")
Back to Journals » International Journal of Nanomedicine » Volume 19
Artificially Modified NK Cell-Based Synergistic Immuno-Gene-Photodynamic Therapy for Cancer
Authors Wu J, Shi K, Chao W, Qin Z, Hu Y, Yang Y, He Y, Hua Y , Zhao Z
Received 2 August 2024
Accepted for publication 3 November 2024
Published 21 November 2024 Volume 2024:19 Pages 12323—12342
DOI https://doi.org/10.2147/IJN.S481368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jie Huang
Jiale Wu,1,2 Kaihong Shi,1,2 Wei Chao,1,2 Zeyu Qin,1,2 Youhui Hu,1,2 Yihua Yang,1,2 Yuan He,1,2 Yabing Hua,1,2 Ziming Zhao1,2
1Department of Pharmacy, Xuzhou Medical University, Xuzhou, People’s Republic of China; 2Jiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, Xuzhou Medical University, Xuzhou, People’s Republic of China
Correspondence: Yabing Hua; Ziming Zhao, Department of Pharmacy, Xuzhou Medical University, Xuzhou, PR China; Jiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, Xuzhou Medical University, Xuzhou, People’s Republic of China, Email [email protected]; [email protected]
Background: The immunotherapeutic approach utilizing Natural Killer (NK) cells for cancer treatment has garnered significant interest owing to its inherent cytotoxicity, immunomodulatory properties, demonstrated safety in in vivo studies. However, multiple immunosuppressive mechanisms in the tumor microenvironment (TME) suppress the anticancer effect of NK cells in the treatment of solid tumors. Herein, a smart NK cell drug delivery system (DDS) with photo-responsive and TME-responsive properties was designed.
Methods: The NK cell DDS consists of two parts: the carrier is living NK cell with pH-low (abbreviated as NKpH) insertion peptide on its surface, the cargo is reductive-responsive nanogel (NG) encapsulated siRNA and photosensitizer (abbreviated as SP-NG), the final carrier was abbreviated as SP-NG@ NKpH. Firstly, pHLip helped artificially modified NK cell target and anchor onto cancer and exert the efficacy of cellular immunotherapy. Then, the strategy of combining photoactivation and bioreduction responsiveness achieved the precise release of cargos in cancer cells. Finally, the DDS combined the effect of the immunotherapy of NK cell, the gene therapy of siRNA, and the photodynamic therapy of photosensitizer.
Results: Under near-infrared laser irradiation, SP-NG@NKpH induced an increase in reactive oxygen species (ROS) within cells, exacerbated cell membrane permeability, and allowed for rapid drug release. Within the tumor microenvironment (TME), NG exhibits highly sensitive reducibility for drug release. The SP-NG released from NK cells can be uptaken by tumor cells. When exposed to near-infrared laser irradiation, SP-NG@NKpH demonstrates significant tumor-targeting specificity and cytotoxicity.
Discussion: The combined effect of the immunotherapy of NK cell, the gene therapy of siRNA, and the photodynamic therapy of photosensitizer obtained a stronger cancer killing effect in vitro and in vivo. Therefore, this versatile NK cell DDS exhibits a good clinical application prospect.
Keywords: natural killer cells, photo-responsive, TME-responsive, anticancer, cellular immunotherapy, nanogel
Graphical Abstract:

Introduction
In recent years, cell-based therapy has been a promising clinical treatment method for various refractory diseases due to its unique advantages.1,2 Along with the development of research, cell-based therapy has been divided into two directions: those serving as enhanced therapeutic agents themselves (cellular immunotherapy), and those serving as delivery vehicles for therapeutic drugs. Cellular immunotherapy has achieved remarkable success in treating blood cancer, lymphoma, and myeloma. However, cellular immunotherapy in the treatment of solid tumors has no breakthrough due to the immunosuppression of tumor microenvironment (TME).3 Drug delivery systems (DDS) based on living cells for solid tumor therapy have attracted great interest from researchers. Compared with synthetic drug carriers, living cells have superior biocompatibility, natural tumor targeting property, and the ability to escape from the immune system. Various types of cells, such as immune cells (macrophages, T cells, neutrophils), nucleated cells (red blood cells and platelets), stem cells, have been used as carriers for anticancer drugs.4–9 Therefore, drug delivery combined with cellular immunotherapy may lead to better outcomes in cancer treatment.
At present, the development of live cell-based DDS is still limited by the adverse effects of drugs and cell carrier on each other.10 How to load drugs without affecting the function of the carrier cells is the main challenge faced by cell-based DDS. One solution is to endocytose drug-loaded nanoparticles into the interior of cellular carriers. However, the premature drug release within the cell can affect the viability and chemotaxis of the carrier cells. In addition, the delivery cells often face challenges in drug release within the cell. In some cases, drugs in organelles such as lysosomes may become inactivate, thereby limiting the efficacy of the medication. Another solution is attaching drug-loaded nanoparticles to the surface of cells through chemical bonding, physical adhesion, or ligand-receptor recognition. This strategy could reduce the damage to the carrier cells, and facilitate the efficient release of drugs at the site of the lesion. While living cell carriers have shown promise in delivering various therapeutic agents, their potential as gene drug carriers remains largely unexplored in current research endeavors.
Natural killer (NK) cells have unique advantages in treating cancer: (1) They have natural killer activity to cancer by releasing perforin and granzyme; (2) NK cells express TNF superfamily members, such as FAS ligand or TRAIL receptor, which induce cancer cell apoptosis; (3) They express Fc receptor (CD16) and exert antibody-dependent cellular cytotoxicity (ADCC); (4) NK cells secrete a large number of cytokines, growth factors, and chemokines, recruiting macrophages, DC cells, T cells to cooperate in fighting cancer.11–14 Engineered cells with a chimeric antigen receptor (CAR), it is possible to recognize tumor-specific proteins and achieve precise tumor killing. Many immune cells, such as T cells, NK cells, and macrophages, have been able to achieve CAR modification and have been shown to have anti-cancer effects in clinical trials. It’s known that NK cells have no significant effect on the treatment of solid tumors. To address this challenge, extensive research has been conducted. For example, the design of modified antibodies or Fc fragments on NK cells to specifically target solid tumors, eliminating the need for specific antigen presentation to the tumor.15 Immunomodulatory nanoparticles have also been developed to modify the activation signals of NK cells on the surface of orthotopic tumor cells. This alteration activates NK cells and facilitates the achievement of safe and effective anti-tumor immunity.16 Zu et al17 developed aptamer-engineered NK cells for targeted immunotherapy. Under the guidance of the surface-anchored aptamer, NK specifically bound to lymphoma cells and subsequently induced apoptosis and cell death. Li et.al 18 engineered NK cell surfaces with two types of framework nucleic acids, serving as artificial receptors (ARs, including AR1 and AR2) on membrane. The recognition capacity of NK cells could potentially be augmented through the multivalent binding phenomenon. Cai et.al 19 construct a bio-orthogonal targeted live-cell nanocarrier (N3-NK-NPs) by coupling with responsive released interleukin-21 (IL-21) nanoparticles (ILNPs) on glyco-engineered NK cell surfaces. The bio-orthogonal strategy effectively promotes specific recognition and migration of NK cells, which show nearly four times deeper infiltration into the tumor than control groups. Pitchaimani, A et.al 20 developed a NK cell membrane camouflage fusion liposome delivery system called “NKsome”. Its excellent safety and strong tumor homing efficiency provide a good direction for NK cell delivery systems. The results also reflect the therapeutic advantages of NK cell biomimetics, which can communicate like immune cells for collaborative drug delivery. Overall, there is limited research on the dual role of NK cells in immunotherapy and drug delivery. Furthermore, there have been no reports on the use of NK cells for gene delivery.
Herein, we engineered NK cells-based DDS with synergistic immuno-gene-photodynamic therapeutic effects for cancer. Firstly, pH-low insertion peptide (pHLip) was modified onto the membrane of NK cell (abbreviated as NKpH) through the mediation of membrane fusion liposome. The next step was to load NG containing PLK1-siRNA and phthalocyanine (abbreviated as SP-NG) into NKpH cell. This artificially modified NK cell-based carrier (abbreviated as SP-NG@NKpH) has the following anticancer abilities. The pHLip can alter its conformation under the pH of TME and recover its cell insertion ability, which helps to bind NK cells and cancer cells, and then trigger ADCC of NK cells. The siRNA and Pc encapsulated in NG in NKpH were able to safely escape from the mononuclear phagocyte system (MPS) in vivo. After penetrating the tumor tissue, Pc inside the carrier would generate reactive oxygen species (ROS) under near-infrared laser irradiation, resulting in the rupture of the NK cell membrane and the release of the SP-NG. Upon the introduction of SP-NG into the tumor cells, a significant concentration of GSH could dismantle the disulfide bond of NG, subsequently liberating siRNA and Pc. Ultimately, upon the application of a secondary near-infrared laser irradiation, the cancer cells succumbed to the synergistic effect of gene and photodynamic therapy. In summary, this multifunctional NK cells-based DDS for tumor treatment would be explored a safe, effective, specific and manageable novel treatment method.
Materials and Methods
Materials
Tris(2-Carboxyethyl) phosphine hydrochloride (TECP), Polyethyleneimine (PEI, 1.8kDa) and Poly(γ-L-glutamate) (γ-PGA) were purchased from Macklin Biochemical (Shanghai, China). 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS) and 3, 3-dithiobispropionic acid was purchased from Macklin Biochemical (Shanghai, China). 5.5’-Dithiobis-(2-nitrobenzoic acid) (DTNB) was purchased from Aladdin (Shanghai, China). Phthalocyanin (Pc, MW:574.62) and L-Cysteine were purchased from Sigma-Aldrich (St. Louis, USA). SiPKL1 targeting Polo-like kinase-1 (siPLK1, sense sequence, 5’-UGAAGAAGAUCACCCUCCUUAdTd-3’) were synthesized by Sinopharm (Shanghai, China). Dioleoylphosphatidylethanolamine (DOPE), 1.2-dioleoyl-3-trimethylammonium-propane (DOTAP), DSPE-PEG2k-pHLip (AAEQNPIYWRYADWLFTTPLLLLDLALLVDEGTGG, supplied at >95% purity) were purchased from Ruixi Co. (Shanxi, China). Cell counting kit-8 (CCK-8), 4’,6-diamidino-2-phenylindole (DAPI), 1.1’-Dioctadecyl-3,3,3’,3’-Tetramethylindodicarbocyanine,4-Chlorobenzenesulfonate Salt (DID) were obtained from Beyotime (Shanghai, China). Dulbecco’s modified Eagle medium (DMEM), fetal bovine serum (FBS) and RPMI-1640 medium were purchased from Gibco (Grand Island, USA). Reactive Oxygen Species Assay Kit (ROS Assay Kit), Lactate Dehydrogenase Assay Kit (LDH Assay Kit) and fluorescein isocyanate (FITC) were purchased from Meilunbio® (Dalian, China). NK92-MI special medium was purchased from Gaining (Shanghai, China). All other chemicals and reagents were of analytical grade.
The cell lines, including NK and human cervical carcinoma cells (HeLa), and human normal liver cells (LO2) were obtained from the cell bank of Chinese Academy of Sciences (Shanghai, China). HeLa cells, and LO2 cells were cultured in a DMEM medium supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin. NK cells were cultured in NK92-MI special medium. All the cells were maintained with 5% CO2 at 37°C under fully humidified atmosphere. Female BALB/c nude mice (5 weeks) were supplied by the Charles River Laboratories (Beijing, China). All animal experiments were carried out according to the National Institutes of Health (NIH) guide for care and use of laboratory animals.
To establish subcutaneous tumor models, 100 μL of HeLa cells suspension were injected subcutaneously with a density of 2×104 cells/μL into the right foreleg armpit of BALB/c nude mice (20 ± 2 g). The tumor-bearing mice were housed with free access to food and water. After 7 d, tumor volumes reached around 100 mm3 and the treatment initiated.
Preparation and Characterization of the NG Loaded siPLK1(siRNA) and Pc (SP-NG)
Preparation of SP-NG
The synthesis of thiolated PEI (PEI-SH): EDC (1.5 g) and NHS (0.9 g) were dissolved in 3, 3-dithiobispropionic acid solution (20 mg/mL) and stirred for 1 h. The pH was adjusted to 5, then PEI (1 g, 1.8 kDa) was added into the mixed solution (The pH was adjusted to 8) with stirring for 24 h. TECP (0.5 mM,1 g) was added to the above resulting reaction mixture and stirred for 24 h. Then, the mixture was dialyzed for 24 h against deionized water using a dialysis membrane (MWCO: 1 kDa) followed by freeze-drying.
The same operation was performed as described above when synthesis of thiolated γ-PGA (γ-PGA-SH).
NG was prepared by suspension method. PEI-SH and γ-PGA-SH were dispersed with different weight ratio of 1:1, 2:1 and 4:1 in 2 mL of HEPES buffer (10 mM HEPES, pH 8.0) followed by mixing totally. Then, siPLK1 and Pc were added, mixing, and incubated at 37°C for 1 h.
The product (SP-NG) was stored at 4 °C for further use.
1H NMR and IR of PEI-SH and γ-PGA-SH
Thiol was detected by Hydrogen nuclear magnetic resonance,1H NMR infrared absorption spectroscopy (IR) and Ellman’s method.
The Morphology of NG and SP-NG
The morphology of NG and SP-NG were characterized by using transmission electron microscopy (TEM, FEI, USA).
Particle Size and Zeta Potential
The particle size and Zeta potential of NG and SP-NG were measured by a Nicomp 380 ZLS dynamic light scattering (DLS) instrument (PSS, USA). The zeta potential of PEI-SH, γ-PGA-SH, NG and SP-NG were determined by zeta potential analyzer (PSS, USA).
Detection of Sulfhydryl Content
In order to confirm the crosslinking effect of formed disulfide for NG, the remaining free thiol groups were quantified using Ellman’s method after preparation of NG.
Bio-Reducible Rupture of SP-NG
The switch (on/off) release performance of SP-NG was evaluated by reduction test. Firstly, freshly prepared 10 mM and 10 μM TECP solution were respectively added into SP-NG whose weight ratio was 1:1, and then visualized after gel electrophoresis. Finally, the changes of particle size and PDI of SP-NG with 10 mM and 10 μM TECP were analyzed by using DLS analyzer after 0.5 h, 3 h, 24 h incubation. The bio-reducible ruptured morphology of SP-NG after adding 10 mM and 10 μM TECP was also characterized by using TEM.
The Vitro Release of siPLK1 from NG
The vitro release profile of siPLK1 from NG was studied by using a dialysis method. Firstly, NG containing 500 μM siPLK1 were suspended in 1.0 mL of PBS solution (pH 7.4, pH 6.8 and pH 5.0, respectively) and PBS solution (pH 6.8) with 5 mM TECP followed by placing into a tightly clamped dialysis bag (MWCO: 10,000 Da).
Then, the NG in the dialysis bag was put at 37°C for different duration with agitation by a shaking table at 100 rpm. At selected predetermined time intervals, 1 mL of dissolution medium was taken out and replaced with 1 mL preheated fresh medium. At desired time intervals, the solution was obtained and free siPLK1 was determined by using the Ultraviolet spectrophotometer (wavelength: 260 nm).
Preparation and Characterization of SP-NG@NKpH
Preparation and Characterization of the Membrane Fusion Liposomes Loaded with pHLip (pHLIP-MFL)
The preparation of the Liposomes loaded with pHLip (pHLip-MFL): Briefly, the lipid mixture composed of DOPE, DOTAP and DSPE-PEG2k-pHLip (molar ratio of 15:4:1) was dissolved in 5 mL of dichloromethane and evaporated at 40°C to near-dryness. The resulting lipid film was hydrated with PBS (pH 7.4) for redispersion with stirring. The final concentration was 2 mg/mL. After hydration, the dispersion was sonicated for 6 min to obtain membrane fusion liposomes loaded with pHLip (pHLip-MFL). Referring to the above method, FITC-labeled pHLip-MFL (FITC-pHLip-MFL) were prepared.
Particle size and stability of pHLip-MFL were measured by using DLS analyzer. The morphology of pHLip-MFL was also observed by using TEM.
Decoration and Characterization of NK Cells
NK cells were co-incubated with pHLip-MFL or FITC-pHLip-MFL for 2 h to obtain NK cells with surface-modified pHLip (NKpH), after discarding the supernatant-free pHLip-MFL or FITC-pHLip-MFL.
The amount of peptide loaded was calculated by fluorescent labeling method, and NKpH were observed with confocal microscopy (Olympus, Japan).
Preparation and Characterization of SP-NG@NKpH
NKpH were co-incubated with SP-NG for 2 h to obtain NKpH with SP-NG (SP-NG@NKpH), after discarding the supernatant-free SP-NG.
Inverted fluorescence microscope (Olympus, Japan) and TEM were used to observe the intracellular distribution of NKpH after uptake of SP-NG. The amount of siPKL1 and Pc was calculated by the standard curve method.
Stability Evaluation of SP-NG@NKpH in vitro
To certify the stability of SP-NG@NKpH, SP-NG@NKpH were resuspended in an isotonic phosphate buffer solution (PBS) at 37°C. The free concentration of Pc and siPKL1 were determined by multifunctional enzyme marker (Thermo, USA) and Ultraviolet-visible Spectrophotometer (Mapada, China) at certain times.
Photoresponsivity and Phototoxicities of SP-NG@NKpH
The photoresponsivity and phototoxicities of Pc were determined by using the ROS Assay Kit and LDH Assay Kit according to the instruction of the manual.
Aliquots of exponentially growing cells (NKpH cells, 1×105 cells/mL) were placed in 96-multiwell plates at a volume of 100 µL per well and incubated for 12 h. The cells were incubated with complete medium with SP-NG loaded various concentration of photosensitizers for 4 h. One control column in the plate was filled with culture medium as a blank. The cells were washed with sterile PBS twice and filled with 200 µL fresh culture medium.
The cells were then irradiated at a light dosage of 1200 mw/cm2 by using 808 nm infrared laser source for 3min. After irradiation, cells were continually cultivated for another 4 h. Cellular viability was then measured by adding 10 µL ROS and LDH detection reagent to each well followed by incubation for 30 min at 37°C. After observation with an inverted fluorescence microscope, the fluorescence intensity of the probe was measured on a multifunctional enzyme marker (Thermo, USA) by recording the absorbance (excitation wavelength 502 nm, emission wavelength 530 nm), and cell membrane damage was characterized by using TEM. The cell viability of the treated samples was then obtained by comparison with the photosensitizer-free columns.
Then, to prove that Pc and siPKL1 were released under laser irradiation, the content of free Pc and siPKL1 in PBS was detected by multifunctional enzyme marker (Thermo, USA) and Ultraviolet-visible Spectrophotometer (Mapada, China).
Laser Irradiation Disruption and Reuptake of SP-NG@NKpH
After co-incubating SP-NG@NKpH with HeLa cells for 2 h, the resulting mixture were irradiated with 1200 mw/cm2 infrared laser (808 nm) for 3 min. The cell membrane fluorescent dye DiD stains HeLa cells with red fluorescence, and phthalocyanine emits green fluorescence under the microscope. The effect of near-infrared laser irradiation on the release of SP-NG from NK cells and reuptake by HeLa cells was investigated by inverted fluorescence microscope (Olympus, Japan).
Cytotoxicity Assessment
In vitro antiproliferative activities of SP-NG@NKpH against HeLa cells were determined by CCK-8 assay. Cells were planted into 96-well plates with a density of 5×103 cells per well and incubated for 12 h.
The cells were co-incubated with various formulations of SP-NG@NKpH at different concentrations (①NK cells; ②S-NG; ③P-NG; ④P-NG+Laser;⑤SP-NG+Laser; ⑥SP-NG@NKpH; ⑦SP-NG@NKpH+Laser) for 24 h. Furthermore, blank NG, siPKL1 and P-NG were tested by HeLa cells to select the most suitable dosing concentration. Then, PI/Calcein-AM apoptosis detection kit was used to detect the apoptosis of SP-NG@NKpH after laser irradiation.
In vivo Imaging
The tissue distribution and in vivo tumor targeting of SP-NG@NKpH was evaluated by an in vivo fluorescence imaging system.
After DiD co-incubated with modified NK cells, DiD labeled SP-NG@NKpH (DiD-SP-NG@NKpH) and DiD labeled SP-NG@NK (DiD-SP-NG@NK) were obtained. BALB/c nude mice were randomly assigned into 2 groups of 6 mice each group (DiD-SP-NG@NKpH and DiD-SP-NG@NK) and injected intravenously via tail vein with DiD labeled NPs.
At 4 h, 12 h, and 24 h post-injection, the living mice were photographed using a NightOWL LB 983 imaging system (Berthold, German). After in vivo images were photographed, the mice were subjected to euthanasia and their major organs (liver, spleen, lung, heart, and kidney) and tumor were photographed too (n=3).
Furthermore, fluorescence sections of tumor tissue were prepared, tumor blood vessels were labeled with CD31 green fluorescence, and NK cell membranes were labeled with DiD red fluorescence. The sections were photographed and recorded with an inverted fluorescence microscope. The targeting effect of cell carrier in tumor tissue was observed after administration.
Antitumor Efficacy Study in vivo
All animal experiments were approved by the Animal Care Committee of Xuzhou Medical university. BALB/c nude mice were randomly assigned into 10 groups: (1)saline group, (2)″NK+NG″group, (3)″siRNA″group, (4)″Pc+Laser″group, (5)″SP+Laser″group (intratumoral injection of siRNA +Soluble Pc), (6) ″P-NG+Laser″group, (7)″P-NG@NK+Laser″group, (8)″P-NG@NKpH+Laser″, (9) ″SP-NG@NKpH+Laser″group, (10) ″SP-NG@NKpH″, n=3. The corresponding formulations were injected into mice via tail vein every three days for 1 time (PLK1-siPKL1 dosage: 200nM, Pc dosage: 20μg/mL, NK cells: 8×105 cells/mL). Tumor volumes were calculated by the equation: V = (L × W2)/2 (L, the largest tumor diameter; W, the smallest tumor diameter). All mice were executed on day 3 after the third injection, and the tumor tissues were dissected and weighed. For histopathologic examination, tumors were fixed with 4% paraformaldehyde, embedded in paraffin, and cut into 3 μm sections. Hematoxylin and eosin (H&E) staining and TdT-mediated dUTP Nick-End Labeling (TUNEL) were used to examine the histopathologic changes of tumors.
Statistical Analysis
All data were provided as mean ± standard deviation (SD). Statistical evaluations were carried out using Student’s t-test. Statistical significance was noted as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Results and Discussion
The ligand of NK cell activation receptor expressed on the surface of tumor cells can activate NK cells, enabling them to identify tumor cells and reach the tumor site. However, tumor cells also could secrete tumor regulatory factors to inhibit the activation of NK cells, therefore relying solely on the migratory ability of NK cells themselves may not generate sufficient targeted anti-tumor effects. By fusing pHLip peptide with NK cell membrane using membrane fusion liposomes, drug delivery occurs selectively into tumor cells within the acidic tumor microenvironment. This responsive self-assembly on the tumor cell membrane enhances the selectivity and targeting of NK cells towards the tumor. Through controlling the switch of cell carriers release, the safety within normal tissues is ensured. Due to the reducing conditions of tumor cells and the overexpression of PLK1, SP-NG@NKpH remains intact until reaching the tumor site, preventing premature drug release.
Synthesis and Characterization of PEI-SH and γ-PGA-SH
Polyethylenimine (PEI), a synthetic cationic polymer characterized with notable charge density, is extensively employed as a gene transfection vector. Since every third atom is a protonated amino nitrogen atom, the polymer network acts as an efficient proton sponge. High Mw PEI (25 kDa) demonstrates high gene transfection efficiency, yet it also exhibits high cytotoxic and poorly biocompatible. Low Mw PEI (1.8 kDa) exhibits low transfection efficiency, but less cytotoxic. Additionally, macromolecular polymer-polyglutamic acid (γ-PGA) was chosen for high gene transfection efficiency, which is an anionic polymer with good water solubility and excellent biodegradability. And γ-PGA is easily cross-linked to form a hydrogel with excellent properties. To increase the charge density and finally improve the transfection efficiency, cross-linking or grafting low-Mw PEI (1.8 kDa) into the main chain of γ-PGA is necessary. Hence, we synthesized thiolated PEI with low Mw (herein after referred to as “thiolated PEI”) and thiolated γ-PGA to form the blank NG through the disulfide bond and electrostatic adsorption.
In Figure 1A (left),1H NMR spectrum of PEI-SH (300 MHz, D2O, ppm) appeared a new proton peak at δ0.95 ppm (labeled 1) due to the introduced thiol protons. The peaks at δ2.01 ppm (labeled 2) corresponds to the protons on the methylene group affected by thiol. In Figure 1A (right),1H NMR spectrum of γ-PGA-SH (300 MHz, D2O, ppm) also appeared new proton peaks at δ0.95 ppm (labeled 1), and the peak at δ1.15ppm (labeled 2) was due to introduced thiol protons and methylene group.
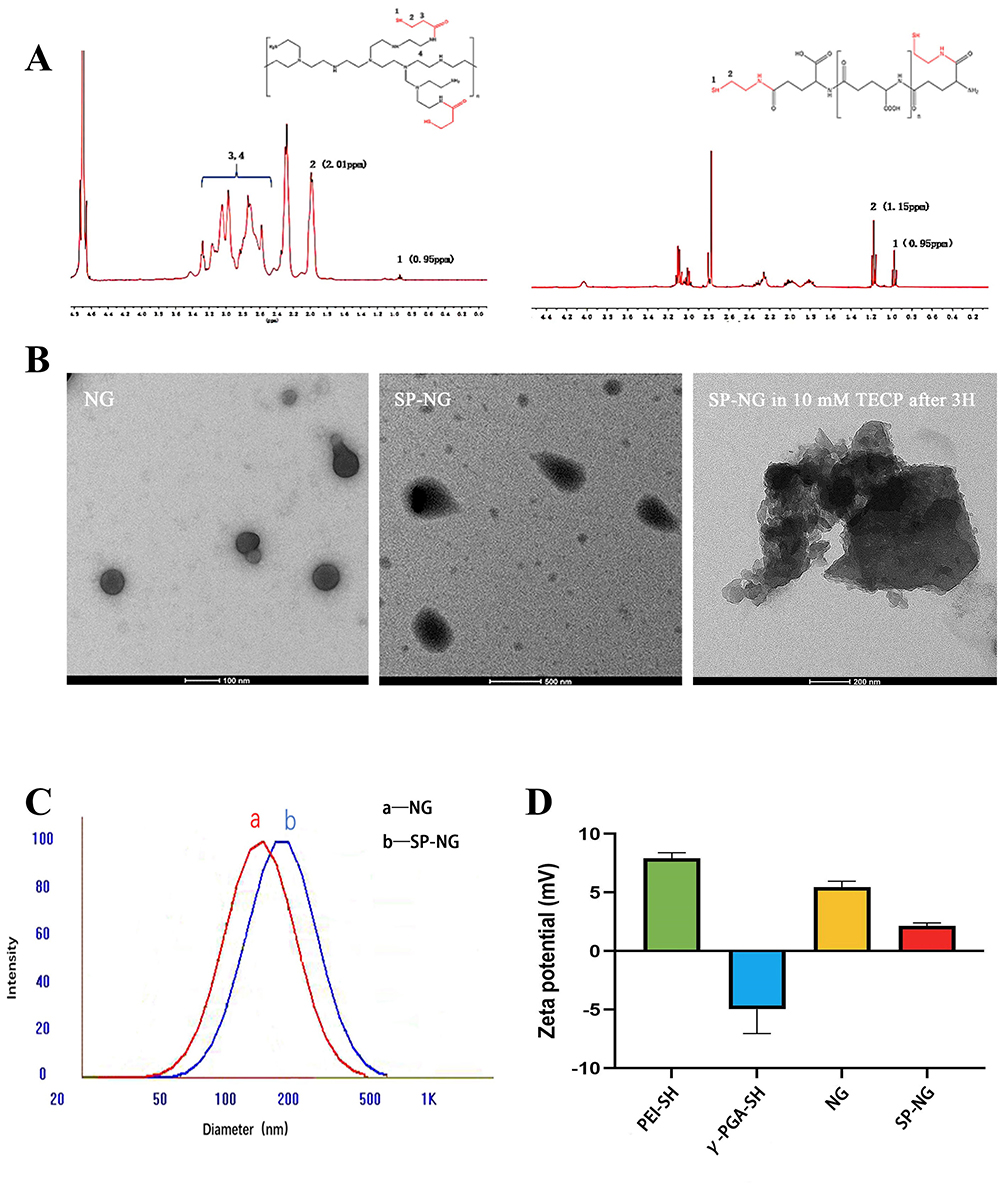 |
Figure 1 (A)1 H NMR spectra of PEI-SH(left) and γ-PGA-SH(right). Label 3 and 4 were the hydrogen peak formed by the methylene group next to the carbonyl group and amino group. (B) TEM images of NG, SP-NG and SP-NG in 10 mM TECP after 3 h. (C) DLS of NG (curve a) and SP-NG (curve b). (D) Zeta potential of PEI-SH, γ-PGA-SH, NG and SP-NG. |
The IR spectrum of PEI-SH for the weak absorption band at 2588 cm−1 was attributed to the introduction of a thiol group, and the characteristic absorption of amide bonds appeared at 1521 and 1699 cm−1 (Figure S1A). The IR spectrum of γ-PGA-SH also shows the weak absorption band at 2689 cm−1 and the characteristic absorption of amide bonds appeared at 1588 and 1639 cm−1 (Figure S1B). And the thiol content was 1.2265 µmol/mL in 10 mg/mL PEI-SH and 0.4480 µmol/mL in 10 mg/mL γ-PGA-SH respectively by Ellman’s examination (Figure S1C).
These results above confirmed the success of the synthesis of PEI-SH and γ-PGA-SH, and laid the foundation of cross-linked structure derived from disulfide bond in the further study of NG preparation.
Preparation and Characterization of SP-NG
Exogenous small interference RNA (siRNA) can selectively silence gene expression and inhibit protein transcription.21 Such as polo-like kinase 1 (PLK1), a member of the Polo-like kinase family, is an enzyme mainly involved in cell cycle progression and differentially expressed in normal and cancer tissues, making it an ideal target for cancer-specific silencing.22 Nevertheless, due to the unsatisfactory stability in the systemic circulation and the low permeability of biofilms, efficient delivery of siRNA to their application sites is one of the major challenges to realize their full potential.23 Non-viral gene platforms such as liposomes and polymers can enhance gene stability and therapeutic effect in vivo.24 However, the widespread clinical application of gene therapy is currently hindered by the demand for higher safety and efficacy of gene delivery vectors. Therefore, PGA and PEI cross-linked NG were used to deliver siRNA. And the NG loaded siRNA and Pc was prepared by a suspension method. Here, the disulfide cross-linking and electrostatic absorption were the driving force for NG formation and loaded gene and Pc.
The sulfhydryl groups were cross-linked at 1:1. Our previous study found the ratio of sulfhydryl groups contained in γ-PGA-SH and PEI-SH at the same concentration was about 1:2 by Ellman’s method, and therefore the ratio of γ-PGA-SH to PEI-SH was 1:1, 2:1, and 3:1. When the ratio of γ-PGA-SH and PEI-SH was 2:1, the particle size of the NG exhibited a favorable particle size (174.24 nm) and polydispersity index (PDI) was more uniform (0.135) (Table S1). Therefore, NG was synthesized at a ratio of 2:1 between γ-PGA-SH and PEI-SH in the subsequent experiments.
Moreover, the residual thiol groups after crosslinking were only 0.0208 μmol/mL, which indicated that most of the disulfide bonds had been used for crosslinking. As shown in Figure 1B and C, the NG was spherical, with a uniform size distribution and regular shape, which consistent with the particle size results. The SP-NG was a non-round spherical shape with divergent edges, which may be attributed to the binding of negatively charged siRNA chains. The particle size and PDI of remained basically stable within 7 d at room temperature (Figure S1D), and no precipitation occurred, which suggested that NG possessed a satisfied stability.
After cross-linking of positively charged PEI-SH and negatively charged γ-PGA-SH, NG were positively charged, making it easier to load negatively charged siRNA and Pc. Zeta potential (shown in Figure 1D), reducing interference with siRNA transcription, applies to passive tumor accumulation of EPR effects. Meanwhile, in gene therapy, intracellularly degraded reducible polycations not only facilitate the disassembly of multimers, but also enable gene-controlled release with better spatial selectivity in the cytoplasm.
Bioreducible Rupture of siRNA/Pc-NG
As illustrated above, disulfide cross-linking played a critical role in the formation of siRNA/Pc-NG. Therefore, we hypothesized that with the disulfide breaking in the siRNA/Pc-NG network, the siRNA/Pc-NG can be bioreducible rupture, especially in tumor tissue. The cancer cells have oxidation defense systems, expressing high concentration of intracellular antioxidant glutathione (GSH, 1–10 mM), and other reducing compounds, while the content in normal tissues is only 2–20 μM. Hence, we used 10 mM TECP to simulate similar reductive conditions in tumor cells for reductive response experiments and 10 μM TECP to simulate similar reductive conditions in normal tissues.
Due to the high glutathione environment in tumor cells, siRNA and Pc can be released by breaking the disulfide bonds in the NG. This study simulated the high glutathione environment in tumor cells in vitro using TECP. As shown in Figure 2A, compared with incubation with low concentration TECP (10 μM) for 24 h (a), the particle size and PDI of the NG significantly increased after incubation with high concentration TECP (10 mM) for only 0.5 h (b) and 3 h (c). The TEM results (Figure 1B) show that in a 10 mM TECP environment, the NG swell in volume until they were completely degraded. This indicates that the redox-triggered switch has advanced assembly and disassembly control to regulate siRNA release, benefiting gene therapy within tumor cells.
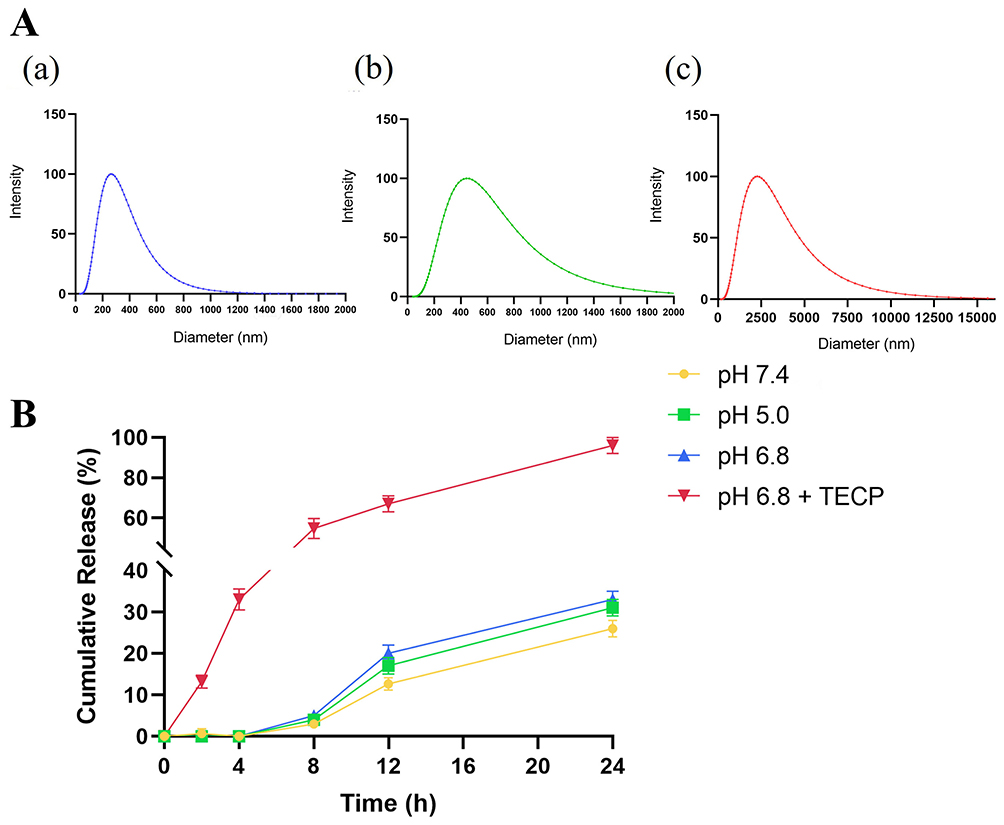 |
Figure 2 (A)Particle size distribution of SP-NG at different time in a reducing environment: (a) 10 μM TECP, 24 h;(b)10 mM TECP, 0.5 h(c) 10 mM TECP, 3 h. (B) The release of siRNA from NG in different pH. |
In vitro Release of siRNA from Nanogels
The release behavior of siRNA from nanogels in different pH release media was investigated. In Figure 2B, after incubation for 24 h under high redox environment (pH 6.8 and 10 mM TECP), approximately 95% of siRNA was released from the nanogels. Under other low redox physiological conditions, only about 25% of siRNA was released. This indicated that nanogels can effectively break the disulfide bonds connecting PEI-SH and γ-PGA-SH only under high redox conditions, thereby releasing siRNA.
Characterization of NK Cells with Surface-Modified pHLip (NKpH)
The accumulation of transforming growth factor-b (TGF-β) in TME could lead to the upregulation of fructose-1,6-bisphosphatase (FBP1) expression on NK cells. And the FBP1 upregulation expression would inhibit NK cell metabolism and survival,25,26 thereby impacting NK cell antitumor activity. Therefore, modification of NK cells is needed for precise targeting and killing of tumor cells. The reactivity self-assembly study of pHLip peptides has been utilized in the therapeutic research of various solid tumors. In the mildly acidic TME, pHLip peptides can transition from a random coil conformation to an α-helix conformation, with its C-terminus located intracellularly, while the N-terminus aggregates extracellularly and anchors to the tumor cell membrane. Herein, the modification of pHLip peptide into NK cells enables them to undergo conformational changes within the TME and selectively target tumor cells.
To modify pHLip peptide onto NK cells, we synthesized pHLip-MFL with an average particle size of 152.0±4.4 nm (Figure S2A). The TEM results in Figure 3A revealed that pHLip-MFL appeared spherical, with a light outer color and a misty appearance, possibly related to the grafting of pHLip peptide on its surface. Meanwhile, the particle size of pHLip-MFL remained stable within 7 days (Figure S2B), showing no precipitation, indicating good stability of pHLip-MFL.
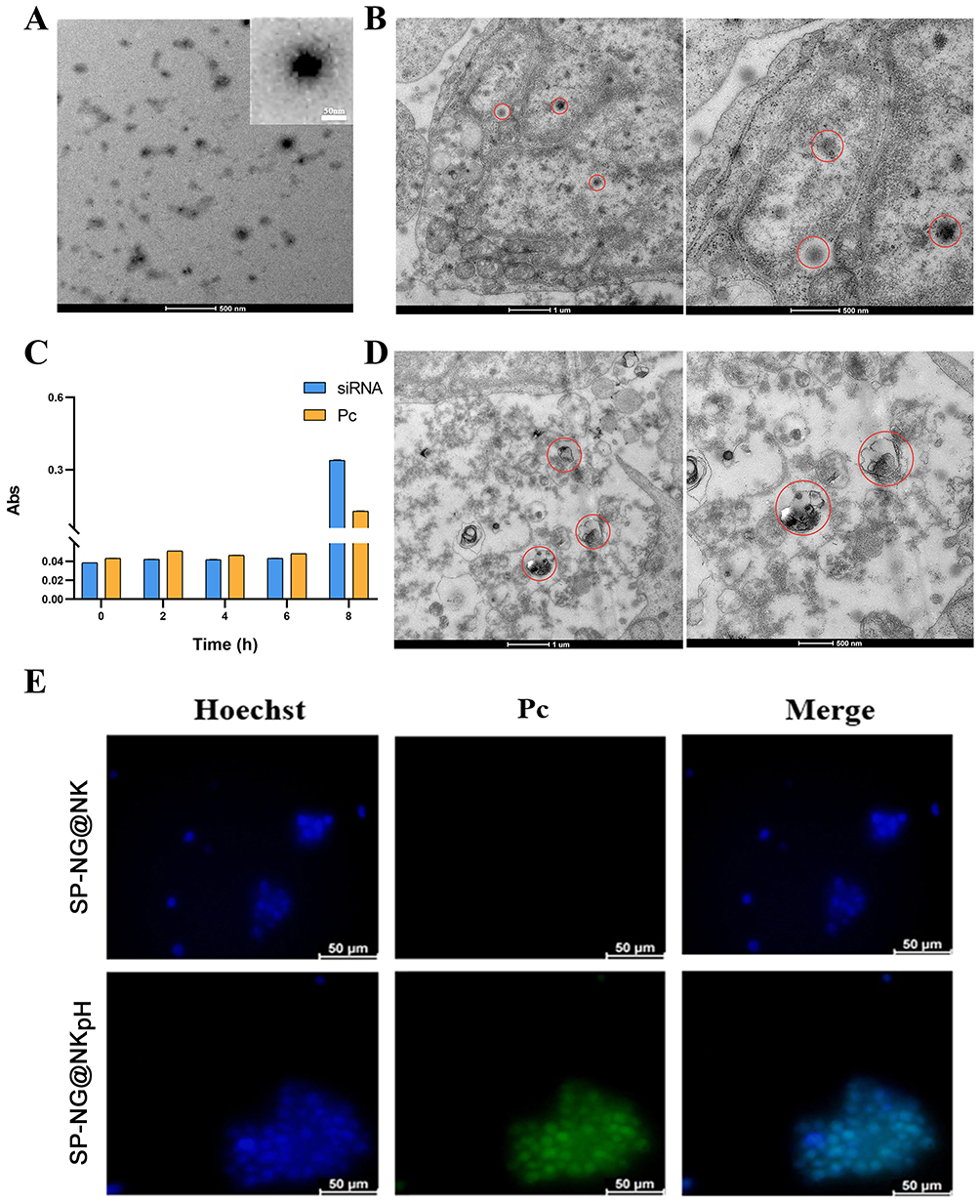 |
Figure 3 (A) TEM of pHLip-MFL liposomes. (B) TEM of the uptake of SP-NG by NKpH cells. (C) The stability of the SP-NG@NKpH. (D) TEM of the destroyed cell structure by laser irradiation. (E) Fluorescence images of the uptake of siRNA/Pc-NG by NKpH cells. The blue represented the nucleus. The green represented the photosensitizer, Pc. (The red circle outlines shown in Figure 3B and D indicated the location of the engulfed vector and the site of NK cell lysis). |
NKpH was obtained through co-incubating NK cells with pHLip-MFL for 2 hours, The adherence of the peptide segment to the NK cell membrane was observed using confocal microscopy (Figure S2C), indicating successful modification of the pHLip peptide on the surface of NK cells. The amount of pHLip in NKpHLip quantified by the fluorescence standard curve was determined to be 6.30 μg/105 cells (Figure S2D).
Preparation and Characterization of NKpH Loaded SP-NG (SP-NG@NKpH)
The SP-NG@NKpH system developed in our study consisted of a reducible nanogel encapsulating siRNA and Pc within NKpH. The pHLip helped artificially modified NK cell target and anchor onto cancer and exert the efficacy of cellular immunotherapy. Then, the strategy of combining photoactivation27 and bioreduction responsiveness achieved the precise release of cargos in cancer cells. Finally, the combined effect of the immunotherapy of NK cell, the gene therapy of siRNA, and the photodynamic therapy of photosensitizer obtained a stronger cancer killing effect. In Figure 1B, NG exhibited a uniform spherical shape with regular dimensions, whereas SP-NG presented an irregular morphology with dispersed edges, which may be associated with the negative charge of the encapsulated siRNA. The overall structure of SP-NG@NKpH was examined by ultrathin sectioning and observed under a TEM, as depicted in Figure 3B. SP-NG enters cells in a diffused spherical state and is widely distributed within cellular mitochondria, suggesting that NKpH could carry a substantial amount of SP-NG.
The uptake of SP-NG by NKpH cells was examined utilizing inverted fluorescence microscopy. The results (Figure 3E) show that compared to the SP-NG@NK group, SP-NG@NKpH group exhibited strong green fluorescence (Pc) inside the cells, indicating successful uptake of SP-NG by NKpH. In addition, the uptake of SP-NG by NKpH after laser irradiation was examined. In Figure 3D, the structure of NKpH cells was significantly disrupted, which may be attributed to NKpH internalizing SP-NG loaded with the photosensitizer Pc. Upon irradiation of Pc with laser, the ensuing generation of ROS resulted in the lysis of NK cells, thereby providing indirect evidence for the phagocytosis by NKpH cells. The contents of siRNA and Pc were detected through UV spectrophotometer and ELISA. The results (Figure 3C) showed that the concentration of siRNA and Pc was 3.95 μg/105 cells and 1.06 μg/105 cells, respectively. Furthermore, the drug leakage was almost non-existent within 6 hours for SP-NG@NKpH, indicating good stability.
Cell membrane injury caused by ROS could lead to the extracellular release of lactate dehydrogenase (LDH). Here, NKpH was incubated with SP-NG containing different concentrations of Pc, and subsequently exposed to an 808 nm near-infrared laser for 3 minutes. These could assess the LDH leakage resulted from cytoplasmic membrane rupture. In Figure 4A and B, as the concentration of Pc increases, there is a gradual rise in LDH leakage, indicating a positive correlation between the membrane damage caused by ROS generated in NK cells and the concentration of Pc. These findings underscored the efficacy of SP-NG in facilitating PDT outcomes through Pc incorporation. In Figure 4C, siRNA and Pc were almost undetectable without laser irradiation. While after laser irradiation, the cumulative release of siRNA and Pc was 43.77% and 38.11%, respectively. And the low release rate of siRNA and Pc may be due to that part of SP-NG was metabolized in lysosomes. However, compared with no laser irradiation, the photoresponse results still showed that the laser irradiation could be used as a “delivery switch” to control the release of SP-NG from NK cells to tumor cells. In addition, SP-NG@NKpH was incubated with HeLa cells for 4h. Subsequently, an 808nm NIR laser was employed for 3 minutes, after which the incubation process was sustained for 2 h, 6 h, and 24 h to evaluate the cellular reuptake of SP-NG. As shown in Figure 4D, SP-NG was released from NKpH. And with the increase of uptake time, there was a large area of overlap between the red fluorescence of HeLa cells and the green fluorescence of Pc, indicating that SP-NG was reuptake by HeLa cells. While SP-NG could not be released from NKpH without laser irradiation. The reuptake findings demonstrated that only upon exposure to 808nm laser irradiation, SP-NG was able released from NKpH and subsequently uptaken by target cells, highlighting the superior responsive “switch” properties of SP-NG@NKpH.
 |
Figure 4 The production of (A) reactive oxygen species (ROS) and (B) LDH in cells at different concentration of Pc after near-infrared laser (808 nm) irradiation for 3 min. (C) The accumulative release rate of siRNA and Pc. ***p < 0.001 (D) The uptake of SP-NG by HeLa cells in 2, 6, 24 h. Green represents SP-NG; Red represents HeLa cells. |
In vitro Drug Release
In our study, the in vitro release of SP-NG@NKpH can be divided into two parts: (1) In vitro release of siRNA from NG, we investigated the drug release behavior of SP-NG in pH 5.0, pH 6.8, pH 7.0, and pH 6.8 buffer with a reducing agent (pH 6.8+TECP). As depicted in Figure 2B, only a small amount of siRNA was detected in the buffer without the reducing agent. In the “pH 6.8+TECP” release medium, under reducing conditions, the disulfide bonds of the nanogel were cleaved, leading to the release of the encapsulated siRNA, with approximately 95% siRNA released cumulatively within 24 hours. These results indicate that under normal tissue conditions, SP-NG exhibits good stability without rupturing to release the encapsulated drug. However, in the reducing environment of tumor cells, the nanogel ruptures to release the drug, demonstrating the responsive drug release behavior of SP-NG. Under other low redox physiological conditions, only about 25% of siRNA was released. This indicated that NG can effectively break the disulfide bonds connecting PEI-SH and γ-PGA-SH only under high redox conditions, thereby releasing siRNA. (2) The result in Figure 4C illustrates the drug release behavior of SP-NG@NKpH within 4 hours. In the absence of laser irradiation, siRNA and Pc were hardly released. However, with laser irradiation, approximately 40% of siRNA and 30% of Pc were released within 4 hours. This indicates that pHLip-modified NK cells could uptake siRNA and Pc and responded to release in the tumor reductive environment.
Cytotoxicity Assay
The cytotoxic effect of SP-NG@NKpH on HeLa cells was examined through the CCK-8 assay. The results indicated a certain level of cytotoxicity of SP-NG towards HeLa cells. Furthermore, it was observed that siRNA and SP-NG@NKpH exhibited a dose-dependent response on the toxicity of HeLa cells (Figure 5A). During the preparation of the NG, a small amount of cytotoxic PEI-SH may remain in the SP-NG solution, and this issue would escalate with increasing NG concentration. Consequently, the 375 μg/mL NG was deemed as the optimal drug concentration to mitigate toxicity and maintain drug-loading efficiency.
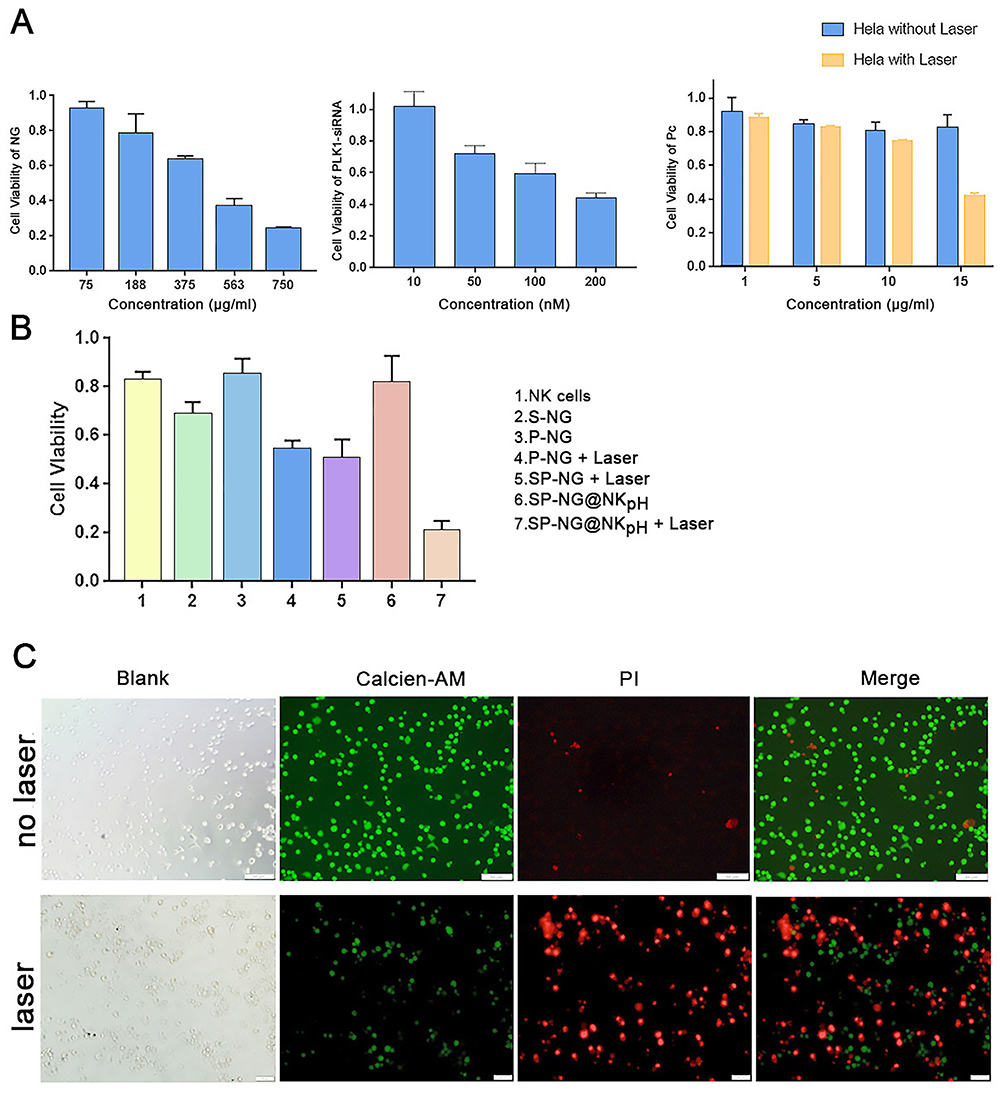 |
Figure 5 (A) Cytotoxicities of NG, siRNA and Pc on HeLa cells. (B) Cytotoxicities of different groups on HeLa cells. (C) Live/Dead analysis of SP-NG@NK after PI/Calcein AM staining with laser irradiation or not. Scale bar is 50 μm. |
The results illustrated in Figure 5A (b) depicted the cytotoxic effects of siRNA. Due to its negative charge, siRNA exhibits limited cellular internalization and toxicity. To facilitate cellular uptake, a liposome transfection reagent (Lip2000) was employed to create a complex with the negatively charged siRNA, enabling efficient endocytosis, cytoplasmic entry, and subsequent release of siRNA. The cytotoxicity was observed to escalate with higher siRNA concentrations, then 200 nM was selected as the optimal siRNA administration concentration.
NG loaded different concentrations of Pc were incubated with Hela cells for photoresponse experiments. In Figure 5A (c), compared with non-laser irradiation, the laser-irradiated NG with 15 μg/mL Pc had the strongest killing effect on HeLa cells. Pc is negatively charged and not easily endocytosed by cells, which make it low cytotoxicity. Herein, the Pc concentration was determined to be 15 μg/mL.
Furthermore, an examination was conducted on the cytotoxicity of various groups on HeLa cells. The results depicted in Figure 5B indicate that NK cells (group 1) exhibit a relatively low cytotoxic effect on Hela cells, approximately 20%. There was about 30% of HeLa cell were killed in the reduced nano-gel loaded with siRNA (S-NG, group 2) due to the gene silencing effect of the transfected siRNA. Pc-loaded NG (P-NG, group 3) showed less than 20% cytotoxicity against HeLa cells. However, the P-NG+Laser (group 4) exerted the Pc photodynamic effect, and more than 40% HeLa cells were killed. The NG simultaneously encapsulated siRNA and Pc with laser irradiation (SP-NG+Laser, group 5) exhibited a slightly stronger cytotoxic effect on HeLa cells compared to group 4. It should be noted that SP-NG@NKpH (group 6) without laser irradiation had only a slight killing effect on HeLa cells. This may be due to the low GSH content in NK cells, which does not cause the NG to rupture and release the drug, and indicated the safety of NK cells as delivery system. However, the SP-NG@NKpH+Laser (group 7) killed more than 80% of HeLa cells, which was much higher than other groups, indicating the precise targeting of NKpH and that laser irradiation of Pc could control the release of drugs from NK cells.
The SP-NG@NKpH Live/Dead staining results were shown in Figure 5C. Compared with no laser irradiation group, the red fluorescence of PI-stained dead cells was significantly increased after laser irradiation, while the green fluorescence of Calcein AM-stained live cells was significantly decreased. This indicates that SP-NG@NKpH combined with laser irradiation has a significant killing effect and a greater ability to induce tumor cell apoptosis.
Antitumor Efficacy Study in vivo
The tumor model was established by subcutaneous injection of HeLa cells in BALB/c nude mice. And the tumor growth curve and tumor weight were observed after multiple injections of different groups of vectors. In Figure 6A, the tumor size was greatly increased in the saline group (1). Due to the immune effect of NK cells, the growth rate of tumor volume in the “NG+NK” group (2) was slower than that in the saline group. Although the gene silencing effect of siRNA can specifically interfere with tumor cells, tumor volume was only slightly reduced after intratumoral injection of siRNA alone (3), which would result from the easy degradation of siRNA in vivo and other reasons. In the “Pc+Laser” group (4), the treatment effect was not obvious, and the tumor volume was not significantly reduced, and the body weight of the mice was significantly reduced. After four days of treatment by the “SP+Laser” group(5), the tumor volume decrease, and the body weight also decreased. In contrast to the “Pc+Laser” group (4), the “P-NG+Laser” group (6) showed weak tumor inhibition, which showed that the laser irradiation could control the release of Pc from the NG.
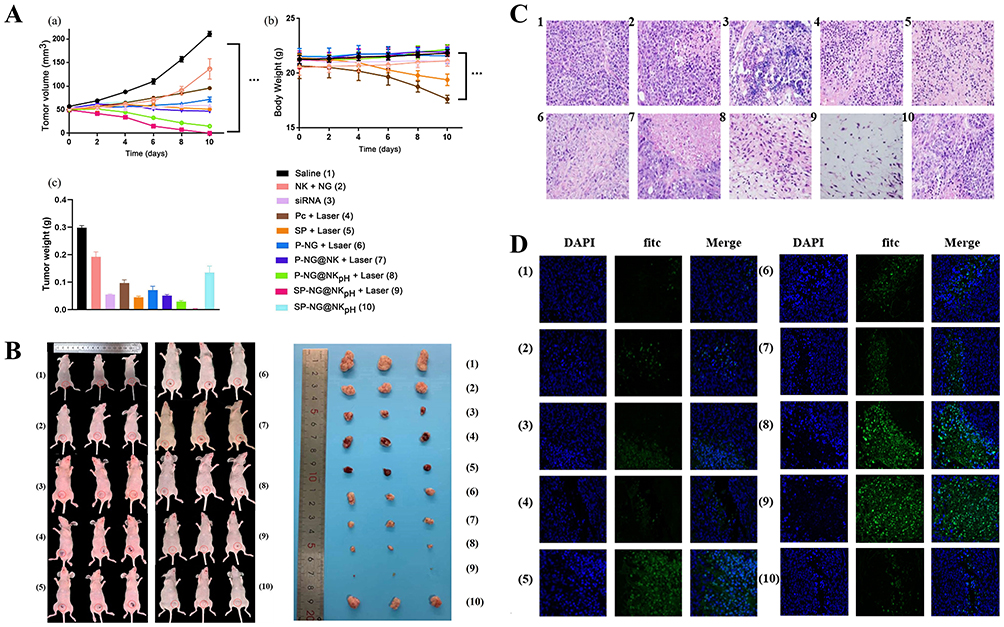 |
Figure 6 The tumor growth curve (A(a)), body weight (A(b)) and the tumor weights(A(c)) after being dissected of BALB/c nude mice after multiple injections of different formulations. (B) The pictures of tumor-bearing Balb/c nude mice and the solid tumor in each group (C)H&E staining of tumor tissue sections in different administration groups (scale: 20 μm). (D) Tunnel staining of tumor tissue sections in different administration groups (scale: 20 μm) (1: Saline, 2: NK+NG, 3. siRNA(Intratumoral injection), 4: Pc+Laser (Soluble Pc), 5: SP+Laser (siRNA intratumoral injection +Soluble Pc), 6: Pc-NG+Laser, 7: P-NG@NK+Laser; 8: P-NG@NKpH+Laser, 9: SP-NG@NKpH+Laser, 10: SP-NG@NKpH). |
The unmodified NK cells loaded with P-NG for tumor treatment and subjected to laser irradiation (“P-NG@NK+Laser” group) (7). The results showed that the “P-NG@NK+Laser” group (7) had a better inhibitory effect on tumor growth. Compared to unmodified NK cells, “P-NG@NKpH+Laser” group (8) showed a stronger tumor inhibitory effect, with no significant change in mouse body weight, indicating that the pHLip-modified NK cell had better target ability. The “SP-NG@NKpH+Laser” group (9) combined with gene therapy had the most significant inhibitory effect on tumor cell growth. This drug delivery system was anchored to the tumor site through responsive conformational switching of pHLip peptides in the TME. Then, the dual-drug release switch of photodynamic (Pc) and reductive response (NG) was used to achieve the precise drug release at the tumor site. Moreover, the “SP-NG@NKpH” group (10) had a high stability due to the absence of laser irradiation and essentially no drug release, which is consistent with the in vitro results. In addition, the pictures of tumor-bearing Balb/c nude mice and the solid tumor in each group were showed in Figure 6B.
The results of H&E staining (Figure 6C) and Tunel fluorescent staining (Figure 6D) showed that tumor cells in the saline group (1) and the “NK+NG” group (2) maintained normal cellular structure and morphology. Varying degrees of karyopyknosis and karyorrhexis were observed in the tumors after treatment with different groups of drugs. What’s more, tumor cells showed the greatest degree of apoptosis and necrosis after treatment with “SP-NG@NKpH+Laser” group (9), which was consistent with the in vitro anti-tumor results.
In vivo Imaging
NK cells would be activated by receptor ligand binding to tumor cells. However, tumor cells secrete tumor regulatory factors to inhibit NK cell activation. Therefore, the migration ability of NK cells alone cannot produce sufficient tumor targeting. To verify the targeting of SP-NG@NKpH in tumor therapy, the biodistribution of DiD-stained SP-NG@NKpH in tumor models was investigated. In vivo biodistribution at specific time intervals was observed using a near-infrared imaging system.
The Figure 7A showed the in vivo imaging results of SP-NG@NKpH and Figure 7B showed the in vivo imaging results of SP-NG@NK. Compared with unmodified NK cells (SP-NG@NK), after the injection of SP-NG@NKpH, the fluorescence intensity at the tumor site was strongest at 4 hours, and relatively weaker at 12 hours. Combined with the biodistribution results in the corresponding tissues and organs, the pHLip modification resulted in superior accumulation of NK cells at the tumor site. In a slightly acidic TME, pHLip could reactively self-assemble on the tumor cell membrane, allowing selective drug entry into the tumor cells, thereby enhancing NK cell selectivity, and targeting to the tumor. This phenomenon contributes to the heightened selectivity of NK cells and promotes their precise localization within the tumor site.
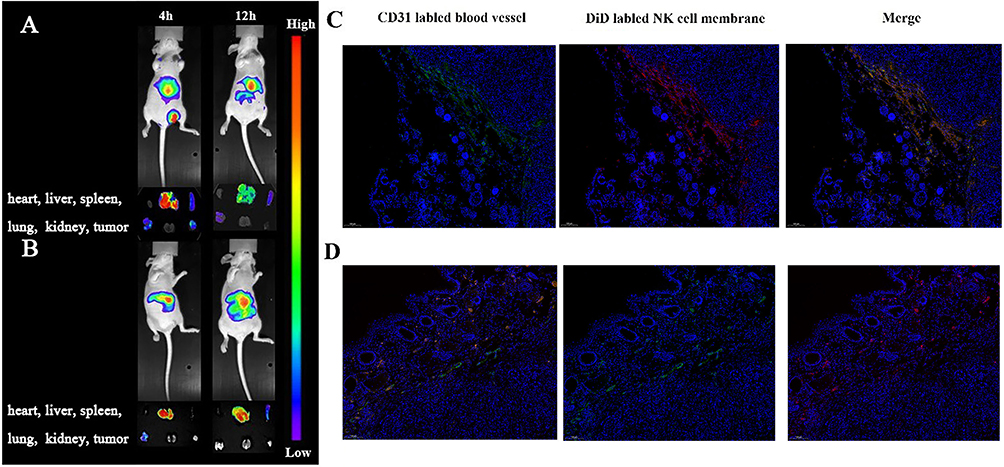 |
Figure 7 In vivo imaging of tumor bearing Balb/c nude mice and tumor tissue after intravenous injection of SP-NG@NKpH group (A and C) and SP-NG@NK group (B and D). |
The fluorescence results of tumor tissue sections were shown in Figure 7C and D. The red fluorescence representing NK cell carriers in the control group “SP-NG@NK” was less collected in the tumor tissue, while the red fluorescence of NK cells in the “SP-NG@NKpH” mostly overlapped with the green fluorescence of tumor blood vessels, which proved that pHLiP-modified NK cells were more able to target the tumor tissue.
Additionally, the effect of SP-NG@NKpH was further compared with others. Individual administration of PLK1-siRNA: the individual administration of PLK1-siRNA was limited by rapid degradation and clearance of PLK1-siRNA, leading to the lack of significant anti-tumor efficacy in vivo; “P-NG@NK+Laser” group: The NK cells were engineered for artificial modification, and encapsulated a NG containing PLK1-siRNA and Pc. The utilization of unmodified NK cells loaded with P-NG (“P-NG@NK+Laser” group) and subsequent laser irradiation has shown some inhibitory effects on tumor growth. However, the NK cells modified with pHLip (“P-NG@NKpH+Laser” group) exhibited a stronger tumor inhibitory effect, indicating that NK cells modified with pHLip enhanced their targeting ability; “SP-NG@NKpH” group: Upon infiltration into tumor tissues, “SP-NG@NKpH” triggers the generation of ROS by the internal Pc when exposed to near-infrared laser light. This process leads to the rupture of NK cell membranes, facilitating the release of NG. In the absence of laser irradiation treatment (“SP-NG@NKpH” group), minimal drug release was observed, demonstrating the photodynamic switch effect. However, when laser irradiation was applied (“SP-NG@NKpH+Laser” group), the photodynamic effect of Pc was triggered, leading to NK cell rupture and subsequent release of internal NG. The NG experienced a reductive-triggered disintegration at the tumor location, liberating Pc and gene silencing, leading to a notable suppression of tumor cell proliferation. This highlights the efficacy of the dual drug release mechanism, which relies on photodynamic and reductive responses, in facilitating targeted drug distribution at the tumor site. Furthermore, we will consider conducting further comparative studies with other outstanding similar entities in the future.
Preliminary Safety Evaluation
The toxicity of NG, siRNA, Pc and SP-NG@NKpH on human normal liver cells (LO2) was studied. As shown in Figure 8, neither siRNA nor Pc-NG exhibited significant cytotoxicity to LO2 cells over the range of doses administered. When siRNA and Pc were administered at concentrations of 250 nM and 12.5μg/mL, respectively, the survival rate of NK cells was still higher than 80% (Figure 8B), and high cytotoxicity was not produced. In Figure 8C, NG, siRNA, Pc, and NK cells incubated with LO2 normal cells in the absence of laser irradiation, which similarly maintained around 80% cell viability. The above results indicated that the combination of NG, siRNA, Pc, and NK cells did not produce significant toxicity to normal cells at the administered dose.
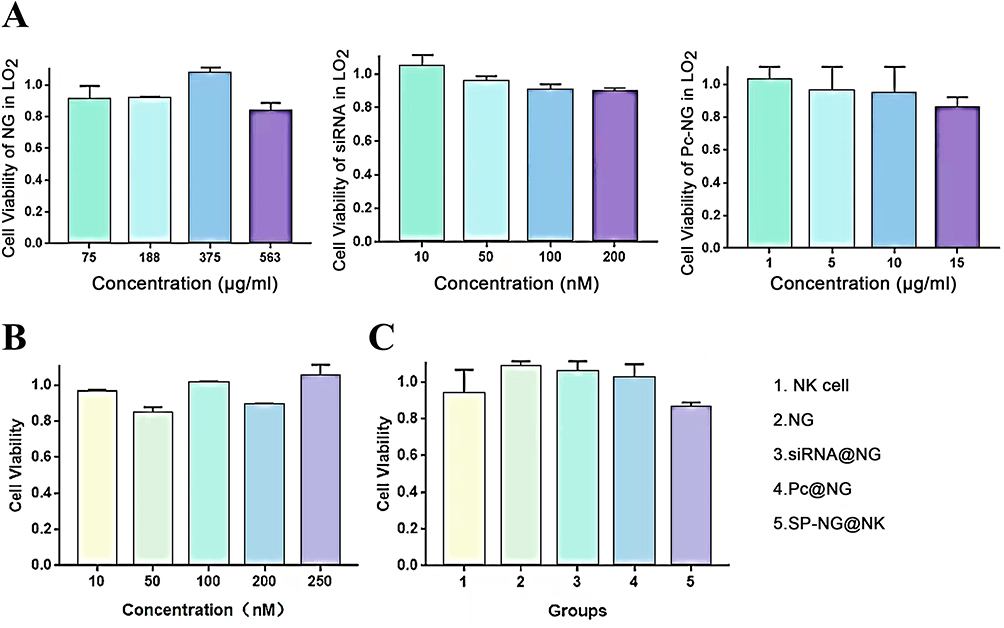 |
Figure 8 (A) Cytotoxicities of NG, siRNA and P-NG in LO2. (B) Cytotoxicity of SP-NG in NK cells. (C) Cytotoxicities of different groups in LO2. |
There is generally no reducing environment in normal cells, it is difficult for NG to be destroyed and release the drug in normal tissues, therefore the reduction reaction of NG may be safe. The siRNA is often overexpressed in various types of human tumors, whereas the expression target of PLK1-siRNA is lacking in normal tissues. Through controlling the release of the cellular carrier switch, could be demonstrated that the carrier and drug were safe in normal tissues. The safety of NK cells can be ensured through the carriers and drugs. Consequently, the complexes containing NK cells that we have developed may exhibit a notable level of safety.
Conclusion
NK cell-based immunotherapy of cancer has gained a lot of attention due to its natural killing activity, immunomodulatory ability, and in vivo safety. However, multiple immunosuppressive mechanisms in the TME suppress the anticancer effect of NK cells in the treatment of solid tumors. The smart NK cell drug delivery system with photo-responsive and TME-responsive properties (SP-NG@NKpH) were designed. And the combined effect of the immunotherapy of NK cell, the gene therapy of siRNA, and the photodynamic therapy of photosensitizer obtained a stronger cancer killing effect in vitro and in vivo. It is expected to be a promising anti-tumor drug delivery system.
Abbreviations
ApEn-NK, Aptamer-engineered NK cells; AR, Artificial receptors; N3-NK-NPs, Bio-orthogonal targeted live-cell nanocarrier; CAR-NK, Chimeric antigen receptor NK cells; FITC-pHLip-MFL, FITC-labeled pHLip-MFL; FBP1, Fructose-1, 6-biphosphatase; FR, Fusion receptor; H&E, Hematoxylin and eosin; HeLa, Human cervical carcinoma cells; HLA, Human leukocyte antigens; LO2, Human normal liver cells; ILNPs, Interleukin-21 nanoparticles; pHLip-MFL, Liposomes loaded with pHLip; MFL, Membrane-fusogenic liposome; MPS, Mononuclear phagocyte system; NIH, National Institutes of Health; NK, Natural killer; pHLip, pH-low insertion peptides; NKpH, pH-low insertion peptides on the surface of NK cells; PDT, photodynamic therapy; Pc, phthalocyanine; PEI, Polyethylenimine; γ-PGA, Polymer-polyglutamic acid; ROS, Reactive oxygen species; SD, Standard deviation; TUNEL, TdT-mediated dUTP Nick-End Labeling; PEI-SH, Thiolated PEI; TGF-β, Transforming growth factor-B; TME, Tumor microenvironment.
Ethical Approval
Ethical approval to conduct the study was obtained from the Ethical Committee on Animal Experiments of Xuzhou Medical University. And the Ethical Number is 202211S032. The guiding principle for the welfare of experimental animals is called the “3R” principle, which refers to replacement, reduction, and refinement.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (grant no. 81773643). This research was also supported by the Start-up Fund for Excellent Talents of Xuzhou Medical University (grant no. D2021055).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hosseini R, Sarvnaz H, Arabpour M, et al. Cancer exosomes and natural killer cells dysfunction: biological roles, clinical significance and implications for immunotherapy. Mol Cancer. 2022;21(1):15. doi:10.1186/s12943-021-01492-7
2. McKenna MK, Ozcan A, Brenner D, et al. Novel banana lectin CAR-T cells to target pancreatic tumors and tumor-associated stroma. Journal for Immunotherapy of Cancer. 2023;11(1):e005891. doi:10.1136/jitc-2022-005891
3. Li J, Hu B, Chen Z, et al. Mn(iii)-mediated carbon-centered radicals generate an enhanced immunotherapeutic effect. Chem. Sci. 2024;15(2):765–777. doi:10.1039/D3SC03635A
4. Zhang W, Wang M, Tang W, et al. Nanoparticle-laden macrophages for tumor-tropic drug delivery. Advanced Materials. 2018;30(50):1805557.1–1805557.11. doi:10.1002/adma.201805557
5. Zhang Y, Li S, Wang Y, et al. A novel and efficient CD22 CAR-T therapy induced a robust antitumor effect in relapsed/refractory leukemia patients when combined with CD19 CAR-T treatment as a sequential therapy. Exp Hematol Oncol. 2022;11(1):15. doi:10.1186/s40164-022-00270-5
6. Yin Y, Tang W, Ma XJCEJ. Biomimetic neutrophil and macrophage dual membrane-coated nanoplatform with orchestrated tumor-microenvironment responsive capability promotes therapeutic efficacy against glioma. Chem Eng J. 2022;433:133848. doi:10.1016/j.cej.2021.133848
7. Ding Y, Xu Q, Chai Z, et al. All-stage targeted red blood cell membrane-coated docetaxel nanocrystals for glioma treatment. Journal of Controlled Release. 2024;369:325–334. doi:10.1016/j.jconrel.2024.03.055
8. Chen M, Geng D, Yang X, et al. In vitro nephrotoxicity induced by herb-herb interaction between radix glycyrrhizae and radix euphorbiae pekinensis. Oxid Med Cell Longev. 2020;2020:6894751. doi:10.1155/2020/6894751
9. Li H, Lu R, Pang Y, et al. Zhen-Wu-Tang protects IgA nephropathy in rats by regulating exosomes to inhibit NF-κB/NLRP3 pathway. Front Pharmacol. 2020;11:1080. doi:10.3389/fphar.2020.01080
10. Li X, Liu R, Su X, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18(1):177. doi:10.1186/s12943-019-1102-3
11. Zheng P, Liu Y, Zhang X, et al. Transcriptomic and metabolomic analysis of the effects of Zhenwu decoction on kidney yang deficiency pattern in chronic kidney disease. J Traditional Chin Med Sci. 2023;10(2):228–243. doi:10.1016/j.jtcms.2023.01.002
12. Tomaipitinca L, Russo E, Bernardini G. NK cell surveillance of hematological malignancies. Therapeutic implications and regulation by chemokine receptors. Mol Aspect Med. 2021;80:100968. doi:10.1016/j.mam.2021.100968
13. Dong W, Wu X, Ma S, et al. The mechanism of Anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discovery. 2019;9(10):1422–1437. doi:10.1158/2159-8290.CD-18-1259
14. Mark C, Czerwinski T, Roessner S, et al. Cryopreservation impairs 3-D migration and cytotoxicity of natural killer cells. Nat Commun. 2020;11(1):5224. doi:10.1038/s41467-020-19094-0
15. Ji T, Lang J, Ning B, et al. Enhanced natural killer cell immunotherapy by rationally assembling Fc fragments of antibodies onto tumor membranes. Adv Materials. 2019;31(6):e1804395.
16. Zheng C, Wang Q, Wang Y, et al. In situ modification of the tumor cell surface with immunomodulating nanoparticles for effective suppression of tumor growth in mice. Adv Materials. 2019;31(32):e1902542. doi:10.1002/adma.201902542
17. Yang S, Wen J, Li H, et al. Aptamer-engineered natural killer cells for cell-specific adaptive immunotherapy. I. 2019;15(22):e1900903.
18. Xiao M, Lai W, Yao X, Pei H, Fan C, Li L. Programming receptor clustering with DNA probabilistic circuits for enhanced natural killer cell recognition. Angewandte Chemie. 2022;61(28):e202203800. doi:10.1002/anie.202203800
19. Meng D, Pan H, He W, et al. In situ activated NK cell as bio-orthogonal targeted live-cell nanocarrier augmented solid tumor. Immunotherapy. 2022.
20. Pitchaimani A, Nguyen TDT, Aryal S. Natural killer cell membrane infused biomimetic liposomes for targeted tumor therapy. Biomaterials. 2018;160:124–137. doi:10.1016/j.biomaterials.2018.01.018
21. Nguyen MK, Huynh CT, Gilewski A, et al. Covalently tethering siRNA to hydrogels for localized, controlled release and gene silencing. Sci Adv. 2019;5(8):eaax0801. doi:10.1126/sciadv.aax0801
22. Abdelfatah S, Berg A, Böckers M, Efferth T. A selective inhibitor of the Polo-box domain of Polo-like kinase 1 identified by virtual screening. J Adv Res. 2019;16:145–156. doi:10.1016/j.jare.2018.10.002
23. Alidori S, Akhavein N, Thorek DL, et al. Targeted fibrillar nanocarbon RNAi treatment of acute kidney injury. Sci, trans med. 2016;8(331):331ra39. doi:10.1126/scitranslmed.aac9647
24. Liu S, Wu F, Gu S, et al. Gene silencing via PDA/ERK2-siRNA-mediated electrospun fibers for peritendinous antiadhesion. Advanced Science (Weinheim, Baden-Wurttemberg, Germany). 2019;6(2):1801217. doi:10.1002/advs.201801217
25. Cong J, Wang X, Zheng X, et al. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metab. 2018;28(2):243–255.e5. doi:10.1016/j.cmet.2018.06.021
26. Jin W, Chen Z, Wang Y, et al. Nano metal-photosensitizer based on Aza-BODIPY-Cu complex for CDT-enhanced dual phototherapy. Chinese Chemical Letters. 2024;35(7):109328. doi:10.1016/j.cclet.2023.109328
27. Feng W, Lv Y, Chen Z, et al. A carrier-free multifunctional nano photosensitizer based on self-assembly of lactose-conjugated BODIPY for enhanced anti-tumor efficacy of dual phototherapy. Chem Eng J. 2021;417:129178. doi:10.1016/j.cej.2021.129178
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.