")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 21
Altered Gut Microbiota and Plasma Metabolome Profiles Characterize Depression Individuals with Ischemic Stroke: A Comparative Analysis
Authors Cao N, Lv D, Liu Y, Zhang H, Zhang X
Received 8 January 2025
Accepted for publication 16 April 2025
Published 29 April 2025 Volume 2025:21 Pages 973—987
DOI https://doi.org/10.2147/NDT.S513364
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Ning Cao,1 Dongsheng Lv,2 Yanbin Liu,3 Huiru Zhang,4 Xingguang Zhang1
1School of Public Health, Inner Mongolia Medical University, Hohhot, Inner Mongolia, 010110, People’s Republic of China; 2Sleep Medicine Center, Mental Health Center of Inner Mongolia Autonomous Region, Hohhot, 010010, People’s Republic of China; 3Community Rehabilitation and Guidance Division, National Center for Mental Health, Beijing, 100013, People’s Republic of China; 4Cadre Healthcare Department, Mental Health Center of Inner Mongolia Autonomous Region, Hohhot, Inner Mongolia, 010010, People’s Republic of China
Correspondence: Huiru Zhang, Cadre Health Section, Mental Health Center of Inner Mongolia Autonomous Region, Hohhot, Inner Mongolia, 010010, People’s Republic of China, Email [email protected] Xingguang Zhang, School of Public Health, Inner Mongolia Medical University, Hohhot, Inner Mongolia, 010110, People’s Republic of China, Email [email protected]
Purpose: Depression has been recognized as a significant risk factor for ischemic stroke (IS). This study aimed to describe gut microbiota differences between depression people with and without IS, thereby establishing the link between gut microbiota and an elevated risk of IS development in people with depression.
People and Methods: This study included 30 hospitalized patients with comorbid depression and IS, and 30 age-/sex-matched patients with depression alone. We used two approaches: (1) genetic analysis techniques (16S rRNA gene sequencing) to map gut microbial ecosystems, and (2) broad-spectrum chemical (nontargeted metabolomics) analysis to detect blood metabolites.
Results: Alpha (α)-diversity and beta (β)-diversity of people with depression, with or without IS, did not show significant differences between the two groups. The IS group showed increased levels of gut bacteria carrying pro-inflammatory molecules, specifically Gram-negative Enterobacteriaceae containing lipopolysaccharide (LPS) components, the Linear discriminant analysis (LDA) value =4.177, P=0.014. Alongside, the IS group reduced populations of beneficial microbes that produce butyric acid important for gut health, such as Acidaminococcaceae (LDA value =4.045, P=0.014), Roseburia (LDA value =3.894, P=0.007), and Fusicatenibacter (LDA value =3.345, P=0.012), compared to the non-IS group. 38 plasma metabolites with significant differences between people with IS and non-IS groups. The abundance of Alloprevotella and Bacteroides massiliensis was correlated with 9 and 4 metabolites, respectively.
Conclusion: This study highlighted that people with depression and IS exhibited distinct alterations in both their gut microbiome and metabolite profiles, in contrast to people with depression without IS. These findings may guide future interventions targeting gut microbiota to identify IS in depression people.
Plain Language Summary: Depression has been identified as a significant risk factor for ischemic stroke (IS); this study aimed to describe gut microbiota differences between depression people with and without IS, thereby establishing the link between gut microbiota and an elevated risk of IS development in people with depression. This study included 30 hospitalized patients with comorbid depression and IS, and 30 age-/sex-matched patients with depression alone. We used two approaches: (1) genetic analysis techniques (16S rRNA gene sequencing) to map gut microbial ecosystems, and (2) broad-spectrum chemical (nontargeted metabolomics) analysis to detect blood metabolites. The results showed that people with depression and IS exhibited a higher prevalence of bacteria with lipopolysaccharide (LPS) structures and reduced presence of butyrate-producing bacteria. This study highlighted that people with depression and IS exhibited distinct alterations in both their gut microbiome and metabolite profiles, in contrast to people with depression without IS.
Keywords: ischemic stroke, depression, gut microbiota, metabolome
Introduction
Ischemic stroke (IS) is a critical condition that imposes substantial burden on both individuals and society, ranking among the primary causes of disability and mortality worldwide.1,2 The estimated global cost of stroke is over US$721 billion.3 In 2019, the global incidence of IS reached 77.63 million cases, resulting 3.23 million deaths and a loss of 63.48 million disability adjusted life years (DALYs), with the bulk of the global stroke burden (86.0% of deaths and 89.0% of DALYs) residing in lower-income and lower-middle-income countries.3,4 In China, IS remains the leading cause of death and disability among adults.5,6
Depression is a prevalent and severe mental illness characterized by persistent feelings of sadness and impaired cognitive functioning, often accompanied by reduced motivation and decelerated thought processes.7 In 2020, depression accounted for 49.4 million DALYs globally, making it one of the leading contributor to the global disease burden.8 Depression poses a considerable public health challenge, resulting in diminished person functionality and elevated mortality rates.9 The 12-month global incidence of depression is estimated at approximately 6%, with over 20% of individuals experiencing at least one depressive episode during their lifetime.10 Moreover, individuals diagnosed with depression exhibit a mortality rate nearly double that of the general population.11
Depression is closely related to IS, but most previous studies have focused on post-stroke depression. However, extensive cohort studies, meta-analyses of cohort studies, and Mendelian randomization studies have consistently identified depression as a substantial risk factor for IS.12–17 Given these statistics, reducing the risk of IS in individuals with depression is crucial for the primary prevention of IS and alleviating the overall disease burden.
Although a significant epidemiological association exists between depression and IS, the precise mechanisms underlying the coexistence of these conditions remain poorly understood. Next-generation sequencing technology is expected to play a crucial role in elucidating the mechanisms underlying the increased risk of IS in people with depression, particularly with respect to the gut microbiota. Often referred to as the “second genome” of humans, the gut microbiota plays a crucial role in regulating physiological processes across various bodily systems through diverse pathways, thereby contributing to the development of several diseases. Conversely, systemic disorders may disrupt the gut microbiota balance through various mechanisms. Recent studies have separately linked gut microbiota to depression and IS pathogenesis, yet no work has directly compared these microbial profiles in comorbid cases. Numerous clinical studies and meta-analyses have reported significant differences in the gut microbiota composition and metabolites between depression people and healthy controls.18–20 Similarly, the gut microbiota has been implicated in the pathogenesis of IS by producing specific microbiota-derived metabolites such as lipopolysaccharide (LPS), short-chain fatty acids (SCFAs), trimethylamine-N-oxide (TMAO), and phenylacetylglutamine (PAGln).21–24 These findings suggest that gut microbiota may play a pivotal role in the development of IS in individuals with depression, although the exact mechanisms remain to be fully characterized.
Based on this, we hypothesized that depression individuals with IS would exhibit distinct gut microbiota and metabolomic profiles compared to those without IS. This study aimed to describing gut microbiota differences between depression people with and without IS, to establish the link between gut microbiota and an elevated risk of IS development in patients with depression. Furthermore, metabolomics offers a novel approach to examine host-microbiota interactions, potentially elucidating the impact of gut microbiota on host metabolic alterations. To this end, we sequenced the gut microbiota and analyzed the plasma metabolome of depression people with and without IS. This is the first study to directly compare gut microbiota and plasma metabolomes between depression people with and without IS. Unlike prior work focusing on post-stroke depression, our findings uniquely identify microbial and metabolic signatures that may precede IS development in high-risk populations, offering a new possibly preventive point of view to IS.
Materials and Methods
Study Subjects and Sample Collection Procedures
This study focused on selected individuals with depression at the Inner Mongolia Autonomous Region Mental Health Center in China between December 2022 and May 2023. The study included hospitalized patients with depression in the IS group who met the following criteria: (i) aged 18 years or older; (ii) previously diagnosed with depression according to International Classification of Diseases, 10th version (ICD-10), the ICD-10 codes were F32 and F33, and the history of depression was 5 years or more, and (iii) had a history of hospitalization for IS within the past 3 years according to the person’s past medical history. And the diagnosis of IS was in accordance with the “Chinese guidelines for the diagnosis and treatment of acute ischemic stroke (2018)”. The exclusion criteria were the presence of other mental disorders, as defined by the ICD-10, treatment-resistant depression, severe suicidal and self-injurious tendencies, schizophrenia, bipolar disorder, neurodegenerative diseases, chronic inflammatory disorders, thyroid disease, or cancer. Other exclusion factors included pregnancy or breastfeeding, recent antibiotics (within the month prior to sampling), significant dietary changes, or specific dietary restrictions (eg, food allergy, food intolerance, and vegetarianism).
Using a matched-pair design, each depression individual combined with IS was paired with another depression individual without a history of concurrent IS. The matching criteria included sex, antidepressant medication use, and an age difference of no more than two years. The individuals without IS were assigned to the non-IS group. The final study sample comprised 60 participants: 30 participants diagnosed with depression and IS, and 30 participants diagnosed with depression but without IS.
The current depression severity was assessed using the 24-item Hamilton Depression Rating Scale (HAMD-24). Individuals were also interviewed regarding their smoking habits, and data on their blood pressure (BP), blood glucose (GLU), blood lipid profiles, and past medication history were recorded. Fecal and blood samples were collected from all participants within 48 h of hospital admission. For fecal collection, participants deposited their samples into clean, dry containers specifically designed for this purpose. A sterile fecal sampler with a 15 mL capacity was used to collect samples from the middle portion of the stool. A minimum of 2 g of fecal material was retained in a tube, and the samples were stored at −80°C until analysis. Whole blood was collected by certified nurses and allowed to coagulate at room temperature for 15 minutes. Blood was then centrifuged at 5000 rpm for 10 min. The resultant plasma was carefully separated and stored at −80°C until further use.
16S rRNA Gene Sequencing of the Gut Microbiome
Fecal samples were thawed prior to DNA extraction using cetyltrimethylammonium bromide (CTAB) or sodium dodecyl sulphonate (SDS). Agarose gel electrophoresis was used to assess the DNA concentration and purity, followed by dilution to 1 ng/µL using sterile water. The V4 region of the 16S rRNA gene was amplified by PCR using specific primers including barcodes (515F: GTGCCAGCMGCCGCGGTAA and 806R: GGACTACHVGGGTWTCTAAT). Amplification was performed using the Phusion® High-Fidelity PCR Master Mix with GC Buffer (New England Biolabs, USA). The PCR products were purified using the Qiagen Gel Extraction Kit (Qiagen, Germany). Sequencing libraries were constructed according to the manufacturer’s guidelines using the TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA). Library quality was evaluated using a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, USA) and qPCR. Sequencing was performed using the NovaSeq 6000 system (Illumina, USA).
Plasma Metabolomic
Frozen plasma samples (−80°C) were thawed on ice, vortexed (10 s), and mixed with 300 μL extraction solution (ACN: Methanol=1:4 v/v) containing internal standards. After 3 min vortexing, samples were centrifuged (12,000 rpm, 10 min, 4°C). The supernatant (200 μL) underwent secondary freezing (−20°C, 30 min) and centrifugation (12,000 rpm, 3 min, 4°C), with 180 μL ultimately collected for LC-MS.
All samples were for two LC/MS methods. Analysis was performed on Waters ACQUITY Premier HSS T3 column (1.8 μm, 2.1×100 mm) under two ion modes. Positive mode: 0.1% formic acid/water (A) vs 0.1% formic acid/acetonitrile (B) Gradient: 5–20% B (0–2 min), 20–60% B (2–5 min), 60–99% B (5–6 min), 99% B (6–7.5 min), 5% B (7.6–10 min). Another aliquot was using negative ion conditions and was the same as the elution gradient of positive mode.
The data acquisition was operated using the information-dependent acquisition (IDA) mode using Analyst TF 1.7.1 Software (Sciex, Concord, ON, Canada). The source parameters were set as follows: (1) Ion source: GAS1/GAS2 50 psi, CUR 25 psi, TEM 550°C; (2) Voltages: DP ±60 V, ISVF ±5000/4000 V (positive/negative) (3) TOF MS: 50–1000 Da, 200 ms accumulation (4) Product ion scan: 25–1000 Da, 40 ms accumulation, CE ±30 V.
Statistical Analysis
All statistical analyses were performed using R software version 4.2.0. Continuous variables were presented as mean ± standard deviation (SD), and statistical comparisons between groups were made using one-way analysis of variance (ANOVA) or t-test. Categorical data were analyzed using the chi-square test. Spearman correlation tests were used to assess the correlation between the gut microbiome and the metabolome. Statistical significance was set at P<0.05.
Gut Microbiome Analysis
After 16S rRNA Gene Sequencing of the gut microbiome, the raw data were processed to generate effective tags. The UPARSE method (UPARSE v7.0.1001, http://www.drive5.com/uparse/) was applied to optical transform unit (OTU) clustering analysis of sequences with a 97% similarity threshold. The resulting representative OTU sequences were aligned with a microbial taxonomy database to identify the representative species.25 OTU analysis results were used to determine the community makeup of both sample groups across all taxonomic levels (phylum, class, order, family, genus, and species).
Mothur software (v1.48) was used to calculate the Shannon diversity index, Simpson’s index of diversity, Chao1 estimator, and abundance-based coverage estimator (ACE), providing insights into species abundance and diversity within each sample. The Bray‒Curtis method was employed for principal coordinate analysis (PCoA) to evaluate variations in species diversity, community composition, and structure across samples. Linear discriminant analysis effect size (LEfSe) analysis was conducted to identify bacterial markers with high-dimensional characteristics among the different groups. Finally, the functional composition profiles of the 16S rRNA sequences were inferred and mapped to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways using Tax4Fun2.
Plasma Metabolomics Analysis
The initial LC‒MS data file was first converted into the mzML format using ProteoWizard, followed by peak extraction, alignment, and retention time correction using XCMS. The matching criteria were set as follows: m/z error <25 ppm and retention time error <6 seconds between primary (Q1 from Full Scan) and secondary (Q1 from Product ion scan) spectra. Post-correction peaks were identified through a comprehensive approach combining in-house laboratory databases, public databases, predictive libraries, and metDNA methodology.26 Database searches employed mass tolerances of 25 ppm for precursor ions (Q1) and 50 ppm for MS2 spectra, with a retention time filter of ±60 seconds. The MS2 scoring system comprised three weighted components: fragment score (10%), forward search score (30%), and reverse search score (60%), requiring a minimum composite score of 0.3.
Peaks with >50% missing values across sample groups were filtered out. Missing values were imputed using either 1/5 of the minimum value (for samples with >50% blanks) or K-nearest neighbors (KNN) imputation (for samples with <50% blanks).27 Peak areas were subsequently normalized using Support Vector Regression (SVR).28 Post-correction peaks were identified through a comprehensive approach combining in-house laboratory databases, public databases, predictive libraries, and metDNA methodology.26 Identified metabolites were selected based on a comprehensive score ≥0.5 and quality control (QC) samples demonstrating coefficient of variation (CV) <0.3. Positive and negative mode results were merged, retaining compounds with the highest identification confidence level and lowest CV values.
Principal component analysis (PCA) and orthogonal partial least-squares discriminant analysis (OPLS-DA) were used for data analysis. Differentially abundant metabolites were identified based on the following criteria: variable importance in projection (VIP) score ≥ 1, P-value ˂ 0.05, and fold change ≥ 2 or ≤ 0.5. LDA effect size plots and volcano plots were generated to visualize metabolite differences across groups. KEGG pathway was used to elucidate the metabolic pathways enriched with differentially abundant metabolites.
Results
Clinical Characteristics of the Participants
This study encompassed a comprehensive analysis of the gut microbiota and plasma metabolome in 60 samples derived from depression individuals with and without IS. No statistically significant differences were observed between the groups in demographic parameters, including HAMD-24 score, blood pressure, blood glucose level, or blood lipid profiles. Demographic parameters showed no differences between groups (Table 1). These well-matched samples facilitated the identification of differences in gut microbiota and plasma metabolome profiles between depression individuals with and without IS.
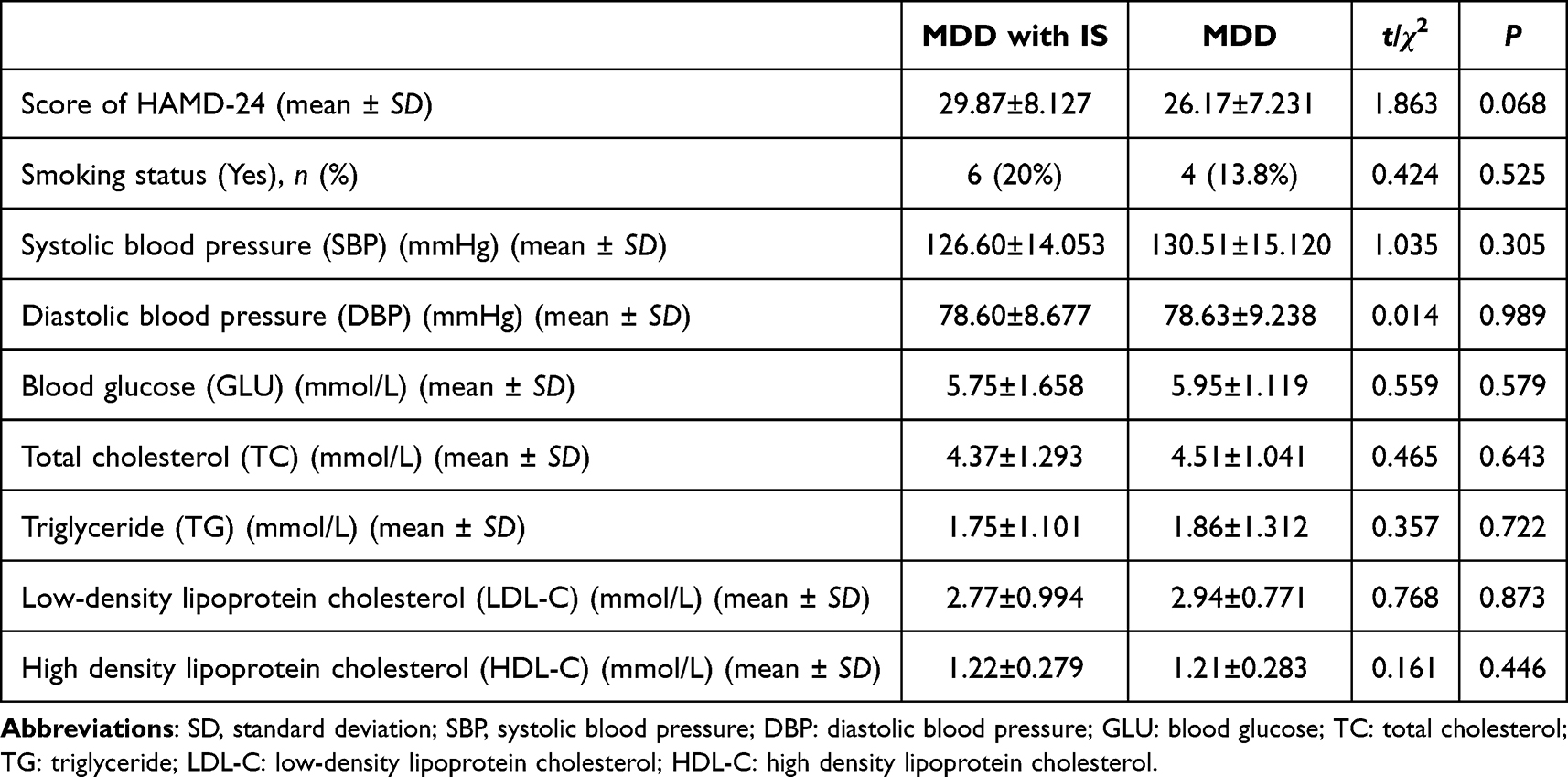 |
Table 1 Clinical Characteristics of the Participants |
There was No Difference in the Diversity of the IS People Compared to the Non-IS People
Gut microbiomes of individuals with depression, with or without IS, were assessed using 16S rRNA gene sequencing. Alpha (α)-diversity, which quantifies both species richness and their relative abundance, was initially assessed. The results suggested a trend toward reduced gut microbiota diversity in people with depression and IS, as indicated by comparatively lower Shannon diversity indices (Figure 1A), Simpson’s indices (Figure 1B), Chao1 (Figure 1C), and ACE indices (Figure 1D). However, none of these differences was statistically significant. Additionally, PCoA was performed to evaluate beta (β)-diversity, but no clear segregation was observed between the two groups. Furthermore, the Adonis test based on Bray-Curtis dissimilarity did not identify significant bacterial differences between the groups (Figure 1E).
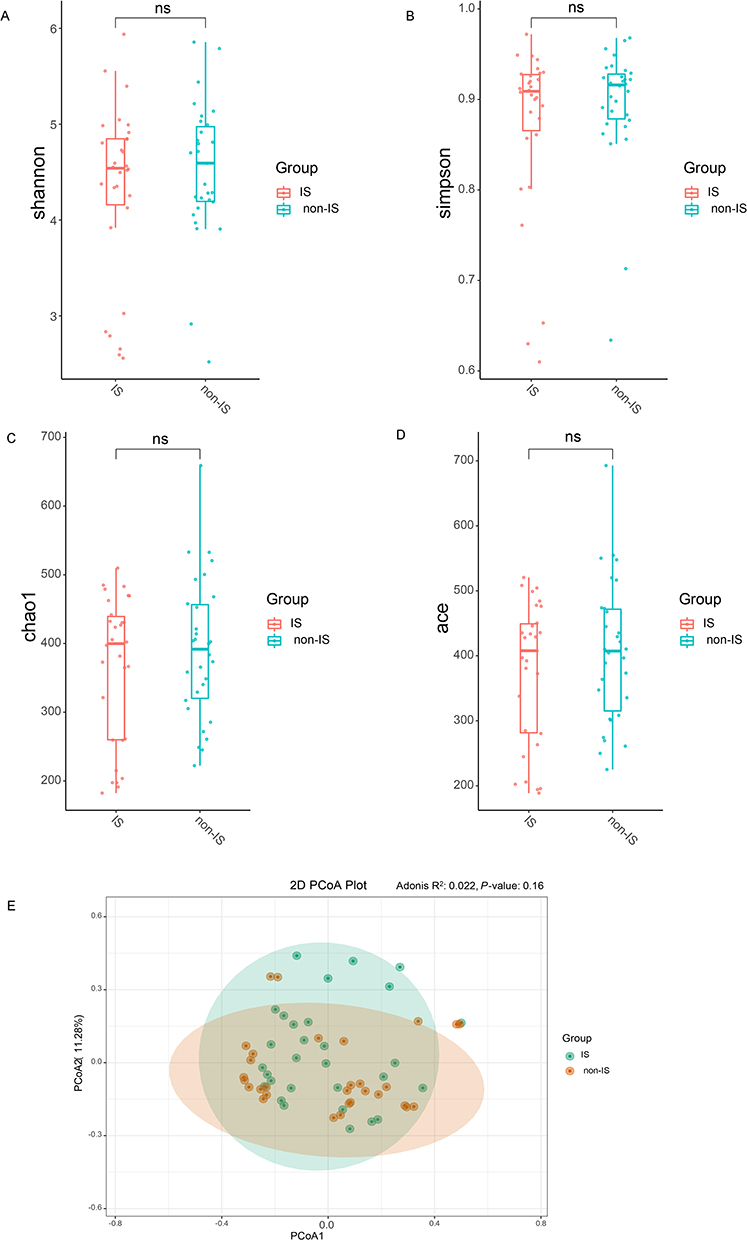 |
Figure 1 Gut microbiome characteristics in people with IS versus non-IS people. The Shannon diversity index (A), Simpson’s index of diversity (B), Chao1 index (C), and ACE index (D) are presented using boxplots for the IS group (red) and the non-IS group (blue). Boxplots represent the median, 25th, and 75th quartiles, as well as the minimum and maximum values. The study consisted of two groups: the IS group, which included 30 participants, and the non-IS group, which included 30 participants. The statistical significance of the alpha diversity was assessed using the Wilcoxon rank-sum test. (E) The bacterial signatures did not show any notable differences between the two groups (Bray-Curtis distance, Adonis test, P = 0.161). Abbreviation: ns, no significance. |
Taxonomic and Functional Characterization of Gut Microbiota in IS and Non-IS People
The gut microbiota composition was analyzed at six taxonomic levels (phylum, class, order, family, genus, and species) to investigate potential differences in taxonomic composition between IS and non-IS people. The LEfSe technique was used to identify taxa that served as biomarkers for each group. Bar plots illustrating relative abundances at the phylum level demonstrate clear differences in the gut microbiota between IS and non-IS people. Firmicutes and Bacteroidetes were the most prevalent phyla in both groups, with relative abundances > 10%. Although Proteobacteria was not the dominant phylum in either group, it exhibited a significantly higher relative abundance in the IS group than in the non-IS group (Figure 2A). At the order level, the IS group showed a significantly greater abundance of Enterobacterales than did the non-IS group (LDA value=4.252, P=0.019). Comparable findings were observed at the family level, with Enterobacteriaceae showing greater abundance in the non-IS group (LDA value=4.177, P=0.014). Enterobacteriaceae, which are gram-negative bacteria possessing LPS structures, were more prevalent in the gut microbiota of the IS group. The distribution of SCFA-producing bacteria in the gut microbiota of people differed markedly between those with and non-IS people. Alloprevotella (LDA value = 2.964, P = 0.00003), a propionic acid-producing bacterium, was significantly more abundant in the IS group. Additionally, the subspecies Prevotella and Prevotella sp. Marseille P2931 (LDA value =2.573, P=0.006) and Prevotella stercorea (LDA value =2.344, P=0.014), were more abundant in the IS group. In contrast, the IS group exhibited was a lower abundance of butyric acid-producing bacteria, such as Acidaminococcaceae (LDA value =4.045, P=0.014), Roseburia (LDA value =3.894, P=0.007, and Fusicatenibacter (LDA value =3.345, P=0.012), compared to the non-IS group. At the species level, the IS group exhibited a lower prevalence of Bacteroides massiliensis (LDA value =4.202, P=0.00015) and Bacteroides finegoldii (LDA value =3.164, P=0.02) than the non-IS group (Figure 2B and C).
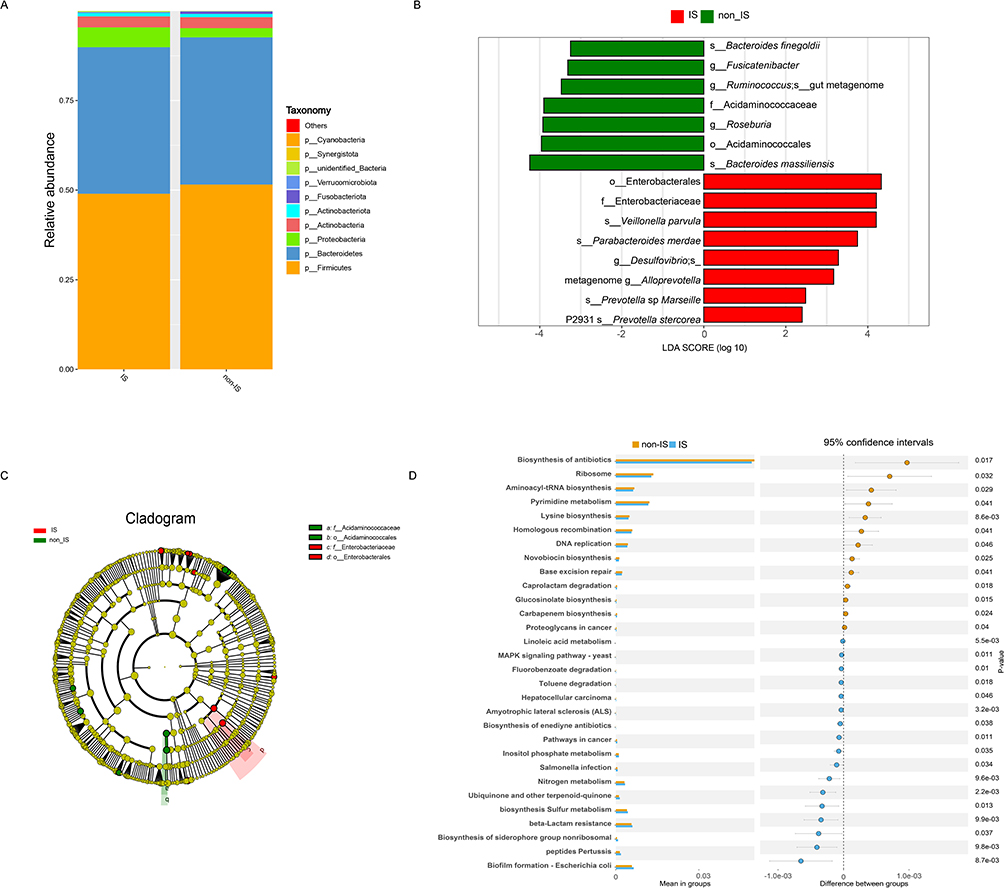 |
Figure 2 Differences in the taxonomic composition between people with and non-IS people. Stacked bar plot indicating the average relative abundance of phyla in IS and non-IS people (A). LDA value coupled with effect size measurements in the IS and non-IS people. Enriched taxa in the IS (red) and non-IS (green) groups are displayed using LDA values. Only taxa with LDA value scores above the threshold of 2.0 are shown (B). Cladogram representation of gut microbiota in IS people versus non-IS people. By 16S rRNA sequencing. The enriched taxa in the IS (red) and non-IS (green) groups are indicated. The brightness of each dot is correlated with its LDA value effect size (C). The functional potential of microbial communities of IS and non-IS people via Tax4Fun2. Statistical analysis of the OTU-based relative abundance of functional annotations in the IS and non-IS groups was performed using Student’s t-test (D). |
To predict the functional capacity of microbial communities in IS and non-IS people, we used Tax4Fun2. This approach involved analyzing the 16S rRNA gene content, which was specifically designed to assess the functional potential of microbial communities identified through 16S rRNA sequencing. The pathways associated with IS primarily involve the metabolism of terpenoids and polyketides, such as the biosynthesis of siderophore group nonribosomal peptides. Additionally, these pathways are linked to energy metabolism, including sulfur and nitrogen metabolisms. Furthermore, genes related to the metabolism of cofactors and vitamins, such as ubiquinone and other terpenoid quinone biosynthesis products, were identified. Carbohydrate metabolism, particularly inositol phosphate metabolism, is another important aspect of this pathway. Conversely, the pathways linked to non-IS participants were predominantly involved in translation (ribosome function and aminoacyl-tRNA biosynthesis), nucleotide metabolism (pyrimidine metabolism), amino acid metabolism (lysine biosynthesis), and processes related to replication and repair (homologous recombination and DNA replication). It is important to note that these functional predictions are speculative, based on the analysis of 16S rRNA composition, and do not provide definitive information on functional capabilities or transcriptional activity (Figure 2D).
Metabolome Differences in IS and Non-IS People
LC–MS–based metabolomic analysis was conducted to assess differences in plasma metabolic profiles between depression individuals with and without IS. An OPLS-DA model was employed to examine the metabolite disparities between the two groups. OPLS-DA results revealed a clear separation between the IS and non-IS groups into distinct clusters (Figure 3A). The goodness-of-fit and predictive ability values (R2X = 0.153, R2Y = 0.988, Q2 = 0.753, P < 0.05) indicated a robust fit and effective predictive power (Figure 3B).
 |
Figure 3 Non-target metabolomes of IS and non-IS people. OPLS-DA was performed to determine the metabolite profiles (IS, n = 30; non-IS, n = 30). Plasma metabolomics OPLS-DA score plot of IS (red) vs non-IS (green) groups. Each dot denotes an individual subject (A). Permutation test for the OPLS-DA model: 999 permutations led to intercepts of R2X = 0.153, R2Y = 0.988, Q2 = 0.753, P < 0.05, implying an acceptable model minus overfitting (B). Volcano plot showing differentially abundant metabolites between the IS and non-IS people. Each dot denotes a metabolite (C). Each significantly selected subpathway is denoted by a circle and described by three parameters. The circle size shows the number of metabolites selected in the subpathway (see the legend in gray to the right of the plot for relative sizes). Circles from purple to red denote selected subpathway significance levels based on −log10 (p-value) (see the legend to the right of the plot for the relative color gradient). Pathways were markedly enriched if P < 0.05, which was comparable to a −log10 (P-value) > 1.3. The circular position along the rich factor axis shows the abundance of selected metabolites from the subpathway against all subpathway metabolites (D). |
OPLS-DA data were further analyzed using VIP as a criterion to identify metabolites with significant intergroup differences. Metabolites were considered differentially abundant if they met the following criteria: VIP ≥1.0, P < 0.05, or fold change ≥ 2 or ≤ 0.5. This analysis revealed 38 metabolites with significant differences between IS and non-IS groups. Specifically,17 metabolites showed significantly higher levels in the IS group, whereas 21 exhibited significantly lower levels than those in the non-IS group (Figure 3C). The differentially abundant metabolites were primarily comprised of amino acids and their derivatives, benzene and its substituted compounds, glycerophospholipids, and heterocyclic compounds.
To explore the metabolic pathways associated with these differentially abundant metabolites, they were compared using KEGG database. The analysis revealed that these metabolites are involved in various metabolic pathways, including amino acid metabolism (specifically tryptophan), lipid metabolism (steroid hormone and sphingolipid), carbohydrate metabolism (ascorbate and aldarate, glycolysis/gluconeogenesis), signal transduction (sphingolipid signaling pathway), and processes related to cell growth and death (necroptosis), as well as pathways linked to the endocrine and nervous systems (eg, adipocytokine signaling, neurotrophin signaling). Notably, tryptophan metabolism and steroid hormone biosynthesis pathways were enriched for two metabolites, whereas the other pathways were enriched with one metabolite each. Glucobrassicin and N-acetylpentetreotide were identified in the tryptophan metabolic pathway; glucobrassicin levels decreased, and N-acetylpentetreotide levels increased in the IS group. The two metabolites enriched in the tryptophan metabolic pathway were 21-Hydroxypregn-4-ene-3,11–20-trione and estradiol-3-sulfate; estradiol-3-sulfate levels decreased and 21-Hydroxypregn-4-ene-3,11–20-trione levels increased in the IS group. These findings suggest that disturbances in multiple metabolic pathways contribute to IS progression (Figure 3D).
Correlation Analysis Between the Gut Microbiota and Metabolites
Spearman correlation coefficient was calculated to examine the functional relationships between alterations in the gut microbiome and plasma metabolites, based on the differences observed between individuals with IS and those without IS. Correlations were considered statistically significant when the absolute value of r was ≥ 0.5 and the P-value was < 0.05. The analysis revealed negative correlations between the levels of several metabolites, including glucobrassicin, daidzein, 5-hydroxypseudobaptigenin, k-strophanthoside, arbutin 6-phosphate, 1-pentadecanoyl-2-lignoceroyl-sn-glycerol, and 1-stearoyl-2-myristoyl-sn-glycero-3-phosphocholine and the abundance of Alloprevotella. The corresponding r values were −0.575, −0.554, −0.553, −0.539, −0.626, −0.508, −0.573, and −0.501, with all P-values being less than 0.001. Additionally, the level of 7(14)-farnesene-9,12-diol was positively correlated with Alloprevotella abundance (r=0.515, P<0.001; Figure 4A). Furthermore, glucobrassicin, daidzein, 5-hydroxypseudobaptigenin, and dinoseb were positively correlated with Bacteroides massiliensis, with r values of 0.635, 0.602, 0.511, and 0.539, respectively, and P-values all less than 0.001 (Figure 4B). These findings indicate that alterations in the gut microbiome may interact with metabolites and potentially influence the development of ischemic stroke.
 |
Figure 4 Correlation analysis between the gut microbiota and metabolites. Chord plots of the correlation analysis between Alloprevotella and related metabolites (A) and between Bacteroides massiliensis and related metabolites (B). The breadth of the link represents the relative coefficient between the differential species and the metabolite. Greater correlation is indicated by a wider link. Pink denotes positive correlation, whereas blue denotes negative correlation. |
Discussion
The relationship between depression and IS has been documented in population level studies; however, the underlying mechanisms that explain their co-occurrence remain unclear. To establish the link between gut microbiota and an elevated risk of IS development in individuals with depression, a comprehensive analysis of the gut microbiota and metabolome of people with depression, with and without IS, was conducted. Our findings revealed significant differences in both the gut microbiota community composition and metabolomic profiles between the two groups. Although causality cannot be established in this observational study, this study offers a novel perspective on the relationship between depression and IS, highlighting the potential role of gut microbiota and critical metabolites in this comorbidity.
Previous research has demonstrated that individuals with depression exhibit disrupted gut microbiota features. Although there is significant variability in the methodologies and reports of these studies,29–32 enriched pro-inflammatory bacteria and depletion of anti-inflammatory bacteria have been the most consistent findings.18–20 However, our study showed that there was a more pronounced disruption of gut microbiota in people with depression and IS than in people with depression alone. Analysis of the taxonomic composition disparity between the two groups revealed a higher proportion of Proteobacteria at the phylum level in depression people with IS than in those non-IS people. This increase in Proteobacteria is recognized as an indicator of microbial dysbiosis.33 The observed elevation in Proteobacteria abundance can be attributed to the enrichment of Enterobacteriaceae, which is consistent with a previous study on individuals with IS but without depression during both the acute and recovery phases.22 Enterobacteriaceae, characterized by LPS structures in their outer membrane, which are essential components of the external layer of the outer membrane of Gram-negative bacteria. LPS has the capacity to trigger immune responses and induce inflammation in the host.34–36 When present in high quantities, particularly in the intestines, Enterobacteriaceae coupled with an impaired gut barrier can rapidly induce systemic inflammation throughout the body by releasing substantial amounts of LPS. This inflammatory cascade may contribute to the development of IS, with higher LPS levels also correlating with poorer outcomes in IS people.22,37–39 These results suggest that depression people with IS may have a higher abundance of pro-inflammatory bacteria than depression people without IS. The distribution of SCFA-producing bacteria in the gut microbiota of people differed significantly between IS and non-IS people. IS people exhibited higher levels of propionic acid-producing bacteria, including Alloprevotella, and two Prevotella subspecies (Prevotella_sp_Marseille_P2931 and Prevotella_stercorea). Conversely, butyric acid-producing bacteria, including Acidaminococcaceae, Roseburia, and Fusicatenibacter, were significantly less abundant in IS people. These findings suggest that different SCFAs may play distinct roles in IS progression in individuals with depression. Earlier studies have demonstrated that individuals with acute IS and diabetes exhibit lower levels of butyric acid-producing bacteria.40 Furthermore, individuals without a history of stroke but at high risk also exhibited decreased levels of these bacteria,41 indicating a potential protective role of butyrate-producing bacteria in both the development and prognosis of IS, regardless of the underlying disease. Overall, these findings suggest that the primary characteristics of gut microbiota of depression people with IS include a more increased presence of LPS-producing bacteria and a more reduced abundance of butyrate-producing bacteria compared with non-IS people.
Bacteroides massiliensis is observed at a relatively low abundance in depression individuals with IS. Previous studies on the gut microbiota of people with various cancers, including prostate cancer,42 colorectal cancer,43 and melanoma,44 have demonstrated that the abundance of Bacteroides massiliensis is elevated in people with tumors. In contrast, it has also been observed that individuals with diabetes, as well as those with diabetes-related cardiovascular complications, exhibit significantly lower levels of Bacteroides massiliensis compared to healthy individuals.45 These contrasting findings highlight the complex relationship between Bacteroides massiliensis and various diseases. Further research is needed to elucidate whether Bacteroides massiliensis plays a role in IS development in people with depression.
Metabolome reflects the combined effects of internal physiological processes and external factors. Significant interactions occur between the human gut microbiota and host through substrate metabolism and metabolite exchange.46 Therefore, we conducted an analysis of people using plasma metabolomics. Our findings revealed the substantial impact of the presence or absence of IS on the metabolites of these strains. Notably, two distinct compounds, daidzein and glucobrassicin, were identified and were strongly associated with the presence of two bacterial species, Alloprevotella and Bacteroides massiliensis. Daidzein is a flavonoid compound commonly found in soybeans and soy-based products and primarily exists as a glucoside.47 The gut microbiota, which includes members such as Bacteroidetes, Firmicutes, Enterococcus, Lactobacillus, and Bifidobacterium, contains genes encoding various glycosidase enzymes, such as β-glucosidase. These enzymes break down soy sapogenins and glycosides into free sapogenins and glucose.48 Subsequently, free sapogenins undergo a range of metabolic transformations including dihydroxylation, reduction, pyrone ring cleavage, and demethylation. These processes result in the formation of bioactive molecules, such as equol, which exhibit potent estrogenic activity, or inactive products, such as O-desmethylangolensin.47,49 Equol has shown considerable promise in cardiovascular disease (CVD) prevention, potentially reducing coronary heart disease risk by enhancing anti-atherogenic properties, improving arterial stiffness, and lowering LDL-C levels in overweight individuals.50 Glucobrassicin, a key secondary metabolite found in cruciferous vegetables, is broken down into indole-3-methanol (I3C). Both I3C and its primary in vivo derivative, 3,3′-diindolylmethane (DIM), have demonstrated efficacy as cancer chemopreventive agents in preclinical models and are promising clinical trials. I3C also exhibited notable neuroprotective effects in a rat model of clonidine-induced depression.51 Additionally, I3C has shown significant protective effects against conditions, such as diabetes,52,53 hypertension,54 and cerebral ischemia/reperfusion injury in rats.55 These therapeutic effects are likely due to its antioxidant properties and ability to inhibit the inflammatory response. Although much of the prior research has been based on animal models, the current study revealed that individuals with depression and IS exhibit reduced glucobrassicin levels. These findings suggest that glucobrassicin may provide protective benefits to people with depression complicated by IS. Moreover, an inverse relationship was observed between daidzein and glucobrassicin levels and Alloprevotella, whereas a positive correlation was found in Bacteroides massiliensis. These findings suggest that daidzein and glucobrassicin may influence depression people through their interactions with these two bacterial species. However, further studies are required to elucidate the precise mechanisms involved.
The distinct microbial signatures observed here—particularly the enrichment of LPS-producing Enterobacteriaceae and depletion of butyrate-generating taxa—suggest actionable targets for intervention. For instance, dietary strategies (eg, high-fiber diets to promote butyrate production) or probiotics targeting Roseburia and Fusicatenibacter could be tested in clinical trials to mitigate IS risk in depression patients. Additionally, plasma metabolites like glucobrassicin, which correlated with microbial shifts, may serve as biomarkers for early risk stratification.
This study has several limitations that warrant consideration. First, the observational design of this study cannot establish the causality between gut microbiota alterations and IS risk in depression individuals. Second, the modest sample size (n=60) may limit statistical power to detect subtle microbial or metabolomic differences. While we employed rigorous matching criteria, larger prospective cohorts are needed to validate these findings. Third, the inclusion of IS patients in the recovery phase (rather than acute phase) may not fully capture dynamic microbiota changes during stroke onset. Future studies should track microbial profiles longitudinally from acute to chronic stages. Finally, while 16S rRNA sequencing provides taxonomic insights, its resolution at the species level is constrained. Shotgun metagenomics and mettranscriptomics would better characterize functional pathways and strain-level variations linked to IS pathogenesis.
These findings provide novel insights into the mechanisms that contribute to the occurrence of IS in depression people. However, the observational design and limited sample size necessitate validation in larger, prospective cohorts to confirm the causal relationship between gut microbiota alterations and IS risk. Future studies should explore whether modulating specific taxa (eg, Roseburia) or metabolites (eg, glucobrassicin) could reduce IS incidence in high-risk depression populations. Additionally, clinical trials targeting gut microbiota interventions—such as probiotics to enhance butyrate producers or dietary modifications to suppress LPS-producing bacteria—may provide novel strategies for IS prevention in individuals with depression.
Conclusion
In conclusion, this study revealed that people with depression and IS exhibit distinct alterations in their gut microbiome and metabolite profiles compared with depression people without IS. Specifically, these changes included an increase in the abundance of bacteria containing LPS structures and a decrease in the abundance of butyrate-producing bacteria. Significant shifts in microbial populations and metabolite profiles may play a role in IS development in people with depression. In addition, the strong association of Alloprevotella and Bacteroides Massiliensis with some metabolites in depressed people with IS warrants further exploration. These findings not only advance our understanding of the gut-brain axis in depression-associated IS but also highlight translational opportunities. Future research should prioritize: (1) Mechanistic validation using animal models to test causality; (2) Clinical trials evaluating microbiota-targeted therapies; (3) Multi-omics integration (metagenomics, metabolomics, and host genomics) to unravel personalized microbial risk profiles.
Abbreviations
IS, ischemic stroke; LPS, lipopolysaccharide; DALY, disability-adjusted life years; SCFAs, short-chain fatty acids; HAMD, Hamilton Depression Rating Scale; BP, blood pressure; GLU, blood glucose; OUT, optical transform unit; ACE, abundance-based coverage estimator; PCoA, Principal Coordinate Analysis; LEfSe, linear discriminant analysis effect size; KEGG, Kyoto Encyclopedia of Genes and Genomes; LC‒MS, liquid chromatography‒mass spectrometry; PCA, principal component analysis; OPLS-DA, orthogonal partial least-squares discriminant analysis; VIP, variable importance in projection; LDA, Local Discriminant Analysis; I3C indole-3-methanol.
Data Sharing Statement
The 16S rRNA sequencing data presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1085864. Metabolomics data are not publicly available due to ongoing analyses but can be requested from the corresponding author: Huiru Zhang ([email protected]).
Study Approval Statement:
This study protocol was reviewed and approved by the Ethics Committee of Mental Health Center of Inner Mongolia Autonomous Region, approval number:2022009.
Consent to Participate Statement:
In the presence of clinicians, people’ families were informed of the study and provided written informed consent. All the research in this study was carried out in compliance with the Declaration of Helsinki, ensuring the protection of people’ privacy rights.
Acknowledgments
We thank all the doctors, nurses and clinical scientists who worked in the hospital during the period of person recruitment as well as the people who were involved in this study. We also thank Wuhan MetWare Biotechnology Co., Ltd., for sample detection. This paper has been uploaded to ResearchSquare as a preprint: https://www.researchsquare.com/article/rs-3948912/v1.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Natural Science Foundation of Inner Mongolia Autonomous Region (2024QN08070); the Inner Mongolia Medical University General Program (YKD2024MS003); the National Natural Science Foundation of China (82160639); the Inner Mongolia Autonomous Region Health Science and Technology Program (202202104); the Science and Technology Project of High-level Clinical Specialties Construction in Public Hospitals of Inner Mongolia Autonomous Region Health Commission (2023SGGZ049); and the Science and Technology Program of the Joint Fund for Scientific Research in Public Hospitals (2023GLLH0160).
Disclosure
The authors declare that they have no competing interests in relation to this manuscript.
References
1. Saini V, Guada L, Yavagal DR. Global epidemiology of stroke and access to acute ischemic stroke interventions. Neurology. 2021;97(20 Suppl 2):S6–S16. doi:10.1212/WNL.0000000000012781
2. Thayabaranathan T, Kim J, Cadilhac DA, et al. Global stroke statistics 2022. Int J Stroke off J Int Stroke Soc. 2022;17(9):946–956. doi:10.1177/17474930221123175
3. Feigin VL, Brainin M, Norrving B, et al. World Stroke Organization (WSO): global stroke fact sheet 2022. Int J Stroke off J Int Stroke Soc. 2022;17(1):18–29. doi:10.1177/17474930211065917
4. Feigin VL, Stark BA, Johnson CO, GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the global burden of disease study 2019. Lancet Neurol. 2021;20(10):795–820. doi:10.1016/S1474-4422(21)00252-0
5. Hu SS, Writing Committee of the Report on Cardiovascular Health and Diseases in China. Epidemiology and current management of cardiovascular disease in China. J Geriatr Cardiol JGC. 2024;21(4):387–406. doi:10.26599/1671-5411.2024.04.001
6. Tu WJ, Wang LD, Special Writing Group of China Stroke Surveillance Report. China stroke surveillance report 2021. Mil Med Res. 2023;10(1):33. doi:10.1186/s40779-023-00463-x
7. Anderson E, Crawford CM, Fava M, et al. Depression - understanding, identifying, and diagnosing. N Engl J Med. 2024;390(17):e41. doi:10.1056/NEJMp2310179
8. Santomauro DF, Mantilla Herrera AM, Shadid J, COVID-19 Mental Disorders Collaborators. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet Lond Engl. 2021;398(10312):1700–1712. doi:10.1016/S0140-6736(21)02143-7
9. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Lond Engl. 2018;392(10159):1789–1858. doi:10.1016/S0140-6736(18)32279-7
10. Malhi GS, Mann JJ. Depression. Lancet. 2018;392(10161):2299–2312. doi:10.1016/S0140-6736(18)31948-2
11. Risks of all‐cause and suicide mortality in mental disorders: a meta‐review - Chesney - 2014 - World Psychiatry - Wiley Online Library. Available from: https://onlinelibrary.wiley.com/doi/10.1002/wps.20128.
12. Wium-Andersen MK, Wium-Andersen IK, Prescott EIB, Overvad K, Jørgensen MB, Osler M. An attempt to explain the bidirectional association between ischaemic heart disease, stroke and depression: a cohort and meta-analytic approach. Br J Psychiatry J Ment Sci. 2020;217(2):434–441. doi:10.1192/bjp.2019.130
13. Harshfield EL, Pennells L, Schwartz JE, et al. Association between depressive symptoms and incident cardiovascular diseases. JAMA. 2020;324(23):2396–2405. doi:10.1001/jama.2020.23068
14. Zhou Y, Kivimäki M, Lim CCW, et al. Bidirectional associations between cardiometabolic multimorbidity and depression and mediation of lifestyles: a multicohort study. JACC Asia. 2024;4(9):657–671. doi:10.1016/j.jacasi.2024.06.004
15. Chen F, Lin H, Zhang Y, Zhang Y, Chen L. The mediating role of sleep disturbance in the relationship between depression and cardiovascular disease. Front Psychiatry. 2024;15:1417179. doi:10.3389/fpsyt.2024.1417179
16. Cai D, Xia M, Chen X, et al. Heartache and Heartbreak: an Observational and Mendelian Randomization Study. Glob Heart. 2024;19(1):19. doi:10.5334/gh.1302
17. Wang M, Su W, Chen H, Li H. Depressive symptoms and risk of incident cardiometabolic multimorbidity in community-dwelling older adults: the China health and retirement longitudinal study. J Affect Disord. 2023;335:75–82. doi:10.1016/j.jad.2023.04.048
18. Liu L, Wang H, Chen X, Zhang Y, Zhang H, Xie P. Gut microbiota and its metabolites in depression: from pathogenesis to treatment. eBioMedicine. 2023;90:104527. doi:10.1016/j.ebiom.2023.104527
19. Liu P, Liu Z, Wang J, et al. Immunoregulatory role of the gut microbiota in inflammatory depression. Nat Commun. 2024;15(1):3003. doi:10.1038/s41467-024-47273-w
20. Simpson CA, Diaz-Arteche C, Eliby D, Schwartz OS, Simmons JG, Cowan CSM. The gut microbiota in anxiety and depression – a systematic review. Clin Psychol Rev. 2021;83:101943. doi:10.1016/j.cpr.2020.101943
21. Nemet I, Saha PP, Gupta N, et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell. 2020;180(5):862–877.e22. doi:10.1016/j.cell.2020.02.016
22. Xu K, Gao X, Xia G, et al. Rapid gut dysbiosis induced by stroke exacerbates brain infarction in turn. Gut. 2021;70(8):1486–1494. doi:10.1136/gutjnl-2020-323263
23. Chen R, Xu Y, Wu P, et al. Transplantation of fecal microbiota rich in short chain fatty acids and butyric acid treat cerebral ischemic stroke by regulating gut microbiota. Pharmacol Res. 2019;148:104403. doi:10.1016/j.phrs.2019.104403
24. Peh A, O’Donnell JA, Broughton BRS, Marques FZ. Gut microbiota and their metabolites in stroke: a double-edged sword. Stroke. 2022;53(5):1788–1801. doi:10.1161/STROKEAHA.121.036800
25. Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590–D596. doi:10.1093/nar/gks1219
26. Shen X, Wang R, Xiong X, et al. Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat Commun. 2019;10(1):1516. doi:10.1038/s41467-019-09550-x
27. Kokla M, Virtanen J, Kolehmainen M, Paananen J, Hanhineva K. Random forest-based imputation outperforms other methods for imputing LC-MS metabolomics data: a comparative study. BMC Bioinf. 2019;20(1):492. doi:10.1186/s12859-019-3110-0
28. Pang Z, Zhou G, Ewald J, et al. Using metaboanalyst 5.0 for LC-HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc. 2022;17(8):1735–1761. doi:10.1038/s41596-022-00710-w
29. Yang J, Zheng P, Li Y, et al. Landscapes of bacterial and metabolic signatures and their interaction in major depressive disorders. Sci Adv. 2020;6(49):eaba8555. doi:10.1126/sciadv.aba8555
30. Liu RT, Rowan-Nash AD, Sheehan AE, et al. Reductions in anti-inflammatory gut bacteria are associated with depression in a sample of young adults. Brain Behav Immun. 2020;88:308–324. doi:10.1016/j.bbi.2020.03.026
31. Lai WT, Deng WF, Xu SX, et al. Shotgun metagenomics reveals both taxonomic and tryptophan pathway differences of gut microbiota in major depressive disorder patients. Psychol Med. 2021;51(1):90–101. doi:10.1017/S0033291719003027
32. McGuinness AJ, Davis JA, Dawson SL, et al. A systematic review of gut microbiota composition in observational studies of major depressive disorder, bipolar disorder and schizophrenia. mol Psychiatry. 2022;27(4):1920–1935. doi:10.1038/s41380-022-01456-3
33. Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33(9):496–503. doi:10.1016/j.tibtech.2015.06.011
34. Gorman A, Golovanov AP. Lipopolysaccharide structure and the phenomenon of low endotoxin recovery. Eur J Pharm Biopharm Off J Arbeitsgemeinschaft Pharm Verfahrenstechnik EV. 2022;180:289–307. doi:10.1016/j.ejpb.2022.10.006
35. Garcia-Vello P, Di Lorenzo F, Zucchetta D, Zamyatina A, De Castro C, Molinaro A. Lipopolysaccharide lipid A: a promising molecule for new immunity-based therapies and antibiotics. Pharmacol Ther. 2022;230:107970. doi:10.1016/j.pharmthera.2021.107970
36. Di Lorenzo F, De Castro C, Silipo A, Molinaro A. Lipopolysaccharide structures of gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol Rev. 2019;43(3):257–272. doi:10.1093/femsre/fuz002
37. Klimiec E, Pasinska P, Kowalska K, Pera J, Slowik A, Dziedzic T. The association between plasma endotoxin, endotoxin pathway proteins and outcome after ischemic stroke. Atherosclerosis. 2018;269:138–143. doi:10.1016/j.atherosclerosis.2017.12.034
38. Wei YH, Bi RT, Qiu YM, et al. The gastrointestinal-brain-microbiota axis: a promising therapeutic target for ischemic stroke. Front Immunol. 2023;14:1141387. doi:10.3389/fimmu.2023.1141387
39. Xu H, Xu Z, Long S, et al. The role of the gut microbiome and its metabolites in cerebrovascular diseases. Front Microbiol. 2023;14:1097148. doi:10.3389/fmicb.2023.1097148
40. Wang H, Song W, Wu Q, et al. Fecal transplantation from db/db mice treated with sodium butyrate attenuates ischemic stroke injury. Microbiol Spectr. 2021;9:e00042–21. doi:10.1128/Spectrum.00042-21
41. Zeng X, Gao X, Peng Y, et al. Higher risk of stroke is correlated with increased opportunistic pathogen load and reduced levels of butyrate-producing bacteria in the gut. Front Cell Infect Microbiol. 2019;9:4. doi:10.3389/fcimb.2019.00004
42. Garbas K, Zapała P, Zapała Ł, Radziszewski P. the role of microbial factors in prostate cancer development—an up-to-date review. J Clin Med. 2021;10(20):4772. doi:10.3390/jcm10204772
43. Hasan R, Bose S, Roy R, et al. Tumor tissue-specific bacterial biomarker panel for colorectal cancer: bacteroides massiliensis, alistipes species, alistipes onderdonkii, bifidobacterium pseudocatenulatum, Corynebacterium appendicis. Arch Microbiol. 2022;204(6):348. doi:10.1007/s00203-022-02954-2
44. Peters BA, Wilson M, Moran U, et al. Relating the gut metagenome and metatranscriptome to immunotherapy responses in melanoma patients. Genome Med. 2019;11(1):61. doi:10.1186/s13073-019-0672-4
45. Chen X, Xue Y, Song X, Zhu B. Gut microbiota in diabetic patients and diabetic patients with cardiovascular complications. Acta Microbiol Sin. 2019;59(9):1660–1673. doi:10.13343/j.cnki.wsxb.20190082
46. Gong X, Liu Y, Liu X, et al. Disturbance of gut bacteria and metabolites are associated with disease severity and predict outcome of NMDAR encephalitis: a prospective case–control study. Front Immunol. 2022;12:791780. doi:10.3389/fimmu.2021.791780
47. Laddha AP, Kulkarni YA. Pharmacokinetics, pharmacodynamics, toxicity, and formulations of daidzein: an important isoflavone. Phytother Res. 2023;37(6):2578–2604. doi:10.1002/ptr.7852
48. Xu J, Chen HB, Li SL. Understanding the molecular mechanisms of the interplay between herbal medicines and gut microbiota. Med Res Rev. 2017;37(5):1140–1185. doi:10.1002/med.21431
49. Mayo B, Vázquez L, Flórez AB. Equol: a bacterial metabolite from the daidzein isoflavone and its presumed beneficial health effects. Nutrients. 2019;11(9):2231. doi:10.3390/nu11092231
50. Usui T, Tochiya M, Sasaki Y, et al. Effects of natural S-equol supplements on overweight or obesity and metabolic syndrome in the Japanese, based on sex and equol status. Clin Endocrinol. 2013;78(3):365–372. doi:10.1111/j.1365-2265.2012.04400.x
51. El-Naga RN, Ahmed HI, Abd Al Haleem EN. Effects of indole-3-carbinol on clonidine-induced neurotoxicity in rats: impact on oxidative stress, inflammation, apoptosis and monoamine levels. NeuroToxicology. 2014;44:48–57. doi:10.1016/j.neuro.2014.05.004
52. Poornima J, Mirunalini S. Regulation of carbohydrate metabolism by indole-3-carbinol and its metabolite 3,3’-diindolylmethane in high-fat diet-induced C57BL/6J mice. mol Cell Biochem. 2014;385(1–2):7–15. doi:10.1007/s11010-013-1808-2
53. Jayakumar P, Pugalendi KV, Sankaran M. Attenuation of hyperglycemia-mediated oxidative stress by indole-3-carbinol and its metabolite 3, 3’- diindolylmethane in C57BL/6J mice. J Physiol Biochem. 2014;70(2):525–534. doi:10.1007/s13105-014-0332-5
54. Pedras MSC, Nycholat CM, Montaut S, Xu Y, Khan AQ. Chemical defenses of crucifers: elicitation and metabolism of phytoalexins and indole-3-acetonitrile in brown mustard and turnip. Phytochemistry. 2002;59(6):611–625. doi:10.1016/s0031-9422(02)00026-2
55. Chichai AS, Popova TN, Kryl’skii ED, Oleinik SA, Razuvaev GA. Indole-3-carbinol mitigates oxidative stress and inhibits inflammation in rat cerebral ischemia/reperfusion model. Biochimie. 2023;213:1–11. doi:10.1016/j.biochi.2023.04.018
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Novel Insight into the Modulatory Effect of Traditional Chinese Medicine on Cerebral Ischemia-Reperfusion Injury by Targeting Gut Microbiota: A Review
Ren Y, Chen G, Hong Y, Wang Q, Lan B, Huang Z
Drug Design, Development and Therapy 2025, 19:185-200
Published Date: 10 January 2025