")
Back to Journals » Cancer Management and Research » Volume 17
ArfGAP with the SH3 Domain, Ankyrin Repeat and PH Domain 1 Inversely Regulates Programmed Death-Ligand 1 Through Negative Feedback of Phosphorylated Epithelial Growth Factor Receptor and Activation of Nuclear Factor-Kappa B in Non-Small Cell Lung Cancer
Authors Chiba N, Menju T , Shimazu Y, Toyazaki T, Sumitomo R, Miyamoto H, Tamari S, Nishikawa S , Date H
Received 31 August 2024
Accepted for publication 7 January 2025
Published 20 January 2025 Volume 2025:17 Pages 91—102
DOI https://doi.org/10.2147/CMAR.S493368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Naohisa Chiba,1 Toshi Menju,1 Yumeta Shimazu,1 Toshiya Toyazaki,1 Ryota Sumitomo,1 Hideaki Miyamoto,1 Shigeyuki Tamari,1,2 Shigeto Nishikawa,1 Hiroshi Date1
1Department of Thoracic Surgery, Graduate School of Medicine, Kyoto University, Kyoto, Japan; 2Department of Thoracic Surgery, Shizuoka City Shizuoka Hospital, Shizuoka, Japan
Correspondence: Toshi Menju, Department of Thoracic Surgery, Graduate School of Medicine, Kyoto University, 54 Shigoin-Kawahara-cho, Sakyo-ku, Kyoto, 606-8507, Japan, Tel +81-757514975, Email [email protected]
Background: Signaling pathways centered on the G-protein ADP-ribosylation factor 6 (Arf6) and its downstream effector ArfGAP with the SH3 Domain, Ankyrin Repeat and PH Domain 1 (AMAP1) drive cancer invasion, metastasis, and therapy resistance. The Arf6-AMAP1 pathway has been reported to promote receptor recycling leading to programmed cell death-ligand 1 (PD-L1) overexpression in pancreatic ductal carcinoma. Moreover, AMAP1 regulates of nuclear factor-kappa B (NF-κB), which is an important molecule in inflammation and immune activation, including tumor immune interaction through PD-L1 regulation. In this study, we investigated the function of AMAP1 on PD-L1 expression using lung cancer cells.
Methods: We used two non-small cell lung cancer cell lines. Protein expression was evaluated by Western blotting. AMAP1 and NF-kB expression were reduced by conventional siRNA methods, and osimertinib was used as an epithelial growth factor receptor (EGFR) inhibitor. Multiple analysis of receptor tyrosine kinases (RTKs) was conducted using a semi-comprehensive RTKs assay.
Results: We found that AMAP1 inversely regulated PD-L1 expression. Based on these results, we examined the activation levels of RTKs associated with both AMAP1 and PD-L1. Following a semi-comprehensive phosphorylated RTK assay, we observed the upregulation of phosphorylated EGFR (pEGFR) led by the downregulation of AMAP1. The inhibition of pEGFR by osimertinib downregulates PD-L1 expression. We investigated the relationships between AMAP1, NF-κB, and PD-L1 expression. AMAP1 knockdown upregulated the expression of both NF-κB and PD-L1. Subsequently, NF-κB knockdown downregulated PD-L1 levels, while double knockdown of AMAP1 and NF-κB, restored PD-L1 expression.
Conclusion: AMAP1 may inversely regulate PD-L1 through negative feedback of pEGFR and activation of NF-κB in NSCLC cell lines.
Keywords: AMAP1, PD-L1, NF-κB, pEGFR
Introduction
Among various solid tumors, lung cancer is the most common cause of death worldwide.1 One reason for this is tendency to involve invasion and metastasis. Programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1) are vital proteins in maintaining immune homeostasis.2,3 The PD-1/PD-L1 pathway inhibits the hyperactivation of immune cells and prevents autoimmune diseases.4 However, in the tumor microenvironment (TME), cancer cells hijack the PD-1/PD-L1 axis to escape immune surveillance.5 Inhibiting PD-1/PD-L1 signaling is a crucial strategy for normalizing the dysregulated TME.6 Recently, PD-1/PD-L1 blockade has shown clinical efficacy in non-small cell lung cancer (NSCLC); therefore, PD-L1 is a useful biomarker, and immune checkpoint inhibitor. Currently, immune checkpoint inhibitors are used for systemically unresectable metastatic cases, post chemoradiation, neoadjuvant chemotherapy and as adjuvant chemotherapy after surgery for NSCLC.7–10
Nuclear Factor-kappa B (NF-κB) consists of a family of transcription factors that play critical roles in inflammation, immunity, cell proliferation, differentiation, and survival.11,12 It generally exists as a homo- or heterodimer in the cytosol that is bound to the inhibitor of κB (IκB).13 In response to a wide variety of stimuli, including inflammatory cytokines, IκB is phosphorylated and degraded via the ubiquitin pathway, leading NF-κB translocation to the nucleus and transcription activation. NF-κB signaling proteins are essential for maintaining the living body, but they have also been implicated in the pathogenesis of many human diseases, including cancer.14–17 Activation of NF-κB controls multiple cellular processes in cancer, including inflammation, transformation, proliferation, angiogenesis, invasion, metastasis, chemoresistance and radioresistance.15,17 A direct link between epidermal growth factor receptor (EGFR) and PD-L1 expression via NF-κB has been described in NSCLC, which is the most common type of lung cancer.18,19
ADP-ribosylation factor 6 (Arf6) is a small molecular weight G-protein primarily involved in the outward flow of plasma membrane components.20 Arf6 and its downstream effector ArfGAP with the SH3 Domain, Ankyrin Repeat and PH Domain 1 (AMAP1) (also called DDEF1 or ASAP1) are involved in invasion metastasis and treatment resistance in cancer.21 The Arf6-AMAP1 pathway is found in lung, breast, renal and pancreatic ductal cancers, and reports indicate that its abnormally high expression significantly differs from the malignant prognosis of patients.22–28 The Arf6-AMAP1 pathway can be activated by different types of receptor tyrosine kinases (RTKs), such as EGFR, via the guanine nucleotide exchanger GEP10025 and by G-protein-coupled receptor via EFA.27 In pancreatic ductal cancer, the Arf6-AMAP1 pathway promotes intracellular to cell-surface recycling of integrins and PD-L1 recycling.21 Furthermore, downregulation of Arf6 and AMAP1 expression, along with recycling, has been reported to significantly downregulate PD-L1 cell surface levels.21
Elucidating the cause of increased PD-L1 expression is important for understanding the reasons for immune escape and the use of immune checkpoint inhibitors in NSCLC. We considered the possibility that PD-L1 expression increase in association with increased AMAP1 expression in NSCLC. In this study, we aimed to investigate the relationship between AMAP1 and PD-L1 expression in NSCLC.
Materials and Methods
Cells and Cell Culture
HCC4006, A549, NCI-H1975 (H1975), NCI-H460 (H460), NCI-H1792 (H1792), NCI-H1650 (H1650) and PANC-1 cells purchased from the American Type Culture Collection (ATCC). HCC4006 harbors an EGFR mutation and a p53 wild type. A549 harbors a KRAS mutation and a p53 wild type. HCC4006, H1650, H1792 and H1975 cells were cultured in RPMI-1640 medium (MilliporeSigma, Germany) supplemented with 10% heat-inactivated fetal bovine serum (Biosera, France), 100units/mL penicillin and 100mg/mL streptomycin (FUJIFILM Wako Pure Chemical Corporation) at 37°C in a humidified atmosphere containing 5% CO2. A549 and Panc-1 cells were cultured in Dulbecco’s modified Eagle’s medium (MilliporeSigma) supplemented with 10% heat-inactivated fetal bovine serum, 100units/mL penicillin and 100mg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Mycoplasma negativity was confirmed in these cell lines before their use. All cells were preserved in liquid nitrogen using a CELLBANKER 1 (Nippon Zenyaku Kogyo Co., Ltd).
Reagents
Osimertinib was purchased from Selleck Chemicals and dissolved in dimethyl sulfoxide (DMSO). Gefitinib was purchased from BLD pharm and dissolved in DMSO. Transforming growth factor β (TGF-β) was purchased from Biolegend and dissolved in BSA/PBS. Interferon-gamma (IFN-γ) was purchased from Peprotech. pEBG-AMAP1 plasmids were kindly provided by Professor Sabe of Hokkaido University.
Western Blot Analysis
To prepare total cell lysates, cells were lysed in radioimmunoprecipitation assay buffer (20 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet p‑40, 1% deoxycholic acid and 0.1% sodium dodecyl sulfate) supplemented with protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 1 mm sodium vanadate, 1 ng/mL aprotinin, 1 ng/mL leupeptin and 1 ng/mL pepstatin A). Protein concentrations were determined using Bio‑Rad Protein Assay Dye Reagent Concentrate (#5000006JA; Bio‑Rad Laboratories, Inc). The following primary antibodies were used: Anti‑phospho-epidermal growth factor receptor (pEGFR) (Cell Signaling Technology; cat. no. 2236 1:1000), anti‑PD-L1 (Cell Signaling Technology; cat. no. 13684 1:1000), anti- phospho-ACK1 (pACK1) (Cell Signaling Technology; cat. no. 3138 1:500), and anti‑phospho-Syk (pSyk) (Cell Signaling Technology; cat. no. 2710 1:1000), anti‑DDEF1 (Santa cruz; cat. sc-374410 1:1000), anti‑NF-κB (Santa cruz; cat. sc-8008 1:1000) and anti‑β‑actin (Millipore Sigma; cat. no. A5441 1:2000). Gel electrophoresis was performed using PowerPac™ HC High‑Current Power Supply (1645052; Bio‑Rad), with a 10% gel used for all antibodies. The polyvinylidene difluoride membranes (IPVH00010; Merck Millipore Ltd). were incubated with 2% bovine serum albumin (A9647‑100G; Millipore Sigma) for 60 min at 20–25°C for blocking. Following incubation with the primary antibodies for 20 hours at 4°C, the polyvinylidene difluoride membranes were incubated with horseradish peroxidase‑conjugated secondary antibodies (1:10,000; 711‑035‑152; donkey anti‑rabbit IgG, 715‑035‑150; donkey anti‑mouse IgG, Jackson ImmunoResearch Laboratories, Inc). for 60 min at 20–25°C. Protein bands were visualized using an Ez West Lumi Plus (ATTO Corporation) and a LuminoGraph II imaging system (ATTO Corporation). We performed Ez West Lumi Plus according to the manufacturer’s protocol. Protein expression was semi-quantified using ImageJ 1.53 software (National Institute of Health). In brief, we measured the intensity of each band using image J. Among them, the band used as the reference was set to 1.0. Then, the relative expression of the other bands were quantified and displayed in a graph for comparison.
Small Interfering RNA‑mediated Gene Silencing, Transfection, and Overexpression
AMAP1 small interfering RNA (siRNA) was purchased from Ajinomoto Bio-Pharma Services Japan GeneDesign, and Ambion’s Silencer™ validated siRNAs (ID nos. s11914 and s11916) targeting NF-κB were purchased from Thermo Fisher Scientific, Inc. Mission(R) siRNA Universal Negative Control#2 was purchased from Merck and used as the negative control. The siRNA sequences are presented in Table S1. The sequence of Mission(R) siRNA Universal Negative Control #2 was not disclosed by the supplier. The cells were seeded at 60–80% confluency in 6-cm dishes with antibiotic-free medium containing 14 µL Lipofectamine 2000® (cat. no. 11668019; Thermo Fisher Scientific, Inc). and 800 µL Opti‑MEM (cat. no. 31985062; Thermo Fisher Scientific, Inc)., which yielded a final siRNA concentration of 20 −75 nM, transfected for 48 h, and then harvested for lysate preparation.
For the overexpression method, A549, cells were seeded at 60–80% confluency in 6-cm dishes with antibiotic-free medium containing 2.4 µg pEBG or pEBG-AMAP1, 4.7 µL Lipofectamine 2000® and 800 µL Opti‑MEM, which gave a final siRNA concentration of 20 nM, transfected for 48 h, and then harvested for lysate preparation. HCC4006, cells were seeded at 80% confluency in 6-cm dishes with antibiotic-free medium containing 3 µg pEBG or pEBG-AMAP1, 7 µL Lipofectamine 3000® (cat. no. L3000015; Thermo Fisher Scientific, Inc). and 800 µL Opti‑MEM, which gave a final siRNA concentration of 50 nM, transfected for 48 h, and then harvested for lysate preparation.
Receptor Tyrosine Kinase (RTK) Assay
The RTK assay was performed using Human RTK Phosphorylation Antibody Array C1 Kit (RayBiotech, Inc., U.S.A) according to the manufacturer’s protocol. Briefly, to prepare total cell lysates, cells were lysed using the lysis buffer provided in the kit. A total of 100 μg of protein extract was incubated with RTK glass slide subarrays spotted with 71 different anti-RTK antibodies for 20 hours. Subsequently, the antibody array chips were washed and biotinylated anti-phospho tyrosine antibodies were added. After incubation labeled-streptavidin was added. All blots were developed using the Ez West Lumi Plus (ATTO Corporation) and the LuminoGraph II imaging system (ATTO Corporation). Protein expression was semi-quantified using ImageJ 1.53 software (National Institute of Health). In brief, first, the positive control (PC) for siRNA and negative control was measured using ImageJ. Subsequently, the spots for anti-RTK antibodies in siRNA and negative control were measured. To compare siRNA and negative control, the positive control was used for normalization to calculate the respective values.
Immunofluorescence Assay
The cells were seeded at 2×104 cells/well in A549 and 3×104 cells/well in HCC4006 on a 2-chamber slide. They were then fixed in 4% paraformaldehyde for 10 min at 37̊C, permeabilized with 0.1% Triton X‑100 for 15 min at room temperature and blocked in 2% bovine serum albumin (Biosera) in phosphate‑buffered saline for 60 min at 20–25°C.
Antigen recognition was performed by incubating the cells with primary antibodies against pEGFR (Cell Signaling Technology; cat. no. 3777 1:1000), PD-L1 (Cell Signaling Technology; cat. no. 13684 1:1000) and DDEF1 (Santa cruz; cat. sc-374410 1:1000) for 20 hours at 4°C, followed by incubation with Alexa Fluor 568‑ (Invitrogen; Thermo Fisher Scientific, Inc. A-10037,1:1000) and 488‑ (Invitrogen; Thermo Fisher Scientific, Inc. A‑21441 1:1000) conjugated secondary antibodies. Nuclei were counter‑stained with ProLong Gold Antifade Mountant containing DAPI (P36931; Invitrogen; Thermo Fisher Scientific, Inc). Fluorescence images were obtained using a BZ-X810 microscope (Keyence). Protein expression was semi-quantified via BZ-X810 analysis. In brief, the fluorescence intensity and the number of cells within an image were measured, and the average fluorescence intensity per cell was calculated.
Results
Upregulation of PD-L1 Concomitantly Occurred with the Downregulation of AMAP1
We initially investigated the expression of PD-L1 when AMAP1 was knocked down by Western blot analysis. Similar expression trends were observed in other NSCLC cell lines (Supplementary Figure 1A-D). We selected two NSCLC cell lines, HCC4006 and A549. Because the original expression of PD-L1 was low in the A549 cell line, they were stimulated with 1 ng/mL of TGF-β to increase its expression. In the subsequent experiments using A549, TGF-β at 1 ng/mL was utilized. The results revealed that in both cell lines, AMAP1 knockdown led to the upregulation of PD-L1 expression (Figure 1A–D).
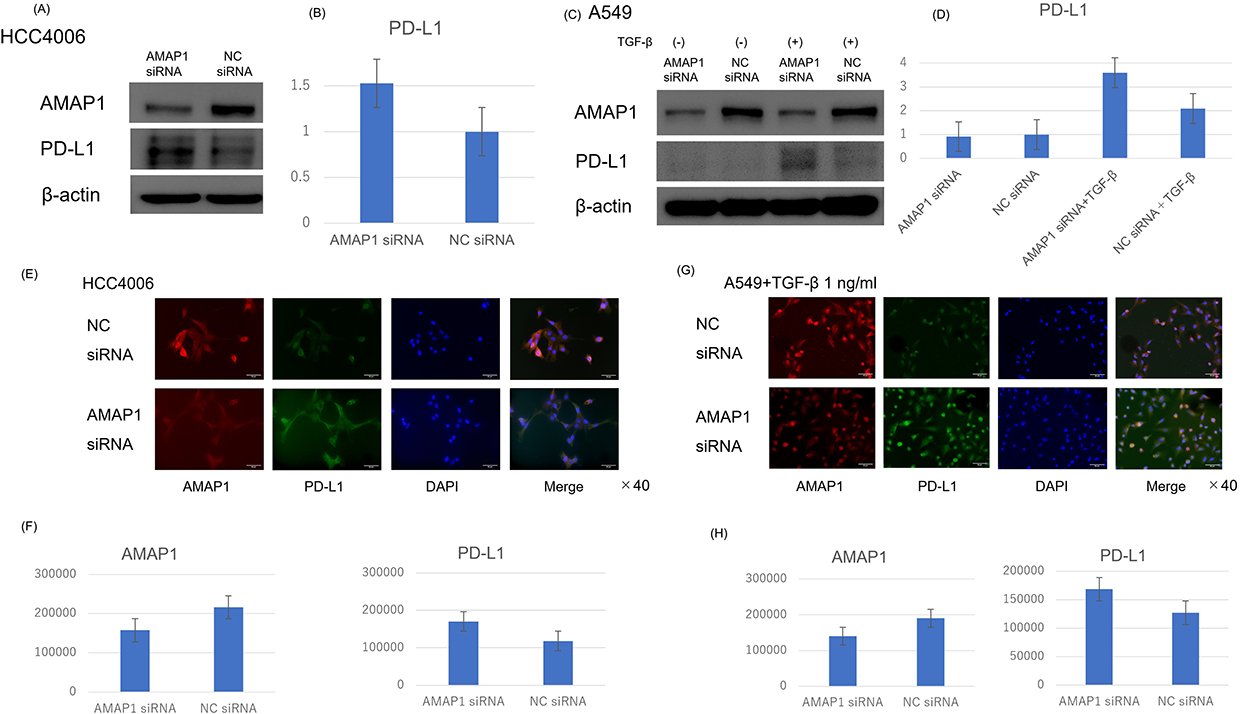 |
Figure 1 Results of AMAP1 knockdown: The impact of AMAP1 knockdown on PD-L1 expression in HCC4006 (A) and A549 (C) cells as observed by Western blot analysis. For A549 cells, owing to the low PD-L1 expression, stimulation was performed with TGF-β 1 ng/mL. Results of immunofluorescence (IF) in HCC4006 (E) and A549 (G) cells, where red represents AMAP1 and green represents PD-L1. Each result was semi-quantified using the ImageJ software (B and D) and the analysis application of BZ-X810 (F and H). The vertical axis of represents the band ratio normalized to the brightness of the negative control (set to 1) (B and D). The vertical axis of the graph represents the cell count, and the brightness per cell was measured (F and H). Abbreviation: NC, negative control. |
Next, we investigated the expression of PD-L1 in HCC4006 and A549 cells through immunofluorescence (IF) following AMAP1 knockdown. In both cases, upregulation of PD-L1 expression was observed following AMAP1 knockdown (Figure 1E–H). Notably, these findings are in contrast with the results of a previous report on pancreatic ductal cancer (Supplementary Figure 1E).
PD-L1 Expression Was Downregulated When AMAP1 Was Overexpressed
Next, we investigated how PD-L1 expression changed with forced overexpression of AMAP1 using the transfected vector, pEBG-AMAP1. In HCC4006 cells, AMAP1 overexpression resulted in a slight downregulation of PD-L1 expression (Figure 2A and B). In A549 cells, AMAP1 overexpression downregulated PD-L1 expression. (Figure 2C and D). Next, we investigated the expression of PD-L1 when both knockdown and overexpression of AMAP1 were simultaneously knocked down and overexpressed. In HCC4006 and A549 cells, AMAP1 knockdown led to the greatest upregulation of PD-L1 expression, whereas AMAP1 overexpression led to the most significant downregulation of PD-L1 expression (Figure 2E-H). For either line, the recovery conditions were similar from those of the negative control (NC). These results indicated, that AMAP1 inversely regulates PD-L1 expression. We investigated the mechanism underlying the inverse correlation between AMAP1 and PD-L1 expression.
 |
Figure 2 Results of AMAP1 overexpression: The overexpression of AMAP1, as observed in Western blot analysis, in both HCC4006 (A) and A549 (C) cells. For A549 cells, owing to the low PD-L1 expression, stimulation was performed with TGF-β 1 ng/mL. The lanes from left to right represented the simultaneous execution of AMAP1 knockdown and overexpression in HCC4006 (E) and A549 (G) cells. The order of lanes was as follows: AMAP1 knockdown and overexpression, AMAP1 overexpression, AMAP1 knockdown, and negative control. The arrowed portion represents a band of overexpressed proteins. Each result was semi-quantified using the ImageJ software (B, D, F and H). The vertical axis represents the band ratio normalized to the brightness of the negative control (set to 1). AMAP1, pEBG-AMAP1. Abbreviation: NC, negative control. |
pEGFR, pACK1 and pSyk Were Detected in a Semi-Comprehensive Phosphorylated RTK Analysis by AMAP1 Knockdown
In this study, we investigated the association between AMAP1 and RTKs upstream of it. We employed a semi-comprehensive assay for activated RTKs that allowed us to examine multiple targets simultaneously. The results revealed the expression of pACK1, pEGFR, and pSyk in HCC4006 cells (Figure 3A and Supplementary Figure 2A). ACK1 is a well-known non-receptor tyrosine kinase that regulates cell proliferation and growth through the activation of intracellular signaling pathways, including the mitogen-activated protein kinase pathway.29 Syk is a non-receptor tyrosine kinase that has been shown to mediate a surprisingly diverse range of biological functions, including cell adhesion, innate immune recognition, osteoclast maturation, platelet activation, and angiogenesis.30 In A549, the expression of pACK1 and pSyk was detected (Figure 3B and Supplementary Figure 2B). We compared the expression of these three activated RTKs upon AMAP1 knockdown by Western blot analysis. In HCC4006 cells, no significant changes were observed in the expression of pSyk, and no detectable alteration in the expression of pACK1 was noted (Figure 3C). However, AMAP1 knockdown led to the upregulation of pEGFR expression (Figure 3C-D). In A549 cells, AMAP1 knockdown resulted in a slight downregulation of pSyk expression, whereas pACK1 showed a low baseline expression (Figure 3E). In pEGFR expression, AMAP1 knockdown resulted in upregulation (Figure 3E and F).
 |
Figure 3 Changes in the expression of activated receptor tyrosine kinases (RTKs) when AMAP1 was knocked down: The investigation of changes in activated RTK expression when AMAP1 was knocked down in HCC4006 (A) and A549 (B) cells using the activated RTKs assay. Each antibody was plotted in pairs, with the first six and last two representing the positive control. For A549 cells, owing to the low PD-L1 expression, stimulation was performed with TGF-β 1 ng/mL. Western blot analyses of pEGFR, pSyk, and pACK1 in HCC4006 (C) and A549 (E) cells. A comparison of pEGFR expression in HCC4006 (G) and A549 (I) cells when AMAP1 was knocked down, as observed through IF. Each result was semi-quantified using the ImageJ software (D and F) and the analysis application of BZ-X810 (H and J). The vertical axis represents the band ratio normalized to the brightness of the negative control (set to 1) (D and F). The vertical axis of the graph represents the cell count, and the brightness per cell was measured (H and J). Abbreviation: NC, negative control. |
From these two results, AMAP1 knockdown led to the upregulation of pEGFR expression in both HCC4006 and A549 cells. Based on the results of the activated RTKs assay and Western blot analysis, which indicated an upregulation of pEGFR expression upon AMAP1 knockdown, we further confirmed these observations using IF. In the IF analysis, we observed an upregulation of pEGFR expression upon AMAP1 knockdown in both HCC4006 and A549 cells (Figure 3G-J).
Inhibition of pEGFR Suppressed PD-L1 Expression
Cumulative results, evidently demonstrate that knockdown of AMAP1 led to the upregulation of both PD-L1 and pEGFR. To explore the association between PD-L1 and pEGFR, we first investigated the mechanism by which downregulation of pEGFR expression affects the expression of PD-L1. Downregulation of pEGFR expression was achieved using the EGFR inhibitor, osimertinib. The concentration of osimertinib in HCC4006 and A549 cells was determined based on the IC50 values (Supplementary Figure 3) and previous reports.31–33 Western blot analysis showed that suppressing pEGFR expression led to the downregulation of PD-L1 expression in both HCC4006 and A549 cells (Figure 4A-D). The results showed that gefitinib suppressed pEGFR expression, leading to the downregulation of PD-L1 expression (Supplementary Figure 4).
 |
Figure 4 Results of inhibiting pEGFR using the EGFR inhibitor osimertinib: Western blot analysis showing the inhibition of pEGFR in HCC4006 (A) cells using osimertinib. The concentration of osimertinib was set at 25 nM. Western blot analysis demonstrating the inhibition of pEGFR in A549 (C) cells using osimertinib. The concentration of osimertinib was set at 2 μM. For A549 cells, owing to the low PD-L1 expression, stimulation was performed with TGF-β 1 ng/mL. Each result was semi-quantified using the ImageJ software (B and D). The vertical axis of the graph represents the band ratio normalized to the brightness of the negative control (set to 1). Abbreviation: NC, negative control. |
AMAP1 and NF-κB Double Knockdown Restored PD-L1 Expression
The downregulation of AMAP1 has been demonstrated to activate EGFR. EGFR activation upregulates PD-L1 expression via the NF-κB pathway.18 However, it is also known that AMAP1 positively regulates NF-κB directly, which is downstream of the EGFR pathway.11 Due to this contradiction, we investigated the expression of AMAP1, pEGFR, NF-κB, and the modulation of PD-L1. AMAP1 and NF-κB were individually knocked down, and their effects were examined by Western blot analysis. The results showed that AMAP1 knockdown upregulated in the expression of NF-κB and PD-L1 (Figure 5A–D). NF-κB knockdown resulted in a downregulation of PD-L1 expression in both HCC4006 and A549.
 |
Figure 5 Results of PD-L1 expression when NF-κB and AMAP1 were simultaneously knocked down: Western blot analysis showing the double knockdown of AMAP1 and NF-κB in HCC4006 (A) and A549 (C) cells. In A549 cells, TGF-β 1 ng/mL was used. From left to right, the lanes represent NF-κB knockdown, AMAP1 + NF-κB double knockdown, AMAP1 knockdown, and negative control. PD-L1 expression when NF-κB and AMAP1 were simultaneously knocked down, and osimertinib was used. Western blot analysis showing the double knockdown of AMAP1 and NF-κB, followed by osimertinib in HCC4006 (E) and A549 (G) cells. From left to right, the lanes represent NF-κB knockdown, AMAP1 + NF-κB double knockdown, AMAP1 knockdown, and the negative control. Each result was semi-quantified using the ImageJ software (B, D, F and H). The vertical axis represents the band ratio normalized to the brightness of the negative control (set to 1). Abbreviation: NC, negative control. |
When performing double knockdowns of NF-κB and AMAP1, the downregulation of PD-L1 expression was more pronounced than when only AMAP1 was knocked down in both HCC4006 and A549 cells.
AMAP1 and NF-κB Double Knockdown+osimertinib Showed No Change in PD-L1 Expression
Subsequently, we investigated the changes in AMAP1 and NF-κB expression upon inhibiting EGFR activation through Western blot analysis for each knockdown. In HCC4006 cells, double knockdown of NF-κB and AMAP1 did not result in a significant downregulation of PD-L1 expression (Figure 5E and F). In A549 cells, the double knockdown of NF-κB and AMAP1 led to the downregulation of PD-L1 expression (Figure 5G and H).
Discussion
In this study, we observed an inverse correlation between AMAP1 and PD-L1 expression in NSCLC. The results suggest associations with RTKs, particularly pEGFR, and NF-κB as potential mechanisms underlying this correlation.
In lung cancer cells, the EGFR-NF-κB-PD-L1 pathway is present. Numerous reports have established the relationship between the EGFR pathway and PD-L1 expression. Zhang. et al reported that the EGFR pathway is involved in PD-L1 expression through IL-6/JAK/STAT3 signaling in EGFR-positive lung cancer.34 Chen. et al reported that EGFR activation is associated with the upregulation of PD-L1 expression through the activation of ERK1/2/c-Jun.35 The results of this study are consistent with previous reports, demonstrating that PD-L1 expression was increased via the EGFR pathway.
Tanaka. et al reported that NF-κB is a crucial downstream signal when EGFR is activated.36 NF-κB is known to be involved in the regulation of PD-L1 expression, and Tien. et al reported that AMAP1 plays a role in negative feedback to NF-κB.11 In this study, we observed that a decrease in AMAP1 expression leads to an increase in NF-κB expression. Consequently, the upregulation of NF-κB resulted in, an increase in PD-L1 expression. These findings were consistent with those of previous studies.
One of the pivotal findings of this study was the activation of the intrinsic pathway of PD-L1 expression following EGFR stimulation. This pathway, distinct from pro-inflammatory external stimuli by immune cells, suggests a potential resistance to treatment with PD-1/PD-L1 inhibitors. In other words, the activation of the EGFR to NF-κB pathway may be a crucial route for PD-L1 treatment, potentially serving as a biomarker for predicting therapeutic efficacy.
Suppression of AMAP1 results in the activation of EGFR, and the mechanism underlying the increase in PD-L1 expression may be attributed to the inhibition of the internalization of activated RTK or negative feedback. Activated EGFR is typically internalized, transported to Early Endosomes, ubiquitinated, and then undergoes proteasome-dependent degradation.37 Consequently, EGFR inactivation occurs, leading to a decrease in signaling pathways. The Cbl plays a crucial role in this internalization process, where the binding of EGFR to the Cbl triggers the internalization signal.38,39 AMAP1 is suggested to function as a crucial adapter molecule in the binding between Cbl and EGFR. Therefore, a decrease in AMAP1 expression leads to an increase in activated EGFR as part of a negative feedback mechanism. The reduction in internalized activated EGFR may result in the suppression of binding with Cbl, subsequently inhibiting degradation and promoting recycling. This was a speculative interpretation of the observed outcomes.
Additionally, AMAP1 is primarily an adapter molecule for receptors and integrins and serves as an effector molecule when Arf1/6 is activated. The recycling of each molecule is activated upon Arf1/6 activation.40 In this study, changes in AMAP1 levels were associated with alterations in EGFR activation and NF-κB, leading to changes in PD-L1 expression. However, it cannot be conclusively determined whether AMAP1 directly regulates these processes.
According to previous reports, the Arf6-AMAP1-PD-L1 pathway in pancreatic ductal cancer cell lines is implicated in KRAS mutations and p53 mutations.21 The two cell lines used in our study ―HCC4006 with EGFR mutations and A549 with KRAS mutations― are known to lack p53 mutations. Therefore, unlike previously reported findings in pancreatic ductal cancer cells, the inverse correlation between the regulation of AMAP1 and PD-L1 observed in our study may be attributed to the absence of p53 mutations. However, in lung cancer, the EGFR-GEP100-Arf6 pathway is associated with malignant progression, including postoperative recurrence and a poor prognosis.41,42 Therefore, factors other than these pathways could potentially lead to opposite outcomes regarding invasion, metastasis, and immune evasion. This study demonstrated that EGFR and NF-κB signals mediated by AMAP1 are involved in modulating PD-L1 expression. Nonetheless, the minimal changes observed in PD-L1 expression when inhibiting the EGFR pathway, AMAP1, and NF-κB with EGFR-TKI suggest the involvement of alternative pathways in the regulation of PD-L1. The involvement of the AMAP1 pathway in the regulation of other co-inhibitory signaling molecules, apart from PD-L1, in terms of their expression, activation, and recycling, is crucial for a comprehensive understanding of cancer immune evasion. This is an important topic for future research.
As a limitation, it was not clear from this study whether EGFR and NF-κB were mutually related or acted separately, and how they influenced PD-L1 expression. Furthermore, the lack of evidence demonstrating direct interaction between AMAP1 and PD-L1 or the presence of PD-L1 in the recycling endosome containing AMAP1. These aspects remain topics for further investigation in future studies.
Summarizing the discussed signaling pathways in this study, inhibiting AMAP1 results in negative feedback on RTKs, particularly EGFR, leading to an increase in the activated form, pEGFR. Consequently, there is a potential pathway through which PD-L1 expression is elevated. Another pathway suggested is that when AMAP1 is inhibited, the feedback to NF-κB is eliminated, resulting in the activation of NF-κB and an increase in PD-L1 expression (Figure 6). There have been few reports on the molecular level relationship between AMAP1 and PD-L1 in lung cancer cell lines, making this study a potentially novel finding. Additionally, our findings suggest that targeting AMAP1 function to regulate activated EGFR and NF-κB holds promise as a strategy for novel cancer therapy, particularly in the context of immune checkpoint blockade.
 |
Figure 6 Proposed model of the signaling pathway: Proposed model schema of the signaling pathway for AMAP1 knockdown. We proposed negative feedback to RTKs and activation of NF-κB. |
Data Sharing Statement
All data generated in this study are available within the article and its Supplementary file or available from the authors upon request.
Acknowledgments
The authors thank Drs. Hisataka Sabe, and Ari Hashimoto at Hokkaido University for kindly providing the pEBG-AMAP1 plasmid. The authors would also like to thank the members of our department for useful discussions.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by a Grant-in Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (no. 20H03770 and no. 24K19432).
Disclosure
The authors declare that they have no competing interests for this work.
References
1. Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med. 2020;1:1–24.
2. Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 in the microenvironment. J Hematol Oncol. 2021;14:10. doi:10.1186/s13045-020-01027-5
3. Cha JH, Chan LC, Li CW, Hsu JL, Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. 2019;76:359–370. doi:10.1016/j.molcel.2019.09.030
4. Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–452.
5. Li X, Shao C, Han W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J Hematol Oncol. 2018;11:31.
6. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175:313–326.
7. Reck M, Delvys RA, Robinson A, et al. Five-year outcomes with pembrolizumab versus chemotherapy for metastatic non-small-cell lung cancer with PD-L1 tumor proportion score≧50. J Clin Oncol. 2021;39:2339–2349.
8. Spigel D, Faivre-Finn C, Gray J, et al. Five-year survival outcomes from the PACIFIC trial: durvalumab after chemoradiotherapy in stage III non-small -cell lung cancer. J Clin Oncol. 2022;40:1301–1311. doi:10.1200/JCO.21.01308
9. Forde P, Spicer J, Lu S, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386:1973–1985. doi:10.1056/NEJMoa2202170
10. Felip E, Altorki N, Zhou C, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (Impower010): a randomized, multicentre, open-label, Phase 3 trial. Lancet. 2021;398:1344–1357. doi:10.1016/S0140-6736(21)02098-5
11. Tien D, Kishihata M, Yoshikawa A, et al. AMAP1 as a negative-feedback regulator of nuclear factor-κB under inflammatory conditions. Sci Rep. 2014;4:5094. doi:10.1038/srep05094
12. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi:10.1101/cshperspect.a000034
13. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi:10.1038/nature04870
14. Agarwal A, Das K, Lerner N, et al. The AKT/I kappa B kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-kappa B and beta-catenin. Oncogene. 2005;24:1021–1031. doi:10.1038/sj.onc.1208296
15. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi:10.1101/cshperspect.a000141
16. Jiang R, Xia Y, Li J, et al. High expression levels of IKKalpha and IKKbeta are necessary for the malignant properties of liver cancer. Int, J, Cancer. 2010;126:1263–1274. doi:10.1002/ijc.24854
17. Chaturvedi M, Sung B, Yadav V, Kannappan R, Aggarwal B. NF-κB addiction and its role in cancer: ‘one size does not fit all’. Oncogene. 2011;30:1615–1630. doi:10.1038/onc.2010.566
18. Lin K, Cheng J, Yang T, Li Y, Zhu B. EGFR-TKI down-regulates PD-L1 in mutant NSCLC through inhibiting NF-κB. Biochem Biophys Res Commun. 2015;463:95–101. doi:10.1016/j.bbrc.2015.05.030
19. Antonangeli F, Natalini A, Garassino M, Sica A, Santoni A, Rosa F. Regulation of PD-L1 expression by NF-κB in cancer. Front Immunnol. 2020;11:584626.
20. Tsutaho A, Hashimoto A, Hashimoto S, et al. High expression of AMAP1, an ARF6 effector, is associated with elevated levels of PD-L1 and fibrosis of pancreatic cancer. Cell Commun Signal. 2020;18:101. doi:10.1186/s12964-020-00608-8
21. Hashimoto S, Furukawa S, Hashimoto A, et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc Natl Sci USA. 2019;116:17450–17459. doi:10.1073/pnas.1901765116
22. Sabe H. Requirement for Arf6 in cell adhesion, migration, and cancer cell invasion. J Biochem. 2003;134:485–489. doi:10.1093/jb/mvg181
23. Hashimoto S, Onodera Y, Hashimoto A, et al. Requirement for Arf6 in breast cancer invasive activities. Proc Natl Sci USA. 2004;101:6647–6652. doi:10.1073/pnas.0401753101
24. Onodera Y, Hashimoto S, Hashimoto A, et al. Expression of AMAP1, an ArfGAP, provides novel targets to inhibit breast cancer invasive activities. EMBO J. 2005;24:963–973. doi:10.1038/sj.emboj.7600588
25. Morishige M, Hashimoto S, Ogawa E, et al. GEP100 links epidermal growth factor receptor signalling to Arf6 activation to induce breast cancer invasion. Nat Cell Biol. 2008;10:85–92. doi:10.1038/ncb1672
26. Sabe H, Hashimoto S, Morishige M, et al. The EGFR-GEP100-Arf6-AMAP1 signaling pathway specific to breast cancer invasion and metastasis. Traffic. 2009;10:982–993. doi:10.1111/j.1600-0854.2009.00917.x
27. Hashimoto S, Mikami S, Sugino H, et al. Lysophosphatidic acid activates Arf6 to promote the mesenchymal malignancy of renal cancer. Nat Commun. 2016;7:10656. doi:10.1038/ncomms10656
28. Hashimoto A, Oikawa T, Hashimoto S, et al. P53- and mevalonate pathway-driven malignancies require Arf6 for metastasis and drug resistance. J Cell Biol. 2016;213:81–95. doi:10.1083/jcb.201510002
29. Lee HW, Choi Y, Lee AR, et al. Hepatocyte growth factor-department antiviral activity of activated cdc42-associated kinase 1 against hepatitis B virus. Front Microbiol. 2021;12:800935. doi:10.3389/fmicb.2021.800935
30. Mócsai A, Ruland J, Tybulewicz V. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402.
31. Truini A, Starrett J, Stewart T, et al. The EGFR exon 19 mutant L747-A750>P exhibits distinct sensitivity to tyrosine kinase inhibitors in lung adenocarcinoma. Clin Cancer Res. 2019;25:6382–6391. doi:10.1158/1078-0432.CCR-19-0780
32. Ohara S, Suda K, Fujino T, et al. Dose-dependence in acquisition of drug tolerant phenotype and high RYK expression as a mechanism of osimertinib tolerance in lung cancer. Lung Cancer. 2021;154:84–91. doi:10.1016/j.lungcan.2021.02.017
33. Hu J, Han Y, Wang J, et al. Discovery of selective EGFR modulator to inhibit L858R/T790M double mutants bearing a N-9-Diphenyl-9H-purin-2-amine scaffold. Bioorg Med Chem. 2018;26:1810–1822. doi:10.1016/j.bmc.2018.02.029
34. Zhang N, Zeng Y, Du W, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016;49:1360–1368.
35. Chen N, Fang W, Zhan J, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10:910–923. doi:10.1097/JTO.0000000000000500
36. Tanaka K, Babic I, Nathanson D, et al. Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance. Cancer Discov. 2011;1:524–538. doi:10.1158/2159-8290.CD-11-0124
37. Kuriyan J, Pinilla-Macua I, Grassart A, Duvvuri U, Watkins S, Sorkin A. EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis in tumors in vivo. Elife. 2017;6:e31993. doi:10.7554/eLife.31993
38. Sorkin A, Goh L. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi:10.1016/j.yexcr.2008.07.029
39. Visser SG, Place T, Cole S, et al. Cbl controls EGFR fate by regulating early endosome fusion. Sci Signal. 2009;2:ra86.
40. Hashimoto S, Hashimoto A, Yamada A, Onodera Y, Sabe H. Assays and properties of the ArfGAPs, AMAP1 and AMAP2, in Arf6 function. Methods Enzymol. 2005;404:216–231.
41. Menju T, Hashimoto S, Hashimoto A, et al. Engagement of overexpressed Her2 with GEP100 induces autonomous invasive activities and provides a biomarker for metastases of lung adenocarcinoma. PLoS One. 2011;6:e25301.
42. Oka S, Uramoto H, Shimokawa H, Yamada S, Tanaka F. Epidermal growth factor receptor-GEP100-Arf6 axis affects the prognosis of lung adenocarcinoma. Oncology. 2014;86:263–270.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.