")
Back to Journals » Infection and Drug Resistance » Volume 18
AI-2 Signaling: A Potential Driver of Bacteremia in Non-Typhoidal Salmonella Infections
Authors Li Y, Lu B, Qiang X, Lin Y, He J, Cai Y
Received 22 November 2024
Accepted for publication 12 March 2025
Published 19 March 2025 Volume 2025:18 Pages 1521—1537
DOI https://doi.org/10.2147/IDR.S507908
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Yu Li,1,2,* Bing Lu,1,3,* Xinhua Qiang,3 Yibin Lin,1 Jie He,2 Yunxiang Cai3
1School of Medicine, Huzhou University, Huzhou, Zhejiang, 313000, People’s Republic of China; 2Department of Infectious Diseases, First Affiliated Hospital of Huzhou University, Huzhou, Zhejiang, 313000, People’s Republic of China; 3Department of Clinical Laboratory, First Affiliated Hospital of Huzhou University, Huzhou, Zhejiang, 313000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jie He, Department of Infectious Diseases, First Affiliated Hospital of Huzhou University, No. 158 Back Square Road, Wuxing District, Huzhou, Zhejiang, 313000, People’s Republic of China, Email [email protected] Yunxiang Cai, Department of Clinical Laboratory, First Affiliated Hospital of Huzhou University, No. 158 Back Square Road, Wuxing District, Huzhou, Zhejiang, 313000, People’s Republic of China, Email [email protected]
Introduction: Non-typhoidal Salmonella (NTS) infections typically present as localized inflammation near the intestinal mucosal epithelium. However, some NTS strains can breach the intestinal barrier and enter the bloodstream, leading to bacteremia and severe systemic infections. The mechanisms by which NTS invades the bloodstream remain unclear.
Methods: In this study, we isolated 36 NTS strains from patients with diarrhea and bacteremia at First Affiliated Hospital of Huzhou University. Strains represented two distinct clinical manifestations, and were subjected to whole-genome sequencing, comparative genomics, and genetic differentiation analysis to identify genes potentially involved in bloodstream invasion. Additionally, we conducted inhibition assays using quercetin, a chemical inhibitor of the identified gene pathways, to validate our findings.
Results: Our analysis revealed that genes distinguishing the bloodstream Salmonella isolates from the fecal Salmonella isolates were primarily involved in the AI-2 quorum sensing pathway and biofilm-associated protein transport. Subsequent biofilm formation assays demonstrated that the bloodstream isolates exhibited significantly higher biofilm formation capacity compared to the fecal isolates. Upon the addition of quercetin, biofilm formation was equally inhibited in both groups. Collectively, these findings suggest that genes involved in the AI-2 pathway and biofilm-associated protein transport may be key factors contributing to the development of bacteremia in NTS infections.
Keywords: invasive non-typhoidal Salmonella, whole genome sequencing, comparative genomics analysis, biofilm, quorum sensing, AI-2
Introduction
Non-typhoidal Salmonella (NTS) infections typically present as self-limiting gastroenteritis, with clinical symptoms primarily including diarrhea, nausea, and vomiting. These infections usually remain confined to the gastrointestinal mucosa.1 However, NTS infections can also lead to bloodstream infections (eg, bacteremia) or secondary systemic focal infections (eg, meningitis, osteomyelitis). Such strains are referred to as invasive non-typhoidal Salmonella (iNTS).2 If not treated promptly, iNTS infections can become life-threatening. iNTS has been identified as a major pathogen responsible for bloodstream infections in sub-Saharan Africa,3,4 particularly affecting vulnerable populations such as HIV-infected individuals, children under five years of age, and adults over 70 years old. The case fatality rate for iNTS can reach up to 20%.5 It is estimated that iNTS causes approximately 3.4 million infections and over 680,000 deaths annually, representing a significant global disease burden.6
The core mechanism by which invasive iNTS breaches the intestinal barrier and causes bloodstream infections involves two key stages: intestinal epithelial colonization and systemic dissemination.7 Previous studies have established that the critical factors for the iNTS and NTS are primarily the type 3 secretion systems (T3SS) encoded by the Salmonella pathogenicity islands 1 and 2 (SPI-1 and SPI-2). T3SS-1, encoded by SPI-1, mediates the invasion of intestinal epithelial cells, while T3SS-2, encoded by SPI-2, facilitates the invasion of Salmonella from gastrointestinal epithelial cells into surrounding tissues.8 T3SS-2 drives the movement of Salmonella-containing vacuoles (SCV) along the host microtubule network to the perinuclear region of non-polarized host cells, where effector proteins such as SseJ and SifA promote bacterial release into the circulatory system.9 In addition to these T3SS-dependent mechanisms, the study by Manon Rosselin et al10 confirmed that Salmonella mediates T3SS-independent host cell invasion through the “Zipper” mechanism involving the Rck outer membrane protein.
The molecular mechanisms underlying the ability of iNTS to breach the intestinal barrier still represent a certain degree of scientific gap. Current research predominantly focuses on the classical functions of the SPI-1/SPI-2, making it difficult to comprehensively explain the multidimensional interaction network involved in the transition from intestinal colonization to bloodstream dissemination.
Therefore, this study conducted whole-genome sequencing (WGS) on 36 NTS and iNTS strains isolated from clinical samples. Comparative genomic analysis was employed to examine their virulence factors, antibiotic resistance genes, and evolutionary relationships, while also identifying unique pathogenic determinants present in bloodstream isolates at the genomic level. The aim of this study is to uncover the potential mechanisms underlying Salmonella-induced systemic infections and provide a theoretical basis for the development of novel antimicrobial agents, as well as strategies for the control and treatment of pathogenic infections.
Materials and Methods
Sample Collection and Salmonella Identification
This study employed a single-center, retrospective cohort design, including 36 Salmonella isolates recovered from biological samples of outpatient and inpatient patients at First Affiliated Hospital of Huzhou University between 2017 and 2023. Based on clinical phenotypes and sample sources, the isolates were categorized into two groups: the fecal group (NTS group, n=30) and the blood group (iNTS group, n=6, A9, A11, A15, A39, A92, A128). The fecal group consisted of strictly selected fecal isolates from patients with acute diarrhea (defined as ≥3 episodes of diarrhea within 24 hours, with abnormal stool characteristics such as loose, semi-formed, or watery stools) and without systemic inflammatory response.11 The blood group comprised isolates from patients diagnosed with bacteremia, based on diagnostic criteria including fever (body temperature >38°C or <36°C), chills, hypotension, oliguria, or elevated lactate levels, with positive blood cultures.12 The bacterial species were identified based on colony morphology, growth characteristics, Gram staining, and MALDI-TOF MS. The isolates were stored at −80°C in a microbiological culture facility. The study protocol (ethics approval number: 2024KYLL008-01) was approved by the Institutional Review Board, and written informed consent was obtained from all participants. The age and sampling time of the corresponding patients were also recorded.
Antimicrobial Susceptibility Testing
Following recommendations from the Clinical and Laboratory Standards Institute (CLSI), the susceptibility of experimental bacterial strains to the following antimicrobial agents was determined using the microbroth dilution method, as outlined in CLSI M100-33 (2023): ampicillin, ampicillin/sulbactam, amoxicillin/clavulanic acid, cefpodoxime, cefuroxime, cefotetan, ciprofloxacin, ertapenem, imipenem, levofloxacin, piperacillin/tazobactam, trimethoprim/sulfamethoxazole, and colistin. The specific experimental procedure involved selecting fresh colonies of Salmonella grown for 18–24 hours. The Salmonella strains were adjusted to a concentration equivalent to a 0.5 McFarland turbidity standard. For the susceptibility testing, an appropriate volume of the bacterial suspension, meeting the requirement of a 1:10,000 ratio of bacterial suspension to the culture medium, was added to the culture media containing gradients of the respective antimicrobial agents. The cultures were then incubated at 35°C ± 2°C for 16–24 hours. Interpretation of susceptibility, intermediate, and resistance was determined according to the breakpoints specified in CLSI M100-33 (2023), with Escherichia coli ATCC 25922 used for quality control purposes.
Whole-Genome Sequencing
DNA templates were extracted using a boiling method. One to two pure bacterial colonies from the MHA plates were picked and dissolved in 200 μL of sterile distilled water in a 1.5 mL EP tube. The mixture was ground thoroughly and then heated in a boiling water bath for 10 minutes. Following heating, the samples were immediately cooled in ice water. After cooling, the samples were centrifuged at 12,000 rpm for 1 minute to pellet cell debris, and the supernatant containing DNA was collected. Qualified DNA samples were randomly sheared into fragments approximately 350 bp in length using a Covaris ultrasonic disruptor. Subsequently, the fragmented DNA underwent library preparation using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, USA), which involved steps such as end repair, A-tailing, adapter ligation, purification, and PCR amplification. Following library construction, preliminary quantification was performed using the Qubit 2.0 fluorometer, and the library was diluted to a concentration of 2 ng/μL. Insert size analysis of the library was conducted using the Agilent 2100 Bioanalyzer, and once the insert size met the expected criteria, the effective concentration of the library was accurately quantified using qPCR to ensure library quality. The Illumina NovaSeq PE150 platform (Illumina, San Diego, CA, United States) was selected for whole-genome sequencing.
Bioinformatics Analysis
We performed bioinformatics analysis of the strains using an in-house Linux-based platform. Initially, the quality of the raw sequencing reads was assessed using FastQC v0.12.1 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), followed by trimming using fastp v0.23.413 to remove low-quality and adapter sequences. Subsequently, clean data were assembled using unicycler v0.5.0,14 with the default “normal” correction mode selected and merging of overlaps and bridges at a multiplicity of 1 to reduce mismatches in the final assembly. The evaluation was conducted using QUAST v.5.2.015 with standard settings. Genome annotation was performed using Prokka v.1.14.6.16 Prophages, genomic islands (GIs), and CRISPRs were predicted using Phigaro, IslandPath-DIOMB, and CRISPRCasTyper software, respectively.17–19
Comparisons were made between the genomes of the blood group and fecal group strains against the reference strain—Salmonella enterica serovar Typhimurium LT2 (University of Washington Genome Sequencing Center, 2016, accession number: SAMN02604315). The average nucleotide identity (ANI) of all genomes was assessed using fastANI (https://github.com/ParBLiSS/FastANI). Phylogenetic analysis of the 36 strains from both groups was conducted based on single nucleotide polymorphisms (SNPs) and achieved by using Snippy v4.6.0 (https://github.com/tseemann/snippy) for genome alignment with default parameters, followed by calculation of the number of SNPs between each isolate using Snp-dists (https://github.com/tseemann/snp-dists). Systematic evolution calculation was performed using Fasttree20 with the generalized time-reversible model, and the resulting tree was visualized using iTOL.21
Whole-genome sequences were aligned against the ResFinder antimicrobial resistance gene database using Abricate v0.7 (available at https://github.com/tseemann/abricate) to identify antimicrobial resistance genes (ARGs). Additionally, Abricate was utilized in conjunction with the Virulence Factors Database (VFDB)22 and PlasmidFinder23 to predict virulence-associated genes and plasmid replicons, respectively, with a similarity threshold set at 90%. Serotype and multilocus sequence typing (MLST) predictions were made using SeqSero2 v1.2.124 and MLST 2.0 v2.23.025 respectively. Additionally, the presence of SPIs was detected using SPIFinder26 against a custom database constructed from publicly available SPIs sequences from the NCBI database.27 Secondary metabolite biosynthesis gene clusters were predicted using AntiSMASH 7.0 (https://antismash.secondarymetabolites.org).
Pan-genome analysis was performed using BPGA v1.3.028 to compare the differences between the core genes of the blood group and the fecal group, as well as the shared core genes. This analysis allowed for the identification of unique genes specific to the blood group and the fecal group. Functional annotation of genes was conducted using the eggNOG database,29 followed by functional classification and homology analysis using the COG database.30 Pathway enrichment analysis was then performed using the KEGG database.31 Except for the phylogenetic tree, all visualization operations were performed using the TBtools32 software and ChiPlot (https://www.chiplot.online).
Biofilm Formation Assay
Biofilm formation in Salmonella isolates from fecal and bloodstream sources was assessed using the crystal violet staining method, and the involvement of quorum sensing (QS) in the bloodstream isolates was evaluated with the addition of a QS inhibitor. Bacterial cultures were initially grown overnight on MHA plates, after which 1–2 pure colonies were transferred into 5 mL of LB broth and incubated until the OD600nm reached 0.6–0.8. For the blank control group, 230 μL of LB medium and 20 μL of bacterial suspension were added to a 96-well plate. The experimental group received 230μL of LB medium containing 80μg/mL quercetin33 (a QS inhibitor, also capable of suppressing the AI-2 signaling pathway) along with 20μL of bacterial suspension. The plates were incubated at 37°C for 48 hours, after which the bacterial cultures were discarded, and the wells were washed three times with 300 μL of sterile water. Subsequently, 250 μL of methanol was added to fix the biofilm for 15 minutes, and after removing the methanol, the plates were air-dried. Each well was then stained with 250 μL of 0.1% crystal violet for 10 minutes, and excess dye was removed by washing with sterile water. After drying the wells, 250 μL of 33% glacial acetic acid was added to solubilize the crystal violet bound to the biofilm. Finally, the optical density at 595 nm (OD595nm) was measured using a microplate reader to quantify the biofilm.
Statistical analysis was performed on at least three independent experiments. Data are presented as the median and compared against the control group. All data were analyzed using analysis of variance (ANOVA), followed by Tukey’s test using SPSS Statistics 26.0. A p-value of <0.05 was considered statistically significant.
Results
Patient and Strain Information
In this study, a total of 36 non-duplicate Salmonella isolates were obtained. Comparative genomics analysis was performed to determine ANI. Evolutionary distances between the genomes of these isolates and the reference genome of Salmonella enterica serovar Typhimurium LT2 exceeded 98%, confirming their identification as Salmonella isolates (Figure S1). The isolates were derived from two distinct patient groups: the fecal group consisted of 30 isolates from fecal samples of patients attending outpatient and inpatient services across pediatrics, emergency medicine, intensive care unit (ICU), infectious diseases, and gastroenterology departments. All patients in this group presented clinical symptoms indicative of gastrointestinal system infections. The blood group included six isolates obtained from blood samples of inpatients from pediatrics, orthopedics, infectious diseases, and ICU departments, all of whom exhibited severe systemic infections. Patients were stratified by age, as illustrated in Figure 1, into the following categories: infants (0–6 years), children (7–15 years), adolescents and adults (16–59 years), elderly (60–75 years), and very elderly (≥75 years). The results indicated that the highest infection rate occurred in the infant group, accounting for 42% (17/36) of all cases, highlighting young children as a particularly vulnerable population. MLST revealed that ST11 accounted for 22% (8/36) and ST34 for 27% (10/36), making these the predominant sequence types in this study. Further serotype analysis identified a diverse distribution among the six bloodstream isolates, which were classified as Goldcoast, Panama, London, Enteritidis, and Typhimurium serotypes. In contrast, isolates from the fecal group were primarily concentrated in Typhimurium and its monophasic variants, as well as Enteritidis.
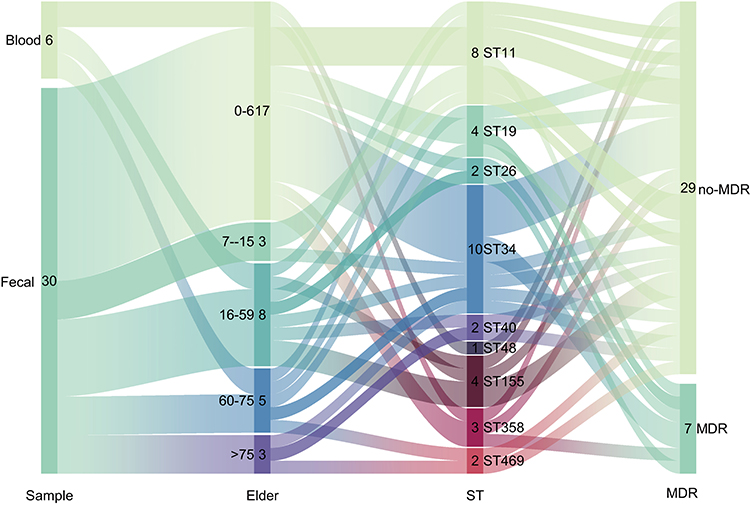 |
Figure 1 Rainbow Plot of Sample Information, this comprehensive dataset visualization includes: Sample Origins, Patient Age Stratification, MLST Profiles, Multi-Drug Resistance Information. |
Genomic Features of the Strains
In this study, the genome sizes of the bloodstream isolates ranged from 4.71 to 4.96 Mb, whereas the fecal isolates exhibited genome sizes ranging from 4.70 to 5.30 Mb, indicating that the bloodstream isolates had slightly smaller genomes compared to the fecal isolates. The GC content of both groups was comparable, ranging from 51.88% to 52.28%. Analysis of coding sequences revealed that the size of coding genes in bloodstream isolates ranged from 4.11 Mb to 4.34 Mb, while that of fecal isolates ranged from 4.10 Mb to 4.62 Mb. Coding sequences accounted for 86.83% to 87.58% of the total genome size in both groups (Table S1). The number of transfer RNA (tRNA) genes in the bloodstream isolates ranged from 76 to 79, with a total length of 5995 bp to 6231 bp. In comparison, fecal isolates contained 76 to 83 tRNA genes, with a total length of 5995 bp to 6526 bp. Ribosomal RNA (rRNA) gene counts and sizes were consistent across both groups (Table S2). All 36 isolates exhibited consistent CRISPR counts (Table S3). In terms of genomic islands (GIs), bloodstream isolates harbored 5 to 10 GIs, whereas fecal isolates contained 5 to 12 GIs (Table S4). Prophage counts also varied, with bloodstream isolates carrying 1 to 7 prophages and fecal isolates harboring 1 to 8 prophages (Table S5). These genomic differences provide a foundational basis for further investigation into the structural variations in Salmonella genomes associated with different infection routes.
Antibiotic Susceptibility and Resistance Gene Analysis in Salmonella Isolates
Antibiotic susceptibility testing was performed on all Salmonella isolates using antibiotics commonly employed in clinical treatment. Multidrug-resistant (MDR) was defined as resistance to three or more classes of antibiotics. Among fecal isolates, six strains (A55, A58, A105, A124, A126, A134) were identified as MDR, corresponding to an MDR rate of 20.0% (6/30). In contrast, only one bloodstream isolate (A128) was classified as MDR, with an MDR rate of 16.7% (1/6). Notably, all isolates demonstrated 100% (36/36) resistance to ampicillin and ampicillin-sulbactam. Resistance rates for polymyxin E, sulfamethoxazole, ciprofloxacin, and ceftriaxone in bloodstream isolates were 50.0% (3/6), 33.3% (2/6), 16.7% (1/6), and 16.7% (1/6), respectively. In fecal isolates, resistance rates for these antibiotics were 16.7% (5/30), 53.3% (16/30), 10.0% (3/30), and 13.3% (4/30), respectively. Both groups exhibited low resistance rates to carbapenems (Table 1).
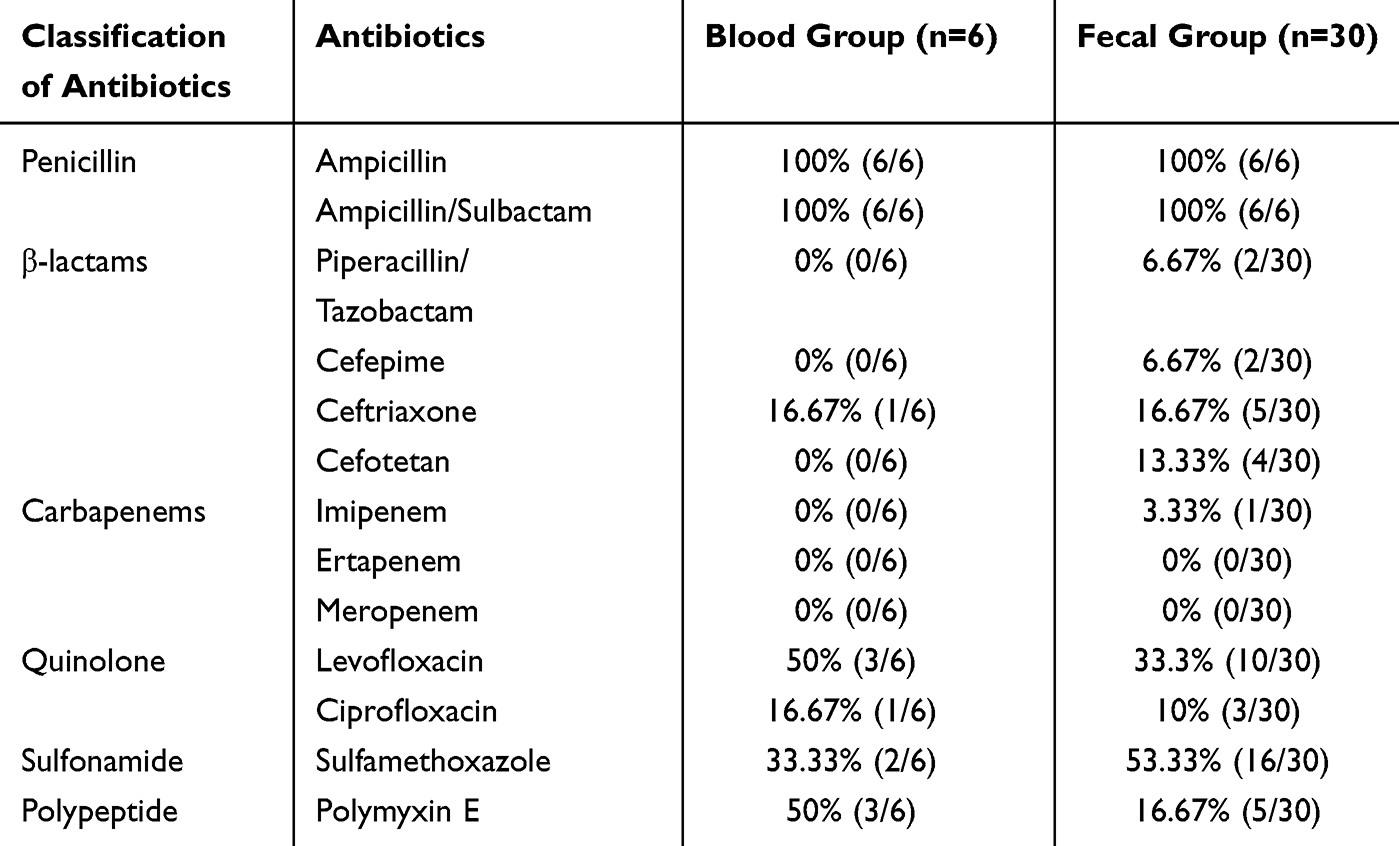 |
Table 1 Antibiotic Susceptibility Profile |
Genomic analysis of all isolates identified a total of 60 antibiotic resistance genes (Figure 2). Every isolate harbored at least one resistance gene. The most prevalent genes were β-lactamase genes (eg, blaCMY, blaCTX, blaDHA, blaOXA, blaTEM), present in all isolates. Chloramphenicol resistance genes (catA, catB) were detected in 91% of isolates (33/36), followed by sulfonamide resistance genes (sul1, sul2, sul3) in 88% (32/36), tetracycline resistance genes (tetA, tetB, tetM) in 77% (28/36), and aminoglycoside resistance genes (aac(3), aac(6’)Ib, aadA12, aadA16, aadA1, aadA2, aadA3, aadA5, aph(3’), aph(4)) in 61% (22/36). Interestingly, phenotypic analysis revealed that 27% (10/36) of the isolates were resistant to polymyxin E, with a higher proportion in the blood group (50%, 3/6) compared to the fecal group. However, no common resistance genes for polymyxin were identified.
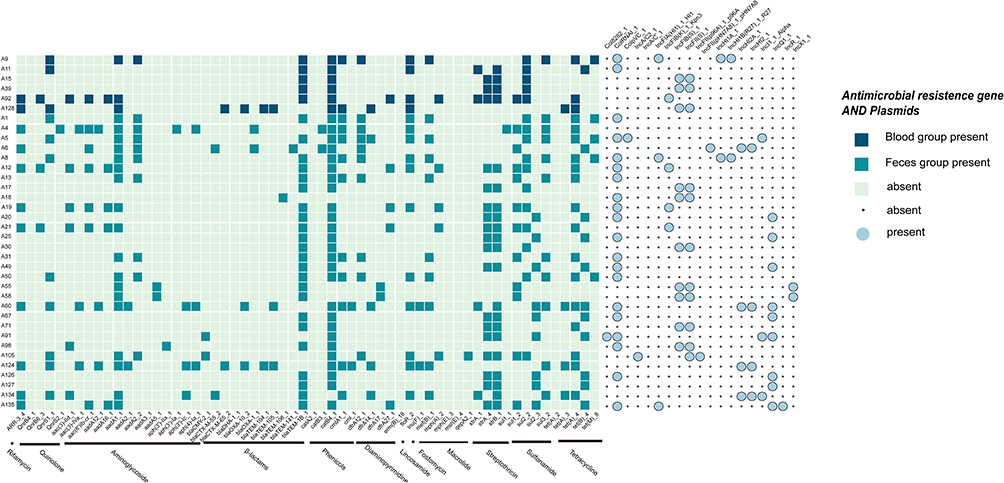 |
Figure 2 The heat map of ARGs and plasmids in the studied Salmonella isolates. The presence of antibiotic resistance genes was marked with dark green color, while the absence was marked with light green color. The presence of plasmids was marked with big blue circles, while the absence was marked with little white circles. |
A total of 25 distinct plasmid replicons were identified across the 36 isolates. The most frequently detected replicon was ColRNAI-1, found in 55% of isolates (20/36), followed by IncFII(S)-1 in 30% (11/36), as shown in Figure 2. The plasmid types IncFII(S) and IncFIB(S)were the primary carriers of the β-lactamase gene blaTEM−1B.
The bloodstream isolates harbored fewer resistance genes (31) compared to fecal isolates (53). Similarly, bloodstream isolates contained fewer plasmids (9) than fecal isolates (23). This observation suggests that bacterial invasion into the bloodstream might not be directly associated with higher antibiotic resistance.
Virulence Factor Analysis of Salmonella Isolates
To comprehensively characterize the virulence profiles of the isolates and investigate the role of virulence factors in bloodstream invasion, we analyzed all isolates using the VFDB database. A total of 161 distinct virulence factors were identified across the isolates. These genes encode proteins with diverse functions, including extracellular enzymes, exotoxins, motility, nutrient/metabolic factors, immune modulation, and transcription regulation. The most abundant virulence factors were associated with the SPI-2 encoded T3SS-2, comprising 16 genes. T3SS-2 is critical during the later stages of infection, where it secretes effector proteins that suppress host immune responses, enabling Salmonella to persist and replicate within host cells.34 Additionally, 24 genes encoding fimbrial adherence determinants were identified. Bloodstream isolates carried 152 virulence factors, compared to 155 in fecal isolates. While the total number of virulence factors differed slightly between the two groups, the difference was not statistically significant (Table 2 and Table S6). However, bloodstream isolates exhibited elevated expression of plasmid-encoded virulence genes, including the pef fimbrial gene cluster, the spvB gene encoding an ADP-ribosylating toxin, the cdtB gene encoding cytolethal distending toxin, and the spvC gene encoding phosphothreonine lyase. These genes are likely associated with Salmonella bloodstream invasion and systemic dissemination.
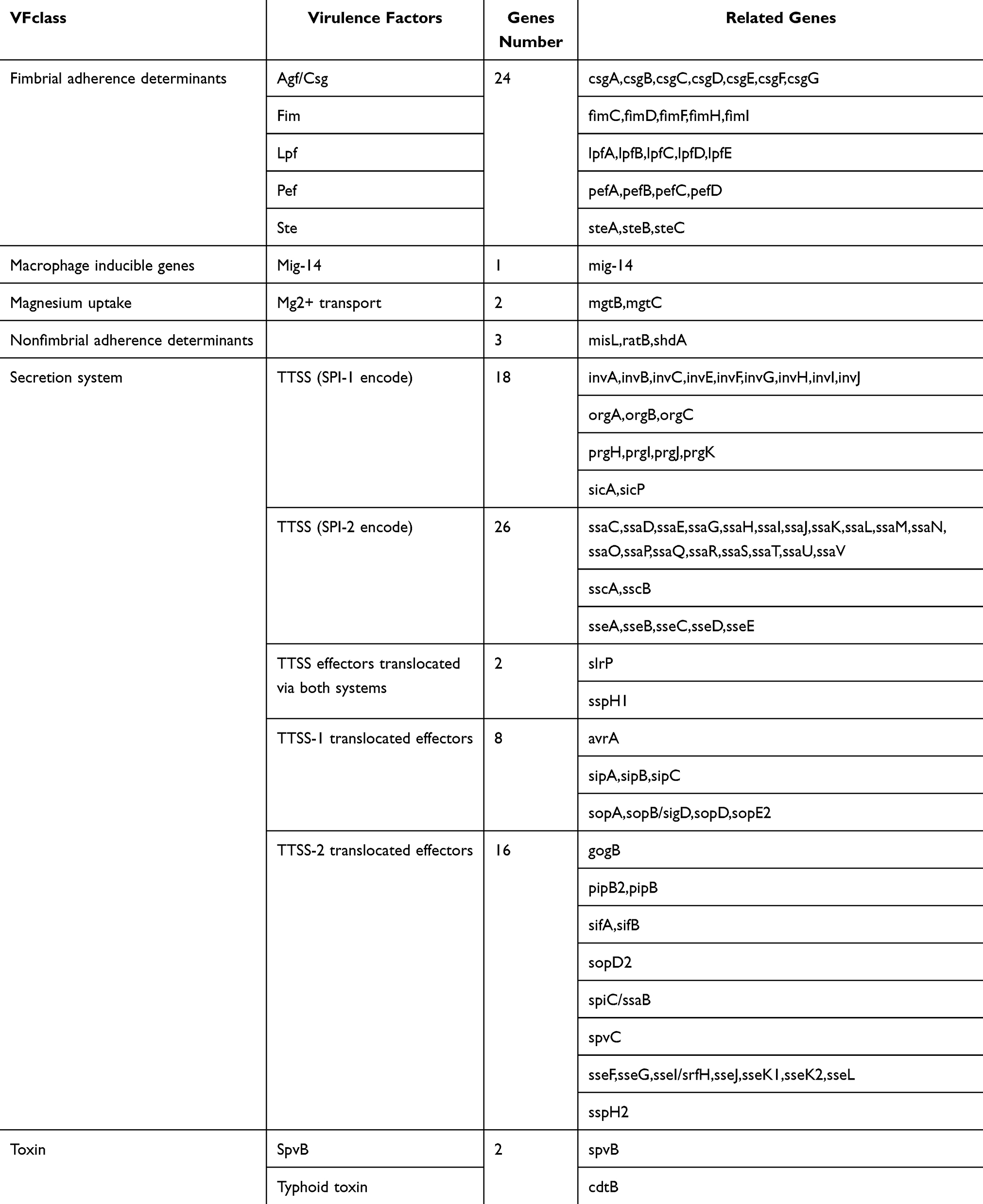 |
Table 2 Virulence Factors of Salmonella Isolates |
To explore the relationship between SPIs and bloodstream infections, we screened all isolates for SPIs. All isolates harbored SPI-1 to SPI-5, SPI-9, and SPI-12 (Figure 3). Notably, SPI-6 was present in all bloodstream isolates, while SPI-8 was detected only in a subset of fecal isolates (A13 and A31).
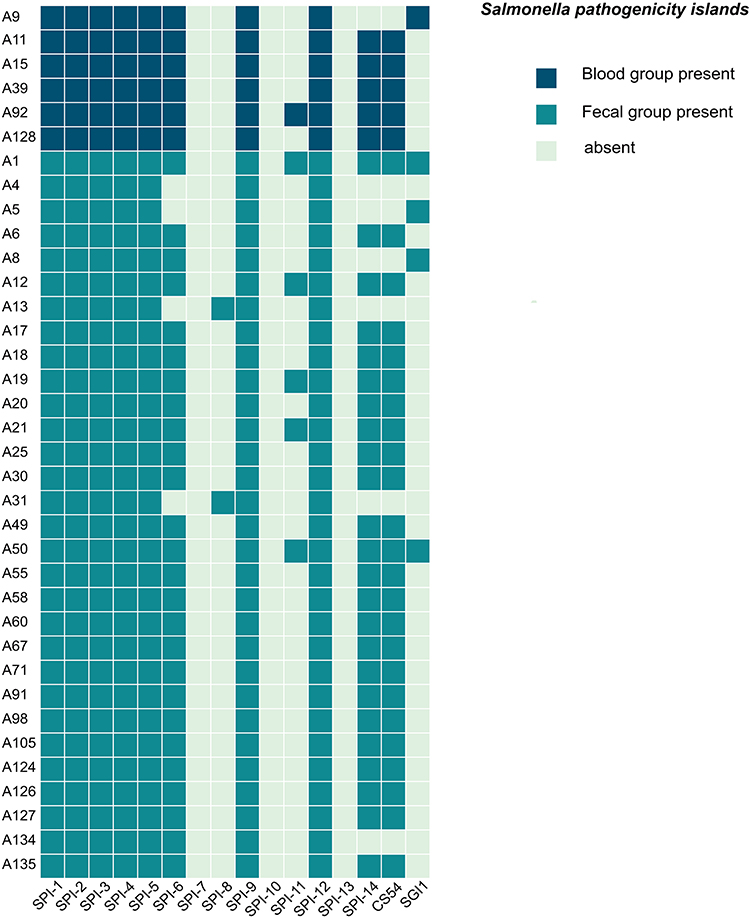 |
Figure 3 The heat map of SPIs in the studied Salmonella isolates. The presence of SPIs was marked with dark green color, while the absence was marked with light green color. |
Using AntiSMASH 7.0 to predict secondary metabolite biosynthetic gene clusters, we identified two clusters across the 36 Salmonella isolates. These clusters primarily encoded non-ribosomal peptide synthetases (NRPS) and thiopeptides, with NRPS being the most abundant. No significant difference was observed in the number of gene clusters between bloodstream and fecal isolates (Figure S2). The identified NRPS cluster encodes Enterobacter, which facilitates iron acquisition and protects intracellular bacteria from host antimicrobial defenses.35
Phylogenetic Analysis of Salmonella Isolates
To investigate the epidemiological relationships among the Salmonella isolates associated with diverse clinical outcomes, we performed core SNPs-based phylogenetic analysis. Using Salmonella enterica serovar Typhimurium LT2 as the reference strain, a phylogenetic tree was constructed (Figure 4). The analysis revealed that Typhimurium and its monophasic variant (I 4,[5],12:i:-) were predominant among the isolates, with the monophasic variant being the most frequently identified. Fourteen isolates of Typhimurium and its monophasic variant were classified into sequence types ST19 and ST34. Among the remaining serovars, a one-to-one correlation between serovar and MLST was observed. Isolates of serovars Goldcoast, Panama, Rissen, and Derby clustered within the same branch, indicating a high degree of genetic relatedness. Similarly, isolates of serovars London and Thompson grouped into another distinct branch, reflecting close genetic relationships. The phylogenetic tree also demonstrated that certain bloodstream isolates (A9, A11, A15, A39, and A92) clustered within the same major branch as several fecal isolates, suggesting relatively similar genomic profiles. In contrast, the bloodstream isolate A128 was positioned outside this cluster, exhibiting distinct genomic characteristics. Intriguingly, A128 was the only MDR isolate identified in the bloodstream group in this study. Overall, iNTS were not restricted to a single serovar and exhibited considerable diversity. Notably, MDR and non-MDR isolates did not form distinct clusters based on phylogenetic analysis, highlighting substantial genomic differences between these groups.
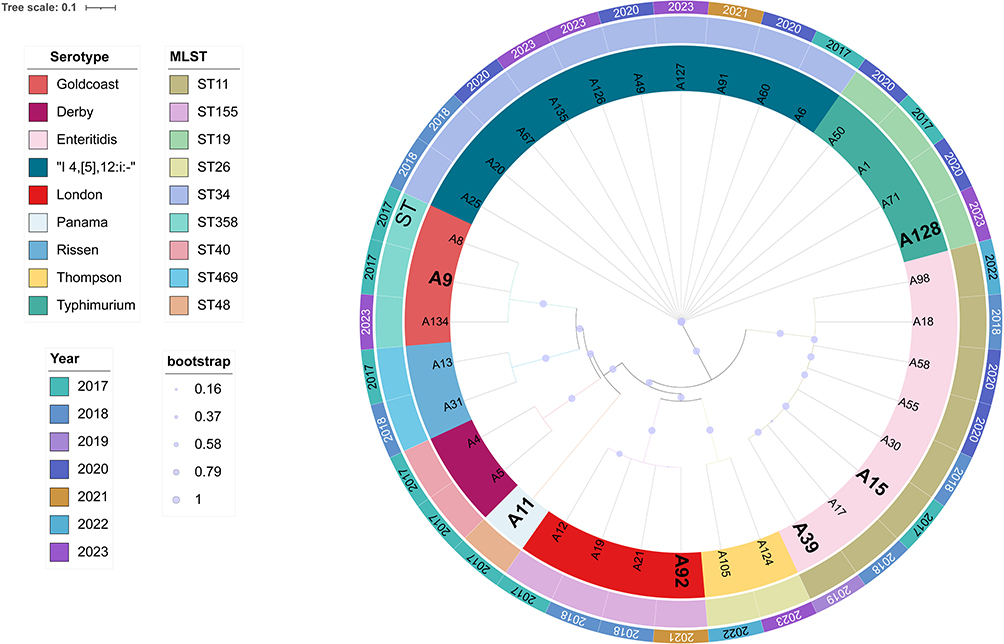 |
Figure 4 The phylogenetic tree, constructed using core genome SNPs and the General Time Reversible (GTR) model, encompasses 36 Salmonella strains and incorporates metadata on year of isolation, serotype, and MLST. The tree is rooted with Salmonella enterica serovar Typhimurium LT2. The black bolded labels represent the blood group, while the non-bolded labels represent the fecal group. |
Bioinformatics Analysis of Differential Genes Between Bloodstream and Fecal Isolates
To further explore the genetic diversity of Salmonella in relation to its clinical outcomes, we focused on core genes and genomic content associated with specific clinical presentations. Orthologous genes present across all isolates were defined as the core genome. We analyzed the genomes of 36 isolates, comprising 30 fecal isolates and 6 bloodstream infection isolates, to identify the genes constituting the core genome. Pan-genome analysis identified a total of 7219 distinct genes across all genomes, with 3564 genes shared by all isolates, representing the core genome library (Figure 5A). Subgroup analysis of the fecal and bloodstream isolates was performed to further elucidate the genetic specificity and commonality associated with different clinical isolates. The pan-genome of the 30 fecal isolates comprised 7086 genes, including 3655 core genes, while the pan-genome of the 6 bloodstream isolates consisted of 5306 genes, with 3656 core genes (Figure 5B and C).
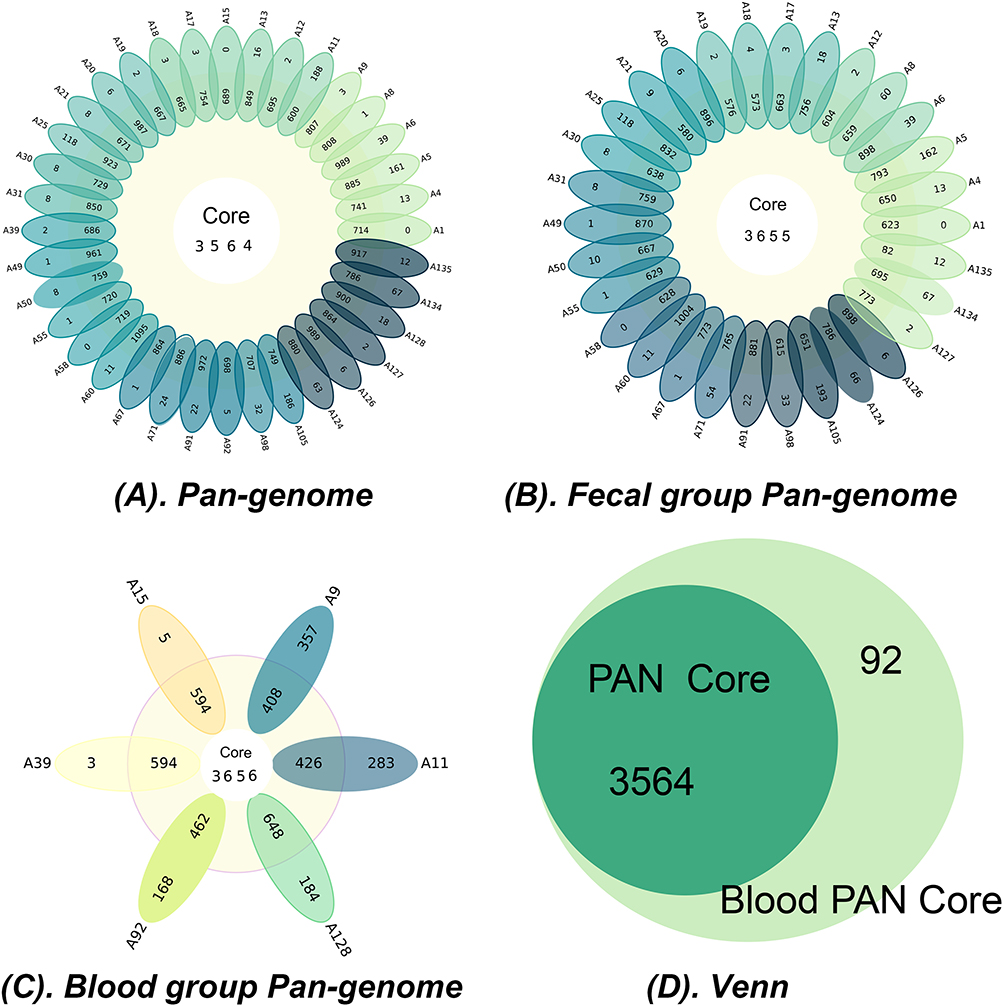 |
Figure 5 Strain pan-genome and differential gene analysis results: (A) The pangenome analysis of the 36 isolates is illustrated as a flower plot, where the central number represents the count of core genes. The proximal part of each petal indicates the number of accessory genes, while the distal part denotes the number of unique genes and exclusively absent genes. (B) The pangenome analysis of the 30 fecal isolates is illustrated as a flower plot, where the central number represents the count of core genes. The proximal part of each petal indicates the number of accessory genes, while the distal part denotes the number of unique genes and exclusively absent genes. (C) The pangenome analysis of the 6 blood isolates is illustrated as a flower plot, where the central number represents the count of core genes. The proximal part of each petal indicates the number of accessory genes, while the distal part denotes the number of unique genes and exclusively absent genes. (D)Venn diagram of differentially expressed genes between blood and fecal core genomes. |
The pan-genome curve (Figure S3) revealed that the size of the pan-genome increased with the addition of new isolates, indicating that Salmonella possesses an open pan-genome and a strong ability to acquire exogenous genes. Functional annotation of the core genes showed enrichment in categories related to energy production (C), carbohydrate transport and metabolism (G), and amino acid transport and metabolism (E), suggesting their roles in fundamental bacterial metabolic processes. In contrast, accessory genes were predominantly enriched in transcription (K), replication, recombination, and repair (L), and uncharacterized functions (S), indicating their potential involvement in the storage and processing of genetic information (Figure 6).
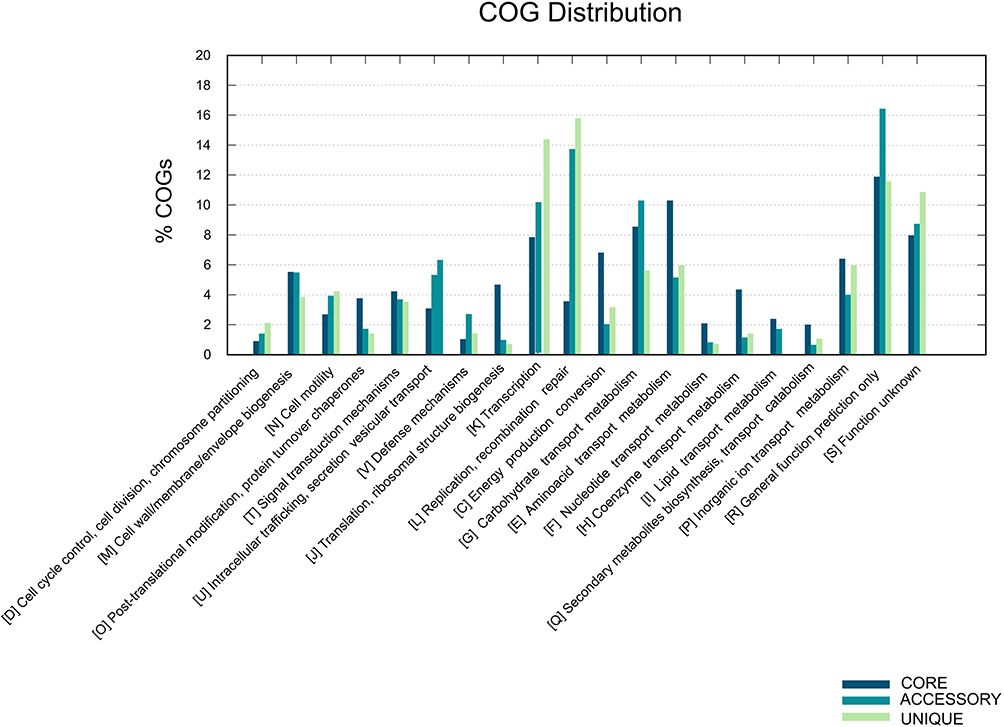 |
Figure 6 The analysis of COG functional annotations. |
We conducted a detailed comparative analysis of core genes between bloodstream and fecal Salmonella isolates. Compared to the core genome of all isolates, 92 core genes were unique to the blood group (Figure 5D), while 88 core genes were unique to the fecal group. These distinct core genes may play critical roles in facilitating Salmonella invasion into the bloodstream and causing systemic infections. The core gene differences between the two groups are summarized in Tables S7 and S8. Among the 92 unique blood core genes, a significant proportion were classified as having unknown functions based on COG functional annotation. For the genes with known functions, the dominant biological processes included carbohydrate transport and metabolism, as well as inorganic ion transport and metabolism. Pathway enrichment analysis using the KEGG database identified the top 10 pathways associated with these blood-specific genes (Figure 7). These pathways primarily involved crucial biological processes such as bacterial density regulation and membrane transport, including quorum sensing (KEGG ID: 02024), prokaryotic cellular communities (B09145), transporters (02000), ABC transporters (02010), protein families related to signaling and cellular processes (B09183), and membrane transport (B09131).
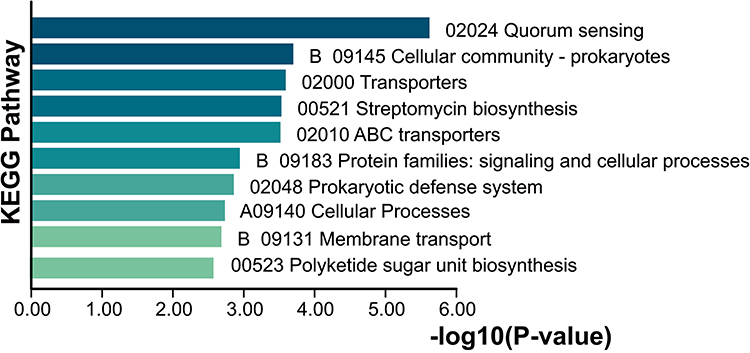 |
Figure 7 KEGG pathway enrichment analysis. |
Notably, quorum sensing (KEGG ID: 02024) was highly enriched among the blood-specific core genes, strongly suggesting that these genes may play a pivotal role in Salmonella bloodstream infection by regulating quorum sensing systems and membrane transport processes. Further analysis of the genes enriched in quorum sensing pathways revealed key contributors such as lsrA, lsrG, lsrF, and lsrK. These genes are predominantly associated with the AI-2 signaling pathway, a critical communication system in bacteria. Based on these findings, we hypothesize that the AI-2 signaling pathway may serve as a central mechanism in Salmonella-induced bloodstream infections. Specifically, this pathway likely facilitates biofilm formation, thereby promoting the systemic dissemination of the pathogen within the host.
Biofilm Formation and Inhibition Assays of Bloodstream and Fecal Isolates
Biofilm formation in bloodstream and fecal Salmonella isolates was quantitatively measured using the crystal violet staining method (Figure 8A). After equal incubation times, bloodstream isolates exhibited significantly higher biofilm-forming capacity compared to fecal isolates (p=0.02 and< 0.05). This finding suggests that genes involved in biofilm formation may play a role in Salmonella bloodstream invasion.
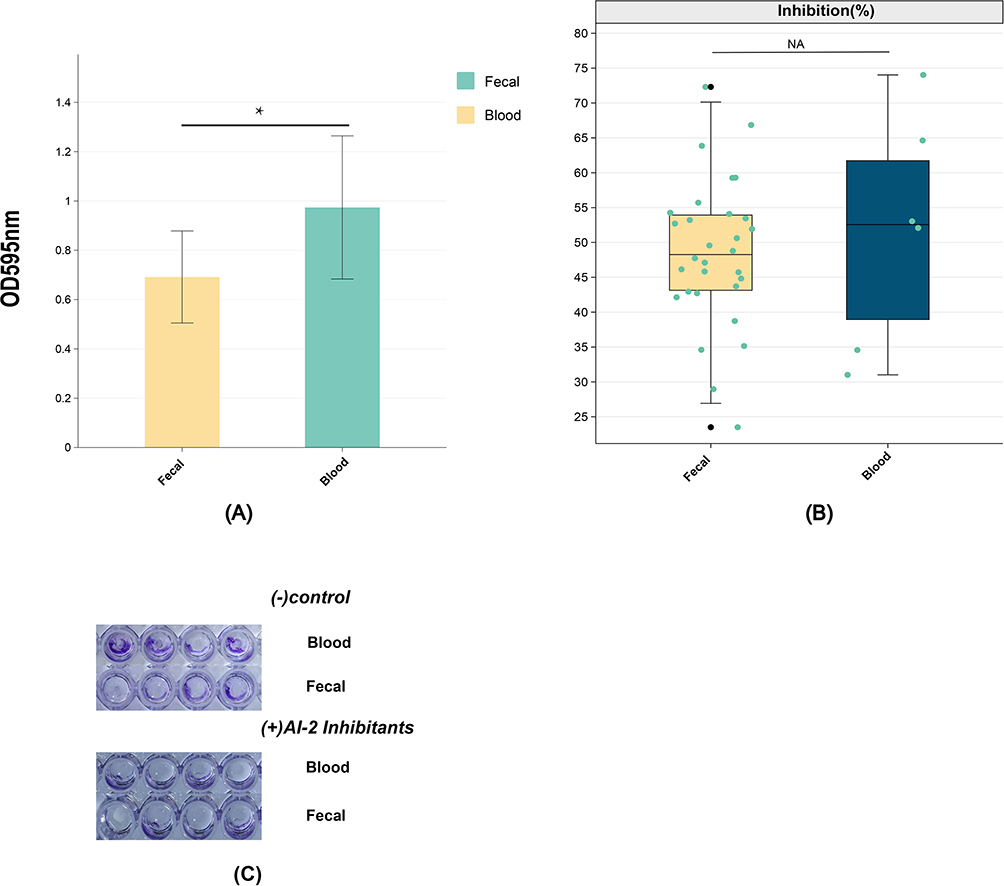 |
Figure 8 (A) Comparison of biofilm staining at OD595nm between blood and fecal groups was conducted, with p< 0.05 considered as the threshold for statistical significance (*). (B) Comparison of biofilm inhibition rates between the blood and fecal groups after the addition of the AI-2 inhibitor. “NA” indicates no statistical significance. (C) The determination of biofilm formation in the microtiter plates is evidenced by the strong blue coloration of the crystal violet staining on the walls and bottom of each well. The positive reaction is evidenced for the Salmonella isolates, validated by the negative control (LB medium only) and positive (AI-2 inhibitor). |
To investigate the regulatory role of the AI-2 signaling pathway in biofilm formation, quercetin, an AI-2 signaling pathway inhibitor, was introduced into experimental groups. Compared to the negative control, most isolates demonstrated a marked reduction in biofilm formation (Figure 8B and C). Calculation of biofilm inhibition rates revealed no significant statistical difference between bloodstream and fecal isolates (p=0.82 and>0.05), as shown in Figure 8. These results indicate that while the AI-2 quorum-sensing pathway is implicated in biofilm regulation, biofilm formation is likely influenced by additional factors beyond the AI-2 pathway. This highlights the complexity of biofilm formation mechanisms and suggests the involvement of multiple regulatory systems in Salmonella pathogenesis.
Discussion
Salmonella is a significant foodborne pathogen, commonly causing self-limiting gastroenteritis. However, the mechanisms underlying its ability to induce systemic infections, such as bloodstream infections, remain incompletely understood. In this study, we employed high-throughput sequencing to perform a comprehensive genomic analysis of Salmonella isolates from blood and fecal samples, aiming to elucidate differences in pathogenic mechanisms. Our findings suggest that the AI-2 signaling pathway may play a pivotal role in Salmonella bloodstream invasion. As a quorum sensing system, the AI-2 pathway likely facilitates bloodstream entry by promoting biofilm formation. Notably, no significant differences were observed between bloodstream and fecal isolates in terms of antimicrobial resistance or virulence factors. This observation has important implications for clinical treatment, providing insights into effective therapeutic strategies for managing Salmonella infections.
In this study, we analyzed the genetic characteristics of non-typhoidal Salmonella isolates from patients with varying clinical manifestations to identify specific genes associated with clinical outcomes. Comparative analysis of core genomes revealed several genes potentially involved in the systemic dissemination of iNTS into the host bloodstream. Our results identified 92 unique core genes in iNTS isolates from blood samples, which were significantly enriched in the QS pathway. QS signaling molecules play critical roles in bacterial density sensing and response, triggering global gene expression changes that regulate biofilm formation, toxin production, and secondary metabolite synthesis. These QS-enriched genes are predominantly involved in the AI-2 signaling pathway and encode membrane transport-related proteins. Interestingly, despite the enrichment of QS-related genes, there were no significant differences in the expression of virulence factors or secondary metabolite genes between the blood and fecal groups. This suggests that biofilm formation mediated by the AI-2 signaling pathway may play a critical role in the ability of Salmonella to cause bloodstream infections. The AI-2/LuxS QS system, widely conserved in both Gram-negative and Gram-positive bacteria, is known to modulate virulence gene expression, biofilm formation, and antimicrobial susceptibility, thereby influencing pathogenicity. Among the identified differential genes, we observed the lsr operon, the only known operon directly regulated by AI-2. This operon encodes an ABC transporter system for AI-2 uptake and degradation.36 Previous studies have shown that inactivating the AI-2/LuxS system significantly enhances the adhesion and invasion capabilities of Salmonella in chicken fibroblast DF-1 and human colon epithelial Caco-2 cells. To further validate our hypothesis, we performed in vitro biofilm formation and inhibition assays to assess the relationship between this pathway and Salmonella invasiveness. Under conditions without an AI-2 inhibitor, biofilm formation in blood isolates was significantly greater than in fecal isolates. Quercetin, a known inhibitor of the AI-2 pathway, effectively suppressed QS system activity. Upon the addition of quercetin, biofilm formation was reduced in both groups, but the inhibition rate showed no significant differences. These results suggest that biofilm formation is regulated not only by the AI-2 signaling pathway but also by other complex factors. In recent years, QS inhibitors have gained considerable attention as novel anti-pathogen strategies. By disrupting bacterial virulence without directly inhibiting growth, QS inhibitors enhance the host immune response and reduce the likelihood of resistance development. These findings underscore the potential of QS inhibitors as therapeutic agents against Salmonella infections. Future studies should explore the combined effects of different signaling pathways and environmental factors on biofilm formation and assess the clinical application potential of novel QS inhibitors to improve treatment outcomes for Salmonella infections.
Through genomic comparisons, we also observed that the genome size of blood isolates was slightly smaller than that of fecal isolates, although the underlying mechanism remains unclear. We hypothesize that this may be linked to the survival strategies of Salmonella within the host. During the transition from intestinal epithelial cells to non-epithelial cells via SCVs, the bacterial genome may undergo dynamic adjustments, potentially involving the loss of non-essential genes for survival in the bloodstream environment. This genomic streamlining may enhance adaptation, but further research is required to elucidate the mechanisms of gene loss and their roles in Salmonella pathogenicity and infection dynamics. Phylogenetic analysis of 36 Salmonella isolates revealed that Salmonella Typhimurium and its monophasic variant I 4,[5],12:i:- were the predominant serotypes. Combined with existing data, these findings suggest that this serotype has likely become the major dominant serotype in Zhejiang Province, China.37–39 Notably, we observed significant resistance to ceftriaxone and ciprofloxacin in S. Typhimurium and its monophasic variant. Considering the clinical and environmental prevalence of this serotype, enhanced surveillance efforts are warranted to mitigate the risk of widespread outbreaks. However, this high transmissibility and notable resistance do not necessarily imply a higher propensity to cause bloodstream infections. Current data do not support a direct association between serotypes and the occurrence of bloodstream infections.
In microbial infection research, virulence factors profiles are often considered key indicators of disease severity. However, based on the findings of this study, the blood group did not show a significant increase in the number of virulence factors. The majority of Salmonella virulence factors are concentrated in the T3SS, one of the most well-studied virulence mechanisms. Despite our comparative analysis across different clinical outcome groups, no significant differences were observed in the expression of this system. This suggests that the T3SS may not be the sole key factor determining the pathogenicity of Salmonella, or that it interacts with other virulence systems, collectively influencing the infection outcome. Additionally, we observed relatively high expression of certain virulence factors in the blood infection group, including the pef gene cluster, spvB, cdtB, and spvC. The Pef operon, which encodes a plasmid-borne fimbriae, is of particular interest. Previous studies have indicated that the QS system can regulate the expression of Salmonella’s rck locus, influencing its resistance to complement and controlling the synthesis and assembly of Pef fimbriae.40 We hypothesize that the Pef fimbriae may be a potential factor facilitating bacterial entry into the bloodstream and the onset of systemic infections. Interestingly, we did not observe high expression of the rck gene, suggesting that its role may involve multiple QS signaling molecules and regulatory networks. Whether AI-2, as one of these molecules, is involved in regulating the expression of the pef gene cluster remains unclear and warrants further investigation. spvB, located on the Salmonella pathogenicity island (pSLT), encodes an ADP-ribosylating toxin that can inhibit receptor-interacting protein kinase 3 (RIPK3) through K48-linked polyubiquitination, leading to RIPK3 degradation in an autophagy-dependent manner. This results in necroptotic cell death in intestinal epithelial cells, allowing Salmonella to breach the intestinal epithelial barrier and invade further into the host, consistent with our observations.41 cdtB encodes the typhoid toxin, which induces DNA damage in host cells.42 Thus, elevated expression of bacterial exotoxins may be one of the contributing factors to Salmonella invasion into the bloodstream. Moreover, the presence of SPI-6 was detected in all blood-derived isolates. This pathogenic island encodes invasion proteins, such as pagN and the CS54 invasion protein sinH, which may play a role in the invasiveness of non-typhoidal Salmonella.40 These findings further suggest that the invasiveness of Salmonella results from the combined action of multiple factors. The mere presence or absence of a single virulence factor is insufficient to fully explain its pathogenic potential. Therefore, further research into these virulence factors and their interactions, particularly their expression and function under different environmental and host conditions, will contribute to a more comprehensive understanding of the invasion mechanisms of Salmonella and provide a basis for developing new therapeutic strategies.
In the treatment of acute gastroenteritis caused by Salmonella, empirical antibiotic therapy typically involves agents such as ceftriaxone or ciprofloxacin.43 In this study, we observed a potential cross-resistance trend between ciprofloxacin and ceftriaxone, which may be attributed to the fact that the resistance genes for these two antibiotics are often located on the same plasmid, thereby increasing their likelihood of co-existence.38 Notably, ciprofloxacin resistance was slightly lower, which may be related to the restricted use of fluoroquinolones in children. When Salmonella causes bloodstream infections, treatment regimens usually favor the use of more advanced antibiotics, such as carbapenems. Analysis of antimicrobial susceptibility testing revealed that the multidrug resistance rate of bloodstream isolates was significantly lower than that of the fecal isolates. Moreover, whole-genome sequencing analysis showed that the number of resistance genes and plasmids in the blood group was markedly lower than in the fecal group. This finding contradicts some previous studies, which typically suggest that Salmonella virulence increases with the rise in antibiotic resistance. Our observation indicates that there is no direct correlation between bacterial invasion of the bloodstream and resistance. This finding has important clinical implications, suggesting that antibiotic selection for Salmonella-induced bloodstream infections should be based on changes in the resistance profile or by adjusting empirical antibiotic dosing, with the goal of reducing unnecessary clinical selection pressure and enabling more precise treatment.
Polymyxin, as the last line of defense against multidrug-resistant Salmonella infections,44 is typically used when other antibiotics fail. In our analysis of antimicrobial susceptibility testing, we observed that Salmonella isolates from the blood group exhibited higher resistance to polymyxin. However, none of the known polymyxin resistance genes were detected in these isolates, suggesting that the resistance mechanism may involve unidentified genes or new regulatory pathways. Biofilm formation plays a crucial role in bacterial pathogenicity and resistance, particularly in clinical settings, where biofilm presence significantly increases the persistence of infections and the difficulty of treatment. Studies estimate that approximately 60% of hospital-acquired infections are closely associated with biofilm formation.44 In this study, Salmonella isolates from the blood group demonstrated higher biofilm formation levels. Biofilms act not only as a physical barrier, delaying antibiotic particles from entering bacterial cells, but also enhance antibiotic failure by activating efflux pump systems that expel antimicrobial agents from the cells. Quorum sensing signals are believed to play a significant role in this process by regulating biofilm formation and bacterial resistance, thereby strengthening the pathogen’s survival capabilities.45 Previous studies have shown that β-adrenergic blockers can enhance the antibacterial activity of polymyxin B against Klebsiella pneumoniae by increasing the thermal stability of LuxS and reducing AI-2 production, thus decreasing biofilm formation.46 Whether the AI-2-mediated quorum sensing system also relies on efflux pump regulation in biofilms to play a key role in Salmonella resistance to polymyxin remains unclear. This warrants further investigation in future studies to elucidate its potential mechanism in Salmonella polymyxin resistance, which could help optimize antimicrobial treatment strategies.
This study has certain limitations. Currently, the analysis is based solely on 36 Salmonella strains isolated from blood and fecal samples from clinical cases in Huzhou. To enhance the representativeness and reliability of the findings, future studies should involve broader and long-term surveillance, incorporating phenotypic and genomic data from a larger number of iNTS and NTS strains. This would provide more accurate and robust information for characterizing and predicting the features of iNTS strains. Additionally, future research should integrate functional transcriptomics and proteomics to elucidate the genetic mechanisms underlying iNTS-induced bloodstream infections.
Conclusion
In summary, our study presents a systematic comparative genomics analysis of invasive non-typhoidal Salmonella (iNTS) and non-typhoidal Salmonella (NTS). Our findings suggest that high levels of antibiotic resistance are not the primary drivers of NTS invasiveness. Rather, Salmonella can modulate biofilm formation through the AI-2 signaling pathway within the quorum sensing system, which subsequently contributes to the development of bloodstream infections. This study provides scientific evidence to guide the rational use of antibiotics in treating Salmonella infections in clinical settings. Additionally, it offers theoretical support for the development of novel therapeutic agents targeting Salmonella and advances the potential for precision medicine in the future.
Data Sharing Statement
The data supporting this study’s findings are available from the corresponding author upon reasonable request.
Ethics Statement
This study was reviewed and approved by First Affiliated Hospital of Huzhou University (approval number: 2024KYLL008-01) and was performed in accordance with the Declaration of Helsinki. Informed consent was obtained from the patient.
Acknowledgments
We thank Researcher Shaoyan Yi for assistance with experimental design and Dr. Qinqin Lou for help with manuscript preparation. We would like to thank the anonymous reviewers for their helpful remarks.
Funding
The study was supported by grants from the public welfare technology application research program of Huzhou (Grant no. 2023GYB19). This research was supported by the National Natural Science Foundation of China (No. 82472291), Zhejiang Provincial Natural Science Foundation of China (No. LTGD24C010001), to Y.L.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gal-Mor O, Boyle EC, Grassl GA. Same species, different diseases: how and why typhoidal and non-typhoidal Salmonella enterica serovars differ. Front Microbiol. 2014;5:102622. doi:10.3389/fmicb.2014.00391
2. Uche IV, MacLennan CA, Saul A. A systematic review of the incidence, risk factors and case fatality rates of invasive nontyphoidal salmonella (iNTS) disease in Africa (1966 to 2014). PLoS Negl Trop Dis. 2017;11(1):e0005118. doi:10.1371/journal.pntd.0005118
3. Deen J, Von Seidlein L, Andersen F, Elle N, White NJ, Lubell Y. Community-acquired bacterial bloodstream infections in developing countries in south and Southeast Asia: a systematic review. Lancet Infect Dis. 2012;12(6):480–487. doi:10.1016/S1473-3099(12)70028-2
4. Reddy EA, Shaw AV, Crump JA. Community-acquired bloodstream infections in Africa: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10(6):417–432. doi:10.1016/S1473-3099(10)70072-4
5. Stanaway JD, Parisi A, Sarkar K, et al. The global burden of non-typhoidal salmonella invasive disease: a systematic analysis for the global burden of disease study 2017. Lancet Infect Dis. 2019;19(12):1312–1324. doi:10.1016/S1473-3099(19)30418-9
6. Kariuki S, Gordon MA, Feasey N, Parry CM. Antimicrobial resistance and management of invasive Salmonella disease. Vaccine. 2015;33:C21–C29.
7. Zha L, Garrett S, Sun J. Salmonella infection in chronic inflammation and gastrointestinal cancer. Diseases. 2019;7(1):28. doi:10.3390/diseases7010028
8. Kaur J, Jain S. Role of antigens and virulence factors of Salmonella enterica serovar Typhi in its pathogenesis. Microbiol Res. 2012;167(4):199–210. doi:10.1016/j.micres.2011.08.001
9. LaRock DL, Chaudhary A, Miller SI. Salmonellae interactions with host processes. Nat Rev Microbiol. 2015;13(4):191–205. doi:10.1038/nrmicro3420
10. Rosselin M, Virlogeux-Payant I, Roy C, et al. Rck of Salmonella enterica, subspecies enterica serovar Enteritidis, mediates Zipper-like internalization. Cell Res. 2010;20(6):647–664. doi:10.1038/cr.2010.45
11. Riddle MS, DuPont HL, Connor BA. ACG clinical guideline: diagnosis, treatment, and prevention of acute diarrheal infections in adults. Am J Gastroenterol. 2016;111(5):602–622. doi:10.1038/ajg.2016.126
12. Vallés J, Ferrer R. Bloodstream Infection in the ICU. Infectious Dis Clin North America. 2009;23(3):557–569. doi:10.1016/j.idc.2009.04.005
13. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi:10.1093/bioinformatics/bty560
14. Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6):e1005595. doi:10.1371/journal.pcbi.1005595
15. Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–1075. doi:10.1093/bioinformatics/btt086
16. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi:10.1093/bioinformatics/btu153
17. Starikova EV, Tikhonova PO, Prianichnikov NA, et al. Phigaro: high-throughput prophage sequence annotation. Bioinformatics. 2020;36(12):3882–3884. doi:10.1093/bioinformatics/btaa250
18. Bertelli C, Brinkman FS. Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics. 2018;34(13):2161–2167. doi:10.1093/bioinformatics/bty095
19. Russel J, Pinilla-Redondo R, Mayo-Muñoz D, Shah SA, Sørensen SJ. CRISPRCasTyper: automated identification, annotation, and classification of CRISPR-Cas loci. CRISPR J. 2020;3(6):462–469. doi:10.1089/crispr.2020.0059
20. Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. doi:10.1371/journal.pone.0009490
21. Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–W296. doi:10.1093/nar/gkab301
22. Chen L. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2004;33(Database issue):D325–D328. doi:10.1093/nar/gki008
23. Carattoli A, Hasman H. PlasmidFinder and in silico pMLST: identification and typing of plasmid replicons in whole-genome sequencing (WGS). In: F DLC editor. Horizontal Gene Transfer. Vol 2075. Methods in Molecular Biology. Springer US; 2020:285–294. doi:10.1007/978-1-4939-9877-7_20
24. Zhang S, Den Bakker HC, Li S, et al. SeqSero2: rapid and improved Salmonella serotype determination using whole-genome sequencing data. Appl Environ Microbiol. 2019;85(23):e01746–19. doi:10.1128/AEM.01746-19
25. Larsen MV, Cosentino S, Rasmussen S, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50(4):1355–1361. doi:10.1128/JCM.06094-11
26. Roer L, Hendriksen RS, Leekitcharoenphon P, et al. Is the evolution of Salmonella enterica subsp. enterica linked to restriction-modification systems? Msystems. 2016;1(3):10–1128. doi:10.1128/mSystems.00009-16
27. Yoon SH, Park YK, Kim JF. PAIDB v2. 0: exploration and analysis of pathogenicity and resistance islands. Nucleic Acids Res. 2015;43(D1):D624–D630. doi:10.1093/nar/gku985
28. Chaudhari NM, Gupta VK, Dutta C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci Rep. 2016;6(1):24373. doi:10.1038/srep24373
29. Huerta-Cepas J, Szklarczyk D, Heller D, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–D314. doi:10.1093/nar/gky1085
30. Tatusov RL, Fedorova ND, Jackson JD, et al. The COG database: an updated version includes eukaryotes. BMC Bioinf. 2003;4(1):1–14. doi:10.1186/1471-2105-4-41
31. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
32. Chen C, Chen H, Zhang Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–1202. doi:10.1016/j.molp.2020.06.009
33. Li Y, Dai J, Ma Y, et al. The mitigation potential of synergistic quorum quenching and antibacterial properties for biofilm proliferation and membrane biofouling. Water Res. 2024;255:121462. doi:10.1016/j.watres.2024.121462
34. Hayward RD, Cain RJ, McGhie EJ, Phillips N, Garner MJ, Koronakis V. Cholesterol binding by the bacterial type III translocon is essential for virulence effector delivery into mammalian cells. Mol Microbiol. 2005;56(3):590–603. doi:10.1111/j.1365-2958.2005.04568.x
35. Saha P, Xiao X, Yeoh BS, et al. The bacterial siderophore enterobactin confers survival advantage to Salmonella in macrophages. Gut Microbes. 2019;10(3):412–423. doi:10.1080/19490976.2018.1546519
36. Xue T, Zhao L, Sun H, Zhou X, Sun B. LsrR-binding site recognition and regulatory characteristics in Escherichia coli AI-2 quorum sensing. Cell Res. 2009;19(11):1258–1268. doi:10.1038/cr.2009.91
37. Chen J, Ed-Dra A, Zhou H, Wu B, Zhang Y, Yue M. Antimicrobial resistance and genomic investigation of non-typhoidal Salmonella isolated from outpatients in Shaoxing city, China. Front Public Health. 2022;10:988317. doi:10.3389/fpubh.2022.988317
38. Shi Q, Ye Y, Lan P, et al. Prevalence and characteristics of ceftriaxone-resistant Salmonella in children’s hospital in Hangzhou, China. Front Microbiol. 2021;12:764787. doi:10.3389/fmicb.2021.764787
39. Ke Y, Lu W, Liu W, Zhu P, Chen Q, Zhu Z. Non-typhoidal Salmonella infections among children in a tertiary hospital in Ningbo, Zhejiang, China, 2012–2019. PLoS Negl Trop Dis. 2020;14(10):e0008732. doi:10.1371/journal.pntd.0008732
40. Suez J, Porwollik S, Dagan A, et al. Virulence gene profiling and pathogenicity characterization of non-typhoidal Salmonella accounted for invasive disease in humans. PLoS One. 2013;8(3):e58449. doi:10.1371/journal.pone.0058449
41. Dong K, Zhu Y, Deng Q, et al. Salmonella pSLT-encoded effector SpvB promotes RIPK3-dependent necroptosis in intestinal epithelial cells. Cell Death Discovery. 2022;8(1):44. doi:10.1038/s41420-022-00841-9
42. Ganji L, Shirazi MH, Ebrahimi-Daryani N, et al. Carriage of cdtB encoding Campylobacter spp. Salmonella enterica, and Yersinia entercolitica in patients with gastroenteritis and irritable bowel syndrome. Dig Dis Sci. 2022;67(12):5522–5528. doi:10.1007/s10620-022-07468-x
43. Haeusler GM, Curtis N. Non-typhoidal Salmonella in children: microbiology, epidemiology and treatment. Hot Topics Infection Immunity Children IX. 2013;13–26.
44. Liu YY, Wang Y, Walsh TR, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16(2):161–168. doi:10.1016/S1473-3099(15)00424-7
45. Aleksandrowicz A, Carolak E, Dutkiewicz A, Błachut A, Waszczuk W, Grzymajlo K. Better together–Salmonella biofilm-associated antibiotic resistance. Gut Microbes. 2023;15(1):2229937. doi:10.1080/19490976.2023.2229937
46. Cui R, Zhang J, Liu X, et al. Dronedarone enhances the antibacterial activity of polymyxin B and inhibits the quorum sensing system by interacting with LuxS. ACS Infect Dis. 2024;10(3):961–970. doi:10.1021/acsinfecdis.3c00591
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.