")
Back to Journals » Drug Design, Development and Therapy » Volume 19
A Phase I Study to Evaluate the Safety, Tolerability, Pharmacokinetics of BGT-002, a Novel ATP-Citrate Lyase Inhibitor, in Healthy Chinese Subjects
Authors Liu Y, Yu C, Zhang Y , Xie Z, Wang Y, Qian H, Liang L, Liu Y, Chen Q, Jia J , Yan S, Lai X, Li W, Li J, Zhang Y, Nan F, Yu C
Received 6 November 2024
Accepted for publication 13 February 2025
Published 11 March 2025 Volume 2025:19 Pages 1783—1794
DOI https://doi.org/10.2147/DDDT.S504814
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Manfred Ogris
Yun Liu,1,2,* Chengyin Yu,1– 3,* Yifan Zhang,3 Zhifu Xie,3 Yating Wang,1,2 Hongjie Qian,1,2 Liyu Liang,1,2 Yanmei Liu,1,2 Qian Chen,1,2 Jingying Jia,1,2 Sai Yan,4 Xiaoyin Lai,4 Wei Li,4 Jingya Li,3 Yangming Zhang,4 Fajun Nan,3 Chen Yu1,2
1Shanghai Xuhui Central Hospital/Zhongshan-Xuhui Hospital, Fudan University, Shanghai, People’s Republic of China; 2Phase I Clinical Research and Quality Consistency Evaluation for Drugs, Shanghai Engineering Research Center, Shanghai, People’s Republic of China; 3State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, People’s Republic of China; 4Burgeon Therapeutics Co., Ltd., Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fajun Nan; Chen Yu, Email [email protected]; [email protected]
Objective: This Phase I study evaluated the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of BGT-002, a novel ATP-citrate lyase (ACLY) inhibitor, in healthy Chinese adults.
Methods: This study included three parts: Part I (single-ascending-dose study), Part II (multiple-ascending-dose study), and Part III (food effect study). A total of 104 healthy subjects were enrolled in the study and were given BGT-002 tablet or placebo per protocol requirements. Blood samples were collected for pharmacokinetic and pharmacodynamic analysis. Safety was assessed by clinical examinations and adverse events.
Results: In Part I, BGT-002 demonstrated rapid absorption with a Tmax of 0.67 to 1.75 hours, and slow elimination with a T1/2 of 24.53 to 72.86 hours, prolonged with increased dosages. Cmax and AUC0-∞ ranged from 1.55 to 48.39 μg/mL, and 31.09 to 2930.69 h·μg/mL, respectively. In Part II, the accumulation index (Rac) of Cmax and AUCtau following 14 days of consecutive administration were 3.53 to 3.62 and 5.29 to 5.59, respectively, with a dose-proportionality PK profile. The levels of total cholesterol (TC), non-high-density lipoprotein cholesterol (non-HDL-C), and low-density lipoprotein cholesterol (LDL-C) were maximally decreased by 15.80%, 18.50%, and 22.37%, respectively. In Part III, the geometric mean ratio (90% CI) of fed to fasting condition in Cmax and AUC0-∞ of BGT-002 were 73.11% and 98.36%, respectively, indicating a minor food effect on the absorption rate. Across the study, two cases of Grade 3 adverse events (elevated blood triglycerides) were reported, both of which were assessed as not related to BGT-002. No serious adverse events were observed.
Conclusion: BGT-002 demonstrated favorable safety, tolerability, and lipid-lowering effects, supporting its potential for further clinical development.
Clinical Trial Registration: ChiCTR2200057793(https://www.chictr.org.cn/showproj.html?proj=160210); ChiCTR2300067474(https://www.chictr.org.cn/showproj.html?proj=182183); ChiCTR2300067472(https://www.chictr.org.cn/showproj.html?proj=184079).
Keywords: safety, tolerability, pharmacokinetics, pharmacodynamics, BGT-002, novel ACLY inhibitor
Introduction
Hypercholesterolemia is a disease characterized by abnormal cholesterol absorption and metabolism caused by the accumulation of various pathogenic factors. This condition leads to abnormally elevated serum cholesterol levels and is often accompanied by chronic metabolic diseases such as inflammation and vascular wall calcification. Hypercholesterolemia is the primary risk factor of atherosclerosis which is the main cause of many cardiovascular diseases.1–3
Guidelines for the prevention and treatment of hypercholesterolemia emphasize that Low-Density Lipoprotein Cholesterol (LDL-C) is the most critical risk factor for atherosclerotic cardiovascular disease and recommend it as the primary intervention target.4 The short-term goal of drug treatment for hypercholesterolemia is to reduce plasma level of LDL-C, and the long-term goal is to delay the occurrence of cardiovascular events so as to prolonging the patients’ survival time.5 Reducing the side effects of drug therapies while maintaining the quality of life is the overall goal of hypercholesterolemia treatment.6,7
Current mainstays for treating hypercholesterolemia include inhibiting hepatic cholesterol synthesis (such as statins), reducing intestinal cholesterol absorption (such as ezetimibe and hyzetimibe), and promoting cholesterol degradation (such as PCSK9 inhibitors).8 Statins are the most prescribed drugs based on their demonstrated clinical benefits. However, some statins associated side effects like adverse musculoskeletal effects and liver function damage lead to a large number of patients intolerant to statins (7–29% of patients) thus demanding novel lipid-lowering treatments.9
ATP-citrate lyase (ACLY) has attracted increasing attention as a novel therapeutic target for treating hypercholesterolemia and other metabolic dysfunction-associated diseases.10,11 In 2020, Nexletol (ETC-1002, bempedoic acid), developed by Esperion, was approved in US and Europe as the first small molecule ACLY inhibitor. In its CLEAR Outcomes study the 4 major adverse cardiovascular events (4-MACE) were significantly reduced by −13% compared with placebo group.9 In contrast to the previous reported ACLY inhibitors, bempedoic acid demonstrated good pharmacokinetic characteristics, favorable membrane permeability, improved cellular activity, and moderate effect in lowering LDL-C both in vitro and in vivo.12–16 However, bempedoic acid is not launched in China.
BGT-002, a novel ACLY inhibitor developed by the scientists of Shanghai Institute of Materia Medica, is being clinically developed as a new modality for treatment of hypercholesterolemia and metabolic dysfunction-associated steatohepatitis.17 The objective of this Phase I study was to assess the safety, tolerability, and pharmacokinetic (PK) profile of BGT-002, as well as to explore its pharmacodynamic (PD) characteristics, in healthy Chinese adults.
Methods
Study Population
Eligible participants included healthy adults aged 18 to 45 years with a body mass index between 19.0 and 30.0 kg/m² and a minimum weight of 50 kg for males and 45 kg for females. All subjects were required to be in good medical condition with no clinically significant abnormalities. This determination was based on a comprehensive evaluation, including detailed medical history, physical examination, clinical laboratory tests, 12-lead electrocardiograms (ECG), and vital sign assessments. The study adhered to the principles outlined in the Declaration of Helsinki and the Guidance on Good Clinical Practice (GCP). Ethical approval was obtained from the Ethic Committee of Shanghai Xuhui Central Hospital (Approval Nos: [2021(045)], [2022(025)], [2022(029)]). Written informed consent was obtained from all participants prior to the initiation of any study-related activities. The study was registered at Chinese Clinical Trial Registry (https://www.chictr.org.cn) under registration numbers ChiCTR2200057793, ChiCTR2300067474, and ChiCTR2300067472, and was conducted in the Phase 1 Clinical Research Unit of Shanghai Xuhui Central Hospital.
Investigational Medicinal Product
BGT-002 was formulated as oral tablets with dosage strength of 50 mg (Lot No: 21091304) and 200 mg (Lot No: 2109132). Placebo tablets, identical in appearance and containing the same inactive ingredients, were also manufactured in 50 mg (Lot No: 21091303) and 200 mg (Lot No: 21091301). All tablets, both the investigational drug and placebo, were supplied by Guangzhou Yipinhong Pharmaceutical Co., Ltd. The 50 mg tablet is a scored tablet. For the 25 mg dose, the 50 mg tablet was split along the score line to obtain a 25 mg portion. Subsequently, the 25 mg portion was further divided and weighed to achieve a 12.5 mg dose.
Study Design
This study employed a single-center, randomized, multicohort design with three parts: Part I (single-ascending-dose study, SAD), Part II (multiple-ascending-dose study, MAD), and Part III (food effect study) (Figure 1). The SAD study was divided into two phases: a preliminary trial phase including one cohort of 12.5 mg with two subjects, and a sequential seven ascending dose cohorts (25 mg, 50 mg, 100 mg, 150 mg, 200 mg, 250 mg, and 300 mg) each with eight subjects (six receiving BGT-002 tablets and two receiving placebo tablets). The decision to proceed to the next dose level was made by both investigator and sponsor according to the dose-escalation criteria in the protocol. The MAD study consisted of three cohorts (25 mg, 50 mg, and 75 mg) each with twelve subjects (nine received BGT-002 tablets and three received placebo tablets). Subjects were given BGT-002 tablets or placebo on the morning of day 1 to day 14. In the food effect study, a two-period crossover design was implemented. Fourteen subjects were divided into two groups with one group receiving a 100 mg dose of BGT-002 under fasting (following a 10-hours fasting) along with 240 mL of water, and the other group receiving the same dosage after a meal consisting of approximately 967.15 kcal. Following a 14-day washout period, the groups were crossed over: participants who initially received BGT-002 in a fasted state were administered the drug after a meal, and vice versa.
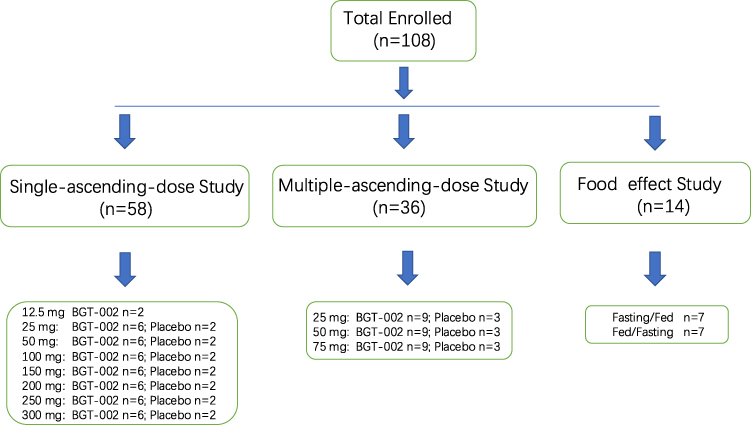 |
Figure 1 Disposition of subjects. |
Safety Assessments
Safety assessments involved the comprehensive evaluation of adverse events (AE), vital signs (including seated blood pressure, pulse rate, respiratory rate, and body temperature), physical examinations, clinical laboratory tests (including blood routine, blood biochemistry, coagulation function, urine routine, stool routine, and occult blood), and triplicate 12-lead ECGs. AEs and serious adverse events (SAE) were assessed following the NCI-CTC AE version 5.0 guidelines.
Descriptive summaries of crucial parameters from laboratory tests, vital signs, and 12-lead ECGs, along with any changes relative to baseline, were provided. Cross-tabulation was used to clinically assess laboratory tests, 12-lead ECGs, and physical examinations, summarizing the number and percentage of clinically significant changes before and after treatment. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 25.1. The analysis of AEs focused on treatment-emergent adverse events (TEAEs), encompassing AEs from the first administration of the investigational drug until the central follow-up on day 10 (±1 day) post-final dose. Treatment-related adverse events (TRAEs) are the TEAEs related to the investigational drugs classified as those with a plausible, likely, or possible causal relationship to the drug. If the association with the drug could not be determined or was unclear, the AE was included in drug-related TEAEs during data compilation. The summary of TEAEs included the count, incidence, and number of occurrences. AE incidence was calculated for each System Organ Class (SOC) and Preferred Term (PT) per participant.
Pharmacokinetic and Pharmacodynamic Sample Collection
In the SAD study, blood samples for PK analysis were collected at various time points: for cohort 1, at 0 hours (60 minutes before dosing) and at 10 min, 20 min, 40 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6 h, 8 h, 12 h, 24 h, 48 h, 72 h, 96 h, and 120 h post-dosing; for cohort 2–6, at 0 hours (60 minutes before dosing) and at 10 min, 20 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h, 96 h, and 120 h post-dosing; for cohort 7–8, at 0 hours (60 minutes before dosing) and at 10 min, 20 min, 40 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6h, 8 h, 12 h, 24 h, 48 h, 72 h, 96 h, 120 h, 168 h, and 240 h post-dosing.
In the MAD study, blood samples for PK analysis were collected on day 1 at 0 hours (60 minutes before dosing) and 10 min, 20 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 4 h, 8 h, and 12 h post-dosing. From day 2 to day 13, blood samples for PK analysis were obtained at 24 h, 144 h, 264 h and 288 h, and on day 14, samples were collected at 0 hours (60 minutes before dosing) and at 10 min, 20 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h, 96 h, 120 h, 168 h (only 75mg cohort), and 240 h (only 75mg cohort), post-dosing. Blood samples for PD analysis were collected at on day 1 at 0 hours, 24 h, 144 h, 360 h, 432 h (for Group 25mg and 50mg), 552 h (for Group 75mg). The PK samples for determining the whole blood-to-plasma concentration ratio were collected at 0 h (pre-dose), 1.5 h, 4 h, 12 h, 24 h, 120 h, and 240 h following the final dose administered on Day 14 in 75mg cohort.
In food effect study, blood samples for PK analysis were collected on day 1 and day 15 at 0 hours (60 minutes before dosing) and at 10 min, 20 min, 40 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6h, 8 h, 12 h, 24 h, 48 h, 72 h, 120 h, 168 h, and 240 h post-dosing.
Blood samples for PK analysis were collected into anticoagulant tubes containing K2EDTA immediately after blood withdrawal. The samples were centrifuged at 1500g for 10 minutes at 4°C within 1 hour of collection. Plasma samples were collected and stored at −80°C for subsequent LC-MS/MS analysis.
Pharmacokinetic and Pharmacodynamics Sample Analysis
BGT-002 and its acyl glucuronide metabolite, ZM326E-M2 in plasma were analyzed according to a previously validated LC-MS/MS method.10
Triglycerides (TG), TC, LDL-C, HDL-C, apolipoprotein A (ApoA), apolipoprotein B (ApoB), high-sensitivity C-reactive protein (hsCRP), β-hydroxybutyrate (β-HB), non-HDL-C, lipoprotein (a)[Lp(a)] were analyzed in the lab of Shanghai Xuhui central hospital.
Statistical Analysis
PK parameters were determined by employing a noncompartmental analysis method. The PK parameters were calculated using WinNonlin (version 8.2), and the other statistical analyses were performed using SAS (version 9.4). PK parameters encompassed Cmax, area under the concentration-time curve from time zero to the last measurable time point (AUC0–t), area under the concentration-time curve from time zero to infinity (AUC0–∞), time to maximum concentration (Tmax), T1/2, terminal clearance (CL/F), volume of distribution (Vz/F). Additionally, the area under the concentration-time curve during the time interval of 24 h (AUCtau), the ratio of the metabolite Cmax or AUC to that of the parent drug after conversion to molar concentrations (metabolic rate, MR), accumulation ratio of AUCtau or Cmax (Rac[AUC], or Rac [Cmax]) were calculated for the MAD, and the lag time (Tlag, the time point before the first measurable concentration corresponds to the time) was calculated for the food effect study.
The dose proportionality of Cmax and AUCs was assessed using a power model, which can be expressed as ln(Y) = α + βln (dose)+ε, where Y represents the evaluated pharmacokinetic parameters, and β is the proportionality coefficient. If the 90% confidence interval (CI) for β was well within the critical interval, the increase in systemic exposure with dose was linear. The critical interval is given as follows: [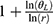 , [
, [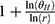 ]. r=H/L, where H represents the highest administered dose, L represents the lowest administered dose;
]. r=H/L, where H represents the highest administered dose, L represents the lowest administered dose; 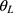 and
and  are the lower and upper confidence intervals, respectively, which were taken as 0.5 and 2 in this study.
are the lower and upper confidence intervals, respectively, which were taken as 0.5 and 2 in this study.
Mixed-effects models were used to assess the effect of food on drug pharmacokinetics. The fixed effects included sequence, treatment (fasting or fed), and period. The random effect was the subject nested within the sequence. The food effect was analyzed by geometric least squares mean (geoLSM) ratios (fed/fasting) and their 90% confidence intervals (CIs) for log-transformed Cmax and AUC values. The absence of a food effect was concluded if the 90% CIs for the test (fed group) vs reference (fasting group) geoLSM ratios fell entirely within the 80% to 125% range for Cmax and AUC values. The differences in Tmax and Tlag under different conditions were analyzed using a non-parametric Wilcoxon rank sum test, with the significance level set at P < 0.05.
Results
Subject Demographics and Baseline Characteristics
In the SAD study, 58 subjects were randomized and evaluated for safety. Among them, 2 participants participated in the preliminary trial, 42 participated in the single-ascending-dose trials and provided pharmacokinetics data, and 14 received a placebo. In the MAD study, 36 subjects were randomized and assessed for safety. Among these subjects, 27 subjects received BGT-002 and provided pharmacokinetics data, while 9 subjects received placebo. In the food effect study, 14 subjects were separated into two crossover groups (fasting, fed). Throughout all the treatment cohorts, subjects demonstrated similar baseline demographics (Table S1). All the subjects completed the study.
Pharmacokinetics
Single-Ascending-Dose Study
In both the preliminary trial and sequential ascending phases, the pharmacokinetics of BGT-002 and its acyl glucuronide metabolite ZM326E-M2 were evaluated. After administering BGT-002 under fasting condition, it was rapidly absorbed, with a Tmax ranging from 0.67 to 1.75 hours. The Cmax ranged from 1.55 to 48.39 μg/mL, and AUC0-∞ spanned from 31.09 to 2930.69 h·μg/mL (Table 1 and Figure 2). It was also found that BGT-002 was eliminated slowly with a half-life (T1/2) ranging from 24.53 to 72.86 hours. The metabolite, ZM326E-M2, showed similar pharmacokinetic characteristics to those of the parent drug. The Cmax for ZM326E-M2 ranged from 0.16 to 12.29 μg/mL, with AUC0-∞ ranging from 5.36 to 797.41 h·μg/mL. The T1/2 of ZM326E-M2 ranged from 24.57 to 79.52 hours.
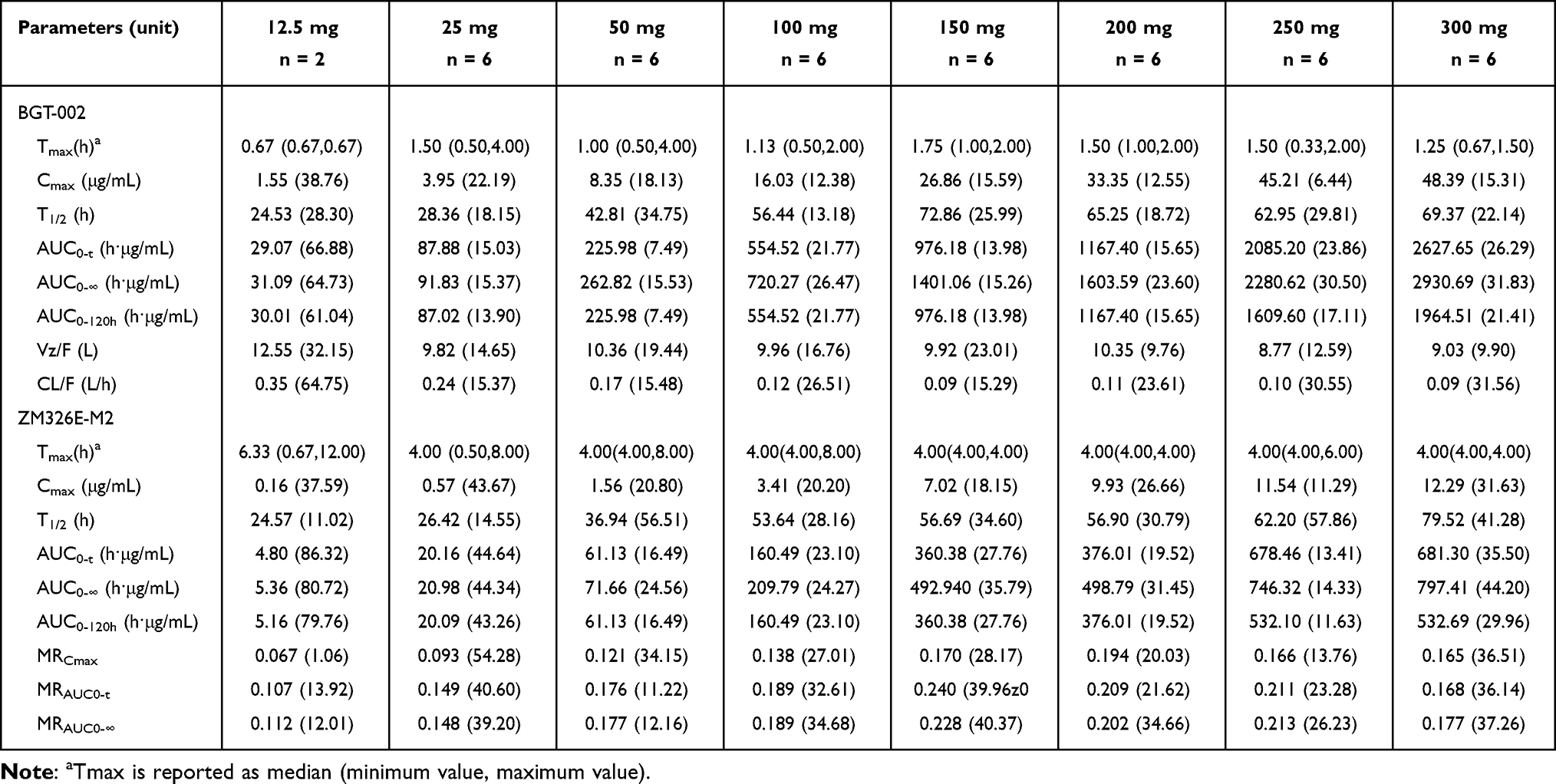 |
Table 1 Geometric Mean (Geometric Standard Deviation) Pharmacokinetic Parameters of BGT-002 and ZM326E-M2 in the SAD Study |
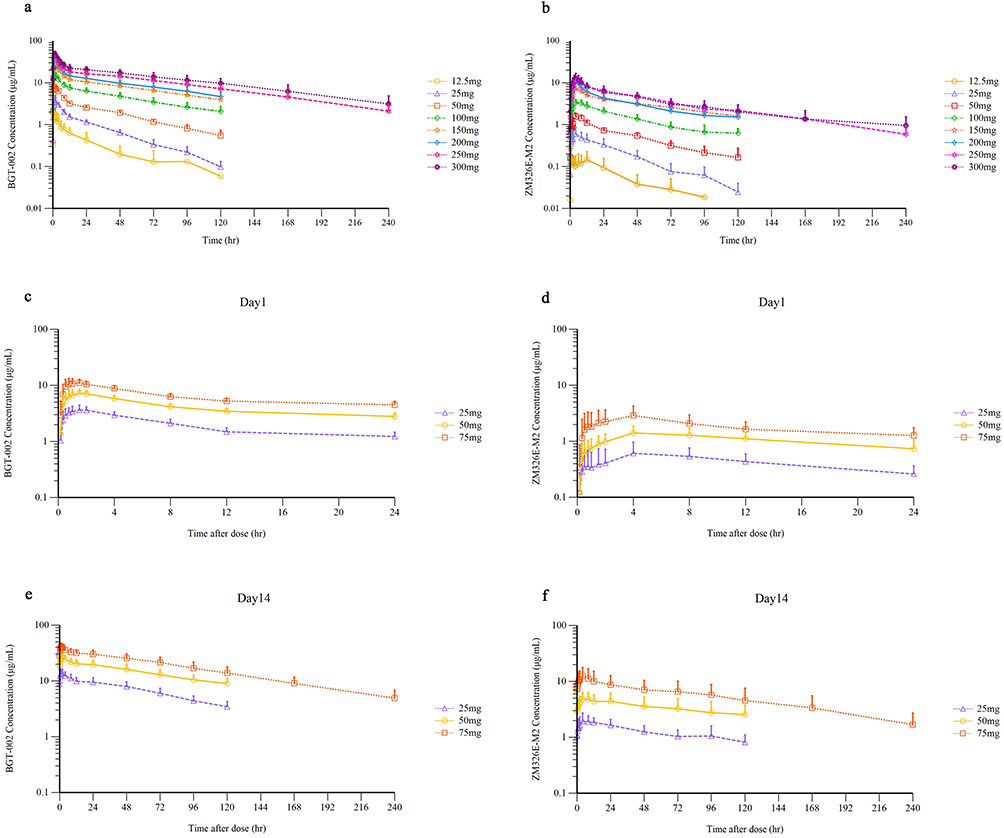 |
Figure 2 Mean (standard deviation) plasma concentration-time profiles (semi-log) of BGT-002 and ZM326E-M2 in the single-ascending-dose study and the multiple-ascending-dose study. PK profiles of (a) BGT-002 concentration in the SAD study. (b) ZM326E-M2 concentration in SAD study. (c) BGT-002 concentration in the MAD on day 1. (d) ZM326E-M2 concentration in the MAD study on day 1. (e) BGT-002 concentration in the MAD study on day 14. (f) ZM326E-M2 concentration in the MAD study on day 14. |
Multiple-Ascending-Dose Study
Healthy Chinese subjects were administered BGT-002 tablets at doses of 25 mg, 50 mg and 75 mg orally once daily for 14 consecutive days (Table 2 and Figure 2). The plasma concentrations of both BGT-002 and ZM326E-M2, reached a steady state by approximately the 12th day. On day 14, at steady state, the Tmax of BGT-002 was between 1.00 h and 2.00 h. Across the dosage range of 25 to 75 mg, the Cmax,ss of BGT-002 ranged from 13.93 to 41.73 h·μg/mL, and the AUCtau,ss ranged from 253.40 to 781.09 h·μg/mL, both increasing proportionally with the dose (Table S2). The BGT-002 exhibited accumulation in the body after multiple administrations, with an accumulation ratio (Rac) for Cmax ranging from 3.53 to 3.62, and Rac for AUCtau from 5.29 to 5.59, based on data collected on Day 1 and Day 14 of the MAD study. The pharmacokinetic characteristics of the metabolite ZM326E-M2 were similar to those of the parent drug after repeated dosing, with plasma exposure levels amounting to 10.53% to 18.77% of the parent drug. The mean of whole blood/plasma partition ratio for BGT-002 was 0.57 for AUCtau (Table S3), indicating that BGT-002 preferentially distributes in plasma.
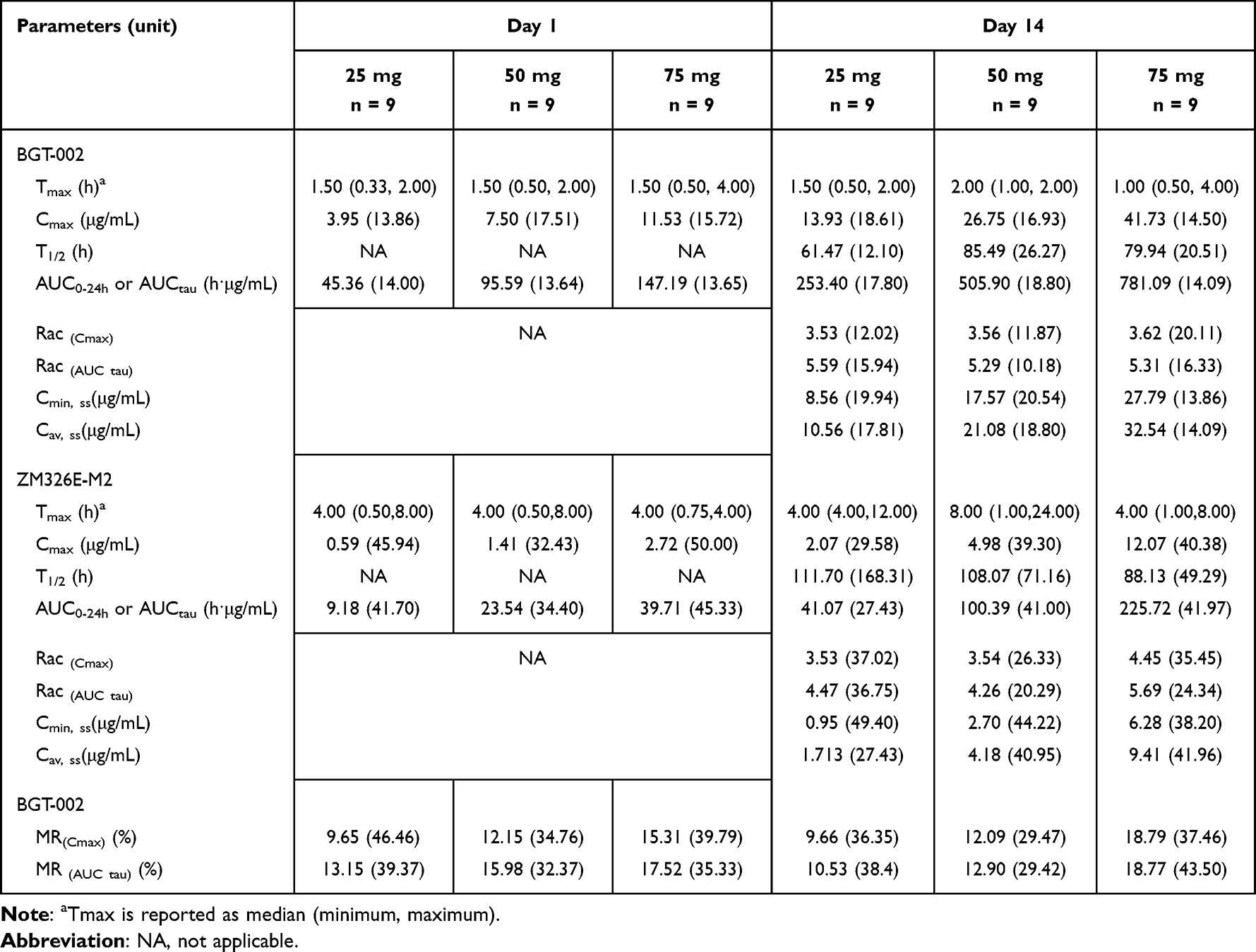 |
Table 2 Geometric Mean (Geometric Standard Deviation) Pharmacokinetic Parameters of BGT-002 and ZM326E-M2 in the MAD Study |
Food Effect Study
In the food effect study, Table 3 summarizes the PK parameters of BGT-002 and ZM326E-M2 under fasting and fed states, whereas primary exposure parameter (Cmax, AUC0-t, and AUC0-∞) estimates are summarized based on a mixed-effects model. In this model, the geometric mean ratio (90% CI) of fed to fasting state in Cmax and AUC0-∞ of BGT-002 were 73.11% (69.24%, 77.20%) and 98.36% (95.47%, 101.34%), respectively (Table 3 and Figure 3). These results suggest that food may have a minor effect on the absorption rate of BGT-002, but does not significantly affect the extent of its absorption. The median (Min-Max) Tmax for fasting and fed conditions was 1.50 hours (0.67–1.50) and 3.50 hours (1.50–4.0), respectively. The difference in Tmax of BGT-002 between the fed and fasting states was statistically significant (P=0.0002).
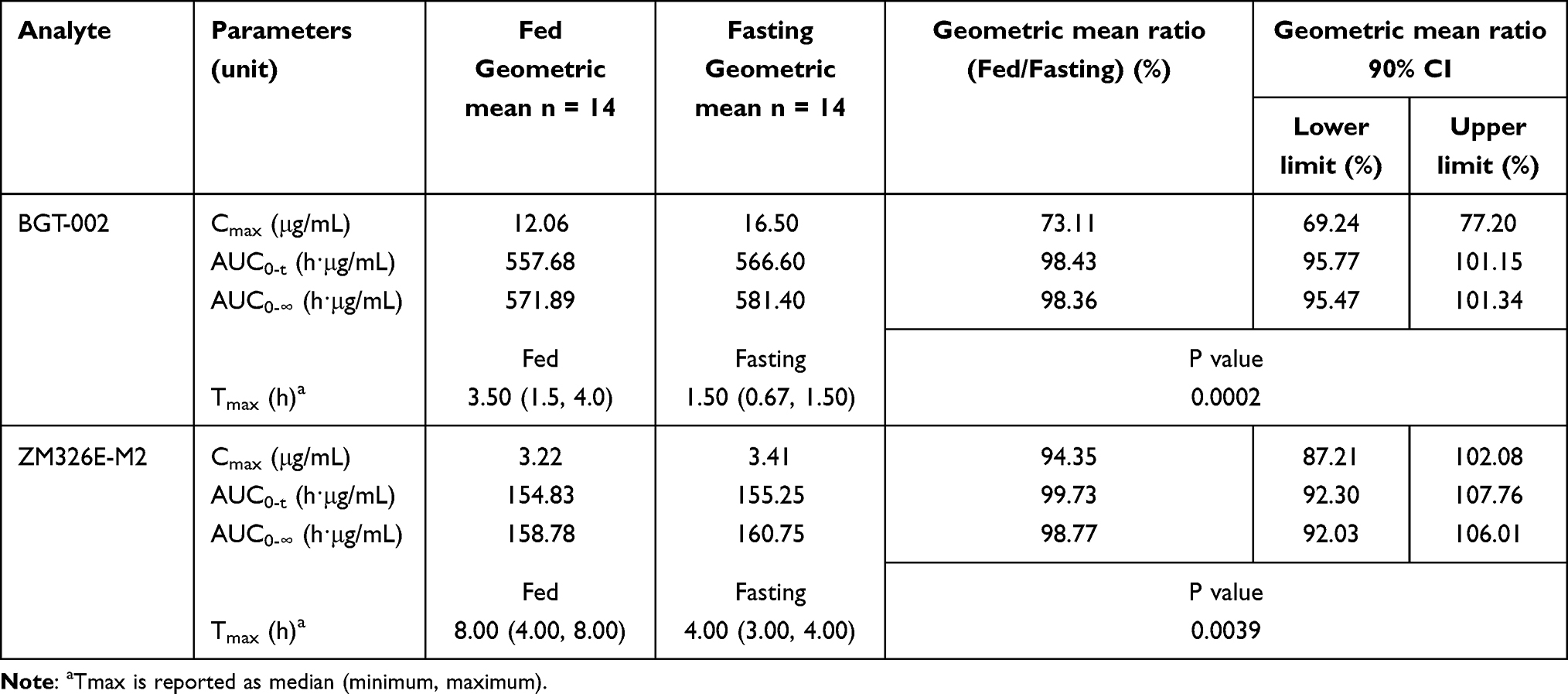 |
Table 3 Geometric Mean Ratios and 90% CI for Key Pharmacokinetic Parameters of BGT-002 and ZM326E-M2 Under Fasting and Fed States |
 |
Figure 3 Mean (standard deviation) plasma concentration-time profiles of BGT-002 under the fasting and fed states. |
Pharmacodynamics
Levels of total cholesterol in the 25 mg, 50 mg, and 75 mg of BGT-002 groups showed a reduction starting from the 7th day after administration, with the reduction percentage increasing over the course of treatment. The maximum percentage reductions from baseline in the 25 mg and 50 mg groups were observed on day 19, at 10.98% and 13.80%, respectively. In the 75 mg group, the maximum reduction occurred on day 14, with a decrease of 15.80%. No significant reduction from baseline was observed in the placebo group (Table S4).
Non-HDL cholesterol levels in the 50 mg and 75 mg BGT-002 groups also began to decrease on the 7th day after administration, with reductions increasing over time. The maximum percentage reductions from baseline in the 50 mg group occurred on day 19 (18.50%), while in the 75 mg group, the maximum reduction was seen on day 14 (13.59%). For the 25 mg group, Non-HDL cholesterol levels decreased by day 14, reaching a maximum reduction of 14.99% on day 19. No significant reduction from baseline was observed in the placebo group.
Low-density lipoprotein cholesterol levels in all three BGT-002 groups (25 mg, 50 mg, and 75 mg) showed decreases by the 7th day after administration. The 25 mg and 75 mg groups exhibited the largest percentage reduction from baseline on day 14, with reduction of 5.88% and 22.37%, respectively. In the 50 mg group, the largest reduction was observed on day 19, at 21.52%. No significant reduction observed in the placebo group.
Safety and Tolerability
In SAD study, a total of 24 (41.4%) subjects experienced at least one AE during the study and the incidence of TRAEs was 37.9% (Table S5). The incidence rates of TEAEs were 50.0%, 33.3%, 33.3%, 33.3%, 50.0%, 33.3%, 33.3%, 50.0%, 50.0% for 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200 mg, 250 mg, 300 mg and placebo group, respectively (Table 4). The most common AEs by organ class were sinus bradycardia, which were reported in the 20.5% participants receiving BGT-002, and 28.6% participants treated with placebo. The second most common AEs by organ class were elevated aspartate aminotransferase, which were reported in the 14.3% participants treated with placebo and 0% participant receiving BGT-002, respectively.
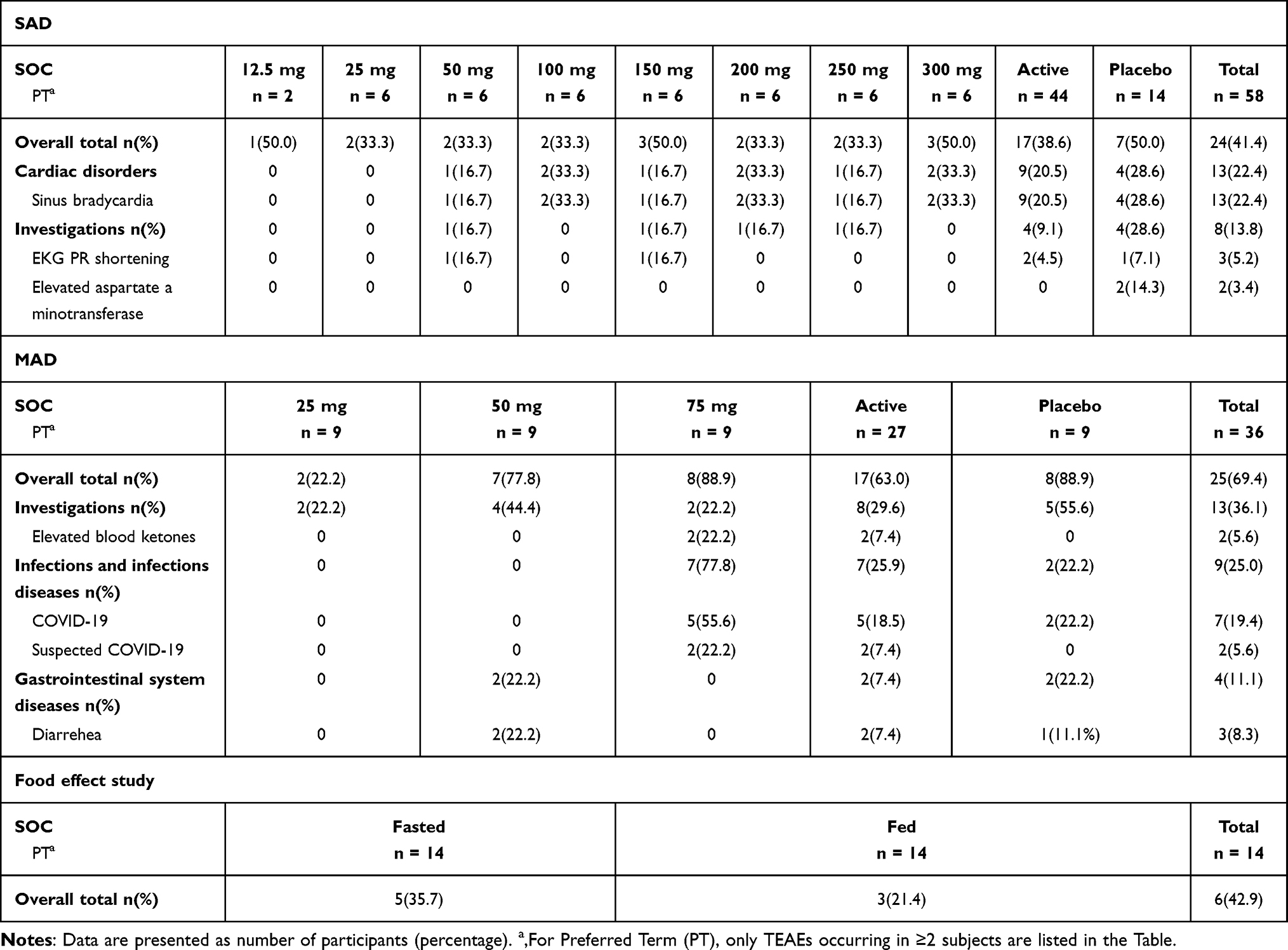 |
Table 4 Treatment-Emergent Adverse Events (TEAEs) per Treatment Group in the Study |
In MAD study, a total of 25 (69.4%) subjects experienced at least one AE during the study and the incidence of AEs related to the investigational drug was 25% (Table S6 and Table 4). The incidence rates of TEAEs were 22.2%, 77.8%, 88.9%, 88.9% for 25 mg, 50 mg, 75 mg and placebo group, respectively. The most common AEs by organ class were COVID-19, which were reported in the 55.6% participants in the 75 mg cohort, and 22.2% participants treated with placebo. The second most common AEs by organ class were elevated blood ketones, which were reported in the 22.2% participants in the 75mg cohort, and 0% participants treated with placebo.
In the food effect study, 14 subjects were included in the safety set. A total of 6 subjects experienced 9 AEs, resulting in an AE incidence rate of 42.9% (Table S6 and Table 4). Of these, 5 AEs in 4 subjects (28.6%) were assessed by investigators as related to the study drug, while the remaining AEs were deemed possibly unrelated. The incidence rates of AEs for fasting and fed administration were 35.7% and 21.4%, respectively, with related AE incidence rates of 21.4% and 7.1%, respectively. The TEAEs and TRAEs were observed more frequently after fasting administration compared to fed administration. All possibly related AEs were of Grade 1 in severity and included positive urine leukocytes (1 case), hematuria (1 case), upper respiratory tract infection (1 case), increased white blood cell count (1 case), and increased neutrophil count (1 case).
The majority of adverse events (AEs) in the study were Grade 1 or Grade 2 in severity. Only one subject in the placebo group and one subject in the fed group which was given 100 mg BGT-002 after a high fat meal experienced a single Grade 3 AE (elevated triglycerides) which were both not related to the investigational drug. All AEs were recovered before the end of the study. There were no deaths, SAEs, or discontinuations during the study.
Discussion
BGT-002 is currently being developed as an oral ACLY inhibitor specifically for the treatment of hypercholesterolemia and has garnered significant interest as an innovative therapeutic option. Its potential as a therapeutic target is further supported by research on ACLY, such as studies on Nexletol.11 BGT-002 has shown favorable safety and effective absorption, with targeted efficacy in the treatment of hypercholesterolemia. When compared to Nexletol, the first ACLY inhibitor approved by FDA, our study revealed that BGT-002 demonstrates higher plasma exposure. Specifically, after a multiple dose of BGT-002 (50 mg), the Cmax reached 27.07±4.18 μg/mL, which is higher than the Cmax of 19.95 μg/mL observed with 180 mg dose of Nexletol. Furthermore, we also identified that administering 75 mg of BGT-002 for 14 days reduced LDL-C by 24.9%, while 180 mg of Nexletol reduced LDL-C by 26% (data of Nexletol was quoted from the submission materials for new drug application). This reduction of LDL-C was comparable between the two drugs. Based on this limited data, it may be suggested that BGT-002 at the same dose could potentially exhibit a more potent lipid-lowering effect compared to Nexletol.
The safety and tolerability of BGT-002 were well-established, with no SAEs reported in the subjects who received with the drug. BGT-002 was safe, well tolerated, and not associated with any dose-limiting side effects, similar to the profile of Nexletol.18 In both single-ascending-dose study and food effect study, one case of Grade 3 AEs (elevated blood triglycerides) was observed in each study. However, in the single-ascending-dose study, there was an increase in blood triglycerides occurred in the placebo group, suggesting that it was unrelated to the drug. Similarly, in the food effect study, the increase in blood triglycerides occurred after a high-fat meal, suggesting it was not attributable to BGT-002.
For single oral doses of BGT-002 in the SAD study, within the dose range of 12.5 mg to 150 mg, the T1/2 of BGT-002 increased with dose escalation, ranging from 24.53 h to 72.86 h. Additionally, the proportional increases in AUCo--t and AUCo-∞ slightly exceeded the proportional increases in dose, indicating characteristics of nonlinear pharmacokinetics. However, at doses above 150 mg, the T1/2 remained largely unchanged. Between 150 mg and 300 mg, the proportional increases in AUC were consistent with the proportional increases in dose. Although the AUC of BGT-002 exhibited certain nonlinear characteristics at lower dose levels, the Cmax demonstrated a good linear relationship with the administered dose within the studied dose range (12.5 mg to 300 mg).
Preliminary studies have shown that glucuronidation is the major metabolic pathway of BGT-002 in the body, with the resulting glucuronide conjugates being excreted via the kidneys or excreted in bile and excreted in feces.10 In this study, the concentration of the glucuronic acid metabolite ZM326E-M2 in plasma was measured and showed that the exposure (AUC0-∞) ratio of ZM326E-M2 to the parent drug increased from 0.112 at the 12.5 mg to 0.228 at 150 mg. At doses range from 50 mg to 300 mg, the ratio remained stable In vitro experiments demonstrated that ZM326E-M2 is a substrate of OATP1B1 and OAT3, and it exhibits an inhibitory effect on both transporters. Therefore, it is speculated that the nonlinear characteristics of drug elimination may be related to transporter saturation or inhibition.
When healthy subjects took 100 mg BGT-002 tablets after a high-fat meal, the Tmax was delayed by 2 hours, and the Cmax decreased by 26.89% compared to fasting condition. There were no significant changes in AUC0-t or AUC0-∞, suggesting that food may slow down the rate of absorption, but does not affect the overall extent of absorption. The lower incidence of adverse events observed under fed state compared to fasting state may also be attributed to a decrease in Cmax following food intake.
In the 12.5 mg to 200 mg dose groups of the SAD study, the sampling duration (0 to 120 h) was insufficient to fully characterize the PK profile of BGT-002 and its metabolite, which represents a limitation. However, additional sampling time points at 168 h and 240 h were included for the 250 mg and 300 mg dose levels. In the food effect study, sampling was extended to 240 h. The PK results indicate that the mean concentration of ZM326E-M2 at 240 h is less than 1/10 of its Cmax, which complies with guideline requirements.
Another limitation of the study is the small sample size used to evaluate the PD effect and food effect of BGT-002. However, it should be noted that this study is a Phase I clinical trial primarily designed to assess the safety, tolerability, and pharmacokinetic (PK) profile of BGT-002 in healthy volunteers. The study was deliberately designed with the smallest sample size necessary to adequately address these primary objectives, thereby facilitating dose-escalation studies. Once sufficient safety data are established, clinical research with larger sample sizes will be conducted. Notably, despite the limited sample size, our study demonstrated the favorable safety profile and potential lipid-lowering effects of BGT-002, which has justified proceeding to a Phase II clinical trial with a larger cohort to further investigate its lipid-lowering efficacy (registration number: CTR20240756, chictr.org.cn). The results of this Phase II study will be reported in due course.
In conclusion, BGT-002 demonstrated favorable safety, pharmacokinetic profiles and promising efficacy characteristics in healthy volunteers. Data from nonclinical studies and current Phase 1 studies support the further clinical development of BGT-002 as a novel therapeutic approach for hypercholesterolemia and other metabolic disorders.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
All procedures performed were reviewed by an independent Ethics Committee, and was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with Good Clinical Practices and applicable regulatory requirements.
All participants were required to provide a signed and dated informed consent prior to the start of the study.
Acknowledgments
BGT-002 is being developed by Burgeon Therapeutics Co., Ltd. We would like to express our sincere gratitude to Professor Chen Xiaoyan, a distinguished pharmacokineticist, for her invaluable contributions to the bioanalytical section of this paper. Her expertise and guidance have greatly enhanced the quality of the research, and her support is deeply appreciated.
Author Contributions
All authors made a significant contribution to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article, gave final approval of the version to be published, have agreed on the journal to which the article has been submitted, and agree to be accountable for all aspects of the work.
Funding
This work was funded by Burgeon Therapeutics Co., Ltd.
Disclosure
Xiaoyin Lai, Wei Li, Yangming Zhang are employees of Burgeon Therapeutics Co., Ltd. The authors report no other conflicts of interest in this work.
References
1. Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438(7068):612–621. doi:10.1038/nature04399
2. Wang HH, Garruti G, Liu M, Portincasa P, Wang DQH. Cholesterol and lipoprotein metabolism and atherosclerosis: recent advances in reverse cholesterol transport. Ann Hepatol. 2017;16(Suppl. 1: s3–105.):s27–s42. doi:10.5604/01.3001.0010.5495
3. Soppert J, Lehrke M, Marx N, Jankowski J, Noels H. Lipoproteins and lipids in cardiovascular disease: from mechanistic insights to therapeutic targeting. Adv Drug Deliv Rev. 2020;159:4–33. doi:10.1016/j.addr.2020.07.019
4. Hu DY. New guidelines and evidence for prevention and treatment of dyslipidemia and atherosclerotic cardiovascular disease in China. Chronic Dis Transl Med. 2017;3(2):73–74. doi:10.1016/j.cdtm.2016.11.001
5. Duell PB, Santos RD, Kirwan BA, Witztum JL, Tsimikas S, Kastelein JJP. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol. 2016;10(4):1011–1021. doi:10.1016/j.jacl.2016.04.013
6. Hu SS; The Writing Committee Of The Report On Cardiovascular Health And Diseases In China. Report on cardiovascular health and diseases in China 2021: an updated summary. J Geriatr Cardiol. 2023;20(6):399–430. doi:10.26599/1671-5411.2023.06.001
7. Millwood IY, Walters RG, Mei XW, et al. Conventional and genetic evidence on alcohol and vascular disease aetiology: a prospective study of 500 000 men and women in China. Lancet. 2019;393(10183):1831–1842. doi:10.1016/S0140-6736(18)31772-0
8. Hartz J, Clauss S. Treatment strategies for hypercholesterolemia. Curr Pediatr Rev. 2017;13(4):243–254. doi:10.2174/1573396314666180111143900
9. Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic acid and cardiovascular outcomes in statin-intolerant patients. N Engl J Med. 2023;388(15):1353–1364. doi:10.1056/NEJMoa2215024
10. Zhu X, Cui S, Liu X, et al. Simultaneous determination of BGT-002 and its acyl glucuronide metabolite ZM326E-M2 in human plasma by liquid chromatography-tandem mass spectrometry and its application to a pharmacokinetic study. J Pharm Biomed Anal. 2024;243(December 2023):116056. doi:10.1016/j.jpba.2024.116056
11. Pinkosky SL, Groot PHE, Lalwani ND, Steinberg GR. Targeting ATP-citrate lyase in hyperlipidemia and metabolic disorders. Trends Mol Med. 2017;23(11):1047–1063. doi:10.1016/j.molmed.2017.09.001
12. Pinkosky SL, Filippov S, Srivastava RAK, et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J Lipid Res. 2013;54(1):134–151. doi:10.1194/jlr.M030528
13. Inc ET. Nexletol. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=211616.
14. Cramer CT, Goetz B, Hopson KLM, et al. Effects of a novel dual lipid synthesis inhibitor and its potential utility in treating dyslipidemia and metabolic syndrome. J Lipid Res. 2004;45(7):1289–1301. doi:10.1194/jlr.M400018-JLR200
15. Zagelbaum NK, Yandrapalli S, Nabors C, Frishman WH. Bempedoic Acid (ETC-1002): ATP citrate lyase inhibitor: review of a first-in-class medication with potential benefit in statin-refractory cases. Cardiol Rev. 2019;27(1):49–56. doi:10.1097/CRD.0000000000000218
16. Ruscica M, Banach M, Sahebkar A, Corsini A, Sirtori CR. ETC-1002 (Bempedoic acid) for the management of hyperlipidemia: from preclinical studies to Phase 3 trials. Expert Opin Pharmacother. 2019;20(7):791–803. doi:10.1080/14656566.2019.1583209
17. Xie Z, Zhang M, Song Q, et al. Development of the novel ACLY inhibitor 326E as a promising treatment for hypercholesterolemia. Acta Pharm Sin B. 2023;13(2):739–753. doi:10.1016/j.apsb.2022.06.011
18. Bilen O, Ballantyne CM. Bempedoic Acid (ETC-1002): an Investigational Inhibitor of ATP Citrate Lyase. Curr Atheroscler Rep. 2016;18(10):61. doi:10.1007/s11883-016-0611-4
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.