")
Back to Journals » International Journal of Nanomedicine » Volume 19
Chitosan Nanoparticles for Targeted Cancer Therapy: A Review of Stimuli-Responsive, Passive, and Active Targeting Strategies
Authors Al-Shadidi JRMH, Al-Shammari S, Al-Mutairi D, Alkhudhair D, Thu HE, Hussain Z
Received 6 April 2024
Accepted for publication 9 August 2024
Published 15 August 2024 Volume 2024:19 Pages 8373—8400
DOI https://doi.org/10.2147/IJN.S472433
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sachin Mali
Jafar RMH Al-Shadidi,1 Shahad Al-Shammari,1 Danah Al-Mutairi,1 Dalal Alkhudhair,1 Hnin Ei Thu,2 Zahid Hussain1,3
1Department of Pharmaceutics and Pharmaceutical Technology, College of Pharmacy, University of Sharjah, Sharjah, 27272, United Arab Emirates; 2Department of Pharmacology, Faculty of Dentistry, Universiti Teknologi MARA, Sungai Buloh Campus, Selangor Branch, Selangor, Malaysia; 3Research Institute for Medical and Health Sciences, University of Sharjah, Sharjah, 27272, United Arab Emirates
Correspondence: Zahid Hussain, Department of Pharmaceutics and Pharmaceutical Technology, College of Pharmacy, University of Sharjah, Sharjah, 27272, United Arab Emirates, Email [email protected]
Abstract: Despite all major advancements in drug discovery and development in the pharmaceutical industry, cancer is still one of the most arduous challenges for the scientific community. The implications of nanotechnology have certainly resolved major issues related to conventional anticancer modalities; however, the undesired recognition of nanoparticles (NPs) by the mononuclear phagocyte system (MPS), their poor stability in biological fluids, premature release of payload, and low biocompatibility have restricted their clinical translation. In recent decades, chitosan (CS)-based nanodelivery systems (eg, polymeric NPs, micelles, liposomes, dendrimers, conjugates, solid lipid nanoparticles, etc.) have attained promising recognition from researchers for improving the pharmacokinetics and pharmacodynamics of chemotherapeutics. However, the specialty of this review is to mainly focus on and critically discuss the targeting potential of various CS-based NPs for treatment of different types of cancer. Based on their delivery mechanisms, we classified CS-based NPs into stimuli-responsive, passive, or active targeting nanosystems. Moreover, various functionalization strategies (eg, grafting with polyethylene glycol (PEG), hydrophobic substitution, tethering of stimuli-responsive linkers, and conjugation of targeting ligands) adapted to the architecture of CS-NPs for target-specific delivery of chemotherapeutics have also been considered. Nevertheless, CS-NPs based therapeutics hold great promise for improving therapeutic outcomes while mitigating the off-target effects of chemotherapeutics, a long-term safety profile and clinical testing in humans are warranted for their successful clinical translation.
Keywords: chitosan nanoparticles, targeted cancer therapy, tumor microenvironment, stimuli-responsive targeting, passive targeting, active targeting
Graphical Abstract:

Cancer: Background, Prevalence, Signs, and Symptoms
The cells grow and divide physiologically, and the deteriorated aged cells undergo apoptosis and replacement by newly generated cells. However, there are instances when an error occurs in the cell regeneration cycle and newly generated cells grow abnormally in an uncontrollable manner, thus leading to tumor formation. These tumors are either benign or malignant. Malignant tumors can metastasize to other tissues of the body, while benign tumors cannot; thus, the naming convention. Although benign tumors can grow large and may cause serious symptoms, they do not typically grow after removal unlike the malignant tumors.1,2
According to the World Health Organization (WHO), cancer accounts for 244.6 million disability-adjusted life years (DALYs) with men and women being almost equally affected at a ratio of 1.37:1.07. For the sake of comparison, the ischemic heart disease and stroke follow closely behind with 203.7 million DALYs and 137.9 million DALYs, respectively.3 Digging deeper in the statistics of the disease, it has been observed that cancer is slightly more prominent in men than women and the majority of DALYs are found in individuals over the age of 60, which concludes almost 124 million out the 244.6 million DALYs. Notably, the most prevalent types of cancer diagnosed at age 14 years and below include lymphomas, brain cancer, and leukemia, with a prevalence of approximately 13%, 16%, and 37%, respectively.4 In contrast, the most prevalent types of cancer in middle-aged patients (15–49 years) include lung, liver, and breast cancers, accounting for approximately 9%, 12%, and 13%, respectively. However, at the ages of 50 to 59 years, the scenario is opposite to that of the previous age group, accounting for approximately 18% lung, 11% liver, and 9% breast cancers, respectively. Finally, at the age of 60 years and above, lung cancer remains the most prevalent type of cancer, accounting for approximately 21% of all cancer types, followed by colorectal, stomach, and liver cancers, each accounting for approximately 9%.3,4
It is usually not possible to know exactly why one person develops cancer and another does not. But research has shown that certain risk factors such as alcohol consumption, smoking, unhealthy diet, obesity, sedentary lifestyle, excessive sun exposure, exposure to radiations, inhalation or ingestion of carcinogen, age, hormonal dysregulation, immunosuppression, and family history of cancer may increase a person’s chances of developing cancer. These factors may provoke transformation of normal/healthy cells into tumor cells, which is a multi-stage process that generally progresses from a pre-cancerous lesion to a malignant tumor.
Although these symptoms may overlap with those of other conditions, several signs and symptoms may indicate cancer. Some of these include fatigue, which is not mitigated by others. Unexplained weight loss or weight gain ≥10 pounds or further, eating and swallowing difficulties, nausea, vomiting, development of lumps, consistent unresolving pain, abnormalities in the skin (eg, scaly or bleeding lumps), development of moles or changes in the existing moles, unhealing sores, and jaundice are among the typical signs and symptoms of cancer. Other symptoms may include persistent cough or hoarseness, spontaneous bleeding, change in stool texture (or runny stool or constipation), change in stool color, painful or bloody urination, sleep hyperhidrosis, visual impairment, mouth sores, and numbness and pain in a specific body area.2
Owing to its complex heterogeneity, patients experiencing specific or generalized symptoms undergo a series of diagnostic tests, including magnetic resonance imaging (MRI), computed tomography (CT) scan, positron emission tomography (PET) scan, ultrasound, X-rays, or tissue biopsies, depending on the type, nature, and location of the tumor or cancer. After successful diagnosis, a team of healthcare professionals consult and prescribe the most suitable therapy for cancer patients. Monotherapy is recommended in some cases, but adjuvant therapy is advisable in most cases. The most commonly used conventional treatments for cancer include chemotherapy, surgery, radiotherapy, immunotherapy, personalized medicine, hormonal therapy, and bone marrow transplantation (Figure 1).
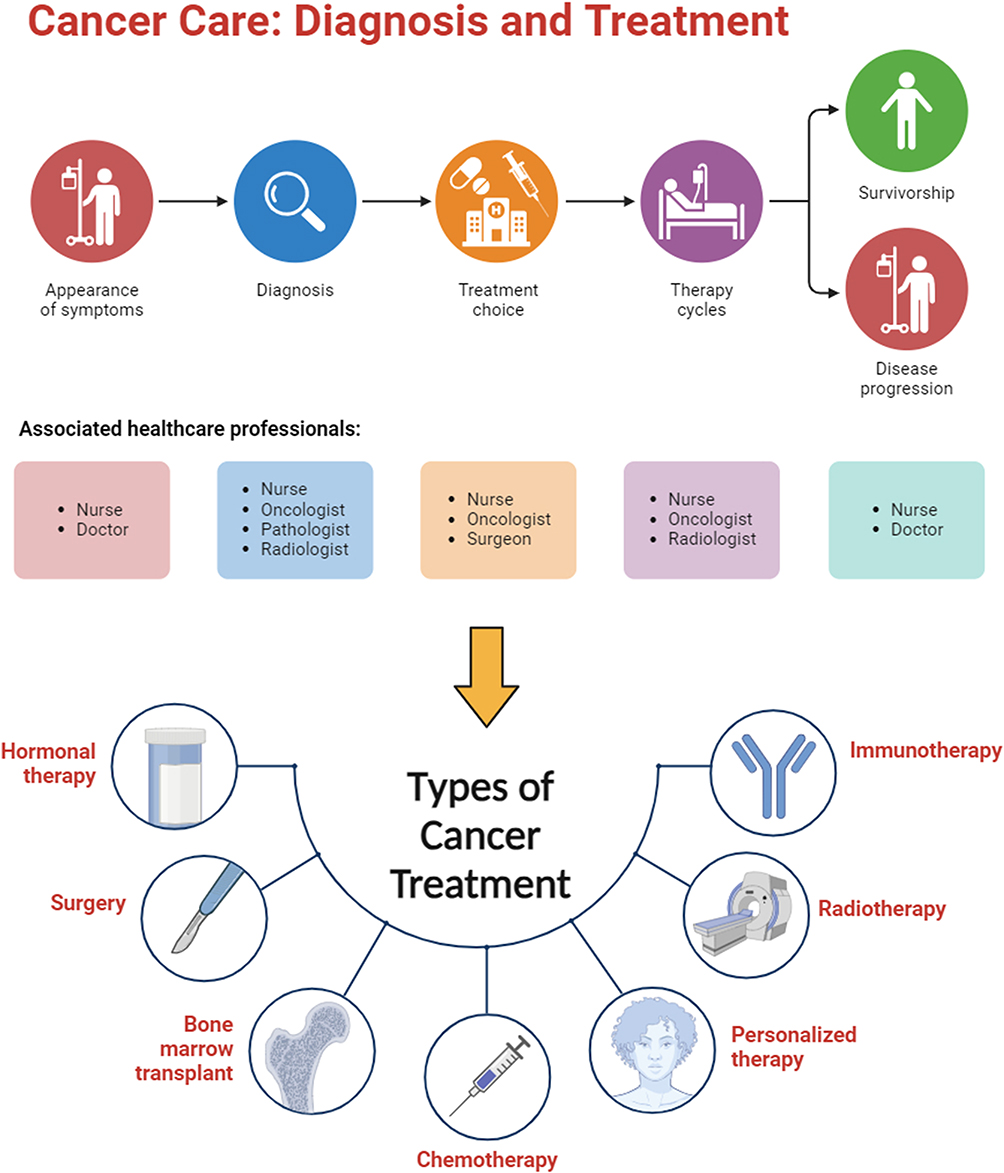 |
Figure 1 Cancer care and types of conventional treatments. Created with BioRender.com. |
Conventional Cancer Treatments and Limitations
Chemotherapy and Limitations
Chemotherapy, which is the optimal option for cancer treatment, is a potent drug that targets and kills rapidly dividing cells in the body. Numerous chemotherapeutic drugs are available and can be used alone or in combination to treat various types of cancer. In some cases, chemotherapy may be used as a primary treatment with the aim of eliminating the cancer and preventing its recurrence, which is known as ”curative chemotherapy”. However, it may be used as either neoadjuvant or adjuvant chemotherapy, by using it prior or post to other treatment options such as surgery. In other situations where even the cancer cannot be cured, chemotherapy may still be used to slow disease progression, provide symptomatic relief, and enhance quality of life. This is called ”palliative chemotherapy, ” which aims to partially shrink tumors, prevent tumor growth, and extend survival for varying durations. Therefore, chemotherapy is a powerful treatment option for cancer that can be used with different goals, including curative, neoadjuvant, adjuvant, and palliative purposes, depending on cancer type and stage. However, it is important to consider the potential side effects and carefully weigh the benefits and risks in consultation with a team of qualified healthcare professional.5
Chemotherapy, as a treatment option for cancer, has several advantages and disadvantages. On the positive side, chemotherapy can shrink the tumor size or slow its growth, which may help the patient live longer and alleviate symptoms. In some cases, chemotherapy may even shrink the cancer sufficiently to enable the surgical procedure to be more effective in removing borderline resectable cancers. Adjuvant chemotherapy lowers the incidence of cancer recurrence in patients who have been successfully treated. Furthermore, patients may have more regular checkups, tests, and contact with their oncologists when undergoing chemotherapy, which some people find reassuring.
While chemotherapy effectively combats various types of cancer, it also carries a wide range of unwanted adverse events that affect a patient’s quality of life and, in some cases, even life-threatening conditions that limit patient acceptability.6 The most common side effects of chemotherapy are bleeding, infections, and hair loss. It can also affect appetite, influence bowel movement behavior, cause nausea and emesis, and cause multiple oral cavity discomforts including ulcers and pharyngitis. Additionally, chemotherapy can affect the nervous system, causing discomfort and tingling sensations, parched skin, discoloration, and deterioration of nail structure. Other undesired adverse events include fluctuations in body weight, mood swings, and weakening of sexual activity and desires. Frequent hospital visits for multiple treatment cycles and regular check-ups and tests also make it difficult and inconvenient for some patients.5,6 A summary of the different classes, mechanisms of action, dosage, type of cancer against which they are most active, and their potential side effects is presented in Table 1.
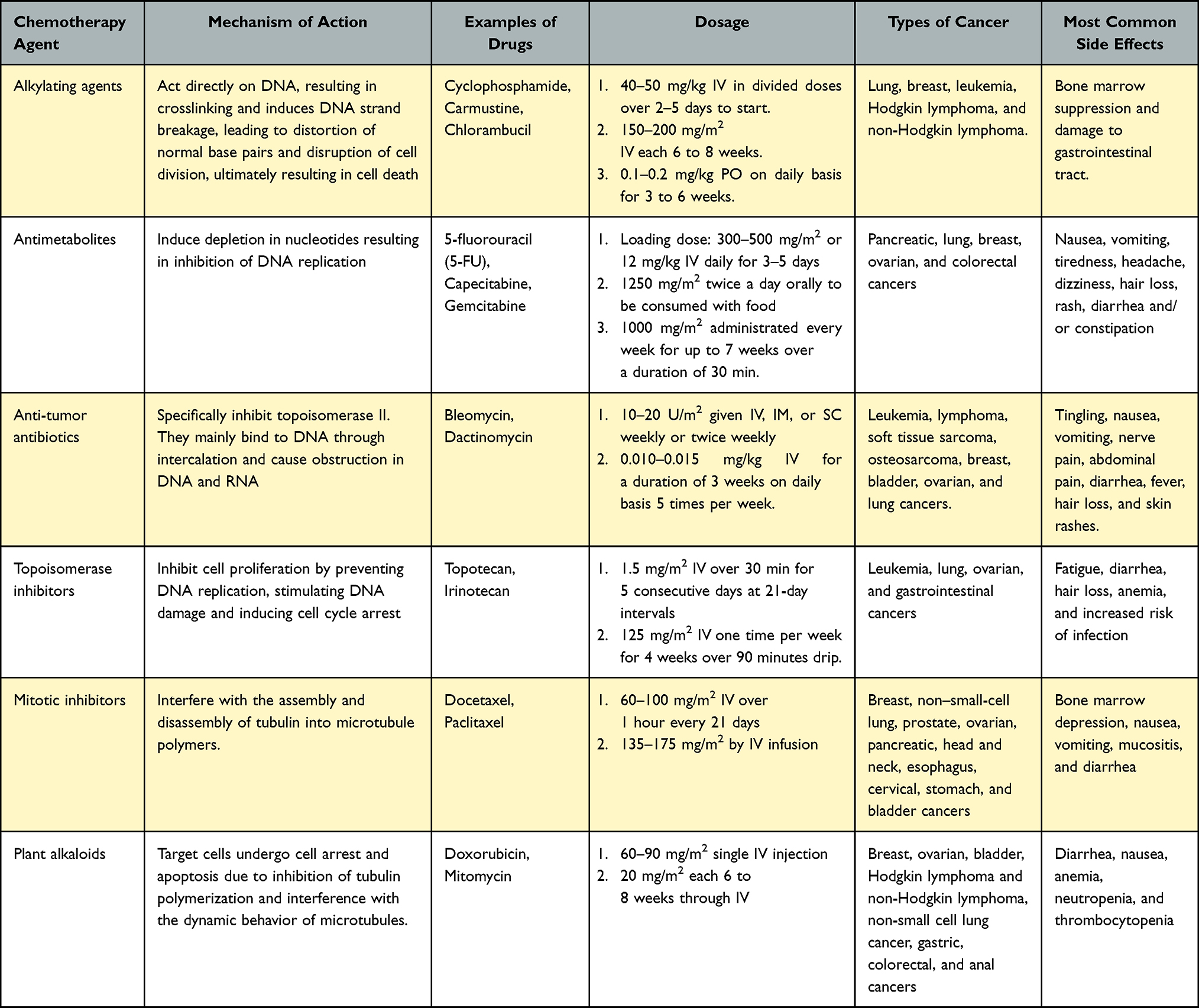 |
Table 1 Mechanism of Action, Examples, Dosages, and Side Effects of Different Classes of Chemotherapy for Treatment of Different Types of Cancers |
Surgery and Limitations
Surgery is also a frequently used approach to treat various types of cancer, especially localized solid tumors. Being classified as a local treatment, it focuses only on the area of the body affected by cancer and is generally not used to treat metastatic cancers or leukemia, a form of blood cancer. Although surgery may be sufficient as a standalone treatment in some scenarios, it is often supplemented with other cancer treatments such as chemotherapy or radiotherapy. There are many surgical techniques, each adapted to the purpose of the intervention, the anatomical site requiring attention, the extent of tissue removal, and patient preference. Surgery can be classified as open or minimally invasive.7
In open surgery, a large incision is made to remove the tumor as well as some surrounding healthy tissue and possibly nearby lymph nodes. In contrast, minimally invasive methods involve making several small incisions, instead of only one large incision. A thin tube equipped with a miniature camera, known as laparoscope, was inserted through a small incision. The camera transmits internal images to the monitor, allowing the surgeon to observe the procedure. Specialized surgical tools are then inserted through other small incisions to surgically remove the tumor and nearby healthy tissues. Compared with open surgery, less invasive surgical procedures are preferred for many patients because they require smaller incisions and a shorter recovery time. Overall, surgical interventions for cancer removal depend on the individual case and patient preferences, and it is vital for oncologists to present all possible options to the patient.7
Surgery is a crucial treatment method that offers several benefits to patients. First, resection of solid tumors lowers the mass effect, thereby leading to an immediate reduction in cancer-related symptoms. Second, surgery can resect cancerous tissues, which lowers the risk of metastasis. Third, surgery can be used to resect tumors from areas where radiation cannot be applied, such as brain cancer. In addition, surgery has the potential to completely resect cancer cells in a confined region, which may result in a complete cure. Furthermore, surgery allows for the examination of cancerous tissue, which can facilitate a confirmatory treatment approach by analyzing the extracted tissue response to treatment types. Finally, surgery is convenient as a one-time intervention that opposes adherence to the treatment course. Surgical intervention is an essential tool in the fight against cancer and can provide significant benefits to patients.7,8
In addition, surgery has certain disadvantages. First, surgical intervention may not completely resect all cancer cells and tissues, resulting in cancer recurrence. Second, not all patients with cancer are fit to undergo surgical procedures because of other medical complications or conditions. Moreover, during surgical intervention, surgeons may need to excise other body tissues or bones to gain access to the localized tumors. Another serious disadvantage is post-surgical complications such as infection. Lastly, surgical intervention is not a valid option for metastatic cancer and, therefore, cannot be considered for all types of cancers. Despite these potential drawbacks, surgical intervention still plays a major role in cancer treatment and can be utilized when its benefits outweigh its potential risks.7,8
Radiotherapy and Limitations
Radiotherapy (RT) is considered as one of an effective cancer treatment that involves the application of beams of intense energy radiation (eg, X-rays) to kill cancerous cells by damaging their genetic material, thereby diminishing their ability to grow and divide. It is also worth mentioning that as a non-selective method, RT may damage the surrounding healthy cells/tissues along with the cancer cells; therefore, it is mainly utilized for specific tumors or cancers in a specific part of the body.9
There are two major forms of RT: 1) External Beam Radiotherapy (EBRT) and 2) internal radiotherapy (IRT), with EBRT being the most common type.9 EBRT works by directing a high-intensity beam of X-rays, protons, or electron rays through line accelerators towards the target tumor with the highest precision to avoid damage to healthy tissues.9 On the other hand, IRT (also known as brachytherapy) involves administration of radio-active material within the cancer tissue or neighboring tissues. This can be done by using either temporary or permanent implants, depending upon the specific needs of the patient, and may require hospitalization. Like other cancer treatments, RT is also associated with several disadvantages, such as damage to neighboring healthy cells/tissues and inability to completely eradicate microaggregates of tumor cells, which can cause cancer reoccurrences as well as their inability to be used against metastatic cancers.9
Hormonal Therapy and Limitations
Hormonal therapy (HT), also known as endocrine therapy, can slow or stop the progression of cancer or block the release or action of hormones used as growth agents by cancer cells.10 HT locates and targets specific hormones throughout the body through multiple mechanisms of action. It either hinders hormone production, prevents hormone adherence to cancer cells, or deactivates the hormone, ultimately suppressing the progression of various cancers (eg, breast, prostate, and thyroid cancers).11 A summary of the roles of hormones in different types of cancers is presented in Table 2.
 |
Table 2 Oncogenic Function of Hormones in the Progression of Different Types of Cancer |
Immunotherapy and Limitations
Immunotherapy (IT) utilizes the body’s ability to identify and destroy foreign materials through several mechanisms. In this section, we discuss some of the different immunotherapies that are used for cancer treatment. First, Chimeric Antigen Receptor (CAR) T-cell therapy, in which a sample of the patient’s cancer-fighting T cells is extracted and functionalized to carry CARS, strengthens the T-cell anti-cancer efficacy. Modified T-cells are reintroduced into the bloodstream to find, attach, and kill cancer cells.11 Immunomodulators represent another type of IT in which a group of drugs is administered to boost the patient’s immune system to fight against different types of cancer. Various immunomodulators have been developed. The first family of drugs is antagonists that block immunosuppressing pathways. The second family of drugs is agonists, which function by stimulating pathways that initiate the action of immune cells. The third family of immunomodulators is checkpoint inhibitors, which are designed to inhibit molecules on immune or cancer cells that trigger the attack of cancer cells. Cytokines represent a fourth group of immunomodulators. The types of cytokines used in anti-cancer therapy include, but are not limited to, interferons and interleukins. Finally, adjuvants, substances that stimulate pathways in the immune system, provide prolonged protection or generate more antibodies. Cancer vaccines constitute a third group of ITs. A vaccine stimulates the immune response of the body against a particular disease. In most cases, they should be administered to healthy individuals to prevent infections. However, some vaccines may prevent or treat cancer. Cancer vaccines stimulate the immune system to attack cancer cells with specific tumor antigens. Many different components, including viruses, vectors, DNA, and proteins, are involved in the formulation of cancer vaccines. Cancer vaccines are used to protect patients by shielding them from bacteria and viruses, which may aid in the development of cancers such as hepatitis B.12,13 Targeted antibodies are proteins that are tailored post-production by the immune system to carry antigens that target cancer cells and diminish their activity. These targeted antibodies can be loaded with anticancer drugs to act as drug delivery systems specific to desired cancer cells. Currently, all targeted antibodies are monoclonal antibodies, which can be very effective in cancer treatment because of their specific and precise targeting capabilities for certain types of cancers.14
Along with several advantages, the disadvantages associated with IT are not less important. A major limitation of IT is that it can induce a reverse effect, where the body’s immune system starts to attack healthy cells within the body, leading to inflammation and undesired side effects, such as nausea and vomiting, generalized fatigue, decreased levels of thyroid hormone, itchy rash, and diarrhea. Overall, it is difficult to predict the body’s reaction to IT, and the scarcity of clinical trials hinders determination of the appropriate dosage and frequency of administration.
Bone Marrow Transplant and Limitations
Bone marrow transplantation is the process of replacing the bone marrow with healthy cells. In addition to obtaining cells from a donor, it is also possible to obtain replacement cells from a patient’s body.15 Bone marrow transplantation is also known as stem cell transplantation or transplantation of hematopoietic stem cells. Hematopoietic stem cell transplantation (HSCT) is beneficial for the treatment of lymphoma, myeloma, and leukemia. In addition, it can be used to treat the immune system and bone marrow diseases. HSCT is beneficial for replacing deteriorated bone marrow after delivering immense doses of chemotherapy or radiation, where newly generated stem cells gain the ability to directly attack and kill cancer cells. With all the aforementioned benefits, there are numerous risks associated with bone marrow transplantation. Complications range from minor to major life-threatening complications that require adequate care and hospitalization. The criteria for bone marrow transplantation are very strict, and many factors contribute to the final decision, such as patient’s age, health status, type of cancer, and type of transplant needed. Possible complications include but are not limited to organ failure, infertility, triggering of immune system and body rejection, and death.15
Personalized Therapy
Personalized medicine is a new form of medicine that uses patient genetic data as a guide to help make decisions regarding the prevention, diagnosis, and treatment of diseases. To craft best possible formulation, doctors must know the genetic data or profile to determine the right dosage regimen for the patient.16
Genes constitute the backend code for cell generation, growth, and development. Many cancers are associated with, and/or affected by, specific genes. Research on the human genome, specifically on the genes involved in different types of cancer, is key to developing personalized cancer medicine. Researchers have used the results of these studies to design more effective treatments. Genetic information has also been used in cancer detection and prevention. The benefit of personalized cancer medicine is that it lowers the chance of adverse side effects, as personalized treatment can have a lower impact on healthy cells, while greatly impacting cancer cells.17 A widely known example of personalized cancer medicine is targeted therapy, which targets specific proteins and genes responsible for the growth and survival of certain types of cancer.
CS-NPs for Targeted Cancer Therapy
Nanotechnology focuses on the design, detection, synthesis, and deployment of nanoscale molecules ranging between 1 and 1000 nanometers in size. Among these nanomaterials, particles with sizes below 100 nm exhibit enhanced qualities that vary according to their size, in contrast to larger particles. Owing to their salient features, various nanomaterials have been extensively used to design different types of nanoconstructs for the targeted delivery of a wide range of diagnostic and therapeutic payloads. Targeted delivery of imaging agents and therapeutics has significantly improved pharmacotherapeutic outcomes and early detection of various diseases, including cancer.
Tumor targeting is one of the most investigated aspects of nanotechnology for early detection and targeted treatment of different types of carcinomas. There are different strategies through which NPs can target the TME. For example, pH-responsive nanodelivery systems specifically respond to acidic TME and release their diagnostic or therapeutic cargo into tumor tissues with limited premature release in other biological tissues. Likewise, the ultrafine particle size of NPs allows for diverse functionalization to improve the plasma circulation time, tumor biodistribution, and uptake into tumor cells, along with the reduced toxicity of chemotherapeutics. Among the various types of nanodelivery systems (eg, polymeric NPs, micelles, liposomes, dendrimers, conjugates, solid lipid nanoparticles, nanostructured lipid carriers, and nanoemulsions), polymeric NPs have been widely recognized because of their unique physicochemical properties, high encapsulation efficiency, stability in biological fluids, ease of formulation, and great flexibility of functionalization.18,19 Because polymeric NPs are synthesized from a wide variety of polymers, fabricating polymeric NPs plays a crucial role in their physicochemical characteristics.
Chitosan (CS) is a natural polyamine polysaccharide obtained from chitin and is a major component of fish scales, fungal cell walls, and insect exoskeletons. Owing to their inherent antimicrobial, antioxidant, wound healing, analgesic, anti-rheumatic, immunomodulatory, mucoadhesive, antiproliferative, and antimetastatic properties, CS and CS-NPs have been extensively investigated for the treatment of various diseases, including cancer. The anticancer potential of CS and CS-NPs was attributed to their antiangiogenic, antioxidant, immunoenhancing, and apoptotic effects.20 The apoptotic effect of CS-NPs is due to the generation of reactive oxygen species (ROS), which induce apoptosis and cause severe stress to the mitochondria and endoplasmic reticulum.21 Moreover, owing to their cationic nature and mucoadhesive properties, CS-NPs improve the rate and extent of absorption of chemotherapeutics from the site of administration owing to their prolonged residence time. CS-NPs have also been well-recognized for the controlled, sustained, and stimuli-responsive release of encapsulated drugs, which prevents their premature release and degradation. Furthermore, their tunable size, high encapsulation efficiency, good stability in biological fluids, biodegradability, and biocompatibility make them promising building materials for a variety of CS-based nanocarriers.22–24
Having been enriched with three distinct functional groups, a variety of CS-based nanospheres and core-shell NPs have been designed for stimuli-responsive, passive, or active delivery of chemotherapeutics to the tumor microenvironment (TME) or in the cytoplasm of tumor cells.25,26 CS-NPs can either passively diffuse to the TME because of their nanoscale size, assisted by an electric or magnetic field, high-intensity focused ultrasound, or ligand–receptor interaction. After penetration into the TME or tumor cells, therapeutic cargo is released from the CS-NPs in response to light, pH, or enzymes, thereby killing tumor cells. To further enhance their targeting efficiency and anticancer potency, hybrid NPs can be synthesized by coupling multiple targeting mechanisms with CS-NPs architecture. Different targeting mechanisms by which CS-NPs can deliver chemotherapeutics to TME have been presented in Figure 2.
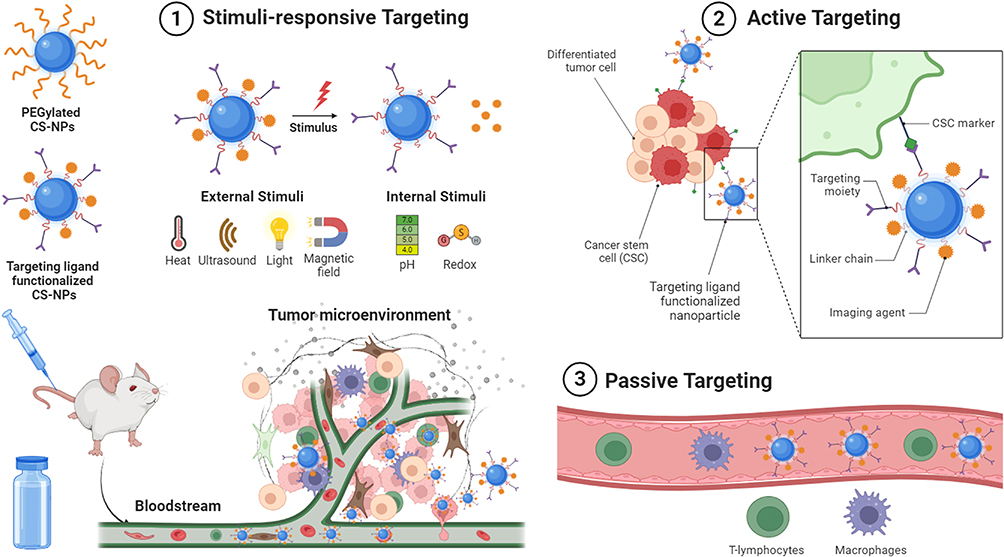 |
Figure 2 Targeted delivery of chemotherapeutics loaded CS-NPs by stimuli-responsive, passive, and active targeting mechanisms. Created with BioRender.com. |
Stimuli-Responsive Targeting
Stimuli-responsive targeting refers to the delivery and release of theranostic payload to a specific region or tissue (eg, TME) by responding to certain endogenous stimuli (eg, pH, enzymes, redox, glucose) or exogenous stimuli (eg, light, temperature, ultrasound, magnetic field, electric field, osmotic pressure).27 Exogenous stimuli (also known as physical stimuli) can be easily manipulated or controlled by the investigator, but endogenous stimuli (also known as chemical stimuli) are always variable, which may lead to inconsistent results.28 A variety of stimuli-responsive CS-NPs based therapeutics have been developed and evaluated for targeted delivery of chemotherapeutics for the treatment of different types of cancer.
Photodynamic therapy (PDT) is a promising light-activated cancer treatment modality that employs photosensitizers or photosensitizing agents to kill tumor cells by generating highly reactive singlet oxygen when irradiated with light of an appropriate wavelength. In addition to generating singlet oxygen, photosensitizers emit fluorescence, which enables easy detection and tracking, both in vitro and in vivo. To achieve effective photodynamic therapy, Lee et al29 investigated the effect of the loading method on the passive delivery of chlorin e6 (Ce6), a hydrophobic photosensitizer for the treatment of squamous cell carcinoma. They utilized a hydrophilic derivative of CS, glycol CS (GCS) (with PEG conjugated to the CS backbone), and chemically conjugated it with 5β-cholanic acid (5β-CA) (a hydrophobic moiety). These amphiphilic conjugates (5β-CA-GCS) self-assembled into core-shell NPs by encapsulating Ce6 in their inner hydrophobic core. They fabricated two different types of 5β-CA-GCS-NPs: the first was physically loaded with Ce6 (HGC-Ce6) and the second was chemically conjugated with Ce6 (GC-Ce6). Both types of GCS-NPs were characterized in terms of particle size (300–350 nm), in vitro drug release, cell uptake efficiency (SCC7 tumor cells), biodistribution to tumor tissues, and anticancer efficacy. The results suggested that HGC-Ce6 displayed a burst release of ~65% of the drug within 6.5 hours compared to only 10% release from GC-Ce6. The rapid drug release from HGC-Ce6 was attributed to the weak hydrophobic interactions between Ce6 and GCS. Although both NPs displayed excellent cell uptake efficiency and cytotoxicity compared to free Ce6, GC-Ce6 was more pronounced than HGC-Ce6, which was attributed to the chemical conjugation of Ce6 to GCS. The biodistribution study showed that after 3 h of injection, fluorescence rapidly declined in the case of free Ce6 owing to fast excretion from the body; however, HGC-Ce6 and GC-Ce6 showed prolonged fluorescence, particularly GC-Ce6, which remained detectable in the tumor tissue even 48 h post-injection. The anticancer efficacy was also superior to case of GC-Ce6 in comparison to HGC-Ce6 and free Ce6, which was attributed to the shrinkage of the tumor volume and minimal variation in the body weight of mice. The overall tumor volume after 20 days of treatment with GC-Ce6 was approximately 160 mm3 which was significantly smaller than that of HGC-Ce6 (560 mm3) and free Ce6 (760 mm3). These results indicate that chemical conjugation of Ce6 with hydrophobically modified GCS is a viable option for effective photodynamic therapy against squamous cell carcinoma.29 Lim et al30 synthesized amphiphilic bioconjugates composed of GCS, Ce6, and densely conjugated diatrizoic acid (3,5-bis(acetamido)-2,4,6-triiodobenzoic acid) as iodine-rich hydrophobic pendants. These amphiphilic conjugates self-assembled into core-shell NPs (GCS-I-Ce6), which were evaluated for their phototoxicity in human breast cancer cells (MDA-MB-231 cells). The fabricated GCS-I-Ce6 core-shell NPs (~20 nm, smooth spherical morphology) displayed promising phototoxicity in MDA-MB-231 cells compared to non-iodinated NPs and plain Ce6.30
Another light-activatable nanosystem was developed by Lee et al,31 in which CS was chemically conjugated with ursodeoxycholic acid (UDCA), a secondary bile acid that can be metabolized by intestinal bacteria. The synthesized amphiphilic conjugates were self-assembled into core-shell NPs by encapsulating Ce6 in the hydrophobic inner core. The fabricated Ce6-UDCA-CS-NPs (200–400 nm, smooth spherical morphology) exhibited stronger phototoxicity in human cholangiocarcinoma cells (HuCC-T1) via generating highly reactive singlet oxygen species, in comparison to the plain Ce6.31 Similarly, Bae et al32 conjugated phenethyl isothiocyanate (PEITC) with CS oligosaccharide (COS), and the resulting amphiphilic conjugates self-assembled into core-shell NPs in an aqueous medium by encapsulating Ce6 in the inner hydrophobic core of the NPs. The prepared Ce6-PEITC-COS-NPs exhibited nanoscale dimensions (200 nm), smooth spherical morphology, good stability in FBS, and sustained continuous release of Ce6 from the nano-photosensitizer for 96 h. Treatment of human colon cancer cells (HCT-116) with Ce6-PEITC-COS-NPs produced dose-dependent phototoxicity and cell uptake compared with free Ce6. Upon IV administration from the tail vein into HCT-116-bearing mice, Ce6-PEITC-COS-NPs showed better biodistribution to tumors and superior anticancer efficacy in terms of suppression of tumor volume compared with free Ce6 and other control formulations.32
On the other hand, Jeong et al33 proposed a simple green synthesis method for preparation of Ce6-loaded CS-NPs (ChitoCe6) via ionic complexation of Ce6 with free –NH2 groups on the contour of water-soluble CS (WSC). ChitoCe6 (<300 nm) displayed potent phototoxicity in cholangiocarcinoma cells (SNU478) via the generation of highly reactive singlet oxygen, which induces DNA and mitochondrial damage. The enhanced phototoxicity induced by ChitoCe6 was attributed to its higher cell uptake efficiency than that of native Ce6. These results were also validated in SNU478-bearing nude mice, which showed a time-mannered enhanced distribution to tumor tissues and reduced tumor volume in mice treated with ChitoCe6 compared to those treated with native Ce6.33
Recently, Al-Nemrawi et al34 proposed another functionalization strategy for fabricating photo-controllable CS-NPs for the targeted delivery of methotrexate (MTX). They prepared MTX-loaded CS-NPs and coated their surfaces with titanium dioxide NPs (TiO2-NPs) to trigger MTX release from the TiO2-NPs-coated MTX-CS-NPs via photolytic degradation when exposed to UV light. In addition to their unique photocatalytic properties, TiO2-NPs have been shown to exert tumor-killing effects by inducing oxidative stress and apoptosis. Hence, TiO2-NPs were used in this study for dual action, photocontrol, and tumor killing. The fabricated TiO2-NPs-coated MTX-CS-NPs exhibited a larger particle size, good polydispersity, higher positive charge, high %EE (80%), and light-sensitive release than the uncoated MTX-CS-NPs. MTX-CS-NPs also displayed higher cytotoxicity in MCF-7 cells than free MTX and plain CS-NPs; however, cytotoxicity was remarkably improved after coating with TiO2-NPs. In summary, the surface coating of MTX-CS-NPs with TiO2-NPs is a promising strategy to enable the light-responsive release of encapsulated drugs at the target site with ultimately stronger anticancer efficacy while minimizing off-target effects.34
Another interesting approach employing high-intensity ultrasound was proposed by Choi et al35 to improve the penetration of CS-NPs into tumor tissues. Briefly, they modified with 5β-CA (a hydrophobic moiety) to produce amphiphilic conjugates, which were self-assembled into core-shell NPs by incorporating doxorubicin (DOX) into the inner hydrophobic core of the CS-NPs. The fabricated DOX-GCS-5β-CA-NPs exhibited nanoscaled size (283 nm), smooth spherical morphology, good stability in FBS, and high-intensity focused ultrasound (HIFU) triggered biphasic release. Interestingly, DOX-GCS-5β-CA-NPs displayed time-mannered cytotoxicity and cell uptake efficiency in A549 cells (lung cancer cells) pretreated with HIFU, compared to A549 cells without HIFU pretreatment and free DOX. These results were also validated in the A549 tumor-bearing animal model, which indicated that pretreatment of xenografted tumors with HIFU resulted in an approximately 2-fold increase in the accumulation of DOX-GCS-5β-CA-NPs in the tumor compared to DOX-GCS-5β-CA-NPs without HIFU (Figure 3). The superior accumulation of DOX-GCS-5β-CA-NPs into HIFU-pretreated tumors was attributed to the breakdown of the dense extracellular matrix (ECM) composed of collagen and hyaluronan, resulting in deeper penetration of DOX-GCS-5β-CA-NPs into A549 tumor tissues.35
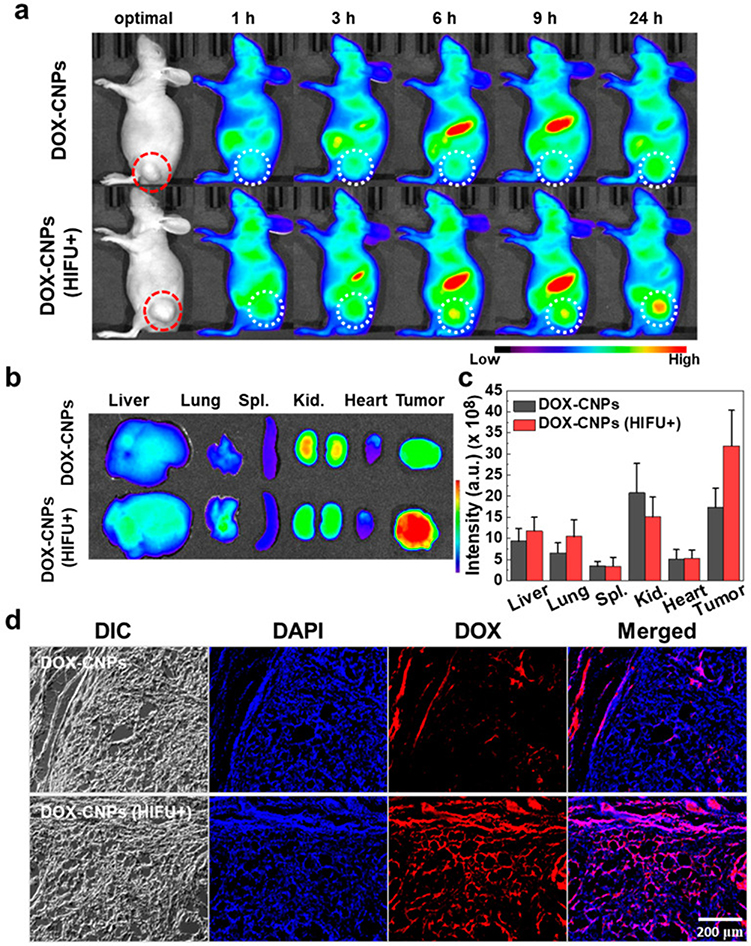 |
Figure 3 In vivo near-infrared fluorescence (NIRF) imaging of Cy5.5-labeled DOX-CNPs in ECM-rich A549 tumor animal model, (a) biodistribution of Cy5.5-labeled DOX-CNPs without and with HIFU treatment (intensity: 5 W/cm2, frequency: 1.5 MHz, duty cycle: 10%, pulse repetition frequency: 1 Hz, time per spot: 30s, interval: 2 mm, expose time: 5 min). The red and white dot circles indicate tumor site, (b) ex vivo NIRF imaging of different organs and tumor at 24 h post-injection. (c) Mean NIRF signal intensity of ex vivo NIRF image, (d) ex vivo NIRF microscopic images of deep tumor penetration of untreated Cy5.5-labeled DOX-CNPs and HIFU-treated Cy5.5-labeled DOX-CNPs in ECM-rich tumor tissues. Reprinted from Choi Y, Han H, Jeon S, et al. Deep tumor penetration of doxorubicin-loaded glycol chitosan nanoparticles using high-intensity focused ultrasound. Pharmaceutics. 2020;12(10):974. Distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.35 |
To provide on-demand drug release, CS-NPs have also been functionalized to respond to different chemical stimuli (eg, pH, redox, and enzymes). Nogueira-Librelotto et al36 proposed the incorporation of a pH-sensitive lysine-based biosurfactant (77KS) as an adjuvant to develop pH-responsive CS-NPs for the targeted delivery of DOX. The developed DOX-CS-77KS-NPs exhibited nanoscale dimensions, smooth spherical morphology, pH-responsive release (accelerated release at acidic pH and minimal release at physiological pH), and pronounced cytotoxicity in MCF-7 and HeLa cells. These findings indicate that the incorporation of 77KS as an adjuvant in the architecture of CS-NPs could be a promising strategy for cytosolic delivery of chemotherapeutics.36,37 Later, the same research group proposed that the incorporation of a pH-sensitive lysine-based surfactant (77KS), with subsequent modification with PEG or poloxamer, could further improve the targetability and cell uptake efficiency of DOX-CS-77KS-NPs. They established that DOX-CS-77KS-NPs modified with poloxamer were more effective for enhancing the cytotoxicity and cell uptake into HeLa cells, compared to PEG-modified CS-77KS-NPs, PEG-CS-NPs, unmodified CS-NPs, and free DOX.38 They also confirmed their findings regarding the pH-responsive release of DOX from CS-NPs for the treatment of cervical and breast cancers.39
Another pH-responsive CS-NPs formulation was proposed by Park et al.40 They conjugated N-acetyl histidine (NAHis) (hydrophobic moiety) with GCS, which self-assembled into core-shell NPs by incorporating paclitaxel (PTX) into the inner hydrophobic core of the GCS-NPs. These hydrophobically modified core-shell NPs (50–250 nm) exhibited a pH-responsive release of PTX with a pronounced release at acidic pH and lower release at physiological pH. This pH-responsive release of PTX from NAHis-modified GCS-NPs was attributed to the hydrophobicity of NAHis at physiological pH, which acts as a barrier to PTX release; however, NAHis-GCS-NPs dissembled at acidic pH because of the breakdown of the hydrophilic/hydrophobic balance due to the protonation of the imidazole group of NAHis. The pH-responsive characteristic of NAHis-GCS-NPs makes them a highly promising nanovehicle for site-specific delivery of chemotherapeutics.40 Similar results were reported by Lee et al,41 who demonstrated that NAHis-modification of CS-NPs resulted in sustained release of all-trans retinoic acid (RA) at acidic pH compared with physiological pH.
pH-responsive behavior can also be introduced into CS-NPs by hydrophobic substitution of water-soluble CS, as proposed by Piroonpan et al.42 They grafted poly(ethylene glycol) monomethacrylate (PEGMA) comb-like brushes onto WCS-deoxycholic acid (DCA) conjugates for site-specific release of PTX. The fabricated DC-WCS-PG-NPs demonstrated a controlled pH-responsive release of PTX (faster release at acidic pH and slower release at physiological pH) and potent anticancer efficacy against breast cancer cells with no noticeable toxicity against human skin fibroblasts, indicating their biocompatibility.42 On the other hand, Ahmadi Nasab et al43 synthesized pH-responsive mesoporous silica nanocarriers encapsulated with curcumin (Cur) and capped with CS for treatment of glioblastoma cells. The developed Cur@CS-MCM-41 was characterized in terms of the particle size (180 nm), %EE (88%), loading capacity (9%), pH-responsive release, and antitumor efficacy against glioblastoma cancer (U87MG). The significantly higher release of Cur at acidic pH compared to physiological pH was attributed to protonation of ionizable amino groups (–NH2) on the contour of CS, which undergo swelling due to repulsion of polymeric chains, leading to increased porosity and drug release from the CS matrix. A cytotoxicity study revealed significantly higher cytotoxicity in U87MG cells treated with Cur@CS-MCM-41 compared to unfunctionalized CS-nanoconstructs and other control groups.43
On the other hand, redox-responsive systems are designed to release their payload in response to redox conditions in the environment. For example, tumors exhibit characteristic oxidizing extracellular and reducing intracellular environments, generating a redox potential that has become the driving force for the development of redox-responsive delivery vectors. Yoon et al44 developed redox-responsive CS-NPs for targeted delivery of DOX. In this study, they chemically conjugated to the CS backbone and then conjugated to the CS backbone through a thioketal linker, which is sensitive to oxidative stress and undergoes cleavage when exposed to the TME. The DOX-mPEG-CS-NPs exhibited nanoscale size (<100 nm), smooth spherical morphology, sustained ROS-responsive release, and dose-dependent cytotoxicity in both DOX-sensitive and DOX-resistant AT84 cells (oral squamous cell carcinoma). The anticancer efficacy of DOX-mPEG-CS-NPs was validated in AT84-tumor bearing mice in terms of enhanced tumor biodistribution, suppressed tumor volume, and improved survival rate.44 Likewise, Jeong et al45 developed mPEG-CS-based redox-responsive nanosystem for the photodynamic therapy of colon cancer. To this end, thiodipropionic acid (TDPA) was conjugated with phenyl boronic acid pinacol ester (PBAP) (TDPA-PBAP), and then the TDPA-PBAP conjugates were chemically conjugated to the CS backbone in the mPEG-CS copolymer. These Ce6-incorporated nanophotosensitizers showed ROS-sensitive triggered release of Ce6, efficient uptake, cytotoxicity in colon cancer cells (CT26), and superior accumulation in irradiated tumor.45
For better targetability and additional control over the release of encapsulated chemotherapeutic payloads, dual- or multiresponsive systems have gained remarkable attention from researchers in recent years. Yoon et al46 developed dual-responsive (GSH + magnetic) CS-NPs for targeted delivery of DOX for the treatment of colon cancer. In this study, mPEG was grafted onto the WSCS backbone, DOX was chemically conjugated to the WSCS backbone via disulfide linkages, and Fe2O3 NPs (IO) were conjugated to the CS backbone for magnetic sensitivity. The developed DOX-IO-mPEG-WSCS-NPs exhibited nanoscale size (100–300 nm), smooth spherical morphology, GSH- and magnetic field-responsive sustained release over a period of four days, promising cell uptake efficiency, and enhanced cytotoxicity in DOX-resistant colon cancer cells (CT26) and glioma cells (U87MG). The magnetic field-responsive behavior of IO-mPEG-WSCS-NPs was also validated in CT26 cell-bearing mice, and the results demonstrated that following IV administration through the tail vein, strong fluorescence intensity was observed in the tumor tissue with magnet (+) with negligible fluorescence in tumor tissue without magnet (−). The superior biodistribution to tumor tissues with magnet (+) resulted in significant suppression of tumor volume compared with the control group.46 Similarly, Yang et al47 conjugated L-histidine methyl ester (HIS) with CS oligosaccharide (CSOS) via disulfide linkage to develop pH and redox dual-responsive CS-NPs for the targeted delivery of DOX. The developed DOX-HIS-CSOS-NPs exhibited nanoscale dimensions (<200 nm), a smooth spherical morphology, and pH- and redox-responsive DOX release. Moreover, dose-dependent cytotoxicity was observed in DOX-resistant HuCC-T1 cells (human cholangiocarcinoma cells) treated with CS-NPs compared with free DOX. The targetability of DOX-HIS-CSOS-NPs was also validated in HuCC-T1 tumor-bearing mice, where dual-responsive HIS-CSOS-NPs demonstrated better tumor biodistribution and accumulation, suppression of tumor volume, and maintenance of body weight.47
Passive Targeting
Passive targeting is a mechanism by which CS-NPs passively diffuse through leaky blood vessels (immature vasculature) that supply cancer tissue. Owing to abnormal lymphatic drainage in tumor tissues, the permeated CS-NPs were retained in tumor tissues for longer periods. This phenomenon is called “Enhanced Permeation and Retention (EPR)” effect. The physicochemical characteristics of CS-NPs, such as particle size, morphology, and surface charge, significantly influence the EPR effect and thus can be optimized by manipulating various formulation ingredients and fabrication processes.48 Moreover, owing to their unique functionalities, CS-NPs can be functionalized to further prolong plasma circulation time and passive permeation of chemotherapeutic agents into tumor tissues.49
Xu et al50 investigated the biopharmaceutical potential of CS-NPs to enhance the aqueous solubility, bioavailability, sustained release, cell uptake efficiency, and anticancer efficacy of PTX. They fabricated PTX-loaded CS-NPs via an emulsification-crosslinking method using glutaraldehyde as the crosslinker. The synthesized PTX-CS-NPs were tested against prostate cancer cells (DU-145), and the results were compared with those of blank CS-NPs. The particle size (496 nm) of PTX-CS-NPs was noticeably larger than that of blank CS-NPs (133 nm), with a promising encapsulation efficiency (~94%) and reasonable loading capacity. The drug release study in PBS (pH 7.4) indicated an initial burst release of PTX (~52%) within the first 12 h, followed by a relatively slow release (~68%) for up to 48 h, which was further sustained for up to seven days (~89%). The faster release was due to the adsorption of PTX on the surface of the CS-NPs, and the subsequent slower release was attributed to the erosion and swelling of the polymeric matrix of the CS-NPs at pH 7.4. PTX-CS-NPs showed dose-dependent (10, 20, 40, and 80 μg/mL) cytotoxicity in human prostate cancer cells (DU-145) within 48 h of treatment, with the highest cytotoxicity observed at 80 μg/mL. The anti-proliferative efficacy of PTX-CS-NPs was also significantly higher than that of blank CS-NPs and native PTX, indicating that CS-NPs are a promising vehicle for improving anticancer efficacy of PTX against prostate cancer.50 Similar findings were reported by Gupta et al.51
CS-NPs have also shown remarkable potential for passive delivery of other chemotherapeutics. For instance, Jain et al52 encapsulated docetaxel (DTX) into CS-NPs via emulsion-solvent evaporation to improve their physicochemical properties, bioavailability, cell uptake efficiency, and anticancer efficacy against breast cancer. The fabricated DTX-CS-NPs exhibited nanoscale particle size (~170 nm), good zeta potential (~32 mV), high encapsulation efficiency (%EE) (~75% EE), loading capacity (%LC) (~13% LC), biphasic release kinetics, and good stability. Dose-dependent and time-mannered cytotoxicity was observed in MDA-MB-231 cells treated with 5.0 μg/mL of DTX-CS-NPs, compared with the low cytotoxicity observed in free DTX and no toxicity in blank CS-NPs. The highest cytotoxicity observed in the DTX-CS-NP group was attributed to the enhanced cellular uptake efficiency of DTX when delivered in the form of CS-NPs.52,53 CS-NPs have also shown promising abilities for the passive delivery of other chemotherapeutics, such as hydroxychloroquine,54 naringenin,55,56 and cromolyn.57
Many natural polyphenols, such as ellagic acid (EA), possess powerful anticancer potential. However, owing to their poor aqueous solubility and low bioavailability, their clinical translation is restricted. Arulmozhi et al58 proposed encapsulation of EA into CS-NPs to improve their aqueous solubility, oral bioavailability, and anticancer potency for the treatment of oral cancer. The fabricated EA-loaded CS-NPs (EA@CS-NP) exhibited an average particle size of ~176 nm with high %EE (~94%) and %LC (~33%). The drug release study in PBS (pH 7.4) showed a biphasic release with burst release in the first 3 h, which was attributed to the adsorbed drug on the CS-NPs surface, followed by sustained release for up to 48 h, which was expected to be due to the liberation of encapsulated EA from the CS matrix. The viability of KB cells (oral cancer cells) was significantly decreased when treated with EA@CS-NP (IC50 = 0.953 μg/mL) compared with that of blank CS-NP and native EA (IC50 = 3.125 μg/mL). Moreover, KB cells treated with EA@CS-NPs underwent enhanced apoptosis compared to free EA, which was attributed to the nanoscale size and cationic charge of CS-NPs, resulting in enhanced uptake and cytotoxicity in KB cells.58 Similarly, Chung et al59 fabricated retinoic acid (RA)-encapsulated GCS-NPs via ionic gelation to improve their physicochemical properties and anticancer efficacy against colon cancer. RA has been extensively investigated for the treatment of different malignancies; however, it has not yet been translated into an effective systemic treatment owing to its poor aqueous solubility and low bioavailability. However, after encapsulation into GCS-NPs with a size of 317 nm and smooth spherical morphology, the aqueous solubility, bioavailability, cellular uptake, and cytotoxicity of RA in cholangiocarcinoma cells (HuCC-T1) dramatically increased. Moreover, HuCC-T1 cells treated with RA-GCS-NPs showed promising inhibition of proliferation and migration compared with free RA and blank GSC-NPs.59
Metformin (MFN) has attracted considerable attention from researchers in the field of oncology owing to its strong anticancer activity. MFN inhibits mTOR (mammalian target of rapamycin) by activating AMP kinase (AMPK), restraining cancer progression. However, its low bioavailability and short plasma half-life block its clinical translation into an effective systemic cancer therapy. Snima et al60 investigated the potential of o-carboxymethyl CS (O-CMC) as a nanocarrier for improving the pharmacokinetics and anticancer efficacy of MFN against pancreatic cancer. MFN-loaded O-CMC-NPs were prepared via ionic gelation and characterized for their particle size (240 nm), morphology (smooth spherical), zeta potential (−18 mV), in vitro drug release, cytotoxicity, cellular uptake, and biocompatibility. Drug release analysis performed in PBS (pH 7.4) showed an initial burst release of ~50% drug content within the initial 10 h, followed by a sustained release of 72% after 70 h. Moreover, a pH-responsive release was exhibited by MFN-loaded O-CMC-NPs, with ~90% of the drug released in the first 1 hour at pH 4.5, which was possibly due to swelling of the O-CMC-NPs matrix due to protonation of –NH2 groups on the backbone of CS. Cytotoxicity assays conducted on both normal (L929 cells) and pancreatic cancer cells (MiaPaCa-2) revealed that neither free MFN nor MNF-loaded O-CMC-NPs produced toxicity in normal cells, indicating their biocompatibility. However, a significantly higher cytotoxicity was noted in MiaPaCa-2 cells treated with MNF-O-CMC-NPs compared to blank O-CMC-NPs, which was attributed to the enhanced inhibition of mTOR in pancreatic cancer cells. Moreover, a hemolytic ratio of <5% induced by MFN, bare O-CMC-NPs, and MFN-O-CMC-NPs indicated their high hemocompatibility and suitability for IV administration.60
CS-NPs have also shown reasonable success in the passive delivery of peptides for the treatment of different types of cancer. For example, Zhang and Hu61 encapsulated alpha-statin (As) (a fragment of human fibrinogen that possesses powerful antiproliferative efficacy against activated endothelial cells) in CS-NPs to improve its aqueous solubility, bioavailability, cell uptake efficiency, and antiangiogenic potency. The fabricated As-CS-NPs were characterized by particle size (~388 nm), zeta potential (~28 mV), %EE (~51%), morphology (spherical), and biocompatibility. The developed As-CS-NPs exhibited an initial burst release (~76%) within the first 6 h, followed by a slower release rate (~45% at 24 h, 60% at 48 h, and 70% at 96 h) for up to 6 days, which was expected to be due to the slow diffusion of the entrapped drug from the CS-NPs matrix. The anti-angiogenic efficacy was assessed using the HUVEC tube formation assay, and the results showed that the As-CS-NPs exhibited a promising ability to repress the formation of tubular structures compared with the free drug and normal saline groups. Mice treated with As-CS-NPs demonstrated a sizable decrease in tumor volume compared with the plain drug and normal saline groups. These results verified that encapsulation of As into CS-NPs improved its anticancer efficacy while mitigating its proteolytic degradation; thus, As-loaded CS-NPs could be a suitable alternative candidate for cancer therapy.61
In addition to monodelivery, CS-NPs have shown tremendous potential for combined delivery of chemotherapeutics. David et al62 encapsulated 5-fluorouracil (5-FU) and quercetin (QUE) in CS-NPs for synergistic anticancer efficacy against pancreatic cancer. The dual-drug-loaded nanocarriers displayed a larger size (~400 nm) than the single-drug (5-FU or QUE)-loaded CS-NPs (~300 nm). The dual-drug-loaded CS-NPs exhibited good %EE for both drugs (QUE ~91% and 5-FU 53%), which was optimized by varying the CS concentration. Evaluation of drug release revealed that dual-drug-loaded CS-NPs exhibited pH-responsive release, with a relatively faster release of both drugs at acidic pH (pH 5.5) than at physiological pH (pH 7.4). The predominant release at acidic pH was attributed to the enhanced protonation of amino groups (–NH3+) on the CS backbone, which caused repulsion between the polymeric chains, resulting in the swelling of the CS matrix. Superior cell uptake and dose-dependent cytotoxicity were observed in MiaPaCa2 cells (pancreatic cancer) treated with dual-drug-loaded CS-NPs compared to single drug-loaded CS-NPs (QUE-CS-NPs or 5-FU-CS-NPs), blank CS-NPs, and free drugs. The nanoscale particle size and cationic charge of the CS-NPs were expected to be responsible for the enhanced cellular uptake and cytotoxicity.62 Similarly, Khan et al63 demonstrated that the encapsulation of curcumin (Cur) into CS-NPs enhanced its aqueous solubility, photostability, bioavailability, plasma circulation time, and anticancer efficacy against cervical cancer. They further discovered that the combination of Cur-loaded CS-NPs with methyl jasmonate (MJ) (a plant stress hormone with anticancer efficacy) was more powerful than Cur-CS-NPs, blank CS-NPs, or plain Cur. The particle size of Cur-CS-NPs (~197 nm) was slightly larger and the zeta potential was slightly lower (~71 mV) than that of the blank CS-NPs (particle size ~190 nm and zeta potential ~76 mV), indicating the successful loading of Cur into CS-NPs. An initial burst release in the first 3 h, followed by a sustained release for up to 120 h, confirmed the biphasic pattern of the Cur-CS-NPs. Moreover, a pH-responsive behavior was observed in Cur-CS-NPs with a relatively higher release at acidic pH than at physiological pH, which was attributed to swelling of the CS matrix due to enhanced protonation of the–NH2 groups of CS at acidic pH. Cellular uptake studies showed that Cur-CS-NPs were more efficient for internalization into cervical cancer cells (SiHa, HeLa, CasKi, and C33A) compared to native Cur, and this was attributed to the nano-size and cationic charge of CS-NPs. More efficient uptake of Cur-CS-NPs resulted in enhanced cytotoxicity and a lower IC50 compared to native Cur and blank CS-NPs. The superior anticancer effect of the combined treatment with Cur-CS-NPs and MJ was attributed to the overexpression of the pro-apoptotic protein (Bax) and downregulation of the anti-apoptotic protein (Bcl-2).63 On the other hand, Xie et al64 encapsulated endostatin (ES) (an antiangiogenic peptide) into CS-NPs for improving its pharmacokinetics and combined it with PTX for treatment of lung cancer. The fabricated ES-CS-NPs were characterized in terms of their particle size (223 nm), zeta potential (+35 mV), %EE (71%), and %LC (10%). The in vitro drug release study showed a pH-responsive biphasic release pattern, with an initial burst release followed by a sustained release. After 21 days of treatment, the combined therapy (ES-NPs-+-PTX) was found to be more potent in reducing tumor volume (1263 mm3) than ES-NPs (1980 mm3), free drugs (ES+PTX) (2772 mm3), PTX (3042 mm3), or the control group (4102 mm3) (Figure 4). Serum levels of vascular endothelial growth factor-α (VEGF-α) were lower in the ES-NPs+PTX group than in the other groups. Moreover, Ki-67 immunohistochemical staining performed on tumors showed that the number of Ki-67 positive cells was lowest in the ES-NPs-+PTX group (30%) compared to the PTX (54%), ES+PTX (36%), ES (73%), ES-NP (57%), and control (82%) groups. In summary, adjuvant therapy with ES-NPs+PTX could be a viable option for the treatment of lung cancer; however, further investigation in humans is highly warranted.64 Combined delivery of chemotherapeutics has also been reported by other researchers. Ahmad et al65 developed CS-NPs for the codelivery of cisplatin (DDPT) and 5-FU for the passive targeting and treatment of colorectal cancer. The developed NPs exhibited nanoscale dimensions (<200 nm), smooth spherical morphology, sustained release of encapsulated drugs, and excellent cytotoxicity against colorectal cancer (HCT-116).65 Similarly, Matalqah et al66 co-encapsulated DDPT and DOX into CS-NPs to improve their pharmacokinetics and synergistic efficacy against breast cancer. The developed NPs exhibited nanoscale dimensions (277 nm), smooth spherical morphology, high EE, pH-responsive release of encapsulated drugs (higher release at acidic pH and lower release at physiological pH), and excellent cytotoxicity against breast cancer cells (MCF-7 BrCA).66 Another co-encapsulation method was proposed by Song et al,67 who prepared CS-NPs via the ionic-gelation method and loaded them with an anti-PD-L1 peptide (PP) and DOX for synergistic immunotherapy against colon cancer. These findings demonstrated that CS-NPs are promising nanovehicles for the codelivery of chemotherapeutics for the treatment of different types of cancers.
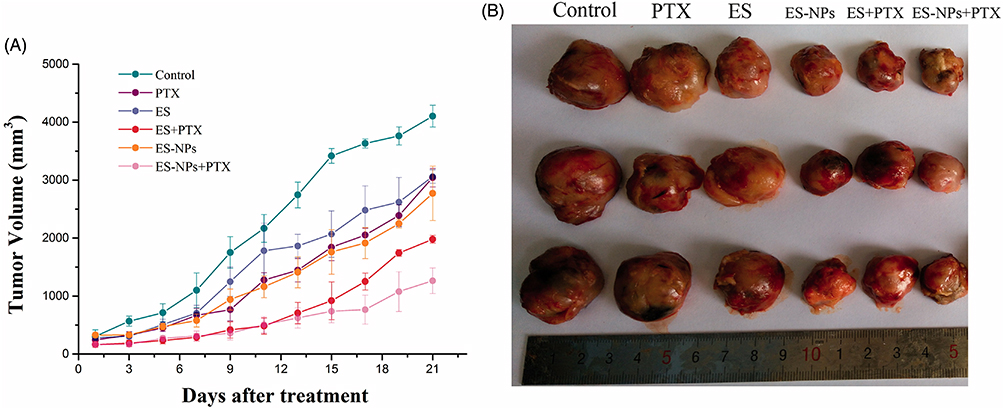 |
Figure 4 Tumor growth in subcutaneous Lewis lung cancer model. (A) Time-mannered growth in tumor volume and (B) final tumor size on day 21 of treatment. Reprinted from Xie F, Ding R-L, He W-F, et al. In vivo antitumor effect of endostatin-loaded chitosan nanoparticles combined with paclitaxel on Lewis lung carcinoma. Drug Deliv. 2017;24(1):1410–1418. Distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.64 |
In addition to optimizing physicochemical properties (ie, particle size, zeta potential, morphology, and sustained release), various functionalization strategies have been adopted in the architecture of CS-NPs to further improve their pharmacokinetic parameters, deeper penetration into tumor tissues, stability in biological fluids, and anticancer efficacy. One of such functionalization strategies is PEGylation, which can be deployed by covalent conjugation of PEG with CS followed by self-assembly to CS-NPs, by coating of PEG on the surface of fabricated CS-NPs, or by using PEG-linker to conjugate CS-NPs with chemotherapeutic drugs. This technique has a long history of credibility in improving the plasma circulation time by suppressing their recognition by MPS and protein binding, which ultimately improves the passive diffusion of PEGylated CS-NPs into the TME.
For instance, Sharma et al68 proposed intravaginal delivery of telmisartan (TEL) (an angiotensin II receptor blocker with anti-proliferative and antimetastatic efficacy) via PEG-grafted CS-NPs to improve its aqueous solubility, residence time in the vagina, and penetration through the thick mucus membrane for the treatment of cervical carcinoma. They fabricated two different types of PEG-grafted CS-NPs: one loaded with soluble telmisartan (S-TEL-PEG-CS-NPs) and the other containing telmisartan (TEL-PEG-CS-NPs). The average particle size (~24 nm) of the S-TEL-PEG-CS-NPs was slightly larger than that of the TEL-PEG-CS-NPs (~16 nm), whereas the zeta potential of the S-TEL-PEG-CS-NPs was slightly lower (~ −22 mV) than that of the TEL-PEG-CS-NPs (~ −24 mV). Evaluation of mucoadhesive properties using female pig vaginal mucosa indicated that S-TEL-PEG-CS-NP demonstrated better mucoadhesion (~40%) than TEL-PEG-CS-NPs (~31%). The drug release profiles of both CS-NPs formulations were investigated at pH 4.2, mimicking vaginal secretions, and the results showed a significantly higher dissolution rate (~99%) and solubility (~93%) of S-TEL-PEG-CS-NPs compared to those of TEL-PEG-CS-NPs (only 9% dissolution and ~32% solubility). Both CS-NPs formulations displayed no cytotoxicity against normal skin cells (HaCaT) indicating their biocompatibility; however, superior cell uptake efficiency, cytotoxicity, and apoptosis were evident in HeLa cells treated with S-TEL-PEG-CS-NPs (IC50 22.3 μM) compared to TEL-PEG-CS-NPs (IC50 = 40.1 μM), plain TEL, and unfunctionalized CS-NP (~IC50 >100 μM). These findings suggest that PEGylated CS-NPs could be a promising platform for improving the passive diffusion of chemotherapeutics through the cervicovaginal mucosal barrier for the treatment of cervical cancer.68 Similarly, Chen et al69 proposed the delivery of MTX via mPEG-CS-NPs prepared by the ionic gelation method. They fabricated MTX-mPEG-CS-NPs with a particle size (213 nm), EE (approximately 87%), LC (approximately 44%), and in vitro drug release. The developed MTX-mPEG-CS-NPs showed better uptake and enhanced cytotoxicity in HeLa cells (cervical cancer cells) than unPEGylated MTX-CS-NPs and free MTX.69 Similar results were reported by Park et al70 for the passive delivery of RA for the treatment of colorectal cancer (CT26).
Jia et al established the ability of mPEG-CS-NPs to synergistically deliver MTX and mitomycin C (MMC) against cervical cancer.71 The developed mPEG-MTX-MMC-CS-NPs exhibited ultrafine particle size (~215 nm), cationic charge (~32 mV), high %EE and %LC, biphasic release with initial burst release followed by sustained release, higher cell uptake and cytotoxicity, and superior tumor-specific biodistribution and localization, compared to mPEG-MMC-CS-NPs, mPEG-CS-NPs, and free drugs.71 Likewise, Hong et al72 grafted mPEG with CS (mPEG-CS), which was then self-assembled into core-shell NPs for the successful encapsulation of PTX and RA in the inner hydrophobic core of self-assembled NPs. The fabricated PTX-RA-mPEG-CS-NPs (~160 nm, smooth spherical morphology) displayed a high cell uptake efficiency, cytotoxicity, and inhibition of invasion (reduction in MMP-2 enzyme) in colon cancer cells (CT26). They demonstrated that mPEG-CS-NPs were promising polymeric nanovehicles for the combined delivery of chemotherapeutics for the treatment of colon cancer.72
The effects of mPEG grafting have been studied using other CS derivatives. Jeong et al73 grafted mPEG with carboxymethyl CS (CMCS), which was then self-assembled to form core-shell NPs encapsulated with DOX via ionic complexation with the carboxymethyl groups of CMCS. The fabricated DOX-mPEG-CMCS-NPs exhibited nanoscale dimensions (<300 nm), a smooth spherical morphology, and pH-responsive release (faster release at acidic pH than at physiological pH). Incubation of these core-shell NPs with glioma cells (C6 cells) showed enhanced cytotoxicity compared with free DOX, which was attributed to the higher cell uptake efficiency of mPEG-CMCS-NPs.73 Jang et al74 grafted mPEG onto the backbone of low-molecular-weight water-soluble (LMWS) CS for the passive delivery of PTX into colon cancer cells (CT26). They demonstrated that encapsulation of PTX into mPEG-LMWSC-NPs resulted in sustained release of PTX, better cell uptake efficiency, and enhanced cytotoxicity in CT26 cells compared with native PTX.74
The grafting density and molecular weight of PEG are important factors that can significantly influence the physicochemical properties, plasma half-life, and in vivo fate of the CS-NPs. Bachir et al75 investigated the effects of different molecular weights (mPEG750, mPEG2000, and mPEG5000) and grafting densities of mPEG on the physicochemical properties, plasma circulation time, and anticancer efficacy of MTX. They demonstrated that an increase in the molecular weight and grafting density of mPEG resulted in a parallel increase in the mean particle size and a decrease in the zeta potential, in vitro drug release rate, plasma circulation time, and cellular uptake efficiency of DOX-mPEG-CS-NPs into macrophages (J774A.1). Interestingly, the molecular weight and grafting density of mPEG did not affect the %EE, %LC, or morphology of MTX-mPEG-CS-NPs. To improve pharmacokinetic parameters and passive targeting efficiency, the use of low molecular weight and low grafting density of mPEG is advisable; however, every nanosystem has its own chemistry and, therefore, demands sufficient optimization of PEGylation before in vivo administration.75
Another innovative approach for improving the passive delivery of chemotherapeutics was proposed by Wang et al.76 They proposed the copolymerization of glycol CS (GCS) with carboxymethyl-β-cyclodextrin (CMβ-CD), self-assembled into polymeric NPs (GCS-CMβ-CD NPs) via ionic gelation. Owing to its hydrophilic exterior and hydrophobic interior, β-CD has gained recognition for improving the aqueous solubility of hydrophobic drugs. The fabricated DOX-loaded CMβ-CD-GCS-NPs exhibited a mean particle size of 100–180 nm, a smooth spherical morphology, good %EE, and pH-responsive release (faster release at acidic pH and minimal release at physiological pH). Treatment of MCF-7 (human breast cancer) and SW480 cells (colon cancer cells) with DOX-CMβ-CD-GCS-NPs showed significantly enhanced cytotoxicity compared with free DOX, which was attributed to the enhanced cellular uptake efficiency of NPs owing to their nanoscale dimensions, cationic charge, and sustained drug release properties.76
The hydrophobic substitution of CS, particularly the hydrophilic derivatives of CS, has received increasing attention for the development of novel amphiphilic derivatives of CS, which significantly affect the physicochemical properties, structure (core-shell), thermodynamic stability, and deformability of CS-NPs. The most promising aspect of this approach is that amphiphilic conjugates of CS can rapidly self-assemble into core-shell NPs in an aqueous medium by incorporating a hydrophobic drug into the inner hydrophobic core of CS-NPs. Thus, aqueous solubility, stability in biological fluids, release kinetics, plasma circulation time, and cellular uptake efficiency of chemotherapeutics can be significantly enhanced. For instance, Zhou et al77 conjugated deoxycholic acid (DCA) (a hydrophobic moiety) and glycidol (G) (a hydrophilic moiety) to the CS backbone. These amphiphilic conjugates rapidly self-assembled into core-shell NPs by encapsulating DOX in the inner hydrophobic core of the NPs. The developed DOX-G-CS-DCA-NPs (160–210 nm) maintained a stable structure for 3 months when stored in PBS (pH 7.4) and showed sustained release of DOX over a week period. Interestingly, the cellular uptake and cytotoxicity of DOX-G-CS-DCA-NPs in MCF-7 cells were lower than those of free DOX after 24 h of incubation; however, the uptake and cytotoxicity were significantly enhanced after 24 h, which was attributed to the sustained release of DOX from the inner hydrophobic core of DOX-G-CS-DCA-NPs.77 Likewise, Jin et al78 conjugated DCA to CMCS, which self-assembled in an aqueous medium to form core-shell NPs incorporated with DOX in the inner hydrophobic core of NPs. The DOX-DCA-CMCS-NPs exhibited nanoscale dimension (87–174 nm), good stability in physiological media, and sustained release over a prolonged period. Time-mannered and dose-dependent cytotoxicity and cell uptake were observed in MCF-7 cells treated with DOX-DCA-CMCS-NPs, which was attributed to the sustained release of DOX from the hydrophobic core of the NPs.78 Similar results have been reported by Zhou et al.79
One of the most studied hydrophobic moieties for synthesizing amphiphilic derivatives of CS is 5β-cholanic acid (5β-CA), which is used to produce core-shell NPs. Notably, the degree of hydrophobic substitution with 5β-CA plays a critical role in the stability, deformability, and tumor targetability of core-shell NPs. To optimize the properties of core-shell NPs, Na et al80 fabricated four different conjugates of 5β-CA and GCS by coupling 7.5%, 12%, 23%, and 35% weight ratios of 5β-CA. The synthesized conjugates were self-assembled into core-shell NPs in an aqueous medium. The obtained results suggested that the degree of substitution did not have any significant effect on the particle size and zeta potential of 5β-CA-GCS-NPs; however, their stability and deformability were greatly dependent on the degree of substitution with 5β-CA. With increasing hydrophobicity (increasing weight ratio of 5β-CA), the formed 5β-CA-GSC-NPs became more rigid and stable. Interestingly, the degree of hydrophobic substitution also affects the tumor targetability and in vivo biodistribution of 5β-CA-GSC-NPs in tumor tissues. The most promising tumor biodistribution was achieved using a weight ratio of 23% %5β-CA. However, every nanosystem has a characteristic chemistry; therefore, it is advisable to optimize the hydrophobic substitution for better targetability and therapeutic outcomes.80 Using this concept, Kim et al81 also fabricated 5β-CA-modified GCS-NPs for the targeted delivery of PTX for the treatment of melanoma. The fabricated PTX-5β-CA-GSC-NPs (~400 nm) remained stable in PBS (pH 7.4) for 10 days and displayed a sustained release of PTX over 8 days period. PTX-5β-CA-GSC-NPs were found to be less cytotoxic to melanoma cells (B16F10) than native PTX after 24 h of incubation; however, they efficiently suppressed the growth of tumors for up to 8 days when injected into B16F10 tumor-bearing mice, compared to native PTX, which did not decrease tumor volume owing to faster elimination from the body.81 Similarly, Min et al82 fabricated 5β-CA-GSC-NPs for the targeted delivery of camptothecin (CPT) for the treatment of breast cancer. The fabricated CPT-5β-CA-GSC-NPs exhibited nanoscale size (280–330 nm), high %EE (80%), protection against enzymatic degradation, and pH-responsive release of encapsulated CPT for one week. The MDA-MB231-tumor bearing mice treated with CPT-5β-CA-GSC-NPs displayed prolonged plasma circulation time, specific tumor biodistribution, and prolonged localization into tumor tissues compared to the control groups.82 Similarly, 5β-CA-mediated hydrophobic modification of GCS has also been adapted by Kim et al83 for the passive delivery of CCDP. The fabricated CCDP-5β-CA-GSC-NPs exhibited nanoscale size (300–500 nm), high %EE (80%), and pH-responsive release over a period of one week because of the slower release of CCDP from the inner hydrophobic core. The hydrophobically modified GSC-NPs were less cytotoxic initially, but over time, their cytotoxicity was enhanced compared to that of free CCDP. Moreover, CCDP-5β-CA-GSC-NPs were more efficient than free CCDP in suppressing tumor growth, maintaining body weight, and improving the survival rate in tumor-bearing mice. The superior anticancer efficacy of CCDP-5β-CA-GSC-NPs was attributed to their prolonged plasma circulation time, specific tumor biodistribution, and prolonged localization in tumor tissues.83 5β-CA-modified GSC-NPs were also evaluated for passive delivery of DTX, as proposed by Hwang et al.84 The developed DTX-5β-CA-GSC-NPs (350 nm) remained stable for two weeks under physiological conditions (pH 7.4, 37°C) and exhibited sustained release over a prolonged period. The developed NPs also showed excellent anticancer efficacy in A549 (lung cancer cells)-bearing mice by reducing tumor volume, maintaining body weight, and higher survival rates in treated animals compared to free DTX.84
These amphiphilic features can also be introduced into CS by chemically conjugating it with stearic acid (SA), which then self-assembles into core-shell NPs in the presence of dextran as a crosslinker, as proposed by Shen et al.85 These core-shell NPs are promising for the high encapsulation of Cur in the inner hydrophobic core of the CS-NPs. The encapsulation of Cur into these core-shell NPs resulted in significant improvements in aqueous solubility, oral bioavailability, and antioxidant and anticancer efficacies.85 Chemical conjugation with hydrotropic agents such as oligomers is another attractive approach for creating amphiphilic features in GCS. For instance, Saravanakumar et al86 chemically conjugated a hydrotropic oligomer (HO) with GCS, which self-assembled into core-shell NPs by incorporating PTX into the inner hydrophobic core. The developed PTX-HO-GCS-NPs exhibited nanoscale size (~313 nm), smooth spherical morphology, promising %EE (97%), good %LC (20%), and excellent stability in PBS (pH 7.4) for 50 d. In in vitro cell culture studies, PTX-HO-GCS-NPs displayed lower toxicity in SCC7 cells compared to free PTX prepared in a 50%/50% Cremophor EL/ethanol mixture; however, the in vivo antitumor study revealed a superior antitumor efficacy of PTX-HO-GCS-NPs in SCC7 tumor-bearing mice compared to free PTX and normal saline groups. The lower toxicity observed in SCC7 cells treated with PTX-HO-GCS-NPs was likely due to the slower initial release of PTX from the inner hydrophobic core of NPs.86 Similar results have been reported by Koo et al.87
Despite possessing several characteristic features of descent, one of the problems encountered with polymeric NPs (eg, CS-NPs) is their recognition by the MPS with ultimate phagocytosis and elimination from the body. To circumvent this problem, several strategies have been adapted in the constructs of CS-NPs such as PEGylation, coupling of hydrophilic moieties, stealthing/camouflaging with cell membranes, conjugation of CD47-derived enzyme-resistant peptide ligands on the surface of NPs which gives “Don’t Eat Me” signals to the immune systems. One such promising adaptation of CS-NPs is their coupling with low-density lipoprotein (LDL). As an endogenous molecule, LDL is nontoxic, biodegradable, biocompatible, and can easily escape RES-mediated phagocytosis. Furthermore, owing to the presence of specific membrane-bound protein transporters that facilitate the diffusion of LDL through cell membranes, coupling LDL with CS-NPs is also considered a promising approach to improve the transcellular permeability of CS-NPs. Moreover, several LDL receptors are heavily expressed in different tumor cells, making them a targeting ligand for the targeted delivery of chemotherapeutics. Using this highly attractive concept, Zhu et al88 developed succinyl-CS (SCS)-NPs encapsulated in DOX. The prepared DOX-SCS-NPs were coupled with SiRNA-LDL via a cholesterol linkage. The developed NPs (DOX-siRNA-LDL-SCS-NPs) exhibited nanoscale dimensions (~206 nm), high %EE (~71%), fair %LC (~12%), and good stability. Moreover, DOX-siRNA-LDL-SCS-NPs showed the highest cell uptake and cytotoxicity in HepG2 cells, low uptake in macrophages (RAW 264.7), and good tumor targetability compared to the control groups.88 Later, the same passive strategy was adapted by Tian et al89 using a simple one-step green mixing of CS solution with LDL suspension under constant magnetic stirring, which underwent agglomeration followed by self-assembly to LDL-CS-NPs. The fabricated DOX-LDL-CS-NPs exhibited nanoscale size (~180 nm), cationic zeta potential (~48 mV), and higher cytotoxicity in gastric cancer cells (SGC7901) than free DOX did. The enhanced cell uptake efficiency and cytotoxicity of DOX-LDL-CS-NPs were attributed to their nanoscale size and cationic charge, which confer their adsorptive endocytosis into tumor cells.89 All these strategies are promising for improving the passive delivery of chemotherapeutics to the TME; however, a comparative evaluation of different strategies is highly warranted to substantiate the existing literature.
Active Targeting
Among the various targeting mechanisms, active targeting is the most selective approach for maximizing therapeutic efficacy while mitigating off-target effects. It refers to the delivery of a chemotherapeutic or diagnostic payload to specific cells without accumulating in non-target cells.90–93 In this approach, specific targeting ligands (eg, folic acid, hyaluronic acid, transferrin, antibodies, peptides, aptamers) are conjugated to CS-NPs to recognize specific receptors or antigens (eg, CD44, FA, transferrin receptors) on cancer cells.28,94,95 Specific binding of ligand-functionalized NPs to cancer cells improves the efficacy of chemotherapeutics.96–98 Being enriched with numerous hydroxyl and amino groups, CS-NPs possess tremendous potential to be functionalized with different targeting ligands for selective targeting of different types of cancer.
One of the most studied cellular targets for selective targeting of tumor cells is FA receptors (FARs), which are highly expressed in a variety of cancer cells and minimally expressed in normal healthy cells. Therefore, many researchers have conjugated FA to the surface of CS-NPs and evaluated their anticancer potential for the treatment of different types of cancers. For instance, Esfandiarpour-Boroujeni et al99 prepared CS-NPs for improving the physicochemical properties and pharmacokinetics of Cur, and for specific targetability, they functionalized the fabricated Cur-CS-NPs with FA to selectively target breast cancer cells having overexpressed FA-receptors (FARs). They prepared FA-modified Cur-CS-NPs via self-assembly and evaluated their particle size (119–127 nm), zeta potential, %EE (96%), morphology (smooth spherical), drug release, and in vitro cytotoxicity in breast cancer cells (MCF-7). The in vitro release study showed pH-responsive release behavior of FA-Cur-CS-NPs, with predominant release at acidic pH (pH 5.0) compared to physiological pH (pH 7.4), which was expected to be due to swelling of the CS-matrix after protonation of the amine groups in the acidic environment. Compared to plain CS-NPs and free Cur, FA-Cur-CS-NPs demonstrated superior cytotoxicity and uptake into MCF-7 cells, with no significant cytotoxicity observed in healthy breast cells (L929) at all incubation times. The enhanced uptake and cytotoxicity displayed by FA-CS-NPs were attributed to their FAR-mediated endocytosis into MCF-7 cells.99
To further improve accumulation into tumor cells, Wang et al100 proposed co-functionalization with PEGylation and FA conjugation on CS-NPs for the treatment of lung cancer. They fabricated gemcitabine (GEM) (a third-generation chemotherapeutic from the taxane family)-loaded CS-NPs with additional functionalization with mPEG and FA. The developed FA-mPEG-GEM-CS-NPs exhibited nanoscaled size (~185 nm), cationic zeta potential (~29 mV), smooth spherical morphology, good %EE (~40%), pH-dependent release (high release at pH 5.8 and lower release at pH 7.4), higher uptake, a dose-dependent cytotoxicity in A549 cells, and better pharmacokinetic (Cmax, mean resident time, MRT, and area-under-the-curve, AUC0−t), compared to mPEG-GEM-CS-NPs and free drug. A tissue biodistribution study in tumor-bearing BALB/c mice showed that the maximum tumor accumulation of FA-mPEG-CS-NPs was attained within 8 h of IV injection through the tail vein, which started decreasing after 24 h, indicating the successful elimination of NPs from the body. An in vivo antitumor study showed significant suppression of tumor volume in tumor-bearing Balb/c mice treated with FA-mPEG-GEM-CS-NPs compared to mPEG-GEM-CS-NPs and free GEM.100 Using the same concept, Zamanvaziri et al101 co-functionalized CS-NPs with PEG and FA for the combined passive and active delivery of sodium butyrate (SB) for better therapeutic outcomes against prostate cancer. The fabricated FA-PEG-SB-CS-NPs exhibited nanoscale size (140–170 nm), cationic zeta potential (~15 mV), narrow PDI, smooth spherical morphology, high %EE (~75%), and pH-dependent release (high release at pH 5.0 and lower release at pH 7.4). To evaluate the selective targetability of FA-PEG-SB-CS-NPs, cellular uptake and cytotoxicity were assessed in two different prostate cancer cell lines: PC3 cells (FARs-negative) and DU145 cells (FARs-positive). Dose-dependent cytotoxicity was observed in both prostate cancer cell lines; however, more potent cytotoxicity was observed in DU145 cells. The higher cytotoxicity observed in FA-PEG-SB-CS-NP-treated DU145 cells was attributed to a significant induction of pro-apoptotic (Caspase-9, Bax) and autophagy genes (ATG5, BECLIN1, and mTORC1) compared to the PEG-SB-CS-NPs, FA-SB-CS-NPs, plain CS, and control groups.101 The biopharmaceutical viability of FA-functionalization on CS-NPs has also been reported by Liang et al102 and Wang et al.103
Owing to their high encapsulation efficiency, thermodynamic stability, and flexibility, Shi et al104 proposed FA functionalization of CS-based core-shell NPs for the targeted delivery of DOX. CS was conjugated with DCA and PEG (PEG-CS-DCA), and these amphiphilic conjugates were then functionalized with FA and self-assembled into core-shell NPs. DOX-loaded FA-PEG-CS-DCA-NPs exhibited nanoscale size (~200 nm), cationic zeta potential, smooth spherical morphology, good %EE, and pH-dependent release kinetics. FA-PEG-CS-DCA-NPs displayed higher cellular uptake and cytotoxicity in FARs-positive HeLa cells, with no significant uptake or cytotoxicity in FARs-negative fibroblasts (3T3 cells).104 Another CS-based nanoconstruct was fabricated by Mi et al105 via ionic gelation of negatively charged CM-β-CD and positively charged 2-hydroxypropyltrimethyl ammonium chloride (HACS) (hydrophilic derivative of CS). FA was conjugated to the HACS backbone for selective targeting. The FA-DOX-CD-HACS-NPs exhibited nanoscale size (~220 nm), cationic zeta potential (~20 mV), smooth spherical morphology, high %EE (~75%), and pH-dependent release (higher release at acidic pH and lower release at physiological pH). A cell cytotoxicity assay revealed that FA-DOX-CD-HACS-NPs displayed stronger cytotoxicity against different types of cancer cells (BGC-823, MCF-7, HEPG-2, and A549) but no significant cytotoxicity against normal healthy cells (L929).105
To optimize targeting efficiency, Dramou et al106 proposed a new hybrid CS-based nanosystem for the targeted delivery of CPT for the treatment of colon cancer. They merged stimuli-responsive (magnet-guided) and active targeting (FA) strategies to achieve better targetability. FA-conjugated CSOS-assembled magnetic halloysite nanotubes (FA-CSOS/MHNTs) encapsulated in CPT were prepared using coprecipitation and solvent exchange methods. The developed FA-CPT-CSOS/MHNTs exhibited a nanoscale size (~147 nm), tubular morphology, and pH-dependent release. The in vitro release study indicated pH-responsive release of CPT from FA-CPT-CSOS/MHNTs, with nearly 83% drug release within 24 h at pH 5.0, compared to a significantly lower release at pHs 6.8 and 7.4. CPT@FA-COS/MHNTs showed excellent receptor-specific targeting of Caco-2 cells, excellent superparamagnetic properties, and exceptional anticancer potential.106 Another novel hybrid design for CS-NPs was proposed by Al-Musawi et al.107 This study aimed to construct CS-polyacrylic acid (PAA)-encapsulated Fe3O4 magnetic core-shell NPs for dual-targeted delivery of 5-FU for the treatment of bladder cancer. The synthesized FA-CS-5-FU-SPION yielded 73% EE, narrow PDI, and spherical morphology with a diameter of ~80 nm. The MTT assay and flow cytometry showed that FA-CS-5-FU-SPION exhibited higher cell uptake and cytotoxicity via the induction of apoptosis in bladder cancer cells (T24 cells) compared to plain CS-NPs and free drug. FA-CS-5FU-SPION exhibited a specific cytotoxic effect on T24 cells with no cytotoxicity in normal healthy cells (HBlEpC cells), indicating its selective targeting ability.107
CD44 receptors are another well-researched cellular target that can be exploited for selective targeting of chemotherapeutics by CS-NPs. To achieve CD44-mediated targeting, Liang et al108 functionalized CS-NPs with hyaluronic acid (HA) for targeted delivery of siRNA into bladder cancer cells. The developed HA-functionalized NPs (siRNA@CS-HAD) exhibited nanoscale size (100–200 nm), cationic zeta potential, smooth spherical morphology, high %EE (~95%), good stability, and biocompatibility. In vitro cell studies indicated that siRNA@CS-HAD displayed higher cellular uptake and cytotoxicity in CD44+ T24 cells, with no significant uptake and cytotoxicity in CD44 − normal healthy cells (L929). Moreover, an in vivo study in T24 tumor-bearing mice also indicated a higher tumor tissue biodistribution of siRNA@CS-HAD NPs compared to control groups.108 Similarly, Kousar et al109 prepared DDPT-loaded thiolated CS (THCS)-NPs using the ionic gelation method and subsequently functionalized them with HA for targeted therapy of cervical cancer. The developed HA-DDPT-THCS-NPs exhibited nanoscale size (~265 nm), cationic zeta potential (~22 mV), smooth spherical morphology, high %EE (~70%), stability for 3 months, and pH-responsive release (higher release at pH 6.8 and lower release at pH 7.4) over 72 h. In vitro cell studies indicated that HA-DDPT-THCS-NPs showed stronger cytotoxicity in CD44+ cervical cancer cells (HeLa) with no significant cytotoxicity in normal cervical epithelial cells (HCK1T) compared to the native drug.109 Later, the same research group confirmed the CD44-mediated targetability of HA-functionalized THCS-NPs for efficient immunotherapy of cervical cancer with the oncolytic Newcastle disease virus (NDV).110 Oncolytic virotherapy has emerged as a promising immunotherapy modality; however, it has several drawbacks, including the rapid clearance of the virus from the body due to immune neutralization in the host. However, encapsulation of NDV into HA-THCS-NPs showed longer stability (3 months) and pH-responsive release (higher release at pH 6.8 and lower release at pH 7.4) for 72 h. Moreover, HA-NDV-THCS-NPs displayed stronger cytotoxicity in CD44+ cervical cancer cells (HeLa) than did the native drug.110 HA-functionalized THCS-NPs were recently developed by Abduh et al111 for the CD44-mediated endocytosis of Cyclosporine A (CsA) for treatment of triple-negative breast cancer. The developed HA-CsA-THCS-NPs exhibited nanoscale size (~192 nm), cationic zeta potential (~39 mV), narrow PDI, smooth spherical morphology, high EE (~85%), and pH-responsive release over 72 h. In vitro cell studies indicated that HA-CsA-THCS-NPs showed stronger dose-dependent cytotoxicity in CD44+ breast cancer cells (MDA-MB-231) than the native drug, with no significant cytotoxicity in normal breast epithelial cells (MCF-10A).111
To further enhance the specificity and selectivity of CS-NPs against cancer, hybridization of multiple targeting strategies has been investigated. For instance, Yang et al112 designed dual-targeting HA-functionalized CMCS-modified graphene oxide (GO) NPs for the targeted delivery of DOX to the TME of cervical cancer. The fabricated NPs (GO-CMCS-FI-HA/DOX) exhibited nanoscale size (<200 nm), anionic zeta potential (~−42 mV), lamellar morphology, good %EE (~95%), and pH-dependent release (higher release at acidic pH (pH 5.8) and lower release at physiological pH (pH 7.4)). A cell cytotoxicity assay revealed that GO-CMCS-FI-HA/DOX displayed stronger cytotoxicity against CD44+ HeLa cells but no significant cytotoxicity against CD44- L929 cells.112 Similarly, Li et al113 developed dual-sensitive triphosphate (ATP)/hyaluronidase (Hyals) dual-sensitive core-shell HA-CS-NPs for the targeted delivery of SNX2112 (SNX, heat shock protein 90 inhibitor) to treat breast cancer. The developed HA-CS-NPs (SNX@HTCC-FPBA/mHA-PEG NPs) exhibited nanoscale size (100–200 nm), smooth spherical morphology, good biocompatibility with BSA, and ATP/Hyals-responsive release (higher release at higher ATP/Hyals concentrations and vice versa). Cell uptake and cytotoxicity studies showed higher uptake and cytotoxicity in both breast cancer cell lines (MDA-MB-453 and MCF-7) and no noticeable uptake or cytotoxicity in normal skin fibroblasts (HSF). These results indicate tumor-specific cytotoxicity via CD44-mediated endocytosis with no harm to normal healthy cells.113 Another dual-targeting CS-NPs were developed by Zhang et al.114 They copolymerized CS and di(ethylene glycol) methyl ether methacrylate (CS-g-PDEGMA), a temperature-sensitive polymer that undergoes swelling or erosion in the TME. The synthesized copolymer (CS-g-PDEGMA) was functionalized with HA, which self-assembled into core-shell NPs by incorporating PTX into the inner hydrophobic core of NPs. The developed HA-functionalized core-shell NPs (HA-CS-g-PDEGMA-PTX) exhibited nanoscale size (~170 nm), high %EE (~77%), smooth spherical morphology, good hemocompatibility, and pH-responsive release. Confocal microscopy showed that HA-CS-g-PDEGMA-PTX was more efficiently internalized into MD-MB-231 cells than unfunctionalized CS-g-PDEGMA-PTX, which was attributed to CD44-mediated endocytosis. The superior uptake of HA-CS-g-PDEGMA-PTX was responsible for its higher cytotoxicity in MDA-MB-231 cells compared to CS-g-PDEGMA-PTX. An in vivo biodistribution study indicated that HA-CS-g-PDEGMA-PTX remained accumulated in tumor tissues for a longer time, which resulted in significant suppression of tumor volume, maintenance of body weight, and higher survival rate compared to the free PTX and saline groups.114
Peptide or aptamer functionalization of CS-NPs has also attracted great attention from researchers for the active targeting of chemotherapeutics to different cancer cells. Qian et al115 designed hybrid CS/poly(N-isopropylacrylamide) NPs functionalized with K237-peptide for the active targeting of PTX to breast cancer cells (MDA-MB-231) via KDR/Flk 1 tyrosine kinase receptors. The prepared NPs exhibited ultrafine particle sizes (≤100 nm), good stability, and smooth spherical morphology. These hybrid NPs showed quicker drug release at acidic pH than at alkaline or neutral pH. K237-functionalized CS-NPs were more harmful to MDA-MB-231 cells than to non-cancerous cells, which was attributed to KDR/Flk-1 receptor-mediated internalization.115
On the other hand, Kurmi et al116 functionalized CS-NPs with estrone (Es) (sex hormone) for selective delivery of DOX to breast cancer cells. The optimized Es-functionalized DOX-CS-NPs exhibited nanoscale dimensions (~199 nm), high %EE (~65%), cationic zeta potential (~30 mV), smooth spherical morphology, good hemocompatibility, and biphasic release, with an initial burst release followed by a sustained release for 80 h. The in vitro cytotoxicity study showed stronger cytotoxicity in MCF-7 cells treated with Es-DOX-CS-NPs compared with free DOX and blank CS-NPs. In vivo studies showed a good pharmacokinetic profile and tumor-specific biodistribution of Es-DOX-CS-NPs compared to free DOX.116
Xu et al117 prepared Cur-loaded CS hydrochloride NPs (Cur-loaded PSNPs) and functionalized them with lactoferrin (Lf) (a mammalian cationic iron-binding glycoprotein belonging to the transferrin family) for targeted delivery to brain tumors via Lf receptors, which are heavily expressed in glioma cells and blood–brain barriers (BBBs). The developed Lf-Cur-PSNPs exhibited nanoscale dimension (220–230 nm), anionic zeta potentials (−20 to −30 mV), high %EE (80–85%), and a smooth spherical morphology. In vitro cytotoxicity and cell uptake studies showed stronger cytotoxicity and higher uptake of Lf-Cur-PSNPs into brain capillary endothelial cells (BCECs) and glioma cells (C6 cells) compared to unfunctionalized Cur-PSNPs, blank PSNPs, and free Cur. An in vivo biodistribution study in ICR mice also indicated that DiR-loaded Lf-PSNPs accumulated more efficiently in the brain than did unfunctionalized DiR-loaded PSNPs.117 On the other hand, Anandhakumar et al118 proposed functionalization of CS-NPs with collagen peptide for targeted delivery DOX to human cervical cancer cells (HeLa).
Active targeting can also be attained by conjugating monoclonal antibodies (MAb) to the surface of CS-NPs. MAb are extremely important vectors for direct NPs towards target cells with overexpressed specific antigens. For instance, Arya et al119 functionalized CS-NPs with anti-human epidermal growth factor receptor-2 (anti-HER2) MAb for the targeted delivery of GEM for the treatment of pancreatic cancer. Time-dependent cellular uptake and dose-dependent cytotoxicity were observed in pancreatic cancer cells (Mia Paca 2 and PANC 1) treated with anti-HER2-FITC-CS-NPs, with no significant uptake or cytotoxicity observed in human embryonic kidney cells (HEK293). Anti-HER2-GEG-CS-NPs also displayed stronger antiproliferative activity by inducing apoptosis than unfunctionalized GEM-CS-NPs and free drug.119
Sun et al120 proposed the functionalization of CS-NPs with galactose for the active targeting of DOX to liver cancer cells through asialoglycoprotein receptor (ASGPR), which is mainly expressed on hepatic cells. They conjugated glycidol (Gly) and DCA or vitamin E with CS, and these amphiphilic conjugates self-assembled into core-shell NPs by encapsulating DOX in the inner hydrophobic core of the NPs. Gly-CS-DCA-NPs and Gly-CS-VE-NPs were functionalized with galactose (Gal) to selectively target HepG2 cells for liver carcinoma treatment. Time-dependent cellular uptake and dose-dependent cytotoxicity were observed in HepG2 cells treated with Gal-Gly-CS-DCA-NPs compared to other formulations. Moreover, HepG2 cell monolayers treated with Gal-Gly-CS-DCA-NPs showed suppressed proliferation and migration compared with other treatments, indicating their powerful anti-metastatic potential. H22 tumor-xenografted BALB/c mice treated with Gal-Gly-CS-DCA-NPs showed superior anticancer efficacy in terms of the lowest tumor volume and minimal variations in body weight (Figure 5).120 Receptor-mediated active targeting has also been achieved for the treatment of different cancers by functionalizing PEG-CS-NPs with glycyrrhetinic acid121 or anisamide (AA) (a high-affinity benzamide derivative) for the treatment of different cancers.122
 |
Figure 5 In vivo anticancer efficacy: (A) in vivo anticancer experimental outline, (B) tumor volume growth curve of each group (**p < 0.01 and ***p < 0.001), (C) photographs of tumors excised from each group at Day 11, (D) tumor weight at Day 11 (**p < 0.01, ***p < 0.001), (E) body weight change curve of each group. (F) H&E staining of tumors excised from each group at Day 11 (scale bar = 50 μm). Reprinted from Sun R, Fang L, Lv X, et al. In vitro and in vivo evaluation of self-assembled chitosan nanoparticles selectively overcoming hepatocellular carcinoma via asialoglycoprotein receptor. Drug Deliv. 2021;28(1):2071–2084. Distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.120 |
Summary
CS is a polycationic linear polysaccharide with three important functional groups: the primary amino group and the primary and secondary hydroxyl groups on the CS backbone. These three functional groups are readily available for modification or grafting with different chemical moieties to produce versatile derivatives of native CS with distinct physicochemical properties (aqueous solubility, biodegradability, and biocompatibility). Moreover, the cationic amino groups on the contour of CS offer great opportunities for ionic complexation with multivalent anions and make it possible to implicate the CS as a promising vehicle for drugs for various biomedical applications. A variety of CS derivatives have been synthesized and exploited to construct different types of nanodelivery systems for targeted delivery of chemotherapeutics to the TME or intracellularly. However, the present review aimed to critically evaluate CS-based polymeric NPs, including nanospheres and nanocapsules (core-shell NPs), for the targeted delivery of chemotherapeutics through the three most important mechanisms: stimuli-responsive, passive, and active targeting approaches.
Having been enriched with three distinct functional groups, a variety of CS-based nanospheres and core-shell NPs have been designed for stimuli-responsive delivery of chemotherapeutics to the TME. They range from encapsulating the photoactivatable payload (eg, Ce6) into the polymeric matrix or hydrophobic core to coating different photoactivatable materials (eg, TiO2-NPs) on the surface of drug-loaded CS-NPs. These NPs are either passively diffused in the TME or can be assisted by an electric or magnetic field or high-intensity focused ultrasound for deeper penetration into tumor tissues. After penetration into the TME, therapeutic cargo is released from the CS-NPs in response to light of a suitable wavelength, resulting in the formation of highly reactive singlet oxygen or ROS, which can kill tumor cells. The therapeutic payload can also be released from CS-NPs in response to certain chemical stimuli such as pH, redox, or enzymes (eg, hyaluronidase). Chemical stimuli-responsive delivery systems can be designed by incorporating an adjuvant sensitive to certain stimuli in the architecture of CS-NPs or can be used as a linker to conjugate the chemotherapeutic drug with the backbone or on the surface of CS-NPs. When exposed to specific stimuli, the therapeutic payload is released because of deagglomeration of the amphiphilic chains of CS or breakage of the sensitive linker.
Passive targeting of chemotherapeutics can be successfully achieved because of the unique physicochemical properties of CS-NPs (nanoscale size, cationic zeta potential, high encapsulation efficiency, thermodynamic stability, biodegradability, and biocompatibility). By optimizing these properties, the aqueous solubility, plasma circulation time, tumor biodistribution and localization, and anticancer efficacy of chemotherapeutics can be maximized. In addition, various functionalization strategies (eg, PEGylation, hydrophobic substitution of CS, conjugation of LDL, and stealthing) have been adapted to the architecture of CS-NPs to further improve their pharmacokinetic parameters, deeper penetration into tumor tissues, stability in biological fluids, and anticancer efficacy.
Moreover, owing to their unique functionalities, CS-NPs have been conjugated with a variety of targeting ligands (eg, folic acid, hyaluronic acid, transferrin, antibodies, peptides, and aptamers) to enable selective intracellular delivery. To further enhance their targeting efficiency and anticancer potency, hybrid NPs can be synthesized by coupling multiple targeting mechanisms with CS-NPs architecture. Wang et al100 prepared hybrid CS-NPs via co-functionalization with PEGylation and folic acid, which extended their plasma circulation time and boosted their selective cellular uptake and cytotoxicity in A549 cells in a dose-dependent manner through FA receptor-mediated endocytosis. In summary, CS-NPs possess tremendous potential for targeted therapy of different types of cancer with better therapeutic outcomes than conventional treatments while mitigating their off-target events.
Despite holding remarkable anticancer potential, clinical development of CS-NPs based theranostics is limited due to several challenges including undesired interactions with non-specific molecules and biological tissues, biocompatibility and safety issues, large-scale manufacturing, ethical and regulatory restrictions, intellectual property (IP), and cost-effectiveness in comparison to current treatments. For instance, owing to their ultrasmall size and significantly higher surface area than traditionally formulated drugs, CS-NPs may interact with non-specific target molecules, cells, or tissues in ways that are not fully understood yet, making it difficult to predict and prevent their potential toxicity. In addition, behavior of NPs may significantly fluctuate based on their route of administration, which signifies testing of NPs before and after administration. For example, evaluation varies based on whether orally administered nanomedicines are found in nano forms or non-nano forms in the gastrointestinal tract. Hence, for successful clinical translation, a thorough in vitro/in vivo evaluations, establishment of safety and toxicity profiles, pharmacokinetic and pharmacodynamic studies, and excretion profile are ethical requirements that are mandatory.
Acknowledgments
This work was supported by SEED grant “Development and evaluation of Novel Multi-functionalized Nanomedicine for Early Diagnosis and efficient Treatment of Breast Cancer” and Postgraduate Research Fund provided by the University of Sharjah, United Arab Emirates.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Debela DT, Muzazu SG, Heraro KD, et al. New approaches and procedures for cancer treatment: current perspectives. SAGE Open Med. 2021;9:20503121211034366. doi:10.1177/20503121211034366
2. Scheel BI, Holtedahl K. Symptoms, signs, and tests: the general practitioner’s comprehensive approach towards a cancer diagnosis. Scand J Prim Health Care. 2015;33(3):170–177. doi:10.3109/02813432.2015.1067512
3. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48. doi:10.3322/caac.21763
4. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12–49. doi:10.3322/caac.21820
5. Scheel BI, Ingebrigtsen SG, Thorsen T, Holtedahl K. Cancer suspicion in general practice: the role of symptoms and patient characteristics, and their association with subsequent cancer. Br J Gen Pract. 2013;63(614):e627–35. doi:10.3399/bjgp13X671614
6. Anand U, Dey A, Chandel AKS, et al. Cancer chemotherapy and beyond: current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2022;10(4):1367–1401. doi:10.1016/j.gendis.2022.02.007
7. Benjamin DJ. The efficacy of surgical treatment of cancer - 20 years later. Med Hypotheses. 2014;82(4):412–420. doi:10.1016/j.mehy.2014.01.004
8. Franceschini G, Di Leone A, Natale M, Sanchez MA, Masett R. Conservative surgery after neoadjuvant chemotherapy in patients with operable breast cancer. Ann Ital Chir. 2018;89:290.
9. Wang W. Radiotherapy in the management of early breast cancer. J Med Radiat Sci. 2013;60(1):40–46. doi:10.1002/jmrs.1
10. Hou N, Hong S, Wang W, Olopade OI, Dignam JJ, Huo D. Hormone replacement therapy and breast cancer: heterogeneous risks by race, weight, and breast density. J National Cancer Inst. 2013;105(18):1365–1372. doi:10.1093/jnci/djt207
11. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–1717. doi:10.1016/S0140-6736(05)66544-0
12. Liu J, Fu M, Wang M, et al. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol. 2022;15(1):28. doi:10.1186/s13045-022-01247-x
13. Grimmett E, Al-Share B, Alkassab MB, et al. Cancer vaccines: past, present and future; a review article. Discov Onc. 2022;13(1):31. doi:10.1007/s12672-022-00491-4
14. Jin S, Sun Y, Liang X, et al. Emerging new therapeutic antibody derivatives for cancer treatment. Sig Transduct Target Ther. 2022;7:39.
15. Welch HG, Mogielnicki J. Presumed benefit: lessons from the American experience with marrow transplantation for breast cancer. BMJ. 2002;324(7345):1088–1092. doi:10.1136/bmj.324.7345.1088
16. Hoeben A, Joosten EAJ, van den Beuken-van Everdingen MHJ. Personalized medicine: recent progress in cancer therapy. Cancers. 2021;13(2):242. doi:10.3390/cancers13020242
17. Gambardella V, Tarazona N, Cejalvo JM, et al. Personalized medicine: recent progress in cancer therapy. Cancers. 2020;12(4):1009. doi:10.3390/cancers12041009
18. Hussain Z, Ei Thu H, Khan S, et al. Phytonanomedicines, a state-of-the-art strategy for targeted delivery of anti-inflammatory phytochemicals: a review of improved pharmacokinetic profile and therapeutic efficacy. J Drug Delivery Sci Technol. 2022;77:103895. doi:10.1016/j.jddst.2022.103895
19. Hanif S, Muhammad Sarfraz R, Ali Syed M, Mahmood A, Hussain Z. Smart mucoadhesive buccal chitosan/ HPMC scaffold for sore throat: in vitro, ex vivo and pharmacokinetic profiling in humans. J Drug Delivery Sci Technol. 2022;71:103271. doi:10.1016/j.jddst.2022.103271
20. Wang H, Yu X, Su C, Shi Y, Zhao L. Chitosan nanoparticles triggered the induction of ROS-mediated cytoprotective autophagy in cancer cells. Artif Cells Nanomed Biotechnol. 2018;46(sup1):293–301. doi:10.1080/21691401.2017.1423494
21. Jiang Y, Yu X, Su C, Zhao L, Shi Y. Chitosan nanoparticles induced the antitumor effect in hepatocellular carcinoma cells by regulating ROS-mediated mitochondrial damage and endoplasmic reticulum stress. Artif Cells Nanomed Biotechnol. 2019;47(1):747–756. doi:10.1080/21691401.2019.1577876
22. Bashir S, Aamir M, Muhammad Sarfaraz R, et al. Fabrication, characterization and in vitro release kinetics of tofacitinib-encapsulated polymeric nanoparticles: a promising implication in the treatment of rheumatoid arthritis. Int J Polym Mater Polym Biomater. 2020. doi:10.1080/00914037.2020.1725760
23. Hussain Z. Nanotechnology guided newer intervention for treatment of osteoporosis: efficient bone regeneration by up-regulation of proliferation, differentiation and mineralization of osteoblasts. Int J Polym Mater Polym Biomater. 2021;70(1):1–13. doi:10.1080/00914037.2019.1683558
24. Hussain Z, Khan S, Imran M, Sohail M, Shah SWA, de Matas M. PEGylation: a promising strategy to overcome challenges to cancer-targeted nanomedicines: a review of challenges to clinical transition and promising resolution. Drug Deliv Transl Res. 2019;9(3):721–734. doi:10.1007/s13346-019-00631-4
25. Fang G, Zhang Q, Pang Y, Thu HE, Hussain Z. Nanomedicines for improved targetability to inflamed synovium for treatment of rheumatoid arthritis: multi-functionalization as an emerging strategy to optimize therapeutic efficacy. J Control Release. 2019;303:181–208. doi:10.1016/j.jconrel.2019.04.027
26. Hussain Z, Pandey M, Thu HE, et al. Hyaluronic acid functionalization improves dermal targeting of polymeric nanoparticles for management of burn wounds: in vitro, ex vivo and in vivo evaluations. Biomed Pharmacother. 2022;150:112992. doi:10.1016/j.biopha.2022.112992
27. Sun Y, Davis E. Nanoplatforms for targeted stimuli-responsive drug delivery: a review of platform materials and stimuli-responsive release and targeting mechanisms. Nanomaterials. 2021;11(3):746. doi:10.3390/nano11030746
28. Rahim MA, Jan N, Khan S, et al. Recent advancements in stimuli responsive drug delivery platforms for active and passive cancer targeting. Cancers. 2021;13(4):670. doi:10.3390/cancers13040670
29. Lee SJ, Koo H, Jeong H, et al. Comparative study of photosensitizer loaded and conjugated glycol chitosan nanoparticles for cancer therapy. J Control Release. 2011;152(1):21–29. doi:10.1016/j.jconrel.2011.03.027
30. Lim CK, Shin J, Kwon IC, Jeong SY, Kim S. Iodinated photosensitizing chitosan: self-assembly into tumor-homing nanoparticles with enhanced singlet oxygen generation. Bioconjug Chem. 2012;23(5):1022–1028. doi:10.1021/bc300012g
31. Lee HM, Jeong YI, Kim DH, et al. Ursodeoxycholic acid-conjugated chitosan for photodynamic treatment of HuCC-T1 human cholangiocarcinoma cells. Int J Pharm. 2013;454(1):74–81. doi:10.1016/j.ijpharm.2013.06.035
32. Bae I, Kim TG, Kim T, et al. Phenethyl isothiocyanate-conjugated chitosan oligosaccharide nanophotosensitizers for photodynamic treatment of human cancer cells. Int J Mol Sci. 2022;23(22):13802. doi:10.3390/ijms232213802
33. Jeong YI, Cha B, Lee HL, et al. Simple nanophotosensitizer fabrication using water-soluble chitosan for photodynamic therapy in gastrointestinal cancer cells. Int J Pharm. 2017;532(1):194–203. doi:10.1016/j.ijpharm.2017.08.128
34. Al-Nemrawi N, Hameedat F, Al-Husein B, Nimrawi S. Photolytic controlled release formulation of methotrexate loaded in Chitosan/TiO2 nanoparticles for breast cancer. Pharmaceuticals. 2022;15(2):149. doi:10.3390/ph15020149
35. Choi Y, Han H, Jeon S, et al. Deep tumor penetration of doxorubicin-loaded glycol chitosan nanoparticles using high-intensity focused ultrasound. Pharmaceutics. 2020;12(10):974. doi:10.3390/pharmaceutics12100974
36. Nogueira-Librelotto DR, Scheeren LE, Vinardell MP, Mitjans M, Rolim CMB. Chitosan-tripolyphosphate nanoparticles functionalized with a pH-responsive amphiphile improved the in vitro antineoplastic effects of doxorubicin. Colloids Surf B Biointerfaces. 2016;147:326–335. doi:10.1016/j.colsurfb.2016.08.014
37. Nogueira DR, Scheeren LE, Macedo LB, et al. Inclusion of a pH-responsive amino acid-based amphiphile in methotrexate-loaded chitosan nanoparticles as a delivery strategy in cancer therapy. Amino Acids. 2016;48(1):157–168. doi:10.1007/s00726-015-2075-1
38. Nogueira-Librelotto DR, Scheeren LE, Macedo LB, Vinardell MP, Rolim CMB. pH-Sensitive chitosan-tripolyphosphate nanoparticles increase doxorubicin-induced growth inhibition of cervical HeLa tumor cells by apoptosis and cell cycle modulation. Colloids Surf B Biointerfaces. 2020;190:110897. doi:10.1016/j.colsurfb.2020.110897
39. Scheeren LE, Nogueira DR, Macedo LB, et al. PEGylated and poloxamer-modified chitosan nanoparticles incorporating a lysine-based surfactant for pH-triggered doxorubicin release. Colloids Surf B Biointerfaces. 2016;138:117–127. doi:10.1016/j.colsurfb.2015.11.049
40. Park JS, Han TH, Lee KY, et al. N-acetyl histidine-conjugated glycol chitosan self-assembled nanoparticles for intracytoplasmic delivery of drugs: endocytosis, exocytosis and drug release. J Control Release. 2006;115(1):37–45. doi:10.1016/j.jconrel.2006.07.011
41. Lee CM, Park JW, Kim J, Kim DW, Jeong HJ, Lee KY. Influence of histidine on the release of all-trans retinoic acid from self-assembled glycol chitosan nanoparticles. Drug Dev Ind Pharm. 2010;36(7):781–786. doi:10.3109/03639040903514812
42. Piroonpan T, Rimdusit P, Taechutrakul S, Pasanphan W. pH-responsive water-soluble chitosan amphiphilic core-shell nanoparticles: radiation-assisted green synthesis and drug-controlled release studies. Pharmaceutics. 2023;15(3):847. doi:10.3390/pharmaceutics15030847
43. Ahmadi Nasab N, Hassani Kumleh H, Beygzadeh M, Teimourian S, Kazemzad M. Delivery of curcumin by a pH-responsive chitosan mesoporous silica nanoparticles for cancer treatment. Artif Cells Nanomed Biotechnol. 2018;46(1):75–81. doi:10.1080/21691401.2017.1290648
44. Yoon K, Jung S, Ryu J, Park HJ, Oh HK, Kook MS. Redox-sensitive delivery of doxorubicin from nanoparticles of poly(ethylene glycol)-chitosan copolymer for treatment of drug-resistant oral cancer cells. Int J Mol Sci. 2023;24(18):13704. doi:10.3390/ijms241813704
45. Jeong YI, Kim T, Hwang EJ, et al. Reactive oxygen species-sensitive nanophotosensitizers of aminophenyl boronic acid pinacol ester conjugated chitosan-g-methoxy poly(ethylene glycol) copolymer for photodynamic treatment of cancer. Biomed Mater. 2020;15(5):055034. doi:10.1088/1748-605X/ab9bb2
46. Yoon HM, Kang MS, Choi GE, et al. Stimuli-responsive drug delivery of doxorubicin using magnetic nanoparticle conjugated poly(ethylene glycol)-g-chitosan copolymer. Int J Mol Sci. 2021;22(23):13169. doi:10.3390/ijms222313169
47. Yang JI, Lee HL, Yun JJ, et al. pH and redox-dual sensitive chitosan nanoparticles having methyl ester and disulfide linkages for drug targeting against cholangiocarcinoma cells. Materials. 2022;15(11):3795. doi:10.3390/ma15113795
48. Zhang J, Sun J, Li C, Qiao H, Hussain Z. Functionalization of curcumin nanomedicines: a recent promising adaptation to maximize pharmacokinetic profile, specific cell internalization and anticancer efficacy against breast cancer. J Nanobiotechnology. 2023;21(1):106. doi:10.1186/s12951-023-01854-x
49. Moti LAA, Hussain Z, Thu HE, Khan S, Sohail M, Sarfraz RM. Multi-functionalization, a promising adaptation to overcome challenges to clinical translation of nanomedicines as nano-diagnostics and nano-therapeutics for breast cancer. Curr Pharm Des. 2021;27(43):4356–4375. doi:10.2174/1381612827666210830092539
50. Xu J, Ma L, Liu Y, Xu F, Nie J, Ma G. Design and characterization of antitumor drug paclitaxel-loaded chitosan nanoparticles by W/O emulsions. Int J Biol Macromol. 2012;50(2):438–443. doi:10.1016/j.ijbiomac.2011.12.034
51. Gupta U, Sharma S, Khan I, et al. Enhanced apoptotic and anticancer potential of paclitaxel loaded biodegradable nanoparticles based on chitosan. Int J Biol Macromol. 2017;98:810–819. doi:10.1016/j.ijbiomac.2017.02.030
52. Jain A, Thakur K, Kush P, Jain UK. Docetaxel loaded chitosan nanoparticles: formulation, characterization and cytotoxicity studies. Int J Biol Macromol. 2014;69:546–553. doi:10.1016/j.ijbiomac.2014.06.029
53. Jain A, Thakur K, Sharma G, Kush P, Jain UK. Fabrication, characterization and cytotoxicity studies of ionically cross-linked docetaxel loaded chitosan nanoparticles. Carbohydr Polym. 2016;137:65–74. doi:10.1016/j.carbpol.2015.10.012
54. Elshami FI, Shereef HA, El-Mehasseb IM, Shaban SY, van Eldik R. Hydroxychloroquine-loaded chitosan nanoparticles induce anticancer activity in A549 lung cancer cells: design, BSA binding, molecular docking, mechanistic, and biological evaluation. Int J Mol Sci. 2023;24(18):14103. doi:10.3390/ijms241814103
55. Kumar SP, Birundha K, Kaveri K, Devi KT. Antioxidant studies of chitosan nanoparticles containing naringenin and their cytotoxicity effects in lung cancer cells. Int J Biol Macromol. 2015;78:87–95. doi:10.1016/j.ijbiomac.2015.03.045
56. Ramya Devi KT, Jaganathan MK, Ganesh MR, Dharshene K. Chitosan-encapsulated naringenin promotes ROS mediated through the activation of executioner caspase-3. Med Oncol. 2023;41(1):3. doi:10.1007/s12032-023-02227-y
57. Motawi TK, El-Maraghy SA, ElMeshad AN, Nady OM, Hammam OA. Cromolyn chitosan nanoparticles as a novel protective approach for colorectal cancer. Chem Biol Interact. 2017;275:1–12. doi:10.1016/j.cbi.2017.07.013
58. Arulmozhi V, Pandian K, Mirunalini S. Ellagic acid encapsulated chitosan nanoparticles for drug delivery system in human oral cancer cell line (KB. Colloids Surf B. 2013;110:313–320. doi:10.1016/j.colsurfb.2013.03.039
59. Chung KD, Jeong YI, Chung CW, Kim DH, Kang DH. Anti-tumor activity of all-trans retinoic acid-incorporated glycol chitosan nanoparticles against HuCC-T1 human cholangiocarcinoma cells. Int J Pharm. 2012;422(1–2):454–461. doi:10.1016/j.ijpharm.2011.10.057
60. Snima KS, Jayakumar R, Unnikrishnan AG, Shantikumar VN, Lakshmanan V-K. O-Carboxymethyl chitosan nanoparticles for metformin delivery to pancreatic cancer cells. Carbohydr Polym. 2012;89(3):1003–1007.
61. Zhang L, Hu Y. Alphastatin-loaded chitosan nanoparticle preparation and its antiangiogenic effect on lung carcinoma. Int J Polym Sci. 2019;2019:1–9.
62. David KI, Jaidev LR, Sethuraman S, Krishnan UM. Dual drug loaded chitosan nanoparticles—sugar-coated arsenal against pancreatic cancer. Colloids Surf B. 2015;135:689–698. doi:10.1016/j.colsurfb.2015.08.038
63. Khan MA, Zafaryab M, Mehdi SH, Ahmad I, Rizvi MMA. Characterization and anti-proliferative activity of curcumin loaded chitosan nanoparticles in cervical cancer. Int J Biol Macromol. 2016;93:242–253. doi:10.1016/j.ijbiomac.2016.08.050
64. Xie F, Ding R-L, He W-F, et al. In vivo antitumor effect of endostatin-loaded chitosan nanoparticles combined with paclitaxel on Lewis lung carcinoma. Drug Deliv. 2017;24(1):1410–1418. doi:10.1080/10717544.2017.1378938
65. Ahmad N, Khan MR, Palanisamy S, Mohandoss S. Anticancer drug-loaded chitosan nanoparticles for in vitro release, promoting antibacterial and anticancer activities. Polymers. 2023;15(19):3925. doi:10.3390/polym15193925
66. Matalqah SM, Aiedeh K, Mhaidat NM, Alzoubi KH, Al-Husein BA. Preparation of modified chitosan-based nanoparticles for efficient delivery of doxorubicin and/or cisplatin to breast cancer cells. Curr Cancer Drug Targets. 2022;22(2):133–141. doi:10.2174/1568009622666220126100532
67. Song S, Shim MK, Yang S, et al. All-in-one glycol chitosan nanoparticles for co-delivery of doxorubicin and anti-PD-L1 peptide in cancer immunotherapy. Bioact Mater. 2023;28:358–375. doi:10.1016/j.bioactmat.2023.05.016
68. Sharma A, Jyoti K, Bansal V, Kumar Jain U, Bhushan B, Madan J. Soluble telmisartan bearing poly (ethylene glycol) conjugated chitosan nanoparticles augmented drug delivery, cytotoxicity, apoptosis and cellular uptake in human cervical cancer cells. Mater Sci Eng. 2017;72:69–76. doi:10.1016/j.msec.2016.11.048
69. Chen J, Huang L, Lai H, et al. Methotrexate-loaded PEGylated chitosan nanoparticles: synthesis, characterization, and in vitro and in vivo antitumoral activity. Mol Pharm. 2014;11(7):2213–2223. doi:10.1021/mp400269z
70. Park JS, Koh YS, Bang JY, Jeong YI, Lee JJ. Antitumor effect of all-trans retinoic acid-encapsulated nanoparticles of methoxy poly(ethylene glycol)-conjugated chitosan against CT-26 colon carcinoma in vitro. J Pharm Sci. 2008;97(9):4011–4019. doi:10.1002/jps.21221
71. Jia M, Li Y, Yang X, et al. Development of both methotrexate and mitomycin C loaded PEGylated chitosan nanoparticles for targeted drug codelivery and synergistic anticancer effect. ACS Appl Mater Interfaces. 2014;6(14):11413–11423. doi:10.1021/am501932s
72. Hong GY, Jeong YI, Lee SJ, Lee E, Oh JS, Lee HC. Combination of paclitaxel- and retinoic acid-incorporated nanoparticles for the treatment of CT-26 colon carcinoma. Arch Pharm Res. 2011;34(3):407–417. doi:10.1007/s12272-011-0308-8
73. Jeong YI, Jin SG, Kim IY, et al. Doxorubicin-incorporated nanoparticles composed of poly(ethylene glycol)-grafted carboxymethyl chitosan and antitumor activity against glioma cells in vitro. Colloids Surf B Biointerfaces. 2010;79(1):149–155. doi:10.1016/j.colsurfb.2010.03.037
74. Jang MK, Jeong YI, Nah JW. Characterization and preparation of core-shell type nanoparticle for encapsulation of anticancer drug. Colloids Surf B Biointerfaces. 2010;81(2):530–536. doi:10.1016/j.colsurfb.2010.07.053
75. Ait Bachir Z, Huang Y, He M, et al. Effects of PEG surface density and chain length on the pharmacokinetics and biodistribution of methotrexate-loaded chitosan nanoparticles. Int J Nanomed. 2018;13:5657–5671. doi:10.2147/IJN.S167443
76. Wang Y, Qin F, Tan H, et al. pH-responsive glycol chitosan-cross-linked carboxymethyl-β-cyclodextrin nanoparticles for controlled release of anticancer drugs. Int J Nanomed. 2015;10:7359–7369. doi:10.2147/IJN.S91906
77. Zhou H, Yu W, Guo X, et al. Synthesis and characterization of amphiphilic glycidol-chitosan-deoxycholic acid nanoparticles as a drug carrier for doxorubicin. Biomacromolecules. 2010;11(12):3480–3486. doi:10.1021/bm100989x
78. Jin YH, Hu HY, Qiao MX, et al. pH-sensitive chitosan-derived nanoparticles as doxorubicin carriers for effective anti-tumor activity: preparation and in vitro evaluation. Colloids Surf B Biointerfaces. 2012;94:184–191. doi:10.1016/j.colsurfb.2012.01.032
79. Zhou H, Liu X, Guo X, et al. Synthesis and characterization of amphiphilic chitosan derivatives as a nano-carrier for paclitaxel delivery. J Control Release. 2011;152 Suppl 1:e124–5. doi:10.1016/j.jconrel.2011.08.166
80. Na JH, Lee SY, Lee S, et al. Effect of the stability and deformability of self-assembled glycol chitosan nanoparticles on tumor-targeting efficiency. J Control Release. 2012;163(1):2–9. doi:10.1016/j.jconrel.2012.07.028
81. Kim JH, Kim YS, Kim S, et al. Hydrophobically modified glycol chitosan nanoparticles as carriers for paclitaxel. J Control Release. 2006;111(1–2):228–234. doi:10.1016/j.jconrel.2005.12.013
82. Min KH, Park K, Kim YS, et al. Hydrophobically modified glycol chitosan nanoparticles-encapsulated camptothecin enhance the drug stability and tumor targeting in cancer therapy. J Control Release. 2008;127(3):208–218. doi:10.1016/j.jconrel.2008.01.013
83. Kim JH, Kim YS, Park K, et al. Antitumor efficacy of cisplatin-loaded glycol chitosan nanoparticles in tumor-bearing mice. J Control Release. 2008;127(1):41–49. doi:10.1016/j.jconrel.2007.12.014
84. Hwang HY, Kim IS, Kwon IC, Kim YH. Tumor targetability and antitumor effect of docetaxel-loaded hydrophobically modified glycol chitosan nanoparticles. J Control Release. 2008;128(1):23–31. doi:10.1016/j.jconrel.2008.02.003
85. Shen D, Chen H, Li M, et al. Effects of different molecular weight oxidized dextran as crosslinkers on stability and antioxidant capacity of curcumin-loaded nanoparticles. Foods. 2023;12(13):2533. doi:10.3390/foods12132533
86. Saravanakumar G, Min KH, Min DS, et al. Hydrotropic oligomer-conjugated glycol chitosan as a carrier of paclitaxel: synthesis, characterization, and in vivo biodistribution. J Control Release. 2009;140(3):210–217. doi:10.1016/j.jconrel.2009.06.015
87. Koo H, Min KH, Lee SC, et al. Enhanced drug-loading and therapeutic efficacy of hydrotropic oligomer-conjugated glycol chitosan nanoparticles for tumor-targeted paclitaxel delivery. J Control Release. 2013;172(3):823–831. doi:10.1016/j.jconrel.2013.08.297
88. Zhu QL, Zhou Y, Guan M, et al. Low-density lipoprotein-coupled N-succinyl chitosan nanoparticles co-delivering siRNA and doxorubicin for hepatocyte-targeted therapy. Biomaterials. 2014;35(22):5965–5976. doi:10.1016/j.biomaterials.2014.03.088
89. Tian J, Xu S, Deng H, et al. Fabrication of self-assembled chitosan-dispersed LDL nanoparticles for drug delivery with a one-step green method. Int J Pharm. 2017;517(1–2):25–34. doi:10.1016/j.ijpharm.2016.11.030
90. Madawi EA, Al Jayoush AR, Rawas-Qalaji M, et al. Polymeric nanoparticles as tunable nanocarriers for targeted delivery of drugs to skin tissues for treatment of topical skin diseases. Pharmaceutics. 2023;15(2):657. doi:10.3390/pharmaceutics15020657
91. Haider M, Elsherbeny A, Pittalà V, et al. Nanomedicine strategies for management of drug resistance in lung cancer. Int J Mol Sci. 2022;23(3):1853. doi:10.3390/ijms23031853
92. Haider M, Zaki KZ, El Hamshary MR, Hussain Z, Orive G, Ibrahim HO. Polymeric nanocarriers: a promising tool for early diagnosis and efficient treatment of colorectal cancer. J Adv Res. 2022;39:237–255. doi:10.1016/j.jare.2021.11.008
93. Hussain Z, Rahim MA, Jan N, et al. Cell membrane cloaked nanomedicines for bio-imaging and immunotherapy of cancer: improved pharmacokinetics, cell internalization and anticancer efficacy. J Control Release. 2021;335:130–157. doi:10.1016/j.jconrel.2021.05.018
94. Gao X, Guo L, Li J, Thu HE, Hussain Z. Nanomedicines guided nanoimaging probes and nanotherapeutics for early detection of lung cancer and abolishing pulmonary metastasis: critical appraisal of newer developments and challenges to clinical transition. J Control Release. 2018;292:29–57. doi:10.1016/j.jconrel.2018.10.024
95. Choudhury H, Pandey M, Chin PX, et al. Transferrin receptors-targeting nanocarriers for efficient targeted delivery and transcytosis of drugs into the brain tumors: a review of recent advancements and emerging trends. Drug Deliv Transl Res. 2018;8(5):1545–1563. doi:10.1007/s13346-018-0552-2
96. Khan S, Imran M, Tahir Butt T, et al. Curcumin based nanomedicines as efficient nanoplatform for treatment of cancer: new developments in reversing cancer drug resistance, rapid internalization, and improved anticancer efficacy. Trends Food Sci Technol. 2018;80:8–22. doi:10.1016/j.tifs.2018.07.026
97. Hussain Z, Arooj M, Malik A, et al. Nanomedicines as emerging platform for simultaneous delivery of cancer therapeutics: new developments in overcoming drug resistance and optimizing anticancer efficacy. Artif Cells Nanomed Biotechnol. 2018;46(sup2):1015–1024. doi:10.1080/21691401.2018.1478420
98. Safdar MH, Hussain Z, Abourehab MAS, Hasan H, Afzal S, Thu HE. New developments and clinical transition of hyaluronic acid-based nanotherapeutics for treatment of cancer: reversing multidrug resistance, tumour-specific targetability and improved anticancer efficacy. Artif Cells Nanomed Biotechnol. 2018;46(8):1967–1980. doi:10.1080/21691401.2017.1397001
99. Esfandiarpour-Boroujeni S, Bagheri-Khoulenjani S, Mirzadeh H, Amanpour S. Fabrication and study of curcumin loaded nanoparticles based on folate-chitosan for breast cancer therapy application. Carbohydr Polym. 2017;168:14–21. doi:10.1016/j.carbpol.2017.03.031
100. Wang F, Wang Y, Ma Q, Cao Y, Yu B. Development and characterization of folic acid-conjugated chitosan nanoparticles for targeted and controlled delivery of gemcitabinein lung cancer therapeutics. Artif Cells Nanomed Biotechnol. 2017;45(8):1530–1538. doi:10.1080/21691401.2016.1260578
101. Zamanvaziri A, Meshkat M, Alazmani S, Khaleghi S, Hashemi M. Targeted PEGylated chitosan nano-complex for delivery of sodium butyrate to prostate cancer: an in vitro study. Technol Cancer Res Treat. 2023;22:15330338231159223. doi:10.1177/15330338231159223
102. Liang J, Cao L, Zhang L, et al. Preparation, characterization, and in vitro antitumor activity of folate conjugated chitosan coated EGCG nanoparticles. Food Sci Biotechnol. 2014;23(2):569–575. doi:10.1007/s10068-014-0078-4
103. Wang W, Tong C, Liu X, et al. Preparation and functional characterization of tumor-targeted folic acid-chitosan conjugated nanoparticles loaded with mitoxantrone. J Cent South Univ. 2015;22(9):3311–3317. doi:10.1007/s11771-015-2871-5
104. Shi Z, Guo R, Li W, et al. Nanoparticles of deoxycholic acid, polyethylene glycol and folic acid-modified chitosan for targeted delivery of doxorubicin. J Mater Sci Mater Med. 2014;25(3):723–731. doi:10.1007/s10856-013-5113-0
105. Mi Y, Zhang J, Tan W, Miao Q, Li Q, Guo Z. Preparation of doxorubicin-loaded carboxymethyl-β-cyclodextrin/chitosan nanoparticles with antioxidant, antitumor activities and ph-sensitive release. Mar Drugs. 2022;20(5):278. doi:10.3390/md20050278
106. Dramou P, Fizir M, Taleb A, et al. Folic acid-conjugated chitosan oligosaccharide-magnetic halloysite nanotubes as a delivery system for camptothecin. Carbohydr Polym. 2018;197:117–127. doi:10.1016/j.carbpol.2018.05.071
107. Al-Musawi S, Jawad A, Hadi S, Hindi N. Preparation and characterization of folated chitosan/magnetic nanocarrier for 5-fluorouracil drug delivery and studying its effect in bladder cancer therapy. J Glob Pharma Technol. 2019;11:628–637.
108. Liang Y, Wang Y, Wang L, et al. Self-crosslinkable chitosan-hyaluronic acid dialdehyde nanoparticles for CD44-targeted siRNA delivery to treat bladder cancer. Bioact Mater. 2021;6(2):433–446. doi:10.1016/j.bioactmat.2020.08.019
109. Kousar K, Naseer F, Abduh MS, et al. Green synthesis of hyaluronic acid coated, thiolated chitosan nanoparticles for CD44 targeted delivery and sustained release of Cisplatin in cervical carcinoma. Front Pharmacol. 2023;13:1073004. doi:10.3389/fphar.2022.1073004
110. Kousar K, Naseer F, Abduh MS, Anjum S, Ahmad T. CD44 targeted delivery of oncolytic Newcastle disease virus encapsulated in thiolated chitosan for sustained release in cervical cancer: a targeted immunotherapy approach. Front Immunol. 2023;14:1175535. doi:10.3389/fimmu.2023.1175535
111. Abduh MS. Anticancer analysis of CD44 targeted cyclosporine loaded thiolated chitosan nanoformulations for sustained release in triple-negative breast cancer. Int J Nanomed. 2023;18:5713–5732. doi:10.2147/IJN.S424932
112. Yang H, Bremner DH, Tao L, Li H, Hu J, Zhu L. Carboxymethyl chitosan-mediated synthesis of hyaluronic acid-targeted graphene oxide for cancer drug delivery. Carbohydr Polym. 2016;135:72–78. doi:10.1016/j.carbpol.2015.08.058
113. Li H, Zhuang S, Yang Y, Zhou F, Rong J, Zhao J. ATP/Hyals dually responsive core-shell hyaluronan/chitosan-based drug nanocarrier for potential application in breast cancer therapy. Int J Biol Macromol. 2021;183:839–851. doi:10.1016/j.ijbiomac.2021.05.020
114. Zhang X, Niu S, Williams GR, et al. Dual-responsive nanoparticles based on chitosan for enhanced breast cancer therapy. Carbohydr Polym. 2019;221:84–93. doi:10.1016/j.carbpol.2019.05.081
115. Qian Q, Niu S, Williams GR, Jianrong W, Zhang X, Zhu L-M. Peptide functionalized dual-responsive chitosan nanoparticles for controlled drug delivery to breast cancer cells. Colloids Surf A. 2019;564:122–130. doi:10.1016/j.colsurfa.2018.12.026
116. Das Kurmi B, Paliwal R, Paliwal SR. Dual cancer targeting using estrogen functionalized chitosan nanoparticles loaded with doxorubicin-estrone conjugate: a quality by design approach. Int J Biol Macromol. 2020;164:2881–2894. doi:10.1016/j.ijbiomac.2020.08.172
117. Xu Y, Asghar S, Yang L, et al. Lactoferrin-coated polysaccharide nanoparticles based on chitosan hydrochloride/hyaluronic acid/PEG for treating brain glioma. Carbohydr Polym. 2017;157:419–428. doi:10.1016/j.carbpol.2016.09.085
118. Anandhakumar S, Krishnamoorthy G, Ramkumar KM, Raichur AM. Preparation of collagen peptide functionalized chitosan nanoparticles by ionic gelation method: an effective carrier system for encapsulation and release of doxorubicin for cancer drug delivery. Mater Sci Eng C Mater Biol Appl. 2017;70(Pt 1):378–385. doi:10.1016/j.msec.2016.09.003
119. Arya G, Vandana M, Acharya S, Sahoo SK. Enhanced antiproliferative activity of Herceptin (HER2)-conjugated gemcitabine-loaded chitosan nanoparticle in pancreatic cancer therapy. Nanomedicine. 2011;7(6):859–870. doi:10.1016/j.nano.2011.03.009
120. Sun R, Fang L, Lv X, et al. In vitro and in vivo evaluation of self-assembled chitosan nanoparticles selectively overcoming hepatocellular carcinoma via asialoglycoprotein receptor. Drug Deliv. 2021;28(1):2071–2084. doi:10.1080/10717544.2021.1983077
121. Tian Q, Zhang CN, Wang XH, et al. Glycyrrhetinic acid-modified chitosan/poly(ethylene glycol) nanoparticles for liver-targeted delivery. Biomaterials. 2010;31(17):4748–4756. doi:10.1016/j.biomaterials.2010.02.042
122. Garg NK, Dwivedi P, Campbell C, Tyagi RK. Site specific/targeted delivery of gemcitabine through anisamide anchored chitosan/polyethylene glycol nanoparticles: an improved understanding of lung cancer therapeutic intervention. Eur J Pharm Sci. 2012;47(5):1006–1014. doi:10.1016/j.ejps.2012.09.012
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.