")
Back to Journals » Journal of Inflammation Research » Volume 18
Chronic Stress Mediates Inflammatory Cytokines Alterations and Its Role in Tumorigenesis
Received 11 August 2024
Accepted for publication 6 January 2025
Published 22 January 2025 Volume 2025:18 Pages 1067—1090
DOI https://doi.org/10.2147/JIR.S485159
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Zhihan Liu,* Meng Lei,* Yanxia Bai
Department of Otorhinolaryngology-Head & Neck Surgery, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanxia Bai, Department of Otorhinolaryngology-Head & Neck Surgery, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China, Tel +86 85323965, Email [email protected]
Introduction: Prolonged psychological stress is closely associated with cancer due to its role in promoting the release of stress hormones through the sustained activation of the sympathetic-adrenal-medullary system. These hormones interact with receptors on inflammatory cells, leading to the activation of key signaling pathways, including the transcription factors signal transducer and activator of transcription 3 (STAT-3) and kappa-light-chain-enhancer of activated B cells (NF-κB). These factors drive the production of pro-inflammatory substances, such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), which can influence the initiation and progression of cancer.
Purpose: This article aims to summarize how the chronic inflammatory environment induced by chronic stress promotes the initiation, progression, and invasion of cancer. By enhancing our understanding of the complex mechanisms through which stress contributes to cancer, we hope to identify new targets for cancer prevention and treatment.
Conclusion: Chronic stress establishes an inflammatory microenvironment by activating STAT-3 and NF-κB in inflammatory cells. This ongoing inflammation further enhances the activity of these transcription factors, which serve multiple roles: they act as pro-inflammatory agents in inflammatory cells, maintaining chronic inflammation; as oncogenic transcription factors in premalignant cells, promoting cancer initiation; and as pro-differentiation transcription factors in tumor-infiltrating immune cells, facilitating cancer progression. Additionally, the impact of chronic stress varies among different cancer types and individual responses to stress, highlighting the complexity of stress-related cancer mechanisms. Ultimately, this dynamic interplay creates a feedback loop involving IL-6, STAT-3, and TNF-α-NF-κB within the tumor microenvironment, mediating the intricate interactions between inflammation, immunity, and cancer.
Keywords: inflammation, cancer, IL-6, TNF-α, STAT-3, NF-κB
Graphical Abstract:

Introduction
Numerous studies have demonstrated that chronic stress is a significant risk factor for cancer, contributing to its initiation, progression, and metastasis.1,2 Chronic stress mediates the release of pro-inflammatory cytokines, such as interleukin (IL)-6 and tumor necrosis factor-alpha (TNF-α), by activating pro-inflammatory transcription factors (TFs) within inflammatory cells. This process contributes to the chronic inflammatory microenvironment (IME), which is characterized by a prolonged pro-inflammatory state in the human body and serves as an important marker of cancer. Epidemiological evidence suggests a strong link between chronic inflammation and cancer. Reports indicate that 15%-20% of cancers are directly triggered by chronic inflammation in the same tissue or organ that precedes cancer initiation. The released pro-inflammatory cytokines further activate TFs in both cancer cells and immune cells infiltrating the tumor environment, amplifying the effects of pro-inflammatory TFs, pro-oncogenic TFs, and pro-differentiation TFs. This creates an autocrine cycle of pro-inflammatory cytokines, leading to a chronic inflammatory tumor microenvironment (TME) that promotes cancer progression.3,4 From the initiation of cancer to diagnosis and treatment, patients often remain in a chronic state of stress for an extended period. This article reviews the mechanisms by which pro-inflammatory TFs induced by chronic stress mediate the secretion of various pro-inflammatory factors. It explores the cycles created by the multiple effects of corresponding TFs in the initiation and progression of cancers, aiming to provide new research targets and therapeutic directions related to stress management in cancer care. (Figure 1)
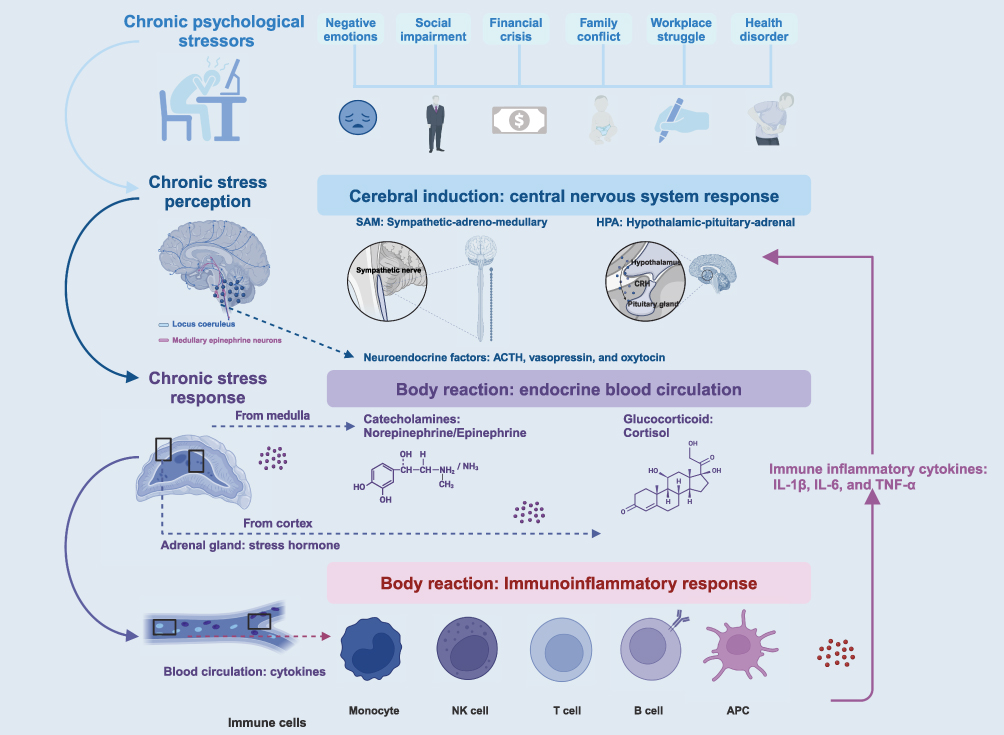 |
Figure 1 Chronic Stress Produces an Axis of Stress Hormones. Stress is a complex reaction in which a stimulus (stressor) activates a brain response (stress perception), leading to a physiological fight or flight reaction (stress response). Chronic stress triggers a sustained stress response mediated by intricate interactions among the nervous, endocrine, and immune systems. |
Mechanism and Physiological Effects of Chronic Stress
Stress is a pervasive and familiar aspect of daily life. For some individuals, it serves as a stimulant; however, for many others, it becomes a burden. In 1936, Selye first described stress as a general adaptation syndrome, which he divided into three phases: alarm response, resistance, and collapse.5 Stress triggers a response mediated by complex interactions among the nervous, endocrine, and immune systems. This physiological response involves both rapid responses, primarily mediated by the sympathetic-adrenomedullary (SAM) axis, and slow responses mediated by the hypothalamic-pituitary-adrenal (HPA) axis within the central nervous system. The signals from the SAM and HPA axes form the body’s overall stress response, which can be classified into acute and chronic stress based on the duration of biological effects. Chronic stress refers to stress that persists for weeks, months, or even years. It is important to note that while the SAM axis plays a significant role in the body’s immediate stress response, its sustained activation occurs during chronic stress. This prolonged activation leads to the continuous release of stress hormones. Specifically, chronic stress results in persistent activation of the SAM axis, which increases the production of norepinephrine (NE) by sympathetic nerve endings and promotes the secretion of both NE and epinephrine (E) by the adrenal medulla. These hormones bind to β-adrenergic receptors (β-ARs) located in various organs throughout the body, prompting the conversion of ATP to cyclic AMP (cAMP). cAMP then activates downstream effectors, including protein kinase A (PKA) and exchange proteins directly activated by cAMP (EPAC). PKA primarily plays a role in inflammation, angiogenesis, and cell invasion processes, while EPAC mainly influences cell morphology, movement, and changes in secretion kinetics. Over time, the sustained release of these cellular molecules and inflammatory factors can adversely affect health. This includes impaired immune function, increased inflammation, and an elevated risk of various stress-related diseases that exceed an individual’s ability to adapt, a phenomenon known as adaptive loading. (Figure 2)
 |
Figure 2 Chronic Stress Primarily Activates SAM-β-AR-cAMP-PKA/EPAC Axis. The persistent activation of the sympathoadrenal medullary (SAM) axis due to chronic stress results in increased production of norepinephrine (NE) by sympathetic nerve endings, as well as the production of both NE and epinephrine (E) by the adrenal medulla. Both NE and E bind to β-adrenergic receptors (β-ARs) located on the cells of various organs throughout the body. This binding acts as a first messenger, initiating the conversion of ATP to cyclic adenosine monophosphate (cAMP). Subsequently, cAMP functions as a secondary messenger in the inflammatory response by targeting downstream effectors, such as protein kinase A (PKA) and exchange protein directly activated by cAMP (EPAC). |
Chronic Stress Promotes the Formation and Maintenance of Chronic IME
Chronic stress activates the cAMP-PKA/EPAC signaling pathway, which primarily mediates the activation of two key transcription factors (TFs): signal transducer and activator of transcription 3 (STAT-3) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). These pro-inflammatory TFs are present in inflammatory and immune cells. Their activation results in the upregulation of promoter activity for target genes containing binding sites for these TFs, leading to an increased secretion of pro-inflammatory cytokines, and contributing to the persistence of chronic IME. In addition to STAT-3 and NF-κB, the cAMP-PKA/EPAC pathway regulates numerous other inflammation-associated TFs that collectively enhance the expression of pro-inflammatory cytokines, further sustaining the long-term presence of chronic IME.6–15 (Table 1, Figure S1)
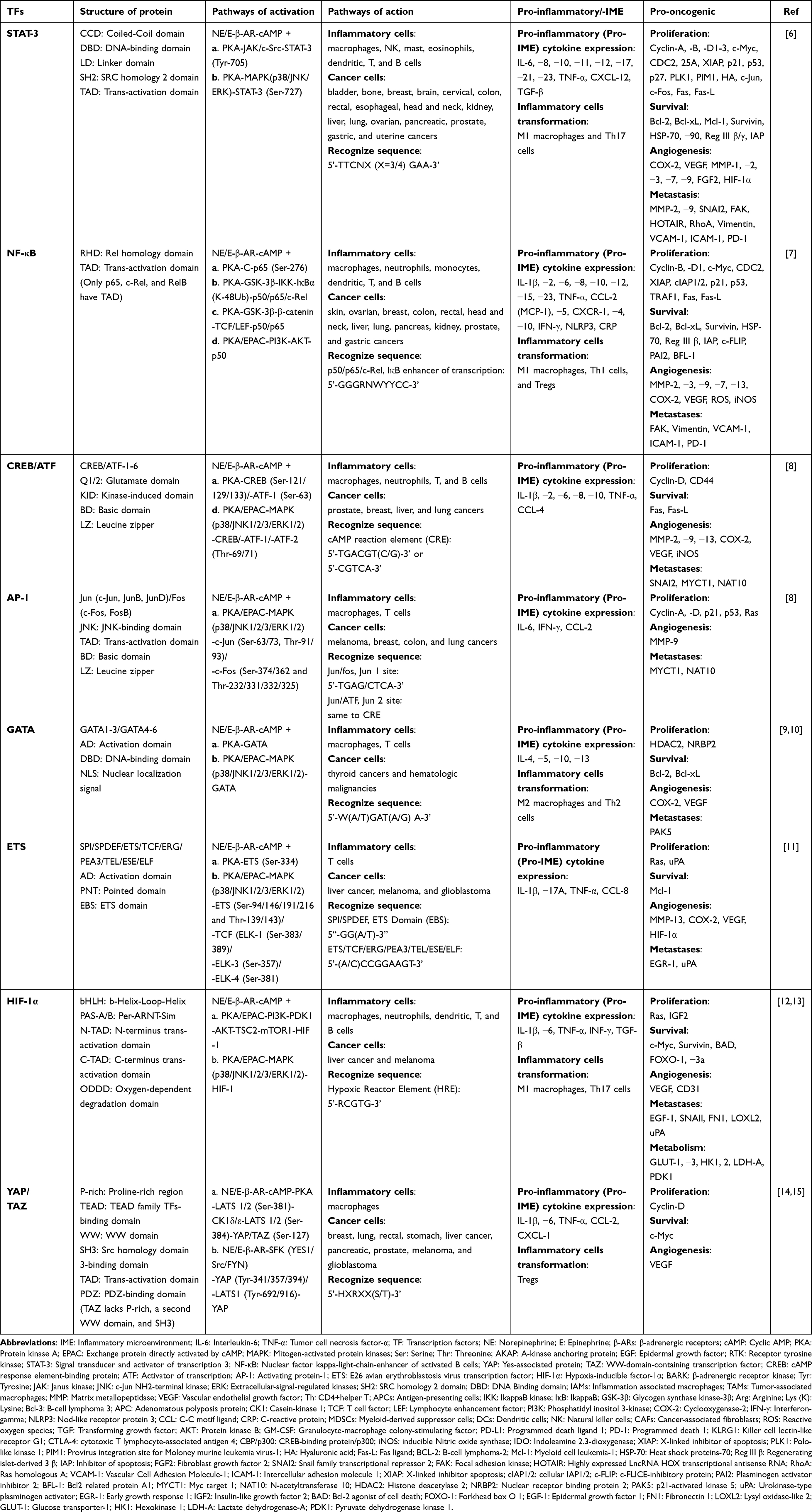 |
Table 1 Pro-Inflammatory and Pro-Oncogenic Effects of Chronic Stress-Activated Pro-Inflammatory Transcription Factors |
Chronic Stress Activates the Pro-Inflammatory TF STAT-3
NE and E have been shown to activate STAT-3 through the β-AR-cAMP-PKA pathway, independent of cytokines such as interleukin-6 (IL-6). This activation facilitates the nuclear translocation of STAT-3. The STAT-3 protein serves a dual role within cells: it acts as a messenger between the cell surface and the nucleus while also playing a direct role in transcriptional regulation.
Chronic Stress Activates STAT-3 Through the cAMP-PKA Pathway
Under chronic stress, cAMP-PKA signaling is activated, which directly or indirectly regulates STAT-3 signal transduction by modulating its phosphorylation state. When extracellular ligands bind to cell membrane receptors, STAT-3 can be phosphorylated directly by PKA in the cytoplasm, or indirectly through the activation of the β-adrenergic receptor kinase (BARK)-c-Src pathway located on the cell membrane. This phosphorylation occurs at two critical sites on STAT-3. The first is the tyrosine (Tyr)-705 site, which is phosphorylated by the non-receptor tyrosine kinase c-Src or Janus kinase (JAK) and is essential for STAT-3 activation. To achieve maximum transcriptional activity, secondary phosphorylation of STAT-3 at the serine (Ser)-727 site is also required, involving p38, c-Jun NH2-terminal kinase (JNK 1/2/3), and extracellular signal-regulated kinases (ERK 1/2) within the mitogen-activated protein kinase (MAPK) cascade. Once phosphorylated,16,17 STAT-3 can form either heterologous or homologous dimers, thereby inducing transcription of corresponding target genes.18
Activated STAT-3 as a Key TF Mediating IME Production
Activated STAT-3 is a critical pro-inflammatory TF that triggers the secretion of pro-inflammatory cytokines by the innate immune cells. Macrophages, as highly plastic innate immune cells, can be classified into two types based on their activation: classically activated (M1) macrophages and alternatively activated (M2) macrophages. They can also be categorized into inflammation-associated macrophages (IAMs) in the context of IME and tumor-associated macrophages (TAMs) in the context of TME based on their distribution. Under the influence of STAT-3 activated by chronic stress, macrophages are often transformed into pro-inflammatory M1 macrophages. These cells play a vital role in IME by secreting various pro-inflammatory cytokines, thereby contributing to the formation and maintenance of chronic inflammation.19,20 STAT-3 remains consistently activated as a pro-inflammatory TF in response to chronic stress, binding directly to genes encoding pro-inflammatory cytokines in M1 macrophages to initiate the transcription of corresponding target genes.
In addition to its role in innate immunity, activated STAT-3 serves as a key pro-differentiation TF that facilitates the differentiation of adaptive immune cells into inflammatory phenotypes. T lymphocytes, the predominant adaptive immune cells in IME, can be classified into CD8+ cytotoxic T (CTL) cells and CD4+ T helper (Th) cells based on their effector functions. Naive CD4+ T cells are activated by two co-stimulatory signals from antigen-presenting cells (APCs) and subsequently differentiate into five major effector T cell subtypes. Among these subtypes, Th1 and Th17 cells are particularly associated with various inflammatory diseases and are generally recognized as inflammatory T cells.21 STAT-3 is essential for mediating the differentiation of naive CD4+ T cells into these inflammatory T cells. Chronic stress induces the high expression of downstream Th cell-specific TFs, including the related orphan receptor-γt (ROR-γt) and related orphan receptor-α (ROR-α), by activating STAT-3 in naive Th cells. ROR-γt acts as the main switch in Th17 cell differentiation, directly regulating the continuous expression of ROR-α and indirectly upregulating the expression of effector cytokines such as IL-17A, IL-17F, IL-21, IL-22, and IL-23. These cytokines are essential for Th17 cell differentiation and serve as key drivers of chronic inflammation. Additionally, STAT-3 can bind directly to the gene promoters of these Th17 hallmark cytokines, marking a critical step required to initiate the differentiation of naive Th cells into Th17 cells.22 Through these mechanisms, the effector molecules associated with Th17 cells sustain a stable production of Th17 cells and influence the differentiation pathway of Th cells, as well as the expression of Th cell-specific cytokines. This creates a feedback loop that regulates the immune function of T cells in the context of IME.23
Chronic Stress Activates the Pro-Inflammatory TF NF-κB
NE and E can also activate NF-κB through the β-AR-cAMP-PKA pathway, independent of cytokines such as TNF-α, thereby promoting its nuclear translocation. NF-κB comprises five structurally similar family members: NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), RelB, and c-Rel.24 These members bind to DNA promoter or enhancer elements as homodimers or heterodimer complexes, with the p65/p50 dimers being the most common NF-κB pro-inflammatory TFs activated by chronic stress. The cAMP-activated protein kinase (PKA) not only directly regulates the transcriptional activity of NF-κB but also indirectly influences the NF-κB signaling pathway by modulating the activity of key metabolic enzymes involved in this pathway.25
Chronic Stress Activates NF-κB Through the cAMP-PKA/EPAC Pathway
Chronic stress primarily activates NF-κB through the cAMP-PKA pathway. Firstly, cAMP-PKA, activated by chronic stress, can directly influence NF-κB-related signal transduction by regulating the phosphorylation state of NF-κB and its inhibitor, I kappa B (IκBα), within cells. Secondly, chronic stress impacts NF-κB signaling by modulating the phosphorylation of glycogen synthase kinase-3β (GSK-3β), a critical component of the NF-κB pathway. In resting cells, GSK-3β exhibits consistently high basal activity due to its susceptibility to autophosphorylation at the Tyr-216 site. However, when cAMP levels rise, GSK-3β activity is significantly affected by inhibitory phosphorylation at the Ser-9 site, which leads to GSK-3β ubiquitination (Ub) and proteasomal degradation.26,27 GSK-3β directly influences NF-κB activity by phosphorylating it; for example, it inhibits the transcriptional activation of p65 by phosphorylating the Ser-468 site of p65. Furthermore, GSK-3β can indirectly regulate NF-κB activity by influencing the phosphorylation status of the IκB kinase (IKK) complex. When IKK phosphorylates IκB, it becomes a target for ubiquitin-dependent degradation, resulting in the lysine (Lys/K)48-Ub degradation of IκB. Then, the typical of the NF-κB family is released to bind to a specific DNA sequence.28 In resting cells, GSK-3β generally acts as a negative regulator of the NF-κB pathway, inhibiting IKK’s phosphorylation of IκBα. Additionally, GSK-3β can affect NF-κB activity through its interactions with Wnt/β-catenin signaling, creating a complex network where NF-κB, GSK-3β, β-catenin, and their target gene products can influence each other. Chronic stress also activates NF-κB via the β-AR-cAMP-EPAC pathway. For example, in prostate cancer cells, the vasoactive intestinal peptide (VIP) has been shown to rely on the cAMP-EPAC-phosphatidylinositol 3-kinase (PI3K)-ERK1/2 signaling pathway to mediate the transactivation and nuclear translocation of p50, thereby increasing the synthesis of inflammation-related cytokines.29
Activated NF-κB as a Key TF Mediating IME Production
Activated NF-κB is a crucial pro-inflammatory TF that triggers the secretion of pro-inflammatory cytokines by innate immune cells. Studies have shown that chronic restraint stress and social frustration stress increase the levels of various pro-inflammatory cytokines through the activation of NF-κB.30,31 As innate immune cells, macrophages, dendritic cells, and neutrophils participate in the inflammatory response via a common signaling event, which is typical of NF-κB activation. Activated NF-κB plays an important role in chronic inflammation by inducing the production of pro-inflammatory cytokines, chemokines, and other inflammatory mediators.32 Chronic stress promotes the transformation of macrophages into pro-inflammatory M1 macrophages by activating the p65/p50 heterodimer, which acts as a pro-differentiation factor in macrophages and sustains the long-term presence of IME in tissues.
Activated NF-κB is also key in facilitating the differentiation of adaptive immune cells into inflammatory phenotypes, particularly regarding the proliferation and differentiation of T lymphocytes. When naive CD4+ T cells differentiate into inflammatory T cells, this process is regulated by pro-differentiation cytokines secreted by APCs and M1 macrophages, alongside the intrinsic activation of NF-κB within T cells. For instance, the c-Rel subunit of NF-κB indirectly influences Th1 cell differentiation by modulating the secretion of IL-12 cytokines in APCs, which in turn affects Th1 cell progression. In addition to its distinct pro-inflammatory role, NF-κB collaborates with the Wnt/β-catenin signaling pathway to further promote chronic inflammation. Anson et al observed that oncogenic activation of Wnt/β-catenin signaling significantly triggers the constitutive activation of the inflammatory NF-κB program in mouse hepatocytes.33 However, in a liver inflammation model associated with liver cancer, NF-κB does not mediate the activation of classical liver inflammation-related cytokines such as IL-1β, IL-6, and TNF-α, as documented in previous studies. Instead, NF-κB mediates the expression of specific pro-inflammatory cytokines, including C-C motif ligands (CCL) 2 and 5, as well as IL-15, under the influence of β-catenin activation. Together, these pathways construct an IME that supports cancer progression.33 (Figure 3)
 |
Figure 3 Chronic Stress-Mediated Activation of Pro-Inflammatory Transcription Factors Leads to Over-Secretion of Pro-Inflammatory Cytokines. Chronic stress increases the transcription of multiple pro-inflammatory transcription factors (TFs) in inflammatory cells through the action of stress hormones. The resultant pro-inflammatory factors are involved in the development and maintenance of chronic inflammation. |
Chronic Inflammation Promotes the Formation of Inflammation-Associated TME
Cancer formation is a continuous process, and the role of chronic stress in this context is still unclear because of individual differences, cancer heterogeneity, and variations among cancer types. Chronic inflammation is a non-specific and complex biological response of the immune system to damage caused by chronic stress. The presence of white blood cells, first observed in cancers by Rudolf Virchow in the 19th century, marked an early indication of a connection between inflammation and cancer.34 A recent population-based cohort study revealed a clear relationship between chronic biological stress—assessed through levels of hair cortisol and hair cortisone—and cancer incidence, after adjusting for potential confounding factors.35 Additionally, another study indicated that chronic stress can worsen the progression of colorectal cancer by decreasing the presence of Lactobacillus johnsonii and its metabolites, underscoring the connection between mental health and colorectal cancer outcomes.36 These studies, validated through animal models and biochemical markers, suggest that chronic stress plays a dual role in both the initiation and progression of cancer. Some research indicates that chronic inflammation can promote the progression of cancer by activating dormant premalignant lesions. Other findings show that chronic inflammation exacerbates cancer progression and affects the biological behaviors of tumor cells, including growth, differentiation, and metastasis. These behaviors significantly depend on direct cellular contact between tumor cells and non-tumor cells (such as inflammatory cells, immune cells, and stromal cells) within the TME. Furthermore, they involve complex dynamic interactions mediated by bioactive cytokine signaling occurring in a paracrine or autocrine manner. The relationship between chronic inflammation and tumors is not a simple linear pathway; instead, it forms a multifaceted, complex, self-regulating, and reinforced feedback loop. (Table 2)
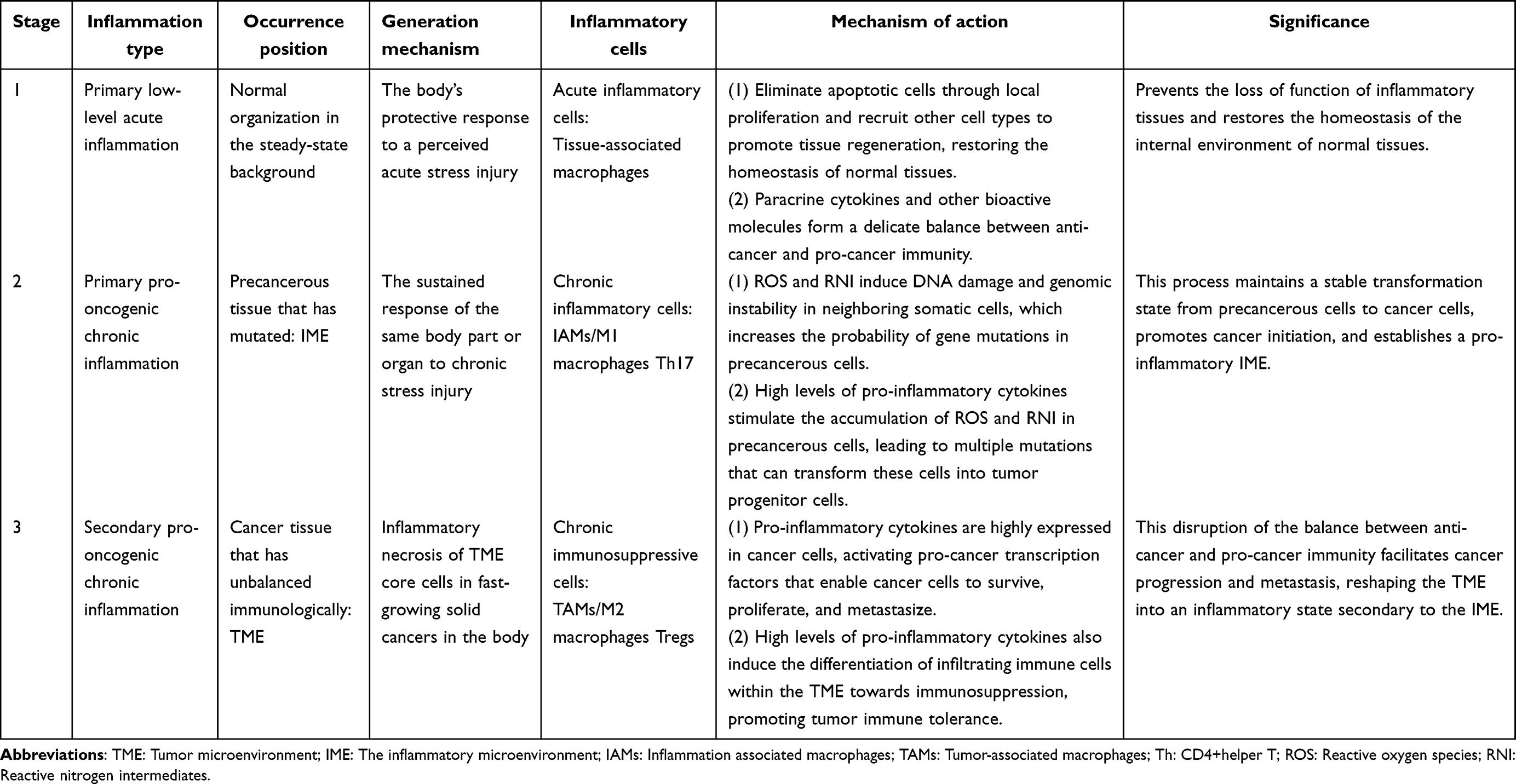 |
Table 2 Classification of Chronic Stress-Mediated Inflammation and Its Role in Cancer Initiation, Progression, and Metastasis |
The IL-6-STAT-3 Positive Feedback Loop Promotes Cancer Initiation and Progression
Chronic stress primarily induces the production of the pro-inflammatory cytokine IL-6 by activating inflammation-related TFs such as STAT-3 and NF-κB in inflammatory cells, thereby triggering the formation of an IL-6-STAT-3 inflammatory feedback loop.37 Under normal physiological conditions, the serum IL-6 level is less than 10 pg/mL, while the salivary IL-6 level is approximately 10 pg/mL. IL-6 is expressed at high levels in both the serum and TME of cancer patients, originating from cancer-infiltrated immune cells (including TAMs, myeloid-derived suppressor cells (MDSCs), monocytes, and granulocytes), stromal cells (such as fibroblasts and adipocytes), and production by cancer cells themselves.38,39 The level of circulating IL-6 in patients with colorectal, cervical, ovarian, breast, and lung cancers is closely related to tumor size, stage, and metastasis, and it has become a prognostic indicator of survival status as well as a predictor of treatment response.40
The receptor complex that mediates the biological activity of IL-6 consists of IL-6 binding to both IL-6R and gp130. IL-6R can be classified into two types: membrane-bound IL-6R (mIL-6R) and soluble IL-6R (sIL-6R). mIL-6R is expressed on the cell membranes of cancer-infiltrating immune cells, including macrophages, monocytes, neutrophils, B cells, and T cells, and is involved in activating the intracellular IL-6 canonical pathway.41 In contrast, sIL-6R is primarily produced by the shedding of mIL-6R-expressing cells and plays a role in activating the IL-6 trans-signaling pathway in cancer cells that have low or no expression of mIL-6R.
Following IL-6 binding, gp130 serves as a docking site for proteins that initiate intracellular signaling pathways. This process activates three main signal transduction pathways: the JAK-STAT-3 pathway, the MAPK pathway (comprising p38, JNK, and ERK), and the PI3K-protein kinase B (AKT) pathway. The IL-6-STAT-3 positive feedback loop in cancer can be categorized into three main types. (Figure 4)
 |
Figure 4 Chronic Inflammation-Mediated Positive Feedback Loop of Pro-Inflammatory Factors Promotes the Initiation and Progression of Cancers. Interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), two key pro-inflammatory cytokines, create an autocrine positive feedback loop among tumor-associated macrophages (TAMs), cancer cells, and immune cells. This loop operates by activating their respective pro-inflammatory TFs and influencing surrounding cells in the tumor microenvironment (TME) through paracrine signaling. These TFs also function as pro-oncogenic TFs in cancer cells, promoting their proliferation, survival, angiogenesis, and metastasis. Additionally, these TFs serve as pro-differentiation TFs in immune cells, enhancing their differentiation and the secretion of signature cytokines. |
IL-6 in the IME Leads to Over-Activation of STAT-3 in Precancerous Cells to Promote Cancer Initiation
IL-6 within the IME promotes the over-activation of the pro-oncogenic TF STAT-3 in precancerous cells through a paracrine pathway, generating an IL-6 positive feedback loop that mediates cancer initiation. In the IME formed by chronic stress, IAMs, which are the main source of IL-6, can bind to mIL-6R on the precancerous cell membrane through the paracrine pathway. This binding triggers the transient phosphorylation and dimerization of STAT-3 at Tyr-705 by recruiting Janus kinase (JAK), promoting the translocation of STAT-3 to the nucleus, and initiating the transcription of the corresponding target genes.42 This IL-6-classical pathway plays a crucial role in the transformation of precancerous cells into cancer cells.43 However, because mIL-6R is expressed only in a limited subset of cells, most STAT-3 activation in precancerous cells is mediated by the IL-6 trans-signaling pathway. To maintain a stable state of transformation, STAT-3 must also act as a pro-inflammatory TF, promoting the continued production of IL-6 within precancerous cells. This autocrine IL-6 production subsequently induces further activation of STAT-3 through the aforementioned mechanisms, establishing an IL-6-STAT-3 positive inflammatory feedback loop. Ultimately, precancerous cells undergo an epigenetic transformation that positions them between untransformed normal cells and transformed cancer cells, stabilizing into motile and aggressive tumorigenic forms.44 This inflammatory feedback regulatory loop initiates epigenetic switching of cells soon after their formation, thereby triggering and maintaining the transformation state of susceptible precancerous cells at the transition stage between primary cells and cancer cells, which is essential for tumorigenicity in a variety of cancer cell lines of different progressional origins, such as colon, prostate, lung, and liver cancer.45–47
IL-6 in the TME Leads to Over-Activation of STAT-3 in Cancer Cells to Promote Cancer Progression
IL-6 in the TME promotes the over-activation of the pro-oncogenic TF STAT-3 in cancer cells through autocrine and paracrine pathways, generating an IL-6 positive feedback loop that mediates cancer progression and invasion. The continuous production of paracrine IL-6 by IAMs within the IME stimulates various cancer cells. These cells utilize distinct combinations of signaling pathways to trigger the JAK-STAT-3 classical signaling pathway or the PI3K/AKT and MEK/ERK signaling pathways, which are associated with mutations in the TME. This results in the formation of a positive feedback loop of IL-6 autocrine signaling in cancer cells.48,49 The activation of the IL-6 trans-signaling pathway, the most important pathway mediating this positive feedback loop, occurs through the binding of sIL-6R to gp130, which is co-expressed by various cancer cells. This binding activates the same JAK-STAT-3 pathway as the classical pathway, leading to the phosphorylation of AKT at Ser-473 and ERK1/2 at the Thr-202/Tyr-204 sites, thereby activating the PI3K/AKT and MEK/ERK pathways. Together, these two pathways serve as the upstream signaling pathways for IL-6 in cancer cells, inducing the expression of pro-inflammatory TFs such as STAT-3. They indirectly increase the autocrine activity of IL-6 in cancer cells and directly enhance IL-6 autocrine signaling by localizing to the IL-6 promoter through the complex formed by the interaction between STAT-3 and NF-κB.50 Similar to circulating IL-6 in serum, IL-6 levels derived from cancer cells in patients increase with cancer stage, primarily due to the over-activation of STAT-3. When cancer cell-derived IL-6 levels rise two to three times, the abnormally activated STAT-3 in cancer cells functions as a pro-oncogenic TF in the TME, establishing an IL-6-STAT-3 autocrine cycle that enhances cancer cell growth, survival, proliferation, epithelial-mesenchymal transition (EMT), invasion, and metastasis, thereby facilitating cancer progression. Moreover, STAT-3 can also act as a pro-inflammatory TF in the TME, further exacerbating the inflammatory response at the cancer site, pre-metastatic sites, and metastatic sites. This occurs through autocrine and paracrine signaling involving pro-inflammatory cytokines such as IL-6 and IL-8, which help shape the TME and provide necessary conditions for cancer growth and proliferation. This process participates in the pathophysiological mechanisms underlying inflammation and inflammation-related cancer initiation and progression. STAT-3 is considered an oncogene, with approximately 70% of human cancers exhibiting abnormally elevated STAT-3 activity. This increase is closely associated with poor clinical outcomes in a variety of cancers, including bladder, bone, breast, brain, cervical, colon, rectal, esophageal, head and neck, kidney, liver, lung, ovarian, pancreatic, prostate, gastric, and uterine solid tumors.42,51
IL-6 in the TME Leads to Over-Activation of STAT-3 in Immune Cells to Promote Cancer Progression
IL-6 in the TME contributes to the over-activation of the pro-inflammatory TF STAT-3 in immune cells through autocrine and paracrine pathways, generating an IL-6 positive feedback loop that mediates the inhibition of anti-cancer immunity. In cancers that develop in the context of chronic inflammation or advanced cancers characterized by secondary chronic inflammatory infiltration, macrophages and CD4+ T cells are activated and polarized into M2 macrophages and Th17 cells by IL-6 secreted by cancer cells or TAMs in the TME, primarily through the IL-6 classical and trans-signaling pathways. Simultaneously, this interaction forms an IL-6-STAT-3 autocrine and paracrine positive feedback loop that promotes the long-term maintenance of a chronic inflammatory TME and facilitates immune dialogue between cancer cells and tumor-infiltrating immune cells. Additionally, CD8+ T cells, plasma cells, and natural killer (NK) cells are activated in response to cancer cells in the TME, which express “abnormal” antigens that signal their status as “altered selves” in the context of chronic stress, thus contributing to immune surveillance and tumor destruction. Consequently, it can be said that within the TME of the same cancer, both cancer-promoting and cancer-suppressing immune states can coexist at different points along the cancer progression path, influencing one another. However, due to the heterogeneity among patients and the diverse types of cancers, the activation status of various immune cells, their relative abundances, and the expression of different cytokines in the TME ultimately determine whether the balance leans toward pro-cancer inflammation or anti-cancer immunity. In most cancers, this balance is significantly tilted in favor of pro-cancer inflammation, and without timely therapeutic intervention, cancer progression can occur rapidly.
(1) Innate immune macrophages in the TME secrete IL-6 to exert their immunosuppressive functions. TAMs, as key players in chronic inflammation-related cancers, comprise more than 50% of the TME. Their presence is closely linked to poor cancer prognosis, and they serve as a primary source of pro-inflammatory cytokines, such as IL-6.52,53 In response to IL-6 secreted by cancer cells, many TAMs become polarized toward the “tissue repair” type. This polarization enhances their ability to express tissue-protective factors and growth factors while diminishing their capacity to participate in phagocytosis, antigen presentation, and the induction of antigen-dependent immune responses.54 Importantly, TAM polarization is influenced by signals generated by adaptive immune cells infiltrating the tumor. For instance, Th17 cells can drive the polarization of TAMs toward a functional phenotype that suppresses anti-cancer immunity through an IL-6-dependent mechanism.55 In the presence of elevated concentrations of IL-6 within the TME, polarized TAMs activate STAT-3 via the p38 MAPK cascade in the IL-6 trans-signaling pathway, thereby forming an IL-6-STAT-3 autocrine positive feedback loop. Paracrine IL-6 in TAMs also upregulates the expression of pro-oncogenic factors related to cell proliferation, angiogenesis, and metastasis by activating the pro-oncogenic TF STAT-3 in the IL-6 trans-signaling pathway in cancer cells.56 Additionally, it acts as a bridge for communication between cancer-associated T cells and cancer cells in the TME. Furthermore, TAMs can regulate the immune microenvironment in the TME by activating the pro-inflammatory TF STAT-3 to secrete a variety of chemokines, including IL-10, IL-23, CCL2, and transforming growth factor (TGF)-β.57
(2) Adaptive immune T cells in the TME secrete IL-6 to exert their immunosuppressive functions. T cells share similarities with TAMs in that their differentiation and immune functions are mediated by pro-inflammatory cytokines within the TME. IL-6 induces the expression of the pro-inflammatory TF STAT-3 in both immature and mature T cell lines derived from the thymus, spleen, and lymph nodes, thereby establishing an IL-6-STAT-3 autocrine positive feedback loop.58 Naive CD4+ T cells exhibit significant plasticity in the presence of elevated levels of IL-6 and various co-existing cytokines in the TME. This environment inhibits differentiation toward Th1 and Treg cells while inducing differentiation toward Th17 cells. Concurrently, CD8+ T cells are also transformed into a specialized subset expressing ROR-γt under the influence of high IL-6 levels, allowing them to secrete Th17-related cytokines that support cancer growth. The differentiation of Treg cells is similarly influenced by high IL-6 levels in the TME, which activate STAT-3 and enable it to compete with STAT-5 for binding to the intron of the forkhead box P3 (Foxp3) gene. This competition reduces STAT-5’s ability to transcribe Foxp3, thereby inhibiting the differentiation of CD4+ T cells into Treg cells.59 Th17 cells can be categorized into pathogenic Th17 cells (p-Th17) and non-pathogenic Th17 cells (non-Th17) based on their immunopathological potential to destroy or protect tissues. Regardless of their specific subtype, fully differentiated Th17 cells can establish an IL-6-STAT-3 autocrine positive feedback loop, continually producing pro-inflammatory cytokines such as IL-6 and IL-17. This sustained activation of pro-inflammatory TF STAT-3 contributes to the formation of a complex immune TME, ultimately impacting the proliferation and metastasis of various cancer cells.60 In turn, these Th17 cells can recruit TAMs and other immune cells to exacerbate secondary IME effects within the TME. As a result, the TME becomes engineered into an environment conducive to cancer invasion and, ultimately, affects cancer proliferation and metastasis (Figure S2).
The TNF-α - NF-κB Positive Feedback Loop Promotes Cancer Initiation and Progression
Like IL-6, TNF-α is a potent pro-inflammatory cytokine produced in response to chronic stress by activating multiple pro-inflammatory TFs, such as NF-κB and STAT-3 within inflammatory cells. This activation triggers the formation of subsequent TNF-α-NF-κB inflammatory positive feedback loops. TNF-α exists in two primary forms: membrane-bound (mTNF-α) and soluble (sTNF-α). Under normal physiological conditions, serum levels of TNF-α are typically below 10 pg/mL. However, in patients with breast, colon, rectal, pancreatic, skin, lung, ovarian, and oral cancers, circulating serum levels of TNF-α are elevated. It can be produced by immune cells infiltrating the TME, including TAMs, NK cells, T cells, monocytes, and granulocytes, as well as by stromal cells such as fibroblasts, endothelial cells, and adipocytes, along with various cancer cells. Current understanding suggests that chronic low-dose TNF-α in the TME can sustain the activation of NF-κB, allowing it to exert both pro-inflammatory and pro-oncogenic effects. Studies indicate that TNF-α levels correlate strongly with tumor size, stage, and metastasis, making them important prognostic indicators of survival and predictors of treatment response.61 Soluble TNF-α primarily binds to TNF receptor 1 (TNF-R1), while mTNF-α mainly binds to TNF receptor 2 (TNF-R2), forming receptor complexes that mediate biological activity.62 TNF-R1 is constitutively expressed in the cell membranes of most tissues in the human body and is involved in activating the classical (canonical) TNF-α pathway within cells. In contrast, TNF-R2 is primarily expressed in immune cells. Although TNF-R2 has been shown to independently mediate signals that promote tissue repair through non-classical (alternative) pathways, it is believed that TNF-R2 enhances the function of TNF-R1 by trapping TNF-α.63 The TNF-α-NF-κB positive feedback loop in cancer can be categorized into three main types.
TNF-α in the IME Leads to Over-Activation of NF-κB in Precancerous Cells to Promote Cancer Initiation
In the IME, TNF-α leads to the over-activation of the pro-oncogenic TF NF-κB in precancerous cells through the paracrine pathway, generating a TNF-α positive feedback loop that mediates cancer initiation. Within chronic stress-induced IMEs, paracrine TNF-α produced by IAMs binds to TNF-R1 on the membranes of precancerous cells. This interaction ultimately induces an IKK-like effect on IκB phosphorylation, resulting in K48-Ub degradation of IκB. This degradation allows the nuclear translocation of NF-κB classical family members, including p50, p65, and c-Rel, effectively activating NF-κB as a cancer-promoting TF and positively regulating the transcription of related pro-oncogenic target genes in precancerous cells.64 Simultaneously, autocrine TNF-α produced by precancerous cells continues to activate NF-κB through these mechanisms, forming a positive feedback loop of TNF-α and NF-κB and thereby maintaining a stable transformation state in precancerous cells.
TNF-α in the TME Leads to Over-Activation of NF-κB in Cancer Cells to Promote Cancer Progression
In the TME, TNF-α promotes the over-activation of the pro-oncogenic TF NF-κB in cancer cells through both autocrine and paracrine pathways, generating a TNF-α positive feedback loop that mediates cancer progression and invasion. TNF-α produced by TAMs within the IME can bind to TNF-R1 on the cell membranes of cancer cells through the paracrine pathway, triggering significant activation of NF-κB as a pro-inflammatory TF. This activation strongly induces cancer cells with mutations and high malignant potential to express pro-inflammatory cytokines such as IL-1β, IL-6, IL-8, and TNF-α at chronically low levels, thereby establishing a positive feedback loop of TNF-α autocrine production by cancer cells in the TME. This signaling pathway is primarily responsible for promoting the formation of secondary IMEs, creating an inflammatory TME that favors cancer cell survival while suppressing anti-cancer immunity. Additionally, aberrantly activated NF-κB in these cells acts as a pro-oncogenic TF that drives cancer progression by promoting growth, survival, proliferation, EMT, invasion, metastasis, and chemotherapy resistance. Moreover, autocrine TNF-α produced by cancer cells can also lead to the translocation of β-catenin from the cytoplasm to the nucleus by phosphorylating GSK-3β. This process induces continuous activation of the Wnt/β-catenin signaling pathway, which enhances the malignant transformation potential of cancer cells, including proliferation, colony formation, chemoresistance, migration, and invasion. It can also induce the dedifferentiation of non-stem cells, thereby playing a critical role in cancer progression and progression.65 Thus, these two pathways function together as upstream signaling mechanisms for the activation of NF-κB in cancer cells, and both are considered oncogenes, with their activation levels closely related to poor clinical outcomes in various cancers, including skin, ovarian, breast, colon, rectal, head and neck, liver, lung, pancreatic, renal, prostate, and gastric cancers.66–68
TNF-α in the TME Leads to Over-Activation of NF-κB in Immune Cells to Promote Cancer Progression
In the TME, TNF-α leads to the over-activation of the pro-inflammatory TF NF-κB in immune cells through autocrine and paracrine pathways, generating a TNF-α positive feedback loop that inhibits anti-cancer immunity. In advanced cancers characterized by chronic inflammation or secondary chronic inflammatory infiltration, paracrine TNF-α can activate NF-κB in TAMs and CD4+ T cells. In these major tumor-infiltrating immune cells, NF-κB acts as a pro-inflammatory TF, forming a positive feedback loop between TNF-α and NF-κB. The TNF-α produced through this mechanism possesses both pro-inflammatory and immunomodulatory biological activities. It not only promotes the sustained presence of chronic inflammation in the TME but also facilitates immune communication between cancer cells and infiltrating immune cells. This interaction enhances the activation, differentiation, and functional secretion of cancer-infiltrating immune cells while tilting the balance of the TME toward pro-oncogenic inflammation.
(1) Innate immune macrophages in the TME secrete TNF-α to exert their immunosuppressive functions. As the primary source of pro-inflammatory cytokines such as TNF-α in the TME, TAMs promote the transformation of macrophages into pro-oncogenic “tissue repair” TAMs through the p50/p50 homodimer of NF-κB in response to TNF-α secreted by cancer cells. Activated TAMs subsequently enhance angiogenesis and inhibit immune responses through various cytokines such as continuously activated NF-κB high expression of TNF-α, which has become an important component of TME.69
(2) Adaptive immune T cells in the TME secrete TNF-α to exert their immunosuppressive functions. In the TME, TNF-α secreted by TAMs is known to guide naive CD4+ T cells to proliferate and differentiate into Tregs, exerting immunosuppressive functions via the activation of both the canonical and non-canonical NF-κB signaling pathways. Tregs typically constitute 5–15% of CD4+ T cells and can be categorized into natural Tregs (nTregs) derived from the thymus and inducible Tregs (iTregs) formed in peripheral tissues. Additionally, iTregs can acquire the ability to migrate to non-lymphoid tissues, such as inflamed tissues and tumors, where they are referred to as effector iTregs (eTregs).70 Tregs differentiate into heterogeneous subsets based on the cytokines and signaling pathways present in the TME. This differentiation process consists of three sequential steps: initiation, selection, and maintenance. In the initiation phase, persistent TNF-α signaling in the TME activates the c-Rel and p65 subunits in naive CD4+ T cells through classical TNF-α signaling.71 During the selection phase, TGF-β in the TME further enhances the expression of Foxp3. Fully activated Foxp3 then inhibits the differentiation of immature CD4+ T cells into Th17 cells by antagonizing ROR-γt, ultimately resulting in their differentiation into Foxp3+ Tregs. In the maintenance phase, differentiated Foxp3+ Tregs in the TME secrete TGF-β, utilizing the continuously activated c-Rel and p65 subunits. TGF-β synergistically binds with IL-2 to activate the transcription factor STAT-5, which is crucial for maintaining the expression of Foxp3. The mature iTregs predominantly exhibit sustained p50/p65 activity under steady-state conditions in the TME. They can exert non-contact immunosuppression by secreting immunosuppressive cytokines such as IL-10, TGF-β, adenosine, prostaglandin E2, and Galectin-1. Additionally, they can induce contact-dependent immunosuppression by expressing apoptosis-related molecules such as CTLA-4, Programmed death 1(PD-1), PD-L1, lymphocyte activation protein 3, CD39/CD73, and neurosecretion 1.72 In summary, TNF-α primarily promotes the proliferation, survival, and differentiation of Tregs by activating the TNF-α classical pathway in the TME of solid cancers, while also maintaining the immunosuppressive homeostasis of mature Tregs. Consequently, the TME becomes an environment conducive to cancer cells escaping immune surveillance, acquiring malignant traits to achieve immune tolerance, inhibiting anti-cancer immune responses, and facilitating the growth and metastasis of various malignant tumors.
Signal Crosstalk of IL-6-STAT-3 and TNF-α-NF-κB Positive Feedback Loops in the TME
The continuous activation of the IL-6-STAT-3 and TNF-α-NF-κB loops in the TME has been observed in most human cancers. These two circulating TFs exert their pro-inflammatory TFs effects through indirect cytokine interactions and jointly promote the production and maintenance of chronic inflammation. For example, TNF-α-induced activation of NF-κB in TAMs is sufficient to stimulate IL-6 expression, and conversely, IL-6-induced activation of STAT-3 is involved in enhancing TNF-α secretion.73,74 Both TFs also control the transcriptional activation of numerous oncogenes with overlapping target genes, which means they can act as pro-oncogenic TFs through direct physical interaction and contribute to cancer initiation and progression. For instance, unphosphorylated STAT-3 in cancer cells can bind to the p50/p65 subunit of the IκB complex, displacing IKK and promoting NF-κB activation. In the nucleus, STAT-3 interacts with the p65 subunit, enhancing its transcriptional activity by recruiting the acetyltransferase CBP/p300 (CREB-binding protein/p300) to acetylate the p65 subunit, allowing it to remain in the nucleus. Moreover, these TFs can synergistically enhance the expression of the same target gene by binding to the same promoter or different promoters of that gene. Anti-apoptotic molecules such as Bcl-xL, Bcl-2, and c-IAP2, along with cell cycle regulators such as cyclin D and c-Myc, are activated by both STAT-3 and NF-κB, thus maintaining the survival and proliferation of precancerous cells. Pro-angiogenic factors, such as VEGF and HIF1-α, which are also activated by both STAT-3 and NF-κB in cancer cells, promote tumor angiogenesis.
Additionally, these two circulating TFs mediate communication between cancer cells and immune cells, regulating the balance of Th17 and Tregs. In early cancer, this balance often shifts toward Th17 cells.75 The imbalance activates STAT-3 through autocrine IL-6 and IL-22, leading to the accumulation of Th17-related cytokines such as IL-22 and IL-23. This forms a pro-inflammatory cytokine cycle that promotes inflammatory cell infiltration into tumors via paracrine effects, thereby shaping the TME into an inflammatory environment that is more conducive to cancer growth and survival. In advanced cancer, the balance typically shifts toward Tregs. Imbalanced Tregs activate NF-κB through autocrine TNF-α, establishing an immunosuppressive homeostasis that converts the TME into an immune-tolerant environment, facilitating immune escape and metastasis.76 IL-6 and TNF-α, two pro-inflammatory cytokines, form an autocrine positive feedback loop in TAMs, cancer cells, and immune cells by activating their corresponding pro-inflammatory TFs and exerting effects on other cells in the TME through paracrine signaling. These TFs also function as pro-oncogenic TFs in cancer cells, promoting their proliferation, survival, angiogenesis, and metastasis, as well as acting as pro-differentiation TFs in immune cells, enhancing their differentiation and the secretion of signature cytokines.
Treatments for Chronic Stress-Related Cancers
The among chronic stress, chronic inflammation, and cancer poses a significant challenge as we seek new cancer prevention and treatment strategies. Most cancers can be addressed through primary prevention measures, which often yield better outcomes than surgical interventions. Effective primary and secondary prevention relies on understanding the causes of inflammation linked to chronic stress and mitigating its progression in high-risk populations. Adopting a healthy lifestyle, including emotional stability and good dietary habits, is crucial in preventing chronic stress-induced inflammation that can lead to cancer. Tertiary prevention involves treatment after a cancer diagnosis, and it is essential to consider how anticancer immunity and inflammation promote cancer progression. Chronic inflammation produces growth factors that support tumor progression. The complex interactions within the tumor microenvironment trigger tumorigenesis and drive malignant progression. This primarily explores two positive feedback loops—IL-6-STAT-3 and TNF-α-NF-κB—which represent just a small fraction of the extensive molecular communication network through which chronic stress influences the initiation and progression of cancer. It discusses the latest research advancements in therapeutic strategies targeting these feedback loops, as well as specific directions and prospects for future research.
Natural Anticancer Agents
Isolating plant-derived natural compounds from foods that possess both anti-inflammatory and anticancer properties present a promising strategy for developing inflammation-related cancer treatments.77 For example, flavonols can inhibit the secretion of IL-6 and TNF-α from colon cancer cells by blocking the NF-κB pathway, and they induce apoptosis in breast cancer cells by suppressing the STAT-3 pathway.78 Myricetin also exhibits both anti-inflammatory and antitumor effects by inhibiting key inflammatory factors like IL-6 and TNF-α through the NF-κB signaling pathway.79,80 While these findings are encouraging, further experimental research is essential to successfully translate them into clinical trials.81
Anti-Inflammatory Treatments
Medications such as nonsteroidal anti-inflammatory drugs (NSAIDs), statins, and metformin have been shown to reduce the risk and incidence of various cancers. NSAIDs primarily exert their anticancer effects by inhibiting cyclooxygenase (COX).82 Clinical studies have identified aspirin as a broad-spectrum cancer preventative agent that inhibits NF-κB activation without disrupting cellular transcription mechanisms. Regular aspirin use has been associated with lower cancer incidence and mortality, as well as reduced metastasis across several cancer types, including colorectal, ovarian, prostate, hepatocellular, skin, esophageal, pancreatic, and breast cancers.83,84 While NSAID therapy—alone or in conjunction with cytotoxic agents and targeted immunotherapies—shows potential as a treatment strategy, the non-specific nature of these drugs may result in limited efficacy and increased risk of adverse effects like bleeding, myocardial infarction, gastrointestinal issues, and renal failure. Therefore, careful evaluation is warranted before use, and further research is needed to clarify the underlying mechanisms of their anticancer effects.85
Targeted Inhibition of Pro-Inflammatory Feedback Mechanisms
Targeted Inhibition of IL-6-STAT-3 Feedback Loop
(1) Anti-IL-6 Monoclonal Antibodies. Agents like Siltuximab, Sirukumab, Olokizumab, Clazakizumab, and MEDI5117 inhibit both classic and trans signaling pathways.86,87 Siltuximab has shown antitumor efficacy in preclinical models across various cancers, though it has not demonstrated significant clinical activity in advanced colorectal and head and neck cancers.88 (2) Anti-IL-6R Monoclonal Antibodies. Tocilizumab and Sarilumab target classic and trans signaling, with promising preclinical studies indicating Tocilizumab’s effectiveness in ovarian, pancreatic, and colitis-associated colorectal cancer.89 gp130-Fc fusion proteins like Olamkicept can inhibit trans-signaling, with evidence showing effectiveness against lung and pancreatic cancer.90 (3) Targeting JAK-STAT-3 Pathway. Small-molecule tyrosine kinase inhibitors like Tofacitinib, Ruxolitinib, and AZD1480 selectively inhibit JAK, preventing the phosphorylation of STAT-3. These JAK inhibitors have demonstrated the ability to suppress tumor growth in various solid tumors, including brain, breast, colorectal, and lung cancers, indicating their potential as effective anticancer agents for clinical trials.91 High-selectivity inhibitors such as Stattic, LL1, and Bruceantinol block the binding of STAT-3 to its target DNA, showcasing promise in preclinical research.92,93 Additionally, Src homology domain 2 (SH2) domain inhibitors, like C188-9 and OPB-51602, disrupt STAT-3 dimerization, inhibiting cancer cell growth. The antisense oligonucleotide AZD9150 binds to STAT-3 mRNA, leading to decreased STAT-3 expression.94 Bazedoxifene, an estrogen receptor ligand, has shown efficacy in clinical trials for breast cancer and may require combination with other treatments for enhanced antitumor effects.95 A range of new inhibitors targeting IL-6, JAK, and STAT-3 are currently under evaluation.
Targeted Inhibition of TNF-α-NF-κB Feedback Loop
(1) Anti-TNF-α Monoclonal Antibodies. Monoclonal antibodies that target TNF-α, including infliximab, adalimumab, and golimumab, are designed to inhibit classic TNF-α signaling. These agents have been evaluated in Phase I and II clinical trials, with infliximab demonstrating good tolerability in patients with advanced cancers and no dose-limiting toxic effects.96 (2) Anti-TNF-R Monoclonal Antibodies. These antibodies inhibit non-classical TNF-α signaling. Studies in colorectal cancer models have shown that anti-TNF-R2 monoclonal antibodies can reduce the infiltration of activated CCR8+ Treg subgroups and decrease TNF-α secretion within the TME. This action can inhibit tumor progression and enhance the effectiveness of anti-PD-1 therapy, making TNF-R2 a promising target for immunotherapy.97 (3) Targeting the TNF-α-NF-κB Pathway. TNF-α Processing Inhibitor-1 (TAPI-1) is a broad-spectrum metalloproteinase inhibitor that prevents the shedding of the TNF-R1 receptor and blocks the NF-κB-p65 signaling pathway. TAPI-1 has shown antitumor efficacy by inhibiting the vitality, migration, and invasion of cancer cells in pancreatic, breast, esophageal, and liver cancers.98 However, it is essential to note that the long-term use of NF-κB inhibitors may lead to immune deficiencies. Therefore, these inhibitors should ideally be used for short-term treatments in cancer therapy. An effective NF-κB inhibitor should specifically target the NF-κB pathway without affecting other signaling pathways.
Despite results in preclinical studies targeting the IL-6-STAT-3 and TNF-α-NF-κB signaling pathways, there is limited evidence of effective single-agent clinical trials for solid tumors. This highlights the challenge of achieving significant prognostic improvements by solely blocking IL-6 or TNF-α in unselected patient populations. To address this, developing effective combination therapies and identifying reliable biomarkers for predicting treatment responses are essential. Combining therapies, particularly with immunotherapy, shows promise as a potential strategy for treating cancers driven by IL-6 and TNF-α signaling. For instance, preclinical evidence suggests that IL-6 and PD-L1 combined immunosuppressive therapies may enhance the Th1 immune response in mice, leading to the recruitment of T cells, production of chemokines, and increased CD4+ T cell infiltration that produces Interferon-gamma (IFN-γ).99 This combination has the potential to enhance the effectiveness of immune checkpoint inhibitors against tumors. However, further extensive preclinical and clinical studies are necessary to validate these findings and explore their therapeutic implications.
Conclusion
When summarizing the relationship between long-term chronic psychological stress and the initiation, progression, and metastasis of cancer, we found that the core hubs connecting chronic stress, chronic inflammation, and cancer converge on two positive feedback loops involving autocrine/paracrine pro-inflammatory cytokines: IL-6-STAT-3 and TNF-α-NF-κB. Catecholamines produced by chronic stress target the downstream cAMP-PKA/EPAC signaling pathway by binding to β-ARs present in various organ cells, which activates STAT-3 and NF-κB as pro-inflammatory TFs in inflammatory cells. This activation promotes the autocrine and paracrine signaling of multiple pro-inflammatory cytokines, such as IL-6 and TNF-α. The paracrine actions of IL-6 and TNF-α establish an IME in vivo, consisting of various inflammation-related cytokines and inflammatory cells, by directly recruiting these cells or indirectly promoting their differentiation. In established IME, the autocrine signaling of IL-6 and TNF-α by inflammatory cells continuously activates STAT-3 and NF-κB as pro-inflammatory TFs, forming a positive feedback loop that connects chronic stress and chronic inflammation while maintaining the long-term existence of IME. Additionally, within the existing inflammation-associated TME, IL-6 and TNF-α paracrine signaling by IAMs or TAMs continuously activates STAT-3 and NF-κB as pro-oncogenic TFs in cancer cells, initiating the transcription of target genes responsible for cancer cell survival, proliferation, invasion, metastasis, angiogenesis, chemotherapy resistance, and EMT. This signaling also reinforces a positive feedback loop within the TME that favors cancer progression and invasion. In late-stage, secondary IME-infiltrated TMEs, IL-6 and TNF-α paracrine signaling by cancer cells or TAMs can continuously activate STAT-3 and NF-κB as pro-differentiation TFs in immune cells, inducing their proliferation, activation, differentiation, and secretion of pro-inflammatory cytokines such as IL-6 and TNF-α. Moreover, autocrine signaling of IL-6 and TNF-α by activated TAMs, Th17 cells, and Tregs can sustain the activation of STAT-3 and NF-κB, disrupting the Th17/Tregs cancer immune homeostasis and exacerbating secondary IME infiltration into the TME. This interplay mediates the immune dialogue between cancer cells and infiltrating immune cells, maintaining the long-term activation of pro-oncogenic TFs within the TME.
This retrospective study is groundbreaking as it connects the cycle of pro-inflammatory cytokines induced by chronic stress, rationally linking stress, inflammation, and tumors while elucidating the molecular relationships among them. These insights could aid in developing more effective strategies for cancer prevention and treatment. Quantifying and modeling the effects of chronic stress on human cancer involves significant uncertainties and challenges. Commonly used animal models in preclinical research often fail to capture the complexity of human stress responses, which encompass various physiological, psychological, and environmental factors. Additionally, variations in age, sex, genetics, and lifestyle further complicate the research process. The inherent heterogeneity, complexity, and plasticity of human cancers and their microenvironments make the experimental design even more challenging. To gain a clear understanding of the relationship between chronic stress and cancer, researchers must develop more sophisticated and representative models, alongside rigorous methodologies that consider diverse biological and environmental factors. This ongoing challenge underscores the need for innovative research approaches to bridge the gap between preclinical findings and human clinical outcomes. Future longitudinal studies are essential for tracking the long-term effects of chronic stress on different individuals. These studies should aim to clarify the specific relationship between chronic stress and cancer progression and evaluate the impact of psychological therapies designed to alleviate chronic stress on cancer progression and prognosis. Moreover, it is crucial to address significant obstacles in effectively targeting chronic stress-mediated chronic inflammation in anti-tumor therapies. This includes considering the bystander effects of anti-inflammatory therapies on non-cancerous tissues. Additionally, identifying predictive biomarkers is vital for rationally incorporating drugs that target inflammatory factors into standard comprehensive cancer treatment regimens, which should be used in conjunction with chemotherapy, radiotherapy, and immune checkpoint inhibitors while closely monitoring the effects of these combined therapies in both preclinical and clinical settings.
Abbreviations
IME, The inflammatory microenvironment; IL-6, Interleukin-6; TNF-α, Tumor cell necrosis factor-α; TF, Transcription factors; TME, Tumor microenvironment; SAM, Sympathetic-adreno-medullary; HPA, Hypothalamic-pituitary-adrenal; NE, Norepinephrine; E, Epinephrine; β-ARs, β-adrenergic receptors; cAMP, Cyclic AMP; PKA, Protein kinase A; EPAC, Exchange protein directly activated by cAMP; MAPK, Mitogen-activated protein kinases; Ser, Serine; Thr, Threonine; AKAP, A-kinase anchoring protein; EGF, Epidermal growth factor; RTK, Receptor tyrosine kinase; STAT-3, Signal transducer and activator of transcription 3; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; YAP, Yes-associated protein; TAZ, WW-domain-containing transcription factor; CREB, cAMP response element-binding protein; ATF, Activator of transcription; AP-1, Activating protein-1; ETS, E26 avian erythroblastosis virus transcription factor; HIF-1α, Hypoxia-inducible factor-1α; BARK, β-adrenergic receptor kinase; Tyr, Tyrosine; JAK, Janus kinase; JNK, c-Jun NH2-terminal kinase; ERK, Extracellular-signal-regulated kinases; SH2, SRC homology 2 domain; DBD, DNA Binding domain; IAMs, Inflammation associated macrophages; TAMs, Tumor-associated macrophages; MMP, Matrix metallopeptidase; VEGF, Vascular endothelial growth factor; TCRs, T cell receptors; CTL, CD8+cytotoxic T; Th, CD4+helper T; APCs, Antigen-presenting cells; ROR-γt, Related orphan receptor; ROR-α, Related orphan receptor-α; IKK, IkappaB kinase; IκB, IkappaB; GSK-3β, Glycogen synthase kinase-3β; GSKIP, GSK-3β interaction protein; Arg, Arginine; Lys (K), Lysine; Ub, Ubiquitination; Bcl-3, B-cell lymphoma 3; APC, Adenomatous polyposis protein; CK1, Casein-kinase 1; TCF, T cell factor; LEF, Lymphocyte enhancement factor; VIP, Vasoactive intestinal peptide; PI3K, Phosphatidyl inositol 3-kinase; COX-2, Cyclooxygenase-2; IFN-γ, Interferon-gamma; NLRP3, Nod-like receptor protein 3; Pro-IL-1β, protoIL-1β; Pro-IL-18, protoIL-18 precursor; CCL, C-C motif ligand; CRP, C-reactive protein; MDSCs, Myeloid-derived suppressor cells; DCs, Dendritic cells; NK, Natural killer cells; CAFs, Cancer-associated fibroblasts; ROS, Reactive oxygen species; TGF, Transforming growth factor; mIL-6R, membrane-bound type; sIL-6R, soluble type; SHP-2, Tyr phosphatase-2; AKT, Protein kinase B; sgp130, Soluble form of gp130; SOCS3, Cytokine signaling inhibitory proteins 3; EMT, Epithelial-mesenchymal transition; GM-CSF, Granulocyte-macrophage colony-stimulating factor; PD-L1, Programmed death ligand 1; PD-1, Programmed death 1; Foxp3, Forkhead box p3; p-Th17, pathogenic Th17 cells; non-Th17, non-pathogenic Th17 cells; TNF-R, TNF receptor; nTregs, natural Tregs from the thymus; iTregs, inducible Tregs from the periphery; KLRG1, Killer cell lectin-like receptor G1; CTLA-4, cytotoxic T lymphocyte-associated antigen 4; CBP/p300, CREB-binding protein/p300; iNOS, inducible Nitric oxide synthase; IDO, Indoleamine 2.3-dioxygenase; XIAP, X-linked inhibitor of apoptosis; PLK1, Polo-like kinase 1; PIM1, Provirus integration site for Moloney murine leukemia virus-1; HA, Hyaluronic acid; Fas-L, Fas ligand; BCL-2, B-cell lymphoma-2; Mcl-1, Myeloid cell leukemia-1; HSP-70, Heat shock proteins-70; Reg III β, Regenerating islet-derived 3 β; IAP, Inhibitor of apoptosis; FGF2, Fibroblast growth factor 2; SNAI2, Snail family transcriptional repressor 2; FAK, Focal adhesion kinase; HOTAIR, Highly expressed LncRNA HOX transcriptional antisense RNA; RhoA, Ras homologous A; VCAM-1, Vascular Cell Adhesion Molecule-1; ICAM-1, Intercellular adhesion molecule 1; PD-1, Programmed death 1; XIAP, X-linked inhibitor apoptosis; cIAP1/2, cellular IAP1/2; c-FLIP, c-FLICE-inhibitory protein; PAI2, Plasminogen activator inhibitor 2; BFL-1, Bcl2 related protein A1; MYCT1, Myc target 1; NAT10, N-acetyltransferase 10; HDAC2, Histone deacetylase 2; NRBP2, Nuclear receptor binding protein 2; PAK5, p21-activated kinase 5; uPA, Urokinase-type plasminogen activator; EGR-1, Early growth response 1; IGF2, Insulin-like growth factor 2; BAD, Bcl-2 agonist of cell death; FOXO-1, Forkhead box O 1; EGF-1, Epidermal growth factor 1; FN1, Fibronectin 1; LOXL2, Lysyl oxidase-like 2; GLUT-1, Glucose transporter-1; HK1, Hexokinase 1; LDH-A, Lactate dehydrogenase-A; PDK1, Pyruvate dehydrogenase kinase 1; ROS, Reactive oxygen species; RNI, Reactive nitrogen intermediates; NLS, Nuclear localization signal; SCF, Skp1-Cullin1-F-box; Dvl/Dsh, Dishevelled; FZD, Frizzled receptors; LRP5/6, Low-density lipoprotein receptor-associated protein; ACTH, Adrenocorticotropic hormone; GPCRs, G protein-coupled receptors; AC, Adenylate cyclase; β-Trcp, β-transducin repeat containing protein; TRADD, Tumor necrosis factor receptor-associated death domain protein; DD, Death domain; RIPK1/RIP1, Receptor-interacting serine/threonine protein kinase 1; cIAP1/2, cellular IAP1/2; TAK1, TGF-β activating kinase 1; Table 2/3, TAK1binding protein 2/3; TRAF, TNF receptor-associated factor.
Data Sharing Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the Declaration of Helsinki and approved by the Medical Ethics Committee of The First Affiliated Hospital of Xian Jiaotong University.
Acknowledgments
We would like to thank all the research participants who participated in this study and the patients who participated in this study for their contributions to science. We would also like to thank BioRender for providing illustration resources.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no competing interests.
References
1. Chida Y, Hamer M, Wardle J, Steptoe A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat Clin Pract Oncol. 2008;5(8):466–475. doi:10.1038/ncponc1134
2. Kruk J, Aboul-Enein BH, Bernstein J, Gronostaj M. Psychological Stress and Cellular Aging in Cancer: a Meta-Analysis. Oxid Med Cell Longev. 2019;2019:1270397. doi:10.1155/2019/1270397
3. Dhabhar FS. Effects of stress on immune function: the good, the bad, and the beautiful. Immunol Res. 2014;58(2–3):193–210. doi:10.1007/s12026-014-8517-0
4. Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10(12):1314–1319. doi:10.1038/embor.2009.243
5. Szabo S, Yoshida M, Filakovszky J, Juhasz G. ”Stress” is 80 Years Old: from Hans Selye Original Paper in 1936 to Recent Advances in GI Ulceration. Curr Pharm Des. 2017;23(27):4029–4041. doi:10.2174/1381612823666170622110046
6. Tolomeo M, Cascio A. The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy. Int J mol Sci. 2021;22(2):603. doi:10.3390/ijms22020603
7. Zhao X, You X, Huang C, Liu G, Cheng Z, Zhang H. Steroid Receptor Coactivator-1 (SRC-1) Promoted Cell Metastasis of Gastric Cancer via VEGFC Activator by NF-κB. Crit Rev Eukaryot Gene Expr. 2022;32(4):21–29. doi:10.1615/CritRevEukaryotGeneExpr.2021040849
8. Degirmenci U, Wang M, Hu J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells. 2020;9(1). doi:10.3390/cells9010198
9. Zhang W, Wang Z, Luo Y, Zhong D, Luo Y, Zhou D. GATA3 expression correlates with poor prognosis and tumor-associated macrophage infiltration in peripheral T cell lymphoma. Oncotarget. 2016;7(40):65284–65294. doi:10.18632/oncotarget.11673
10. Li M, Jiang H, Chen S, Ma Y. GATA binding protein 1 recruits histone deacetylase 2 to the promoter region of nuclear receptor binding protein 2 to affect the tumor microenvironment and malignancy of thyroid carcinoma. Bioengineered. 2022;13(4):11320–11341. doi:10.1080/21655979.2022.2068921
11. Glidewell-Kenney CA, Trang C, Shao PP, et al. Neurokinin B induces c-fos transcription via protein kinase C and activation of serum response factor and Elk-1 in immortalized GnRH neurons. Endocrinology. 2014;155(10):3909–3919. doi:10.1210/en.2014-1263
12. Su X, Yang Y, Guo C, et al. NOX4-Derived ROS Mediates TGF-β1-Induced Metabolic Reprogramming during Epithelial-Mesenchymal Transition through the PI3K/AKT/HIF-1α Pathway in Glioblastoma. Oxid Med Cell Longev. 2021;2021:5549047. doi:10.1155/2021/5549047
13. Malekan M, Ebrahimzadeh MA, Sheida F. The role of Hypoxia-Inducible Factor-1alpha and its signaling in melanoma. Biomed Pharmacother. 2021;141:111873. doi:10.1016/j.biopha.2021.111873
14. Zhao D, Yin Z, Soellner MB, Martin BR. Scribble sub-cellular localization modulates recruitment of YES1 to regulate YAP1 phosphorylation. Cell Chem Biol. 2021;28(8):1235–1241.e1235. doi:10.1016/j.chembiol.2021.02.019
15. Hsu PC, Yang CT, Jablons DM, You L. The Crosstalk between Src and Hippo/YAP Signaling Pathways in Non-Small Cell Lung Cancer (NSCLC). Cancers. 2020;12(6):1361. doi:10.3390/cancers12061361
16. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402. doi:10.1038/s41392-021-00791-1
17. Carpenter RL, Lo HW. STAT3 Target Genes Relevant to Human Cancers. Cancers. 2014;6(2):897–925. doi:10.3390/cancers6020897
18. Kim M, Morales LD, Jang IS, Cho YY, Kim DJ. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int J mol Sci. 2018;19(9).
19. Szebeni GJ, Vizler C, Kitajka K, Puskas LG. Inflammation and Cancer: extra- and Intracellular Determinants of Tumor-Associated Macrophages as Tumor Promoters. Mediators Inflamm. 2017;2017:9294018. doi:10.1155/2017/9294018
20. Hedrich CM, Rauen T, Apostolidis SA, et al. Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc Natl Acad Sci U S A. 2014;111(37):13457–13462. doi:10.1073/pnas.1408023111
21. Oh H, Ghosh S. NF-κB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev. 2013;252(1):41–51. doi:10.1111/imr.12033
22. Hall JA, Pokrovskii M, Kroehling L, et al. Transcription factor RORα enforces stability of the Th17 cell effector program by binding to a Rorc cis-regulatory element. Immunity. 2022;55(11):2027–2043.e2029. doi:10.1016/j.immuni.2022.09.013
23. Perez LG, Kempski J, McGee HM, et al. TGF-β signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat Commun. 2020;11(1):2608. doi:10.1038/s41467-020-16363-w
24. Thoma A, Lightfoot AP. NF-kB and Inflammatory Cytokine Signalling: role in Skeletal Muscle Atrophy. Adv Exp Med Biol. 2018;1088:267–279.
25. Lu Q, Tong B, Luo Y, et al. Norisoboldine suppresses VEGF-induced endothelial cell migration via the cAMP-PKA-NF-κB/Notch1 pathway. PLoS One. 2013;8(12):e81220. doi:10.1371/journal.pone.0081220
26. McCubrey JA, Fitzgerald TL, Yang LV, et al. Roles of GSK-3 and microRNAs on epithelial mesenchymal transition and cancer stem cells. Oncotarget. 2017;8(8):14221–14250. doi:10.18632/oncotarget.13991
27. Failor KL, Desyatnikov Y, Finger LA, Firestone GL. Glucocorticoid-induced degradation of glycogen synthase kinase-3 protein is triggered by serum- and glucocorticoid-induced protein kinase and Akt signaling and controls beta-catenin dynamics and tight junction formation in mammary epithelial tumor cells. Mol Endocrinol. 2007;21(10):2403–2415. doi:10.1210/me.2007-0143
28. Polley S, Huang DB, Hauenstein AV, et al. A structural basis for IκB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol. 2013;11(6):e1001581. doi:10.1371/journal.pbio.1001581
29. Fernández-Martínez AB, Carmena MJ, Bajo AM, Vacas E, Sánchez-Chapado M, Prieto JC. VIP induces NF-κB1-nuclear localisation through different signalling pathways in human tumour and non-tumour prostate cells. Cell Signal. 2015;27(2):236–244. doi:10.1016/j.cellsig.2014.11.005
30. Fu X, Zunich SM, O’Connor JC, Kavelaars A, Dantzer R, Kelley KW. Central administration of lipopolysaccharide induces depressive-like behavior in vivo and activates brain indoleamine 2,3 dioxygenase in murine organotypic hippocampal slice cultures. J Neuroinflammation. 2010;7:43. doi:10.1186/1742-2094-7-43
31. Gárate I, García-Bueno B, Madrigal JL, et al. Origin and consequences of brain Toll-like receptor 4 pathway stimulation in an experimental model of depression. J Neuroinflammation. 2011;8:151. doi:10.1186/1742-2094-8-151
32. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi:10.1038/sigtrans.2017.23
33. Anson M, Crain-Denoyelle AM, Baud V, et al. Oncogenic β-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J Clin Invest. 2012;122(2):586–599. doi:10.1172/JCI43937
34. Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441(7092):431–436. doi:10.1038/nature04870
35. Pham AT, van Dijk BAC, van der Valk ES, van der Vegt B, van Rossum EFC, de Bock GH. Chronic Stress Related to Cancer Incidence, including the Role of Metabolic Syndrome Components. Cancers (Basel). 2024;16(11):2044. doi:10.3390/cancers16112044
36. McCollum SE, Shah YM. Stressing Out Cancer: chronic Stress Induces Dysbiosis and Enhances Colon Cancer Growth. Cancer Res. 2024;84(5):645–647. doi:10.1158/0008-5472.CAN-23-3871
37. Popko K, Gorska E, Demkow U. Influence of interleukin-6 and G174C polymorphism in IL-6 gene on obesity and energy balance. Eur J Med Res. 2010;2(Suppl 2):123–127. doi:10.1186/2047-783X-15-S2-123
38. Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–248. doi:10.1038/nrclinonc.2018.8
39. Ataie-Kachoie P, Pourgholami MH, Richardson DR, Morris DL. Gene of the month: interleukin 6 (IL-6). J Clin Pathol. 2014;67(11):932–937. doi:10.1136/jclinpath-2014-202493
40. López RV, Zago MA, Eluf-Neto J, et al. Education, tobacco smoking, alcohol consumption, and IL-2 and IL-6 gene polymorphisms in the survival of head and neck cancer. Braz J Med Biol Res. 2011;44(10):1006–1012. doi:10.1590/S0100-879X2011007500097
41. Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70(1):11–20. doi:10.1016/j.cyto.2014.05.024
42. Španko M, Strnadová K, Pavlíček AJ, et al. IL-6 in the Ecosystem of Head and Neck Cancer: possible Therapeutic Perspectives. Int J mol Sci. 2021;22(20):11027. doi:10.3390/ijms222011027
43. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448–457. doi:10.1038/ni.3153
44. Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39(4):493–506. doi:10.1016/j.molcel.2010.07.023
45. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139(4):693–706. doi:10.1016/j.cell.2009.10.014
46. Schetter AJ, Leung SY, Sohn JJ, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299(4):425–436. doi:10.1001/jama.299.4.425
47. Tavazoie SF, Alarcón C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451(7175):147–152. doi:10.1038/nature06487
48. Tang CH, Chuang JY, Fong YC, Maa MC, Way TD, Hung CH. Bone-derived SDF-1 stimulates IL-6 release via CXCR4, ERK and NF-kappaB pathways and promotes osteoclastogenesis in human oral cancer cells. Carcinogenesis. 2008;29(8):1483–1492. doi:10.1093/carcin/bgn045
49. Chang Q, Bournazou E, Sansone P, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013;15(7):848–862. doi:10.1593/neo.13706
50. Huang WL, Yeh HH, Lin CC, et al. Signal transducer and activator of transcription 3 activation up-regulates interleukin-6 autocrine production: a biochemical and genetic study of established cancer cell lines and clinical isolated human cancer cells. mol Cancer. 2010;9:309. doi:10.1186/1476-4598-9-309
51. Berishaj M, Gao SP, Ahmed S, et al. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007;9(3):R32. doi:10.1186/bcr1680
52. Kobawala TP, Trivedi TI, Gajjar KK, Patel DH, Patel GH, Ghosh NR. Significance of Interleukin-6 in Papillary Thyroid Carcinoma. J Thyroid Res. 2016;2016:6178921. doi:10.1155/2016/6178921
53. Hill BS, Sarnella A, D’Avino G, Zannetti A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin Cancer Biol. 2020;60:202–213. doi:10.1016/j.semcancer.2019.07.028
54. Verdeil G, Lawrence T, Schmitt-Verhulst AM, Auphan-Anezin N. Targeting STAT3 and STAT5 in Tumor-Associated Immune Cells to Improve Immunotherapy. Cancers (Basel). 2019;11(12). doi:10.3390/cancers11121832
55. Fang W, Zhou T, Shi H, et al. Progranulin induces immune escape in breast cancer via up-regulating PD-L1 expression on tumor-associated macrophages (TAMs) and promoting CD8(+) T cell exclusion. J Exp Clin Cancer Res. 2021;40(1):4. doi:10.1186/s13046-020-01786-6
56. Radharani NNV, Yadav AS, Nimma R, et al. Tumor-associated macrophage derived IL-6 enriches cancer stem cell population and promotes breast tumor progression via Stat-3 pathway. Cancer Cell Int. 2022;22(1):122. doi:10.1186/s12935-022-02527-9
57. Peng G, Wang C, Wang H, et al. Gankyrin-mediated interaction between cancer cells and tumor-associated macrophages facilitates prostate cancer progression and androgen deprivation therapy resistance. Oncoimmunology. 2023;12(1):2173422. doi:10.1080/2162402X.2023.2173422
58. Narimatsu M, Maeda H, Itoh S, et al. Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. mol Cell Biol. 2001;21(19):6615–6625. doi:10.1128/MCB.21.19.6615-6625.2001
59. Yao Z, Kanno Y, Kerenyi M, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109(10):4368–4375. doi:10.1182/blood-2006-11-055756
60. Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32(5):605–615. doi:10.1016/j.immuni.2010.05.003
61. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9(5):361–371. doi:10.1038/nrc2628
62. Fanzani A, Conraads VM, Penna F, Martinet W. Molecular and cellular mechanisms of skeletal muscle atrophy: an update. J Cachexia, Sarcopenia Muscle. 2012;3(3):163–179. doi:10.1007/s13539-012-0074-6
63. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15(6):362–374. doi:10.1038/nri3834
64. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev mol Cell Biol. 2014;15(2):135–147. doi:10.1038/nrm3737
65. Kafri P, Hasenson SE, Kanter I, et al. Quantifying β-catenin subcellular dynamics and cyclin D1 mRNA transcription during Wnt signaling in single living cells. Elife. 2016;5.
66. Wilson W 3rd, Baldwin AS. Maintenance of constitutive IkappaB kinase activity by glycogen synthase kinase-3alpha/beta in pancreatic cancer. Cancer Res. 2008;68(19):8156–8163. doi:10.1158/0008-5472.CAN-08-1061
67. Chen WC, Li QL, Pan Q, et al. Xenotropic and polytropic retrovirus receptor 1 (XPR1) promotes progression of tongue squamous cell carcinoma (TSCC) via activation of NF-κB signaling. J Exp Clin Cancer Res. 2019;38(1):167. doi:10.1186/s13046-019-1155-6
68. Wang Y, Lin Z, Sun L, et al. Akt/Ezrin Tyr353/NF-κB pathway regulates EGF-induced EMT and metastasis in tongue squamous cell carcinoma. Br J Cancer. 2014;110(3):695–705. doi:10.1038/bjc.2013.770
69. Ju X, Zhang H, Zhou Z, Chen M, Wang Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-ɑ signaling. Exp Cell Res. 2020;396(2):112315. doi:10.1016/j.yexcr.2020.112315
70. Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14(3):154–165. doi:10.1038/nri3605
71. Vahl JC, Drees C, Heger K, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 2014;41(5):722–736. doi:10.1016/j.immuni.2014.10.012
72. Erfani N, Mehrabadi SM, Ghayumi MA, et al. Increase of regulatory T cells in metastatic stage and CTLA-4 over expression in lymphocytes of patients with non-small cell lung cancer (NSCLC). Lung Cancer. 2012;77(2):306–311. doi:10.1016/j.lungcan.2012.04.011
73. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–489. doi:10.1146/annurev-immunol-030409-101212
74. McFarland BC, Hong SW, Rajbhandari R, et al. NF-κB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PLoS One. 2013;8(11):e78728. doi:10.1371/journal.pone.0078728
75. Duan MC, Han W, Jin PW, et al. Disturbed Th17/Treg Balance in Patients with Non-small Cell Lung Cancer. Inflammation. 2015;38(6):2156–2165. doi:10.1007/s10753-015-0198-x
76. Marshall EA, Ng KW, Kung SH, et al. Emerging roles of T helper 17 and regulatory T cells in lung cancer progression and metastasis. mol Cancer. 2016;15(1):67. doi:10.1186/s12943-016-0551-1
77. Yin Y, Wang Z, Hu Y, Wang J, Wang YI, Lu Q. Caffeic acid hinders the proliferation and migration through inhibition of IL-6 mediated JAK-STAT-3 signaling axis in human prostate cancer. Oncol Res. 2024;32(12):1881–1890. doi:10.32604/or.2024.048007
78. Han M, Song Y, Zhang X. Quercetin Suppresses the Migration and Invasion in Human Colon Cancer Caco-2 Cells Through Regulating Toll-like Receptor 4/Nuclear Factor-kappa B Pathway. Pharmacogn Mag. 2016;12(Suppl 2):S237–244. doi:10.4103/0973-1296.182154
79. Ward AB, Mir H, Kapur N, Gales DN, Carriere PP, Singh S. Quercetin inhibits prostate cancer by attenuating cell survival and inhibiting anti-apoptotic pathways. World J Surg Oncol. 2018;16(1):108. doi:10.1186/s12957-018-1400-z
80. Wu TC, Chan ST, Chang CN, Yu PS, Chuang CH, Yeh SL. Quercetin and chrysin inhibit nickel-induced invasion and migration by downregulation of TLR4/NF-κB signaling in A549 cells. Chem Biol Interact. 2018;292:101–109. doi:10.1016/j.cbi.2018.07.010
81. Hu A, Li K. Erianin Impedes the Proliferation and Metastatic Migration Through Suppression of STAT-3 Phosphorylation in Human Esophageal Cancer Cells. Appl Biochem Biotechnol. 2024;196(9):5859–5874. doi:10.1007/s12010-023-04829-8
82. Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13(5):518–527. doi:10.1016/S1470-2045(12)70112-2
83. Kolawole OR, Kashfi K. NSAIDs and Cancer Resolution: new Paradigms beyond Cyclooxygenase. Int J mol Sci. 2022;23(3). doi:10.3390/ijms23031432
84. Michels N, van Aart C, Morisse J, Mullee A, Huybrechts I. Chronic inflammation towards cancer incidence: a systematic review and meta-analysis of epidemiological studies. Crit Rev Oncol Hematol. 2021;157:103177. doi:10.1016/j.critrevonc.2020.103177
85. Bosetti C, Santucci C, Gallus S, Martinetti M, La Vecchia C. Aspirin and the risk of colorectal and other digestive tract cancers: an updated meta-analysis through 2019. Ann Oncol. 2020;31(5):558–568. doi:10.1016/j.annonc.2020.02.012
86. Haq ATA, Yang PP, Jin C, et al. Immunotherapeutic IL-6R and targeting the MCT-1/IL-6/CXCL7/PD-L1 circuit prevent relapse and metastasis of triple-negative breast cancer. Theranostics. 2024;14(5):2167–2189. doi:10.7150/thno.92922
87. Hu Z, Sui Q, Jin X, et al. IL6-STAT3-C/EBPβ-IL6 positive feedback loop in tumor-associated macrophages promotes the EMT and metastasis of lung adenocarcinoma. J Exp Clin Cancer Res. 2024;43(1):63. doi:10.1186/s13046-024-02989-x
88. Angevin E, Tabernero J, Elez E, et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2014;20(8):2192–2204. doi:10.1158/1078-0432.CCR-13-2200
89. Dijkgraaf EM, Santegoets SJ, Reyners AK, et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann Oncol. 2015;26(10):2141–2149. doi:10.1093/annonc/mdv309
90. Brooks GD, McLeod L, Alhayyani S, et al. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016;76(4):866–876. doi:10.1158/0008-5472.CAN-15-2388
91. Reilley MJ, McCoon P, Cook C, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J Immunother Cancer. 2018;6(1):119. doi:10.1186/s40425-018-0436-5
92. Cortés-Ballinas L, López-Pérez TV, Rocha-Zavaleta L. STAT3 and the STAT3‑regulated inhibitor of apoptosis protein survivin as potential therapeutic targets in colorectal cancer (Review). Biomed Rep. 2024;21(6):175. doi:10.3892/br.2024.1863
93. Méndez‑Clemente A, Bravo‑Cuellar A, González‑Ochoa S, et al. Dual STAT‑3 and IL‑6R inhibition with stattic and tocilizumab decreases migration, invasion and proliferation of prostate cancer cells by targeting the IL‑6/IL‑6R/STAT‑3 axis. Oncol Rep. 2022;48(2). doi:10.3892/or.2022.8349
94. Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting Interleukin-6 Signaling in Clinic. Immunity. 2019;50(4):1007–1023. doi:10.1016/j.immuni.2019.03.026
95. Shi C, Bopp T, Lo HW, Tkaczuk K, Lin J. Bazedoxifene as a Potential Cancer Therapeutic Agent Targeting IL-6/GP130 Signaling. Curr Oncol. 2024;31(10):5737–5751. doi:10.3390/curroncol31100426
96. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5(1):209. doi:10.1038/s41392-020-00312-6
97. Guo Y, Xie F, Liu X, et al. Blockade of TNF-α/TNFR2 signalling suppresses colorectal cancer and enhances the efficacy of anti-PD1 immunotherapy by decreasing CCR8+T regulatory cells. J mol Cell Biol. 2024;16(6).
98. Gao L, Li L, Zhang D, Qiu J, Qian J, Liu H. TAPI-1 Exhibits Anti-tumor Efficacy in Human Esophageal Squamous Cell Carcinoma Cells via Suppression of NF-κB Signaling Pathway. Dig Dis Sci. 2024;69(1):81–94. doi:10.1007/s10620-023-08181-z
99. Mace TA, Shakya R, Pitarresi JR, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67(2):320–332. doi:10.1136/gutjnl-2016-311585
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Prognostic Roles of Inflammation- and Nutrition-Based Indicators for Female Patients with Cancer
Yang M, Zhang Q, Ge Y, Tang M, Hu C, Wang Z, Zhang X, Song M, Ruan G, Zhang X, Liu T, Xie H, Zhang H, Zhang K, Li Q, Li X, Liu X, Lin S, Shi H
Journal of Inflammation Research 2022, 15:3573-3586
Published Date: 17 June 2022
Sarcopenia and COVID-19 Outcomes
Wang Y, Tan S, Yan Q, Gao Y
Clinical Interventions in Aging 2023, 18:359-373
Published Date: 9 March 2023
Response to Article “Combined Silver Sulfadiazine Nanosuspension with Thermosensitive Hydrogel: An Effective Antibacterial Treatment for Wound Healing in an Animal Model” [Letter]
Intan PR, Noviantari A, Alegantina S
International Journal of Nanomedicine 2023, 18:2257-2259
Published Date: 1 May 2023
Association of Body Composition and Handgrip Strength with Interleukin-6 (IL-6) and Vitamin D Level in Cancer Patients
Sutandyo N, Cintakaweni DMW, Setiawan L, Hariani R, Utami N
International Journal of General Medicine 2023, 16:1995-2001
Published Date: 23 May 2023
JAK-STAT Signaling in Autoimmunity and Cancer
Parveen S, Fatma M, Mir SS, Dermime S, Uddin S
ImmunoTargets and Therapy 2025, 14:523-554
Published Date: 11 May 2025