")
Back to Journals » International Journal of Nanomedicine » Volume 20
Combined Oxaliplatin with 5-Fluorouracil for Effective Chemotherapy Against Gastric Cancer in Animal Model
Authors Ren J, Song M, Ding D, Lan T, Li Y , Liang R, Huang S, Jiang G, You J , Yang J , Chen C, Luan W, Abdullaev B, Huang H , Zhao Y, Wei B
Received 26 February 2025
Accepted for publication 26 May 2025
Published 17 June 2025 Volume 2025:20 Pages 7763—7780
DOI https://doi.org/10.2147/IJN.S520603
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Farooq A. Shiekh
Jiannan Ren,1,* Menglin Song,1,* Dongbing Ding,1,* Tianyun Lan,2 Yiquan Li,1 Rongpu Liang,1 Shengxin Huang,1 Guangchun Jiang,1 Jiarong You,1 Jianming Yang,1 Chi Chen,1 Weiyi Luan,3 Bekhzod Abdullaev,4 He Huang,1 Yang Zhao,1 Bo Wei1
1Department of Gastrointestinal Surgery, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, 510630, People’s Republic of China; 2Central Laboratory, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, 510630, People’s Republic of China; 3Shaoguan University, Shaoguan, 512005, People’s Republic of China; 4Research Department of Biotechnology, New Uzbekistan University, Tashkent, Uzbekistan
*These authors contributed equally to this work
Correspondence: Bo Wei, Department of Gastrointestinal Surgery, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, 510630, People’s Republic of China, Tel +86 20 85252228, Email [email protected]
Introduction: Oxaliplatin (OXA) and 5-fluorouracil (5-Fu) are standard chemotherapy agents used to treat advanced gastric cancer (GC). However, their clinical efficacy is often limited by systemic toxicity, poor tumor selectivity, and suboptimal therapeutic outcomes when administered as monotherapy. These limitations underscore the need for innovative approaches to improve chemotherapy sensitivity and treatment efficacy.
Methods: We developed a glucose-responsive, RGD peptide-functionalized biporous silica nanocarrier (R-BSN). This system integrates hollow glucose oxidase (hGOx)-modified micelles onto hollow mesoporous silica nanoparticles functionalized with RGD peptides, enabling targeted, sustained drug release and inducing a starvation effect in tumor cells.
Results: The glucose-triggered biporous release strategy significantly prolongs the drug release profile, ensuring sustained chemotherapy delivery while simultaneously depleting intratumoral glucose to enhance therapeutic efficacy. This strategy not only increases chemotherapy potency but also exacerbates oxidative stress in tumor cells, leading to the induction of immunogenic cell death (ICD). Furthermore, R-BSN exhibits robust anti-tumor activity in both subcutaneous tumor models and peritoneal metastasis models, supporting its potential for multi-pathway tumor eradication.
Conclusion: The glucose-triggered biporous silica nanocarrier offers a promising strategy to enhance chemotherapy outcomes in advanced gastric cancer, integrating sustained drug release, starvation therapy, and amplification of oxidative stress. This approach holds significant potential for clinical translation in gastric cancer treatment.
Keywords: biporous silica nanocarrier, glucose-responsive, sequential sustained chemotherapy, starvation therapy, advanced gastric cancer
Introduction
Gastric cancer (GC) is one of the most lethal malignancies worldwide, characterized by delayed diagnosis, rapid progression, and a high mortality rate.1,2 Surgical intervention remains the primary treatment strategy for early-stage GC, though the majority of patients are diagnosed at an advanced stage, often with complications such as peritoneal or distant metastases, which limit the eligibility for curative surgery and reduce treatment options.3 In advanced GC, chemotherapy is a critical treatment approach.4 Oxaliplatin (OXA)- and 5-fluorouracil (5-Fu)-based chemotherapies are commonly employed as first-line chemotherapies, and their sequential combination has shown improved therapeutic outcomes, resulting in better overall efficacy in cancer treatment.5–8 Despite initial effectiveness, the long-term outcomes of conventional chemotherapy regimens are often limited by suboptimal drug delivery, drug resistance, limited therapeutic efficacy, and significant side effects.9–11 These challenges underscore the urgent need for novel strategies that effectively target tumor cells, minimize adverse effects, and enhance therapeutic efficacy.
With rapid advancements in nanotechnology, nanomaterials have emerged as promising tools in biomedicine, primarily due to their superior physicochemical properties.12,13 Various nanoformulations have been extensively investigated for applications in drug delivery,14 disease diagnosis,15 and treatment.16,17 Compared with their larger-scale counterparts, nanomaterials offer distinct advantages, including enhanced bioavailability, reduced toxicity, and greater selectivity within biological systems.11,18 Mesoporous silica nanomaterials (MSNs) have emerged as a pivotal class of materials in various scientific and technological domains since their discovery in the late 1990s.19,20 Their distinctive, organized pore structures, typically characterized by pore sizes ranging from 2 to 50 nanometers, offer high surface areas, biocompatibility, and adjustable properties.21 These qualities facilitate a broad range of applications, from drug delivery systems in biomedicine to catalysts in chemical processes.22–25 Among the various types of MSNs, hollow mesoporous silica nanoparticles (HMSNs) stand out due to their unique structures, featuring hollow interiors and mesoporous shells of varying dimensions, which result in low density and a high surface-to-volume ratio.19,26 The mesoporous channels within the shell facilitate the diffusion of guest molecules between the interior and exterior, making HMSNs highly promising pharmaceutical carriers.27 However, challenges remain with traditional HMSN drug carriers, including non-responsive release, drug leakage, insufficient targeting, rapid release, and limited killing efficiency.
To address these issues, we propose a biporous drug delivery system based on hollow porous glucose oxidase micelles that block the pores of drug-loaded HMSNs, preventing premature drug leakage. Our previous research revealed that αvβ3 integrin is significantly overexpressed in GC cells.28 The functionalization of HMSNs with targeting ligands such as RGD peptides can further increase their specificity for tumor cells.29,30 Upon reaching the tumor, the RGD-modified biporous silica nanocarrier (R-BSN) triggers an enzyme-catalyzed reaction between glucose and glucose oxidase in the small porous micelles. This reaction gradually unblocks the nanopores, enabling extended and controlled drug release. Additionally, the substantial glucose consumption during this process induces starvation in tumor cells, further enhancing the efficacy of chemotherapy (scheme 1). Both in vitro and in vivo experiments demonstrated that the R-BSN increases oxidative stress through glucose depletion, inducing immunogenic cell death while ensuring precise drug delivery. Compared with free drugs, sequential administration of OXA and 5-Fu via the R-BSN significantly suppressed tumor growth and peritoneal metastasis, indicating superior antitumor effects. This nanomedicine shows significant promise as a potential treatment for advanced GC.
Materials and Methods
Cell Line and Animal Models Used in the Study
The HGC-27 cells, obtained from BIOSPECIES, were cultured in DMEM supplemented with 10% FBS and 1% penicillin‒streptomycin. Additionally, BALB/C nude mice were obtained from the SPF (Beijing) biotechnology.co.Ltd. The animal experiments were conducted in compliance with ethical standards as approved by the Animal Experimental Ethics Committee of Sun Yat-sen University (SYXK2022-0081), under approval number SYSU-IACUC-2024-B1560. Ethical guidelines were strictly followed during the entire experimental process.
The Development of Biporous Silica Nanocarriers
Synthesis of Hollow Mesoporous Silica Nanoparticles
First, 480 mL of deionized water was heated to 80°C in a round-bottom flask equipped with a magnetic stirrer. Then, 1.0 g of CTAB was added, followed by 3.5 mL of NH4OH, and the mixture was stirred at 600 rpm for 30 minutes. Next, 5 mL of TEOS was added dropwise while stirring vigorously for 2 hours to facilitate the formation of silica nanoparticles. The resulting silica nanoparticles were collected by washing three times and drying them for further use.
One gram of dried silica nanoparticles was dispersed in 100 mL of cyclohexane in a round-bottom flask, and 1.0 g of PVA and 0.1 g of NaOAc were added. The mixture was stirred at 60°C at 600 rpm to allow the PVA to form a soft template around the silica cores. After 6 hours, 5 milliliters of TEOS was slowly added, and the mixture was stirred for another 2 hours. Cyclohexane was then removed by evaporation under reduced pressure. The resulting material was calcined at 550°C for 6 hours to remove the PVA template and create a hollow structure.
The calcined HMSN was dispersed in 100 mL of an ethanol-HCl solution and refluxed for 6 hours to remove the CTAB surfactant. The purified HMSN was collected by centrifugation and washed multiple times with ethanol and deionized water, and then dried under vacuum at 60°C overnight.
Amino Functionalization of Hollow Mesoporous Silica Nanoparticles
HMSN (1.0 g) was dispersed in 100 mL of anhydrous ethanol. To this suspension, 2 mL of APTES was added, resulting in a volume-to-mass ratio of 2 mL APTES per gram of HMSN. The mixture was stirred (300 rpm) at room temperature for 12 hours to ensure complete functionalization. Finally, the amino-functionalized HMSN was collected and washed three times to remove any unreacted APTES.
Chemotherapeutic Drug Loading of Hollow Mesoporous Silica Nanoparticles
To load OXA and 5-Fu into HMSNs, HMSN was dispersed in deionized water, and the chemotherapeutic agents were added separately. The chemotherapeutic agent-to-HMSN mass ratio was set at 3:1 for both OXA and 5-Fu to achieve optimal loading efficiency and therapeutic efficacy. The mixture was stirred at room temperature for 12 hours to allow drug adsorption into the mesopores of the HMSNs. After the loading process, the drug-loaded HMSNs were separated from the solution by ultrafiltration to remove any unbound drugs. The drug loading capacity (DLC) and encapsulation efficiency (EE) were calculated according to the following formulas:
DLC (%) = (Drug mass) / (Carrier material mass + Drug mass) × 100%.
EE (%) = (Encapsulated drug mass) / (Total drug mass) × 100%.
Synthesis of Hollow Micelles Loaded with Glucose Oxidase
To synthesize the hollow micelles, 0.3 g of Pluronic F127, 0.2 g of potassium fluoride (KF), 2 mg of GOx from Aspergillus niger, and 500 μL of TMB were added to 15 mL of deionized water. The mixture was stirred vigorously at room temperature at 1000 rpm for 2 hours. Next, 1.5 g of TMOS was added dropwise while stirring the mixture at 1000 rpm. After reacting for 1 hour at room temperature, the mixture was dialyzed in deionized water for 48 hours using a dialysis membrane with a molecular weight cutoff of 200,000 to remove byproducts and unreacted reagents.
Synthesis of RGD Peptide-Functionalized Biporous Silica Nanocarriers
The amino-functionalized HMSN was mixed with the pre-synthesized H-micelle-GOx and incubated for 12 hours, followed by centrifugation at 10,000 rpm for 5 minutes to separate the BSN nanoassemblies. In the NHS/EDC coupling reaction, the RGD peptide was allowed to react with GOx/HMSN-NH2 in PBS buffer at 25°C for 12 hours. Finally, the R-BSN was washed and collected by ultrafiltration centrifugation at 10000 rpm.
Characterization of Biporous Silica Nanocarriers
The size and zeta potential of the R-BSN were measured via a Zetasizer Nano ZS90 (Malvern, UK), with each sample undergoing three measurements of 12 runs. The morphology of the R-BSN was assessed through transmission electron microscopy (TEM) (HT7700, Hitachi, Japan) after the samples were diluted 100-fold with distilled deionized water. Additionally, high-angle annular dark field-scanning transmission electron microscopy was employed to further evaluate the morphology. Elemental analysis of the R-BSN was performed using energy dispersive spectroscopy (EDS) for both elemental mapping and spectral analysis.
The cumulative release of the R-BSN under different conditions was measured using the dialysis method (MWCO Da) at 37°C in a water bath shaker. At predetermined time intervals, the release medium was collected and analyzed using a UV‒vis spectrophotometer to measure R-BSN at a specific wavelength.
Hemolysis Assay for Biocompatibility Evaluation
To assess the biocompatibility of the HMSN and H-micelles, a hemolysis assay was performed using blood from BALB/C nude mice. Briefly, fresh blood was collected and was then anticoagulated using heparin. The collected blood was diluted with PBS to a final concentration of 2%. Different concentrations of HMSN and H-micelles (5, 10, 20, 50, and 100 µg/mL) were incubated with the diluted blood for 1 hour at 37°C. As a positive control, water was used, which causes complete hemolysis, while PBS was used as a negative control, ensuring that no hemolysis occurs under normal conditions. After incubation, the samples were centrifuged at 3,000 rpm for 5 minutes, and the absorbance of the supernatant was measured at 540 nm using a microplate reader.
Cellular Uptake of Biporous Silica Nanocarriers
HGC-27 cells were seeded at a density of 1×104 cells per well in 35 mm dishes for confocal laser scanning microscopy (CLSM). The cells were subsequently coincubated with Nile red-loaded nanoparticles for various durations. After incubation, the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime, Shanghai, China) for visualization. The cells were then fixed with glutaraldehyde and washed thoroughly with PBS. Finally, the cellular uptake of Nile Red was observed and assessed using CLSM. Additionally, flow cytometry was employed to quantify the cellular uptake of Nile red-loaded nanoparticles. The cells were harvested after incubation, and the fluorescence intensity was measured using flow cytometry, providing a quantitative analysis of the nanoparticle uptake levels across different treatment groups.
Cell Counting Kit-8 Assay for Cytotoxicity Evaluation
CCK-8 assays were conducted to evaluate the cytotoxicity of the nanodrugs. Briefly, HGC-27 cells were seeded into 96-well plates at a density of 10,000 cells per well. The cells were categorized into the following groups: PBS, 5-Fu, OXA, BSN(5-Fu), BSN(OXA), R-BSN(5-Fu), and R-BSN(OXA). Following incubation, 10 µL of CCK-8 reagent was added to each well, and the plates were further incubated at 37°C for 20 minutes. The absorbance was measured at 450 nm using a microplate reader (ELX800, Biotek, USA). Cell viability was calculated by using the control group of cells incubated in the medium as a reference.
Live/Dead Viability Assay for Cytotoxicity Evaluation
HGC-27 cells (3×104 cells per dish) were seeded into 24-well plates. The following treatments, all at equivalent concentrations of 5-Fu(4µg/mL) or OXA(1µg/mL), were added to different dishes: PBS, 5-Fu, OXA, BSN(5-Fu), BSN(OXA), R-BSN(5-Fu), R-BSN(OXA), and the sequential treatment group (R-BSN(OXA) + R-BSN(5-Fu)). For the sequential treatment group, R-BSN(OXA) was administered for 24h, followed by PBS washes, and then incubated with R-BSN(5-Fu) for an additional 24h. After incubation, the cells were stained with calcein acetoxymethyl ester (AM) and propidium iodide (PI) (KeyGEN BioTECH, China). The cells were then observed using an inverted fluorescence microscope.
Wound Healing Assay for Cell Migration Evaluation
HGC-27 cells were cultured in 6-well plates (1×105 cells per dish) and incubated for 24 hours. The following treatments were then applied: PBS, 5-Fu, OXA, BSN(5-Fu), BSN(OXA), R-BSN(5-Fu), R-BSN(OXA), and the sequential treatment group with R-BSN(OXA) followed by R-BSN(5-Fu). After the incubation period, HGC-27 cells were collected and reseeded into fresh 6-well plates (1×105 cells per dish) to form a monolayer. A wound was then created in the monolayer using a 200µL pipette tip, and the detached cells and debris were washed away with fresh media. Cell migration into the wound area was observed and monitored over time via microscopy.The wound closure rate was quantified by comparing the area of the wound at different time points.
Transwell Assay for Cell Invasion Evaluation
HGC-27 cells were cultured in 6-well plates (1×105 cells per dish) for 24 hours, after which the following treatments were administered: PBS, 5-Fu, OXA, BSN(5-Fu), BSN(OXA), R-BSN(5-Fu), R-BSN(OXA), and sequential treatment with R-BSN(OXA) followed by R-BSN(5-Fu). The cells were then collected and resuspended in serum-free medium, then added to the Transwell chambers for a 24-hour incubation. After 24 hours of incubation, the cells were fixed with 4% paraformaldehyde and stained with 1% crystal violet. The remaining cells on the upper surface of the transwell membrane were gently removed, and the cells that migrated to the lower surface were counted under a microscope.The invasion rate was calculated by comparing the number of migrated cells in treated groups versus the control group.
Apoptosis Assay and Western Blot Analysis
HGC-27 cells were seeded into 12-well plates (5×104 cells per dish) and cultured for 24 hours. After incubation, the cells were treated with PBS, 5-Fu, OXA, BSN(5-Fu), BSN(OXA), R-BSN(5-Fu), R-BSN(OXA), or R-BSN(OXA) followed by R-BSN(5-Fu), all at IC50s of 5-Fu and OXA. Following treatment, the cells were harvested, gently washed, resuspended in binding buffer, stained with Annexin V-FITC and PI. After a 15-minute incubation at room temperature in the dark, the samples were immediately analyzed by flow cytometry to assess the percentage of apoptotic cells.
For Western blot analysis, the treated cells were collected, and whole-cell proteins were extracted using a cell lysis buffer. The protein concentrations were determined using a BCA protein assay. Equal amounts of protein (20 µg) were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were incubated with primary antibodies against apoptosis-related proteins, including caspase3, cleaved caspase3, Bcl-2, Bax, and caspase9 at 4°C overnight. After washing, the membranes were incubated with secondary antibodies for 1 hour at room temperature. Protein bands were detected using ECL chemiluminescent substrate and analyzed using a chemi-doc imaging system.
Detection of Reactive Oxygen Species
HGC-27 cells were seeded into 6-well plates (5×104 cells per dish) and incubated for 24 hours. Following that, each group was treated with PBS, GOx, H-micelle-GOx, BSN, or R-BSN, respectively. After treatment, the cells were washed with PBS, resuspended in serum-free medium containing 10 µM DCFH-DA, and incubated at 37°C in the dark for 30 minutes to allow dye uptake and oxidation. After incubation, the excess dye was removed, and ROS levels were assessed using flow cytometry.
Determination of Immunogenic Cell Death Biomarkers
To detect calreticulin (CRT) expression, HGC-27 cells were seeded into 12-well plates at a density of 3×104 cells per well and incubated for 24 hours. The cells were then treated with PBS, HMSN, BSN, OXA, BSN(OXA), or R-BSN(OXA). After 24 hours, the cells were harvested, stained with a CRT antibody (1 µg/1x106 cells) at 4°C overnight, and then incubated with a fluorescent secondary antibody (1:1000 dilution) at room temperature for 1 hour. CRT expression levels in each group were then detected using flow cytometry.
We further validated CRT expression, HGC-27 cells were seeded onto glass coverslips at a density of 1×104 cells per well. The cells were treated with PBS, HMSN, BSN, Oxaliplatin (OXA), BSN(OXA), or R-BSN(OXA) and incubated for 24 hours. Afterward, the cells were fixed with 4% paraformaldehyde for 15 minutes, stained with a CRT antibody (1 µg/mL) at 4°C overnight, and then incubated with a fluorescent secondary antibody (1:1000 dilution) at room temperature for 1 hour. The cells were then counterstained with DAPI for 10 minutes, and CRT expression was observed using CLSM.
HMGB1 and ATP are also important markers of ICD. To assess their release, HGC-27 cells were seeded into 24-well plates at a density of 1×104 cells per well and incubated for 24 hours. After incubation, the cells were treated with PBS, HMSN, BSN,OXA, BSN(OXA), or R-BSN(OXA). After 24 hours of treatment, the cell supernatants were collected and analyzed for HMGB1 and ATP levels using ELISA kits according to the manufacturer’s instructions.
The in-vivo Studies Using Subcutaneous Animal Model
Male BALB/c nude mice, aged 4–6 weeks, were obtained from SPF (Beijing) Biotechnology Co., Ltd. All animal procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Sun Yat-sen University. To establish the subcutaneous tumor model, HGC-27 cells were injected subcutaneously into the right anterior axillary region of the mice. Tumor size and body weight of the mice were measured regularly. Tumor volume was calculated using the formula: length × width² × 0.5.
To verify the in vivo targeting ability of R-BSN, we employed a subcutaneous tumor model for in vivo fluorescence imaging. Briefly, DiR-labeled R-BSN were intravenously injected into tumor-bearing mice via the tail vein. Fluorescence imaging was performed using the Carestream IS 4000 system (USA) at designated time points post-injection. At 48 hours after administration, in vivo fluorescence imaging was conducted to capture images of the tumors and main organs.
Finally, in vivo antitumor assays and toxicity evaluation were conducted. When the tumor volume reached approximately 250 mm³, the mice bearing tumors were randomly assigned to eight different groups, with six mice in each group. Prior to group assignment, a consistency assessment was performed to ensure uniform tumor size across the groups. Each group received 200 µL of PBS, 5-Fu, OXA, BSN(5-Fu), BSN(OXA), R-BSN(5-Fu), R-BSN(OXA) or R-BSN(OXA) + R-BSN(5-Fu) every three days, with 5-Fu and OXA doses of 40 mg/kg and 25 mg/kg body weight, respectively. After five cycles of treatment, the mice were sacrificed. Tumors were then dissected for H&E, Ki67, TUNEL, and CRT staining.
To assess the in vivo toxicity of nanodrugs, serum samples from the tumor-bearing mice were analyzed for myelosuppression (WBC, RBC, PLT), liver function (AST, ALT) and renal function (CREA) at the end of the treatment period. Additionally, the major organs were harvested and subjected to H&E staining to assess any possible organ damage.
The in-vivo Studies Using Peritoneal Metastasis Model
To assess the therapeutic efficacy of nanodrugs in treating peritoneal metastasis, male BALB/c nude mice were intraperitoneally injected with HGC-27 cells. Ten days post-inoculation, the mice were randomly divided into six treatment groups, with six mice in each group. Each group received PBS, 5-Fu, OXA, R-BSN(5-Fu), R-BSN(OXA) or R-BSN(OXA) + R-BSN(5-Fu), with 5-Fu or OXA doses of 40 mg/kg or 25 mg/kg body weight, respectively. Treatments were administered every 3 days for a total of five cycles. Instances of tumor nodules exceeding 0.5 mm in diameter within the abdominal cavity were documented, and the nodules were further assessed.
Statistical Analysis
The data are presented as the means ± standard deviations (SDs). Differences between two groups were assessed via an unpaired Student’s t test, whereas comparisons among multiple groups were analyzed via one-way analysis of variance (ANOVA) followed by Tukey’s HSD post-hoc test (GraphPad Prism 9, San Diego, CA, USA). Statistical significance is denoted as *p < 0.05, **p < 0.01, ***p < 0.001, and ****P < 0.0001.
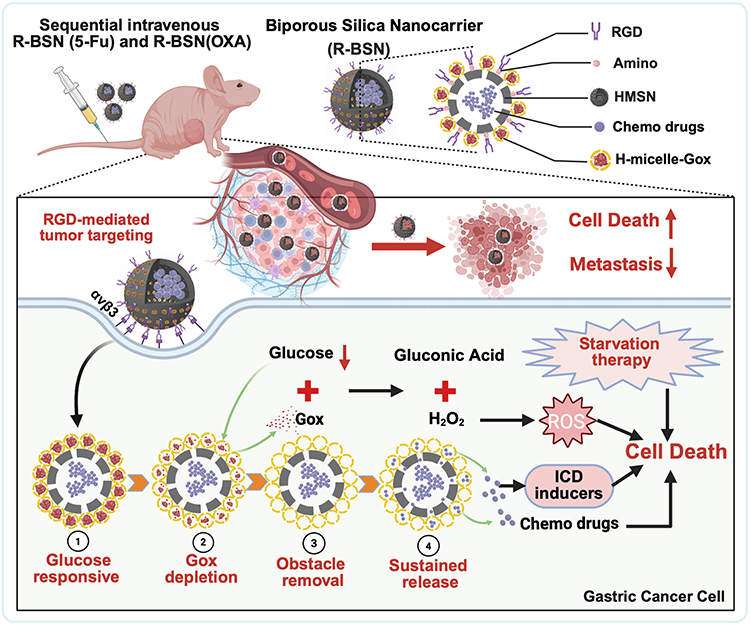 |
Scheme 1 Schematic diagram of a glucose-responsive biporous silica nanocarrier designed to against advanced GC and peritoneal metastasis. |
Results and Discussion
Preparation and Characterization of Nanocarrier
To enhance the bioavailability, biosafety, and therapeutic efficacy of OXA and 5-Fu, a biporous silica nanocarrier (R-BSN) was developed. This innovative system uses hollow mesoporous silica nanoparticles (HMSNs) as the core, with an externally anchored glucose oxidase (GOx)-based porous micelle. The HMSNs enable excellent drug-loading capacity. However, the presence of mesopores may lead to premature drug leakage, which can reduce drug delivery efficacy. To address this issue, hollow GOx silica micelles (H-millce-GOx) were synthesized and anchored onto the surface of HMSNs through electrostatic interactions. To further improve the active targeting ability of these nanocarriers, RGD peptides were functionalized onto the surface, resulting in the formation of an RGD-modified biporous silica nanocarrier. Figure 1A schematically illustrates the drug loading and release process, exemplified by the R-BSN(OXA), showcasing the drug synthesis and controlled release mechanisms.
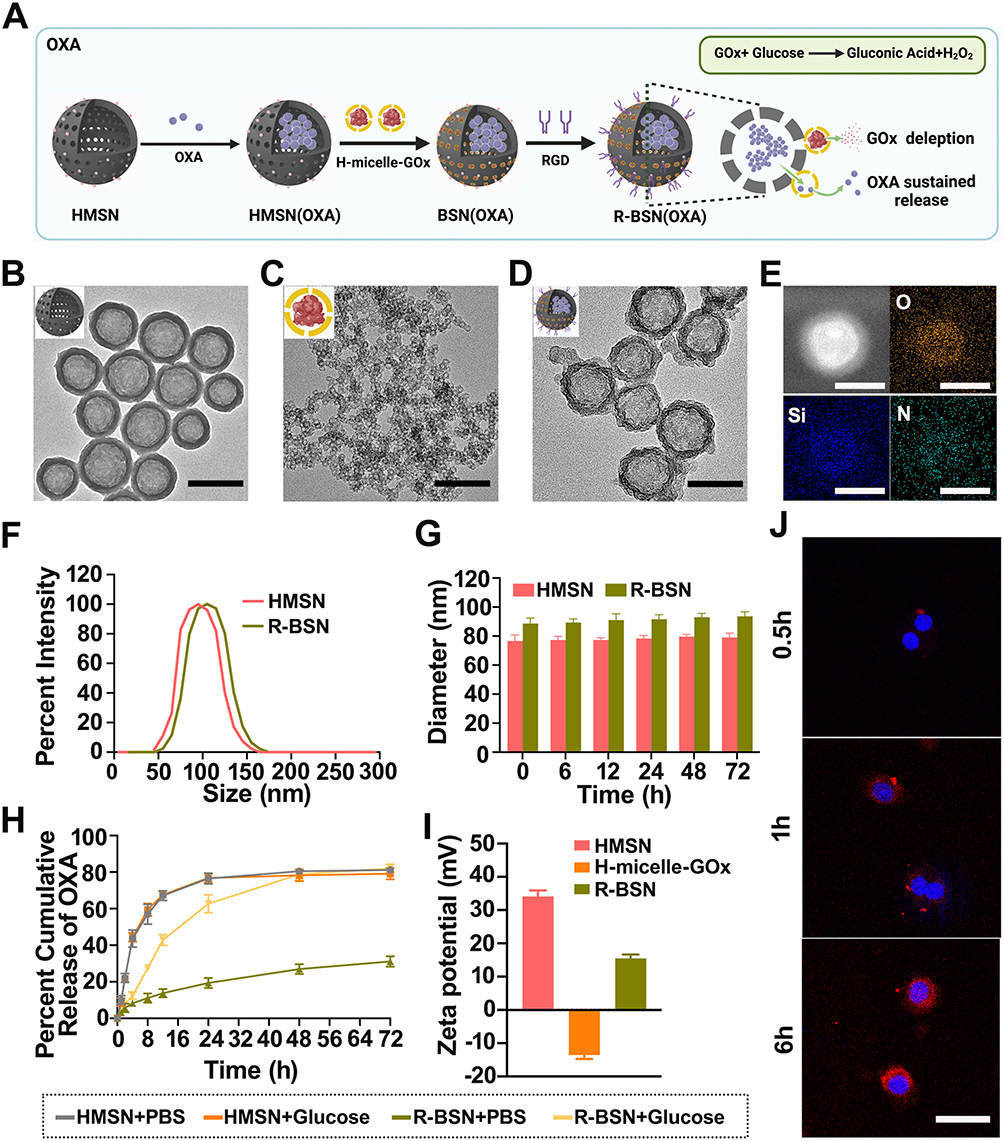 |
Figure 1 Characterization of the RGD-modified biporous silica nanocarrier (R-BSN). (A) Schematic illustration showing the formulation of OXA in RGD- and H-micelle-Gox-based HMSNs. (B–D) TEM images of the HMSN (B), H-micelle-Gox (C), and R-BSN (D), scale bar, 100 nm. (E) HAADF-STEM image and the corresponding EDS elemental mappings of one typical R-BSN nanoparticle; scale bar: 100 nm. (F) Particle size distribution of the nanodrugs. (G) Stability of the nanodrugs in vitro. (H) OXA release profiles of R-BSNs. (I) Zeta potential distribution of the nanodrugs. (J) In vitro cellular uptake of the nanodrugs. Scale bar, 50 µm. |
The morphology of the nanocarriers was analyzed via TEM. As shown in Figure 1B and C, both HMSNs and H-millce-GOx exhibited nearly spherical shapes and were evenly distributed. The hollow structures of HMSNs were clearly visible. Following the grafting of H-millce-GOx to HMSNs, a thin outer layer was observed (Figure 1D). Furthermore, HAADF-STEM and EDS elemental mapping (Figure 1E) confirmed the distribution of Si, O, and N elements within the R-BSN, demonstrating the successful functionalization of the RGD peptide onto the nanocarrier surface. Furthermore, DLS measurements revealed that the particle sizes of HMSNs and R-BSN were 76.03±0.34 nm and 97.62±0.57 nm, respectively (Figure 1F). The particle size was monitored continuously for 72 hours to assess the stability of both HMSN and R-BSN in PBS containing 10% FBS. Throughout the observation period, neither nanoparticle exhibited significant changes in particle size, indicating good stability (Figure 1G).
To evaluate the drug loading capacity (DLC) and encapsulation efficiency (EE%) of the R-BSN, we found that R-BSN(OXA) and R-BSN(5-Fu) had drug loading capacities of 33.9 ± 1.2% and 31.5 ± 0.9%, respectively. The encapsulation efficiencies for R-BSN(OXA) and R-BSN(5-Fu) were 49.3 ± 0.7% and 41.6 ± 0.6%, respectively. Furthermore, we examined the release profiles of the R-BSN under both PBS and glucose conditions. As shown in Figures 1H, Figure S1A–D, and Figure S2, the chemotherapy drugs OXA and 5-Fu loaded in HMSN were rapidly released in PBS, reaching their maximum release levels. In contrast, drug release from the R-BSN system was minimal, indicating that H-millce-GOx effectively blocked the HMSN pores. However, in a glucose-rich environment, due to GOx consumption, the cumulative release of OXA and 5-Fu gradually increased, with both reaching nearly 80% after 72 hours, demonstrating a controlled and sustained drug release profile. These results suggest that H-millce-GOx can effectively block the pores of HMSNs, thereby reducing premature drug leakage and allowing for more prolonged and controlled release of therapeutic drugs.
Moreover, the zeta potentials of HMSNs and H-millce-GOx were 36.83 ± 2.19 mV and −14.34 ± 1.26 mV, respectively (Figure 1I), showing opposite surface charges. Based on electrostatic attraction, hollow micelle-GOx adsorbs onto the HMSN core, enabling BSN synthesis. The average zeta potential of R-BSN was 13.21 ± 1.04 mV, indicating a positive charge that facilitates strong electrostatic interaction with the negatively charged tumor cell membranes, promoting drug accumulation in tumor cells. As shown in Figure 1J and Figure S3, R-BSN was efficiently absorbed by HGC-27 tumor cells, confirming its potential for targeted drug delivery. Upon blocking αvβ3 integrin with a specific antibody, the uptake of R-BSN was significantly reduced compared to BSN, confirming successful RGD peptide modification.
In vitro Antitumor Effects of the Nanomedicine
OXA and 5-Fu are key chemotherapeutic agents used to treat GC because of their potent tumoricidal effects.31 OXA interferes with DNA replication by forming platinum–DNA adducts, while 5-Fu inhibits thymidylate synthase, disrupting DNA synthesis and inducing cell death. The combination of these drugs offers enhanced therapeutic efficacy by complementing each other’s actions, promoting tumor cell death and reducing resistance.32–34 However, side effects, resistance, and limited efficacy remain challenges.
In this study, the cytotoxicity of HMSNs and H-micelles was evaluated using CCK-8 assays. The results showed that both HMSNs and H-micelles exhibited no significant cytotoxicity, indicating good biocompatibility (Figure 2A and Figure S4), which was further confirmed by hemolysis assay results (Figure S5). CCK-8 assays were also used to assess the cytotoxicity of various treatments. The results demonstrated that the nanodrugs loaded with chemotherapeutic agents displayed significantly greater tumoricidal effects than traditional chemotherapy drugs. These findings underscore the potential of the R-BSN platform as an effective strategy for enhancing the efficacy of chemotherapeutic agents.
 |
Figure 2 In vitro antitumor effects of the nanodrugs. (A) The cytotoxicity of HMSN and H-micelles to HGC-27 cells in vitro was evaluated via CCK-8 assays, which revealed the inhibitory effects of the nanodrugs on cell viability. (B) Cytotoxicity of different drug formulations to HGC-27 cells in vitro evaluated via CCK-8 assays; the antitumor effects of various treatments were compared. (C and D) Transwell invasion assays illustrating the changes in the invasive capacity of HGC-27 cells after treatment with different drug formulations. Scale bar, 50 µm. (E and F) Wound healing assays were used to detect the migration ability of HGC-27 cells after treatment with different drugs, indicating the effectiveness of the treatments in inhibiting cell migration. Scale bar, 50 µm. (G) Live/dead staining of HGC-27 tumor cells after different treatments, providing a visual representation of cell viability and cytotoxicity. Scale bar, 50 µm. (H and I) Flow cytometry analysis of apoptosis in HGC-27 cells treated with various drugs. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
Invasion and migration capabilities are crucial indicators of tumor activity and malignancy. As shown in Figure 2C and D, transwell invasion assays indicated that treatment with nanodrugs significantly reduced cell invasion, with the sequential treatment group exhibiting the greatest reduction. Similarly, a scratch assay demonstrated a significant reduction in cell migration in the R-BSN drug-loaded treatment group (Figure 2E–F). These results collectively highlight the efficacy of the nanodrugs in inhibiting both cell invasion and migration, underscoring their potential to effectively control tumor progression and metastasis.
The cytotoxic effects of the nanodrugs on GC cells were assessed using calcein-AM/PI double staining and apoptosis assays. The R-BSN (OXA) and R-BSN (5-Fu) sequential treatment group exhibited significantly higher levels of cell death compared to the conventional treatment group and single nanodrug treatment groups (Figure 2G). The percentage of apoptotic cells in R-BSN (OXA) and R-BSN (5-Fu) sequential treatment group was 59.9%, showing a similar trend (Figure 2H and I). Furthermore, we explored the underlying apoptotic mechanisms, and Western Blot analysis revealed that the R-BSN nanocarriers enhanced the mitochondrial (intrinsic) pathway, as evidenced by the activation of Bax and cleaved caspase3, alongside the downregulation of the anti-apoptotic protein Bcl-2 (Figure S6). These findings underscore that R-BSN significantly enhances the overall anti-tumor effect of conventional chemotherapy drugs, with even more pronounced effects in sequential treatment.
Starvation Effect and Oxidative Stress Levels Induced by the Nanocarrier
Tumor cells exhibit a high metabolic rate and glucose uptake to sustain their rapid growth and proliferation.35 Starvation therapy targets this by restricting glucose uptake, limiting the resources needed for tumor cell proliferation, and enhancing the effectiveness of traditional cancer treatments.36,37 One promising approach involves using glucose oxidase (GOx) to deplete intracellular glucose, thereby limiting the energy and nutrients essential for tumor survival.38 In addition to disrupting tumor metabolism, glucose depletion produces gluconic acid, lowering the local pH and promoting chemotherapeutic drug release.39 GOx-induced glucose oxidation generates H2O2, which reacts with glutathione (GSH) in tumor cells, leading to oxidative stress, cell damage, and apoptosis.40 This dual mechanism both kills tumor cells and increases their sensitivity to chemotherapy and immunotherapy, providing a comprehensive approach to cancer treatment.
Since GOx is insoluble and prone to aggregation, delivering sufficient amounts to tumor cells remains challenging.41 In this study, we synthesized H-millce-GOx and coated them on the surface of HMSNs, which prevented premature drug release while allowing efficient delivery of GOx to the tumor site. As shown in Figure 3A and B, when R-BSN was dissolved in a glucose solution, both the glucose concentration and pH of the solution significantly decreased compared to the HMSN and PBS groups. This indicates that the R-BSN effectively depletes glucose, functioning as intended.
 |
Figure 3 Multiplex induction of tumor cell death by the R-BSN. (A) Time-dependent variations in the concentration of glucose in the solution under different conditions. (B) pH variation of the solution under different conditions. (C and D) Flow cytometry analysis of ROS levels in HGC-27 cells treated with the R-BSN, including quantitative analysis of MFI. (E and F) Flow cytometry analysis of CRT expression on the membrane of HGC-27 cells treated with R-BSN, including quantitative analysis of the MFI. (G) The release of HMGB1 from HGC-27 cells following different treatments. (H) ATP release from HGC-27 cells following different treatments. (I) Movement of CRT to the outer surface of HGC27 cells following different treatments. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Abbreviation: ns, not significant. |
We further investigated the production of ROS in HGC-27 cells treated with different drugs. As shown in Figure 3C and D, the mean fluorescence intensity (MFI) of DCF in the PBS and HMSN groups was similar, with no statistically significant difference. However, the MFI of DCF in the R-BSN and R-BSN(OXA) groups significantly increased. These results demonstrate that R-BSN-mediated starvation therapy can induce the production of substantial amounts of ROS within HGC-27 tumor cells, contributing to tumor cell death and highlighting the role of oxidative stress in the therapeutic mechanism.
Previous studies have shown that interfering with tumor metabolism triggers ICD in tumor cells, leading to the release of damage-associated molecular patterns (DAMPs) that modulate the tumor immune microenvironment, with ROS further enhancing this effect.37,42 Additionally, OXA has been shown to induce ICD, further activating immune cells to target and attack cancer cells.43 In this study, we observed a significant increase of CRT on the cell membrane in the OXA and R-BSN treatment groups, with the effect being most pronounced in the R-BSN(OXA) group (Figure 3I). Flow cytometry further confirmed these findings, revealing that the MFI of CRT was significantly higher in the OXA, R-BSN, and R-BSN(OXA) groups compared to the PBS and HMSN groups (P < 0.0001) (Figure 3E and F). Additionally, the ELISA results revealed substantial increases in the levels of ATP and HMGB1 in the OXA, R-BSN, and R-BSN (OXA) treatment groups compared with those in the other groups (Figure 3G and H), which is consistent with the CRT expression data. These findings suggest that R-BSN can enhance the ICD-inducing effect of OXA, similar to strategies that utilize dendritic nanomedicines to remodel the tumor stroma and improve drug delivery, as well as those that modulate the tumor immune microenvironment to boost chemo-immunotherapy.44,45 This highlights a promising combination strategy to improve the efficacy of immunotherapy.
In vivo Antitumor Efficacy of Nanomedicines
Nanoparticles of appropriate size can exploit the enhanced permeability and retention (EPR) effect observed in tumors, allowing them to pass through the leaky vasculature characteristic of tumor tissues and remain there for extended periods.46,47 However, the EPR effect offers only modest specificity, typically increasing drug concentrations at tumor sites by 20–30% compared to vital normal organs.47 RGD peptides are known to specifically bind to integrins, such as αvβ3, which are overexpressed on the surface of GC cells and their vasculature.28,48 This active targeting enhances the selective delivery of nanoparticles to the tumor, improving therapeutic efficacy.
As shown in Figure 4A, the fluorescence intensity of DiR-labeled R-BSN in the tumor area progressively increased, this highlights the superior tumor-targeting capability of R-BSN. These results demonstrate that R-BSN effectively accumulates at the tumor site in in vivo, which is essential for the therapeutic efficacy of the nanomedicine.
 |
Figure 4 Evaluation of the therapeutic efficacy of the nanodrug against GC in vivo. (A) Fluorescence imaging of HGC-27 tumor-bearing mice at different time points and fluorescence imaging of major organs and tumors 48 hours after tail vein injection of DiR-labeled R-BSN, demonstrating the nanodrug’s in vivo distribution and tumor-targeting efficiency. (B) Schematic illustration of the therapeutic strategy employed in the subcutaneous tumor model, providing a visual summary of the treatment protocol. (C) Photographs of subcutaneous tumors excised from mice subjected to various treatments, highlighting the comparative reduction in tumor size across different treatment groups. (D) Comparison of tumor weight among the different treatment groups revealed a significant reduction in tumor mass with nanodrug treatment. (E) Subcutaneous tumor growth curves for mice receiving different treatments (n = 6), with data presented as the mean ± standard error of the mean, showing the efficacy of the nanodrug in inhibiting tumor growth. (F) Changes in the body weights of the mice during the treatment period (n = 6), reflecting the systemic effects and potential toxicity of the treatments. (G) H&E staining, Ki-67 staining, and TUNEL staining of tumor slices from different groups, providing histological and molecular evidence of cell proliferation, apoptosis, and overall treatment efficacy. Scale bar: 50 μm. (H) IHC staining of CRT in tumors post-treatment to assess the impact of the nanodrug on the ICD of the tumor. Scale bar: 50 μm. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
Building on the significant antitumor effects of the nanodrugs observed in vitro, we further evaluated their efficacy in a subcutaneous tumor model. The establishment and treatment protocol for the mouse model are shown in Figure 4B. Tumor volume and mouse body weight were monitored throughout the treatment period. At the end of the treatment, subcutaneous tumors were excised from the mice (Figure 4C), and their diameters and masses were measured. As shown in Figure 4D and E, tumors in the PBS group grew rapidly, while mice treated with various drug formulations exhibited significant tumor growth inhibition. A comparison between the nanocarrier-treated groups and conventional chemotherapy groups demonstrated that R-BSN had superior efficacy. Additionally, the sequential treatment yielded the best results, highlighting the significant synergistic antitumor effects of the nanoparticles. Furthermore, RGD-modified nanoparticles outperformed the non-RGD-modified groups, emphasizing the excellent tumor targeting and precise drug delivery capabilities of the R-BSN platform.These findings reinforce the effectiveness of R-BSN nanocarriers in preclinical tumor models and suggest a promising path for clinical translation. The improved tumor targeting and enhanced chemotherapy efficacy make R-BSN a strong candidate for further development.
Although there was a slight decrease in body weight in the nanoparticle-treated mice during treatment, the difference was not statistically significant compared to the other groups (Figure 4F), indicating no significant systemic toxicity. The efficacy of the nanoparticles was further validated through H&E staining analysis of tumor samples (Figure 4G), Ki67 staining was used to assess cell proliferation, and TUNEL staining was employed to evaluate apoptosis induction. Both markers exhibited patterns consistent with previously reported findings.
To verify the induction of ICD by the nanoparticles in vivo, CRT immunofluorescence staining was performed on tumor tissue sections. Consistent with the in vitro results, CRT expression was significantly elevated in the R-BSN treatment group, and even higher levels were observed in the R-BSN(OXA) group (Figure 4H).
In vivo Safety Evaluation of R-BSN
Effectiveness and safety are critical in designing antitumor nanomedicines. To assess the biosafety of the R-BSN in vivo, blood samples from each treatment group were subjected to clinical analysis. The use of nanoparticles did not alter the levels of alanine aminotransferase (ALT) (Figure 5A), aspartate aminotransferase (AST) (Figure 5B), or serum creatinine (CREA) (Figure 5C), indicating the absence of hepatotoxicity or nephrotoxicity. Additionally, complete blood cell count analysis revealed no significant differences in the numbers of red blood cells (RBCs) (Figure 5D), white blood cells (WBCs) (Figure 5E), or platelets (PLTs) (Figure 5F) among the eight groups. Furthermore, H&E staining of major organs, including the heart, liver, spleen, lung, and kidney, revealed no severe organ damage in the free drug-treated groups or the nanoparticle-treated groups (Figure 5G). Collectively, these results confirm the biocompatibility of R-BSN.
 |
Figure 5 Detection of the systemic toxicity of R-BSN treatment. (A and B) Liver function assessment of serum, evaluating the levels of ALT (A) and AST (B). (C) Renal function assessment of serum and evaluation of CREA levels. (D–F) Hematopoietic function assessment of bone marrow, analysis of cell counts and differentiation status. (G) H&E staining of major organs, including the heart, liver, spleen, lung, and kidney. Scale bar: 50 µm. Statistical significance: ns, not significant. |
Inhibition of Peritoneal Metastasis by Nanomedicines
Peritoneal metastasis is a common and serious complication of gastric cancer (GC), affecting nearly one-third of patients at diagnosis and accounting for 53–66% of cases with distant metastasis.49,50 This form of metastasis is particularly challenging to treat due to the widespread dissemination of tumor cells across the peritoneal surface, often leading to a poor prognosis.
To validate the ability of nanomedicine to inhibit peritoneal metastasis, we established peritoneal metastasis model in nude mice. After completing the fifth treatment cycle, the mice were sacrificed, and the nodules in the peritoneal cavity and mesentery were counted. The numbers of peritoneal nodules in the PBS, 5-Fu, OXA, R-BSN(5-Fu), R-BSN(OXA), and sequential treatment groups were 44.0±15.0, 31.8±16.4, 33.8±19.4, 19.6±12.6, 21.8±14.7, and 6±1.6, respectively. Both the conventional drug groups and the nanomedicine groups showed significantly fewer peritoneal metastases compared to the PBS group, with the sequential treatment group exhibiting the fewest tumor nodules (Figure 6A). A similar trend was observed for mesenteric tumor nodules (Figure S7). Representative gross anatomy images are shown in Figure 6B, with additional details available in Figures S8 and Figure S9. Additionally, no significant difference in body weight was observed among the groups (Figure 6C), suggesting that R-BSN significantly enhances the ability of chemotherapeutic drugs to inhibit tumor metastasis without causing severe systemic toxicity. Furthermore, tumor tissues from all groups were subjected to H&E staining and immunohistochemical analysis for Ki67 and TUNEL to assess the antitumor effects at the tissue level. The results were consistent with those observed in the subcutaneous tumor model (Figure 6D). These findings suggest that the R-BSN holds great potential for effectively targeting and inhibiting peritoneal metastasis in GC, offering a promising strategy to improving the treatment outcomes of patients with metastatic GC.
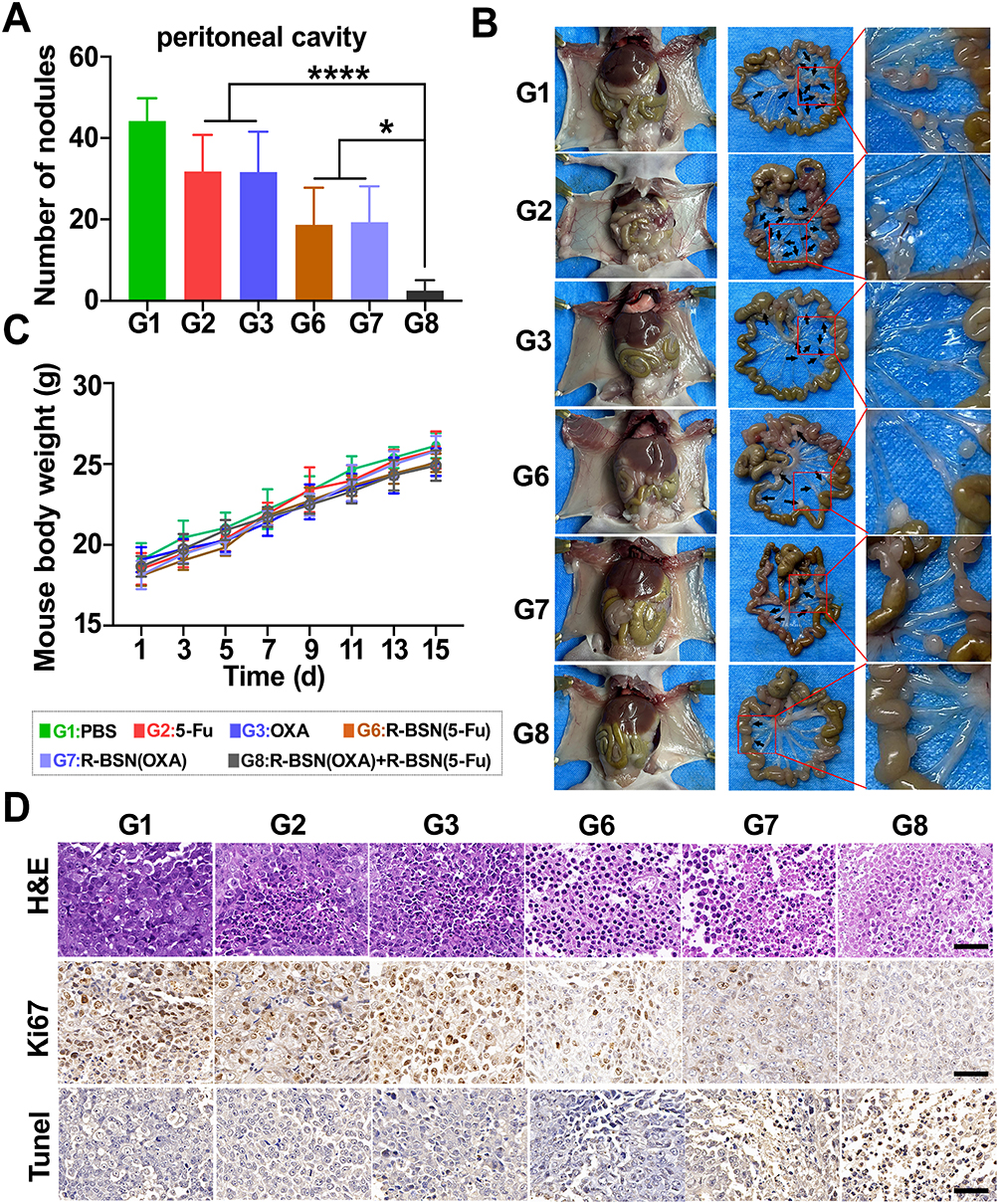 |
Figure 6 The R-BSN increased OXA and 5-Fu efficacy in a peritoneal metastasis mouse model of GC. (A) Quantification of peritoneal metastases in different treatment groups. (B) Photographs of mesenteric metastases in mice subjected to various treatments. Metastatic nodules are indicated by black arrows. (C) Trends in body weight changes in mice in different treatment groups after drug administration. (D) H&E staining, Ki-67 staining, and TUNEL staining of tumor slices from different groups, providing histological and molecular evidence of cell proliferation, apoptosis, and overall treatment efficacy. Scale bar: 50 μm. The values represent the means ± SDs. *: P < 0.05, ****P < 0.0001. |
Conclusion
The RGD peptide-modified biporous silica nanocarrier (R-BSN) demonstrates exceptional performance as a multifunctional nanomedicine platform, combining RGD-mediated active tumor targeting with glucose-responsive drug release for precise therapy. Its innovative design synergistically integrates chemotherapy, starvation therapy, and oxidative stress induction, resulting in enhanced antitumor efficacy while maintaining excellent in vivo biosafety. This comprehensive approach positions R-BSN as a highly promising therapeutic strategy for the treatment of advanced and metastatic gastric cancer.
Data Sharing Statement
Data will be made available on request.
Acknowledgments
This study was financially supported by the Natural Science Foundation of Guangdong Province (2024A151501148), the Key Program of Science and Technology of Guangdong Province (2021A0505030020), the Special Foundation Project of GuangDong Province (0620220203), the Key Science & Technology Project of Guangzhou City (SL2023A03J00413), the Sun Yat-sen University Clinical Research ‘5010’ Plan Project (2023011), the Guangdong Basic and Applied Basic Research Foundation (2022A1515111195), the Five-Five Engineering Projects of the Third Affiliated Hospital of Sun Yat-sen University (2023WW502), the Science and Technology Program of Guangzhou, China (Grant No. 2025A04J4346) and the 100 Talents Programme of Sun Yat-sen University (Grant No. P02673).
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12–49. doi:10.3322/caac.21820
3. Li GZ, Doherty GM, Wang J. Surgical management of gastric cancer: a review. JAMA Surg. 2022;157(5):446–454. doi:10.1001/jamasurg.2022.0182
4. Wang FH, Zhang XT, Tang L, et al. The Chinese Society of Clinical Oncology (CSCO): clinical guidelines for the diagnosis and treatment of gastric cancer, 2023. Cancer Commun. 2024;44(1):127–172. doi:10.1002/cac2.12516
5. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396(10251):635–648. doi:10.1016/S0140-6736(20)31288-5
6. Guan WL, He Y, Xu RH. Gastric cancer treatment: recent progress and future perspectives. J Hematol Oncol. 2023;16(1):57. doi:10.1186/s13045-023-01451-3
7. Kim R, An M, Lee H, et al. Early tumor-immune microenvironmental remodeling and response to first-line fluoropyrimidine and platinum chemotherapy in advanced gastric cancer. Cancer Discov. 2022;12(4):984–1001. doi:10.1158/2159-8290.CD-21-0888
8. Al-Batran SE, Homann N, Pauligk C, et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, Phase 2/3 trial. Lancet. 2019;393(10184):1948–1957. doi:10.1016/S0140-6736(18)32557-1
9. Guo J, Yu Z, Sun D, Zou Y, Liu Y, Huang L. Two nanoformulations induce reactive oxygen species and immunogenetic cell death for synergistic chemo-immunotherapy eradicating colorectal cancer and hepatocellular carcinoma. Mol Cancer. 2021;20(1):10. doi:10.1186/s12943-020-01297-0
10. Chen L, Zhao R, Kang Z, et al. Delivery of short chain fatty acid butyrate to overcome Fusobacterium nucleatum-induced chemoresistance. J Control Release. 2023;363:43–56. doi:10.1016/j.jconrel.2023.09.028
11. Lammers T. Nanomedicine tumor targeting. Adv Mater. 2024;36(26):e2312169. doi:10.1002/adma.202312169
12. Xiao Z, Li Y, Xiong L, et al. Recent advances in anti-atherosclerosis and potential therapeutic targets for nanomaterial-derived drug formulations. Adv Sci. 2023;10(29):e2302918. doi:10.1002/advs.202302918
13. Ding D, Liang R, Li T, et al. Nanodrug modified with engineered cell membrane targets CDKs to activate aPD-L1 immunotherapy against liver metastasis of immune-desert colon cancer. J Control Release. 2024;369:309–324. doi:10.1016/j.jconrel.2024.03.052
14. Eygeris Y, Gupta M, Kim J, Sahay G. Chemistry of lipid nanoparticles for RNA delivery. Acc Chem Res. 2022;55(1):2–12. doi:10.1021/acs.accounts.1c00544
15. Zhang H, Guo Y, Jiao J, et al. A hepatocyte-targeting nanoparticle for enhanced hepatobiliary magnetic resonance imaging. Nat Biomed Eng. 2023;7(3):221–235. doi:10.1038/s41551-022-00975-2
16. Kim H, Park Y, Stevens MM, Kwon W, Hahn SK. Multifunctional hyaluronate - nanoparticle hybrid systems for diagnostic, therapeutic and theranostic applications. J Control Release. 2019;303:55–66. doi:10.1016/j.jconrel.2019.04.003
17. Huang S, Ding D, Lan T, et al. Multifunctional nanodrug performs sonodynamic therapy and inhibits TGF-beta to boost immune response against colorectal cancer and liver metastasis. Acta Biomater. 2023;164:538–552. doi:10.1016/j.actbio.2023.04.001
18. Zhang J, Zhou J, Tang L, et al. Custom-design of multi-stimuli-responsive degradable silica nanoparticles for advanced cancer-specific chemotherapy. Small. 2024;20(35):e2400353. doi:10.1002/smll.202400353
19. Sun L, Lv H, Feng J, et al. Noble-metal-based hollow mesoporous nanoparticles: synthesis strategies and applications. Adv Mater. 2022;34(31):e2201954. doi:10.1002/adma.202201954
20. Li Y, Shi J. Hollow-structured mesoporous materials: chemical synthesis, functionalization and applications. Adv Mater. 2014;26(20):3176–3205. doi:10.1002/adma.201305319
21. Xu B, Li S, Shi R, Liu H. Multifunctional mesoporous silica nanoparticles for biomedical applications. Signal Transduct Target Ther. 2023;8(1):435. doi:10.1038/s41392-023-01654-7
22. Jahnke JP, Kim D, Wildemuth DJ, et al. Mesostructured materials with controllable long-range orientational ordering and anisotropic properties. Adv Mater. 2023;35(51):e2306800. doi:10.1002/adma.202306800
23. Song Y, Sun Q, Luo J, et al. Cationic and anionic antimicrobial agents co-templated mesostructured silica nanocomposites with a spiky nanotopology and enhanced biofilm inhibition performance. Nanomicro Lett. 2022;14(1):83. doi:10.1007/s40820-022-00826-4
24. Nguyen TL, Choi Y, Kim J. Mesoporous silica as a versatile platform for cancer immunotherapy. Adv Mater. 2019;31(34):e1803953. doi:10.1002/adma.201803953
25. Escriche-Navarro B, Escudero A, Lucena-Sanchez E, Sancenon F, Garcia-Fernandez A, Martinez-Manez R. Mesoporous silica materials as an emerging tool for cancer immunotherapy. Adv Sci. 2022;9(26):e2200756. doi:10.1002/advs.202200756
26. Perez-Garnes M, Gutierrez-Salmeron M, Morales V, et al. Engineering hollow mesoporous silica nanoparticles to increase cytotoxicity. Mater Sci Eng C Mater Biol Appl. 2020;112:110935. doi:10.1016/j.msec.2020.110935
27. Lee JY, Kim MK, Nguyen TL, Kim J. Hollow mesoporous silica nanoparticles with extra-large mesopores for enhanced cancer vaccine. ACS Appl Mater Interfaces. 2020;12(31):34658–34666. doi:10.1021/acsami.0c09484
28. Shao J, Zheng X, Feng L, et al. Targeting fluorescence imaging of RGD-modified indocyanine green micelles on gastric cancer. Front Bioeng Biotechnol. 2020;8:575365. doi:10.3389/fbioe.2020.575365
29. Liang HB, Chen X, Zhao R, et al. Simultaneous ischemic regions targeting and BBB crossing strategy to harness extracellular vesicles for therapeutic delivery in ischemic stroke. J Control Release. 2024;365:1037–1057. doi:10.1016/j.jconrel.2023.12.021
30. Shao J, Liang R, Ding D, et al. A smart multifunctional nanoparticle for enhanced near-infrared image-guided photothermal therapy against gastric cancer. Int J Nanomed. 2021;16:2897–2915. doi:10.2147/IJN.S289310
31. Luo Y, Zheng S, Wu Q, et al. Long noncoding RNA (lncRNA) EIF3J-DT induces chemoresistance of gastric cancer via autophagy activation. Autophagy. 2021;17(12):4083–4101. doi:10.1080/15548627.2021.1901204
32. Pan Z, Zheng J, Zhang J, et al. A novel protein encoded by exosomal CircATG4B induces oxaliplatin resistance in colorectal cancer by promoting autophagy. Adv Sci. 2022;9(35):e2204513. doi:10.1002/advs.202204513
33. Li Q, Sun H, Luo D, et al. Lnc-RP11-536 K7.3/SOX2/HIF-1alpha signaling axis regulates oxaliplatin resistance in patient-derived colorectal cancer organoids. J Exp Clin Cancer Res. 2021;40(1):348. doi:10.1186/s13046-021-02143-x
34. Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther. 2020;206:107447. doi:10.1016/j.pharmthera.2019.107447
35. Shi Q, Shen Q, Liu Y, et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell. 2022;40(10):1207–1222e10. doi:10.1016/j.ccell.2022.08.012
36. Yu S, Chen Z, Zeng X, Chen X, Gu Z. Advances in nanomedicine for cancer starvation therapy. Theranostics. 2019;9(26):8026–8047. doi:10.7150/thno.38261
37. Liu J, Li X, Li Y, Gong Q, Luo K. Metformin-based nanomedicines for reprogramming tumor immune microenvironment. Theranostics. 2025;15(3):993–1016. doi:10.7150/thno.104872
38. Xu Y, Liu SY, Zeng L, et al. An enzyme-engineered nonporous copper(I) coordination polymer nanoplatform for cuproptosis-based synergistic cancer therapy. Adv Mater. 2022;34(43):e2204733. doi:10.1002/adma.202204733
39. Li SY, Cheng H, Xie BR, et al. Cancer cell membrane camouflaged cascade bioreactor for cancer targeted starvation and photodynamic therapy. ACS Nano. 2017;11(7):7006–7018. doi:10.1021/acsnano.7b02533
40. Cui Q, Wang JQ, Assaraf YG, et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1–25. doi:10.1016/j.drup.2018.11.001
41. He T, Xu H, Zhang Y, et al. Glucose oxidase-instructed traceable self-oxygenation/hyperthermia dually enhanced cancer starvation therapy. Theranostics. 2020;10(4):1544–1554. doi:10.7150/thno.40439
42. Yang J, Ren B, Yin X, et al. Expanded ROS generation and hypoxia reversal: excipient-free self-assembled nanotheranostics for enhanced cancer photodynamic immunotherapy. Adv Mater. 2024;36(30):e2402720. doi:10.1002/adma.202402720
43. Zheng L, Wang H, Zhou J, et al. Off-the-shelf CAR-NK cells targeting immunogenic cell death marker ERp57 execute robust antitumor activity and have a synergistic effect with ICD inducer oxaliplatin. J Immunother Cancer. 2024;12(7). doi:10.1136/jitc-2024-008888
44. Zhang Y, Fang Z, Pan D, et al. dendritic polymer-based nanomedicines remodel the tumor stroma: improve drug penetration and enhance antitumor immune response. Adv Mater. 2024;36(25):e2401304. doi:10.1002/adma.202401304
45. Li Y, Wu Y, Fang Z, et al. Dendritic nanomedicine with boronate bonds for augmented chemo-immunotherapy via synergistic modulation of tumor immune microenvironment. Adv Mater. 2024;36(2):e2307263. doi:10.1002/adma.202307263
46. Wang L, Huang J, Chen H, et al. Exerting enhanced permeability and retention effect driven delivery by ultrafine iron oxide nanoparticles with T(1)-T(2) switchable magnetic resonance imaging contrast. ACS Nano. 2017;11(5):4582–4592. doi:10.1021/acsnano.7b00038
47. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4(1):81–89. doi:10.7150/thno.7193
48. Yang P, Xu Y, Zhi X, et al. Photodynamically tumor vessel destruction amplified tumor targeting of nanoparticles for efficient chemotherapy. ACS Nano. 2024;18(20):12933–12944. doi:10.1021/acsnano.4c00833
49. Dong D, Tang L, Li ZY, et al. Development and validation of an individualized nomogram to identify occult peritoneal metastasis in patients with advanced gastric cancer. Ann Oncol. 2019;30(3):431–438. doi:10.1093/annonc/mdz001
50. Gwee YX, Chia DKA, So J, et al. Integration of genomic biology into therapeutic strategies of gastric cancer peritoneal metastasis. J Clin Oncol. 2022;40(24):2830. doi:10.1200/JCO.21.02745
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.