")
Back to Journals » Journal of Inflammation Research » Volume 18
Biological Functions and Clinical Implications of CFLAR: From Cell Death Mechanisms to Therapeutic Targeting in Immune Regulation
Received 28 January 2025
Accepted for publication 1 April 2025
Published 9 April 2025 Volume 2025:18 Pages 4911—4928
DOI https://doi.org/10.2147/JIR.S519885
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Haiyang Zhang,1– 3 Xin Li,1– 3 Liangkang Lin1,4
1Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, People’s Republic of China; 2Key Laboratory of Birth Defects and Related Diseases of Women and Children, Sichuan University, Ministry of Education, Chengdu, People’s Republic of China; 3NHC Key Laboratory of Chronobiology, Sichuan University, Chengdu, People’s Republic of China; 4Department of Pediatrics, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, People’s Republic of China
Correspondence: Liangkang Lin, Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, People’s Republic of China, Tel/Fax +86 15361908128, Email [email protected]
Abstract: Since its initial functional characterization in the late 1990s, CASP-8 and FADD-like apoptosis regulator (CFLAR) has been recognized as a crucial regulator of both apoptosis and immune responses. CFLAR inhibits caspase-8 activation by forming heterodimers with procaspase-8 at the death-inducing signaling complex (DISC), thereby preventing its proteolytic maturation. In addition to its role in cell death, CFLAR is integral to immune regulation, modulating NF-κB-dependent cytokine production (eg, IL-1β, TNF-α) and effector functions of T cells and macrophages. Recent studies underscore the pathological significance of dysregulated CFLAR expression in a variety of diseases, including cancers and inflammatory conditions. Within the tumor microenvironment, elevated CFLAR expression confers resistance to therapy, while in infectious and inflammatory diseases, its expression levels modulate the magnitude and direction of the immune response. This review provides an in-depth exploration of CFLAR’s structural and functional properties, focusing on its involvement in apoptosis, autophagy, and immune modulation. Moreover, we examine its translational potential as a therapeutic target, evidenced by ongoing preclinical studies targeting CFLAR isoforms in cancer immunotherapy. By synthesizing recent advances in CFLAR’s dual roles in cell death and immune surveillance, this review highlights actionable targets for overcoming therapy resistance and immune dysregulation.
Keywords: CFLAR, apoptosis, autophagy, immunomodulation, signal path
Introduction
Caspase-8 and FADD-like apoptosis regulator (CFLAR), commonly referred to as cellular FLICE-inhibitory protein (c-FLIP), has been recognized as a critical inhibitor of apoptosis since its discovery in 1997.1 CFLAR exerts its anti-apoptotic effects by binding to Fas-associated death domain protein (FADD), thereby competitively inhibiting the interaction between caspase-8 and FADD. This prevents the assembly of the death-inducing signaling complex (DISC) and thereby impedes the transmission of extrinsic apoptotic signals. Beyond apoptosis regulation, CFLAR is also critical in immune responses and inflammation. It modulates cytokine secretion and immune cell activation, thus influencing the host’s immune response to pathogens. These properties position CFLAR as a key molecule in the study of autophagy, immunity, and inflammation. For instance, CFLAR regulates key stages of autophagy, including autophagosome formation and maturation, which in turn affect the differentiation and migration of effector cells.
In recent years, the role of CFLAR in a range of diseases has gained significant attention. In cancer, upregulated CFLAR expression is strongly associated with immune evasion mechanisms, such as the inhibition of apoptosis and the modulation of immune checkpoint pathways, leading to enhanced tumor cell resistance to treatment. In the context of infection and inflammation, CFLAR expression directly influences immune cell function and the intensity of inflammatory responses. TThus, a more comprehensive understanding of the mechanisms by which CFLAR regulates autophagy and immune modulation may provide valuable insights into its potential as both a disease mediator and a therapeutic target. This review focuses on exploring the mechanistic involvement of CFLAR in these processes, offering a framework for future research into targeted therapeutic strategies.
Overview of the Structure and Function of CFLAR
In 1997, Thome et al first identified the viral FLICE-inhibitory protein (vFLIP), which contains two death effector domains (DEDs) and can inhibit apoptotic signaling by binding to death receptors.1 Building upon this, Irmler et al subsequently identified a gene in the human genome homologous to vFLIP, which was named CFLAR.2 Although CFLAR shares a high degree of homology with vFLIP, it differs significantly in terms of its functional roles and regulation of expression, particularly in how it influences apoptosis and immune signaling. The CFLAR gene is located on the human chromosome at region 2q33-34, adjacent to the genes encoding caspase-8 and caspase-10, a positioning that is likely related to its functional interactions with these key regulators of apoptosis. The CFLAR gene consists of 14 exons and generates multiple mRNA isoforms through alternative splicing. These isoforms encode different forms of the protein, collectively known as c-FLIP, which can vary in their ability to regulate apoptosis and immune responses.
In humans, CFLAR has three primary isoforms: c-FLIP_L, c-FLIP_S, and c-FLIP_R. These isoforms perform distinct functions in various immune and tumor cell types. c-FLIP_L is a 55 kDa protein that contains two N-terminal DEDs and a C-terminal caspase-like domain. Despite structural similarities to procaspase-8, the catalytic sites within the caspase-like domain are not conserved in c-FLIP_L, thus it lacks proteolytic activity. Despite the structural similarity of this domain to that of procaspase-8, the catalytic sites are not conserved in c-FLIP_L, and thus it lacks caspase-like proteolytic activity.3–7 c-FLIP_S is a 27 kDa protein that contains only two DEDs and lacks a caspase-like domain. Unlike c-FLIP_L, c-FLIP_S is absent in mice, highlighting a potential species-specific difference in isoform expression.8,9 c-FLIP_R is a 25 kDa protein that is specifically expressed in certain cell lines, such as Raji and SKW6.4, as well as in human primary T cells. Its expression pattern suggests a role in specific immune responses and cell signaling in these contexts.10,11 In several signaling pathways, both c-FLIP_L and c-FLIP_S play crucial roles in promoting cell survival by activating or upregulating key pro-survival proteins, such as Akt, ERK, and nuclear factor-kappa B (NF-κB), often through their interaction with death receptors and modulation of caspase activity. All CFLAR isoforms are known to interact with caspase-8 via DED-DED interactions to form heterodimers. Moreover, caspase-8-mediated proteolysis at the Asp376 and Asp196 sites generates truncated c-FLIP fragments, known as p43-FLIP and p22-FLIP, respectively. These fragments are thought to have distinct functional roles, including modulating the apoptotic response and influencing immune signaling.12 The biological functions of the major CFLAR isoforms in various immune and tumor cell types, including their roles in apoptosis and immune regulation, are summarized in Table 1.
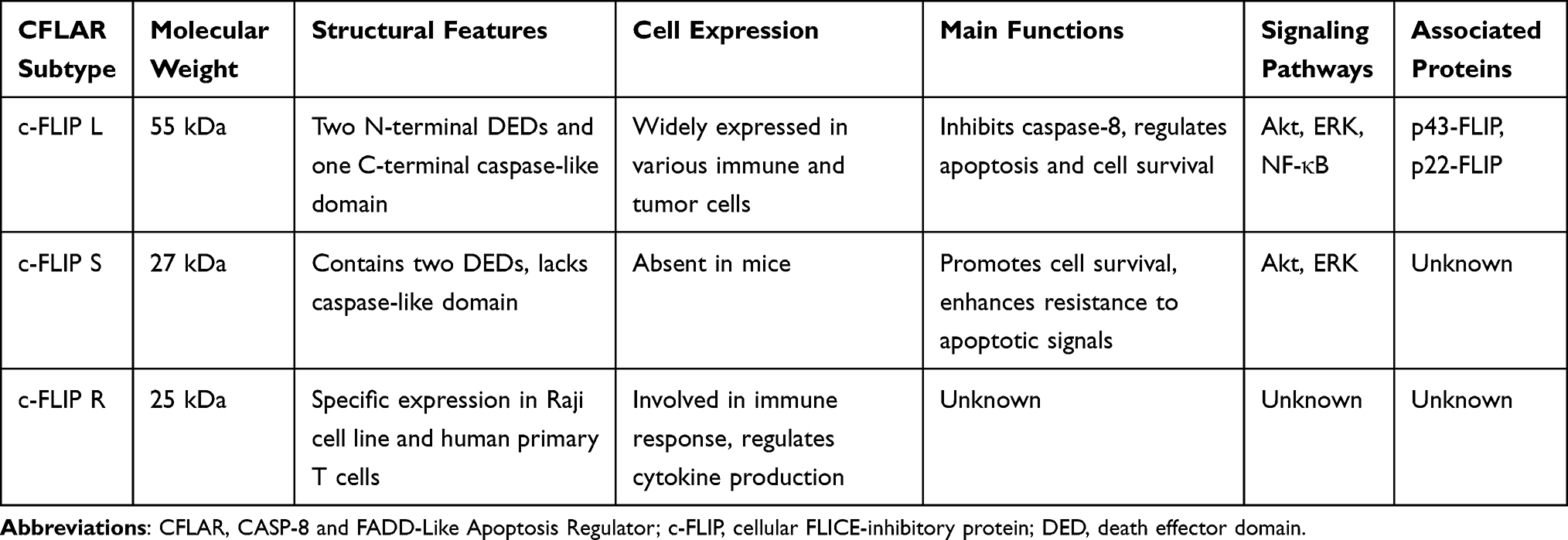 |
Table 1 Summary of CFLAR Subtypes and Their Biological Functions |
CFLAR, as a multifunctional protein, plays a pivotal role in regulating caspase-8 activity, which in turn modulates key cellular processes such as apoptosis, necroptosis, and autophagy.13 CFLAR can interact with FADD, caspase-8, caspase-10, and death receptor 5 (DR5), either in a ligand-dependent or independent manner, leading to the formation of an apoptosis inhibitory complex (AIC). This interaction prevents the assembly of the DISC and inhibits the activation of caspases. Furthermore, studies have demonstrated that caspase-8 and FADD can be recruited to specialized complexes at the endoplasmic reticulum (ER) and mitochondria, facilitating signal exchange between these organelles.14–21 At the ER-mitochondria interface, several molecular platforms have been identified, which consist of membrane-bound proteins and cytosolic apoptosis regulators. These platforms are involved in regulating key processes such as membrane anchoring, lipid metabolism, Ca²⁺ signaling, and the coordination of apoptosis between the ER and mitochondria.22,23
Role of CFLAR in Cell Death Mechanisms
CFLAR is involved in both the classical death receptor-mediated extrinsic apoptotic pathway and the regulation of non-canonical, pattern recognition receptor (PRR)-dependent apoptotic pathways. Furthermore, CFLAR is crucial for the formation of the RIP1-dependent, non-death receptor apoptotic platform, termed the ripoptosome. Figure 1 illustrates the regulatory mechanisms of CFLAR in apoptosis and autophagy.
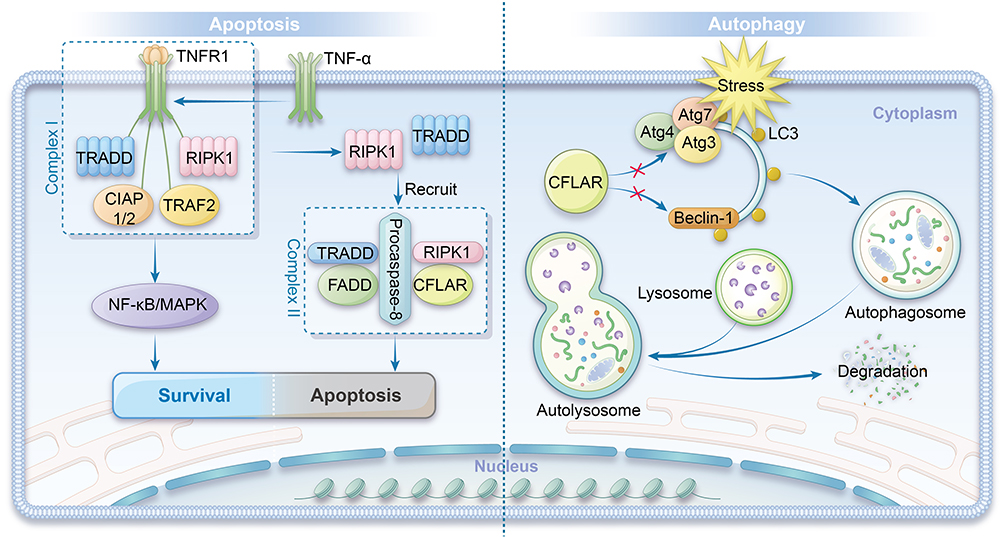 |
Figure 1 Regulatory Mechanisms of CFLAR in Apoptosis and Autophagy. Left panel: CFLAR modulates apoptosis and autophagy by interacting with key molecular complexes. TNF-α activates TNFR1, forming Complex I, which recruits TRADD, RIPK1, cIAP1/2, and TRAF2, thereby activating the NF-κB and MAPK signaling pathways. Additionally, Complex I can transition into a secondary Complex II, which associates with FADD, CFLAR, and procaspase-8 to further regulate cellular signaling events. Right panel: CFLAR inhibits autophagy by preventing the interaction between the Atg family proteins and LC3, and by suppressing Beclin-1 activity, thereby blocking autophagosome formation and protecting cells. Under stress conditions, autophagic membranes gradually form, eventually completing the autophagosome, which then fuses with lysosomes for the degradation of cellular components. |
CFLAR and Autophagy
Autophagy is an intracellular degradation process in which cells sequester cytoplasmic proteins into autophagosomes, which are then transported to lysosomes for subsequent degradation.24 This process is essential for maintaining cellular homeostasis by eliminating unnecessary proteins, dysfunctional complexes, and damaged organelles, thus preventing cellular stress and damage.25 Under stress conditions, autophagy is activated to maintain cellular homeostasis. However, it is tightly regulated, and its dysregulation is strongly associated with various pathological conditions, including neurodegenerative diseases, cancer, and metabolic disorders. The formation of autophagosomes depends on a family of proteins known as autophagy-related gene (Atg), with Atg3 playing a crucial role in conjugating microtubule-associated protein light chain 3 (LC3) to the autophagosome membrane and facilitating its subsequent lipidation and processing. LC3, particularly its lipidated form LC3-II, is a critical component of the autophagosome membrane, where it plays a vital role in autophagosome maturation and cargo recognition.26–28
Studies have shown that CFLAR can directly bind to Atg3, inhibiting its interaction with LC3, thereby disrupting the lipidation process of LC3 and impairing the formation of autophagosomes.29 In addition, CFLAR indirectly regulates autophagy by interacting with procaspase-10, potentially influencing its activation and subsequent downstream effects on autophagic signaling. Beclin-1, which is crucial for autophagosome formation, promotes the localization of Atg to the pre-autophagosomal membrane, a process facilitated by its interaction with the Vps34 complex. Beclin-1’s activity is regulated by its release from the anti-apoptotic protein Bcl-2.30 The interaction between Bcl-2 and Beclin-1 serves as a critical point of convergence between apoptosis and autophagy, as Bcl-2 prevents Beclin-1 from interacting with essential autophagy proteins. As an anti-apoptotic protein, Bcl-2 can suppress Beclin-1’s autophagic function by interacting with it, potentially preventing autophagy-dependent cell death.
In multiple myeloma cells, the presence of c-FLIP_L inhibits the activation of Beclin-1, potentially through interference with the Beclin-1 activation complex or by preventing the dissociation of Bcl-2 from Beclin-1. The complex formed by the DED-mediated heterodimerization of procaspase-10 and c-FLIP_L exhibits proteolytic activity, which cleaves the Bcl-2 interacting protein BCLAF1. This cleavage prevents BCLAF1 from displacing Bcl-2, thereby blocking the activation of Beclin-1 and inhibiting Beclin-1-mediated autophagy.31 Myeloma cells require basal levels of autophagy for survival; however, caspase-10 attenuates this autophagic response to prevent cell death, with c-FLIP_L promoting caspase-10 activation in multiple myeloma cells. This dysregulation of autophagy may contribute to the survival of tumor cells. Therefore, drugs that disrupt this balance may hold therapeutic potential for myeloma treatment.
It is important to note that the role of c-FLIP_L in these mechanisms is independent of caspase-8. However, in the context of procaspase-10 heterodimerization, caspase-10 acts as a classic pseudo-caspase, regulating the activity of its enzymatically active homologs and modulating apoptotic signaling.
CFLAR and Apoptosis
Necroptosis is a form of programmed cell death that is independent of caspase activation and distinct from the apoptosis pathway, which relies on caspases and TNFRSF signaling. It primarily involves the activation of receptor-interacting protein kinase 1 (RIPK1) and RIPK3, which subsequently activate mixed lineage kinase domain-like (MLKL), leading to cellular membrane rupture. Necroptosis can occur when apoptotic pathways are blocked.13 Under specific conditions, such as in the absence of external death ligands or death receptor interactions, cells can autonomously activate caspase-8, a process that may involve intracellular signaling pathways or cellular stress responses.3 RIPK1 was initially identified as a protein interacting with the tumor necrosis factor receptor 1 (TNF-R1) signaling complex.32,33 It has since been shown to play a crucial role in regulating cell survival, apoptosis, and autophagy. RIPK1 mediates cell death through the formation of the necrosome complex, which involves its interaction with RIPK3 and MLKL, leading to necroptosis. Additionally, RIPK1’s role in apoptosis is modulated through its kinase activity and interactions with other signaling molecules. The function of RIPK1 is modulated by phosphorylation and ubiquitination, which are critical for determining cell fate. Cellular inhibitor of apoptosis proteins (cIAPs), functioning as E3 ubiquitin ligases, can ubiquitinate RIPK1, thereby promoting its activation or preventing its degradation. This modification also extends to cIAPs themselves, influencing their own stability and interactions within cell death signaling complexes.34
To investigate the self-ubiquitination of cIAPs and its impact on cell death pathways, Tenev et al used the cytotoxic drug etoposide to induce genotoxic cell death.35 Similarly, Feoktistova et al employed IAP antagonists (Smac mimetics) in combination with dsRNA poly I:C to induce immune-mediated cell death via Toll-like receptor 3 (TLR3) activation.36 Both studies observed the spontaneous formation of the ripoptosome, a 2 MDa complex consisting of RIPK1, FADD, caspase-8, and CFLAR. This complex plays a pivotal role in mediating both caspase-dependent apoptosis and RIP kinase-dependent necroptosis, with its formation and function varying in a cell type-dependent manner. Both genotoxic stress and TLR3 stimulation require ripoptosome formation to induce cell death. According to the current model, procaspase-8 integrated into the ripoptosome is activated via homodimerization and processed into its active form, leading to the cleavage and inactivation of RIPK1, and disintegration of the ripoptosome. This process predominantly leads to caspase-8-mediated apoptosis.37
When c-FLIP_L is incorporated into the ripoptosome, it activates procaspase-8 through heterodimerization. However, procaspase-8 activated by c-FLIP_L cleaves only a limited subset of substrates, such as RIPK1, and upon disintegration of the ripoptosome, procaspase-8 is inactivated again. In this manner, c-FLIP_L effectively prevents apoptosis and promotes cell survival by controlling caspase-8 activation. In contrast, c-FLIP_S promotes the assembly of the ripoptosome but does not activate procaspase-8 to inactivate RIPK1. This prevents the execution of apoptosis while still facilitating the formation of the ripoptosome complex.38 Furthermore, other studies suggest that the balance between the functional RIPK1-RIPK3-MLKL axis and CFLAR isoforms is a crucial determinant in the regulation of cell survival, apoptosis, and necroptosis. This balance influences the cellular decision to undergo either caspase-mediated apoptosis or RIP kinase-dependent necroptosis, depending on the specific cellular context.39–41 Figure 2 illustrates the formation and mechanism of action of the ripoptosome.
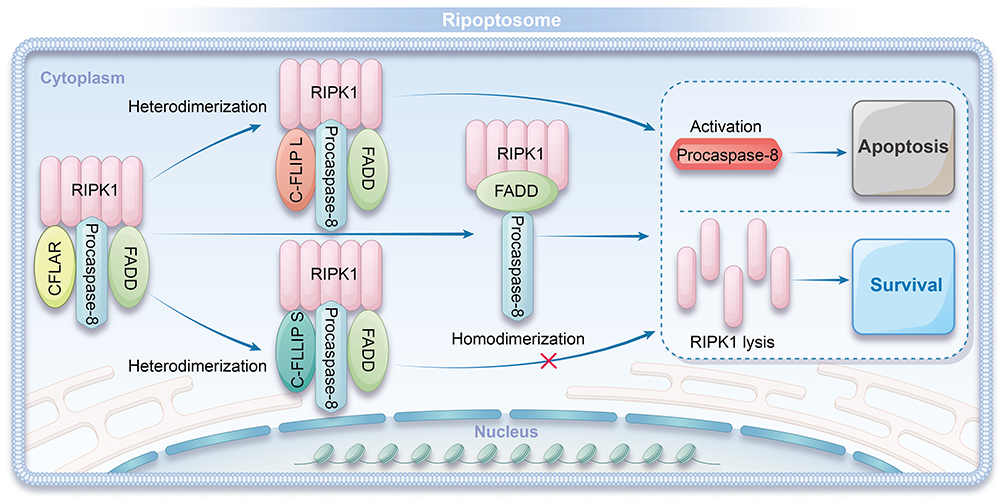 |
Figure 2 Formation and Mechanism of Ripoptosome. RIPK1 forms a complex with procaspase-8, FADD, and CFLAR isoforms to create the ripoptosome. c-FLIP L activates procaspase-8 through heterodimerization, leading to the inactivation of RIPK1 and subsequent disassembly of the ripoptosome, promoting apoptosis. In contrast, c-FLIP S facilitates ripoptosome assembly but inhibits the activation of procaspase-8 and prevents the inactivation of RIPK1, thereby modulating the apoptotic response. |
Regulation of Other Death-Inducing Protein Complexes
CFLAR plays a significant role in the formation of several protein complexes associated with cell death. c-FLIP_L plays a significant role in regulating the signaling pathways of several death receptors, including Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Receptors (TRAIL), Fas/CD95, and TNF-R1. In particular, c-FLIP_L plays a critical role downstream of the TNF-R1 signaling pathway. TNF-R1, a member of the TNF receptor superfamily (TNFRSF), forms Complex I upon binding to its ligand TNF-α. This process recruits TNFR1-associated death domain (TRADD) protein, followed by RIPK1, TNF receptor-associated factor 2 (TRAF2), and cellular inhibitors of apoptosis (cIAP1/2). The assembly of this complex is driven by the formation of linear ubiquitin chains, a process dependent on RIPK1 ubiquitination. The resulting complex activates both the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways.42,43
Following the formation of Complex I, RIPK1 undergoes deubiquitination by CYLD, a lysine deubiquitinase, which triggers the formation of the secondary TNFR1 Complex II. This complex consists of TRADD and RIPK1 dissociated from Complex I and can further recruit FADD, procaspase-8, and CFLAR. Ultimately, these molecules form a structure similar to the DISC.44 In Complex II, the ratio of homodimers to heterodimers of c-FLIP_L and procaspase-8 plays a critical role in regulating signal transduction. Sufficient homodimerization of procaspase-8 is required to activate caspase-3/7, while a higher proportion of procaspase-8/c-FLIP_L heterodimers inhibits this activation, thus impeding apoptosis. Furthermore, TNFR1 Complex I enhances the expression of CFLAR, which, in turn, modulates the signaling output from Complex II, influencing the balance between cell survival and apoptosis.45,46
Research by Day et al demonstrated that in MCF-7 breast cancer cells, removal of c-FLIP_L from the AIC, which includes DR5, FADD, and caspase-8, triggers spontaneous, ligand-independent cell death. The precise role of RIPK1 in this process remains unclear and requires further investigation to determine its involvement in regulating spontaneous cell death in the absence of ligands.47 In addition, Estornes et al48 identified an atypical death complex induced by double-stranded RNA, which includes TLR3, the TIR domain-containing adaptor protein (TRIF), and caspase-8. In contrast to the ripoptosome, the TLR3-dependent complex requires RIPK1, rather than FADD, for the activation of procaspase-8, highlighting a distinct mechanism of caspase-8 activation in response to RNA-induced stress. These findings suggest that various death-inducing stimuli can give rise to distinct apoptosis-inducing complexes, which share similarities with the ripoptosome, a key regulator of cell death in response to specific cellular stress signals.
Biochemical Properties of CFLAR
c-FLIP_L is considered a more potent activator of procaspase-8 compared to its isoform c-FLIP_S, particularly in terms of substrate specificity.49 Forced dimerization experiments have demonstrated that c-FLIP_L can activate procaspase-8 in the absence of inter-domain cleavage, leading to changes in substrate specificity. This finding highlights an alternative mechanism by which c-FLIP_L modulates procaspase-8 activity.50 Yu et al51 found that the p43-FLIP fragment, generated by procaspase-8-mediated cleavage, displayed significantly higher affinity for heterodimerization with procaspase-8 compared to the uncleaved c-FLIP_L. This enhanced interaction suggests a potential regulatory mechanism for procaspase-8 activation. This finding suggests a potential mechanism by which p43-FLIP may enhance procaspase-8 activity, possibly through stabilization of the procaspase-8/p43-FLIP complex or by altering the conformational dynamics of procaspase-8. In related studies, Dickens52 and Schleich53 quantitatively analyzed the DISC initiated by TRAIL and Fas ligand (CD95L), respectively. They discovered that the number of procaspase-8 molecules in the DISC was significantly greater than the number of FADD molecules, while the content of CFLAR in the natural DISC was relatively low. These results indicate that multiple procaspase-8 molecules can simultaneously bind to a single FADD or c-FLIP_L molecule, suggesting a potential cooperative mechanism that enhances DISC stability and procaspase-8 activation.
Earlier studies have suggested that overexpression of procaspase-8, FADD, and CFLAR in cells leads to the formation of filamentous structures known as “death effector filaments”, which implies that procaspase-8 in the DISC may form multimeric complexes through DED-DED interactions.54 However, quantitative Western blot analysis by Majkut et al revealed that the highest procaspase-8 to CFLAR ratio in the DISC induced by DR5 agonist antibodies was 2:1, a result that challenges the predictions of the DED-chain model.55 Majkut et al further proposed a two-step DISC model based on site-directed mutagenesis and molecular modeling, in which the incorporation of c-FLIP_L facilitates heterodimerization with procaspase-8, thereby activating procaspase-8 without the need for inter-domain cleavage. However, their findings also indicated that the procaspase-8 to CFLAR ratio in the DISC did not exceed 2:1, which is inconsistent with the predictions made by the DED-chain model.55
Kallenberger et al used compartment-specific fluorescence probes to establish a mathematical model for procaspase-8 activation kinetics, concluding that procaspase-8 is initially cleaved at the pro-domain in a dimeric form within a single DISC complex.56 However, these models were trained using cell population-level measurements, and may not fully capture the detailed dynamics at the single-cell resolution. Subsequently, the aggregation of multiple DISC complexes induces procaspase-8 cleavage at the catalytic domain through inter-dimer interactions, resulting in the release of fully processed caspase-8 into the cytoplasm. In contrast, the c-FLIP_S and c-FLIP_R isoforms within the DISC can bind to procaspase-8 but fail to induce its activation. Schleich’s study further showed that the N-terminal pro-domain of procaspase-8, generated upon full processing, structurally associates with c-FLIP_S/R within the DISC, forming a negative feedback loop that terminates procaspase-8 activation.57
Initially, these short isoforms were thought to primarily promote cell survival by inhibiting apoptosis. However, it is now recognized that they play a more complex role, not only in regulating cell survival but also in modulating multiple signaling pathways involved in both cell survival and cell death.
Role of CFLAR in Inflammatory Mediators and Signaling Pathways
Inflammatory Mediators and Signaling Pathways
The transcription of the CFLAR gene can be activated by various stimuli, including TNF ligands such as TNF-α, epidermal growth factor (EGF), interleukin-6 (IL-6), chemokines, and certain chemotherapy agents (eg, doxorubicin).58 Several transcription factors, such as NF-κB, p63, and EGR1, are involved in regulating CFLAR gene expression during this process.59 Notably, several transcription factors promote CFLAR transcription, including NF-κB, p63, and early growth response 1 (EGR1). Other factors, such as nuclear factor of activated T-cells cytoplasmic 2 (NFATc2), heterogeneous nuclear ribonucleoprotein K (hnRNP K), androgen receptor (AR), and specificity protein 1 (SP1), also contribute to its regulation. On the other hand, c-myc, forkhead box O3a (FOXO3a), c-Fos, interferon regulatory factor 5 (IRF5), and SP3 suppress CFLAR transcription.58,59 Research suggests that p53 may upregulate CFLAR gene expression by modulating the levels of specific transcription factors, such as NF-κB, while simultaneously promoting CFLAR degradation through the proteasomal pathway. This indicates that p53 plays a critical role in balancing cell death and survival.60 CFLAR participates in regulating cell survival, proliferation, and tumorigenesis by activating various cell-protective signaling pathways, such as NF-κB and ERK, which are crucial for preventing apoptosis and promoting cellular transformation.
CFLAR facilitates the transmission of survival signals by interacting with key proteins, particularly in the NF-κB and extracellular signal-regulated kinase (ERK) pathways, where it acts as a molecular scaffold to facilitate the assembly of signaling complexes. For example, c-FLIP_L interacts with TNF receptor-associated factor 1 (TRAF1), TRAF2, RIP1, and Raf-1 to facilitate NF-κB activation, promoting cell survival and inflammation.12,61 TRAF1 and TRAF2 are crucial regulators in the TNF signaling pathway, modulating cell survival and apoptosis through their interactions with other molecules. Additionally, the N-terminal fragment of c-FLIP_L, the caspase-8 processed p43-CFLAR, recruits TRAF2 and RIP1 more effectively than the full-length c-FLIP_L, leading to stronger NF-κB activation.12,62–64
In viable cells, CFLAR forms a heterodimer with procaspase-8, resulting in the generation of a novel N-terminal fragment of CFLAR (p22-FLIP), which plays a pivotal role in regulating survival signaling by modulating key downstream effectors. The p22-FLIP fragment interacts with the IKK complex, facilitating its activation and thereby playing a critical role in the subsequent NF-κB activation.65 In the c-Jun N-terminal kinase (JNK) signaling pathway, TNF-α-mediated JNK activation promotes CFLAR transcription by inducing NF-κB, which acts as a key mediator in this process.66 This process is not directly driven by CFLAR phosphorylation but relies on JNK-mediated phosphorylation, which in turn activates the E3 ubiquitin ligase Itch, leading to the ubiquitination and degradation of CFLAR. E3 ubiquitin ligase Itch specifically ubiquitinates CFLAR, targeting it for proteasomal degradation, thus reducing its cellular levels. Therefore, JNK contributes to TNF-α signaling by promoting the proteasomal degradation of c-FLIP_L, thereby reducing its ability to inhibit caspase activation, which in turn enhances NF-κB signaling and promotes apoptosis. These findings underscore the intricate and context-dependent interplay between the JNK and NF-κB pathways, which together regulate cell survival and apoptosis in response to various stress signals.66
In the calcium/calmodulin-dependent signaling pathway, calcium/calmodulin-dependent protein kinase II (CaMKII) upregulates CFLAR expression by phosphorylating key transcription factors, thereby protecting cancer cells from TRAIL-induced apoptosis. Treatment with the CaMKII inhibitor KN-93 in resistant cells effectively suppresses CaMKII activity, leading to reduced CFLAR expression and phosphorylation, which in turn enhances the cells’ sensitivity to Fas agonist antibody (CH-11) and promotes apoptosis.67,68
Inflammasomes
Inflammasomes are multiprotein complexes that primarily mediate the activation of caspases, particularly caspase-1, and are critical for recognizing damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), thereby initiating inflammatory immune responses, such as the production of pro-inflammatory cytokines. In inflammasomes, procaspase-1 assembles into a multi-protein complex, where it undergoes autocleavage at specific sites to activate caspase-1, which is a critical step in the inflammasome-mediated inflammatory response. The activated caspase-1 processes the inactive precursors of pro-inflammatory cytokines, such as pro-IL-1β and pro-IL-18, by cleaving them at specific sites, thereby generating their biologically active forms, IL-1β and IL-18.69–71 Studies have shown that procaspase-8 plays a role in regulating inflammasome activity, but this regulation is primarily mediated through a specific splice variant of c-FLIP_L, rather than via the activation of caspase-8 itself. This underscores the non-canonical role of c-FLIP_L in modulating inflammasome function.72 This finding underscores the significant role of c-FLIP_L as a “pseudocaspase” in mediating non-canonical functions, particularly its involvement in inflammasome regulation independent of caspase activation.
Interaction with the NLRP3 Inflammasome
c-FLIP_L directly interacts with NLR family pyrin domain-containing 3 (NLRP3) and procaspase-1, modulating the assembly and activation of the NLRP3 inflammasome. This interaction is essential for the efficient activation of caspase-1 and subsequent cytokine processing. This interaction promotes the processing and secretion of IL-1β, a critical mediator of inflammation, while the absence of c-FLIP_L impairs pyroptotic cell death, suggesting its essential role in regulating both inflammasome activation and cell death pathways. Upon the recognition of DAMPs and PAMPs, the NLRP3 inflammasome recruits procaspase-1 through the adapter protein ASC (apoptosis-associated speck-like protein containing a CARD), which facilitates the formation of the inflammasome complex and the activation of caspase-1. By interacting with the NLRP3 inflammasome, c-FLIP_L modulates the inflammatory response to infections and tissue damage, thus playing a crucial role in maintaining immune system homeostasis during acute and chronic inflammatory conditions, such as sepsis or autoimmune diseases.72
Interaction with the AIM2 Inflammasome
c-FLIP_L is known to influence the activity of the absent in melanoma 2 (AIM2) inflammasome. AIM2 inflammasome activation is triggered by the recognition of cytosolic DNA, either from host cells or pathogens, which serves as DAMPs or PAMPs. AIM2 contains an N-terminal pyrin domain (PYD), which mediates protein-protein interactions, and a C-terminal HIN domain, which is responsible for recognizing and binding to cytosolic DNA. Similar to NLRP3, AIM2 activation leads to the recruitment and activation of procaspase-1 via ASC, resulting in the secretion of IL-1β and IL-18, as well as the cleavage of gasdermin D, which is pivotal for initiating pyroptotic cell death. Studies have demonstrated that the caspase-like domain of c-FLIP_L directly interacts with the C-terminal HIN domain of AIM2, facilitating inflammasome assembly and ensuring the full activation of AIM2, which is crucial for effective immune responses.73
Wnt Signaling Pathway
c-FLIP_L is a key regulator of the Wnt signaling pathway, primarily by modulating the stability and transcriptional activity of β-catenin. Interestingly, this regulation occurs independently of its well-established role in procaspase-8 activation. Upon binding to the extracellular domains of Frizzled receptors, Wnt proteins initiate a cascade of signaling events involving the activation of dishevelled proteins and the stabilization of β-catenin, which subsequently translocates to the nucleus to regulate gene expression.74,75
Inhibition of β-Catenin Ubiquitination
c-FLIP_L inhibits the ubiquitination of β-catenin by interfering with the activity of E3 ubiquitin ligases, leading to a significant accumulation of β-catenin in the cytoplasm. This process prevents β-catenin degradation, allowing its translocation to the nucleus, where it interacts with TCF/LEF transcription factors to activate target genes associated with Wnt signaling. This c-FLIP_L-mediated regulation is closely linked to the development of various cancers, particularly colorectal cancer, by promoting the activation of Wnt/β-catenin signaling, which drives tumorigenesis and cancer cell proliferation.
Interaction with TIP49
Studies have shown that c-FLIP_L interacts with the nuclear protein TIP49 via its DED, modulating Wnt signaling by regulating the activity of the β-catenin-TCF complex. TIP49 is a regulatory factor that modulates the stability and transcriptional activity of the β-catenin-TCF complex in the Wnt pathway. cc-FLIP_L enhances the transcriptional activity of β-catenin by facilitating the accumulation of TIP49 at the promoter of immunoglobulin transcription factor 2 (ITF-2), thereby promoting the activation of downstream Wnt target genes.76 This process is crucial for activating downstream Wnt target genes, which are involved in regulating cell proliferation, differentiation, and tumorigenesis.
Cellular Localization and Functional Regulation of c-FLIP_L
The C-terminal region of c-FLIP_L contains both a nuclear localization signal (NLS) and a nuclear export signal (NES), facilitating its shuttling between the cytoplasm and the nucleus in a highly regulated manner. The cellular localization of c-FLIP_L is critical for its regulatory role in the Wnt signaling pathway, as its subcellular distribution influences its interactions with key signaling components and transcription factors. If the NLS of c-FLIP_L is mutated, preventing its entry into the nucleus, its ability to modulate Wnt signaling is significantly impaired, including its influence on β-catenin stabilization and transcriptional regulation. This indicates that c-FLIP_L’s role in Wnt signaling depends not only on its interactions with other proteins but also on its nuclear localization, where it enhances the transcriptional activity of key transcription factors involved in Wnt signaling.
Through its inhibition of β-catenin degradation, interaction with TIP49, and nuclear localization, c-FLIP_L exerts a multifaceted regulatory function in the Wnt signaling pathway, influencing both cytoplasmic and nuclear processes. It not only stabilizes β-catenin, promoting its translocation to the nucleus and activation of target genes, but also directly interacts with transcription factor complexes, such as TCF/LEF, to further amplify Wnt signaling at the transcriptional level. These mechanisms demonstrate that c-FLIP_L is not merely a passive participant in the Wnt pathway but actively regulates multiple levels of signaling, influencing processes such as cell proliferation, differentiation, and tumorigenesis.
Role of CFLAR in Immune Cells
CFLAR plays a complex and critical role in immune cells, including T and B cells, by regulating cellular processes such as apoptosis and immune responses through modulation of multiple signaling pathways.
Role of CFLAR in T Cells
CFLAR, particularly its long form c-FLIP_L, plays a crucial role in regulating T cell survival, proliferation, and activation, primarily by modulating key signaling pathways involved in these processes.64,77 During T cell activation, c-FLIP_L not only acts as an initiator of caspase-8 activation but also serves as a critical regulator of downstream signaling events essential for T cell activation. The p43FLIP complex, formed by c-FLIP_L and caspase-8, stabilizes caspase-8 activity and facilitates the activation of signaling pathways that promote T cell proliferation.77 Studies have shown that T cells lacking CFLAR exhibit significantly reduced proliferation and activation in response to T cell receptor (TCR) stimulation, accompanied by impaired activation of the ERK signaling pathway and other key downstream effectors.78,79 CFLAR protects mature T cells from exogenous apoptotic signals by inhibiting TCR-mediated apoptosis, thereby promoting T cell survival under stress conditions.80 In resting T cells, CFLAR effectively inhibits endogenous apoptosis by preventing the release of cytochrome c from the mitochondria, thus blocking the initiation of the intrinsic apoptotic pathway.81 In the context of T cell apoptosis, CFLAR plays a key negative regulatory role during T cell maturation, primarily by suppressing intrinsic apoptotic pathways through inhibition of pro-apoptotic molecules such as Bim.82 Resting T cells deficient in CFLAR are more susceptible to apoptosis mediated by Bcl-2 interacting mediator (Bim), accompanied by a decrease in reactive oxygen species (ROS) levels and a weakened inflammatory response, which further supports CFLAR’s role as a negative regulator of endogenous apoptosis during immune responses.82 Additionally, studies have shown that in the context of bacterial infections, c-FLIP_L protects T cells from apoptosis induced by inflammatory mediators, whereas c-FLIP_S promotes T cell death and inhibits their proliferation, potentially through modulation of caspase activation and inflammatory cytokine signaling.83,84
CFLAR plays a central role in regulatory T cells, regulating their survival, proliferation, and immunosuppressive function through modulation of key signaling pathways. Research has found that Tregs with low CFLAR expression exhibit a heightened inflammatory state. Mice deficient in CFLAR develop fatal autoimmune responses, marked by a dramatic reduction in peripheral Treg cell numbers, hyperactivation of effector T cells, and extensive multi-organ infiltration of immune cells. This phenotype is associated with impaired Treg-mediated suppression of effector T cell responses.85 These findings underscore the crucial role of CFLAR in preserving Treg function and preventing autoimmune pathology, likely through its regulation of Treg survival, differentiation, and suppression of effector T cell activation.
Role of CFLAR in B Cells
In CD40L-activated naïve B cells, CFLAR is recruited to the signaling complex, significantly delaying the onset of apoptosis by inhibiting caspase activation, thus preventing premature cell death.86 In contrast, CFLAR-deficient B cells exhibit increased susceptibility to Fas-induced apoptosis and altered responses to proliferation signals from TLRs and B cell receptors (BCRs), suggesting a disruption in key signaling pathways. These studies underscore the critical regulatory role of CFLAR in B cell immune function, including its involvement in immune tolerance and antibody production.87 Abnormal activation of the p38 MAPK and JNK pathways in CFLAR-deficient B cells further suggests that CFLAR plays a pivotal role in regulating these critical signaling networks. The absence of CFLAR leads to a reduction in peripheral B cell numbers, impaired proliferative responses, and reduced recruitment to sites of immune activation and inflammation.88 However, CFLAR deletion does not impact the key developmental processes of B cells, such as differentiation and class switching, but significantly impairs their function in activated immune responses, including antigen presentation and antibody production. These findings emphasize the importance of CFLAR in regulating B cell function.
CFLAR in Other Immune Cells
In myeloid cells, Gordy et al89 generated a myeloid-specific CFLAR knockout mouse model by expressing Cre recombinase under the control of the lysozyme 2 (Lyz2) promoter in a c-FLIP F/F background, aiming to investigate the specific role of CFLAR in myeloid cell differentiation and immune responses. These mice exhibited increased circulating neutrophils and spleen enlargement, which were directly associated with macrophage differentiation defects and impaired neutrophil clearance, indicating the key regulatory role of CFLAR in immune cell development. In addition, Huang et al independently developed CFLAR-floxed mice and generated myeloid-specific CFLAR knockout mice using the same strategy, further confirming the critical role of CFLAR in myeloid cells.90
In dendritic cells (DCs), Huang et al specifically deleted CFLAR by expressing Cre recombinase under the control of the integrin alpha X (Itgax) promoter.91 These mice exhibited spontaneous inflammatory arthritis, accompanied by an increase in autoreactive CD4+ T cells and a decrease in Tregs, further confirming CFLAR’s pivotal role in immune regulation and inflammatory responses. The roles of CFLAR in immune cells are summarized in Table 2.
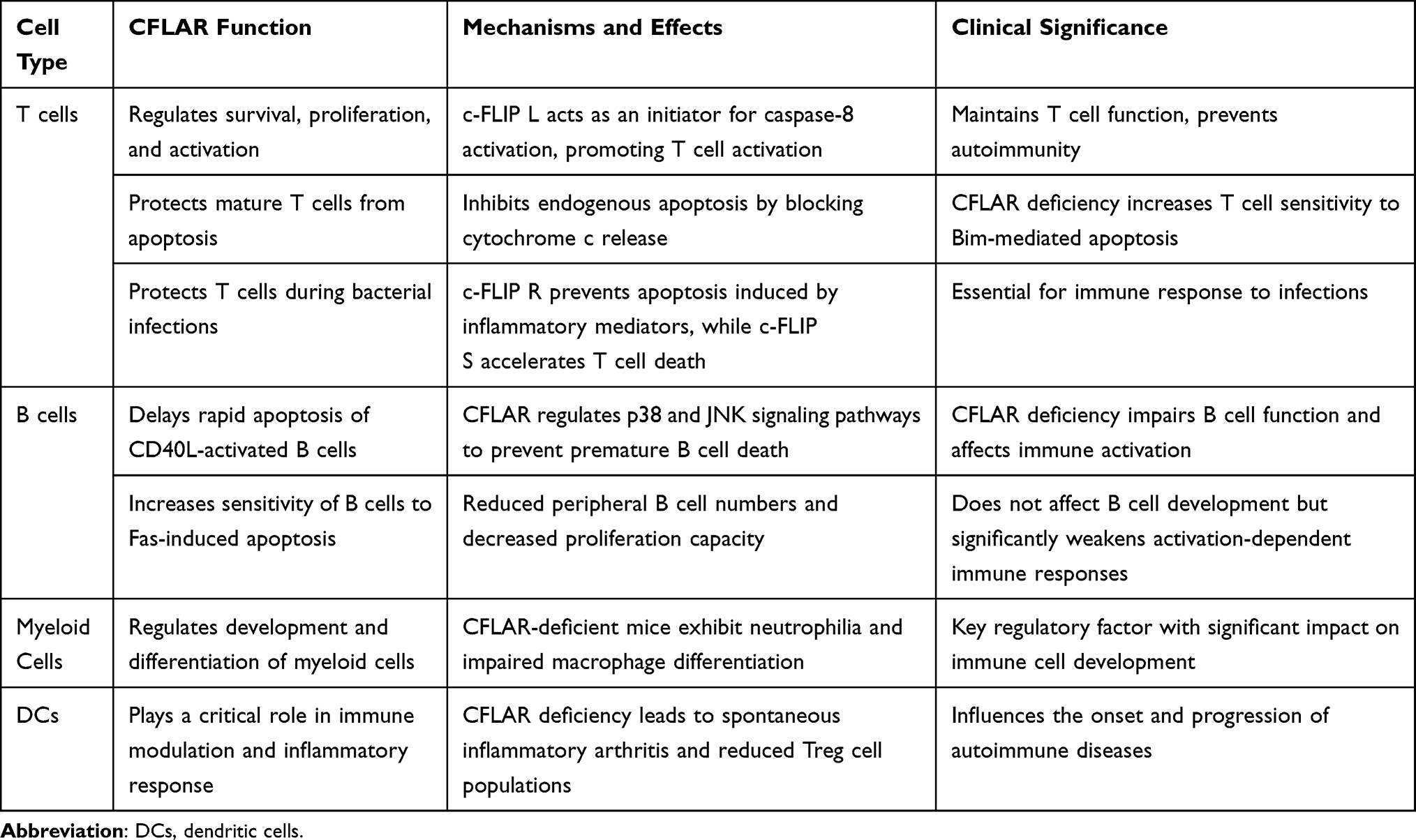 |
Table 2 Overview of CFLAR Functions in Immune Cells |
CFLAR and Cancer
CFLAR plays a critical role in cancer biology, particularly in regulating tumor cell survival and anti-apoptotic mechanisms, which are essential for cancer cell immune evasion and resistance to therapy.92–94 CFLAR inhibits apoptosis by binding to the DISC and suppressing the activation of caspase-8, thus blocking downstream signaling pathways mediated by death receptors (eg, DR5). This mechanism is crucial for cancer cells to evade immune surveillance and contributes to the resistance of cancer cells to apoptotic signals, highlighting CFLAR as a potential target for developing caspase-8-based targeted therapies. Elevated CFLAR expression is often observed in various cancers, including breast, colorectal, and lung cancer, and is associated with tumor initiation, metastasis, and poor prognosis by enabling cancer cells to resist apoptotic signals and chemotherapy-induced cell death. CFLAR blocks the apoptosis process by binding to caspase-8, preventing its activation and subsequent execution of apoptotic signaling, thus enhancing tumor cell survival. Due to its structural similarity to caspase-8, selective inhibitors targeting CFLAR must be carefully designed to avoid off-target effects that could inadvertently inhibit caspase-8, potentially leading to further suppression of apoptosis and resistance to treatment. RRecent studies have identified small molecules and RNA-based therapeutics that downregulate CFLAR expression, thereby enhancing tumor cell sensitivity to targeted therapies and improving the efficacy of chemotherapeutic agents.95,96 For instance, agonist antibodies targeting DR5 promote tumor cell apoptosis and simultaneously downregulate CFLAR expression through caspase-dependent signaling, thus increasing the sensitivity of tumor cells to chemotherapy-induced apoptosis.97 Furthermore, specific CFLAR variants hinder the activation of JNK and p38 MAPK by blocking late-phase death signals, which underscores its critical inhibitory role in regulating cell death and inflammation pathways.98 Recent studies have further revealed that CFLAR regulates the expression of forkhead box M1 (FoxM1), a key transcription factor that plays a pivotal role in enhancing the resistance of non-small cell lung cancer (NSCLC) to chemotherapy and promoting tumor progression.99 This finding suggests that CFLAR not only regulates apoptosis but also modulates additional survival pathways, including cell cycle progression and DNA repair mechanisms, which contribute to tumor cell resistance to therapy. Consequently, CFLAR’s multifaceted roles—such as inhibiting apoptosis, enhancing resistance to cancer therapies, and promoting tumor cell survival—make it a complex and vital therapeutic target in cancer treatment.
Potential Implications of CFLAR in Sepsis Immune Regulation and Treatment
Following our discussion of the role of CFLAR in cancer and its involvement in regulating cell death pathways, it is equally important to explore CFLAR’s functions in other pathological contexts, such as sepsis. Sepsis represents a complex inflammatory condition where immune dysregulation plays a crucial role. Future research should aim to elucidate the specific role of CFLAR in immune regulation within the context of sepsis, particularly its contribution to immune dysregulation. A deeper understanding of CFLAR’s regulatory effects across different immune cell subsets (eg, T cells, B cells, and myeloid cells) and its involvement in sepsis pathophysiology will be pivotal in identifying targeted therapeutic strategies for the personalized treatment of sepsis patients.100–102 Moreover, further investigation is needed to explore the dynamic expression patterns of CFLAR and its regulatory mechanisms under various pathological conditions, particularly in the context of sepsis-induced inflammation and immune dysregulation. This research could provide the necessary foundation for developing precise intervention strategies targeting CFLAR, offering the potential for more effective treatments for sepsis patients.103
In drug development, targeting CFLAR with specific inhibitors, such as small molecules, gene-editing technologies like Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated protein 9 (CRISPR-Cas9), and RNA interference approaches, should be explored. These strategies must be designed to selectively modulate CFLAR without inducing off-target effects on caspase-8 activity, which could lead to unwanted apoptosis inhibition.104,105 Targeting CFLAR in sepsis may help restore immune homeostasis by modulating immune cell activation and tolerance, thereby reducing excessive inflammatory responses and pathological inflammation. Combining CFLAR-targeted therapies with immune checkpoint inhibitors, TRAIL agonists, or ER stress inducers should also be investigated. Such combination treatments may synergistically enhance immune response and apoptotic sensitivity in sepsis, improving therapeutic outcomes. These approaches could enhance the apoptotic sensitivity of immune cells by restoring CFLAR-mediated regulation, as well as modulating cytokine release to mitigate the hyperinflammatory response that characterizes sepsis.106,107 CFLAR not only serves as a promising target for anti-cancer drug development but also holds significant therapeutic potential in treating acute inflammatory diseases such as sepsis, particularly in cases unresponsive to conventional therapies. Continued research into the basic biology of CFLAR and its clinical translational potential in the context of sepsis may open new therapeutic avenues, ultimately improving patient outcomes in this challenging disease. Figure 3 provides a brief overview of the potential applications of CFLAR in immunotherapy for sepsis.
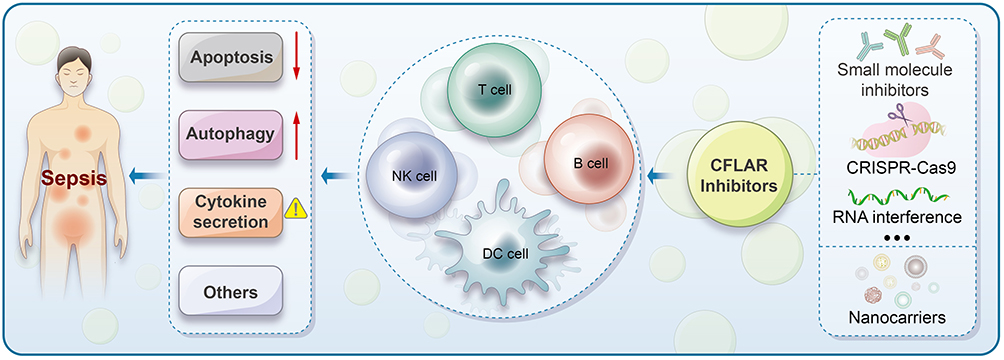 |
Figure 3 Potential Applications of CFLAR in Sepsis Immunotherapy. Sepsis is characterized by dysregulated inflammation, involving complex immune responses that include alterations in apoptosis, autophagy, and cytokine secretion. Immune cells, including T cells, B cells, NK cells, and DCs, play critical roles in the progression of sepsis. CFLAR inhibitors represent a promising therapeutic strategy, and through the application of CRISPR-Cas9, RNA interference, and nanoparticle-based delivery systems, these inhibitors can be engineered to restore immune homeostasis by targeting key processes such as apoptosis, autophagy, and cytokine secretion. |
CFLAR in Autoimmune Diseases and Chronic Inflammatory Conditions
CFLAR may also play a crucial regulatory role in the onset and progression of autoimmune and chronic inflammatory diseases. In chronic inflammation, the expression of Fas, FasL, and FLIP is upregulated, and these molecules are crucial in inflammatory cells. Studies indicate that apoptotic inflammatory cells accumulate in inflamed tissues, and the upregulation of FLIP in these cells may inhibit Fas-mediated apoptosis, contributing to chronic inflammation persistence. During the resolution of granulomatous experimental autoimmune thyroiditis (G-EAT), the increased expression of FLIP in thyroid cells may suppress their apoptosis, promoting cell survival. Conversely, the expression of FasL in thyroid cells may induce apoptosis in inflammatory cells, aiding in the resolution of inflammation.108
Maria Feoktistova et al demonstrated that c-FLIP negatively regulates TNF-dependent apoptosis and localized epidermal inflammation. In c-FLIP-deficient mice, the epidermis exhibits excessive proliferation, impaired differentiation in epidermal cells, and increased apoptosis. Interestingly, the absence of TNF delays the progression of chronic inflammatory skin diseases.109 Death receptor-mediated apoptosis plays a critical role in controlling immune responses, and dysregulation of this pathway may lead to the onset of autoimmune diseases. F. Ewald et al further revealed that transgenic mice with forced expression of c-FLIP R (a variant of c-FLIP) develop a systemic lupus erythematosus (SLE)-like phenotype with aging, indicating that c-FLIP R is a significant regulator of apoptosis.110
In active ulcerative colitis (UC), signaling through the Fas receptor mediating apoptosis in intestinal epithelial cells (IECs) is impaired. Moreover, the expression of c-FLIP S is elevated in IECs of patients with active UC. The MEK-ERK pathway is recognized as a key signaling pathway that regulates IEC apoptosis during UC flare-ups and plays a critical role in the induction of c-FLIP S.111 In c-FLIP L transgenic mice, activated CD4⁺ T cells exhibit enhanced secretion of Th2 cytokines, and the production of Th1 cytokines is reduced. Furthermore, the Th2 bias in these c-FLIP L transgenic CD4⁺ T cells is associated with impaired NF-κB activity and upregulation of GATA-3, leading to a decrease in IFN-γ levels and an increase in Th2 cytokines. This Th2 skewing significantly enhances the susceptibility of these mice to the OVA-induced asthma model.112 Overall, c-FLIP plays a crucial role in regulating apoptosis and immune regulation in chronic inflammation and autoimmune diseases. Its involvement in disease progression and immune homeostasis highlights its potential as a therapeutic target in these conditions.
Challenges and Limitations of CFLAR-Targeted Therapies
CFLAR-targeted therapies offer significant potential for modulating cell death and immune responses in cancer and inflammatory diseases, but several challenges must be addressed before their clinical application. One major concern is the risk of off-target effects due to CFLAR’s involvement in multiple signaling pathways, including apoptosis, necroptosis, and NF-κB activation. Non-specific targeting of CFLAR could lead to unintended immune dysregulation, excessive activation, or suppression, which may ultimately jeopardize therapeutic safety.
The context-dependent duality of CFLAR further complicates its therapeutic targeting. CFLAR can exert both protective and pro-inflammatory effects, depending on disease type, cellular context, and microenvironment. For example, inhibiting CFLAR may sensitize certain cancers to apoptosis while exacerbating inflammatory responses in conditions like sepsis or autoimmune diseases113,114. This duality underscores the need for targeted strategies that consider disease-specific mechanisms.
Resistance mechanisms significantly challenge the efficacy of CFLAR-targeted therapies in cancer treatment. Cancer cells frequently exhibit phenotypic plasticity through the activation of alternative survival pathways that enable them to evade CFLAR inhibition. A study by Yongping Shao et al demonstrated that c-FLIP is crucial for TNFα-induced protection against PLX4720, a selective inhibitor which is used to study vemurafenib. Overexpression of c-FLIP confers protection to melanoma cells against PLX4720-induced apoptosis. Thus, c-FLIP may play a role in RAF inhibitor resistance by preventing the activation of caspase-8, a key mediator of apoptosis, in response to RAF inhibitors.115 This suggests that monotherapies targeting CFLAR are unlikely to achieve long-term efficacy due to the development of resistance, which highlights the need for combination strategies with other apoptotic regulators or immune modulators.
Moreover, the development of selective CFLAR modulators with optimal pharmacokinetics and bioavailability remains challenging. Only a few small-molecule inhibitors or peptide-based agents can specifically target CFLAR activity without affecting other components of cell death pathways. Ongoing research is crucial to improve drug design (eg, isoform-specific modulators) and enhance the specificity and efficacy of CFLAR-based therapies.
Given these complexities, future studies should focus on elucidating the molecular mechanisms that regulate CFLAR in diverse disease contexts and establishing patient-specific biomarkers to predict therapeutic responses. Such efforts will be vital for developing safer, more effective treatment strategies.
Conclusion
CFLAR, a critical anti-apoptotic regulator, plays a pivotal role in immune cell survival, immune regulation, and tumorigenesis by modulating apoptosis pathways and influencing cellular responses to stress. Future research should focus on developing targeted inhibitors of CFLAR, utilizing advanced molecular techniques such as CRISPR-Cas9, RNA interference, and small molecule screening. These approaches could unlock CFLAR’s therapeutic potential in clinical immunotherapy, providing novel strategies for the treatment of sepsis, autoimmune diseases, and cancer. As our understanding of the underlying biological processes deepens, several promising research directions are emerging, including the precision targeting of apoptosis pathways, personalized medicine strategies, the exploration of combination therapies, the identification of new biomarkers, the investigation of immune evasion mechanisms, and the integration of systems biology approaches. Ultimately, these advancements could facilitate the effective translation of research findings into clinical applications, thereby enhancing the therapeutic landscape.
Abbreviations
CFLAR, CASP-8 and FADD-like apoptosis regulator; FADD, Fas-associated death domain; c-FLIP, cellular FLICE-inhibitory protein; DISC, death-inducing signaling complex; vFLIP, viral FLICE-inhibitory protein; DED, death effector domain; NF-κB, nuclear factor-kappa B; DR5, death receptor 5; ER, endoplasmic reticulum; AIC, apoptosis inhibitory complex; PRR, pattern recognition receptor; Atg, autophagy-related gene; LC3, light chain 3; RIPK1, receptor-interacting protein kinase 1; MLKL, mixed lineage kinase domain-like; TNF-R1, tumor necrosis factor receptor 1; cIAPs, cellular inhibitor of apoptosis proteins; TLR3, Toll-like receptor 3; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand receptors; TNFRSF, TNF receptor superfamily; TRADD, TNFR1-associated death domain; TRAF2, TNF receptor-associated factor 2; MAPK, mitogen-activated protein kinase; TRIF, TIR domain-containing adaptor protein; EGF, epidermal growth factor; EGR1, early growth response 1; SP1, specificity protein 1; NFATc2, nuclear factor of activated T-cells cytoplasmic 2; FOXO3a, forkhead box O3a; IRF5, interferon regulatory factor 5; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; CaMKII, calcium/calmodulin-dependent protein kinase II; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; NLRP3, NLR family pyrin domain-containing 3; AIM2, absent in melanoma 2; PYD, pyrin domain; ITF-2, immunoglobulin transcription factor 2; NLS, nuclear localization signal; NES, nuclear export signal; TCR, T cell receptor; ROS, reactive oxygen species; Lyz2, lysozyme 2; DCs, dendritic cells; Itgax, integrin alpha X; FoxM1, forkhead box M1; NSCLC, non-small cell lung cancer; CRISPR-Cas9, CRISPR-associated protein 9; G-EAT, granulomatous experimental autoimmune thyroiditis; SLE, systemic lupus erythematosus; UC, ulcerative colitis; IECs, intestinal epithelial cells.
Acknowledgments
The authors thank all the clinical staff contributed to the study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
All authors declare that the research is conducted in the absence of any commercial relations or financial relationships of interest that might be a constant source of interest.
References
1. Thome M, Schneider P, Hofmann K, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386(6624):517–521. doi:10.1038/386517a0
2. IrmLer M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388(6638):190–195. PMID: 9217161. doi:10.1038/40657
3. Shu H, Halpin D, Goeddel D. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6(6):751–763. doi:10.1016/s1074-7613(00)80450-1
4. Hu S, Vincenz C, Ni J, et al. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J Biol Chem. 1997;272(28):17255–17257. doi:10.1074/jbc.272.28.17255
5. Srinivasula S, Ahmad M, Ottilie S, et al. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J Biol Chem. 1997;272(30):18542–18545. doi:10.1074/jbc.272.30.18542
6. Goltsev Y, Kovalenko A, Arnold E, et al. CASH, a novel caspase homologue with death effector domains. J Biol Chem. 1997;272(32):19641–19644. doi:10.1074/jbc.272.32.19641
7. Inohara N, Koseki T, Hu Y, et al. CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc Natl Acad Sci USA. 1997;94(20):10717–10722. doi:10.1073/pnas.94.20.10717
8. Han D, Chaudhary P, Wright M, et al. MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc Natl Acad Sci USA. 1997;94(21):11333–11338. doi:10.1073/pnas.94.21.11333
9. Rasper D, Vaillancourt J, Hadano S, et al. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5(4):271–288. doi:10.1038/sj.cdd.4400370
10. Golks A, Brenner D, Fritsch C, et al. c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem. 2005;280(15):14507–14513. doi:10.1074/jbc.M414425200
11. Ueffing N, Keil E, Freund C, et al. Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ. 2008;15(4):773–782. doi:10.1038/sj.cdd.4402314
12. Ueffing N, Singh KK, Christians A, et al. A single nucleotide polymorphism determines protein isoform production of the human c-FLIP protein. Blood. 2009;114(3):572–579. doi:10.1182/blood-2009-02-204230
13. He MX, He YW. CFLAR/c-FLIPL. Autophagy. 2014;9(5):791–793. doi:10.4161/auto.23785
14. Ng F, Nguyen M, Kwan T, et al. p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8-associated protein in the endoplasmic reticulum. J Cell Biol. 1997;139(2):327–338. doi:10.1083/jcb.139.2.327
15. Breckenridge D, Nguyen M, Kuppig S, et al. The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc Natl Acad Sci USA. 2002;99(7):4331–4336. doi:10.1073/pnas.072088099
16. Xiang R, Liu Y, Zhu L, et al. Adaptor FADD is recruited by RTN3/HAP in ER-bound signaling complexes. Apoptosis. 2006;11(11):1923–1932. doi:10.1007/s10495-006-0082-0
17. Gonzalvez F, Schug ZT, Houtkooper RH, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183(4):681–696. doi:10.1083/jcb.200803129
18. Schug ZT, Gonzalvez F, Houtkooper RH, et al. Bid is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 2010;18(3):538–548. doi:10.1038/cdd.2010.135
19. Breckenridge D, Stojanovic M, Marcellus R, et al. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160(7):1115–1127. doi:10.1083/jcb.200212059
20. Simmen T, Aslan JE, Blagoveshchenskaya AD, et al. PACS-2 controls endoplasmic reticulum–mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005;24(4):717–729. doi:10.1038/sj.emboj.7600559
21. Chandra D, Choy G, Deng X, et al. Association of active caspase 8 with the mitochondrial membrane during apoptosis: potential roles in cleaving BAP31 and Caspase 3 and mediating mitochondrion-endoplasmic reticulum cross talk in etoposide-induced cell death. mol Cell Biol. 2023;24(15):6592–6607. doi:10.1128/mcb.24.15.6592-6607.2004
22. Decker ST, Funai K. Mitochondrial membrane lipids in the regulation of bioenergetic flux. Cell Metab. 2024;36(9):1963–1978. doi:10.1016/j.cmet.2024.07.024
23. de Brito O, Scorrano L. An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 2010;29(16):2715–2723. doi:10.1038/emboj.2010.177
24. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev mol Cell Biol. 2018;19(6):349–364. doi:10.1038/s41580-018-0003-4
25. Reggiori F, Ungermann C. Autophagosome maturation and fusion. J mol Biol. 2017;429(4):486–496. doi:10.1016/j.jmb.2017.01.002
26. Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev mol Cell Biol. 2013;14(12):759–774. doi:10.1038/nrm3696
27. Weidberg H, Shpilka T, Shvets E, et al. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell. 2011;20(4):444–454. doi:10.1016/j.devcel.2011.02.006
28. Fujita N, Hayashi-Nishino M, Fukumoto H, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. mol Biol Cell. 2008;19(11):4651–4659. doi:10.1091/mbc.e08-03-0312
29. Lee JS, Li Q, Lee JY, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11(11):1355–1362. doi:10.1038/ncb1980
30. Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi:10.1016/j.cell.2005.07.002
31. Lamy L, Ngo Vu N, Emre NCT, et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell. 2013;23(4):435–449. doi:10.1016/j.ccr.2013.02.017
32. Varfolomeev E, Boldin M, Goncharov T, et al. A potential mechanism of “cross-talk” between the p55 tumor necrosis factor receptor and Fas/APO1: proteins binding to the death domains of the two receptors also bind to each other. J Exp Med. 1996;183(3):1271–1275. doi:10.1084/jem.183.3.1271
33. Hsu H, Huang J, Shu H, et al. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4(4):387–396. doi:10.1016/s1074-7613(00)80252-6
34. Weinlich R, Green D. The two faces of receptor interacting protein kinase-1. mol Cell. 2014;56(4):469–480. doi:10.1016/j.molcel.2014.11.001
35. Tenev T, Bianchi K, Darding M, et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. mol Cell. 2011;43(3):432–448. doi:10.1016/j.molcel.2011.06.006
36. Feoktistova M, Geserick P, Kellert B, et al. cIAPs block ripoptosome formation, a RIP1/Caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. mol Cell. 2011;43(3):449–463. doi:10.1016/j.molcel.2011.06.011
37. Schilling R, Geserick P, Leverkus M. Characterization of the ripoptosome and its components: implications for anti-inflammatory and cancer therapy. Methods Enzymol. 2014;545:83–102. doi:10.1016/B978-0-12-801430-1.00004-4
38. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–320. doi:10.1038/nature14191
39. He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell. 2009;137(6):1100–1111. doi:10.1016/j.cell.2009.05.021
40. Feoktistova M, Geserick P, Panayotova-Dimitrova D, et al. Pick your poison: the ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle. 2014;11(3):460–467. doi:10.4161/cc.11.3.19060
41. Mandal P, Berger Scott B, Pillay S, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. mol Cell. 2014;56(4):481–495. doi:10.1016/j.molcel.2014.10.021
42. Hsu H, Xiong J, Goeddel D. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81(4):495–504. doi:10.1016/0092-8674(95)90070-5
43. Nikoletopoulou V, Markaki M, Palikaras K, et al. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833(12):3448–3459. doi:10.1016/j.bbamcr.2013.06.001
44. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–190. doi:10.1016/s0092-8674(03)00521-x
45. Kreuz S, Siegmund D, Scheurich P, et al. NF-κB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. mol Cell Biol. 2023;21(12):3964–3973. doi:10.1128/mcb.21.12.3964-3973.2001
46. Micheau O, Lens S, Gaide O, et al. NF-κB signals induce the expression of c-FLIP. mol Cell Biol. 2023;21(16):5299–5305. doi:10.1128/mcb.21.16.5299-5305.2001
47. Day TW, Huang S, Safa AR. c-FLIP knockdown induces ligand-independent DR5-, FADD-, caspase-8-, and caspase-9-dependent apoptosis in breast cancer cells. Biochem Pharmacol. 2008;76(12):1694–1704. doi:10.1016/j.bcp.2008.09.007
48. Estornes Y, Toscano F, Virard F, et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012;19(9):1482–1494. doi:10.1038/cdd.2012.22
49. Boatright K, Deis C, Denault J, Sutherlin D, Salvesen G. Activation of caspases-8 and −10 by FLIP(L). Biochem J. 2004;382(Pt 2):651–657. doi:10.1042/BJ20040809
50. Pop C, Oberst A, Drag M, et al. FLIPL induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433(3):447–457. doi:10.1042/bj20101738
51. Yu J, Jeffrey P, Shi Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc Natl Acad Sci USA. 2009;106(20):8169–8174. doi:10.1073/pnas.0812453106
52. Dickens Laura S, Boyd Robert S, Jukes-Jones R, et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. mol Cell. 2012;47(2):291–305. doi:10.1016/j.molcel.2012.05.004
53. Schleich K, Warnken U, Fricker N, et al. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. mol Cell. 2012;47(2):306–319. doi:10.1016/j.molcel.2012.05.006
54. Siegel R, Martin D, Zheng L, et al. Death-effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. J Cell Biol. 1998;141(5):1243–1253. doi:10.1083/jcb.141.5.1243
55. Majkut J, Sgobba M, Holohan C, et al. Differential affinity of FLIP and procaspase 8 for FADD’s DED binding surfaces regulates DISC assembly. Nat Commun. 2014;5(1). doi:10.1038/ncomms4350
56. Kallenberger S, Beaudouin J, Claus J, et al. Intra- and interdimeric caspase-8 self-cleavage controls strength and timing of CD95-induced apoptosis. Sci Signal. 2014;7(316):ra23. doi:10.1126/scisignal.2004738
57. Schleich K, Buchbinder JH, Pietkiewicz S, et al. Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2015;23(4):681–694. doi:10.1038/cdd.2015.137
58. Safa AR, Day T, Wu C. Cellular FLICE-like inhibitory protein (C-FLIP): a novel target for cancer therapy. Curr Cancer Drug Targets. 2008;8(1):37–46. doi:10.2174/156800908783497087
59. Bagnoli M, Canevari S, Mezzanzanica D. Cellular FLICE-inhibitory protein (c-FLIP) signalling: a key regulator of receptor-mediated apoptosis in physiologic context and in cancer. Int J Biochem Cell Biol. 2010;42(2):210–213. doi:10.1016/j.biocel.2009.11.015
60. Safa AR, Pollok KE. Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers. 2011;3(2):1639–1671. doi:10.3390/cancers3021639
61. Chaudhary P, Eby M, Jasmin A, et al. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene. 2000;19(39):4451–4460. doi:10.1038/sj.onc.1203812
62. Kump E, Ji J, Wernli M, et al. Gli2 upregulates cFlip and renders basal cell carcinoma cells resistant to death ligand-mediated apoptosis. Oncogene. 2008;27(27):3856–3864. doi:10.1038/onc.2008.5
63. Fang LW, Tai TS, Yu WN, et al. Phosphatidylinositide 3-kinase priming couples c-FLIP to T cell activation. J Biol Chem. 2004;279(1):13–18. doi:10.1074/jbc.M303860200
64. Dohrman A, Kataoka T, Cuenin S, et al. Cellular FLIP (Long Form) regulates CD8+ T cell activation through caspase-8-dependent NF-κB activation. J Immunol. 2005;174(9):5270–5278. doi:10.4049/jimmunol.174.9.5270
65. Golks A, Brenner D, Krammer PH, et al. The c-FLIP–NH2 terminus (p22-FLIP) induces NF-κB activation. J Exp Med. 2006;203(5):1295–1305. doi:10.1084/jem.20051556
66. Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIPL turnover. Cell. 2006;124(3):601–613. doi:10.1016/j.cell.2006.01.021
67. Yang BF, Xiao C, Roa WH, et al. Calcium/Calmodulin-dependent protein kinase II regulation of c-FLIP Expression and phosphorylation in modulation of fas-mediated signaling in malignant glioma cells. J Biol Chem. 2003;278(9):7043–7050. doi:10.1074/jbc.M211278200
68. Xiao C, Fengyang B, Song J, et al. Inhibition of CaMKII-mediated c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Exp Cell Res. 2005;304(1):244–255. doi:10.1016/j.yexcr.2004.11.002
69. Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci. 2017;130(23):3955–3963. doi:10.1242/jcs.207365
70. Yang X, Chang H, Baltimore D. Autoproteolytic activation of pro-caspases by oligomerization. mol Cell. 1998;1(2):319–325. doi:10.1016/s1097-2765(00)80032-5
71. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. mol Cell. 2002;10(2):417–426. doi:10.1016/s1097-2765(02)00599-3
72. Wu YH, Kuo WC, Wu YJ, et al. Participation of c-FLIP in NLRP3 and AIM2 inflammasome activation. Cell Death Differ. 2013;21(3):451–461. doi:10.1038/cdd.2013.165
73. Lugrin J, Martinon F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol Rev. 2017;281(1):99–114. doi:10.1111/imr.12618
74. Cong F, Schweizer L, Varmus H. Wnt signals across the plasma membrane to activate the β-catenin pathway by forming oligomers containing its receptors, Frizzled and LRP. Development. 2004;131(20):5103–5115. doi:10.1242/dev.01318
75. Bhanot P, Brink M, Samos C, et al. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382(6588):225–230. doi:10.1038/382225a0
76. Zhang J, Jiang HY, Zhang LK, et al. C-FLIPL modulated Wnt/β-catenin activation via association with TIP49 protein. J Biol Chem. 2017;292(6):2132–2142. doi:10.1074/jbc.M116.753251
77. Koenig A, Buskiewicz IA, Fortner KA, et al. The c-FLIPL cleavage product p43FLIP promotes activation of extracellular signal-regulated kinase (ERK), nuclear factor κB (NF-κB), and caspase-8 and T cell survival. J Biol Chem. 2014;289(2):1183–1191. doi:10.1074/jbc.M113.506428
78. Chau H, Wong V, Chen N, et al. Cellular FLICE-inhibitory protein is required for T cell survival and cycling. J Exp Med. 2005;202(3):405–413. doi:10.1084/jem.20050118
79. Zhang N, He Y. An essential role for c-FLIP in the efficient development of mature T lymphocytes. J Exp Med. 2005;202(3):395–404. doi:10.1084/jem.20050117
80. Zhang N, Hopkins K, He YW. c-FLIP protects mature T lymphocytes from TCR-mediated killing. J Immunol. 2008;181(8):5368–5373. doi:10.4049/jimmunol.181.8.5368
81. He W, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–1298. doi:10.1038/cr.2015.139
82. He MX, He YW. c-FLIP protects T lymphocytes from apoptosis in the intrinsic pathway. J Immunol. 2015;194(7):3444–3451. doi:10.4049/jimmunol.1400469
83. Telieps T, Ewald F, Gereke M, et al. Cellular‐FLIP, Raji isoform (c‐FLIPR) modulates cell death induction upon T‐cell activation and infection. Eur J Immunol. 2013;43(6):1499–1510. doi:10.1002/eji.201242819
84. Hinshaw-Makepeace J, Huston G, Fortner K, et al. c-FLIP(S) reduces activation of caspase and NF-kappaB pathways and decreases T cell survival. Eur J Immunol. 2008;38(1):54–63. doi:10.1002/eji.200636956
85. Plaza-Sirvent C, Schuster M, Neumann Y, et al. c-FLIP expression in Foxp3-expressing cells is essential for survival of regulatory T cells and prevention of autoimmunity. Cell Rep. 2017;18(1):12–22. doi:10.1016/j.celrep.2016.12.022
86. Hennino A, Berard M, Casamayor-Pallejà M, et al. Regulation of the Fas death pathway by FLICE-inhibitory protein in primary human B cells. J Immunol. 2000;165(6):3023–3030. doi:10.4049/jimmunol.165.6.3023
87. Zhang H, Rosenberg S, Coffey FJ, et al. A role for cFLIP in B cell proliferation and stress MAPK regulation. J Immunol. 2009;182(1):207–215. doi:10.4049/jimmunol.182.1.207
88. Coffey F, Manser T. Expression of cellular FLIP by B cells is required for their participation in an immune response. J Immunol. 2010;184(9):4871–4879. doi:10.4049/jimmunol.0903506
89. Gordy C, Pua H, Sempowski GD, et al. Regulation of steady-state neutrophil homeostasis by macrophages. Blood. 2011;117(2):618–629. doi:10.1182/blood-2010-01-265959
90. Huang Q, Perlman H, Huang Z, et al. FLIP: a novel regulator of macrophage differentiation and granulocyte homeostasis. Blood. 2010;116(23):4968–4977. doi:10.1182/blood-2009-11-252841
91. Huang QQ, Perlman H, Birkett R, et al. CD11c-mediated deletion of Flip promotes autoreactivity and inflammatory arthritis. Nat Commun. 2015;6(1). doi:10.1038/ncomms8086
92. Mukherjee N, Schwan JV, Fujita M, et al. Alternative treatments for melanoma: targeting BCL-2 family members to de-bulk and kill cancer stem cells. J Invest Dermatol. 2015;135(9):2155–2161. doi:10.1038/jid.2015.145
93. Ivanisenko NV, Seyrek K, Hillert-Richter LK, et al. Regulation of extrinsic apoptotic signaling by c-FLIP: towards targeting cancer networks. Trends Cancer. 2022;8(3):190–209. doi:10.1016/j.trecan.2021.12.002
94. Yoon MJ, Kang YJ, Kim IY, et al. Monensin, a polyether ionophore antibiotic, overcomes TRAIL resistance in glioma cells via endoplasmic reticulum stress, DR5 upregulation and c-FLIP downregulation. Carcinogenesis. 2013;34(8):1918–1928. doi:10.1093/carcin/bgt137
95. Zhang T, Wen F, Wu Y, et al. Cross-talk between TGF-beta/SMAD and integrin signaling pathways in regulating hypertrophy of mesenchymal stem cell chondrogenesis under deferral dynamic compression. Biomaterials. 2015;38:72–85. doi:10.1016/j.biomaterials.2014.10.010
96. Safa AR. Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy. J Carcinog Mutagen. 2013;Suppl 6:003. doi:10.4172/2157-2518.S6-003
97. Wang L, Jin H, Jochems F, et al. cFLIP suppression and DR5 activation sensitize senescent cancer cells to senolysis. Nat Cancer. 2022;3(11):1284–1299. doi:10.1038/s43018-022-00462-2
98. Kavuri S, Geserick P, Berg D, et al. Cellular FLICE-inhibitory protein (cFLIP) isoforms block CD95- and TRAIL death receptor-induced gene induction irrespective of processing of caspase-8 or cFLIP in the death-inducing signaling complex. J Biol Chem. 2011;286(19):16631–16646. doi:10.1074/jbc.M110.148585
99. Wang W, Shang Y, Wang C, et al. c-FLIP promotes drug resistance in non-small-cell lung cancer cells via upregulating FoxM1 expression. Acta Pharmacol Sin. 2022;43(11):2956–2966. doi:10.1038/s41401-022-00905-7
100. Lee AR, Park YK, Dezhbord M, et al. Interaction between the hepatitis B virus and cellular FLIP variants in viral replication and the innate immune system. Viruses. 2022;14(2):373. doi:10.3390/v14020373
101. Nanson JD, Rahaman MH, Ve T, et al. Regulation of signaling by cooperative assembly formation in mammalian innate immunity signalosomes by molecular mimics. Semin Cell Dev Biol. 2020;99:96–114. doi:10.1016/j.semcdb.2018.05.002
102. Wu YJ, Wu YH, Mo S-T, Hsiao H-W, He Y-W, Lai M-Z. Cellular FLIP inhibits myeloid cell activation by suppressing selective innate signaling. J Immunol. 2015;195(6):2612–2623. doi:10.4049/jimmunol.1402944
103. Sun CL, Chao CCK. Cross-resistance to death ligand-induced apoptosis in cisplatin-selected hela cells associated with overexpression of DDB2 and subsequent induction of cFLIP. mol Pharmacol. 2005;67(4):1307–1314. doi:10.1124/mol.104.008797
104. Kuehnle N, Osborne SM, Liang Z, et al. CRISPR screens identify novel regulators of cFLIP dependency and ligand-independent, TRAIL-R1-mediated cell death. Cell Death Differ. 2023;30(5):1221–1234. doi:10.1038/s41418-023-01133-0
105. Meng Q, Chen Y, Lian B, et al. miR-218 promotes apoptosis of SW1417 human colon cancer cells by targeting c-FLIP. Oncol Rep. 2018;40(2):916–922. doi:10.3892/or.2018.6460
106. Perez LE, Parquet N, Shain K, et al. Bone marrow stroma confers resistance to Apo2 Ligand/TRAIL in multiple myeloma in part by regulating c-FLIP. J Immunol. 2005;180(3):1545–1555. doi:10.4049/jimmunol.180.3.1545
107. Mora-Molina R, Stöhr D, Rehm M, et al. cFLIP downregulation is an early event required for endoplasmic reticulum stress-induced apoptosis in tumor cells. Cell Death Dis. 2022;13(2):111. doi:10.1038/s41419-022-04574-6
108. Wei Y, Chen K, Sharp GC, et al. FLIP and FasL expression by inflammatory cells vs thyrocytes can be predictive of chronic inflammation or resolution of autoimmune thyroiditis. Clin Immunol. 2003;108(3):221–233. doi:10.1016/s1521-6616(03)00146-3
109. Feoktistova M, Makarov R, Leverkus M, et al. TNF is partially required for cell-death-triggered skin inflammation upon acute loss of cFLIP. Int J mol Sci. 2020;21(22):8859. doi:10.3390/ijms21228859
110. Ewald F, Annemann M, Pils MC, et al. Constitutive expression of murine c-FLIPR causes autoimmunity in aged mice. Cell Death Dis. 2014;5(4):e1168. doi:10.1038/cddis.2014.138
111. Seidelin JB, Coskun M, Vainer B, et al. ERK controls epithelial cell death receptor signalling and cellular FLICE-like inhibitory protein (c-FLIP) in ulcerative colitis. J mol Med. 2013;91(7):839–849. doi:10.1007/s00109-013-1003-7
112. Wu W, Rinaldi L, Fortner KA, et al. Cellular FLIP long form-transgenic mice manifest a Th2 cytokine bias and enhanced allergic airway inflammation. J Immunol. 2004;172(8):4724–4732. doi:10.4049/jimmunol.172.8.4724
113. Shao Y, Wang Z, Wu J, et al. Unveiling immunogenic cell death-related genes in colorectal cancer: an integrated study incorporating transcriptome and Mendelian randomization analyses. Funct Integr Genomics. 2023;23(4):316. doi:10.1007/s10142-023-01238-2
114. Zhu H, Shi J, Li W. Bioinformatics analysis of ceRNA network of autophagy-related genes in pediatric asthma. Medicine (Baltimore). 2023;102(48):e36343. doi:10.1097/MD.0000000000036343
115. Shao Y, Le K, Cheng H, et al. NF-κB regulation of c-FLIP promotes TNFα-Mediated RAF inhibitor resistance in melanoma. J Invest Dermatol. 2015;135(7):1839–1848. doi:10.1038/jid.2015.91
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Metformin Inhibits HaCaT Cell Proliferation Under Hyperlipidemia Through Reducing Reactive Oxygen Species via FOXO3 Activation
Zhang L, Liu X, Huang M, Wang R, Zhu W, Li Y, Shen L, Li C
Clinical, Cosmetic and Investigational Dermatology 2022, 15:1403-1413
Published Date: 22 July 2022
ULK1 Depletion Protects Mice from Diethylnitrosamine-Induced Hepatocarcinogenesis by Promoting Apoptosis and Inhibiting Autophagy
Duan T, Yang X, Kuang J, Sun W, Li J, Ge J, Zhang M, Cai X, Yu P, Yang J, Zhu X
Journal of Hepatocellular Carcinoma 2023, 10:315-325
Published Date: 27 February 2023
Ginsenoside Rg1 Ameliorates Pancreatic Injuries via the AMPK/mTOR Pathway in vivo and in vitro
Chen J, Zhu G, Xiao W, Huang X, Wang K, Zong Y
Diabetes, Metabolic Syndrome and Obesity 2023, 16:779-794
Published Date: 15 March 2023
Antitumor Activity of Berberine by Activating Autophagy and Apoptosis in CAL-62 and BHT-101 Anaplastic Thyroid Carcinoma Cell Lines
Shi XZ, Zhao S, Wang Y, Wang MY, Su SW, Wu YZ, Xiong C
Drug Design, Development and Therapy 2023, 17:1889-1906
Published Date: 26 June 2023
Programmed Cell Death in Liver Fibrosis
Gao R, Tang H, Mao J
Journal of Inflammation Research 2023, 16:3897-3910
Published Date: 1 September 2023