate tumor immunotherapy. Aggregate. 2024;5:e484. doi:10.1002/agt2.484
5. Song D, Ding Y. A new target of radiotherapy combined with immunotherapy: regulatory T cells. Front Immunol. 2024;14. doi:10.3389/fimmu.2023.1330099
6. Barker HE, Paget JTE, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15(7):409–425. doi:10.1038/nrc3958
7. Wirsdörfer F, De Leve S, Jendrossek V. Combining radiotherapy and immunotherapy in lung cancer: can we expect limitations due to altered normal tissue toxicity? Int J mol Sci. 2019;20(1):24. doi:10.3390/ijms20010024
8. Yahyapour R, Motevaseli E, Rezaeyan A, et al. Reduction–oxidation (redox) system in radiation-induced normal tissue injury: molecular mechanisms and implications in radiation therapeutics. Clin Transl Oncol. 2018;20(8):975–988. doi:10.1007/s12094-017-1828-6
9. Gong LY, Zhang YJ, Liu CC, Zhang MZ, Han SX. Application of radiosensitizers in cancer radiotherapy. Int j Nanomed. 2021;16:1083–1102. doi:10.2147/IJN.S290438
10. Toulany M. Targeting DNA double-strand break repair pathways to improve radiotherapy response. Genes. 2019;10(1):25. doi:10.3390/genes10010025
11. Hopkins JL, Lan L, Zou L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. 2022;36(5–6):278–293. doi:10.1101/gad.349431.122
12. Maede Y, Shimizu H, Fukushima T, et al. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. mol Cancer Ther. 2014;13(1):214–220. doi:10.1158/1535-7163.MCT-13-0551
13. Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109(7):2894–2902. doi:10.1021/cr900097c
14. Bulin A-L, Broekgaarden M, Chaput F, et al. Radiation dose-enhancement is a potent radiotherapeutic effect of rare-earth composite nanoscintillators in preclinical models of glioblastoma. Adv Sci. 2020;7(20):2001675. doi:10.1002/advs.202001675
15. Zang Y, Gong L, Mei L, Gu Z, Wang Q. Bi2WO6 semiconductor nanoplates for tumor radiosensitization through high-Z effects and radiocatalysis. ACS Appl Mater Interfaces. 2019;11(21):18942–18952. doi:10.1021/acsami.9b03636
16. Haume K, Rosa S, Grellet S, et al. Gold nanoparticles for cancer radiotherapy: a review. Cancer Nanotechnol. 2016;7(1):8. doi:10.1186/s12645-016-0021-x
17. Javani S, Barsbay M, Ghaffarlou M, et al. Metronidazole conjugated bismuth sulfide nanoparticles for enhanced X-ray radiation therapy. J Drug Delivery Sci Technol. 2022;71:103336. doi:10.1016/j.jddst.2022.103336
18. Liu J, Liu P, Duan J, et al. Macrophages-mediated tumor accumulation and deep penetration of bismuth/manganese biomineralized nanoparticles for enhanced radiotherapy. Chin Chem Lett. 2024;35:109632. doi:10.1016/j.cclet.2024.109632
19. Colak B, Ertas YN. Implantable, 3D-printed alginate scaffolds with bismuth sulfide nanoparticles for the treatment of local breast cancer via enhanced radiotherapy. ACS Appl Mater Interfaces. 2024;16(13):15718–15729. doi:10.1021/acsami.3c17024
20. Liu H, Cheng R, Dong X, et al. BiO2–x nanosheets as radiosensitizers with catalase-like activity for hypoxia alleviation and enhancement of the radiotherapy of tumors. Inorg Chem. 2020;59(6):3482–3493. doi:10.1021/acs.inorgchem.9b03280
21. Lee HR, Kim DW, Jones VO, et al. Sonosensitizer-functionalized graphene nanoribbons for adhesion blocking and sonodynamic ablation of ovarian cancer spheroids. Adv Healthcare Mater. 2021;10(13):2001368. doi:10.1002/adhm.202001368
22. Wang Z, Yu N, Zhang J, Ren Q, Li M, Chen Z. Nanoscale Hf-hematoporphyrin frameworks for synergetic sonodynamic/radiation therapy of deep-seated tumors. J Colloid Interface Sci. 2022;626:803–814. doi:10.1016/j.jcis.2022.06.174
23. Son S, Kim JH, Wang X, et al. Multifunctional sonosensitizers in sonodynamic cancer therapy. Chem Soc Rev. 2020;49(11):3244–3261. doi:10.1039/C9CS00648F
24. Zhou Y, Yu L, Dong C, et al. Two-dimensional semiconductor heterojunction nanostructure for mutually synergistic sonodynamic and chemoreactive cancer nanotherapy. Chem Eng J. 2022;431:134017. doi:10.1016/j.cej.2021.134017
25. Zhao Y, Liu J, He M, et al. Platinum–titania schottky junction as nanosonosensitizer, glucose scavenger, and tumor microenvironment-modulator for promoted cancer treatment. ACS Nano. 2022;16(8):12118–12133. doi:10.1021/acsnano.2c02540
26. Pu Y, Pons T. Gold nanorod/titanium dioxide hybrid nanoparticles for plasmon-enhanced near-infrared photoproduction of hydroxyl radicals and photodynamic therapy. ACS Appl Mater Interfaces. 2023;15(43):49943–49952. doi:10.1021/acsami.3c05566
27. Qiu M, Wang D, Huang H, et al. A regioselectively oxidized 2D Bi/BiOx lateral nano-heterostructure for hypoxic photodynamic therapy. Adv Mater. 2021;33(49):2102562. doi:10.1002/adma.202102562
28. Chen G, Du J, Gu L, et al. Metal-sensitized Au-Bi2O3 nanoheterojunction for immunogenic cell death-boosted sono-immuno cancer therapy. Chem Eng J. 2024;482:148953. doi:10.1016/j.cej.2024.148953
29. Tian Y, Sang W, Tian H, et al. A two-step flexible ultrasound strategy to enhance tumor radiotherapy via metal–phenolic network nanoplatform. Adv Funct Mater. 2022;32(36):2205690. doi:10.1002/adfm.202205690
30. Zhang M, Dong L, Li D, et al. Sonocatalytic in situ induced oxygen storm precision enhanced reactive oxygen therapy for pancreatic cancer. Adv Funct Mater. 2023;33(41):2303451. doi:10.1002/adfm.202303451
31. He Y, Wan J, Yang Y, et al. Multifunctional polypyrrole-coated mesoporous TiO2 nanocomposites for photothermal, sonodynamic, and chemotherapeutic treatments and dual-modal ultrasound/photoacoustic imaging of tumors. Adv Healthcare Mater. 2019;8(9):1801254. doi:10.1002/adhm.201801254
32. Bagwasi S, Niu Y, Nasir M, Tian B, Zhang J. The study of visible light active bismuth modified nitrogen doped titanium dioxide photocatlysts: role of bismuth. Appl Surf Sci. 2013;264:139–147. doi:10.1016/j.apsusc.2012.09.145
33. Luo J, Cao J, Ma G, et al. Collagenase-loaded H-TiO2 nanoparticles enhance ultrasound imaging-guided sonodynamic therapy in a pancreatic carcinoma xenograft model via digesting stromal barriers. ACS Appl Mater Interfaces. 2022;14(36):40535–40545. doi:10.1021/acsami.2c08951
34. Gonbeau D, Guimon C, Pfister-Guillouzo G, Levasseur A, Meunier G, Dormoy R. XPS study of thin films of titanium oxysulfides. Surf Sci. 1991;254(1):81–89. doi:10.1016/0039-6028(91)90640-E
35. Morgan WE, Stec WJ, Van Wazer JR. Inner-orbital binding-energy shifts of antimony and bismuth compounds. Inorg Chem. 1973;12(4):953–955. doi:10.1021/ic50122a054
36. Wang Y, Williams GR, Zheng Y, et al. Polydopamine-cloaked Fe-based metal organic frameworks enable synergistic multidimensional treatment of osteosarcoma. J Colloid Interface Sci. 2023;651:76–92. doi:10.1016/j.jcis.2023.07.146
37. Song Y, Zhu P, Xu Z, Chen J. Dual-responsive dual-drug-loaded bioinspired polydopamine nanospheres as an efficient therapeutic nanoplatform against drug-resistant cancer cells. ACS Appl Bio Mater. 2020;3(9):5730–5740. doi:10.1021/acsabm.0c00512
38. Liu Y, Zhang Y, Wang J, Yang H, Zhou J, Zhao W. Doxorubicin-loaded walnut-shaped polydopamine nanomotor for photothermal-chemotherapy of cancer. Bioconjugate Chem. 2022;33(4):726–735. doi:10.1021/acs.bioconjchem.2c00100
39. Li W-Q, Wang Z, Hao S, et al. Mitochondria-targeting polydopamine nanoparticles to deliver doxorubicin for overcoming drug resistance. ACS Appl Mater Interfaces. 2017;9(20):16793–16802. doi:10.1021/acsami.7b01540
40. Wang Z, Chen Y, Zhang H, et al. Mitochondria-targeting polydopamine nanocomposites as chemophotothermal therapeutics for cancer. Bioconjugate Chem. 2018;29(7):2415–2425. doi:10.1021/acs.bioconjchem.8b00325
41. Lv L, Cheng H, Wang Z, et al. “Carrier–drug” layer-by-layer hybrid assembly of biocompatible polydopamine nanoparticles to amplify photo-chemotherapy. Nanoscale. 2022;14(37):13740–13754. doi:10.1039/D2NR03200G
42. Xing Y, Zhang J, Chen F, Liu J, Cai K. Mesoporous polydopamine nanoparticles with co-delivery function for overcoming multidrug resistance via synergistic chemo-photothermal therapy. Nanoscale. 2017;9(25):8781–8790. doi:10.1039/C7NR01857F
43. Yin Z, Zhang X, Yuan X, Wei W, Xiao Y, Cao S. Constructing TiO2@Bi2O3 multi-heterojunction hollow structure for enhanced visible-light photocatalytic performance. J Cleaner Prod. 2022;375:134112. doi:10.1016/j.jclepro.2022.134112
44. Chen Q, Zhang M, Huang H, et al. Single atom-doped nanosonosensitizers for mutually optimized sono/chemo-nanodynamic therapy of triple negative breast cancer. Adv Sci. 2023;10(6):2206244. doi:10.1002/advs.202206244
45. Deng J, Xu S, Hu W, Xun X, Zheng L, Su M. Tumor targeted, stealthy and degradable bismuth nanoparticles for enhanced X-ray radiation therapy of breast cancer. Biomaterials. 2018;154:24–33. doi:10.1016/j.biomaterials.2017.10.048
46. Xu J, Liu Y, Wang H, Hao J, Cao Y, Liu Z. Titanium boride nanosheets with photo-enhanced sonodynamic efficiency for glioblastoma treatment. Acta Biomater. 2024;188:344–357. doi:10.1016/j.actbio.2024.09.025
47. Liang S, Xiao X, Bai L, et al. Conferring Ti-Based MOFs with defects for enhanced sonodynamic cancer therapy. Adv Mater. 2021;33(18):2100333. doi:10.1002/adma.202100333
48. Feng L, Yang D, Gai S, et al. Single bismuth tungstate nanosheets for simultaneous chemo-, photothermal, and photodynamic therapies mediated by near-infrared light. Chem Eng J. 2018;351:1147–1158. doi:10.1016/j.cej.2018.06.170
49. Feng L, Gai S, He F, Yang P, Zhao Y. Multifunctional bismuth ferrite nanocatalysts with optical and magnetic functions for ultrasound-enhanced tumor theranostics. ACS Nano. 2020;14(6):7245–7258. doi:10.1021/acsnano.0c02458
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.