")
Back to Journals » International Journal of Nanomedicine » Volume 20
Combined Mulberry Leaf Polysaccharide-Caged Liposomes for Effective Oral Drug Delivery in Rat Model
Authors Chen X, Zhou H , Chua An Wen A, Wang S, Zhang L, Yang H, Mao Y, Jia J, Wang D, Wang J, Cao Z, Xu B , Xu Y, Shen Y, Zhang W, Zheng Y
Received 26 December 2024
Accepted for publication 17 April 2025
Published 25 April 2025 Volume 2025:20 Pages 5377—5391
DOI https://doi.org/10.2147/IJN.S514455
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Farooq A. Shiekh
Xiaolan Chen,1,* Han Zhou,2,* Asly Chua An Wen,2 Shengyi Wang,2 Lei Zhang,2 Haifeng Yang,1 Yujuan Mao,1 Jiping Jia,1 Dada Wang,1 Jing Wang,1 Zhaoli Cao,1 Bohui Xu,3 Ying Xu,4 Yan Shen,2 Wenli Zhang,2 Yi Zheng1
1Jiangsu Agri-Animal Husbandry Vocational College, Taizhou, Jiangsu, People’s Republic of China; 2Department of Pharmaceutics, China Pharmaceutical University, Jiangsu, 210009, People’s Republic of China; 3School of Pharmacy, Nantong University, Jiangsu, Nantong, People’s Republic of China; 4College of Pharmacy, Jiangsu University, Zhenjiang, 212013, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wenli Zhang, Department of Pharmaceutics, China Pharmaceutical University, 639 Longmian Avenue, Nanjing, Jiangsu, 211198, People’s Republic of China, Email [email protected] Yi Zheng, Jiangsu Agri-Animal Husbandry Vocational College, 8 Fenghuang East Road, Taizhou, Jiangsu, 225300, People’s Republic of China, Email [email protected]
Introduction: Mulberry leaf polysaccharide (MLP) has gained attention as a potential anti-diabetic agent for lowering blood glucose and improving insulin sensitivity. However, the low gastrointestinal stability and oral bioavailability limit its clinical application. To address this issue, a novel drug-caged liposomes (MLP-CL) was developed to enhance oral delivery efficiency of MLP compared to conventional drug-encapsulated liposomes (MLP-L).
Methods: MLP-L and MLP-CL were prepared by the thin-film hydration method. Subsequently, the structural integrity of these liposomes was assessed via in vitro release test and confocal laser microscopy (CLSM) analysis. Madin-Darby canine kidney (MDCK) cells were employed to investigate the cellular uptake mechanisms and transcellular transport efficiency. Finally, the biodistribution profiles and transport mechanisms of liposomes were evaluated through in vivo fluorescence imaging and pharmacokinetic studies in Sprague Dawley rats.
Results: Compared to MLP-L, which released 80% of MLP within 4 hours, MLP-CL showed sustained release with only 40% released in the same period. MLP-CL also enabled more effective co-delivery of MLP and liposomes to MDCK cells, indicating improved structural integrity and cellular uptake. Transcellular transport assay confirmed that MLP-CL was transported across cells more efficiently. In vivo, MLP-CL increased intestinal accumulation and raised plasma MLP concentration by 50%. Additionally, by comparing the discrepancy between the lymphatic-suppression model and the normal model, it was found that 63.56% of MLP-CL was absorbed through the lymphatic pathway compared to 18.05% for MLP-L.
Conclusion: Compared to conventional MLP-L, conjugation of polysaccharide improves the structural integrity of MLP-CL in the gastrointestinal tract, which in turn improves lymphatic uptake and bioavailability. This provides an effective strategy for the design of polysaccharide delivery systems.
Keywords: bioavailability, diabetes management, liposome, mulberry leaf polysaccharides, oral drug delivery
Graphical Abstract:

Introduction
Mulberry leaf polysaccharide (MLP), a bioactive compound derived from Morus alba,1,2 has garnered significant interest in recent years for its potential therapeutic applications, particularly in diabetes management.3–6 Studies highlight its ability to lower blood glucose levels, enhance insulin sensitivity, and exert anti-inflammatory effects, largely attributed to its rich composition of flavonoids and 1-deoxynojirimycin (DNJ).7 Despite these promising properties, the hydrophilia of MLP8 pose a significant barrier to in vivo absorption, as only a limited percentage of the administered dose is absorbed via pinocytosis and passive diffusion.9 Additionally, the main active compounds included in MLP such as dietary phenolics would progress to the colon and be susceptible to undergoing microbial transformations and degradation due to the poor intestinal absorption.10,11 Although encapsulation strategies have been explored to improve bio-accessibility of MLP, they often diminished bio-efficiency.12,13
Lipid-based nanocarriers, such as nanoemulsions, solid lipid nanoparticles (SLNs), and nanostructured lipid carriers (NLCs), have emerged as a promising solution for enhancing the oral delivery of hydrophilic drugs, including protein and polysaccharide.14 These nanocarriers enhance solubility, protect against enzymatic degradation, and promote uptake through lymphatic transport.15 Among these, liposomes are particularly prominent due to their biocompatibility, biomimetic properties, and the ability to dual encapsulation of hydrophilic and lipophilic compounds. However, conventional liposomes face some challenges which restrict their clinical translation. For example, the gastrointestinal tract with its stringent conditions, such as harsh acidic environments, digestive enzymes, and the dynamic interplay of bile salts, presents a significant barrier for liposomes to overcome and can collectively compromise the structural integrity of liposomes, leading to premature drug release or degradation before reaching the intended site of action.16
Polysaccharide-coated liposomes offer promising advantages for oral drug delivery. These systems combine a drug-encapsulating lipid bilayer with a biocompatible polysaccharide coating, enhancing stability, controlled release, and cellular uptake.17 Polysaccharide coatings can achieve sustained release and protect liposomes from aggregation and degradation in the gut, potentially improving mucoadhesion and retention time.18,19 Interestingly, polysaccharide-based nanocapsules exhibit lymphatic uptake and retention capabilities, effectively bypassing the liver first-pass effect.20 Additionally, The biomimetic membrane structure enhances the lipophilicity of polysaccharides, thereby facilitating efficient cellular uptake and improving bioavailability.21 Inspired by these benefits, conjugating MLP to liposomes via surface modifications may address limitations of conventional liposomes and enhance MLP absorption.
In summary, this study introduces a novel MLP-caged liposome (MLP-CL) system, where MLP is covalently conjugated to the liposomal surface, and compares its performance against conventional MLP-encapsulated liposomes (MLP-L). This was followed by subsequent physicochemical characterization and investigation into the mechanism of cellular uptake and uptake efficiency in Madin-Darby canine kidney (MDCK) cells, and in vivo pharmacokinetic evaluation. Specifically, this study positions MLP-CL as a viable alternative to MLP-L, addressing challenges associated with bioavailability and metabolic degradation. Consequently, it offers a promising platform for chronic disease management and polysaccharide compound delivery.
Materials and Methods
Preparation of Mulberry Leaf Polysaccharide- Encapsulated Liposomes (MLP-L)
The MLP-L were prepared using the thin-film hydration method.22 Specifically, Soy lecithin (SPC), 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (DSPE), and cholesterol (supplied by AVT Pharmaceutical Tech Co., Ltd, Shanghai) were dissolved in chloroform-methanol (4:1, v/v) at a molar ratio of 8:1:1.6. The solvent was subsequently removed through rotary evaporation at 45 °C for 30 minutes to form a lipid thin film. Subsequently, 2 mg of MLP (provided by Zhenghe Pharmaceutical Bioengineering Co., Ltd, Shanxi) was dissolved in 8 mL of PBS and used to rehydrate the lipid film at 50 °C for 1 hour. After rehydration, the solution was subjected to ultrasonication (300 w) for 5 minutes. Finally, the resulting solution was extruded through a 0.45 μm polycarbonate membrane to obtain MLP-L.
Preparation of Mulberry Leaf Polysaccharide-Coated Liposomes (MLP-CL)
Blank liposomes were prepared using the thin-film hydration method. Subsequently, A solution of MLP (1 mg/mL) in water was mixed with 10 mg of 1-ethyl -(3-dimethylaminopropyl) carbamide diimide (EDC) and 5 mg of N-hydroxysuccinimide (NHS). The pH of the mixture was adjusted to 4.0 with continuous stirring at room temperature for 1 hour. Thereafter, the activated MLP solution was titrated to pH 8.5 using 0.1 M NaOH, followed by the dropwise addition of blank liposomes into the solution. The reaction was carried out under stirring conditions at room temperature for 12 hours. After the reaction, the solution was centrifuged at 40,000 rpm for 1 hour to isolate unincorporated MLP from the MLP-CL.
Preparation of Fluorescein Isothiocyanate (FITC)-Labeled Mulberry Leaf Polysaccharide (FITC-MLP)
To evaluate the cellular uptake of MLP in cell-based experiments, FITC (provided by AnnJi Pharmaceutical Co. Ltd.) was covalently conjugated to MLP. Specifically, 100 mg of MLP was dissolved in 2 mL of DMSO and reacted with 10 mg of FITC and dibutyltin dilaurate (provided by AnnJi Pharmaceutical Co. Ltd.) under constant stirring at 90°C for 2 hours. The resulting reaction mixture was precipitated three times using 80% ethanol and subsequently purified via dialysis (Spectra/Por Dialysis Membrane, MWCO: 3500 kDa) for 3 days at room temperature to obtain FITC-MLP. This conjugate was then incorporated into liposome preparation protocols to conduct subsequent experiments, including cell viability, intracellular localization, and the determination of endocytic pathways for cellular internalization.
Quantification of Mulberry Leaf Polysaccharide
Phenolic sulfuric acid method was utilized to determine the total polysaccharide content of the MLP. In short, 2 mL of the liposome sample was added to 1mL of 6% phenol and 5 mL of concentrated sulfuric acid, and then incubated at 70 °C for 20 minutes, and determined by UV spectrophotometry at 490 nm.
Characterization of Liposomes
Particle Size and Polydispersity Index
MLP-L and MLP-CL were diluted in PBS (7.4) and measured in triplicate using a NanoBrook series particle size analyzer (Brookhaven Instruments) at 25 °C. The particle size and polydispersity index (PDI) were recorded to assess size homogeneity. Various formulations were screened by adjusting lipid ratios to identify the most stable liposomal formulation. The optimal formulation, selected based on the lowest PDI and highest encapsulation efficiency (EE%), was used for further characterization.
Encapsulation Efficiency and Drug Loading
The encapsulation efficiency (EE) and drug loading (DL) of both MLP-L and MLP-CL liposomal formulations were quantified through ultrafiltration method. After ultra-high-speed centrifugation, the supernatant and precipitate of each formulation were analyzed using the phenol-sulfuric acid method to determine the concentration of MLP (n=3). The encapsulation efficiency (EE%) and drug loading efficiency (DL%) of formulations were calculated based on the following formula:
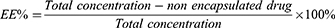

Microstructure Analysis
The microstructures of the liposomes were characterized using transmission electron microscopy (TEM) with negative staining. Briefly, 10 mL aliquots of each liposome solution were diluted with Milli-Q water (1:10) and drop-cast onto carbon-coated copper grids. Following negative staining with 1% (w/v) sodium phosphotungstate solution, the grids were air-dried at room temperature. Subsequently, the samples were analyzed and imaged using a TEM.
In vitro Drug Release Tests
The release behavior of FITC-labeled MLP-L and MLP-CL was systematically evaluated under simulated gastrointestinal conditions. Artificial gastric fluid (SGF, pH 1.2) was prepared by adjusting the pH of ultrapure water to 1.2 using concentrated hydrochloric acid (3.8 mL/L). Artificial intestinal fluid (SIF, pH 6.8) was formulated by dissolving monopotassium phosphate (13.6 g/L) in ultrapure water and subsequently adjusting the pH to 6.8 with 0.1 M NaOH. 3 mL FITC-labeled liposome suspensions were encapsulated in dialysis membranes (300 kDa MWCO) and submerged in 1 L of either SGF or SIF. The dialysis membranes were incubated at 37 °C under constant magnetic stirring at 100 rpm for 24 hours. At predetermined time intervals (0, 0.5, 1, 2, 4, 6, 8, 12, and 24 hours), 1 mL aliquots were withdrawn from the external medium and replaced with fresh medium. The fluorescence intensity of the collected samples was quantified using a microplate reader and normalized to the initial fluorescence intensity (at 0 h) to determine the cumulative release kinetics of FITC-labeled MLP over time.
Cell Viability Assay on Madin-Darby Canine Kidney Cells
The cytotoxicity of FITC-MLP, FITC-MLP-L, and FITC-MLP-CL towards Madin-Darby canine kidney (MDCK) cells was evaluated using the MTT assay.23 Specifically, MDCK cells (ATCC catalog number: CCL-34) were seeded in 96-well plates at a density of 3.0×104 cells per well and incubated for 24 hours in culture medium under standard conditions. After washes with PBS, the cells were exposed to varying concentrations of each respective formulation dissolved in phenol-red-free culture medium for 24 hours at 37 °C and 5% CO2. Following incubation, the medium was removed, and the cells were washed again with PBS. Cell viability was subsequently assessed using the MTT assay. To each well, 100 µL of MTT solution (0.5 mg/mL) was added and incubated for 4 hours. Thereafter, 150 µL of dimethyl sulfoxide (DMSO) was added, and the plate was gently shaken for 10 minutes to dissolve the formazan crystals. The absorbance at 490 nm was measured using a microplate reader. Control groups included cells treated with empty liposomes or DMSO at equivalent concentrations. Cell viability was calculated using the following formula:
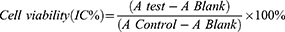
Whereby, “A control” refers to the absorbance value obtained by culturing cells in a drug-free medium, “A test” refers to the absorbance value obtained by culturing cells in a treatment with either free MLP or either liposome formulation, and “A blank” refers to the absorbance value obtained by treating the well without cells by the same method as MTT.
Cellular Uptake Assays
MDCK cells were employed to examine the cellular uptake of liposomes and free MLP. Specifically, MDCK cells (4×105 cells/well) were seeded in 24-well plates and incubated overnight under standard culture conditions. Subsequently, the cells were washed and treated with FITC-labeled MLP, MLP-L, or MLP-CL solutions for 2, 4, and 8 hours, respectively. Following each incubation period, the cells were lysed, centrifuged at a defined speed and duration, and the supernatants were carefully collected. The fluorescence intensity (excitation/emission wavelengths: 490/520 nm) was then measured to quantify the internalized FITC-labeled MLPs, thereby reflecting the relative cellular uptake efficiency of each formulation at each specified time point.
Uptake Mechanism Studies
To elucidate the pathways involved in the cellular uptake of MLP, MLP-L, and MLP-CL, MDCK cells were incubated with specific endocytic inhibitors.24 Briefly, cells were seeded at 4×105 cells/well in 24-well plates and incubated for 24 hours at 37 °C and 5% CO2. Following incubation, they were pretreated with the following inhibitors diluted in phenol-red-free culture medium for 1 hour at 37 °C: amiloride hydrochloride (13 µg/mL) for macropinocytosis inhibition, chlorpromazine (10 µg/mL) for clathrin-mediated endocytosis inhibition, and genistein (54 µg/mL) for caveolae-mediated endocytosis inhibition. After completing pre-incubation, the inhibitors were thoroughly removed via aspiration and the cells were washed twice with PBS. Subsequently, cells were treated with FITC-labeled MLP, MLP-L, or MLP-CL formulations for 4 hours at 37 °C. Following incubation, the culture medium was aspirated, the cells were washed twice with PBS, and then lysed using a 10% Triton X-100 solution. The lysate was centrifuged at 2000 rpm. The collected supernatants were analyzed using a microplate reader with excitation and emission wavelengths set at 490 nm and 520 nm, respectively. This allowed for quantifying the internalized FITC-labeled MLPs and comparing the relative uptake efficiencies of each formulation under varied inhibition conditions.
Laser Confocal Microscopy Imaging
Firstly, FITC/Cy5 dual-labeled liposomes were obtained by labeling liposomes with FITC and MLP with cy5 and were used to study the intracellular trafficking mechanism and stability of MLP-CL. Then, MDCK cells (1×105 cell/well) were cultured in confocal plates for 24 hours. FITC/Cy5 dual-labeled liposomes (2 mg/mL) were then added, and cells were incubated at 37 °C for 2, 4, and 8 hours. Following washes and fixation, DAPI staining visualized nuclei. Confocal microscopy (366, 488, and 647 nm excitation) allowed simultaneous observation of FITC-labeled intact liposomes and Cy5-labeled total MLP. This approach provided insights into intracellular trafficking and potential cargo leakage from MLP-CL formulations.
Transcellular Transport Assessments
MDCK cell monolayer integrity was assessed by transcellular permeability. Inserts seeded with 2.5×104 cells were cultured and washed (n=3). Fluorescein solution (0.01 mg/mL) was added to the upper chamber, with PBS in the lower chamber. After 1, 2, 3, and 4 hours of incubations, lower chamber PBS was analyzed for fluorescence intensity (reflecting tight junction integrity). Fresh Transwell inserts were seeded with MDCK cells (2.5 ×105/well) and cultured for 3–4 days. Following the established protocol, 0.3 mL DMEM medium filled the upper chamber and 0.5 mL PBS filled the lower chamber. Both chambers were subsequently rinsed with PBS three times. FITC-labeled MLP-L or MLP-CL solutions (0.2 mg/mL) were then added to the upper chambers, while the lower chambers received PBS. After 8 hours, the lower chamber PBS was collected and analyzed for fluorescence intensity at 490/520 nm excitation/emission using a spectrofluorometer (n=3). FITC-MLP served as a non-specific interaction control. This design allowed for the comparison of transcellular transport efficiencies between formulations based on measured fluorescence intensity, reflecting the amount of FITC-labeled MLPs traversing the MDCK cell monolayer.
In vivo Studies Using Animal Models
Male Sprague-Dawley rats (280–320 g) were sourced from the reputable Laboratory Animal Center of Jiangxi University of Traditional Chinese Medic and housed individually in polycarbonate cages (45 x 35×18 cm) under controlled conditions. A 12-hour light-dark cycle, a temperature range of 20 ± 2 °C, and a humidity of 50–60% was maintained. The rats had ad libitum access to standard chow and water. Environmental enrichment was provided in the form of nesting materials. Prior to oral administration, a 12-hour fast ensured optimal conditions for the procedure. All animal care and experimental protocols strictly adhered to established ethical guidelines and received prior approval from the China Pharmaceutical University Animal Ethical Experimentation Committee.
In vivo Distribution Assays
To compare the in vivo distribution of MLP-CL and MLP-L, both liposomes were fluorescently labeled with FITC (5 mg/kg) and orally administered to Sprague-Dawley rats (n=3). After 1 hour, the animals were euthanized, and intestinal tissues as well as selected organs (heart, liver, spleen, lung, kidney) were harvested. Biodistribution patterns of each formulation were visualized using fluorescence imaging conducted on a Bruker In Vivo FX Pro system. Besides, the duodenum, jejunum, and ileum segments were excised. Tissue sections (8 µm thick) were prepared and stained with DAPI and rhodamine-phalloidin to delineate cellular architecture.
Pharmacokinetics and Lymphatic Transport Studies
Pharmacokinetic Studies
To investigate the in vivo absorption of MLP formulations, pharmacokinetic studies were conducted in male Sprague-Dawley rats. After an overnight fast, the rats were randomly allocated into three groups and orally administered FITC-labeled MLP, FITC-labeled MLP-L, or FITC-labeled MLP-CL at a dose of 30 mg/kg. Subsequently, blood samples were collected from retro-orbital sinus at 0.5, 1, 2, 3, 4, and 5 hours post-administration. The blood samples were centrifuged at 1000 rpm for 3 minutes, and the resulting plasma supernatants were separated. Each 20 µL plasma supernatants was diluted with 140 µL of saline, vortexed, and then centrifuged at 3000 rpm for 5 minutes. The resultant supernatants (150 µL) were transferred to a 96-well plate for fluorescence intensity measurement using a microplate reader with excitation/emission wavelengths set at 490 nm/520 nm. The measured fluorescence intensities were corrected for background plasma fluorescence and converted to drug concentrations using a pre-established standard curve. Pharmacokinetic parameters were subsequently calculated using a non-compartmental analysis approach.
Lymphatic Transport Evaluation
To evaluate the lymphatic uptake of MLP formulations, a non-invasive cycloheximide-induced chylomicron flow blocking model was utilized in male Sprague-Dawley rats. After an overnight fast, the rats were randomly allocated into three groups and received intraperitoneal injections of cycloheximide solution (Sigma-Aldrich, Germany) (3 mg/kg) to suppress chylomicron production. Twenty minutes later, each group was orally administered 30 mg/kg of either free FITC-labeled MLP, FITC-labeled MLP-L, or FITC-labeled MLP-CL via gastric lavage. Pre-dosing blood samples served as blank controls. Plasma samples were subsequently collected into heparinized tubes from the retro-orbital sinus at 0.5, 1, 2, 3, and 5 hours post-administration. Following centrifugation at 1000 rpm for 3 minutes, plasma supernatants were separated. The plasma drug concentration was then quantified using the fluorescence measurement protocol described in the previous section, including background correction and conversion to concentration via a standard curve.
Results
Drug Loading and Encapsulation Efficiency
Various concentrations of MLP and lipids were evaluated to optimize drug loading and conjugation in liposomal formulations. For MLP-encapsulated liposomes (MLP-L), lipid dispersion was introduced drop-wise into an MLP solution (1 mg/mL) under magnetic stirring at 50°C for 60 minutes. The optimal conjugation rate (8.61%) and drug loading (1.89%) were achieved with a lipid-to-MLP ratio of 2:1:8:1.6. MLP-CL was prepared similarly, except the MLP solution was reacted with EDC and NHS prior to lipid addition. For MLP-CL, the optimal conjugation rate was 8.21%, with drug loading at 1.22% (Table 1).
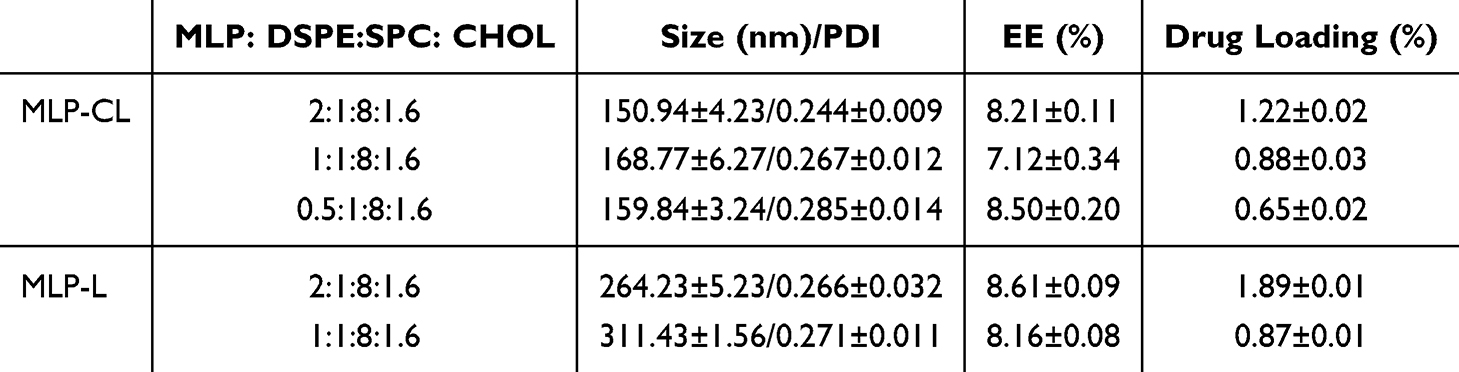 |
Table 1 Physicochemical Properties of MLP-CL and MLP-L (Means ± S.D., n = 3) |
Characterization of Liposomes
Particle size is a crucial factor in liposome stability. Smaller particles (eg, <200 nm) typically exhibit enhanced stability against aggregation due to stronger electrostatic repulsion from higher surface charge density.25 The particle size and PDI of MLP-L and MLP-CL were analyzed using DLS to confirm the stability and uniformity of the formulations. TEM plays a crucial role in verifying the morphology, structural integrity, and vesicular uniformity of liposomes, which can further verify the results of DLS and the microscopic morphology of the liposomes. MLP-L particles exhibited an average size of 264.23 nm and a spherical morphology, confirmed by DLS and TEM (Figure 1A and Table 1). In contrast, MLP-CL had a smaller average particle size of 150 nm and a spherical shape (Figure 2B and Table 1). The successful conjugation of MLP to both liposomal formulations was evident from the significant increase in average particle size compared to blank liposomes. The PDI values of both MLP-L and MLP-CL remained below 0.3, indicating uniform dispersion of particles.
 |
Figure 1 Characterization of MLP-L and MLP-CL. (A and B) TEM images of MLP-L (A) and MLP-CL (B); (C) Drug release curves of MLP-L and MLP-CL in SGF and SIF. |
 |
Figure 2 Cell culture experiments with FITC-MLP, FITC-MLP-L, and FITC-MLP-CL. (A) MTT assay results of liposomal formulations (n=6). Statistical significance was calculated via two-tailed Student’s t-test. **Indicates P < 0.01, ***Indicates P < 0.001, while ns indicates no significant difference between the two groups. (B) Cell uptake of FITC-MLP, FITC-MLP-L, and FITC-MLP- CL. (C) Transcytosis of FITC-MLP, FITC-MLP-L, and FITC-MLP-CL (n=4). Statistical significance was calculated via two-tailed Student’s t-test. ***Indicates P < 0.001, while ns indicates no significant difference between the two groups. (D and E) Mechanism of cell uptake of MLP-L (D) and MLP-CL (E). Statistical significance with “without inhibitor” group was calculated via two-tailed Student’s t-test. *Indicates P < 0.05, **Indicates P < 0.01, ***Indicates P < 0.001, while ns indicates no significant difference between the two groups. |
In vitro Drug Release
The drug release profiles of MLP from MLP-L and MLP-CL were assessed under simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) conditions. MLP release was quantified using UV spectrophotometry at 490 nm. MLP-L exhibited rapid release, reaching 80% within 4 hours in SGF, while MLP-CL showed a slower and sustained release, with only 40% of the drug released in the same timeframe (Figure 1C). This trend was consistent in SIF, indicating that MLP-CL may have enhanced stability and maintain the basic structure of liposomes.
Cell Viability Assays
MTT assay was employed to evaluate the impact of various formulations on the viability of MDCK cells. All groups, including FITC-MLP, FITC-MLP-L, and FITC-MLP-CL, exhibited viability approaching or exceeding 100% (Figure 2A), which suggests that these formulations did not induce significant cytotoxicity relative to the control group. In contrast to free FITC-MLP, both MLP-L and MLP-CL significantly enhanced cell viability at higher concentrations (50 µg/mL) (p < 0.05), potentially due to the advantageous effects conferred by phospholipids.26
Cellular Uptake Assays
Cellular uptake of the FITC-labeled formulations was quantified by measuring the fluorescence intensity of the FITC-labeled preparations in MDCK cells. All formulations exhibited a time-dependent decrease in fluorescence, suggesting potential drug efflux. However, compared with MLP-L, MLP-CL had higher fluorescence intensity at all time points, indicating higher cellular uptake (Figure 2B). The 8-hour groups displayed diminished fluorescence, indicating potential liposome degradation within MDCK cells, leading to the release of encapsulated drugs into the cytoplasm.
Uptake Mechanism Studies
Endocytic inhibitors, including amiloride, genistein, and chlorpromazine, as well as 4°C pretreatment, were employed to suppress macropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and energy-dependent endocytosis in MDCK cells, respectively. The results demonstrated that treatment with amiloride, genistein, or 4°C significantly diminished the cellular uptake of FITC-MLPL, indicating that its internalization mechanism relies on caveolae-mediated and macropinocytosis pathways and appears to be energy-dependent (Figure 2D). In contrast, chlorpromazine markedly affected the uptake of FITC-MLPCL, underscoring clathrin-mediated endocytosis as the predominant internalization pathway.
Laser Confocal Microscopy Imaging
Then, the stability of various formulations and their uptake by MDCK cells were systematically investigated using a confocal laser scanning microscope (CLSM). Intact liposomes were labeled with FITC, while MLP was labeled with Cy5 to prepare FITC/Cy5 dual-labeled liposomes. Following administration, MLP-CL exhibited consistent colocalization of FITC and Cy5 signals, indicating intact delivery into the cytoplasm (Figure 3B). In contrast, inconsistent localization patterns were observed in the MLP-L groups, suggesting potential lysosomal degradation (Figure 3A). When combined with the findings from in vitro release experiments, the colocalized fluorescence of dual-labeled MLP-CL further confirms the structural stability of the liposomes and the sustained intracellular release of the encapsulated cargo.
 |
Figure 3 Intracellular integrity and in vivo imaging. (A) CLSM imaging of MDCK cells treated with MLP-L after 2, 4, and 8 h. MLP (red), liposomes (green), nucleus (blue). (B) CLSM imaging of MDCK cells treated with MLP-CL after 2, 4, and 8 h. MLP (red), liposomes (green), nucleus (blue). (C) In vivo fluorescence images of the heart, liver, spleen, kidneys, lungs, and intestines after administration of MLP-L and MLP-CL. (D) CLSM imaging of intestines after administration of MLP-L and MLP-CL. |
Transcellular Transport Assessments
The MDCK cell transcellular transport assay was employed to evaluate the transcellular transport capability of various formulations (Figure 2C). The results demonstrated that MLP-CL exhibited markedly stronger FITC fluorescence in the lower chamber compared to free MLP, indicating a significant enhancement in the transcellular transport efficiency of MLP. MLP-L slightly improved the transcellular transport capacity of MLP; however, no statistically significant difference was observed. Notably, MLP-CL showed significantly greater accumulation compared to MLP-L (p < 0.05), suggesting that the caged-liposome structure facilitates enhanced transcytosis. This observation, coupled with the previous findings on cellular uptake, shows that MLP-CL may expedite overall cellular translocation and transcytosis across the epithelial barrier. While MLP-CL displayed higher internalization, potential intracellular degradation warrants further investigation to assess its bioavailability.
Distribution of Liposomes in the Intestines
Utilizing in vivo imaging systems (IVIS), the intestinal uptake of both MLP-L and MLP-CL was further validated (Figure 3C). One hour post-administration, both formulations exhibited significant accumulation within the gastrointestinal tract and demonstrated comparable fluorescence intensity. However, in vitro studies highlighted that MLP-CL exhibited higher cellular uptake and enhanced transcellular transport compared to MLP-L, as demonstrated by increased fluorescence intensity and significant accumulation in the lower chamber of the transwell assay (Figure 2C). This indicates that the MLP-CL formulation may facilitate more efficient transcytosis across the epithelial barrier, thereby improving its overall bioavailability.
Then, CLSM analysis of intestinal sections revealed distinct differences in the distribution patterns of MLP-CL and MLP-L (Figure 3D). Intense and widespread fluorescent signals were observed in the intestinal villi and submucosal regions for MLP-CL, significantly exceeding the fluorescence intensity of MLP-L. Combined with the results of cellular uptake assays and transcellular transport assessments, these findings indicate that the caged-MLP structure may enhance both intestinal uptake and penetration efficiency. Additionally, the absence of detectable fluorescence in the liver and kidney suggests that the lymphatic system predominantly governs the in vivo distribution of these formulations (Figure 3C).
In vivo Evaluation of Pharmacokinetics and Lymphatic Transport
Pharmacokinetic Studies
Pharmacokinetic studies of MLP, MLP-L, and MLP-CL were conducted in healthy rats, and the pharmacokinetic parameters for each formulation are summarized in Table 2. The pharmacokinetic data further underscore the superior bioavailability of MLP-CL (Figure 4A and Table 2). The area under the curve (AUC) for MLP-CL was significantly higher compared to the free MLP and MLP-L, indicating improved absorption. Besides, all groups exhibited a plasma half-life (t1/2) of less than 1 hour (Figure 4A). This rapid absorption and elimination may represent inherent characteristics of MLP, consistent with existing literature demonstrating similar behavior for chitosan-based delivery systems. Notably, compared to free MLP, both liposomal formulations, particularly MLP-CL, achieved significantly higher AUC values, indicating a marked enhancement in absorption. The improved absorption is likely attributable to the caged structure of MLP-CL, which enhances the overall structural integrity. This protected MLP from degradation in the gastrointestinal microenvironment, including gastric acid, microbial activity, and metabolic enzymes, ultimately bypassing the gastrointestinal first-pass effect. In addition, lymphatic transport is likely to play a significant role in this process.
 |
Table 2 Pharmacokinetic Parameters of Each Group of Preparations Under Two Conditions (Means ± S.D., n = 3) |
 |
Figure 4 In vivo pharmacokinetic properties of liposomes. (A) Pharmacokinetic properties of FITC-MLP, FITC-MLP-L, and FITC-MLP-CL in healthy rats (n=3). (B-D) Evaluation of lymphatic transport of FITC-MLP (B), FITC-MLP-L (C), FITC-MLP-CL (D) in healthy (without treatment of inhibitor) and in cycloheximide-inhibited rats (n=3). (E, F) Statistical analysis of AUC0-5h (E) and t1/2 (F) of FITC-MLP, FITC-MLP-L, and FITC- MLP-CL. Statistical significance was calculated via two-tailed Student’s t-test. *Indicates P < 0.05, **Indicates P < 0.01, ***Indicates P < 0.001, while ns indicates no significant difference between the two groups. |
Lymphatic Transport Evaluation
To evaluate the contribution of lymphatic transport, a non-invasive cycloheximide-induced chylomicron flow blocking model was employed and the pharmacokinetic procedure was conducted subsequently. By comparing changes in AUC0-5h, the efficiency of lymphatic transport for different formulations could be quantified. The results demonstrated different MLP formulations possessed distinct absorption pathways (Figure 4B–D). Free MLP maintained its AUC0-5h, indicating that it primarily relied on intestinal absorption. MLP-L exhibited an 18.05% reduction in AUC0-5h (Table 2), suggesting that ~18% of its absorption depends on lymphatic transport. In contrast, MLP-CL demonstrated a 63.56% AUC0-5h reduction, indicating that lymphatic transport accounts for >60% of its total absorption (Table 2). This different absorption patterns suggest that the caged liposome alters the absorption mechanism. This may be attributed to the fact that MLP-CL forms an amphiphilic structure that preferentially associates with chylomicrons, thereby enhancing lymphatic uptake and avoiding hepatic first-pass metabolism through the portal circulation. Additionally, treatment with the pathway inhibitor markedly prolonged the t1/2 of MLP across all groups (Figure 4F). This prolongation likely corresponds to the increased AUC0-5h observed in the free MLP group, implying that the inhibitor may have influenced overall circulation or metabolism. Although the extended t1/2 partially compensated for the AUC0-5h reduction in the MLP-L and MLP-CL groups, it might underestimate the actual lymphatic transport rate.
Discussion
In this study, a novel drug-caged liposome (MLP-CL) was developed to investigate its potential in enhancing the oral delivery efficiency of MLP compared with conventional drug-encapsulated liposomes (MLP-L). The drug loading and conjugation efficiency of MLP-CL were optimized, the drug loading and conjugation efficiency of MLP-CL were optimized, achieving conjugation efficiencies of 8.61% and drug loading of 1.22%. Both formulations exhibited particle sizes that were consistent with the expectations for stability and drug delivery. Specifically, MLP-L demonstrated a larger average size of 264.23 nm, while MLP-CL showed a smaller size of 150 nm, which may enhance tissue penetration. Additionally, both liposomal formulations displayed uniform dispersion with PDI below 0.3, ensuring reliable and consistent performance.27
In vitro release profiles under simulated gastric and intestinal conditions revealed that MLP-L released approximately 80% of its drug within four hours in simulated gastric fluid (SGF), while MLP-CL showed a significantly slower release (40%). This sustained release profile ensures that MLP remains caged within liposomes during gastrointestinal transit, facilitating absorption in the form of lipidic nanoparticles via lymphatic pathways. Due to its inherent hydrophilicity, released MLP would otherwise face challenges in transmembrane absorption. Furthermore, the controlled release of MLP from liposomes ensures prolonged circulating time and therapeutic efficacy, addressing the limitations of rapid clearance associated with conventional formulations. This could potentially reduce the frequency of dosing, thereby improving patient compliance, especially in chronic therapies.28 Besides, this steady release could result in fewer fluctuations in drug concentration, which not only enhances therapeutic efficacy but also reduces the risk of adverse side effects that are commonly associated with rapid drug release. Previous studies have consistently shown that slower-release formulations can lead to prolonged therapeutic effects and better control over drug levels in circulation.29
Both MLP-L and MLP-CL exhibited negligible cytotoxicity in MDCK cells, indicating their potential for safe clinical use. Notably, both formulations significantly enhanced cell viability compared to free MLP, likely due to increased stability and bioavailability from liposome encapsulation. MLP-CL demonstrated higher cellular uptake than MLP-L, which may be attributed to the conjugation method facilitating more efficient interaction with cell membranes, promoting improved internalization. The cellular uptake and transcellular transport of MLP-L and MLP-CL were influenced by different internalization pathways. MLP-L uptake was inhibited by amiloride and colchicine, indicating caveolae-mediated endocytosis and macropinocytosis, whereas MLP-CL uptake was significantly reduced by chlorpromazine, pointing to clathrin-mediated internalization.30 This difference likely accounts for the higher uptake of MLP-CL, as supported by MDCK cell transcellular transport assay, which showed significantly greater accumulation of MLP-CL in the lower chamber, suggesting enhanced transcytosis. MLP-CL’s greater intracellular stability and efficient transcellular transport may lead to more consistent and sustained drug delivery, improving drug absorption across epithelial barriers.31
First-pass metabolism, a significant barrier in oral drug delivery, often leads to reduced bioavailability by decreasing the drug amount that reaches systemic circulation. By circumventing first-pass metabolism, MLP-CL not only enhances systemic availability but also enables more precise control over the therapeutic effect, particularly for compounds that are prone to rapid degradation or metabolism in the gastrointestinal tract. Firstly, the caged structure of MLP-CL enhances the overall structural integrity, thereby protecting MLP from degradation in the gastrointestinal microenvironment, including gastric acid, microbial activity, and metabolic enzymes, ultimately bypassing the gastrointestinal first-pass effect. Similar findings have been reported in studies investigating the use of lipid-based carriers to enhance drug absorption and reduce the limitations of gastrointestinal degradation and metabolic clearance.32 Further examination of pharmacokinetics indicated that MLP-CL demonstrated a significantly lesser reduction in AUC values compared to free MLP and MLP-L within the cycloheximide-induced chylomicron blockade model, suggesting that its preferential lymphatic absorption. This could prevent MLP from entering the portal vein, thereby reducing hepatic first-pass metabolism and improving bioavailability.33 These findings are aligned with previous studies on liposomal formulations, which demonstrate the benefits of lymphatic transport in improving bioavailability and reducing the impact of first-pass metabolism.34
This study underscores the potential of liposomal drug delivery systems, particularly MLP-CL, in enhancing the oral bioavailability and therapeutic efficacy of natural compounds like MLP. MLP-CL offers significant advantages over conventional formulations, including enhanced structural integrity, controlled release, and efficient lymphatic uptake. These attributes offer a potential strategy to improve therapeutic outcomes and reduce the challenges of gastrointestinal degradation and metabolic clearance.
Conclusion
This study demonstrates that MLP-caged liposomes (MLP-CL) exhibit significant advantages over conventional encapsulated liposomes (MLP-L) in oral drug delivery. By covalently conjugating MLP to the liposomal surface, MLP-CL achieves enhanced structural stability in the gastrointestinal tract, ensuring intact absorption through lymphatic pathways. This mechanism bypasses hepatic first-pass metabolism, thereby substantially improving systemic bioavailability. Notably, the polysaccharide-lipid conjugation enhances MLP’s lipophilicity, facilitating efficient lymphatic uptake (63.56% for MLP-CL vs 18.05% for MLP-L), which is the basis of its superior pharmacokinetic performance. In summary, MLP-CL represents an effective strategy for oral delivery of hydrophilic polysaccharides. By integrating lymphatic targeting with sustained release, this system addresses challenges in bioavailability and metabolic degradation, offering a promising platform for chronic disease management and polysaccharide compound delivery.
However, this work did not investigate how varying conjugation ratios influence the physicochemical properties or drug release kinetics of MLP-CL, which may affect therapeutic outcomes. Excessive conjugation might impede drug release, while insufficient binding could compromise stability and absorption. Future studies should focus on optimizing conjugation efficiency to obtain more favorable release rate for the optimum absorption. Additionally, evaluating long-term safety and scalability of MLP-CL will be essential for clinical translation.
Ethical Approval Statement
All protocols in this study were approved by the Committee on the Ethics of China Pharmaceutical University Animal Ethical Experimentation Committee, Nanjing, China (2023-04-021), and the authors strictly followed the ethical guidelines by the International Council for Laboratory Animal Science (ICLAS) to minimize animal suffering and ensure humane treatment throughout the experimental process. In order to study the real mechanism of absorption and transport in vivo, SD rats, which are commonly used in pharmacokinetic experiments, were selected as the animal model. All rats housed individually in polycarbonate cages (45 x 35×18 cm) under controlled conditions. A 12-hour light-dark cycle, a temperature range of 20 ± 2 °C, and a humidity of 50-60% was maintained. The rats had ad libitum access to standard chow and water. Environmental enrichment was provided in the form of nesting materials. Isoflurane gas was used for anesthesia in the experiments to reduce the suffering of animals. The mercy endpoint, which is implemented by carbon dioxide inhalation, was administered if the rats lost 20-25% of their body weight, lost their appetite for 24 hours, or ate less than 50% of their normal food intake.
Acknowledgments
This work was financially supported by Taizhou Science and Technology Supporting Agriculture Project (TN202413), Project of Qinglan Engineering in Jiangsu Province (2022), Students Innovation and Entrepreneurship Training Program of Vocational College in Jiangsu Province (G-2023-0726), Jiangsu Agri-animal Husbandry Vocational College Project (NSF2025JB01).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen R, Zhou X, Deng Q, et al. Extraction, structural characterization and biological activities of polysaccharides from mulberry leaves: a review. Int J Biol Macromol. 2024;257(Pt 2):128669. doi:10.1016/j.ijbiomac.2023.128669
2. Chen X, Sheng Z, Qiu S, et al. Purification, characterization and in vitro and in vivo immune enhancement of polysaccharides from mulberry leaves. PLoS One. 2019;14(1):e0208611. doi:10.1371/journal.pone.0208611
3. Zhang L, Su S, Zhu Y, et al. Mulberry leaf active components alleviate type 2 diabetes and its liver and kidney injury in db/db mice through insulin receptor and TGF-β/Smads signaling pathway. Biomed Pharmacothe. 2019;112:108675. doi:10.1016/j.biopha.2019.108675
4. Ai J, Bao B, Battino M, et al. Recent advances on bioactive polysaccharides from mulberry. Food Funct. 2021;12(12):5219–5235. doi:10.1039/D1FO00682G
5. Asai A, Nakagawa K, Higuchi O, et al. Effect of mulberry leaf extract with enriched 1-deoxynojirimycin content on postprandial glycemic control in subjects with impaired glucose metabolism. J Diabet Invest. 2011;2(4):318–323. doi:10.1111/j.2040-1124.2011.00101.x
6. Zhang Y, Miao R, Ma K, et al. Effects and mechanistic role of mulberry leaves in treating diabetes and its complications. Am J Chin Med. 2023;51(7):1711–1749. doi:10.1142/S0192415X23500775
7. Hu TG, Wen P, Shen WZ, et al. Effect of 1-deoxynojirimycin isolated from mulberry leaves on glucose metabolism and gut microbiota in a streptozotocin-induced diabetic mouse model. J Natural Prod. 2019;82(8):2189–2200. doi:10.1021/acs.jnatprod.9b00205
8. Xie Q, Liu X, Xiao S, et al. Effect of mulberry leaf polysaccharides on the baking and staling properties of frozen dough bread. J Sci Food Agric. 2022;102(13):6071–6079. doi:10.1002/jsfa.11959
9. Li Z, Wang L, Lin X, Shen L, Feng Y. Drug delivery for bioactive polysaccharides to improve their drug-like properties and curative efficacy. Drug Deliv. 2017;24(sup1):70–80. doi:10.1080/10717544.2017.1396383
10. Peanparkdee M, Iwamoto S, Borompichaichartkul C, Duangmal K, Yamauchi R. Microencapsulation of bioactive compounds from mulberry (Morus alba L.) leaf extracts by protein–polysaccharide interactions. Int J Food Sci Technol. 2016;51:649–655. doi:10.1111/ijfs.13032
11. Yu Y, Zhang B, Xia Y, et al. Bioaccessibility and transformation pathways of phenolic compounds in processed mulberry (Morus alba L.) leaves after in vitro gastrointestinal digestion and faecal fermentation. J Funct Foods. 2019;60:103406. doi:10.1016/j.jff.2019.06.008
12. Lown M, Fuller R, Lightowler H, et al. Mulberry-extract improves glucose tolerance and decreases insulin concentrations in normoglycaemic adults: results of a randomised double-blind placebo-controlled study. PLoS One. 2017;12(2):e0172239. doi:10.1371/journal.pone.0172239
13. Tchabo W, Kaptso GK, Ngolong Ngea GL, et al. In vitro assessment of the effect of microencapsulation techniques on the stability, bioaccessibility and bioavailability of mulberry leaf bioactive compounds. Food Biosci. 2022;47:101461. doi:10.1016/j.fbio.2021.101461
14. Lu H, Zhang S, Wang J, Chen Q. A review on polymer and lipid-based nanocarriers and its application to nano-pharmaceutical and food-based systems. Front Nutr. 2021;8:783831. doi:10.3389/fnut.2021.783831
15. Gangavarapu A, Tapia-Lopez LV, Sarkar B, Pena-Zacarias J, Badruddoza AZM, Nurunnabi M. Lipid nanoparticles for enhancing oral bioavailability. Nanoscale. 2024;16(39):18319–18338. doi:10.1039/D4NR01487A
16. Wang Y, Zhou Q, Zheng J, et al. Fabricating pectin and chitosan double layer coated liposomes to improve physicochemical stability of beta-carotene and alter its gastrointestinal fate. Int J Biol Macromol. 2023;247:125780. doi:10.1016/j.ijbiomac.2023.125780
17. Peng P, Chen Z, Wang M, Wen B, Deng X. Polysaccharide-modified liposomes and their application in cancer research. Chem. Biol. Drug Des. 2023;101(4):998–1011. doi:10.1111/cbdd.14201
18. Pellá MCG, Lima-Tenório MK, Tenório-Neto ET, Guilherme MR, Muniz EC, Rubira AF. Chitosan-based hydrogels: from preparation to biomedical applications. Carbohydr Polym. 2018;196:233–245. doi:10.1016/j.carbpol.2018.05.033
19. Xian J, Zhong X, Gu H, et al. Colonic delivery of celastrol-loaded layer-by-layer liposomes with pectin/trimethylated chitosan coating to enhance its anti-ulcerative colitis effects. Pharmaceutics. 2021;13(12):2005. doi:10.3390/pharmaceutics13122005
20. Vicente S, Goins BA, Sanchez A, Alonso MJ, Phillips WT. Biodistribution and lymph node retention of polysaccharide-based immunostimulating nanocapsules. Vaccine. 2014;32(15):1685–1692. doi:10.1016/j.vaccine.2014.01.059
21. Kesharwani P, Chadar R, Sheikh A, Rizg WY, Safhi AY. CD44-targeted nanocarrier for cancer therapy. Front Pharmacol. 2021;12:800481. doi:10.3389/fphar.2021.800481
22. Castañeda-Reyes ED, Perea-Flores MJ, Davila-Ortiz G, Lee Y, Gonzalez E. Development, characterization and use of liposomes as amphipathic transporters of bioactive compounds for melanoma treatment and reduction of skin inflammation: a review. Int j Nanomed. 2020;15:7627–7650. doi:10.2147/IJN.S263516
23. Zhao S, Dai W, He B, et al. Monitoring the transport of polymeric micelles across MDCK cell monolayer and exploring related mechanisms. J Control Release. 2012;158(3):413–423. doi:10.1016/j.jconrel.2011.12.018
24. Shen P, Zhang R, McClements DJ, Park Y. Nanoemulsion-based delivery systems for testing nutraceutical efficacy using Caenorhabditis elegans: demonstration of curcumin bioaccumulation and body-fat reduction. Food Res Int. 2019;120:157–166. doi:10.1016/j.foodres.2019.02.036
25. Ohshima H. Chapter 9 - DLVO theory of colloid stability. In: Ohshima H, editor. Interface Science and Technology. Vol. 37. Elsevier; 2024:217–244.
26. Ye J, Gao Y, Ji M, et al. Oral SMEDDS promotes lymphatic transport and mesenteric lymph nodes target of chlorogenic acid for effective T-cell antitumor immunity. J Immunol Ther Cancer. 2021;9(7):e002753. doi:10.1136/jitc-2021-002753
27. Danaei M, Dehghankhold M, Ataei S, et al. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics. 2018;10(2):57. doi:10.3390/pharmaceutics10020057
28. Bhattacharya N, Cahill DM, Yang W, Kochar M. Graphene as a nano-delivery vehicle in agriculture – current knowledge and future prospects. Crit Rev Biotechnol. 2023;43(6):851–869. doi:10.1080/07388551.2022.2090315
29. Teklehaimanot Y. Nanotechnology-enhanced controlled release systems in topical therapeutics. Precis NManomed. 2024;7(4). doi:10.33218/001c.127336
30. Gong J, Wang HX, Leong KW. Determination of cellular uptake and endocytic pathways. Bio-Protocol. 2019;9(4):e3169. doi:10.21769/BioProtoc.3169
31. Hansen ME, Ibrahim Y, Desai TA, Koval M. Nanostructure-mediated transport of therapeutics through epithelial barriers. Int J mol Sci. 2024;25(13):7098. doi:10.3390/ijms25137098
32. Mahor AK, Singh PP, Gupta R, et al. Nanostructured lipid carriers for improved delivery of therapeutics via the oral route. J Nanotechnol. 2023;2023(1):4687959. doi:10.1155/2023/4687959
33. Pandya P, Giram P, Bhole RP, Chang H-I, Raut SY. Nanocarriers based oral lymphatic drug targeting: strategic bioavailability enhancement approaches. J Drug Deliv Sci Technol. 2021;64:102585. doi:10.1016/j.jddst.2021.102585
34. Preeti SS, Malik R. Lipid horizons: recent advances and future prospects in LBDDS for oral administration of antihypertensive agents. Int J Hypertens. 2024;2024:2430147. doi:10.1155/2024/2430147
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.