")
Back to Journals » International Journal of Nanomedicine » Volume 19
A Biomimetic Nanocarrier Strategy Targets Ferroptosis and Efferocytosis During Myocardial Infarction
Authors Song L, Yuan X, Zhao Z, Wang P, Wu W, Wang J
Received 24 January 2024
Accepted for publication 10 August 2024
Published 13 August 2024 Volume 2024:19 Pages 8253—8270
DOI https://doi.org/10.2147/IJN.S461212
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Lin Song,* Xiaosu Yuan,* Zhonghao Zhao, Peiyan Wang, Weiwei Wu, Jianxun Wang
School of Basic Medicine, Qingdao University, Qingdao, 266071, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianxun Wang, School of Basic Medicine, Qingdao University, Qingdao, People’s Republic of China, Tel +86 532 82991791, Email [email protected]
Background: Myocardial infarction (MI) is characterized by irreversible cardiomyocyte death resulting from an inadequate supply of oxygenated blood to the myocardium. Recent studies have indicated that ferroptosis, a form of regulated cell death, exacerbates myocardial injury during MI. Concurrently, the upregulation of CD47 on the surface of damaged myocardium following MI impairs the clearance of dead cells by macrophages, thereby hindering efferocytosis. In this context, simultaneously inhibiting ferroptosis and enhancing efferocytosis may represent a promising strategy to mitigate myocardial damage post-MI.
Methods: In this study, we engineered platelet membrane-coated hollow mesoporous silicon nanoparticles (HMSN) to serve as a drug delivery system, encapsulating ferroptosis inhibitor, Ferrostatin-1, along with an anti-CD47 antibody. We aimed to assess the potential of these nanoparticles (designated as Fer-aCD47@PHMSN) to specifically target the site of MI and evaluate their efficacy in reducing cardiomyocyte death and inflammation.
Results: The platelet membrane coating on the nanoparticles significantly enhanced their ability to successfully target the site of myocardial infarction (MI). Our findings demonstrate that treatment with Fer-aCD47@PHMSN resulted in a 38.5% reduction in cardiomyocyte ferroptosis under hypoxia, indicated by decreased lipid peroxidation and increased in vitro. Additionally, Fer-aCD47@PHMSN improved cardiomyocyte efferocytosis by approximately 15% in vitro. In MI mice treated with Fer-aCD47@PHMSN, we observed a substantial reduction in cardiomyocyte death (nearly 30%), decreased inflammation, and significant improvement in cardiac function.
Conclusion: Our results demonstrated that the cooperation between the two agents induced anti-ferroptosis effects and enhanced dead cardiomyocyte clearance by macrophage as well as anti-inflammation effects. Thus, our nanoparticle Fer-aCD47@PHMSN provides a new therapeutic strategy for targeted therapy of MI.
Keywords: myocardial infarction, ferroptosis, efferocytosis, HMSN
Graphical Abstract:

Introduction
Myocardial Infarction (MI) is one of the most severe forms of cardiovascular disease and stands as the leading cause of morbidity and mortality worldwide. Acute MI leads to ischemic cardiomyocyte death within hours due to inadequate oxygen supply to the myocardium.1,2 Although clinical interventions for MI, such as primary percutaneous coronary intervention and coronary artery bypass grafting, have significantly reduced both morbidity and mortality, heart failure remains a prevalent outcome.3–5 While reperfusion is essential for salvaging injured myocardium, it can also exacerbate injury through a surge of oxidative stress, resulting in increased cardiomyocyte death and further damage. Current therapies focusing on alleviating left ventricular dysfunction have limited efficacy and remain intrinsically non-curative, failing to address the essential loss of cardiomyocytes and vasculature.3 Consequently, improving the viability of cardiomyocytes is directly correlated with patients’ prognosis due to the negligible cardiomyocyte regeneration ability.
Ferroptosis is a novel form of cell death that operates through mechanisms distinct from classical forms of cell death such as apoptosis, necrosis, and autophagy. It is defined as an iron-dependent process characterized by the peroxidation of membrane lipids.6,7 Two primary mechanisms that can induce ferroptosis include iron overload and the inactivation of Glutathione Peroxidase 4 (GPX4), which plays a crucial role in cellular defense against oxidative stress.8,9 Evidence has shown that ferroptosis is implicated in various pathological conditions, including tumors, neurodegenerative diseases, and ischemic injury.10–12 In the context of myocardial infarction (MI), studies have highlighted the occurrence and detrimental effects of ferroptosis.11 Our proposed approach employs Ferrostatin-1, a lipid reactive oxygen species (ROS) scavenger to mitigate lipid peroxidation and inhibit ferroptosis during MI.
Effective efferocytosis by macrophages to promptly clear necrotic cardiomyocytes could prevent secondary cardiomyocyte death, activate tissue repair signaling pathways, and facilitate inflammation resolution. Inefficient efferocytosis is closely associated with myocyte loss, infarct expansion, and adverse ventricular remodeling.13,14 Studies have demonstrated that following MI, there is an increased expression of the integrin-associated protein CD47 on cardiomyocytes, which impairs efferocytosis and subsequent cardiac repair. CD47 acts as an anti-phagocytic molecule, serving as a “don’t eat me” signal during the engulfment of dying cells.15 Acute blockade of CD47 through intramyocardial injection has been shown to enhance the phagocytosis of cardiomyocytes at the infarct site, resulting in a reduction of infarct size and promoting cardiac repair.13
Intravenous injection for heart drug delivery is a minimally invasive, low-risk, and cost-effective method compared to alternatives such as intramyocardial or intrapericardial injections. The advancement of nanomaterials has facilitated substantial and targeted drug delivery to the heart.16–18 Functionalized nanoparticles can evade clearance by the kidneys and reticuloendothelial system, promoting sustained release at lower dosages and reducing the risk of overdose-related side effects.19,20 Thus, nanomaterials offer significant advantages over traditional free drugs, which suffer from rapid clearance, solubility issues, and systemic toxicity due to untargeted delivery. Mesoporous silica nanoparticles (MSNs) are widely utilized as nanocarriers for targeted drug delivery owing to their biocompatibility, large surface area, tunable pore size, modifiable surfaces, and stability.21,22 Hollow mesoporous silica nanoparticles (HMSN), a second-generation MSN variant, exhibit lower density and higher drug-loading capacity due to their hollow interiors compared to conventional MSNs.23–25
Recently, membrane-coated nanomedicines have emerged as a promising platform for therapeutic, preventive, and detection modalities. By harnessing naturally derived cell membranes, coating nanoparticles with cell membranes offers enhanced biointerfacing capabilities. These include prolonged circulation time, high biocompatibility, and disease-specific targeting, thereby enhancing both the efficacy and safety of nanoparticles.26,27 Cell membranes sourced from various cell types such as red blood cells (RBCs), white blood cells (WBCs), platelets, cancer cells, and stem cells28–30 have been utilized. Initially, entire cell membranes were employed for nanoparticle coating, but advancements such as functionalization techniques have enabled additional surface functionalities. Despite challenges related to denaturation, membrane-coated nanoparticles have seen extensive development in applications ranging from drug delivery and imaging to detoxification, immune modulation, and detection. Here, taking advantage of platelets’ natural ability to target injuries, we leverage platelet membranes to enhance the targeting and aggregation of nanoparticles in zones affected by ischemic heart injuries.
In this study, platelet membrane-coated hollow mesoporous silicon nanoparticles (PHMSN) were investigated as a targeted drug delivery system for treating myocardial infarction (MI). The combination therapy of Ferrostatin-1 and CD47 antibody was employed for post-MI treatment. Briefly, Ferrostatin-1 and CD47 antibodies were encapsulated within hollow mesoporous silicon nanoparticles, which were subsequently coated with platelet membranes (Figure 1). The resulting Fer-aCD47@PHMSN formulation aimed to inhibit ferroptosis, facilitate efferocytosis of deceased cardiomyocytes at the MI site, and reduce inflammation, thereby enhancing the effectiveness of MI treatment.
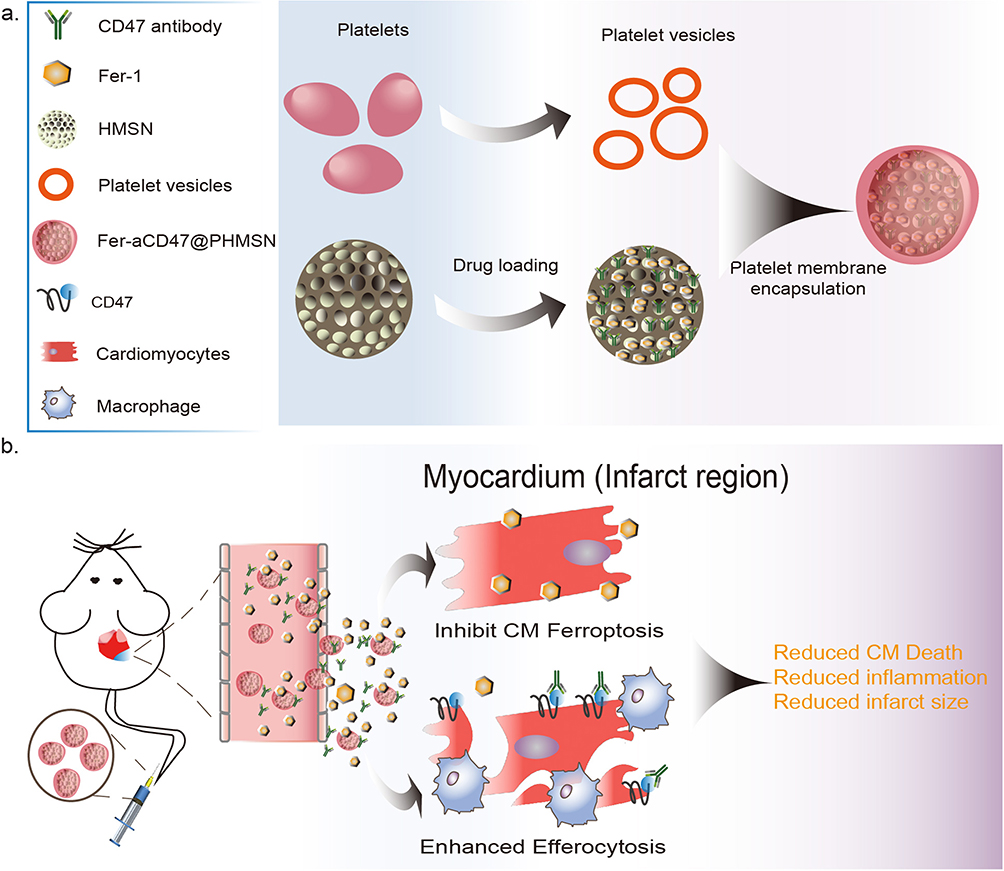 |
Figure 1 Schematic illustration of Fer-aCD47@PHMSN preparation and therapeutic strategy. (a) The efferocytosis enhancer (CD47 antibody) with ferroptosis inhibitor (Ferrostatin-1) were unified into platelet membrane-coated hollow mesoporous silicon nanoparticles (Fer-aCD47@PHMSN). (b) With intravenous administration released Ferrostatin-1 and CD47 antibodies inhibit cardiomyocyte ferroptosis and promote efferocytosis, resulting in reduced inflammation and reduced infarct size after MI. |
Materials and Methods
Materials
Dulbecco’s Modified Eagle Medium (DMEM), RPMI 1640, Fetal bovine serum (FBS), trypsin-EDTA (0.25%) solution, and Penicillin-streptomycin solution were purchased from Life Technologies (Carlsbad, CA, USA). Indocyanine green (ICG) was purchased from Dojindo Molecular Technologies. Ferrostatin-1 was purchased from Cayman Chemical. Anti-CD47 antibody was purchased from BioXcell. Mouse ELISA kits were obtained from Beijing Solarbio Science & Technology.
Synthesis of Fer-aCD47@HMSNs
Hollow mesoporous silicon nanospheres (HMSN) were acquired from XFNANO Technology Co. To synthesize Fer-aCD47@HMSN, 1 mL of 1 mg/mL anti-CD47 antibody solution and 0.2 mg of Ferrostatin-1 were mixed with 1 mL of 2 mg/mL hollow mesoporous silicon nanoparticles. The mixture was vibrated overnight at room temperature, and Fer-aCD47@HMSN was obtained by centrifugation at 12,000 rpm for 10 minutes. For fluorescent tracking in vivo, Fer-aCD47@HMSN was also labeled with the near-infrared dye Indocyanine green (ICG). Briefly, 0.1 mg of ICG was mixed with 1 mL of 2 mg/mL Fer-aCD47@HMSN in PBS (pH 7.4) overnight. Unbound ICG was removed by centrifugation.
Preparation of Platelet Membrane Vesicles
Blood from Balb/C mice was collected into an EDTA tube. The tube was centrifuged at 100g for 20 minutes at room temperature. The supernatant was collected, and 1 μM prostaglandin E1 was added to prevent platelet activation. The solution was centrifuged again at 800g for 20 minutes at room temperature. After removing the supernatant, the pellet containing platelets was suspended in PBS supplemented with 1 mM EDTA and 100x protease inhibitor.
Preparation of Platelet Membrane Camouflaged Fer-aCD47@HMSN
Fer-aCD47@PHMSN was prepared by coating Fer-aCD47@HMSN with a platelet membrane. Initially, platelet membranes were sonicated in a sonicator bath (42 kHz, 100 W) for 5 minutes and then mixed with Fer-aCD47@HMSN. The mixture underwent another round of sonication under the same frequency and power for an additional 2 minutes. The final product, Fer-aCD47@PHMSN, was obtained by centrifugation at 12,000 rpm for 10 minutes. Subsequently, the samples were purified using a 14 kDa dialysis bag over a period of 3 days.
Characterization of Nanoparticles
The size, size distribution, and zeta potential of HMSN and Fer-aCD47@PHMSN were characterized using dynamic light scattering (DLS). Nanoparticle morphology was observed by transmission electron microscopy operating at 200 kV. UV-Vis absorption spectra were measured using a Multiskan Go microplate reader. Polyacrylamide gel electrophoresis (SDS-PAGE) was employed to analyze the membrane proteins of Fer-aCD47@PHMSN. Membrane proteins from platelets and Fer-aCD47@PHMSN were extracted using Cell Total Protein Extraction kits. The extracted proteins were separated on a 10% SDS-PAGE gel using a Bio-Rad electrophoresis system at 80 V for 0.5 hours followed by 120 V for 1 hour. Subsequently, the gel was stained using a quicksilver dye kit for visualization.
Antibody and Drug Loading and in vitro Release Study
The weight of Ferrostatin-1 was determined based on its absorbance measured at 280 nm using a UV/Vis spectrophotometer. The weight of the CD47 antibody was quantified using a Mouse IgG total ELISA kit. The loading capacity and encapsulation efficiency of Fer-aCD47@HMSN and Fer-aCD47@PHMSN were calculated using the following formulas:
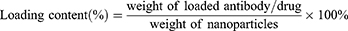
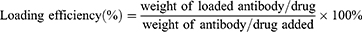
The release dynamics of antibodies and drugs from Fer-aCD47@HMSN and Fer-aCD47@PHMSN were studied separately using a dialysis method. Specifically, 1 mL of Fer-aCD47@PHMSN (2 mg/mL) was placed into a dialysis bag and immersed in 10 mL of PBS solution (pH 7.4) at 37°C. At designated time intervals (1, 12, 24, 36, 48, 60, and 72 hours), 1 mL of solution was sampled and replaced with fresh PBS. The amount of released Ferrostatin-1 was quantified at 280 nm using a UV/Vis spectrophotometer. The release of anti-CD47 antibody was measured using a Mouse IgG total ELISA kit.
DPPH Assay
The free radical scavenging capacity of Fer-aCD47@PHMSN was assessed using the DPPH Assay Kit (Solarbio, Beijing, China, BC4755). Initially, Fer-aCD47@PHMSN was diluted in the provided solution at a ratio of 1:9. Subsequently, the compounds were incubated for 0, 2, 6, and 12 hours at 37°C. Following the respective incubation periods, the mixture was centrifuged at 10,000 rpm for 10 minutes at room temperature, and the supernatant was collected. For the assay, 10 µL of the testing supernatant was thoroughly mixed with 190 µL of DPPH working solution in a 96-well plate and incubated at room temperature in the dark for 30 minutes. After incubation, the absorbance of the reaction mixture was measured at a wavelength of 515 nm using an automatic microplate reader.
Cell Culture and Colocalization Study
H9C2 cell lines and Raw264.7 cells were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). H9C2 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% antibiotic solution, while Raw264.7 cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 1% antibiotic solution. Both cell lines were incubated at 37°C in a humidified chamber with 95% air and 5% CO2. For cell experiments, 4 μg of Fer-aCD47@PHMSN was added to cells seeded in 24-well plates. For cell internalization experiments, H9C2 cells were seeded in confocal dishes and incubated the next day with either free ICG or ICG-labeled Fer-aCD47@PHMSN at 37°C for 4 hours. Following incubation, cells were washed thoroughly with cold PBS, fixed in cold ethanol for 15 minutes at −20°C, and mounted with a medium containing DAPI. Cell internalization of Fer-aCD47@PHMSN was observed using a fluorescent microscope (Olympus, USA), with excitation and emission wavelengths set at 780 nm and 800 nm for ICG, respectively.
Cell Viability, Apoptosis Detection
Cell viability was assessed using a cell counting kit-8 (CCK-8) assay (C0037; Beyotime, Shanghai, China) following the manufacturer’s instructions. Briefly, H9C2 cells were seeded in 96-well plates at a density of 5×104 cells per well. After coculturing with Fer-aCD47@PHMSN under hypoxic conditions, cytotoxicity was evaluated by adding 10 μL of CCK-8 reagent per well and incubating for 1 hour at 37°C in 5% CO2. The absorbance was measured at 450 nm using a microplate reader (Olympus). The cell viability = (Abs of experimental group-Abs of a blank group)/ (Abs of control group-Abs of a blank group) × 100%. Cell apoptosis was tested by Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, cells were seeded at a density of 5×105 cells in 6 cm dishes. After treatment, cells were collected and incubated with 5 µL of annexin V conjugate and 1 µL of PI working solution at room temperature for 15 minutes. Apoptosis and necrosis were analyzed using a FACS Calibur flow cytometer (Becton-Dickinson, USA), and the data were processed with BD Accuri C6 Software (Becton-Dickinson, USA).
Lipid Peroxidation by MDA Assay
The lipid peroxidation was determined by the detection of MDA contents in cell and heart tissue lysates using a lipid peroxidation (MDA) assay kit (BC0020, Solarbio Life Sciences, Beijing, China). The method is based on the reaction of MDA with thiobarbituric acid (TBA) and generates an MDA-TBA adduct which can be determined spectrophotometrically at 532nm using a microplate reader.
GSH Assay
The relative concentration of glutathione (GSH) in cells and heart tissues was determined using the GSH Detection Assay Kit (Cat#A006-2-1, Nanjing Jiancheng Bioengineering Institute, China). This kit utilizes an enzymatic method where GSH reacts with 5.5′-dithiobis-(2-nitrobenzoic acid) (DTNB) to produce a 2-nitro-5-thiobenzoic acid (TNB) chromophore. The reduction of TNB releases a strong yellow signal, which was quantified spectrophotometrically at 405 nm using a microplate reader.
Western Blot
Total proteins were extracted from fresh murine hearts or cell lysates using RIPA buffer supplemented with protease inhibitors. The proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes. Membranes were then blocked with 5% defatted milk at room temperature for 1 hour. Following blocking, membranes were probed with primary antibodies diluted in 5% blocking buffer and incubated overnight at 4°C. The primary antibodies used were anti-rabbit SLC7A11 (#A2413, 1:1000, ABclonal, China), anti-rabbit GPX4 (#A13309, 1:1000, ABclonal, China), and anti-rabbit Actin (#A17910, 1:10000, ABclonal, China). After primary antibody incubation, membranes were washed with PBST and then incubated with goat anti-rabbit secondary antibody (#7074, 1:5000, Cell Signaling Technology, USA) at room temperature for 1 hour. Protein bands were visualized using an enhanced chemiluminescence solution. Actin was used as an internal control to normalize the data.
In vitro Efferocytosis Assay
Raw264.7 cells were labeled with Cell Tracker Red and starved in a serum-free medium overnight. H9C2 cells were labeled with Cell Tracker Green and subjected to hypoxic conditions (1% oxygen) overnight to induce cell death. Fer-aCD47@PHMSN nanoparticles were then added and co-cultured with macrophages at 37°C for 1 hour. Subsequently, H9C2 cells were introduced to macrophages at a ratio of 1:5 in a serum-free medium and co-cultured for an additional 2 hours. Cells were fixed with 4% paraformaldehyde, and efferocytosis was visualized and recorded using fluorescence microscopy.
Animal MI Model and Analyses
All procedures involving laboratory animals were approved and conducted in accordance with the Ethics Committee of Qingdao University. Male C57BL/6 mice aged 7–8 weeks were used in this experiment. The myocardial infarction (MI) model was induced by permanent ligation of the left anterior descending coronary artery (LAD). The sham operation group underwent all surgical procedures for MI except for the ligation of the coronary artery. Two weeks post-surgery, Doppler echocardiography was performed under 1.5% isoflurane anesthesia to assess left ventricular (LV) function in mice. Transthoracic B-mode and 2D M-mode images of the mouse LV were acquired using Doppler echocardiography equipment (VINNO 6 Lab, VINNO, China). LV ejection fraction (LVEF) and LV fractional shortening (LVFS) were calculated using the system. To label dead cardiomyocytes, 100 µL of PI dye (10 mg/kg) was injected into the mouse pulmonary vein before the mice were euthanized by neck dislocation. Hearts were rapidly frozen in liquid nitrogen for tissue sectioning. Sections were mounted with DAPI-containing mounting medium and examined under a fluorescence microscope (Olympus BX51). Masson’s trichrome staining was performed to assess cardiac fibrosis. Hearts were fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 5 μm-thick sections. Mouse ELISA kits were utilized to quantify Monocyte Chemoattractant Protein-1 (MCP-1) and Interleukin-1β (IL-1β) according to the manufacturer’s instructions. In vivo fluorescence imaging was conducted using IVIS Lumina II (Excitation Filter: 780 nm, Emission Filter: 800 nm) one hour after intravenous injection of ICG-labeled Fer-aCD47@PHMSN. In situ efferocytosis was evaluated using mouse heart slices dual-stained with PI and a macrophage (Mac3) antibody. PI-positive nuclei surrounded by or in contact with Mac3-positive macrophages were identified as macrophage-associated PI-positive cells.
In vivo Toxicity Assay
All mice were sacrificed on day 15. Blood serum was collected to quantify concentrations of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (CRE) using an automated analyzer platform (Rayto Life and Analytical Sciences Co., Ltd). Tissues were collected, fixed in 4% paraformaldehyde for 24 hours, and processed using conventional histological techniques. Sections were stained with hematoxylin and eosin (H&E), and images were captured using a Nikon optical microscope (Tokyo, Japan). Analysis of the images was performed using Nikon NIS-Elements software.
Statistical Analysis
Statistical analysis was conducted using GraphPad Prism software. Image processing followed standard guidelines using Image J, Photoshop CS, and Illustrator CS software. Data are presented as mean ± SD and were analyzed using two-tailed Student’s t-test, one-way ANOVA, or two-way ANOVA as appropriate. Statistical significance was set at P < 0.05.
Results and Discussion
Synthesis and Characterization of Fer-aCD47@PHMSN
Fer-aCD47@PHMSNs were synthesized by initially loading Ferrostatin-1 and CD47 antibodies into hollow mesoporous silica nanoparticles (HMSNs), followed by camouflaging with platelet vesicles. Transmission electron microscopy (TEM) revealed that HMSNs were hollow spheres with an average diameter of approximately 110 ± 5 nm. Fer-aCD47@PHMSNs appeared uniformly spherical with a diameter of 119 ± 5 nm, exhibiting a coated membrane layer on their surface (Figure 2a). The zeta potential of Fer-aCD47@PHMSNs varied from −15.1 mV (HMSN) to −20.7 mV (Figure 2b). Dynamic light scattering (DLS) measurements indicated mean diameters of 119 ± 7 nm for HMSNs and 165.3 ± 12 nm for Fer-aCD47@PHMSNs, confirming good dispersibility without aggregation (Figure 2c and d). The hydrodynamic diameter of Fer-aCD47@PHMSNs increased from 129 to 165.3 nm compared to uncoated counterparts, attributed to the presence of the membrane coating.
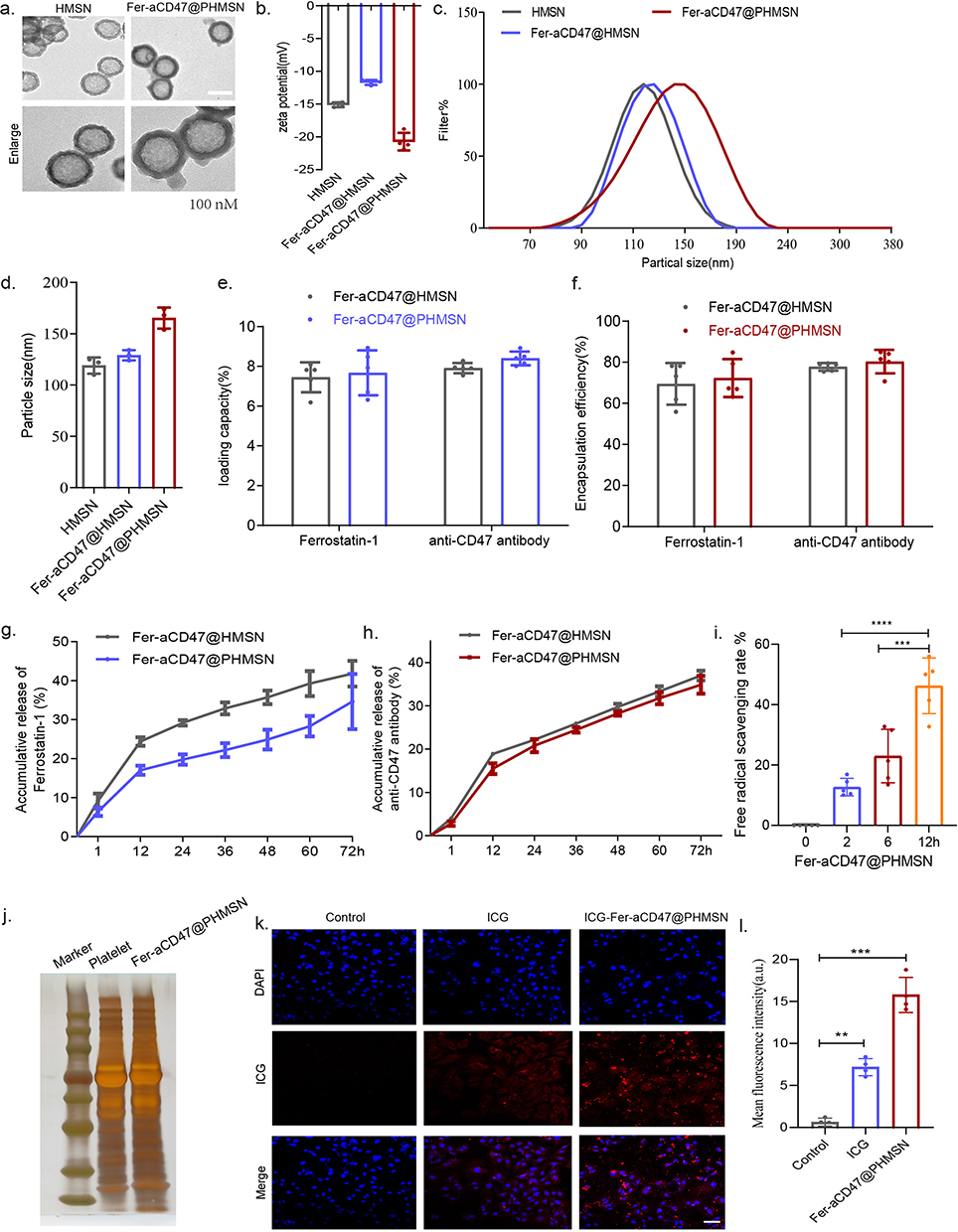 |
Figure 2 Fer-aCD47@PHMSN Characterization. (a) Transmission electron microscopy (TEM) image (scale bar:100 nm). Images were representative of three experiments. (b) Zeta potentials analysis of HMSN, Fer-aCD47@HMSN, and Fer-aCD47@PHMSN. Data represent the mean ± SD from 3 independent experiments. (c and d) Dynamic light scattering size distribution profiles and summary data for HMSN, Fer-aCD47@HMSNand Fer-aCD47@PHMSN. Data represent the mean ± SD from 3 independent experiments. (e and f) The loading capacity and encapsulation efficiency in Fer-aCD47@HMSN and Fer-aCD47@PHMSN (n=5). (g and h) In vitro drug release in Fer-aCD47@HMSN and Fer-aCD47@PHMSN. (i)Free radical scavenging rate of Fer-aCD47@PHMSN. Data represent the mean ± SD from 5 independent experiments. ***p < 0.001, ****p < 0.0001. (j)Proteins in platelet vesicles and Fer-aCD47@PHMSN were characterized by polyacrylamide gel electrophoresis. (k and l) Cell uptake of Fer-aCD47@PHMSN labeling with ICG. Scale bar: 50 μm. Images were representative of three experiments. **p < 0.01, ***p < 0.001. |
Entrapment efficiencies for Ferrostatin-1 and CD47 antibody in Fer-aCD47@PHMSNs were 72.3% and 80.3%, respectively, with corresponding drug loading rates of 7.6% and 8.4% (Figure 2e and f), confirming successful loading. Drug release profiles in PBS simulating extracellular conditions showed slower release rates for Fer-aCD47@PHMSNs compared to Fer-aCD47@HMSNs, with cumulative releases of 34.65% and 34.86% for Ferrostatin-1 and CD47 antibodies after 72 hours (Figure 2g and h), indicating potential for sustained release. The free radical scavenging capacity of Fer-aCD47@PHMSN was assessed using the DPPH Assay Kit. The scavenging rate of free radicals increased significantly with longer incubation times of Fer-aCD47@PHMSN, consistent with the release profile of Ferrostatin-1 from Fer-aCD47@PHMSN (Figure 2i). SDS-PAGE analysis of protein profiles in Fer-aCD47@PHMSNs confirmed retention of key platelet membrane proteins, crucial for immune escape and myocardial infarction (MI) targeting (Figure 2j). In vitro cellular uptake studies using ICG-labeled Fer-aCD47@PHMSNs demonstrated successful accumulation in H9C2 cardiomyocytes after 4 hours (Figure 2k and L).
Fer-aCD47@PHMSN Inhibits Cardiomyocyte Ferroptosis and Enhances Efferocytosis in vitro
Next, the efficacy of Fer-aCD47@PHMSNs in mitigating cardiomyocyte ferroptosis and enhancing efferocytosis in vitro was investigated using H9C2 cells under hypoxic conditions (1% oxygen) to simulate the microenvironment of myocardial infarction (MI). PI staining was employed to assess cardiomyocyte viability (Figure 3a and b). In the normoxia treatment groups, minimal red fluorescent signals were detected, whereas significant increases in red fluorescence were observed in the control group, PHMSN group, Fer-1+aCD47 group, and Fer-aCD47@PHMSN group under hypoxia conditions. This suggests heightened cell death in response to hypoxia. Notably, the inclusion of PHMSN alone did not influence cardiomyocyte death. Both Fer-1+aCD47 and Fer-aCD47@PHMSN attenuated cell death by approximately 41% and 41.8%, respectively, with no significant difference observed in their protective effects. These results indicate that Fer-aCD47@PHMSN effectively inhibits hypoxia-induced cardiomyocyte death in vitro. Cell viability was further assessed using a CCK-8 assay (Figure 3c). Hypoxia treatment markedly reduced cell viability, while treatment with Fer-1+aCD47 and Fer-aCD47@PHMSN significantly mitigated cell death by approximately 36.1% and 43.5%, respectively, compared to the control group. PHMSN treatment alone had no discernible effect on cell viability. To confirm the inhibition of Fer-aCD47@PHMSN on ferroptosis-related cell death, levels of lipid reactive oxygen species (ROS) and glutathione (GSH) were measured. Hypoxia induced substantial lipid peroxidation, whereas both Fer-1+aCD47 and Fer-aCD47@PHMSN reduced lipid peroxidation by approximately 31.5% and 38.5%, respectively, compared to the control group (Figure 3d). Furthermore, Fer-aCD47@PHMSN elevated intracellular GSH levels by 16.1% compared to the hypoxia-only group, indicating potent antioxidant effects (Figure 3e). We also investigated the effects of Fer-aCD47@PHMSNs treatment over different durations. H9C2 cells were exposed to hypoxic conditions (1% oxygen) overnight, followed by treatment with Fer-aCD47@PHMSNs for 2h, 6h, and 12h. Notably, the 12-hour treatment duration aligns with the hypoxic exposure time. We assessed cell viability, lipid peroxidation, and GSH levels. The results indicated that the ability of Fer-aCD47@PHMSNs to salvage cell viability increased with longer treatment durations, with the 12-hour treatment demonstrating the most pronounced effects (Supplementary Figure 1a). Similarly, the impact of Fer-aCD47@PHMSNs on lipid peroxidation and GSH levels also intensified with longer treatment times (Supplementary Figure 1b and c).
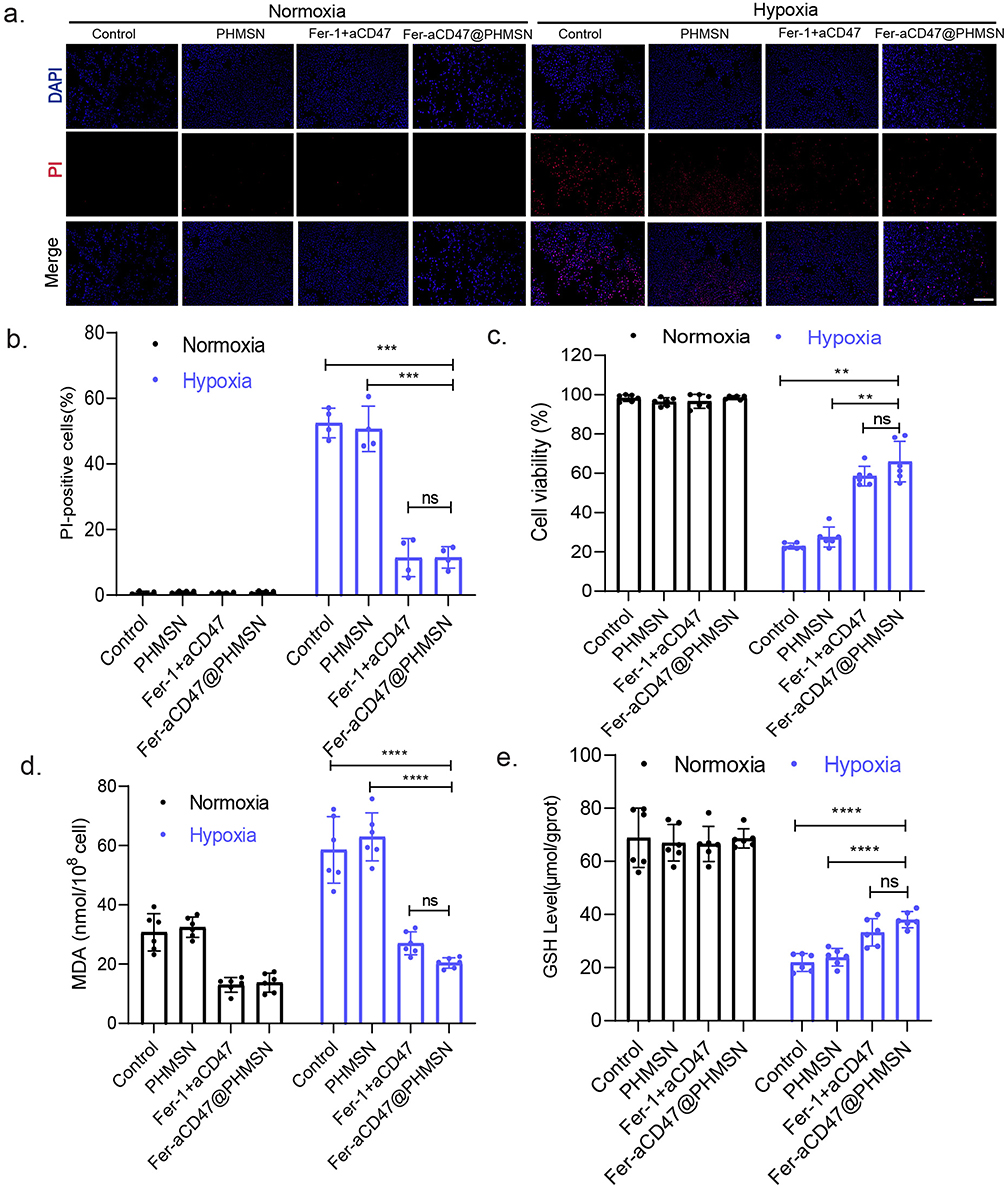 |
Figure 3 Fer-aCD47@PHMSN Inhibits cardiomyocyte death in vitro. Cells were treated with Fer-aCD47@PHMSN with/without hypoxia treatment (1% oxygen) overnight. (a and b) Evaluation of cardiomyocyte death following different treatments was done by propidium iodide (PI) staining. Data represent mean ± SD from 4 independent experiments. ns, not significant, ***p < 0.001. (c) Cell viability was determined by CCK-8. Data represent the mean ± SD from 6 independent experiments. ns, not significant, **p < 0.01. (d) Lipid ROS was quantified through MDA assay. Data represent mean ± SD from 6 independent experiments. ns, not significant, ****p < 0.0001. (e) GSH levels were measured in vitro. Data represent mean ± SD from 6 independent experiments. ns, not significant, ****p < 0.0001. |
The expression of ferroptosis-associated proteins was assessed, revealing that Fer-aCD47@PHMSNs rescued the decrease in GPX4 and SLC7A11 expression induced by hypoxia in H9C2 cells (Figure 4a–c). Furthermore, the impact of Fer-aCD47@PHMSNs on efferocytosis was examined. Figure 4d and e showed that CD47 blockade by Fer-aCD47@PHMSNs significantly enhanced apoptotic cardiomyocyte efferocytosis by macrophages compared to the isotype control. In conclusion, the fabrication of Fer-aCD47@PHMSNs does not compromise its efficacy. Fer-aCD47@PHMSNs could effectively inhibit ferroptosis in H9C2 cells and promote apoptotic cardiomyocyte efferocytosis in vitro.
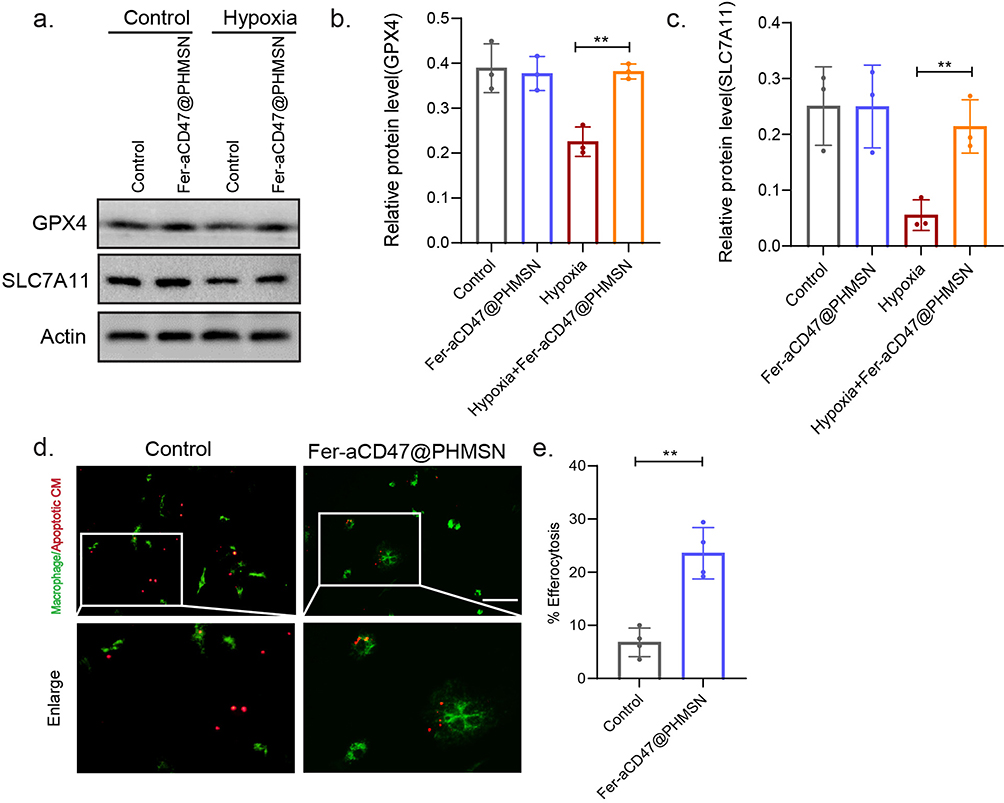 |
Figure 4 Fer-aCD47@PHMSN Inhibits cardiomyocyte ferroptosis and enhances efferocytosis in vitro. (a–c) The expression of ferroptosis-related proteins GPX4 and SLC7A11 were detected by Western blot. Images were representative of three experiments. **p < 0.01. (d and e) Fluorescent images of Dio-labeled macrophages (green) with Dil-labeled apoptotic cardiomyocytes (red). Percent efferocytosis was calculated as the percentage of macrophages with engulfed CM cells to total macrophages. Scale bar, 100 μm. Data represent mean ± SD from 4 independent experiments. **p < 0.01. |
Fer-aCD47@PHMSN Targets the MI Site Specially in vivo
We investigated the MI homing capability of intravenously injected Fer-aCD47@PHMSNs in vivo. ICG-labeled Fer-aCD47@PHMSNs (100 μL) were administered via the tail vein to both control and MI-operated animals (Figure 5a). NIRF imaging of the heart was conducted one-hour post-injection, revealing significant fluorescence in the hearts of MI mice compared to controls, where fluorescence intensity was approximately half (Figure 5b and c). To further confirm the enhanced retention of ICG-labeled Fer-aCD47@PHMSNs at the MI site within the heart, heart slices were stained with DAPI (blue) for confocal imaging of cell nuclei. Minimal red fluorescence indicative of ICG was observed in heart sections of control mice, whereas strong red fluorescence was evident in MI heart slices (Figure 5d and e), demonstrating successful localization of Fer-aCD47@PHMSNs at the injured myocardium. These findings demonstrate that Fer-aCD47@PHMSNs effectively evade immune clearance and accumulate prominently in the MI heart, highlighting their MI-targeted delivery ability in vivo.
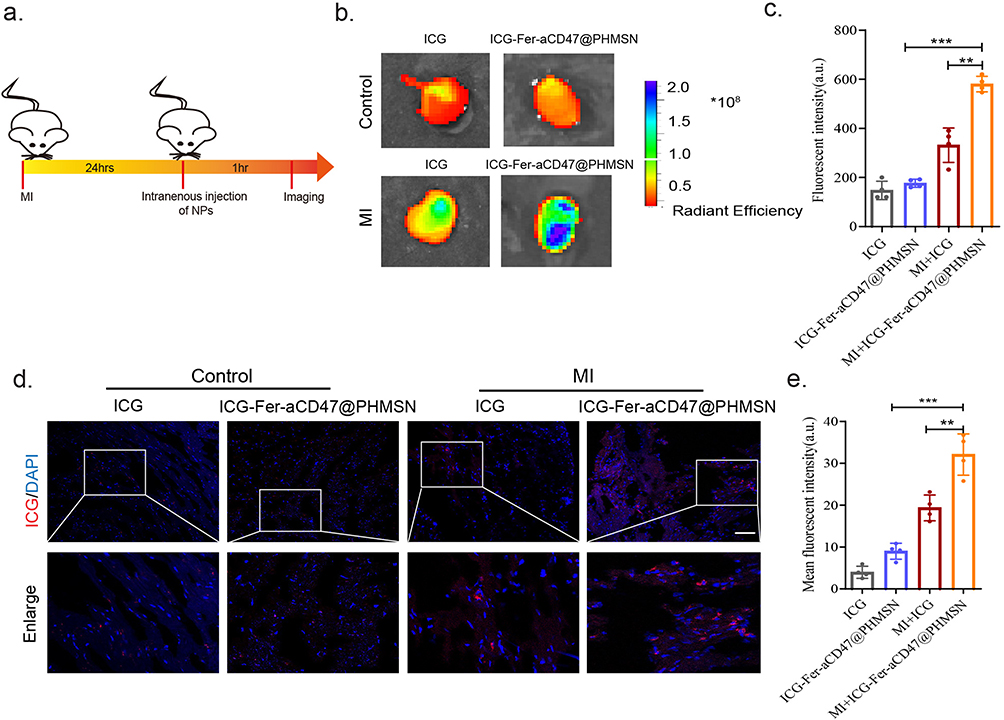 |
Figure 5 Fer-aCD47@PHMSN targets the MI site specially in vivo. (a) Scheme for in vivo design to test the targeting ability of Fer-aCD47@PHMSN to myocardial infarction site. (b) Representative images of ex vivo NIR fluorescent imaging of mouse hearts after intravenous injection of Fer-aCD47@PHMSN in MI/control mice. (c) Quantitative analysis of fluorescent intensity one-hour post-injection. Bars represent the mean ± SD from 4 independent experiments. **p < 0.01, ***p < 0.001. (d) Representative fluorescent microscopic images of heart slices showing the targeting of ICG labeled Fer-aCD47@PHMSN (red) to the MI area (DAPI, nuclei). Scale bars, 50 μm. (e) Quantitative analysis of fluorescent intensity of heart slices. Bars represent the mean ± SD from 4 independent experiments. **p < 0.01, ***p < 0.001. |
Intravenous Injection of Fer-aCD47@PHMSN Inhibits Cardiomyocyte Death and Improves the Cardiac Function of Mice After MI
We subsequently evaluated the impact of Fer-aCD47@PHMSNs on cardiac function in vivo. Mice received intravenous injections of Fer-aCD47@PHMSNs (100 μL) every three days for two weeks, followed by echocardiography (Figure 6a). As shown in Figure 6b and d, administration of Fer-aCD47@PHMSNs increased the ejection fraction (EF) in MI mice by 16.4% and 16% compared to injections of PBS and PHMSNs, respectively. Similarly, Fer-aCD47@PHMSNs administration increased fractional shortening (FS) in MI mice by 9.4% and 11% compared to PBS and PHMSN injections, respectively. Importantly, Fer-aCD47@PHMSNs administration significantly improved EF and FS compared with pure Fer-1 + aCD47 injection in vivo, highlighting the advantages of Fer-aCD47@PHMSNs as nanomedicine for enhancing cardiac function over free drugs. Fer-aCD47@PHMSNs injection did not significantly affect cardiac function in normal mice. Cardiomyocyte death was assessed using PI staining (Figure 6e and f). While Fer-aCD47@PHMSNs had minimal impact on cardiomyocyte death in normal mice, its injection reduced cardiomyocyte death by nearly 18.2% and 18.5% in MI mice compared to PBS and PHMSN injections, respectively. Fer-aCD47@PHMSNs also demonstrated superior efficacy compared with Fer-1 + aCD47 injection in mitigating cardiomyocyte death. These findings demonstrate that Fer-aCD47@PHMSNs effectively enhance cardiac function and reduce cardiomyocyte death in MI mice. Notably, the efficacy of Fer-aCD47@PHMSNs surpasses that of pure Fer-1 + aCD47 injection in vivo.
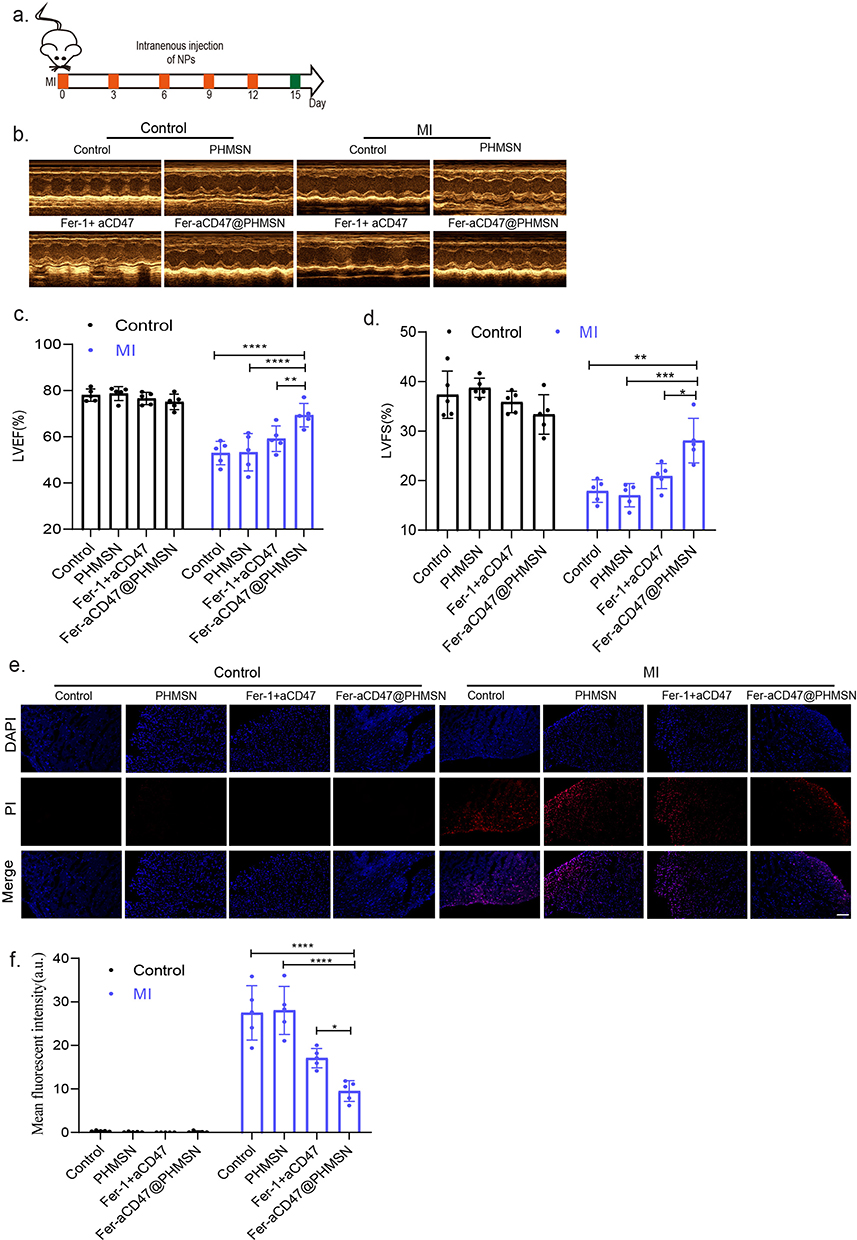 |
Figure 6 Fer-aCD47@PHMSN inhibits cardiomyocyte death after MI in vivo. (a) Scheme of in vivo treatment. PBS or Fer-aCD47@PHMSN was intravenously injected every three days into mice after MI operation for two weeks, mice were further analyzed on day 14. (b–d) Representative images of mouse echocardiography (n=5). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (e and f) Evaluation of cardiomyocyte death following different treatments was done through propidium iodide (PI) staining. Data represent mean ± SD from 5 independent experiments. *p < 0.05, ****p < 0.0001. |
Intravenous injection of Fer-aCD47@PHMSN reduces ferroptosis, enhances phagocytosis, and inhibits inflammation after MI in vivo.
Masson’s trichrome staining was employed to assess the infarct area and collagen deposition in the heart. For subsequent experiments, PBS injection was used as the control group. As illustrated in Figure 7a–c, MI mice exhibited significant infarct areas and collagen deposition, both of which were effectively reduced by approximately 9.2% with Fer-aCD47@PHMSNs injection. The effects of Fer-aCD47@PHMSNs on the normal heart were negligible, with fibrotic conditions in the Fer-aCD47@PHMSNs group comparable to the control group. The in vivo efficacy of Fer-aCD47@PHMSN in alleviating cardiomyocyte death by inhibiting ferroptosis was evaluated. As illustrated in Figure 7d, cardiac lipid ROS levels significantly increased in MI mice compared to controls, whereas treatment with Fer-aCD47@PHMSN reduced lipid ROS levels by nearly 40%. Similarly, Fer-aCD47@PHMSN treatment restored depleted GSH levels induced by MI operation (Figure 7e). Furthermore, the expression of ferroptosis-related proteins in cardiac tissue was assessed. Figure 7f and h demonstrate a notable decrease in SLC7A11 and GPX4 in the MI group, which was effectively inhibited by Fer-aCD47@PHMSN treatment. These findings indicate that intravenous injection of Fer-aCD47@PHMSN successfully mitigates cardiac ferroptosis following MI. To assess the inflammatory response, plasma levels of MCP-1 (monocyte chemoattractant protein 1) and Interleukin-1β (IL-1β) were measured. Figure 8a and b show a significant decrease of approximately 39.8% in MCP-1 and 20.3% in IL-1β levels in MI mice treated with Fer-aCD47@PHMSN compared to those treated with PBS. Additionally, the effect of Fer-aCD47@PHMSN on in vivo efferocytosis was evaluated. The efferocytosis rate, calculated as the ratio of macrophage-associated PI-positive cells to free PI-positive cells, was significantly higher in the Fer-aCD47@PHMSN treatment group by approximately 6% (Figure 8c and d), suggesting enhanced cardiac efferocytosis with Fer-aCD47@PHMSN injection.
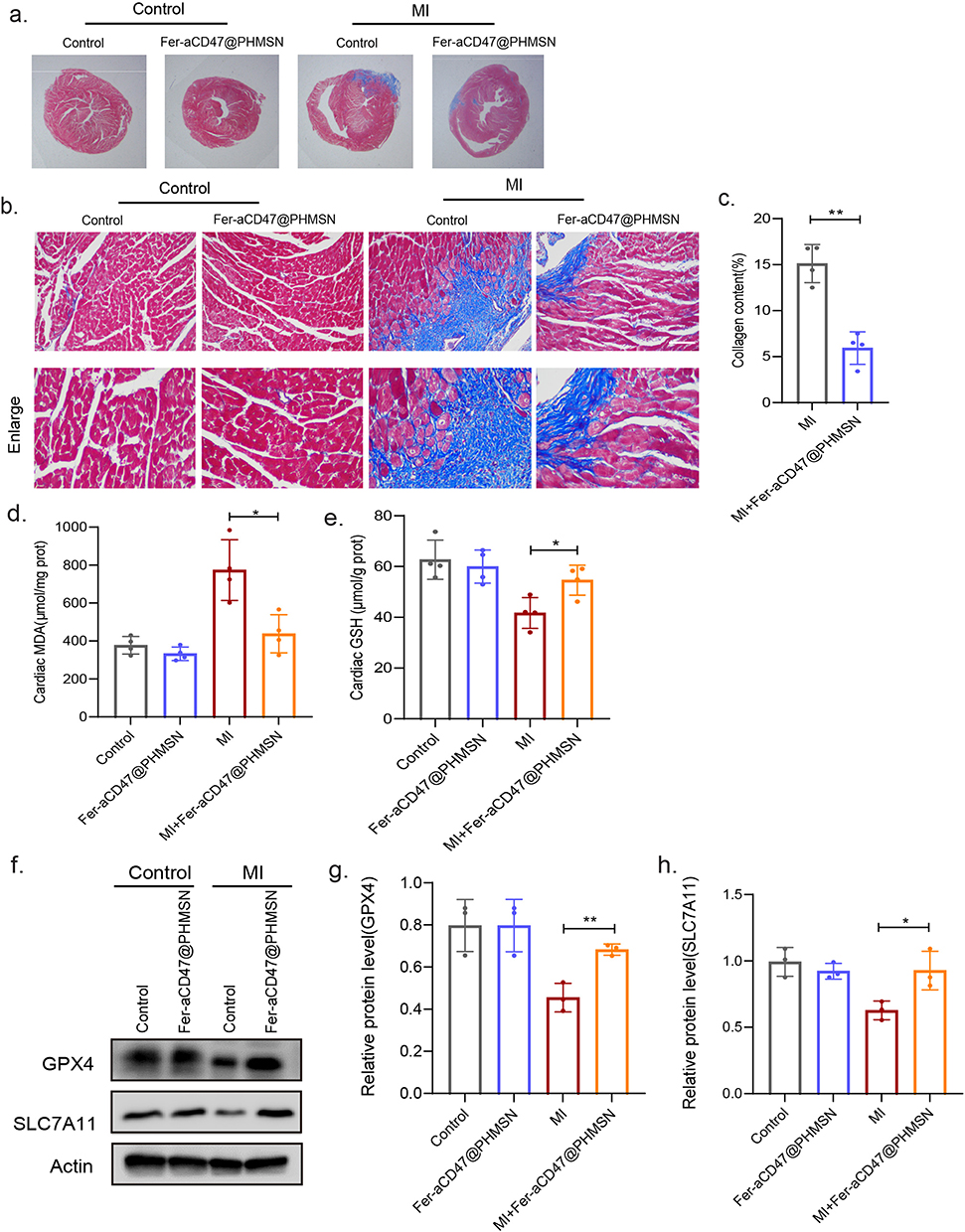 |
Figure 7 Fer-aCD47@PHMSN reduces ferroptosis, and enhances phagocytosis after MI in vivo. (a–c) Representative Masson’s trichrome staining sections of the heart on day 14. Scale bars, 100 μm. **p < 0.01. (d) Cardiac lipid ROS was quantified through MDA assay in vivo. Data represent mean ± SD from 4 independent experiments. *p < 0.05. (e) Cardiac GSH levels were measured. Data represent mean ± SD from 4 independent experiments. *p < 0.05. (f and h) The expression of ferroptosis-related proteins GPX4 and SLC7A11 of the heart were detected by Western blot. Images were representative of three experiments. *p < 0.05, **p < 0.01. |
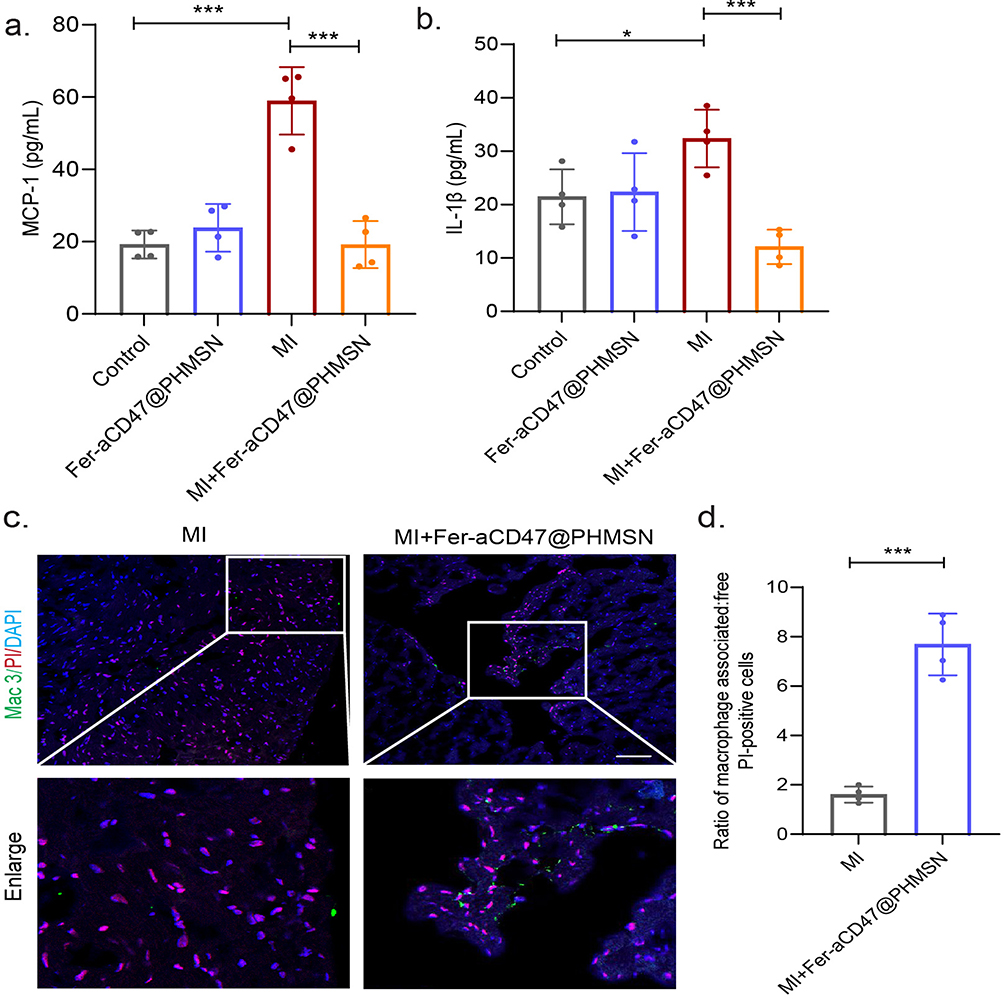 |
Figure 8 Fer-aCD47@PHMSN inhibits inflammation after MI in vivo. (a and b) Quantification of MCP-1 and IL-1β levels in serum (n=4). *p < 0.05, ***p < 0.001. (c and d) Determination of efferocytosis in situ. Efferocytosis percentage equals the ratio of macrophage-associated PI-positive vs free PI-positive cells in heart slices (n=4). Scale bar, 50 µm. ***p < 0.001. |
In vitro and in vivo Toxicity Test
Finally, the toxicity of HMSN and Fer-aCD47@PHMSN was assessed to evaluate their biocompatibility. Cell viability assays demonstrated that both HMSN and Fer-aCD47@PHMSN did not significantly affect cell viability at either 24 hours or 48 hours compared to the control (Figure 9a). Additionally, as depicted in Figure 9b, neither HMSN nor Fer-aCD47@PHMSN showed significant effects on cell apoptosis or necrosis. Throughout a two-week monitoring period, the body weights of mice in both the HMSN and Fer-aCD47@PHMSN groups (Figure 9c) exhibited no substantial changes, indicating low systemic toxicity. Analysis of serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and creatinine (CRE) further confirmed that neither HMSN nor Fer-aCD47@PHMSN affected liver and kidney functions (Figure 9d–g). Moreover, examination of tissue slices from major organs (heart, liver, spleen, lungs, and kidneys) using Hematoxylin and eosin (H&E) staining revealed no significant pathological lesions for either HMSN or Fer-aCD47@PHMSN compared to the control group (Figure 9h). Consequently, both HMSN and Fer-aCD47@PHMSN demonstrated negligible toxicity in vitro and in vivo.
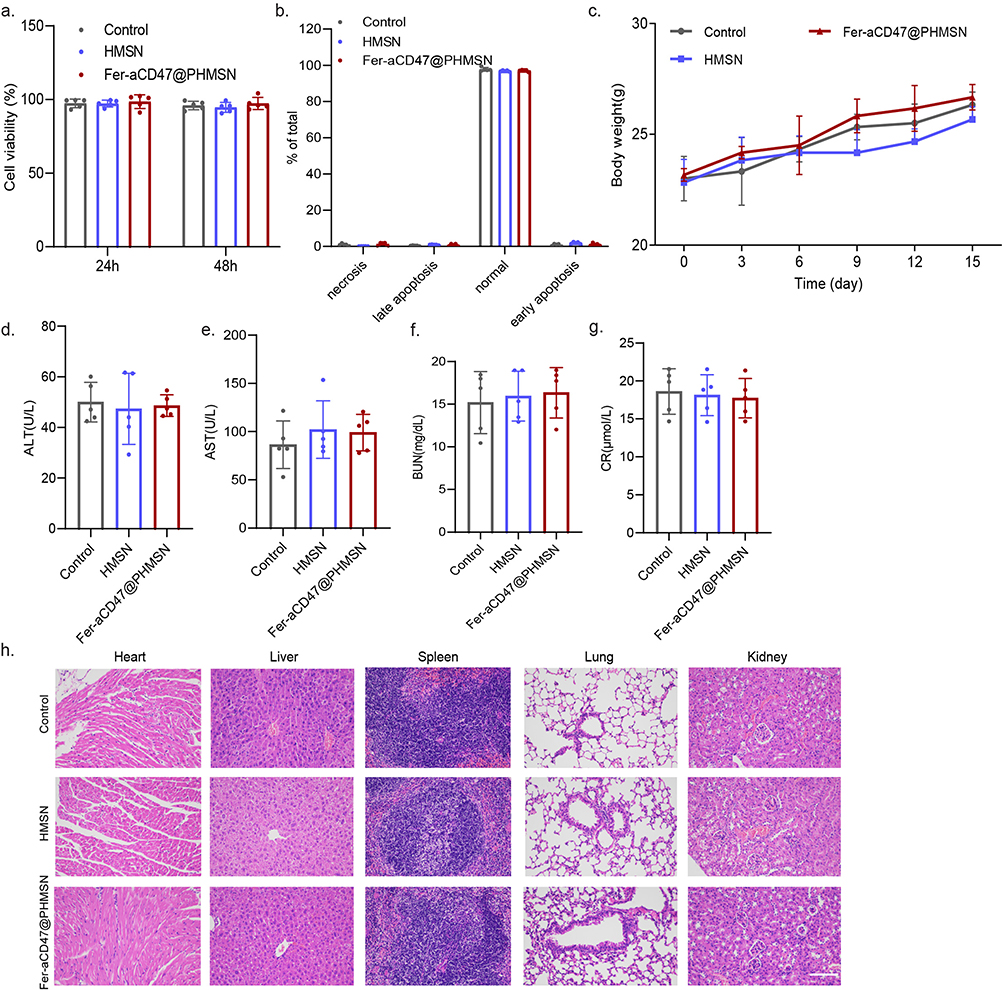 |
Figure 9 In vitro and in vivo toxicity test. (a) Viability assay of H9C2 cells after treatment with HMSN or Fer-aCD47@PHMSN for different times (n=5). (b) Cell apoptosis and necrosis of H9C2 cells after treatment with HMSN or Fer-aCD47@PHMSN for different times (n=5). (c) Body weights of mice after various treatments at different time points. (d–g) Biochemical analysis of ALT, AST, BUN, and CRE relevant to hepatic and kidney function (n=5). (h) Representative H&E sections of the vital organs (heart, liver, spleen, lung, and kidney) after HMSN or Fer-aCD47@PHMSN treatment (200 μL of 20 nM, tail injection) on day 14. n=5, Scale bars, 100 μm. |
Conclusions
In this study, we developed HMSN-based nanoplatforms (Fer-aCD47@PHMSN) for targeted therapy of myocardial infarction (MI) aimed at inhibiting cardiomyocyte ferroptosis and enhancing macrophage-mediated efferocytosis. Both in vitro and in vivo experiments confirmed that Fer-aCD47@PHMSNs effectively mitigated ferroptosis-associated cardiomyocyte death. Fer-aCD47@PHMSNs reduced lipid ROS levels increased GSH content, and upregulated key ferroptosis-associated proteins such as GPX4 and SLC7A11 in cardiomyocytes. Furthermore, Fer-aCD47@PHMSNs enhanced macrophage efferocytosis, as evidenced by increased clearance of dead cardiomyocytes both in vitro. Fer-aCD47@PHMSNs could successfully accumulate in the MI heart, improve cardiac function, and reduce inflammation post-MI in vivo. Importantly, neither cellular apoptosis nor systemic toxicity was observed with HMSN or Fer-aCD47@PHMSN treatment, consistent with previous reports of HMSN safety. Future studies should focus on optimizing the heart-specific accumulation and pharmacokinetics of Fer-aCD47@PHMSNs, determining the optimal dosage and timing for maximal therapeutic efficacy. Our findings underscore the promise of Fer-aCD47@PHMSNs as a promising therapeutic strategy for post-MI therapy.
Acknowledgments
This work was financially supported through the National Natural Science Foundation of China (82100284, 81970253) and Natural Science Foundation of Shandong Province, Youth Program (ZR2021QH051), China Postdoctoral Science Foundation (2022M721746). The authors would like to thank the technical support from the University Research Facility in Basic Medicine of Qingdao University for this work.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Gong FF, Vaitenas I, Malaisrie SC, Maganti K. Mechanical Complications of Acute Myocardial Infarction: a Review. JAMA Cardiol. 2021;6(3):341–349. doi:10.1001/jamacardio.2020.3690
2. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. 2020;116(6):1101–1112. doi:10.1093/cvr/cvz336
3. Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective.. Nat Rev Cardiol. 2020;17(12):773–789. doi:10.1038/s41569-020-0403-y
4. Damluji AA, van Diepen S, Katz JN, et al. Mechanical Complications of Acute Myocardial Infarction: a Scientific Statement From the American Heart Association. Circulation. 2021;144(2):e16–e35. doi:10.1161/cir.0000000000000985
5. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth Universal Definition of Myocardial Infarction. J Am Coll Cardiol. 2018;72(18):2231–2264. doi:10.1016/j.jacc.2018.08.1038
6. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
7. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
8. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82(12):2215–2227. doi:10.1016/j.molcel.2022.03.022
9. Mou Y, Wang J, Wu J, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12(1):34. doi:10.1186/s13045-019-0720-y
10. Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. doi:10.1073/pnas.1821022116
11. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. 2021;11(7):3052–3059. doi:10.7150/thno.54113
12. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381–396. doi:10.1038/s41568-022-00459-0
13. Zhang S, Yeap XY, DeBerge M, et al. Acute CD47 Blockade During Ischemic Myocardial Reperfusion Enhances Phagocytosis-Associated Cardiac Repair. JACC Basic Transl Sci. 2017;2(4):386–397. doi:10.1016/j.jacbts.2017.03.013
14. Xu LN, Wang SH, Su XL, et al. Targeting Glycogen Synthase Kinase 3 Beta Regulates CD47 Expression After Myocardial Infarction in Rats via the NF-κB Signaling Pathway. Front Pharmacol. 2021;12(662726). doi:10.3389/fphar.2021.662726
15. Chen L, Zhou Z, Hu C, et al. Platelet Membrane-Coated Nanocarriers Targeting Plaques to Deliver Anti-CD47 Antibody for Atherosclerotic Therapy. Research (Wash D C). 2022;2022(9845459). doi:10.34133/2022/9845459
16. Pan Q, Xu J, Wen CJ, Xiong YY, Gong ZT. Nanoparticles: promising Tools for the Treatment and prevention of Myocardial Infarction. Int J Nanomedicine. 2021;16:6719–6747. doi:10.2147/ijn.S328723
17. Lv Q, Ma B, Li W, Fu G, Wang X, Xiao Y. Nanomaterials-Mediated Therapeutics and Diagnosis Strategies for Myocardial Infarction. Front Chem. 2022;10(943009). doi:10.3389/fchem.2022.943009
18. Shi HT, Huang ZH, Xu TZ, Sun AJ, Ge JB. New diagnostic and therapeutic strategies for myocardial infarction via nanomaterials. EBioMedicine. 2022;78(103968):103968. doi:10.1016/j.ebiom.2022.103968
19. Chen J, Zhang X, Millican R, et al. Recent advances in nanomaterials for therapy and diagnosis for atherosclerosis. Adv Drug Deliv Rev. 2021;170:142–199. doi:10.1016/j.addr.2021.01.005
20. Cheng Z, Li M, Dey R, Chen Y. Nanomaterials for cancer therapy: current progress and perspectives. J Hematol Oncol. 2021;14(1):85. doi:10.1186/s13045-021-01096-0
21. Hu Y, Wang J, Zhi Z, Jiang T, Wang S. Facile synthesis of 3D cubic mesoporous silica microspheres with a controllable pore size and their application for improved delivery of a water-insoluble drug. J Colloid Interface Sci. 2011;363(1):410–417. doi:10.1016/j.jcis.2011.07.022
22. Maleki A, Kettiger H, Schoubben A, Rosenholm JM, Ambrogi V, Hamidi M. Mesoporous silica materials: from physico-chemical properties to enhanced dissolution of poorly water-soluble drugs. J Control Release. 2017;262:329–347. doi:10.1016/j.jconrel.2017.07.047
23. Yin H, Yan Q, Liu Y, et al. Co-encapsulation of paclitaxel and 5-fluorouracil in folic acid-modified, lipid-encapsulated hollow mesoporous silica nanoparticles for synergistic breast cancer treatment. RSC Adv. 2022;12(50):32534–32551. doi:10.1039/d2ra03718a
24. Cai X, Hua S, Deng J, et al. Astaxanthin Activated the Nrf2/HO-1 Pathway to Enhance Autophagy and Inhibit Ferroptosis, Ameliorating Acetaminophen-Induced Liver Injury. ACS Appl Mater Interfaces. 2022;14(38):42887–42903. doi:10.1021/acsami.2c10506
25. Galabova BB. Mesoporous silica nanoparticles: synthesis, functionalization, drug loading and release - A review. Tropical Journal of Pharmaceutical Research. 2022;20(5):1091–1100. doi:10.4314/tjpr.v20i5.30
26. Feng L, Dou C, Xia Y, et al. Neutrophil-like Cell-Membrane-Coated Nanozyme Therapy for Ischemic Brain Damage and Long-Term Neurological Functional Recovery. ACS Nano. 2021;15(2):2263–2280. doi:10.1021/acsnano.0c07973
27. Jiang Y, Krishnan N, Zhou J, et al. Engineered Cell-Membrane-Coated Nanoparticles Directly Present Tumor Antigens to Promote Anticancer Immunity. Adv Mater. 2020;32(30):e2001808. doi:10.1002/adma.202001808
28. Bao L, Dou G, Tian R, et al. Engineered neutrophil apoptotic bodies ameliorate myocardial infarction by promoting macrophage efferocytosis and inflammation resolution. Bioact Mater. 2022;9:183–197. doi:10.1016/j.bioactmat.2021.08.008
29. Wang Y, Zhang K, Li T, et al. Macrophage membrane functionalized biomimetic nanoparticles for targeted anti-atherosclerosis applications. Theranostics. 2021;11(1):164–180. doi:10.7150/thno.47841
30. Tang J, Su T, Huang K, et al. Targeted repair of heart injury by stem cells fused with platelet nanovesicles. Nat Biomed Eng. 2018;2(1):17–26. doi:10.1038/s41551-017-0182-x
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.