")
Back to Journals » Journal of Inflammation Research » Volume 17
Associations Between Neuropsychiatric Symptoms and Inflammation in Neurodegenerative Dementia: A Systematic Review
Authors Swann P, Mirza-Davies A, O'Brien J
Received 15 April 2024
Accepted for publication 30 August 2024
Published 7 September 2024 Volume 2024:17 Pages 6113—6141
DOI https://doi.org/10.2147/JIR.S385825
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Peter Swann,* Anastasia Mirza-Davies,* John O’Brien
Department of Psychiatry, University of Cambridge, School of Clinical Medicine, Cambridge, UK
*These authors contributed equally to this work
Correspondence: John O’Brien, Department of Psychiatry, University of Cambridge School of Clinical Medicine, Box 189, Level E4 Cambridge Biomedical Campus, Cambridge, CB2 0SP, United Kingdom, Email [email protected]
Background: Neuropsychiatric symptoms are common in dementia and linked to adverse outcomes. Inflammation is increasingly recognized as playing a role as a driver of early disease progression in Alzheimer’s disease (AD) and related dementias. Inflammation has also been linked to primary psychiatric disorders, however its association with neuropsychiatric symptoms in neurodegenerative dementias remains uncertain.
Methods: We conducted a systematic literature review investigating associations between inflammation and neuropsychiatric symptoms in neurodegenerative dementias, including AD, Lewy body, Frontotemporal, Parkinson’s (PD) and Huntington’s disease dementias.
Results: Ninety-nine studies met our inclusion criteria, and the majority (n = 59) investigated AD and/or mild cognitive impairment (MCI). Thirty-five studies included PD, and only 6 investigated non-AD dementias. Inflammation was measured in blood, CSF, by genotype, brain tissue and PET imaging. Overall, studies exhibited considerable heterogeneity and evidence for specific inflammatory markers was inconsistent, with lack of replication and few longitudinal studies with repeat biomarkers. Depression was the most frequently investigated symptom. In AD, some studies reported increases in peripheral IL-6, TNF-a associated with depressive symptoms. Preliminary investigations using PET measures of microglial activation found an association with agitation. In PD, studies reported positive associations between TNF-a, IL-6, CRP, MCP-1, IL-10 and depression.
Conclusion: Central and peripheral inflammation may play a role in neuropsychiatric symptoms in neurodegenerative dementias; however, the evidence is inconsistent. There is a need for multi-site longitudinal studies with detailed assessments of neuropsychiatric symptoms combined with replicable peripheral and central markers of inflammation.
Keywords: dementia, immune, depression, psychosis, biomarkers
Introduction
Dementia is a major cause of morbidity and mortality.1 Neurodegenerative dementias, including Alzheimer’s disease (AD), dementia with Lewy Bodies (DLB), Parkinson’s disease dementia (PDD) and frontotemporal dementia, are all associated with abnormal protein folding, protein aggregation and neuronal loss. Their etiology is complex and poorly understood, but increasing evidence suggests that inflammation may represent a common pathway contributing to pathological protein aggregation and neuronal loss across different neurodegenerative dementias.2,3
AD is defined by the presence of amyloid plaques and neurofibrillary tangles and is the most common cause of dementia.4 It is a clinical syndrome of progressive cognitive decline,5 usually beginning with a prodromal Mild Cognitive Impairment (MCI) stage.6 Neuroinflammation is well recognized in AD. In response to amyloid-β plaques, microglia secrete inflammatory cytokines7,8 and upregulate interferon response genes.9 Imaging microglia in vivo with positron emission tomography (PET) of the translocator protein (TSPO) receptor has identified increased ligand binding, interpreted as microglial activation, in AD.10 TSPO binding correlates with amyloid and tau pathology, as measured by 18-[F]-flumetamol and 18-[F]-AV1451 PET imaging.11 TSPO binding has also been associated with tau spreading12 and subsequent cognitive decline.13
DLB, the second most common cause of dementia,14 is characterized by visual hallucinations, parkinsonism, rapid eye movement (REM) sleep behavior disorder and cognitive fluctuations.15 α-synuclein, a hallmark of DLB pathology,16 induces an immune response in vitro and in animal models.17 There is evidence for cortical recruitment of T-lymphocytes,18 higher levels of peripheral cytokines including interleukin (IL)-6 and tumor necrosis factor (TNF)-α,19 and PET imaging of microglial activation in early DLB.20
FTD presents with behavioral-executive and language (primary progressive aphasia, including semantic variant and non-fluent) variants. Peripheral and central inflammation are well recognized across the FTD spectrum, with alterations in serum cytokines, CSF cytokines, and T-cell phenotypes in patients compared to controls.21
Neuropsychiatric symptoms (NPS) are prevalent in dementia and associated with carer stress and poor outcomes. Common NPS include depression, apathy, anxiety, delusions, hallucinations, and agitation among others.22 They often precede dementia diagnosis as they can occur in the prodromal stages of AD23 and DLB.24–26 NPS are associated with patient outcomes. This varies by neurodegenerative disease and symptom domain. For example, in longitudinal studies of Alzheimer’s dementia psychosis, agitation, and depression were associated with cognitive progression and increased dependence.27 Psychosis, affective symptoms, and agitation/aggression were associated with earlier death.28 A recent meta-analysis in PD has found that psychosis and depression are associated with cognition and disease progression.29 In DLB, higher scores on a composite NPS scale were associated with caregiver burden and depression.4 In AD, total neuropsychiatric symptoms are more closely associated with caregiver distress than cognition,30 with studies also reporting a bidirectional relationship between NPS and distress.31 Apathy in gene carriers at the presymptomatic stage of FTD predicts cognitive decline.32 Across dementia subtypes, a factor including a range of BPSD was associated with hospital admission.33 NPS are predictive of nursing home placement in AD,34 specifically with depression, delusions, and agitation increasing the risk of institutionalization across cognitive disorders.35 In PD, hallucinations are a predictor of future nursing home placement.36
NPS have been associated with specific imaging,37 neurotransmitter,38–40 and pathological changes,41,42 but there is a need for a greater understanding of the mechanisms of these symptoms to develop effective therapies,43 especially in dementias apart from AD where the evidence base in more limited.44,45 Current treatments primarily target neurotransmitters and are of limited effectiveness46 or associated with significant risks.47,48
NPS are common in inflammatory diseases.49 There is evidence that anti-inflammatory or immunomodulatory treatments may play a role in relieving depression comorbid autoimmune conditions.50 There is increasing evidence that inflammation plays a causal role in the development51 and symptoms52 of psychiatric disorders, and trials of anti-inflammatory of immune modulatory treatments are ongoing in depression.53 Several mechanistic pathways have been proposed for the relationship between inflammation and NPS. In depression, chronic stress has been linked to the release of pro-inflammatory cytokines, which has been linked to increased glutamate in magnetic resonance imaging (MRS) studies. This leads to decreased activity in corticostriatal reward networks and symptoms such as anhedonia.54 Cytokines also regulate indoleamine 2.3-dioxygenase (IDO), which metabolizes tryptophan to kynurenine, leading to quinolinic acid production, an N-methyl-D-aspartate (NMDA) receptor agonist, with this pathway linked to depression in animal models and clinical studies.55 In NMDA receptor (NMDAR)-antibody encephalitis, antibody mediated internalisation of the NMDAR leads to reduced signals in inhibitory neurons, which has been proposed as a mechanism for the psychotic symptoms.56
There are several potential mechanisms linking Alzheimer’s disease and the immune response. Firstly, as a core part of the disease pathology – microglial genes represent a large proportion of the risk for AD,57 specific microglial response to amyloid-beta and tau pathology associated with cytokine release, and microglial activation in vivo as measured by PET imaging of the translocator protein (TSPO) linked to amyloid-β58 and tau.59 This central inflammation could lead to peripheral inflammation via the release of cytokines that cross the blood–brain barrier, however there is a paucity of studies with paired measures of central and peripheral inflammation.58,60
Secondly, there is evidence that chronic diseases, such as diabetes61 and cardiovascular disease,62 are associated with risk of AD, or more rapid progression. One proposed mechanism is that the chronic low-grade inflammation associated with chronic disease or acute inflammatory events such as sepsis influence central immune responses and increase the risk of neurodegeneration.63
Mid and late life depression are risk factors for dementia.64 The mechanisms of this association are not fully understood. It could represent prodromal symptoms of neurodegenerative disease, or alternatively the chronic inflammation associated with depression could increase dementia risk. In support of this, individuals with MCI and a history of depression treated with serotonin selective reuptake inhibitors (SSRIs) showed reduced rate of progression to AD,65 and meta-analysis has shown that treatment with SSRIs is associated with a reduction in proinflammatory cytokines.66
Whether the inflammatory response is generated as part of the underlying neurodegenerative process, or secondary to other chronic disease or associated with depression, if this response is causally associated with the development of NPS, this could represent a potential therapeutic target for symptomatic relief.
Therefore, a greater understanding of the biological mechanisms underpinning NPS and the associations with inflammation is needed to develop more effective therapies. Identifying immune biomarkers associated with NPS could guide treatment trials, including repurposing of existing immunomodulatory medications and allowing stratification of clinical trials, to identify which patients are most likely to benefit from these drugs.67 Previous reviews have specifically looked at cytokines with behavioral and psychological symptoms in dementia (BPSD) in dementia,68 apathy in AD,69 and depression in Parkinson’s disease.70 We undertook a systematic review exploring peripheral and central measures of inflammation across different neurodegenerative disorders.
As there is evidence that common pathways such as protein aggregation and neuronal death are associated with common immune pathways, such as toll-like receptor signalling3 and microglial activation2 across neurodegenerative diseases. There is also evidence for distinct immune responses that depend on specific protein pathologies – such as microglial states depending on amyloid-β or tau pathology,71 and the association of CD4 cells with α-synuclein.72 For this reason, we were broad in our inclusion of neurodegenerative dementias, including AD, LBD, FTD, PSP, and their prodromes but excluding non-neurodegenerative causes, which lack these shared features.73
In order to capture the full extent of the literature, we were broad in our definition of an inflammatory marker: we aimed to identify studies that measured a biomarker74 that was related to an immune or inflammatory process. This will include cytokines, chemokines, immune cell subsets, and PET imaging of microglia. As there is evidence for both peripheral and central inflammation in dementia,63 our intention is to include markers measured in both compartments. We have also been broad in our definition of NPS as symptoms associated with changes in perception, thought, mood and/or behaviour,75 including psychosis, agitation, apathy, depression, and sleep disturbance.76
Methods
We searched MEDLINE, Embase, and PsycINFO databases through OVID on 25/07/23 with the terms:
((microglia) OR (TSPO) OR (interleukin) OR (“translocator protein”) OR (“tumour necrosis factor”) OR (TNF) OR (“c-reactive protein”) OR (CRP) OR (cytokine)) AND ((Dementia) OR (“Alzheimer’s disease”) OR (Lewy) OR (“Parkinson’s disease”) OR (“mild cognitive impairment”) OR (MCI) OR (“progressive supranuclear palsy”)) AND ((depression) OR (psychosis) OR (apathy) OR (anxiety) OR (hallucination) OR (delusions) OR (neuropsychiatric) OR (agitation))
We included no publication date restrictions but limited the search to articles in the English language. Therefore, the timeframe of the search was as follows: 1966–2023 for MEDLINE, 1947–2023 for EMBASE and 1967–2023 for PsychINFO. We included additional articles identified from the citations or those known to the authors. Our inclusion criteria were as follows: Firstly, studies of a neurodegenerative dementia (including but not limited to Alzheimer’s disease, Lewy body dementias, frontotemporal dementia or progressive supranuclear palsy) or their prodromal stages (such as mild cognitive impairment due to Alzheimer’s disease or mild cognitive impairment due to Lewy Bodies) or Parkinson’s disease. Secondly, an outcome measure of peripheral or central inflammation (such as inflammatory markers in plasma, serum, or cerebrospinal fluid (CSF), immune cell subsets, or PET imaging of immune markers). Thirdly, an outcome measure of neuropsychiatric symptoms, and finally a reported test for association or relationship between the inflammatory markers and neuropsychiatric symptoms.
Our exclusion criteria were case reports (or small cohorts with less than five participants), fundamental science research, animal studies, or non-neurodegenerative dementia. Our primary outcome was the association between an inflammatory or immune marker and neuropsychiatric symptoms in neurodegenerative dementia groups. Duplicates were removed in Endnote, and the search results were then imported into Rayyan. Articles were then screened by title and abstract, then for potentially suitable papers full text of articles was obtained. These were further screened against the inclusion and exclusion criteria. Reasons for exclusion were recorded and reported in Figure 1. From included articles we extracted from the full text into Excel the study design, setting, diagnosis (and diagnostic criteria), number of participants, demographics, inflammatory marker measured, assay source and details, psychiatric symptom measurement and by which scale, and the overall findings of the study. We did not exclude studies based on formal quality criteria (beyond the lower limit of number of participants). Our aim was to be inclusive, and extract data that allowed the quality of studies to be compared qualitatively, including the diagnostic criteria used (case definition), the source of recruitment, inclusion of controls, the specific assays used for biomarker measurement and scales used to measure NPS.
 |
Figure 1 Flow diagram describing the systematic review process. Notes: **Publication type included abstracts or conference proceedings with insufficient details, book chapters, editorials/letters without new data, or protocol papers. Study design describes reviews with no original data, case reports, animal studies, or fundamental science papers. Study population describes studies with no neurodegenerative disease group. Study outcome describes studies with no psychiatric outcome, no inflammatory measure, or no testing for association between psychiatric and inflammatory measures. This flow diagram is based on PRISMA reporting guidelines.162 |
Results
Our search identified 3208 articles after duplicates were removed. A total of 3074 articles were excluded by title and abstract, with 135 articles sought for full-text retrieval. These were supplemented with 5 articles known to the authors or identified from the citations. Ninety-nine articles were included in the final review (Figure 1). Many excluded studies recorded an inflammatory measure and psychiatric outcome but did not report testing for an association.
 |
Figure 2 Graphical summary of number of studies by diagnostic group. Notes: Bar chart showing the number of studies by diagnosis. Different bars represent diagnostic groups and are identified with the color key on the right. The height of the bar represents the number of studies identified by the systematic review studying each diagnostic group. |
Firstly, we will describe four key themes that emerged across studies, regardless of diagnosis, inflammatory marker, or neuropsychiatric symptom. These were the number and selection of participants, the study design, the choice of outcome measure, and the quality of the assay.
Secondly, we will report the results from each neurodegenerative diagnosis in order from most to least studied, broken down firstly by neuropsychiatric outcome, and within each outcome, assay type.
Participants
Most articles included participants with either AD (including mild cognitive impairment, n = 58) or Parkinson’s disease (n = 35). Very few articles included participants with DLB (n = 3), FTD (n = 2) or Huntington’s disease (n = 2). One study78 did not specify dementia subtype, and one study included a mixed group.79 Comparator groups included cognitively unimpaired controls, major depressive disorder (MDD), delirium, subjective cognitive impairment (SCI), vascular dementia (VD), or other neurodegenerative diseases. Whilst studies frequently included unspecified MCI or MCI due to AD (MCI-AD), there was only one study that included MCI-LB (alongside MCI-AD).80 A wide range of diagnostic criteria were used. Ten studies did not describe standardized diagnostic criteria. A graphical summary of the distribution of studies by diagnostic group is given in Figure 2. The overall study sizes ranged from 10 participants to 3073, with several large multicenter research cohorts with paired blood and CSF.81–83 However, many studies were small including less than 20 participants per group.84,85 Full descriptions of the diagnostic criteria provided in each study are summarized in Supplementary Table 1.
Study Design
Most studies were cross-sectional (n = 72), with many fewer (n = 13) including longitudinal follow-up. These studies primarily focused on baseline biomarkers on disease progression rather than measuring biomarkers at multiple timepoints.86,87 Studies were focused on outpatients, with five studies including participants as inpatients (including nursing homes).
Outcome
The most common symptom domains studied were depression and anxiety (PD, n = 19, AD/MCI-AD, n = 26, DLB/MCI-LB, n = 1). Less frequently studied were psychosis, apathy, agitation, and sleep disturbance. Many studies used a composite measure of NPS (AD/MCI-AD, n = 20, DLB/MCI-LB n = 3) with fewer studies separating the symptom domains.88 A graphical summary of the number of studies by neuropsychiatric symptom outcome is given in Figure 3. Controlling for disease severity was variable. Those with NPS often had lower cognitive scores,89,90 or studies identified groups with increased inflammation, more psychiatric symptoms, and more cognitive or motor symptoms.91,92
 |
Figure 3 Graphical summary showing the proportions of studies by neuropsychiatric symptom outcome measure. Notes: Tree plot showing the proportions of studies exploring each neuropsychiatric symptom. The size of the area within the grid represents the number of studies, and each colored area represents a different neuropsychiatric symptom, as described by the key on the right. Composite symptoms represent those using an outcome with multiple symptom domains summed together, like the total NPI score. |
Assays
Most studies used blood biomarkers of inflammation (n = 58), with fewer having paired CSF (n = 9), or CSF alone (n = 8). Studies on blood were primarily performed on serum and plasma, with some using peripheral blood mononuclear cells (PBMCs). Less commonly used were genetic, epigenetic or RNA sequencing. There were only two imaging studies, both in AD.93,94 Both used PET imaging of the TSPO receptor (one with 11[C]-DPA-713 and one with 11-[C]-PBR28). A summary of number of studies by assay source and sample size is given in Figure 4. Some studies provided detailed descriptions of the assay used, and the parameters around sample collection,95 whilst others provided minimal details.96 Individual studies mostly measured single or small numbers of select inflammatory markers using separate ELISAs, with multiplex methods becoming more common over time (Figure 5).97 Major immune biomarkers like C-reactive protein (CRP), IL1-β, IL-6 and TNF-α were frequently assayed. Across studies there was a spread of biomarkers assessed, with a range of in-house and commercial assays, and limited consistency between studies. These include, but are not limited to, interleukins (such as IL-2, IL-4, IL-6, IL-8, IL-10, and IL-12), interferons (such as IFN-γ), immune cell subsets as measured by flow cytometry (including CD3, CD4, CD8 subsets, and dendritic cell subsets) either stimulated or unstimulated, and mRNA expression from whole blood. There were several studies that looked at immune gene single nucleotide polymorphisms (SNPs) in association with psychiatric symptoms98–107 in small cohorts. Full descriptions of the assays are provided in Supplementary Table 1.
 |
Figure 4 Bar chart of number of participants in each study. Notes: Bar chart showing the number of participants in each study, colored by the assay source. The x-axis displays the number of studies, and each bar on the y-axis represents the number of participants in each study. The assay source is shown by the color key on the right. |
 |
Figure 5 Bar chart showing the change in assay type for studies measuring fluid inflammatory markers. Notes: Each bar represents an epoch of years, and the y-axis shows the number of studies in each year measuring fluid inflammatory markers, split into those using single ELISAs (purple) and those using multiplex assays (yellow). |
Alzheimer’s Disease and MCI-AD
Studies in AD predominantly examined the relationship between inflammation and depression (n = 26), followed by those using composite scores (n = 20). Few studies looked at other individual symptom domains (agitation, apathy, psychosis, and sleep disturbance). All studies looked at sporadic AD or did not specify whether genetic cases were included. Full descriptions of the studies are included in Supplementary Table 2.
AD-Depression
Neuropathology
Two studies used brain tissue. The first108 focused on neutrophil gelatinase-associated lipocalin (NGAL), an innate immune protein associated with Alzheimer’s pathology and depression. They identified increased NGAL levels in the hippocampus of AD patients with depression. This relationship varied by region, with areas of the cingulate having higher NGAL levels in those without depression. This study combined neuropathology with serum and CSF. There were no differences in serum levels of NGAL, but a significant decrease in CSF NGAL. A second study109 used a range of microglial, cell adhesion, and cytokine markers and identified IL-4 as significantly increased in AD-depression compared to AD cases without depression.
Paired CSF and Blood
A large study81 examined a panel of immune biomarkers in CSF, plasma and serum focusing on the link between inflammation and the metabolism of tryptophan via the kynurenine (Kyn) pathway, which has been linked to depression.110 This study identified a higher Kyn/5-HT ratio associated with peripheral immune markers, and this ratio was associated with increased depression and anxiety scores. This study identified a possible shared mechanism between primary depression and depression in AD.
CSF
Total depression scores were not associated with CSF IL-6 or brain-derived neurotrophic factor (BDNF), but there were associations with individual symptom domains (psychomotor slowing and nighttime disturbance).111 Although a single exploratory study, it is of interest as inflammation-driven reductions in BDNF have been implicated in depression112 and psychomotor slowing.113 One study found a micro-RNA (miR-451a) that regulates toll-like receptor (TLR) 4 was lower in AD with depression and correlated with depressive symptom scores.114
Serum and Plasma
There are three studies that measured IL-6 in AD with depression and found an association.89,115,116 This is not without caveats; AD with depression groups often had lower cognitive scores,89,116 or findings had wide confidence intervals and limited details of assays.115 One large study in AD did not find any associations with IL-6 and depression scores,117 and another in MCI118 does not does not report similar findings; however, this was in outpatients with diabetes with MCI diagnosed based on cognitive scores.119 One study measured IL-6 with a composite outcome measure including depression and is detailed in the section below.86 Details of studies measuring either IL-6 or TNF-α in association with depression or anxiety in AD are given in Table 1.
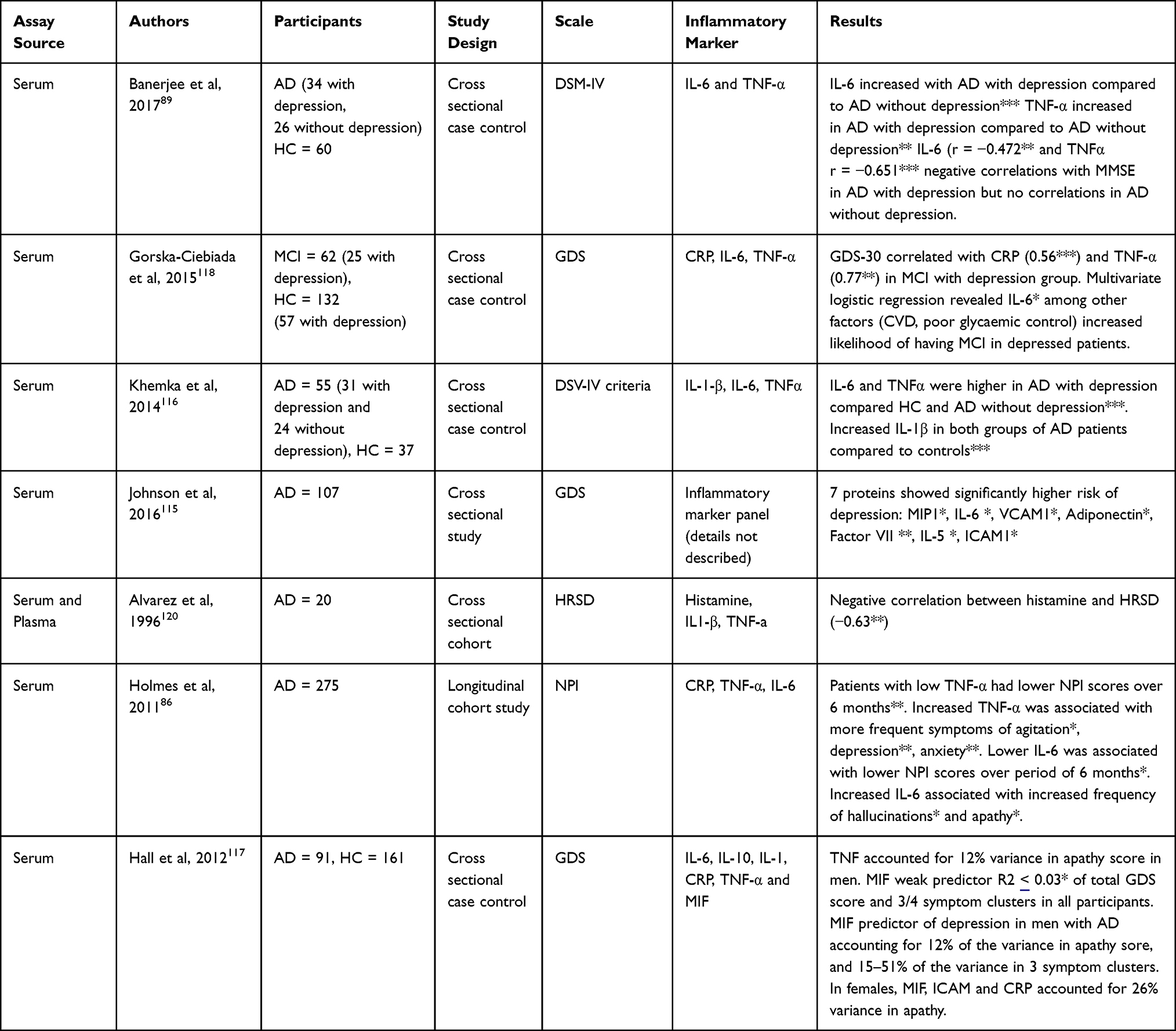 |
Table 1 Studies Measuring Peripheral IL-6 or TNF-α in Association with Depression or Anxiety in Alzheimer’s Disease. Number of Asterisks Denote Significance Level (* P<0.05, ** P<0.01, *** P<0.001) |
Two studies from a large multi-centre research cohort (the Texas Alzheimer’s Research and Care Consortium) have studied the impact of inflammatory biomarkers on depressive symptoms. CRP was a significant predictor of the apathy domain of depression scores, and in combination with MIF and ICAM accounted for 26% of the variance in apathy scores in one study.121 In another study from this cohort, this effect appeared to be driven by female AD cases.122 Another study123 found no association between CRP, IL-1β, or transforming growth factor (TGF)-β and total geriatric depression scale (GDS) scores, despite significant group differences of IL-1β and TGF-β compared to controls, and correlations with cognitive scores.
Two studies89,116 also found higher levels of TNF-α in AD cases with depression, with the second finding a correlation between TNF-α and depressive symptoms, and a further study finding TNF-α contributed to an apathy in men.117 One study did not find an association,120 but this was a small study (n = 20) and did not include details of disease stage or cognitive scores. One study measured TNF-α with a composite outcome measure including depression and is detailed in the section below86 (Table 1 and Figure 6).
 |
Figure 6 Graphical summary of studies testing the association of TNF-α or IL-6 in association with depression or anxiety in AD. Notes: Studies measuring either TNF-α or IL-6 in association with depression or anxiety in AD. Size of the point and the number represent the study size, and color whether the study demonstrated broadly positive (blue) or negative (red) results. Studies in the figure are Alvarez et al 1996,120 Banerjee et al, 2017,89 Gorska-Ciebiada et al, 2015,118 Hall et al, 2012, Holmes et al, 2011,86 Johnson et al, 2012115 and Khemka et al, 2014116 (see “AD-Depression” section in the main text and Supplementary Table 2 for full details of individual studies). |
Studies found higher levels of vascular endothelial growth factor (VEGF)124,125 in AD cases with depression compared to AD cases without depression, or controls, including two large studies81,95 as part of a multiplex panel, and significantly lower levels of the associated protein endocan.126
One study127 found associations between IL-7 and depressive symptoms, but these correlations were inverted depending on the assay source (positive correlation in serum and negative correlation in plasma), and differential effects in males compared to females. This highlights the need to consider assay source. A study that measured messenger ribonucleic acid (mRNA) by reverse transcription polymerase chain reaction (RT-PCR) in PBMCs128 found that whilst a number of cytokines had increased transcripts (interferon (IFN)-γ, TNF-α, IL-1R2) were increased in late life depression, those with AD and depression had downregulation of IL1-β. Measuring triggering receptor expressed on myeloid cells (TREM)1 mRNA in blood identified a significant negative correlation with depressive scores.129 TREM1 is related to TREM2, identified as a key inflammatory risk gene in AD.
AD- Composite
Imaging
Two studies93,94 looked at the association between neuropsychiatric inventory (NPI) scores and regional bindings of PET ligands to the TSPO receptor. The first93 found agitation associated with increased TSPO binding in temporal regions, not explained by cognition or AD pathology (although could potentially be explained by differences in TSPO binding between controls and those with MCI/AD). This study was from a large well-characterized cohort with multimodal PET imaging. Interestingly, the second94 also identified agitation associated with TSPO binding, again excluding confounders.
Paired CSF and Blood
A longitudinal study measured CSF and serum inflammatory markers in cognitively unimpaired older adults, those with MCI, and those with AD, in association with NPS measured by the NPI-Questionnaire (NPI-Q).92 This study identified markers in both the CSF (IL-8, IL-15, soluble intracellular adhesion molecule (sICAM)-1 positively, and inducible protein (IP)10 and CRP negatively) and serum (macrophage inflammatory protein (MIP)-1α, IL-6, Eotaxin-3 positively and CRP negatively) associated with the presence of NPS, suggesting different pathways may be involved peripherally and centrally. These relationships varied when controlling for cognitive impairment and AD biomarker profiles, with CRP levels appearing the most robust. sICAM-1 was associated with longitudinal decline, highlighting how central nervous system (CNS) inflammation contributes to both NPS and cognitive decline.
CSF
There was an inverse relationship between IL-10 and NPS in one study.78 This study specifically targeted recruitment towards those with NPS, capturing a greater range of severity. Studies also identified inflammatory markers associated with cognition (IL-1β, IL-6, and IL-12p70)130 or disease state (gasdermin, a pyroptosis marker)131 but not NPS.
Serum and Plasma
A number of small studies found no relationship between single peripheral cytokines measured by ELISA (TNF-α or IL-1β) and NPS.132,133 Others using multiplex assays134 found no significant relationships; however, one study135 found that relationships depending on APOE status, with lower IL-15 being a significant predictor of NPS in APOE4 carriers, and lower IL-18 and TNF-α were significant predictors in non carriers, although this study did not control for disease stage or severity. A large86 (n = 275) longitudinal study with repeat biomarker measurement found that lower TNF-α predicted lower NPI scores over six months, and higher TNF-α was associated with increased agitation, depression and anxiety, with raised IL-6 associated with hallucinations and apathy. There were no associations between CRP and NPI scores.
Homocysteine, involved in a number of biological processes correlated with inflammatory markers, was associated with increased risk of NPS in a single study.136
PBMCs
Two studies used stimulated PBMCs84,137 which overcomes some challenges of measuring cytokines in serum,138 and found that IL-6 expression was associated with total NPI scores, and psychosis subscale scores. An unstimulated PBMC study found an association between NPI depression subscale and myeloid dendritic cells.139
AD-Agitation
Four studies looked specifically at agitation in AD disease, and each of these studies included an inpatient setting. One study included CSF,140 finding a relationship between the TSPO ligand diazepam-binding inhibitor (DBI) and agitation. DBI was correlated between CSF and serum, and this study is particularly notable as agitation and irritability were both the primary NPS associated with PET imaging of the TSPO receptor. Other studies using serum or plasma identified Il1-β, IL-6, natural killer cell activity (NKCA), and TNF-α associated with agitation.141–143
AD-Apathy
One of the largest studies identified82 (n = 1319) measuring a panel of inflammatory markers identified IL-6 and IL-10 as significant predictors of apathy scores, whilst two small studies identified associations with soluble TNF receptors,85 ICAM-1, and IL-1.144
AD-Psychosis
One study identified reduced IL1β from stimulated PBMCs as associated with psychosis in AD,145 and another found significantly higher IL-8146 and decreases in other cytokines in AD patients without psychosis, which may relate to antipsychotic treatment. A large study that included AD among dementia subtypes found no relationship between CRP and psychosis.79 Although this study grouped together a wide range of dementia diagnosis, it was one of the few performed on an inpatient ward.
AD-Sleep
Two studies by the same author96,147 looked at the relationship between inflammation and sleep in AD using actigraphy, and found no associations in MCI, and IL-6 associated with increased total sleep time and reduced sleep latency in dementia. There were limited details described in these studies, but they are unique in using a biosensor to measure outcomes.
AD-Summary
In summary, there are some potential associations between peripheral and central markers of inflammation and NPS in AD, but primarily without replication or with divergent results between studies. There is interest in how these relationships may be affected by the assay source127 or mediated by APOE status.135 Studies included potential mechanisms for associations, such as tryptophan metabolism81 and reductions in BDNF.111 The most studied combination is peripheral IL-6 and depression, with replication across three studies (although with a high degree of uncertainty). A large study found an association with IL-6 and apathy.82 The other largest studies identified positive associations with NPS and markers including homocysteine,136 TNF-a,86 and CRP,148 or combinations of markers in multiplex panels.81,95 Whilst there are only two PET imaging studies, both found associations between microglial activation and agitation93,94 suggesting there may be both central and peripheral immune associations with NPS.
Parkinson’s Disease
Thirty-five studies explored the relationship between inflammation and NPS in PD. Similarly to AD, the majority of studies looked at the relationship with depression and anxiety. A few studies explored hallucinations. The majority were in idiopathic PD, however two studies included participants with the LRRK2 mutation.91,149 The most common diagnostic criteria used was the UK Brain Bank criteria. Full descriptions of the studies are included in Supplementary Table 3.
PD-Depression
CSF
There were eight studies testing the relationship between inflammatory markers in the CSF and depression in PD.150–157 A variety of markers were examined; however, the most common were IL-6, TNF-α, and IL-1β. The only markers with replicated findings were CRP and IL-10, showing positive associations in two studies.150,153,154,156
Serum and Plasma
Eight studies examined serum inflammatory markers and symptoms of depression or anxiety87,91,149,157–160 with one161 looking at depression as part of a composite measure. Four studies examined inflammatory markers in plasma.77,162,163 The most consistent positive associations were for IL-6 and TNF-α, although results were generally mixed. Details of studies measuring either IL-6 or TNF-α in association with depression or anxiety in PD are given in Table 2 and displayed in Figure 7.
 |
Table 2 Studies Measuring Peripheral IL-6 or TNF-α in Association with Depression or Anxiety in Parkinson’s Disease. Number of Asterisks Denote Significance Level (* P<0.05, ** P<0.01, *** P<0.001) |
 |
Figure 7 Graphical summary of studies testing the association of TNF-α or IL-6 in association with depression or anxiety in PD. Notes: Studies measuring either TNF-α or IL-6 in association with depression or anxiety in PD. Size of the point and number represent the study size, and color whether the study demonstrated broadly positive (blue) or negative (red) results. Studies in the figure are Fu et al, 2023,158 Karpenko et al, 2018,153 Kim et al, 2022,166 Lindqvist et al, 2012,161 Menza et al, 2010,163 Rastegar et al, 2019,149 Selikhova et al, 2002,162 Veselý et al, 2018,87 Wan et al, 2023,164 and Wang et al, 2016165 (see main text and Supplementary Table 3 for full details of individual studies). |
The most studied individual biomarker was IL-6, with nine studies examining the association with peripheral IL-6 and depression or anxiety in PD. Four studies found an association with depression or anxiety. Two studies158,162 found a positive correlation with IL-6 and depression scores, and two longitudinal cohorts87,149 found that IL-6 positively correlated with an increase in depression scores over 2 years, suggesting that increased inflammation predicts subsequent NPS. Other studies165 found no association, or did not directly test the association directly but via miRNA intermediaries.164 The largest studies found positive associations,149,158 except for one which was stratified based on a range of variables beyond NPS.91 The discrepancies between the studies may also be explained by the use of plasma or serum, assay choice, depression scale, and inclusion of covariates. In studies that reported cognitive scores, IL-6 was also associated with worse cognition and more rapid disease progression.158,162
Three studies149,161,163 found an association between depression and peripheral TNF-α, with one showing that along with 13 other cytokines, TNF-α correlated with increases in GDS scores over two years. Four studies found no association between depression and TNF-α. Here there were similar sized studies, with differing assay methods and depression scales finding divergent results,149,153 and differences between studies including both idiopathic PD and LRRK2 cases.91,149
A large study91 stratified participants into three groups based on clinical variables and found that IL-8 (involved in neutrophil recruitment) was higher in a “malignant” subtype of PD patients with the LRRK2 mutation. This subgroup had higher levels of inflammatory markers, including monocyte chemoattractant protein (MCP)-1, and more severe symptoms, including higher levels of depression. One other study found no association with MCP-1149 and another found an association with MCP-1 but in the CSF, but do not report serum results,161 with a separate study finding an association with plasma MCP-1 and depressive symptoms.167
Two studies examined IL-1091,149 finding positive correlations with baseline and longitudinal GDS scores.
Three studies measuring CRP in serum87,160,161 found no association with depression, representing the larger cohorts, whilst two studies77,165 found higher rates of depressive symptoms were associated with elevated CRP. sIL-2r was found to be associated with depression in two studies.161,165 Other studies measuring single inflammatory markers (VIP, S100β, miR-218-5p) found potential associations that would need replication.
PBMCs
Three papers examined PBMCs in relationship to depression in PD.168–170 These studies used different panels looking at different immune cell populations and activation markers. Two168,170 found an association between natural killer (NK) cell populations (one an association between %CD56+ve cells, and one higher levels of p11, a protein involved in endo/exocytosis and linked to mood disorders). The third found an association with depression and myeloid dendritic cells, an association found in AD by the same group.169
Genetics
The largest study identified by our search83 incorporated genetics with disease phenotype in three independent longitudinal cohorts. This identified a cluster where anxiety and depression were associated with a severe non-motor phenotype, rapid disease progression, and AD pathology. This group was associated with increased risk genes expressed in microglia, implicating an inflammatory driver of a severe, neuropsychiatric disease phenotype.
An epigenetics study171 found 35 CpG regions enriched in immune pathways (IFN- γ signaling and T-cell receptor signaling) associated with depression in PD, and the PD with depression group had higher neutrophils, lower CD4 cells, and a higher neutrophil-to-lymphocyte ratio.
PD-Psychosis
Three studies looked at the relationship between inflammatory markers and psychosis in PD. One study172 found increased CRP in PD participants with either illusions or hallucinations, but these participants were also older and had lower cognitive scores, one found no associations with a range of cytokines,173 and one found an association with an IL-6 polymorphism and visual hallucinations. This study looked specifically at the development of visual hallucinations during dopaminergic drug treatment, and measuring a single genetic polymorphism in a small sample size risks a false positive result.
PD-Composite
A longitudinal study using the non-motor symptom scale (NMSS) found no correlations between individual cytokines and symptoms scores either at baseline or follow-up, but using principal components analysis identified a latent variable, representing IL-6 and IL-2, that was associated with worsening NMSS scores, mood, and apathy symptoms over time.166 One study of CSF155 found that lower TNF-α was associated with lower neuropsychiatric symptom scores. Another174 found no association between CRP and non-motor symptom scores.
PD- Fatigue and Sleep
Two studies looked at fatigue in PD, finding associations with sIL-2R161 and IL-6.90 Studies in sleep157 found lower CSF TNF-α and higher prostaglandin E2 (PGE2) in those with sleep disturbance, and no association between CRP174 and sleep quality.
PD - Summary
In summary, most studies measured the association between inflammatory markers and depression in PD. There was a replicated finding of peripheral IL-6 and depression, including increased depression scores at follow-up. IL-6 was also associated with composite NPS and fatigue. A large genetic study identified a cluster that linked inflammation to an overall more severe and rapidly progressive phenotype associated with NPS,83 as did a clinical cohort including LRRK2 participants.91 This suggests peripheral inflammation may be associated with NPS in PD, or may represent a more aggressive phenotype. Further studies are needed to draw conclusions about the impact of inflammation on other neuropsychiatric symptoms, like hallucinations and sleep disturbance, in PD.
Dementia with Lewy Bodies
Two studies specifically focused on DLB, with one study including those with MCI-LB. One used flow cytometry paired with a serum cytokine panel found no relationship between either immune cell subsets or cytokines with depression and NPI scores.175 Another study looked specifically at the relationship of domains of the NPI and cytokines (TNF-α and IL-6) in DLB.88 They identified increased TNF-α (but not IL-6) was associated with overall NPI score. Hallucinations, depression, sleep disturbance and eating disturbance domains were associated with higher TNF-α, although some of these associations could be due to age as a confounder. One study included DLB in a mixed dementia group that, as discussed, found no relationship between CRP and psychosis.79 In the only study of MCI-LB, there were no associations between a multiplex panel of cytokines and NPS.80 Full descriptions of the studies are included in the Supplementary Table 4.
Frontotemporal Dementia
One study looked specifically at the role of inflammation in apathy, a key neuropsychiatric symptom in FTD176 and found an association with B-Cell activating factor (BAFF) with apathy in behavioral variant FTD (bvFTD).
Huntington’s Disease
Two longitudinal studies177,178 investigated the role of peripheral inflammation in apathy, irritability, and depressive symptoms in HD using the problem behavior assessment scale. One found no associations with a cytokine panel, and the other found an association with CRP and apathy, but queried whether this was due to the initiation of antipsychotic medication rather than a disease process.
Full descriptions of the studies including frontotemporal dementia and Huntington’s disease are included in the Supplementary Table 5.
Conclusions and Future Directions
This systematic review identified a number of studies reporting associations between inflammatory biomarkers and NPS in neurodegenerative dementia. However, the results are inconsistent with limited replication. Disease severity is a common confounder, making it difficult to identify whether there is a direct role for inflammation in NPS, or inflammation and NPS are epiphenomena of a more severe disease phenotype. Firstly, we will briefly discuss the heterogeneity, study design and methodological challenges that may contribute to the inconsistent findings and propose areas for further study. Secondly, despite these challenges, there are some tentative mechanisms that may overlap between psychiatric disorders and dementia we shall discuss, and finally we will suggest some recommendations for future research.
Heterogeneity
Our inclusion criteria were broad in order to capture the range of inflammatory markers measured both peripherally and centrally across neurodegenerative diseases, but despite this there was still considerable heterogeneity in study participants, design, outcome measures, and assays. These differences may contribute to the discrepancies in results. For example, a range of different diagnostic criteria were used for AD, with some using standardized criteria including biomarkers,93 and others a single score on a cognitive test.118 Some studies recruit a specific stage of disease, such as mild-moderate,132 although the majority of studies do not specify this. One potential source of heterogeneity is the use of clinical criteria in the selection of participants for cohorts. There is an increasing shift in neurodegenerative diseases to move to biological definitions rather than clinical criteria, where diagnosis depends on the detection of disease-specific biomarkers.179,180 Although diagnostic criteria are validated against neuropathological diagnosis181 and some of the studies included biomarkers of amyloid-beta and tau in AD93 or specific genetic diagnosis of PD,91 a major potential source of bias is the reliance of clinical criteria, unsupported by biomarkers, as case definitions. This may lead to participants with a variety of underlying neuropathology under the same category and fail to capture cases with mixed pathology. As biomarkers for major neurodegenerative diseases become more widely accessible,182,183 future studies could incorporate cohorts with biomarker evidence of pathology, or examine the relationships between pathology, immune response and NPS independently of clinical diagnosis. Measures of outcomes also vary, for instance, depression can be measured with a clinical diagnosis,89 a self-reported measure such as the GDS81 or as a composite in an informant-related scale such as the NPI.135 Finally, for the same inflammatory marker, there are multiple assays used for quantification. TNF-α is measured using ELISAs from different manufacturers,132,147 as part of multiplex panels,175 with a range of preanalytical variables inconsistently reported.
Study Participants
Small studies, without defined diagnostic criteria, can lead to inconsistent results. Larger studies,81,86,92 including participants from multiple sites, and replicating in independent cohorts83 can allow standardization of recruitment, diagnostic criteria, and increase power to detect or refute effects. This is especially important for non-AD dementias, such as DLB and FTD. Despite prominently being associated with NPS and with few treatment options, there is limited research in these groups.
A combination of validated diagnostic criteria supported by biomarkers can improve reliability of results, and this is increasingly possible with novel blood biomarkers.184
It is also important to study the spectrum of disease, including the early and prodromal stages, such as MCI-AD and the more recently recognized MCI-LB,24 where inflammation is increasingly recognized as important.20,185 This is with an awareness that NPS are most common in inpatient settings,186 and whilst research can be more complex in these settings, there may also be a greater need for novel interventions.
Study Design
The role of inflammation develops as disease progresses.187 For example, triggering receptor on myeloid cells 2 (TREM2), a lipid sensing receptor on microglia and macrophages, rises and falls with disease progression.188 Therefore, the relationship between inflammation and NPS may change over time, which would require longitudinal studies to detect. There is an advantage to longitudinal studies with biomarker sampling at multiple time points, alongside cognitive and NPS scales, to assess disease progression.86 This would allow the dynamic changes in immune response across a disease course to be captured, and disentangle relationships between inflammation and symptoms that may be confounded by disease stage or severity. In the studies identified, there are very few longitudinal studies, and those with repeat biomarkers are even rarer. For example, there is evidence for microglial markers188 changing with disease progression in AD and microglial activation as an early event in DLB.20 Other factors such as neurodegeneration may contribute increasingly to neuropsychiatric symptoms over time. This raises the question as to whether the associations between microglial or other inflammatory markers and NPS vary across the disease continuum. There was evidence from some studies in MCI92,118 of associations between CSF and peripheral inflammatory markers and NPS, although some studies found no relationship in MCI.96 Others, such as the relationship between dendritic cell subsets and depression, found this relationship only emerged with the progression of dementia from MCI.139 One study found that the mediating impact of cytokine pathways on the relationship between blood metabolites and depression varied depending on amyloid-β status.81 Other studies did not report the differences depending on MCI or AD diagnosis.95 We would recommend longitudinal studies from the prodromal stages including microglial markers, and measures of neurodegeneration to answer these questions.
Many studies did not incorporate other markers of pathology, neurodegeneration, disease stage, severity, or heterogeneity (such as APOE4 status in AD) that may explain the apparent relationships between NPS and inflammation. Studies that explored the relationship between clinical symptoms91 and disease progression92 found inflammatory biomarkers associated with a more severe disease phenotype. For example, the neuropathology of AD has been associated with psychosis, with evidence for higher Braak stage, increased neuritic plaques, and the presence of Lewy body pathology.189 Two studies incorporated neuropathology in AD in this review108,109 and whilst they used Braak staging to confirm the diagnosis, they did not measure the links between pathology, inflammation and NPS. Therefore, the differences in inflammatory response could reflect different severity of pathology, or the presence of co-pathology, rather than be the direct cause of NPS. Incorporating biomarkers of pathology, alongside markers of inflammation, could help delineate these respective causes.
Another challenge in the studies identified is the limited replication. Multisite studies have the advantage of being able to recruit larger number of participants, which is associated with more robust results,82 and other studies made use of independent replication cohorts to support findings.83 These study designs are essential to progress beyond the limited replication in many smaller studies.
Due to the importance of both peripheral and central inflammation,8 studies were powerful when they measured biomarkers in both blood and CSF, and future studies could capture this by combining blood biomarkers with neuroimaging. Only two imaging studies were identified in this review, both in AD. This is despite numerous studies on primary psychiatric disorders such as major depression and schizophrenia, and studies focusing on dementia and cognition.190 PET ligands in development191 may better capture the spectrum of microglial states,192 and future studies could better capture how microglial phenotype relates to NPS across neurodegenerative diseases. We must acknowledge the challenges associated with CSF collection and PET imaging in terms of patient acceptability and cost, particularly for longitudinal studies, and the ultimate goal would be to validate widely accessible peripheral markers that capture neuroinflammation. One such approach is the use of brain-derived exosomes, which are emerging as potential biomarkers in neurodegenerative disease.193 This could include microglial-derived exosomes detected in blood, although there are challenges in extracting these due to the shared surface markers between microglia and peripheral immune cells.194
Genetic studies reported single SNPs98,99 in small cohorts. Future studies could build on this, such as GWAS of psychiatric symptoms in dementia195 or Mendelian randomisation.196
Outcomes
It can be challenging to accurately measure NPS in dementia, with overlap between depression scales and those for behavioral and psychological symptoms. There are also multiple biopsychosocial mechanisms that contribute to symptoms,43 making it challenging to identify single contributors in cohort studies. Looking at individual symptom domains alongside total scores or using data-driven clustering may help identify patterns of NPS associated with inflammation.197
Assays
Various details around sample handing198 can impact cytokine results, as can the choice between serum and plasma,199 and choice of anticoagulant.200 Factors such as time of day,201 diet, alcohol and smoking202 can impact cytokine levels. Whilst not all of these factors can be controlled for, studies can report the minimum standards of information and where possible organize consistent pre-analytical variables. This makes it challenging to draw firm conclusions, and inconsistent results could be explained by choice of inflammatory marker, preanalytical variability, assay source or variability, before even considering disease stage or heterogeneity.
Studies are increasingly moving from measuring single markers by ELISA to multiplex assays. This has the advantage of capturing the range and interrelationships of inflammatory marker patterns, including using principal component analysis to identify latent variables of inflammation associated with psychiatric symptoms83,166 or cluster clinical phenotypes with inflammatory marker profiles.91 Multiplex or proteomic studies have the advantage of capturing a greater range and complexity of immune dynamics, however clusters or latent variables may be challenging to replicate in independent cohorts. Using proteomics for discovery, followed by creating more selective biomarker panels and replication in independent cohorts could be a way forwards.203
Shared Mechanisms
Despite these limitations, there are a few areas of overlap between studies identified by our search, and the literature on psychiatric disorders.
Peripheral Inflammatory Markers
Several studies found associations between IL-6 and depression or apathy in AD82,89,115,116 or PD.87,149,151,153,158,161,162 IL-6 levels are associated with depression in population studies,52 and there is genetic evidence that this effect is potentially causal.204 IL-6 inhibition in inflammatory diseases reduces depressive symptoms, and there are ongoing trials in major depressive disorder (MDD).205 Similarly, the AD and PD studies found associations with TNF-α and depression89,116,149,161,163 or agitation,86 although results were not consistent.132 TNF-α levels are raised in MDD, and trials for TNF-α inhibitors in a post-hoc analysis showed an improvement in depressive symptoms in those with MDD and higher levels of baseline inflammation.67 Similarly, inconsistent results were found for the association of CRP in neurodegeneration with some studies in AD121 and PD77,165 finding positive results. Low-grade inflammation, at least in a proportion of patients, is associated with the diagnosis of depression.206 A possible mechanism that links peripheral cytokine changes to depressive symptoms is the impact on functional connectivity207
Tryptophan Metabolism
One study81 found links between peripheral inflammation, tryptophan metabolism, and depression in AD. In MDD, studies have shown peripheral inflammation associated with TNF-α, leading to a shift in tryptophan metabolism via the kynurenine pathway,208 and this is linked to hippocampal and amygdala volumes.209
Neutrophils
In AD, neutrophils were associated with depression in a neuropathology study108 and in PD, with increased IL-891 and higher neutrophil-to-lymphocyte ratio.171 Neutrophil infiltration has been linked to depressive behavior in animal models.210 Genes regulating neutrophil function are differentially expressed in MDD.211 Neutrophils are increased in the blood of MDD patients and associated with symptom scores.212 In animal models, stress-induced neutrophils accumulate in the brain and are associated with blood–brain barrier breakdown and alterations in the nucleus accumbens leading to anhedonia.213
TSPO Pet
Two PET studies found TSPO binding in AD to be associated with NPS, such as irritability and agitation. TSPO binding in the anterior cingulate is increased in depression.214 Some studies showing this normalizes following treatment, and baseline TSPO expression predicts depressive symptom response to anti-inflammatory drugs.215 Animal models of AD pathology have shown that sleep deprivation is associated with microglial reactivity, so it is important to consider that NPS that disrupt sleep may themselves be the cause of increased neuroinflammation, rather than the result.216
Conclusion and Recommendations for Future Research
This systematic review identified a number of papers that tested for associations between inflammatory biomarkers and NPS in neurodegeneration. There are a number of studies that report associations, suggesting potential for a targetable mechanism of this crucial contributor to distress, poor outcomes and disease progression. In order to make progress, we would suggest several recommendations. In the design of studies, we would recommend using standardized clinical criteria, supported by diagnostic biomarkers for the selection of participants. We have made a case for longitudinal studies with multiple biomarker assessments and detailed neuropsychiatric symptom measures, where possible incorporating multicenter cohorts with an aim to replicate in independent samples. Detailed information about disease stage and comorbidity would reduce the impact of confounding on results. Pilot studies could be used to identify the optimum assays that are disease relevant and replicable, or robust biomarkers that are not assay dependent. This could lead to standardization of immune biomarkers across studies. We would recommend studies that aim to have, at least in subgroups, measures of both peripheral and central inflammation. This would support the identification of accurate peripheral markers of neuroinflammation, but also to separately assess the relative contributions of peripheral and central immune responses. There is also a lack of studies of non-AD dementias, which may have shared or distinct immune mechanisms. These studies could assess the relative contribution of inflammation, pathology, and neurodegeneration to NPS, identify mechanisms behind associations, and link peripheral to central inflammation. Longitudinal, multi-site studies are most likely to identify (or refute) associations robustly and explore how relationships vary with disease stage and progression, ultimately leading to translatable biomarkers or novel therapeutic targets.
Abbreviations
5-HT, 5-hydroxytryptamine; AD, Alzheimer’s disease; AFP, Alpha-fetoprotein; AMDP, Amsterdam Dementia Screening Test; APOE, Apolipoprotein e; ASA, Activities-specific Balance Confidence Scale; BA24, Brodmann area 24; BAFF, B cell-activating factor; BDI, Beck Depression Inventory; BDNF, Brain-derived neurotrophic factor; bFGF, Basic fibroblast growth factor; BPRS, Brief Psychiatric Rating Scale; BPSD, Behavioral and psychological symptoms in dementia; bvFTD, Behavioral variant frontotemporal dementia; C3, Complement component 3; C4, Complement component 4; CCL11, C-C motif chemokine ligand 11; CCL18, Chemokine ligand 18; CCL2, C-C motif chemokine ligand 2; CCL24, C-C motif chemokine ligand 24; CCL5, C-C motif chemokine ligand 5; CCR2, C-C motif chemokine ligand 2; CCR7, C-C chemokine receptor type 7; CCR7+, C-C chemokine receptor type 7-positive; CD14, Cluster of Differentiation 14; CD19, Cluster of Differentiation 19; CD2, Cluster of Differentiation 2; CD3, Cluster of Differentiation 3; CD4, Clusters of differentiation 4; CD45, Cluster of Differentiation 45; CD45RA, Cluster of Differentiation 45RA; CD45RA+, Cluster of Differentiation 45RA-positive; CD56, Cluster of Differentiation 56; CD64, Cluster of differentiation 64; CD68, Cluster of differentiation 68; CD8, Cluster of Differentiation 8; CDR, Clinical Dementia Rating; CDR-SoB, Clinical Dementia Rating Scale-Sum of Boxes; CMAI, Cohen-Mansfield Agitation Inventory; CRP, C-reactive protein; CSDD, Cornell scale for depression in dementia; CSF, Cerebrospinal fluid; CXCL10, C-X-C motif chemokine ligand 10; DBI, Diazepam binding inhibitor; DLB, Dementia with Lewy Body; DSM-III, Diagnostic and Statistical Manual of Mental Disorders; DSM-IV, The diagnostic and Statistical Manual of Mental Disorders; EDTA, Ethylenediaminetetraacetic acid; EEG, Electroencephalogram; ELISA, Enzyme linked immunosorbent assay; ENA-78, Epithelial-derived neutrophil-activating peptide 78; ESS, Epworth Sleepiness Scale; ET-1, Endothelin-1; FABP, Fatty acid-binding protein; FACIT-f, Functional Assessment of Chronic Illness Therapy-Fatigue; FTD, Frontotemporal dementia; FU, Follow-up; GCSF, Granulocyte colony stimulating factor; Geriatric depression scale, GDS; GH, Growth hormone; GSDMD, Gasdermin-D; GWAS, Genome wide association study; HADS, Hospital Anxiety and Depression Scale; HAMD/HAM-D, Hamilton depression rating scale; HAMD17, Hamilton Depression Rating Scale (17-item version); HAMD24/HAMD-24, Hamilton Depression Rating Scale-24; HC, Healthy control; HD, Huntington’s disease; Histamine: HPLC, Histamine measured using High-Performance Liquid Chromatography; HLA-DR, Human Leukocyte Antigen-DR isotype; HLA-DR+, Human Leukocyte Antigen-DR-positive; HRSD, Hamilton rating scale for depression; hsCRP, High sensitivity CRP; Iba1, Ionised calcium-binding adaptor molecule 1; ICAM, Intercellular adhesion molecular; ICD-10, International Classification of Diseases; IFN-γ, Interferon gamma; IGF-1, Insulin-like Growth Factor 1; IL-10, Interleukin-10; IL12p40, Interleukin 12 p40; IL-12p70, Interleukin-12p70; IL-13, Interleukin-13; IL-15, Interleukin-15; IL-17, Interleukin 17; IL-17A, Interleukin-17A; IL-18, Interleukin 18; IL-18BP, Interleukin-18 binding protein; IL-1B-511, Interleukin 1 Beta (gene); IL1-R2, Interleukin 1 receptor type 2; IL-1R2, Interleukin 1 receptor, type II; IL-1ra, Interleukin 1 receptor antagonist; IL-1-α, Interleukin 1 Alpha; IL1-β, Interleukin-1β; IL-2, Interleukin-2; IL-23, Interleukin 23; IL-3, Interleukin 3; IL-37, Interleukin-37; IL-4, Interleukin-4; IL-5, Interleukin 5; IL-6, Interleukin-6; IL-7, Interleukin-7; IL-8, Interleukin-8; IL-9, Interleukin-9; IP-10, Interferon gamma-induced protein 10; Kyn, Kynurenine; LOAD, Late-Onset Alzheimer’s Disease; LP, Lumbar Puncture; LRRK2, Leucine rich repeat kinase 2; MADRS, Montgomery-Åsberg Depression Rating Scale; MAES, Multiple Abilities Self-Report Questionnaire; MCI, Mild cognitive impairment; MCI-LB, Mild cognitive impairment with Lewy Body; MCP-1, Monocyte chemoattractant protein-1; MCP-4, Monocyte Chemoattractant Protein 4; M-CSF-1, Macrophage colony stimulating factor 1; MDC, Macrophage-Derived Chemokine; MDD, Major depressive disorder; MDS-PD, Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; MIF, Macrophage migration inhibitory factor; MIP1, Macrophage inflammatory protein 1; MIP-1 α, Macrophage Inflammatory Protein 1 Alpha; MIP1a, Macrophage Inflammatory Protein-1; MIP1-β, Macrophage Inflammatory Protein 1-beta; MMP-3, Matrix metalloproteinase-3; MMP-9, Matrix metalloproteinase-9; MMPS, Matrix metallo-proteinase 3; MMSE, Mini-Mental Sate Examination; MOAS, Modified overt aggression score; MoCA, Montreal Cognitive Assessment; mRNA, Messenger ribonucleic acid; MSA, Multiple System Atrophy; NGAL, Neutrophil gelatinase-associated lipocalin; NIA-AA, National Institute on Aging and Alzheimer’s Association; NINCDS-ADRDA, National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association; NINDS, National Institute of Neurological Disorders and Stroke; NINDS-AIREN, National Institute of Neurological Disorders and Stroke and Association; NK, Natural killer; NKCA, Natural killer cell activity; NLR, Neutrophil-to-lymphocyte ratio; NMSS, Non-motor symptoms scale; NPI, Neuropsychiatric inventory; NPI-D, Neuropsychiatric Inventory-Distress; NPI-Q, Neuropsychiatric Inventory Questionnaire; NPS, Neuropsychiatric symptoms; OAS, Overt aggression score; PACAP, Pituitary adenylate cyclase-activating polypeptide; PBA, Problem Behaviors Assessment; PBMCs, Peripheral blood mononuclear cells; PCR, Polymerase chain reaction; PD, Parkinson’s disease; PDD, Parkinson’s disease dementia; PDSS-2, Parkinson’s Disease Sleep Scale; PET, Positron emission tomography; PFS-16, Parkinson Fatigue Scale-16; PGE2, Prostaglandin E2; PPQ-B, Parkinson’s Psychosis Questionnaire-Brief.; PPRS, Parkinson’s Psychosis Rating Scale; PSP, Progressive Supranuclear Palsy; PSQI, Pittsburgh Sleep Quality Index; RANTES, Regulated on activation Normal T Expressed and Secreted; RBM, Rules-based medicine; RNA, Ribonucleic acid; RT-PCR, Reverse transcriptase – polymerase chain reaction; S100β, S100 calcium-binding protein B; SAA, Serum Amyloid A; SCF, Stem cell factor; SCI, Subjective cognitive impairment; SCID-D, Structured Clinical Interview for DSM-IV Dissociative Disorders; SCOPA-S, Scales for Outcomes in Parkinson’s Disease-Sleep; sE-selectin, Soluble endothelial molecule; sFLT-1, Soluble Fms-like Tyrosine Kinase 1; sICAM-1, Soluble intercellular adhesion molecule-1; sIL-2R, Soluble interleukin-2 receptor; SNP, Single nucleotide polypeptide; SORL1, Sortilin-Related Receptor 1; STAI, State-Trait Anxiety Inventory; sTNFR1, Soluble Tumor Necrosis Factor Receptor 1; sTNFR2, Soluble Tumor Necrosis Factor Receptor 2; sVCAM-1, Circulating vascular cell adhesion molecule; TARC, Thymus and Activation-Regulated Chemokine; TF, Tissue factor; TGF-β, Transforming Growth Factor Beta; TGF-β1, Transforming Growth Factor Beta 1; TIE-2, Tyrosine Kinase with Immunoglobulin-like and EGF-like domains 2; TIMP 1, Tissue inhibitor of metalloproteinases 1; TNF RII, Tumor Necrosis Factor Receptor 2; TNF-R1, Tumor Necrosis Factor Receptor 1; TNF-R2, Tumor Necrosis Factor Receptor 2; TNF-α, Tumour Necrosis Factor alpha; TPO, Thrombopoietin; TRAIL, TNF-related apoptosis-inducing ligand; TREM2, Triggering receptor on myeloid cells 2; TSPO, Translocator protein; TTR, Transthyretin; UPDRS, Unified Parkinson’s Disease Rating Scale; VaD, Vascular Dementia; VCAM1, Vascular cell adhesion molecule 1; VD, Vascular dementia; VEGF, Vascular endothelial growth factor; VEGF-C, Vascular Endothelial Growth Factor C; VEGF-D, Vascular Endothelial Growth Factor D; VIP, Vasoactive intestinal peptide.
Funding
JOB and PS are funded by a Program grant by the Medical Research Council as part of the Dementias Platform UK. This research was supported by the NIHR Cambridge Biomedical Research Centre (NIHR203312). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
Disclosure
John O’Brien has acted as a consultant for TauRx, Novo Nordisk, Biogen, Roche, Lilly and GE Healthcare and received grant or academic support from Avid/Lilly, Merck and Alliance Medical.
The other authors report no conflicts of interest in this work.
References
1. Long S, Benoist C, Weidner W. Reducing dementia risk: never too early, never too late. Lond Engl Alzheimers Dis Int. 2023;2023:1.
2. Kwon HS, Koh S-H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9:42. doi:10.1186/s40035-020-00221-2
3. Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023;8:267. doi:10.1038/s41392-023-01486-5
4. Cao Q, Tan -C-C, Xu W, et al. The prevalence of dementia: a systematic review and meta-analysis. J Alzheimers dis. 2020;73:1157–1166. doi:10.3233/JAD-191092
5. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging‐Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi:10.1016/j.jalz.2011.03.005
6. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging‐Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–279. doi:10.1016/j.jalz.2011.03.008
7. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32. doi:10.1186/s13024-019-0333-5
8. Heneka MT, Carson MJ, Khoury JE, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi:10.1016/S1474-4422(15)70016-5
9. Prater KE, Green KJ, Mamde S, et al. Human microglia show unique transcriptional changes in Alzheimer’s disease. Nat Aging. 2023;3:894–907. doi:10.1038/s43587-023-00424-y
10. Suridjan I, Pollock BG, Verhoeff NPLG, et al. In-vivo imaging of grey and white matter neuroinflammation in Alzheimer’s disease: a positron emission tomography study with a novel radioligand, [18F]-FEPPA. Mol Psychiatry. 2015;20:1579–1587. doi:10.1038/mp.2015.1
11. Dani M, Wood M, Mizoguchi R, et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain. 2018. doi:10.1093/brain/awy188
12. Pascoal TA, Benedet AL, Ashton NJ, et al. Microglial activation and tau propagate jointly across Braak stages. Nat Med. 2021;27:1592–1599. doi:10.1038/s41591-021-01456-w
13. Malpetti M, Kievit RA, Passamonti L, et al. Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain. 2020;143:1588–1602. doi:10.1093/brain/awaa088
14. Vann Jones SA, O’Brien JT. The prevalence and incidence of dementia with Lewy bodies: a systematic review of population and clinical studies. Psychol Med. 2014;44:673–683. doi:10.1017/S0033291713000494
15. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89:88–100. doi:10.1212/WNL.0000000000004058
16. Spillantini MG, Schmidt ML, Lee VM-Y, et al. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi:10.1038/42166
17. Amin J, Erskine D, Donaghy PC, et al. Inflammation in dementia with Lewy bodies. Neurobiol Dis. 2022;168:105698. doi:10.1016/j.nbd.2022.105698
18. Amin J, Holmes C, Dorey RB, et al. Neuroinflammation in dementia with Lewy bodies: a human post-mortem study. Transl Psychiatry. 2020;10:267. doi:10.1038/s41398-020-00954-8
19. King E, O’Brien JT, Donaghy P, et al. Peripheral inflammation in prodromal Alzheimer’s and Lewy body dementias. J Neurol Neurosurg Psychiatry. 2018;89:339–345. doi:10.1136/jnnp-2017-317134
20. Surendranathan A, Su L, Mak E, et al. Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain. 2018;141:3415–3427. doi:10.1093/brain/awy265
21. Bright F, Werry EL, Dobson-Stone C, et al. Neuroinflammation in frontotemporal dementia. Nat Rev Neurol. 2019;15:540–555. doi:10.1038/s41582-019-0231-z
22. Tampi RR, Jeste DV. Dementia is more than memory loss: neuropsychiatric symptoms of dementia and their nonpharmacological and pharmacological management. Am j Psychiatry. 2022;179:528–543. doi:10.1176/appi.ajp.20220508
23. Martin E, Velayudhan L. Neuropsychiatric symptoms in mild cognitive impairment: a literature review. Dementia and Geriatric Cognitive Disorders. 2020;49(2):146–155. doi:10.1159/000507078
24. McKeith IG, Ferman TJ, Thomas AJ, et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology. 2020;94:743–755. doi:10.1212/WNL.0000000000009323
25. Singh-Manoux A, Dugravot A, Fournier A, et al. Trajectories of depressive symptoms before diagnosis of dementia: a 28-year follow-up study. JAMA Psychiatry. 2017;74:712. doi:10.1001/jamapsychiatry.2017.0660
26. Mo M, Zacarias-Pons L, Hoang MT, et al. Psychiatric Disorders Before and After Dementia Diagnosis. JAMA Network Open. 2023;6:e2338080. doi:10.1001/jamanetworkopen.2023.38080
27. Zahodne LB, Ornstein K, Cosentino S, Devanand DP, Stern Y. Longitudinal relationships between Alzheimer disease progression and psychosis, depressed mood, and agitation/aggression. Am J Geriatr Psychiatry. 2015;23:130–140. doi:10.1016/j.jagp.2013.03.014
28. Peters ME, Schwartz S, Han D, et al. Neuropsychiatric symptoms as predictors of progression to severe alzheimer’s dementia and death: the cache county dementia progression study. Am j Psychiatry. 2015;172:460–465. doi:10.1176/appi.ajp.2014.14040480
29. Burchill E, Watson CJ, Fanshawe JB, et al. The impact of psychiatric comorbidity on Parkinson’s disease outcomes: a systematic review and meta-analysis. Lancet Reg Health - Eur. 2024;39:100870. doi:10.1016/j.lanepe.2024.100870
30. Kaufer DI, Cummings JL, Christine D, et al. Assessing the impact of neuropsychiatric symptoms in alzheimer’s disease: the neuropsychiatric inventory caregiver distress scale. J Am Geriatr Soc. 1998;46:210–215. doi:10.1111/j.1532-5415.1998.tb02542.x
31. Isik AT, Soysal P, Solmi M, Veronese N. Bidirectional relationship between caregiver burden and neuropsychiatric symptoms in patients with Alzheimer’s disease: a narrative review. Int J Geriatr Psychiatry. 2019;34:1326–1334. doi:10.1002/gps.4965
32. Malpetti M, Jones PS, Tsvetanov KA, et al. Apathy in presymptomatic genetic frontotemporal dementia predicts cognitive decline and is driven by structural brain changes. Alzheimers Dement. 2021;17:969–983. doi:10.1002/alz.12252
33. Matsuoka T, Manabe T, Akatsu H, et al. Factors influencing hospital admission among patients with autopsy‐confirmed dementia. Psychogeriatrics. 2019;19:255–263. doi:10.1111/psyg.12393
34. Tun S-M, Murman DL, Long HL, Colenda CC, Von Eye A. Predictive validity of neuropsychiatric subgroups on nursing home placement and survival in patients with Alzheimer disease. Am J Geriatr Psychiatry. 2007;15:314–327. doi:10.1097/01.JGP.0000239263.52621.97
35. Okura T, Plassman BL, Steffens DC, et al. Neuropsychiatric symptoms and the risk of institutionalization and death: the aging, demographics, and memory study: Neuropsychiatric symptoms, institutionalization, and death. J Am Geriatr Soc. 2011;59:473–481. doi:10.1111/j.1532-5415.2011.03314.x
36. Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in parkinson’s disease: a population‐based, prospective study. J Am Geriatr Soc. 2000;48:938–942. doi:10.1111/j.1532-5415.2000.tb06891.x
37. Chen Y, Dang M, Zhang Z. Brain mechanisms underlying neuropsychiatric symptoms in Alzheimer’s disease: a systematic review of symptom-general and –specific lesion patterns. Mol Neurodegener. 2021;16(38). doi:10.1186/s13024-021-00456-1
38. Rosenberg PB, Nowrangi MA, Lyketsos CG. Neuropsychiatric symptoms in Alzheimer’s disease: what might be associated brain circuits? Mol Aspects Med. 2015;43–44:25–37. doi:10.1016/j.mam.2015.05.005
39. Lanari A, Amenta F, Silvestrelli G, Tomassoni D, Parnetti L. Neurotransmitter deficits in behavioural and psychological symptoms of Alzheimer’s disease. Mech Ageing Dev. 2006;127:158–165. doi:10.1016/j.mad.2005.09.016
40. Khundakar AA, Hanson PS, Erskine D, et al. Analysis of primary visual cortex in dementia with Lewy bodies indicates GABAergic involvement associated with recurrent complex visual hallucinations. Acta Neuropathol Commun. 2016;4:66. doi:10.1186/s40478-016-0334-3
41. Ballard CG, Jacoby R, Del Ser T, et al. Neuropathological substrates of psychiatric symptoms in prospectively studied patients with autopsy-confirmed dementia with Lewy bodies. Am j Psychiatry. 2004;161:843–849. doi:10.1176/appi.ajp.161.5.843
42. Gibson LL, Grinberg LT, Ffytche D, et al. Neuropathological correlates of neuropsychiatric symptoms in dementia. Alzheimers Dement. 2023;19:1372–1382. doi:10.1002/alz.12765
43. Costello H, Roiser JP, Howard R. Antidepressant medications in dementia: evidence and potential mechanisms of treatment-resistance. Psychol Med. 2023;53:654–667. doi:10.1017/S003329172200397X
44. Taylor J-P, McKeith IG, Burn DJ, et al. New evidence on the management of Lewy body dementia. Lancet Neurol. 2020;19:157–169. doi:10.1016/S1474-4422(19)30153-X
45. Buoli M, Serati M, Caldiroli A, et al. Pharmacological management of psychiatric symptoms in frontotemporal dementia: a systematic review. J Geriatr Psychiatry Neurol. 2017;30:162–169. doi:10.1177/0891988717700506
46. Dudas R, Malouf R, McCleery J, Dening T. Antidepressants for treating depression in dementia. Cochrane Database Syst Rev. 2018;2018:1.
47. Katz I, de Deyn -P-P, Mintzer J, et al. The efficacy and safety of risperidone in the treatment of psychosis of Alzheimer’s disease and mixed dementia: a meta‐analysis of 4 placebo‐controlled clinical trials. Int J Geriatr Psychiatry. 2007;22:475–484. doi:10.1002/gps.1792
48. Wang J, Yu J-T, Wang H-F, et al. Pharmacological treatment of neuropsychiatric symptoms in Alzheimer’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2015;86:101–109. doi:10.1136/jnnp-2014-308112
49. Sloan M. Prevalence and identification of neuropsychiatric symptoms in systemic autoimmune rheumatic diseases: an international mixed methods study. Rheumatology. 2023:kead369. doi:10.1093/rheumatology/kead369
50. Nerurkar L, Siebert S, McInnes IB, Cavanagh J. Rheumatoid arthritis and depression: an inflammatory perspective. Lancet Psychiatry. 2019;6:164–173. doi:10.1016/S2215-0366(18)30255-4
51. Khandaker GM. Commentary: causal associations between inflammation, cardiometabolic markers and schizophrenia: the known unknowns. Int J Epidemiol. 2019;48:1516–1518. doi:10.1093/ije/dyz201
52. Milaneschi Y, Kappelmann N, Ye Z, et al. Association of inflammation with depression and anxiety: evidence for symptom-specificity and potential causality from UK Biobank and NESDA cohorts. Mol Psychiatry. 2021;26:7393–7402. doi:10.1038/s41380-021-01188-w
53. Drevets WC, Wittenberg GM, Bullmore ET, Manji HK. Immune targets for therapeutic development in depression: towards precision medicine. Nat Rev: Drug Discov. 2022;21:224–244. doi:10.1038/s41573-021-00368-1
54. Haroon E, Miller AH. Rewiring the brain: inflammation’s impact on glutamate and neural networks in depression. Neuropsychopharmacology. 2024;s41386–024–01921–3. doi:10.1038/s41386-024-01921-3
55. Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12:988–1000. doi:10.1038/sj.mp.4002006
56. Al‐Diwani AAJ, Pollak TA, Irani SR, Lennox BR. Psychosis: an autoimmune disease? Immunology. 2017;152:388–401. doi:10.1111/imm.12795
57. Kunkle BW, Grenier-Boley B, Sims R; Alzheimer Disease Genetics Consortium (ADGC), et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 51;2019:414–430. doi:10.1038/s41588-019-0358-2
58. Zhang M, Qian X-H, Hu J, et al. Integrating TSPO PET imaging and transcriptomics to unveil the role of neuroinflammation and amyloid-β deposition in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging. 2024;51:455–467. doi:10.1007/s00259-023-06446-3
59. Su L, Surendranathan A, Huang Y, et al. Relationship between tau, neuroinflammation and atrophy in Alzheimer’s disease: the NIMROD study. Inf Fusion. 2021;67:116–124. doi:10.1016/j.inffus.2020.10.006
60. Cisbani G, Koppel A, Knezevic D, et al. Peripheral cytokine and fatty acid associations with neuroinflammation in AD and aMCI patients: an exploratory study. Brain Behav Immun. 2020;87:679–688. doi:10.1016/j.bbi.2020.02.014
61. Ott A, Stolk RP, van Harskamp F, et al. Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology. 1999;53:1937–1937. doi:10.1212/WNL.53.9.1937
62. Femminella GD, Taylor-Davies G, Scott J, Edison P; the Alzheimer’s Disease Neuroimaging Initiative. Do cardiometabolic risk factors influence amyloid, tau, and neuronal function in APOE4 carriers and non-carriers in alzheimer’s disease trajectory? J Alzheimers dis. 2018;64:981–993. doi:10.3233/JAD-180365
63. Bettcher BM, Tansey MG, Dorothée G, Heneka MT. Peripheral and central immune system crosstalk in Alzheimer disease — a research prospectus. Nat Rev Neurol. 2021;17:689–701. doi:10.1038/s41582-021-00549-x
64. Livingston G. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396:413–446.
65. Bartels C, Wagner M, Wolfsgruber S, et al. Impact of SSRI therapy on risk of conversion from mild cognitive impairment to alzheimer’s dementia in individuals with previous depression. Am j Psychiatry. 2018;175:232–241. doi:10.1176/appi.ajp.2017.17040404
66. Wang L, Wang R, Liu L, et al. Effects of SSRIs on peripheral inflammatory markers in patients with major depressive disorder: a systematic review and meta-analysis. Brain Behav Immun. 2019;79:24–38. doi:10.1016/j.bbi.2019.02.021
67. Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70:31. doi:10.1001/2013.jamapsychiatry.4
68. Harrison F, Aerts L, Seeher KM, et al. P3-225 systematic review of associations between behavioral and psychological symptoms of dementia and cytokines. Alzheimers Dement. 2017;13:1024–P1024. doi:10.1016/j.jalz.2017.06.1438
69. Azocar I, Rapaport P, Burton A, Meisel G, Orgeta V. Risk factors for apathy in Alzheimer’s disease: a systematic review of longitudinal evidence. Ageing Res Rev. 2022;79:101672. doi:10.1016/j.arr.2022.101672
70. Ramanzini LG, Camargo LFM, Silveira JOF, Bochi GV. Inflammatory markers and depression in Parkinson’s disease: a systematic review. Neurol Sci. 2022;43:6707–6717. doi:10.1007/s10072-022-06363-7
71. Gerrits E, Brouwer N, Kooistra SM, et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol (Berl). 2021;141:681–696. doi:10.1007/s00401-021-02263-w
72. Gate D, Tapp E, Leventhal O, et al. CD4 + T cells contribute to neurodegeneration in Lewy body dementia. Science. 2021;374:868–874. doi:10.1126/science.abf7266
73. Wilson DM, Cookson MR, Van Den Bosch L, et al. Hallmarks of neurodegenerative diseases. Cell. 2023;186:693–714. doi:10.1016/j.cell.2022.12.032
74. Biomarker definitions working group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi:10.1067/mcp.2001.113989
75. Kales HC, Gitlin LN, Lyketsos CG. Assessment and management of behavioral and psychological symptoms of dementia. BMJ. 2015;350:h369–h369. doi:10.1136/bmj.h369
76. Lanctôt KL, Amatniek J, Ancoli‐Israel S, et al. Neuropsychiatric signs and symptoms of Alzheimer’s disease: new treatment paradigms. Alzheimers Dement Transl Res Clin Interv. 2017;3:440–449. doi:10.1016/j.trci.2017.07.001
77. Hassin-Baer S, Cohen OS, Vakil E, et al. Is C-reactive protein level a marker of advanced motor and neuropsychiatric complications in Parkinson’s disease? J Neural Transm. 2011;118:539–543. doi:10.1007/s00702-010-0535-z
78. Holmgren S, Hjorth E, Schultzberg M, et al. Neuropsychiatric symptoms in dementia—A role for neuroinflammation? Brain Res Bull. 2014;108:88–93. doi:10.1016/j.brainresbull.2014.09.003
79. Kunschmann R, Busse S, Frodl T, Busse M. Psychotic symptoms associated with poor renal function in mild cognitive impairment and dementias. J Alzheimers dis. 2017;58:243–252. doi:10.3233/JAD-161306
80. King E, O’Brien JT, Donaghy P, et al. Peripheral inflammation in mild cognitive impairment with possible and probable Lewy body disease and Alzheimer’s disease. Int Psychogeriatr. 2019;31:551–560. doi:10.1017/S1041610218001126
81. Willette AA, Pappas C, Hoth N, et al. Inflammation, negative affect, and amyloid burden in Alzheimer’s disease: insights from the kynurenine pathway. Brain Behav Immun. 2021;95:216–225. doi:10.1016/j.bbi.2021.03.019
82. Teixeira AL, Salem H, Martins LB, et al. Factors associated with apathy in alzheimer’s disease: a cross-sectional analysis of the texas alzheimer’s research and care consortium (TARCC) Study. J Alzheimers dis. 2022;86:403–411. doi:10.3233/JAD-215314
83. Sandor C, Millin S, Dahl A, et al. Universal clinical Parkinson’s disease axes identify a major influence of neuroinflammation. Genome Med. 2022;14:1–15. doi:10.1186/s13073-022-01132-9
84. Kaplin A, Carroll KAL, Cheng J, et al. IL-6 release by LPS-stimulated peripheral blood mononuclear cells as a potential biomarker in Alzheimer’s disease. Int Psychogeriatr. 2009;21:413–414. doi:10.1017/S1041610208008107
85. Guimarães HC, Caramelli P, Fialho PPA, et al. Serum levels of soluble TNF-α receptors but not BDNF are associated with apathy symptoms in mild Alzheimer’s disease and amnestic mild cognitive impairment. Dement Neuropsychol. 2013;7:298–303. doi:10.1590/S1980-57642013DN70300011
86. Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 2011;77:212–218. doi:10.1212/WNL.0b013e318225ae07
87. Veselý B, Dufek M, Thon V, et al. Interleukin 6 and complement serum level study in Parkinson’s disease. J Neural Transm. 2018;125:875–881. doi:10.1007/s00702-018-1857-5
88. Clough Z, Jeyapaul P, Zotova E, Holmes C. Proinflammatory Cytokines and the Clinical Features of Dementia With Lewy Bodies. Alzheimer Dis Assoc Disord. 2015;29:97–99. doi:10.1097/WAD.0b013e3182969905
89. Banerjee A, Khemka VK, Roy D, et al. Role of pro-inflammatory cytokines and vitamin D in probable alzheimer’s disease with depression. Aging Dis. 2017;8:267. doi:10.14336/AD.2016.1017
90. Pereira JR, Santos LVD, Santos RMS, et al. IL-6 serum levels are elevated in Parkinson’s disease patients with fatigue compared to patients without fatigue. J Neurol Sci. 2016;370:153–156. doi:10.1016/j.jns.2016.09.030
91. Brockmann K, Schulte C, Schneiderhan‐Marra N, et al. Inflammatory profile discriminates clinical subtypes in LRRK2 -associated Parkinson’s disease. Eur J Neurol. 2017;24:427–e6. doi:10.1111/ene.13223
92. Clark C, Richiardi J, Maréchal B, et al. Systemic and central nervous system neuroinflammatory signatures of neuropsychiatric symptoms and related cognitive decline in older people. J Neuroinflammation. 2022;19:127. doi:10.1186/s12974-022-02473-3
93. Schaffer Aguzzoli C, Ferreira PCL, Povala G, et al. Neuropsychiatric symptoms and microglial activation in patients with Alzheimer disease. JAMA Network Open. 2023;6:e2345175. doi:10.1001/jamanetworkopen.2023.45175
94. Yasuno F, Kimura Y, Ogata A, et al. Involvement of inflammation in the medial temporal region in the development of agitation in Alzheimer’s disease: an in vivo positron emission tomography study. Psychogeriatrics. 2023;23:126–135. doi:10.1111/psyg.12915
95. Arnold S, Xie SX, Leung -Y-Y, et al. P2‐017: biochemical biomarkers of depressive symptoms in ADNI. Alzheimers Dement. 2011;7:S312–S312. doi:10.1016/j.jalz.2011.05.907
96. Basta M. 1157 inflammation is associated with excessive daytime sleepiness and impaired cognitive performance in patients with mild cognitive impairment (MCI). J Sleep Sleep Disord Res. 2017;40:A432–A432.
97. Leng SX, McElhaney JE, Walston JD, et al. ELISA and multiplex technologies for cytokine measurement in inflammation and aging research. J Gerontol a Biol Sci Med Sci. 2008;63:879–884. doi:10.1093/gerona/63.8.879
98. Caraci F, Bosco P, Signorelli M, et al. The CC genotype of transforming growth factor-β1 increases the risk of late-onset Alzheimer’s disease and is associated with AD-related depression. Eur Neuropsychopharmacol. 2012;22:281–289. doi:10.1016/j.euroneuro.2011.08.006
99. Serretti A, Olgiati P, Politis A, et al. Lack of association between interleukin-1 alpha rs1800587 polymorphism and Alzheimer’s disease in two Independent European samples. J Alzheimers dis. 2009;16:181–187. doi:10.3233/JAD-2009-0946
100. Craig D, Hart DJ, McCool K, McIlroy SP, Passmore AP. The interleukin 1β gene promoter polymorphism (−511) acts as a risk factor for psychosis in Alzheimer’s dementia. Ann Neurol. 2004;56:121–124. doi:10.1002/ana.20120
101. Grana D. Behavioural anomalies in neurodegenerative diseases: role of the immune response. Poster Neuromi. 2016;2016:51.
102. Porcelli S, Salfi R, Politis A, et al. Association between Sirtuin 2 gene rs10410544 polymorphism and depression in Alzheimer’s disease in two independent European samples. J Neural Transm. 2013;120:1709–1715. doi:10.1007/s00702-013-1045-6
103. McCulley MC, Day INM, Holmes C. Association between interleukin 1‐β promoter (−511) polymorphism and depressive symptoms in Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2004;124B:50–53. doi:10.1002/ajmg.b.20086
104. Redenšek S, Jenko Bizjan B, Trošt M, Dolžan V. Clinical and clinical-pharmacogenetic models for prediction of the most common psychiatric complications due to dopaminergic treatment in parkinson’s disease. Int J Neuropsychopharmacol. 2020;23:496–504. doi:10.1093/ijnp/pyaa028
105. Surathi P. Genetic polymorphism (rs6971) in translocator protein (TSPO) and its clinical relevance in Parkinson’s disease. Mov Disord. 2019;34:1.
106. Gao L, Tang H, Nie K, et al. MCP-1 and CCR2 gene polymorphisms in Parkinson’s disease in a Han Chinese cohort. Neurol Sci. 2015;36:571–576. doi:10.1007/s10072-014-1990-3
107. Pesic M, Maksimovic N, Aleksic A, et al. Polymorphisms in genes for proinflammatory cytokines IL-6, IL-1 ss, andTNF-alpha in relation with Parkinson’s disease progression. Eur J Hum Genet. 2020;28:1.
108. Dekens DW, Naudé PJW, Engelborghs S, et al. Neutrophil gelatinase-associated lipocalin and its receptors in Alzheimer’s disease (AD) brain regions: differential findings in AD with and without depression. J Alzheimers dis. 2017;55:763–776. doi:10.3233/JAD-160330
109. Lin J. The neuroinflammatory environment in depression of alzheimer’s disease: a human post-mortem study. Brain Neurosci Adv. 2023;7:115.
110. Marx W, McGuinness AJ, Rocks T, et al. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: a meta-analysis of 101 studies. Mol Psychiatry. 2021;26:4158–4178. doi:10.1038/s41380-020-00951-9
111. Enache D, Aarsland D, Eyjolfsdottir H, et al. 777 Markers of inflammation and depressive symptoms in the elderly with and without Alzheimer’s disease. Eur Neuropsychopharmacol. 2019;29:S520–S521. doi:10.1016/j.euroneuro.2019.09.808
112. Porter GA, O’Connor JC. Brain-derived neurotrophic factor and inflammation in depression: pathogenic partners in crime? World J Psychiatry. 2022;12:77–97. doi:10.5498/wjp.v12.i1.77
113. Primo De Carvalho Alves L, Sica Da Rocha N. Lower levels of brain‐derived neurotrophic factor are associated with melancholic psychomotor retardation among depressed inpatients. Bipolar Disord. 2018;20:746–752. doi:10.1111/bdi.12636
114. Feng H, Hu P, Chen Y, et al. Decreased miR-451a in cerebrospinal fluid, a marker for both cognitive impairment and depressive symptoms in Alzheimer’s disease. Theranostics. 2023;13:3021–3040. doi:10.7150/thno.81826
115. Johnson LA, Weiser B, Gamboa A, et al. P3‐176: biomarkers of a Depressive Endophenotype. Alzheimers Dement. 2016;12:888–P888. doi:10.1016/j.jalz.2016.06.1836
116. Khemka VK, Ganguly A, Bagchi D, et al. Raised serum proinflammatory cytokines in Alzheimer’s disease with depression. Aging Dis. 2014;5:170. doi:10.14336/AD.2014.0500170
117. Hall J, Johnson L, Wiechmann A, et al. P1‐030: biomarkers of depressive symptoms in Alzheimer’s disease: gender differences. Alzheimers Dement. 2012;8. doi:10.1016/j.jalz.2012.05.305
118. Gorska-Ciebiada M, Saryusz-Wolska M, Borkowska A, Ciebiada M, Loba J. Serum levels of inflammatory markers in depressed elderly patients with diabetes and mild cognitive impairment. PLoS One. 2015;10:e0120433. doi:10.1371/journal.pone.0120433
119. Gorska-Ciebiada M, Saryusz-Wolska M, Borkowska A, Ciebiada M, Loba J. Serum soluble adhesion molecules and markers of systemic inflammation in elderly diabetic patients with mild cognitive impairment and depressive symptoms. BioMed Res Int. 2015;2015:1–8. doi:10.1155/2015/826180
120. Alvarez XA, Franco A, Fernández-Novoa L, Cacabelos R. Blood levels of histamine, IL-1β, and TNF-α in patients with mild to moderate Alzheimer disease. Mol Chem Neuropathol. 1996;29:237–252. doi:10.1007/BF02815005
121. Hall JR, Wiechmann AR, Johnson LA, et al. Biomarkers of vascular risk, systemic inflammation, and microvascular pathology and neuropsychiatric symptoms in Alzheimer’s disease. J Alzheimers dis. 2013;35:363–371. doi:10.3233/JAD-122359
122. Hall J, Johnson L, Barber R, O’Bryant S. P1‐418: CRP and depressive symptoms in Alzheimer’s disease. Alzheimers Dement. 2011;7:S246–S246. doi:10.1016/j.jalz.2011.05.699
123. Park JK, Lee KJ, Kim JY, Kim H. The association of blood-based inflammatory factors IL-1β, TGF-β and CRP with cognitive function in Alzheimer’s disease and mild cognitive impairment. Psychiatry Invest. 2021;18:11. doi:10.30773/pi.2020.0205
124. Jung J, Kim S, Yoon K, et al. The effect of depression on serum VEGF level in Alzheimer’s disease. Dis. Markers. 2015;2015. doi:10.1155/2015/742612
125. Kim DH, Moon YS, Yoon KH. P2‐086: the effect of depression on the serum levels of vascular factors in Alzheimer’s disease. Alzheimers Dement. 2015;11:516–P516. doi:10.1016/j.jalz.2015.06.623
126. Kim D, Moon YS, Yoon KH. P.5.b.011 Vascular factors in Alzheimer’s disease with depressive symptoms. Eur Neuropsychopharmacol. 2014;24:S638. doi:10.1016/S0924-977X(14)71025-9
127. Hall JR, Wiechmann A, Edwards M, Johnson LA, O’Bryant SE. IL-7 and depression: the importance of gender and blood fraction. Behav Brain Res. 2016;315:147–149. doi:10.1016/j.bbr.2016.08.026
128. Viganotti M, Ho A, Gallagher R. Immune gene expression is different in Alzheimer disease patients with comorbid depression vs. psychogeriatric depression. J Alzheimers dis. 2012;29:1–109. doi:10.3233/JAD-2012-111424
129. Sao T, Yoshino Y, Yamazaki K, et al. TREM1 mRNA expression in leukocytes and cognitive function in Japanese patients with Alzheimer’s disease. J Alzheimers dis. 2018;64:1275–1284. doi:10.3233/JAD-180418
130. Jacobs HI, Riphagen JM, Ramakers IH, Verhey FR. Alzheimer’s disease pathology: pathways between central norepinephrine activity, memory, and neuropsychiatric symptoms. Mol Psychiatry. 2021;26:897–906. doi:10.1038/s41380-019-0437-x
131. Shen H, Han C, Yang Y, et al. Pyroptosis executive protein GSDMD as a biomarker for diagnosis and identification of Alzheimer’s disease. Brain Behav. 2021;11:e02063. doi:10.1002/brb3.2063
132. Anton Alvarez X, Sampedro C, Cacabelos R, et al. Reduced TNF-α and increased IGF-I levels in the serum of Alzheimer’s disease patients treated with the neurotrophic agent Cerebrolysin. Int J Neuropsychopharmacol. 2009;12:867. doi:10.1017/S1461145709990101
133. Bulut O. Examination of IL-1 beta level as an inflammasome marker in Alzheimer’s disease; 2019.
134. Mahdavi M, Karima S, Rajaei S, et al. Plasma cytokines profile in subjects with Alzheimer’s disease: interleukin 1 alpha as a candidate for target therapy. Galen Med J. 2021;10:e1974. doi:10.31661/gmj.v10i0.1974
135. Hall JR, Wiechmann AR, Johnson LA, et al. The impact of APOE status on relationship of biomarkers of vascular risk and systemic inflammation to neuropsychiatric symptoms in Alzheimer’s disease. J Alzheimers dis. 2014;40:887–896. doi:10.3233/JAD-131724
136. Lauriola M, D’Onofrio G, Ciccone F, et al. Relationship of homocysteine plasma levels with mild cognitive impairment, Alzheimer’s disease, vascular dementia, psychobehavioral, and functional complications. J Alzheimers dis. 2021;82:235–248. doi:10.3233/JAD-210166
137. Olgiati P, Politis A, Malitas P, et al. APOE epsilon‐4 allele and cytokine production in Alzheimer’s disease. Int J Geriatr Psychiatry J Psychiatry Late Life Allied Sci. 2010;25:338–344. doi:10.1002/gps.2344
138. Lee SM, Te S, Breen EC, et al. Circulating versus lipopolysaccharide-induced inflammatory markers as correlates of subthreshold depressive symptoms in older adults. World J Biol Psychiatry. 2020;21:634–641. doi:10.1080/15622975.2019.1671608
139. Ciaramella A, Salani F, Bizzoni F, et al. Myeloid dendritic cells are decreased in peripheral blood of Alzheimer’s disease patients in association with disease progression and severity of depressive symptoms. J Neuroinflammation. 2016;13:18. doi:10.1186/s12974-016-0483-0
140. Aprea V, Conti E, Sala G, et al. Neuroinflammation and behavioral dysfunction: exploring the role of Diazepam binding inhibitor. J Neurol Sci. 2021;429:118986. doi:10.1016/j.jns.2021.118986
141. Noto H. Fasting Blood Interleukin-1ß and Interleukin-6 Levels as Predictors of Agitation in Patients with Alzheimer’s Disease. Int J Gerontol. 2021;15:1.
142. Higuchi M, Hatta K, Honma T, et al. Association between altered systemic inflammatory interleukin‐1β and natural killer cell activity and subsequently agitation in patients with Alzheimer disease. Int J Geriatr Psychiatry J Psychiatry Late Life Allied Sci. 2010;25:604–611. doi:10.1002/gps.2381
143. Ruthirakuhan M, Herrmann N, Andreazza AC, et al. Agitation, oxidative stress, and cytokines in Alzheimer disease: biomarker analyses from a clinical trial with nabilone for agitation. J Geriatr Psychiatry Neurol. 2020;33:175–184. doi:10.1177/0891988719874118
144. Padala PR, Sarkar S, Dennis RA, Lensing SY, Padala KP. P4‐678: Identification of neurovascular damage and neuroinflammatory markers in blood and their correlation with severity of apathy in alzheimer’s dISEASE. Alzheimers Dement. 2019;15:1591–P1591. doi:10.1016/j.jalz.2019.09.043
145. Olgiati P, Politis A, Albani D, et al. Effects of SORL1 gene on Alzheimer’s disease. Focus on gender, neuropsychiatric symptoms and pro-inflammatory cytokines. Curr Alzheimer Res. 2013;10:154–164. doi:10.2174/1567205011310020005
146. Vakilian A, Razavi-Nasab S, Ravari A, et al. Vitamin B12 in association with antipsychotic drugs can modulate the expression of pro-/anti-inflammatory cytokines in Alzheimer disease patients. Neuroimmunomodulation. 2017;24:310–319. doi:10.1159/000486597
147. Basta M, Vgontzas A, Vogiatzi E, et al. 1156 Inflammation is associated with increased daytime and nighttime sleep in patients with dementia. Sleep. 2017;40:A431–A432. doi:10.1093/sleepj/zsx050.1155
148. Barber R. Inflammatory signaling in Alzheimer disease and depression. Cleve Clin J Med. 2011;78:S47–9. doi:10.3949/ccjm.78.s1.08
149. Ahmadi Rastegar D, Ho N, Halliday GM, Dzamko N. Parkinson’s progression prediction using machine learning and serum cytokines. NPJ Park Dis. 2019;5:14. doi:10.1038/s41531-019-0086-4
150. Hall S, Janelidze S, Surova Y, et al. Cerebrospinal fluid concentrations of inflammatory markers in Parkinson’s disease and atypical parkinsonian disorders. Sci Rep. 2018;8:13276. doi:10.1038/s41598-018-31517-z
151. Lian T, Guo P, Zhang Y-N, et al. Parkinson’s disease with depression: the correlations between neuroinflammatory factors and neurotransmitters in cerebrospinal fluid. Front Aging Neurosci. 2020;12:574776. doi:10.3389/fnagi.2020.574776
152. Pålhagen S, Qi H, Mårtensson B, et al. Monoamines, BDNF, IL-6 and corticosterone in CSF in patients with Parkinson’s disease and major depression. J Neurol. 2010;257:524–532. doi:10.1007/s00415-009-5353-6
153. Karpenko M, Vasilishina A, Gromova E, Muruzheva Z, Bernadotte A. Interleukin-1β, interleukin-1 receptor antagonist, interleukin-6, interleukin-10, and tumor necrosis factor-α levels in CSF and serum in relation to the clinical diversity of Parkinson’s disease. Cell Immunol. 2018;327:77–82. doi:10.1016/j.cellimm.2018.02.011
154. Lindqvist D, Hall S, Surova Y, et al. Cerebrospinal fluid inflammatory markers in Parkinson’s disease – associations with depression, fatigue, and cognitive impairment. Brain Behav Immun. 2013;33:183–189. doi:10.1016/j.bbi.2013.07.007
155. Li D, Lian T-H, Zhang W-J, et al. Potential roles of oxidative distress on neurodegeneration in Parkinson’s disease with neuropsychiatric symptoms. Front Aging Neurosci. 2022;14:875059. doi:10.3389/fnagi.2022.875059
156. Modugno N, Morgante F, Marcuzzo E. CSF inflammatory cytokines and correlation with clinical features in early, untreated PD patients. Mov Disord Clin Pract. 2020;7. doi:10.1002/mdc3.13077
157. Yu S, Sun L, Liu Z, et al. Sleep disorders in parkinson’s disease: clinical features, iron metabolism and related mechanism. PLoS One. 2013;8:e82924. doi:10.1371/journal.pone.0082924
158. Fu J, Chen S, Liu J, et al. Serum inflammatory cytokines levels and the correlation analyses in Parkinson’s disease. Front Cell Dev Biol. 2023;11:1104393. doi:10.3389/fcell.2023.1104393
159. Hu S, Huang S, Ma J, et al. Correlation of decreased serum pituitary adenylate cyclase-activating polypeptide and vasoactive intestinal peptide levels with non-motor symptoms in patients with Parkinson’s disease. Front Aging Neurosci. 2021;13:689939. doi:10.3389/fnagi.2021.689939
160. Jiang G, Sheng C, Yan L, et al. Increased serum S100β concentration is associated with depression in parkinson’s disease. Neuropsychiatr Dis Treat. 2023;19:1865–1873. doi:10.2147/NDT.S423312
161. Lindqvist D. Non-motor symptoms in patients with Parkinson’s disease–correlations with inflammatory cytokines in serum; 2012.
162. Selikhova M, Kushlinskii N, Lyubimova N, Gusev E. Impaired production of plasma interleukin-6 in patients with Parkinson’s disease. Bull Exp Biol Med. 2002;133:81–83. doi:10.1023/A:1015120930920
163. Menza M, Dobkin RD, Marin H, et al. The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease. Psychosomatics. 2010;51:474–479. doi:10.1176/appi.psy.51.6.474
164. Wan Z, Rasheed M, Li Y, et al. miR-218-5p and miR-320a-5p as biomarkers for brain disorders: focus on the major depressive disorder and parkinson’s disease. Mol Neurobiol. 2023;60:5642–5654. doi:10.1007/s12035-023-03391-y
165. Wang X-M, Zhang Y-G, Li A-L, et al. Relationship between levels of inflammatory cytokines in the peripheral blood and the severity of depression and anxiety in patients with Parkinson’s disease. Eur Rev Med Pharmacol Sci. 2016;20:3853–3856.
166. Kim R, Kim H-J, Shin JH, et al. Serum inflammatory markers and progression of nonmotor symptoms in early Parkinson’s disease. Mov Disord. 2022;37:1535–1541. doi:10.1002/mds.29056
167. Rocha NP, Scalzo PL, Barbosa IG, et al. Cognitive Status Correlates with CXCL10/IP-10 Levels in Parkinson’s Disease. Park Dis. 2014;2014:1–7.
168. Boika A, Ponomarev V, Aleinikava N, Ivanchik H. Is CD56+ a Possible Laboratory Biomarker of Depressive Symptoms at Parkinson Disorder (PD)? Vol. 27. WILEY 111 RIVER ST, HOBOKEN 07030-5774, NJ USA; 2020:1102–1102.
169. Ciaramella A, Salani F, Bizzoni F, et al. Blood dendritic cell frequency declines in idiopathic parkinson’s disease and is associated with motor symptom severity. PLoS One. 2013;8:e65352. doi:10.1371/journal.pone.0065352
170. Green H, Brodin L, Berg L, Greengard P, Svenningsson P. P11 Protein Levels in Peripheral Blood Mononuclear Cells from Parkinson’s Disease Patients with and without Depression Correlate with Disease Severity. Vol. 41. (NATURE PUBLISHING GROUP MACMILLAN BUILDING, 4 CRINAN ST, LONDON N1 9XW, ENGLAND; 2016:S174–S175.
171. Paul KC, Kusters C, Furlong M, et al. Immune system disruptions implicated in whole blood epigenome-wide association study of depression among Parkinson’s disease patients. Brain Behav Immun -Health. 2022;26:100530. doi:10.1016/j.bbih.2022.100530
172. Sawada H, Oeda T, Umemura A, et al. Subclinical Elevation of Plasma C-Reactive Protein and Illusions/Hallucinations in subjects with parkinson’s disease: case–control study. PLoS One. 2014;9:e85886. doi:10.1371/journal.pone.0085886
173. Debnath M. Impact of Th17 Pathway-Related Immuno-Inflammatory Cytokines in the Risk and Severity of Parkinson’s Disease. Vol. 37. WILEY 111 RIVER ST, HOBOKEN 07030-5774, NJ USA; 2022:S584–S584
174. Kaur J. Blood Biomarkers and Their Association with Non-Motor Symptoms in Early Parkinson’s Disease. Vol. 37. (WILEY 111 RIVER ST, HOBOKEN 07030-5774, NJ USA; 2022:S629–S629
175. Amin J, Boche D, Clough Z, et al. Peripheral immunophenotype in dementia with Lewy bodies and Alzheimer’s disease: an observational clinical study. J Neurol Neurosurg Psychiatry. 2020;91:1219–1226. doi:10.1136/jnnp-2020-323603
176. Chu M, Wen L, Jiang D, et al. Peripheral inflammation in behavioural variant frontotemporal dementia: associations with central degeneration and clinical measures. J Neuroinflammation. 2023;20:65. doi:10.1186/s12974-023-02746-5
177. Bouwens J, Hubers AAM, van Duijn E, et al. Acute-phase proteins in relation to neuropsychiatric symptoms and use of psychotropic medication in Huntington׳ s disease. Eur Neuropsychopharmacol. 2014;24:1248–1256. doi:10.1016/j.euroneuro.2014.05.004
178. Bouwens JA, van Duijn E, Cobbaert CM, et al. Plasma cytokine levels in relation to neuropsychiatric symptoms and cognitive dysfunction in Huntington’s disease. J Huntingt Dis. 2016;5:369–377. doi:10.3233/JHD-160213
179. Jack CR. Revised criteria for diagnosis and staging of Alzheimer’s disease: alzheimer’s Association Workgroup. Alzheimers Dement. 2024;13859. doi:10.1002/alz.13859
180. Höglinger GU, Adler CH, Berg D, et al. A biological classification of Parkinson’s disease: the SynNeurGe research diagnostic criteria. Lancet Neurol. 2024;23:191–204. doi:10.1016/S1474-4422(23)00404-0
181. McKeith IG, Ballard CG, Perry RH, et al. Prospective validation of Consensus criteria for the diagnosis of dementia with Lewy bodies. Neurology. 2000;54:1050–1058. doi:10.1212/WNL.54.5.1050
182. Ashton NJ, Brum WS, Di Molfetta G, et al. Diagnostic accuracy of a plasma phosphorylated tau 217 immunoassay for Alzheimer disease pathology. JAMA Neurol. 2024;81:255. doi:10.1001/jamaneurol.2023.5319
183. Fernandes Gomes B, Farris CM, Ma Y, et al. α-Synuclein seed amplification assay as a diagnostic tool for parkinsonian disorders. Parkinsonism Relat Disord. 2023;117:105807. doi:10.1016/j.parkreldis.2023.105807
184. Hansson O, Blennow K, Zetterberg H, Dage J. Blood biomarkers for Alzheimer’s disease in clinical practice and trials. Nat Aging. 2023;3:506–519. doi:10.1038/s43587-023-00403-3
185. Thomas AJ, Hamilton CA, Donaghy PC, et al. Prospective longitudinal evaluation of cytokines in mild cognitive impairment due to AD and Lewy body disease. Int J Geriatr Psychiatry. 2020;35:1250–1259. doi:10.1002/gps.5365
186. Hessler JB, Schäufele M, Hendlmeier I, et al. Behavioural and psychological symptoms in general hospital patients with dementia, distress for nursing staff and complications in care: results of the General Hospital Study. Epidemiol Psychiatr Sci. 2018;27:278–287. doi:10.1017/S2045796016001098
187. Fan Z, Brooks DJ, Okello A, Edison P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain. 2017;aww349. doi:10.1093/brain/aww349
188. Zetterberg H, Bendlin BB. Biomarkers for Alzheimer’s disease—preparing for a new era of disease-modifying therapies. Mol Psychiatry. 2021;26:296–308. doi:10.1038/s41380-020-0721-9
189. Murray PS, Kumar S, DeMichele-Sweet MAA, Sweet RA. Psychosis in Alzheimer’s Disease. Biol. Psychiatry. 2014;75:542–552. doi:10.1016/j.biopsych.2013.08.020
190. De Picker LJ, Morrens M, Branchi I, et al. TSPO PET brain inflammation imaging: a transdiagnostic systematic review and meta-analysis of 156 case-control studies. Brain Behav Immun. 2023;113:415–431. doi:10.1016/j.bbi.2023.07.023
191. Narayanaswami V, Dahl K, Bernard-Gauthier V, et al. Emerging PET radiotracers and targets for imaging of neuroinflammation in neurodegenerative diseases: outlook beyond TSPO. Mol Imaging. 2018;17:153601211879231. doi:10.1177/1536012118792317
192. Beaino W, Janssen B, Vugts DJ, De Vries HE, Windhorst AD. Towards PET imaging of the dynamic phenotypes of microglia. Clin Exp Immunol. 2021;206:282–300. doi:10.1111/cei.13649
193. Chatterjee M, Özdemir S, Fritz C, et al. Plasma extracellular vesicle tau and TDP-43 as diagnostic biomarkers in FTD and ALS. Nat Med. 2024;30:1771–1783. doi:10.1038/s41591-024-02937-4
194. Dutta S, Hornung S, Taha HB, Bitan G. Biomarkers for parkinsonian disorders in CNS-originating EVs: promise and challenges. Acta Neuropathol (Berl). 2023;145:515–540. doi:10.1007/s00401-023-02557-1
195. Hollingworth P, Sweet R, Sims R, et al. Genome-wide association study of Alzheimer’s disease with psychotic symptoms. Mol Psychiatry. 2012;17:1316–1327. doi:10.1038/mp.2011.125
196. Perry BI, Upthegrove R, Kappelmann N, et al. Associations of immunological proteins/traits with schizophrenia, major depression and bipolar disorder: a bi-directional two-sample Mendelian randomization study. Brain Behav Immun. 2021;97:176–185. doi:10.1016/j.bbi.2021.07.009
197. Franklyn SI, Stewart J, Beaurepaire C, Thaw E, McQuaid RJ. Developing symptom clusters: linking inflammatory biomarkers to depressive symptom profiles. Transl Psychiatry. 2022;12:133. doi:10.1038/s41398-022-01900-6
198. De Jager W, Bourcier K, Rijkers GT, Prakken BJ, Seyfert-Margolis V. Prerequisites for cytokine measurements in clinical trials with multiplex immunoassays. BMC Immunol. 2009;10(52). doi:10.1186/1471-2172-10-52
199. Liu C, Chu D, Kalantar‐Zadeh K, et al. Cytokines: from clinical significance to quantification. Adv Sci. 2021;8:2004433. doi:10.1002/advs.202004433
200. Banks RE. Measurement of cytokines in clinical samples using immunoassays: problems and pitfalls. Crit Rev Clin Lab Sci. 2000;37:131–182. doi:10.1080/10408360091174187
201. Altara R, Manca M, Hermans KC, et al. Diurnal rhythms of serum and plasma cytokine profiles in healthy elderly individuals assessed using membrane based multiplexed immunoassay. J Transl Med. 2015;13:129. doi:10.1186/s12967-015-0477-1
202. D’Esposito V, Di Tolla MF, Lecce M, et al. Lifestyle and dietary habits affect plasma levels of specific cytokines in healthy subjects. Front Nutr. 2022;9:913176. doi:10.3389/fnut.2022.913176
203. Pereira JB, Kumar A, Hall S, et al. DOPA decarboxylase is an emerging biomarker for Parkinsonian disorders including preclinical Lewy body disease. Nat Aging. 2023;3:1201–1209. doi:10.1038/s43587-023-00478-y
204. Kelly KM, Smith JA, Mezuk B. Depression and interleukin-6 signaling: a Mendelian Randomization study. Brain Behav Immun. 2021;95:106–114. doi:10.1016/j.bbi.2021.02.019
205. Khandaker GM, Oltean BP, Kaser M, et al. Protocol for the insight study: a randomised controlled trial of single-dose tocilizumab in patients with depression and low-grade inflammation. BMJ Open. 2018;8:e025333. doi:10.1136/bmjopen-2018-025333
206. Osimo EF, Baxter LJ, Lewis G, Jones PB, Khandaker GM. Prevalence of low-grade inflammation in depression: a systematic review and meta-analysis of CRP levels. Psychol Med. 2019;49:1958–1970. doi:10.1017/S0033291719001454
207. Aruldass AR, Kitzbichler MG, Morgan SE, et al. Dysconnectivity of a brain functional network was associated with blood inflammatory markers in depression. Brain Behav Immun. 2021;98:299–309. doi:10.1016/j.bbi.2021.08.226
208. Haroon E, Welle JR, Woolwine BJ, et al. Associations among peripheral and central kynurenine pathway metabolites and inflammation in depression. Neuropsychopharmacology. 2020;45:998–1007. doi:10.1038/s41386-020-0607-1
209. Paul ER, Schwieler L, Erhardt S, et al. Peripheral and central kynurenine pathway abnormalities in major depression. Brain Behav Immun. 2022;101:136–145. doi:10.1016/j.bbi.2022.01.002
210. Aguliar-Valles A, Kim J, Jung S, Woodside B, Luheshi GN. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol Psychiatry. 2014;19:599–606. doi:10.1038/mp.2013.137
211. Wittenberg GM, Greene J, Vértes PE, Drevets WC, Bullmore ET. Major depressive disorder is associated with differential expression of innate immune and neutrophil-related gene networks in peripheral blood: a quantitative review of whole-genome transcriptional data from case-control studies. Biol. Psychiatry. 2020;88:625–637. doi:10.1016/j.biopsych.2020.05.006
212. Lynall M-E, Turner L, Bhatti J, et al. Peripheral blood cell–stratified subgroups of inflamed depression. Biol. Psychiatry. 2020;88:185–196. doi:10.1016/j.biopsych.2019.11.017
213. Meng L, Zhou M, Wang Y, et al. CD177 on neutrophils engages stress-related behavioral changes in male mice. Brain Behav Immun. 2024;120:403–412. doi:10.1016/j.bbi.2024.06.011
214. Schubert JJ, Veronese M, Fryer TD, et al. A Modest Increase in 11C-PK11195-Positron Emission Tomography TSPO Binding in depression is not associated with serum c-reactive protein or body mass index. Biol Psychiatry Cogn Neurosci Neuroimaging. 2021;6:716–724. doi:10.1016/j.bpsc.2020.12.017
215. Gritti D, Delvecchio G, Ferro A, Bressi C, Brambilla P. Neuroinflammation in major depressive disorder: a review of PET imaging studies examining the 18-kDa translocator protein. J Affect Disord. 2021;292:642–651. doi:10.1016/j.jad.2021.06.001
216. Parhizkar S, Gent G, Chen Y, et al. Sleep deprivation exacerbates microglial reactivity and Aβ deposition in a TREM2 -dependent manner in mice. Sci Transl Med. 2023;15:eade6285. doi:10.1126/scitranslmed.ade6285
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.