")
Back to Journals » Journal of Inflammation Research » Volume 18
Combined Transcriptomic and Mendelian Randomisation Explores the Diagnostic Value of Ubiquitination-Related Genes in Sepsis
Authors Bai X, Liu R, Tang Y, Yang L, Niu Z , Hu Y , Zhang L, Chen M
Received 19 August 2024
Accepted for publication 4 March 2025
Published 4 April 2025 Volume 2025:18 Pages 4709—4724
DOI https://doi.org/10.2147/JIR.S489077
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xue Bai,1,* RuXing Liu,2,* Yujiao Tang,2,* LiTing Yang,1 Zesu Niu,2 Yi Hu,2 Ling Zhang,1 MengFei Chen1
1Department of Emergency, People’s Hospital of Ningxia Hui Autonomous Region, Yinchuan, People’s Republic of China; 2Department of Emergency, The Third Clinical Medical College of Ningxia Medical University, Yinchuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ling Zhang, Email [email protected] MengFei Chen, Email [email protected]
Purpose: Sepsis is the 10th leading cause of death globally and the most common cause of death in patients with infections. Ubiquitination plays a key role in regulating immune responses during sepsis. This study combined bioinformatics and Mendelian randomization (MR) analyses to identify ubiquitin-related genes (UbRGs) with unique roles in sepsis.
Methods: Relevant genes were obtained from the GSE28750 dataset and GSE95233, weighted gene co-expression network analyses were performed to identify gene modules, and differentially expressed UBRGs (DE-UBRGs) were generated by differentially expressed genes (DEGs) crossover with key modular genes and UBRGs in sepsis and normal samples. Causal relationships between sepsis and UbRGs were analysed using MR, performance diagnostics were performed using subject work characteristics (ROC) curves, and an artificial neural network (ANN) model was developed. On this basis, immune infiltration was performed and the expression of key genes was verified in animal models.
Results: 3022 DEGs were found between sepsis and normal. A total of 2620 genes were obtained as key modular genes. Crossing DEGs, key modular genes and UBRGs yielded 93 DE-UBRGs. MR results showed WDR26 as a risk factor for sepsis (OR> 1) and UBE2D1 as a protective factor for sepsis (OR< 1), which was reinforced by scatterplot and forest plot. ROC curves showed that WDR26 and UBE2D1 could accurately differentiate between sepsis and normal samples. Confusion matrix and ROC curve results indicate that the artificial neural network model has strong diagnostic ability. The results of immune infiltration showed that.
WDR26 was negatively correlated with plasma cells, while UBE2D1 was positively correlated with CD4 naïve T cells. Significant differences between sepsis and normal were obtained between UBE2D1 and WDR26 in the animal model.
Conclusion: There appeared to be a causal relationship between sepsis, WDR26 and UBE2D1. The insights were of value for effective clinical diagnosis and treatment in sepsis.
Keywords: sepsis, ubiquitin-related genes, Mendelian randomisation
Introduction
Sepsis, a life-threatening condition characterized by organ dysfunction resulting from a dysregulated host response to infection, is a crucial issue society faces today. Approximately 49 million cases of sepsis are reported annually, with 11 million deaths, accounting for approximately 20% of all annual deaths worldwide. Sepsis imposes a substantial burden on families and society1 Recent studies highlight the complex pathogenesis of sepsis, involves imbalanced inflammatory responses, immune dysfunction, mitochondrial damage, coagulation dysfunction, abnormalities in the neuroendocrine immune network, endoplasmic reticulum stress, and other pathophysiological processes that ultimately lead to organ dysfunction. Despite advancements in anti-infective therapy and organ function support, the intricate mechanisms underlying sepsis contribute to its high morbidity and mortality rate. Therefore, to identify novel therapeutic targets and provide a theoretical basis for the clinic applications, exploring the mechanisms underlying sepsis in greater depth is crucial.
Post-translational modifications (PTMs) play a crucial role in regulating various cellular processes, including phosphorylation, ubiquitination, and acetylation. Among these, dysregulation of ubiquitination is associated with a wide range of diseases. Ubiquitin (Ub), a small protein consisting of 76 amino acids with a molecular weight of approximately 8.5 kDa, is ubiquitously present in living organisms. It is localized in the cytoplasm, nucleus, and on membrane proteins of eukaryotic cells Ub mediates diverse physiological activities within cells, by binding to and labeling proteins destined for degradation. Its primary function is to facilitate the hydrolysis of target proteins into small peptides. Additionally, Ub can label transmembrane proteins, such as receptors, to facilitate their removal from the cell membrane and participates in the vesicular transport of proteins2 Ubiquitination-mediated regulation of protein stability, localization, and function influences numerous essential biological processes, including autophagy, DNA damage repair, the cell cycle, signal transduction, gene expression, inflammation, immunity, and tumorigenesis.3–5 The Ubiquitination involves a series of enzymatic steps mediated by ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). Studies have demonstrated that these enzymes modulate sepsis-induced injury by regulating innate and adaptive immunity. For example, the E3 ligase HECTD3 promotes IFN-I production and sepsis development by catalyzing the ubiquitination of TRAF3 the tumor necrosis factor (TNF) receptor-associated factor family6. Similarly, Qian et al7 identified that the E3 ligase TRIM47 promotes LPS-induced lung inflammation and acute lung injury by facilitating the K63-linked ubiquitination of TRAF2, a key component of the TNFα signaling pathway, activates the NF-κB and MAPK signaling cascades, triggering inflammatory responses in endothelial cells. Xiong et al8 reported that irisin, a cleavage product of fibronectin type III domain-containing protein 5, protects the heart from ischemia/reperfusion injury by upregulating mitochondrial ubiquitin ligase (MITOL). Overall, growing evidence indicates strong association between ubiquitination sepsis. However, the role of UbRGs has not been systematically investigated, and their diagnostic and therapeutic potential in sepsis remains undefined. Therefore, this study aimed to investigate the potential association between UbRGs and the underlying mechanisms of sepsis.
Mendelian randomization (MR) is a statistical method that uses genetics to establish causal relationships between intermediate phenotypes, such as plasma proteins, and clinical phenotypes. In sepsis, numerous studies have demonstrated significant associations between proteins, metabolites, other biomarkers, and outcomes. However, the causal role of these biomarkers is highly uncertain, potentially due to confounding factors, reverse causation, or the fact that inflammatory marker, for instance, are often non-specific collateral phenomena. MR provides a robust approach for identifying which intermediate features (including plasma proteins, metabolites, and even radiological characteristics) contribute to a disease. Studies have investigated the causality associated with the risk of sepsis mortality. For example, low-density lipoprotein (LDL), a complex of proteins, cholesterol, and phospholipids that primarily functions as a carrier of cholesterol in the bloodstream has been examined. Walley KR9 used MR to investigate whether LDL levels increase sepsis mortality. Their results suggested that low LDL increase sepsis mortality and are strongly associated with the Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) genotype. Therefore, MR holds promise for advancing precision sepsis trials. Overall, this study identified UbRGs in patients with sepsis by combining common transcriptome and MR, and analyzed the causal relationship between these genes and sepsis, along with their prognostic value, to further explore the mechanisms of UbRGs in sepsis.
Materials and Methods
Data Preparation
First of all, the GSE28750, regarded as training set, was downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) and it comprised whole blood samples from 10 Sepsis and 20 normal. Besides, the GSE95233, treated as validation set, contained 51 Sepsis and 22 normal whole blood samples. Furthermore, Ubiquitin-related genes (UbRGs) were extracted from integrated annotations for Ubiquitin and Ubiquitin-like Conjugation Database (iUUCD) (http://iuucd.biocuckoo.org/). The flowchart of the relevant study was at Figure S1.
Recognition and Analysis of Differentially Expressed UbRGs (DE-UbRGs)
The ‘Limma’ package (3.57.11)10 was exploited to retrieve differentially expressed genes (DEGs) between Sepsis and normal in GSE28750. The selection conditions of DEGs were |log2FC| > 0.5, adj.P value < 0.0510 The ‘ggVolcano’ (version 0.0.2)11 and ‘ComplexHeatmap’ package (version 2.17.0)12 were employed to visualize the outcomes in the form of volcano map and heat map, respectively. To prospect for modules of co-expressed genes and to inspect the relationship between gene networks and corresponding phenotype, a Weighted Gene Co-expression Network Analysis (WGCNA) network was assembled and modules were characterised in the training set using the ‘WGCNA’ package (version 1.72–1)13 At first explicit outliers were eliminated. Then a soft threshold was determined via the pickSoftThreshold function. The minimum number of genes per genome (minModuleSize) and MergeCutHeight was set to 30 and 0.15 respectively. After that, a gene module was extracted with the help of hierarchical clustering combined with the dynamic tree cutting function. It was finally analysed the correlation between sepsis and modules to pick key modules and key module genes. DE-UbRGs were produced by crossing DEGs, key module genes and UbRGs. For exploring the correlation between DE-UbRGs, Spearman correlation analysis was carried out for DE-UbRGs in the training set via ‘Corrplot’ package (version 0.92)14 Gene Ontology (GO) and Kyoto Encyclopedia of Genes Genomes (KEGG) enrichment analysis for DE-UbRGs were performed using ‘clusterProfiler’ package (version 4.4.9)15 Protein-Protein Interaction (PPI) network was designed using STRING database (https://cn.string-db.org/) (high confidence was set at 0.4), results were visualised in Cytoscape (version 3.7.1).16
Mendel Randomization (MR) Analysis
Initially, a total of 90 expression quantitative trait Loci (eQTL) data of DE-UbRGs were collected from the IEU Open Genome-wide Association Studies (GWAS) database (https://gwas.mrcieu.ac.uk/) as exposure factors, and sepsis was assumed as the outcome. Exposure factors were read and instrumental variables (IVs) were screened through the ‘TwoSampleMR’ package (version 0.5.7)17 The screening standard was P<5*10-6. Then IVs significantly related to exposure factors were found. Clump was set as ‘TRUE’. IVs of linkage disequilibrium analysis (LDA) were discarded. MR analysis was undertaken by combining 5 algorithms: MR Egger18 Weighted median (WM)19 Inverse variance weighted (IVW)20 Simple mode and Weighted mode21 The IVW method was adopted as the main benchmark of the findings. Afterwards, a scatter diagram was constructed to appreciate the correlation between the exposure factors and the outcome. A funnel diagram was plotted to judge randomness. A forest diagram was generated to assess the effectiveness of each he single nucleotide polymorphism (SNP) site in predicting exposure factors for outcome diagnosis. Ultimately, a sum of 3 validation methods were implemented to evaluate the credibility of the analysis: Heterogeneity test, the Horizontal pleiotropy, and Leave-one-out analysis. Eventually, inverse MR analyses were performed.
Construction of Diagnosis Model by Artificial Neural Network (ANN)
The candidate genes analysed using MR were utilised as biomarkers for further analysis. To further evaluate the biomarkers’ capabilities to distinguish sepsis samples from normal samples, ROC curves were analysed applying ‘pROC’ package (version 1.18.4)22 in training and validation sets between Sepsis and normal samples. An ANN model of diagnosis was then constructed to judge the diagnostic value of the biomarkers via ‘neuralnet’ package (version 1.44.2)23 Later on, a three-fold cross-validation method was deployed to enable more effective evaluation of the results of the neural network model. The dataset was randomly separated into training and validation datasets. Eventually the classification score of the neural network model of the disease was extracted.
Function Analysis of Biomarkers and Immune Infiltration
In the training set, ‘c2.cp.kegg.v7.5.1.entrez.gmt’ in the Molecular Signatures Database (MSigDB) database was taken as a reference gene set. The biomarkers were analysed by ‘clusterProfiler’ package (version 4.4.9)15 and P<0.05 was deemed as significant enrichment. Furthermore, genes associated with the function of biomarkers and their corresponding functions were predicted by GeneMANIA (http://www.genemania.org/). The proportion of 22 immune cells in each sample (human blood samples in the database) was then calculated via the Cell-type Identification By Estimating Relative Subsets of RNA Transcripts (CIBERSORT). The distinctions of the 22 immune cell types between samples with sepsis and normal samples were contrasted. Spearman analysis was utilised to explore the correlation between different immune cells and correlation between biomarkers and different immune cells. Afterwards, the biomarkers were entered into miRDB (https://mirdb.org/), Tarbase (https://www.tarbase.com/) and TargetscanHuman (https://www.targetscan.org/) databases, respectively. Then the generated miRNAs were crossed. The received miRNAs were entered into miRNet (https://www.mirnet.ca/) and starbase (https://rnasysu.com/encori/) databases, crossed with lncRNAs. Afterwards the Competing endogenous (ceRNA) network was constructed. Lastly, the differences in biomarker expression in sepsis and normal samples in the training and validation sets were investigated.
The Expression of Biomarkers in Mice Was Verified by Experiments
Construction of Animal Models
First, a total of 12 male 6-8-week-old C57 mice were purchased from Specific (Beijing) Biotechnology Co. Ltd (Production License No.: SCXK (Beijing) 2019–0010; Use License No.: SYXK (Yunnan) K2020-0006). Thereafter, 6 of them were randomly taken for animal model construction, mice were fasted for 12h. Next, mice were anesthetized by intraperitoneal injection of 4% chloral hydrate according to the dose of 0.1 mL/10g. Then, the animals were fixed supine on a surgical board, and their abdominal surgical area was routinely sterilized and depilated. Under aseptic conditions, the abdominal wall of the mice was incised 1 cm with a scalpel. After entering the abdomen through the incision, the cecum was separated at the distal end of the ileocecal valve, the cecum was ligated with a No. 4 silk thread, an 18-gauge injection needle was perforated twice at the ligated end, a small amount of feces was extruded before the peritoneum and skin were closed intermittently with a No. 4 silk suture. The remaining 6 mice were kept normally without surgical manipulation.
Western Blot Assay
For mice in the model and control groups, 50–100mg of lung tissue was respectively weighed and added to 500–1000ul of RIPA lysate and homogenized on ice using a tissue homogenizer. The tissue was centrifuged at 4°C, 16000g for 15min and the supernatant was taken and dispensed into 80ul. Subsequently, the protein concentration was determined by BCA protein quantification kit. The 80ul protein samples were quantified and mixed with the appropriate 5× protein sampling buffer and boiled in a boiling water bath for 10 min, cooled to room temperature and stored at −80°C. After SDS-PAGE electrophoresis, the membrane was transferred and placed in 5% BSA for closure. The closed PVDF membrane was transferred into primary antibodies (UBE2D1, 1:2000, Affinity, DF6715 and WDR26, 1:1000, Bioss, bs-0932R) and incubated at 4°C overnight. On the following day, it was rewarmed for 1h and washed three times with TBST for 5 min each time, followed by incubation of secondary antibody for 60 min at room temperature (secondary antibody: 5% skimmed milk prepared at 1:5000). After the secondary antibody incubation, it was washed 3 times with TBST for 5 min each. Finally, the protein level was assessed on a gel imager using ECL chemiluminescent substrate.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
In order to deeply investigate the role of biomarkers in Sepsis, RNA was extracted from whole blood samples (whole blood from animal models) of models and controls using Trizol reagent according to the manufacturer’s protocol. This study was approved by the Ethics Committee of The People’s Hospital of the Ningxia Hui Autonomous Region [Approval No.: Yinchuan, China; accession No. [2019] Luncheon Review [Scientific] No. (053)]. The cDNA was extracted by reverse transcription reaction through SureScript First Strand cDNA Synthesis Kit. RT-qPCR reaction conditions were as follows: pre-denaturation at 95°C for 2 min, denaturation at 95°C for 10s, annealing at 60°C for 30s, and extension at 60°C for 30s. The reaction conditions were as follows. A total of 40 cycles were performed. Each sample was tested in triplicate and each sample was subjected to unchaining curve analysis to check the specificity of amplification. GAPDH-M was considered as the internal reference gene. qPCR primer details have been listed in Table S1. We used the 2-ΔΔct method to calculate the relative expression levels of key genes.
Statistical Analysis
All analysis were executed in the R software and the comparison of data of different groups were accomplished according to the Wilcoxon test. Except where otherwise stated, the result of statistically significant difference was defined as P<0.05.
Results
A Variety of 2,620 Key Module Genes Were Yielded
The GSE28750 dataset was employed to differential analysis, generating 3,022 DEGs, comprising 1,626 up-regulated and 1,396 down-regulated genes. The results were plotted on volcanic maps and heat maps (Figure 1A and B). Following this, WGCNA was executed. To begin with, all samples were subjected to hierarchical clustering, and obviously distinguished samples were removed (Figure 1C). A scale-free network was then constructed and a soft threshold of 14 was detected (Figure 1D). Hierarchical clustering combined with a dynamic tree cutting function was then applied to determine the gene modules (Figure 1E). Sepsis was then treated as a phenotype and the correlation between sepsis and modules was analyzed. In addition, a heat map was employed to show the correlation between modules and sepsis. According to cor>0.5 and P < 0.05, Sepsis-related modules were selected as key modules, and genes in key modules were screened to obtain MEbrown, MElightcyan, MElightyellow, and MEtan modules (cor> 0.7 and P< 0.05). Moreover, a sum of 2,620 genes were achieved as key module genes (Figure 1F). DEGs, key module genes and UbRGs were taken for intersection and a total of 93 DE-UbRGs were acquired. (Figure 1G).
 |
Figure 1 A variety of 2,620 key module genes were yielded. Notes: (A) Differential volcano plot of the GSE89632 dataset, points within the dashed line in the upper left corner represent down-regulated genes obtained from the screen, and points within the dashed line in the upper right corner represent up-regulated genes obtained from the screen. (B) Differential heatmap of the GSE89632 dataset, red to blue color represents the expression level from high to low in the heatmap. (C) Sample level clustering plot. (D) Soft threshold selection plot, the red line indicates the value of the scale-free fit index taken for subjective selection, and the determination of the optimal soft threshold is mainly referred to the left plot. (E) Gene clustering tree diagram. (F) Module correlation heat map, the darker the red color, the more positive correlation; the darker the blue color, the more negative correlation. (G) DEGs, Module_genes and UbRGs are merged to take the intersection. |
GO Enrichment Analysis Pointed Out That DE-UbRGs Enriched in 33 GO Terms
For the purpose of investigating the relationship among DE-UbRGs, Spearman correlation analysis was performed. The heatmap demonstrated that RLIM was positively correlated with UBA6 and GRAP was negatively associated with UHRF1 (P < 0.01) (Figure 2A). Moreover, to explore the functions of these DE-UbRGs, GO and KEGG enrichment analyses were performed. Interestingly, KEGG enrichment analysis disclosed that 93 DE-UbRGs were significantly enriched in 3 pathways, namely ‘ubiquitin-mediated proteolysis’, ‘autophagy’, and ‘protein processing in the endoplasmic reticulum’ (Figure 2B and C). The findings of GO enrichment analysis pointed out that DE-UbRGs enriched in 33 GO terms, such as ‘regulation of protein catabolic processes’ (Figure 2D and E). PPI network revealed that RNF7 and UBE2D1 have strong collaboration with other genes (Figure 2F).
 |
Figure 2 GO enrichment analysis pointed out that DE-UbRGs enriched in 33 GO terms. Notes: (A) Heat map of top 10 gene correlations. (B) KEGG enrichment analysis bagua diagram. (C) KEGG enrichment analysis string diagram. (D) GO enrichment analysis bagua diagram. (E) GO enrichment analysis string diagram. (F) PPI interaction network diagram, the size and color of the circle indicate its degree, the larger the circle, the greater the degree, the purpler the circle, the greater the degree, and the thickness of the line indicates the degree of gene co-expression, the thicker the line, the greater the degree of co-expression. |
WDR26 Was a Risk Factor and UBE2D1 Was a Protective Factor
In contrast to clinical observational studies, MR analyses provided the advantage of circumventing the effects of suspected reverse causality and confounding factors. As depicted in the table, WDR26 was a risk factor (OR>1) and UBE2D1 was a protective factor for sepsis (OR<1) (Table 1). The scatter diagram clearly indicated that the slope of the WDR26 line was positive, which was a risk factor. The slope of the UBE2D1 line was negative, which was a protective factor (Figure 3A). At the same time the forest map also reinforced this conclusion. The MR effect size for WDR26 was greater than 0 and the MR effect size for UBE2D1 was less than 0 (Figure 3B). By the way, the funnel plot illustrated that both genes were symmetrically allocated along the IVW line, evidencing that the results were in compliance with Mendel’s second law (Figure 3C). Heterogeneity test revealed that there appeared to be no heterogeneity of results (Q>0.05) (Table 2). The horizontal pleiotropy test suggested no level (P>0.05) (Table 3). The Leave-one-out analysis suggested that there were no serious biases for the 2 exposure factors, reflecting the validity of the results and the fact that no single SNP had a large impact on the outcome (Figure 3D). Eventually, the results of inverse Mendelian randomization analysis revealed that there was no causal relationship between Sepsis as an exposure factor and WDR26 and UBE2D1 as endpoints when they were considered as endpoints (P>0.05) (Figures S2–5, Tables S2–4). That is to say, WDR26 and UBE2D1 could affect sepsis, while sepsis did not affect these two genes, which demonstrated the reliability of the causal results we obtained.
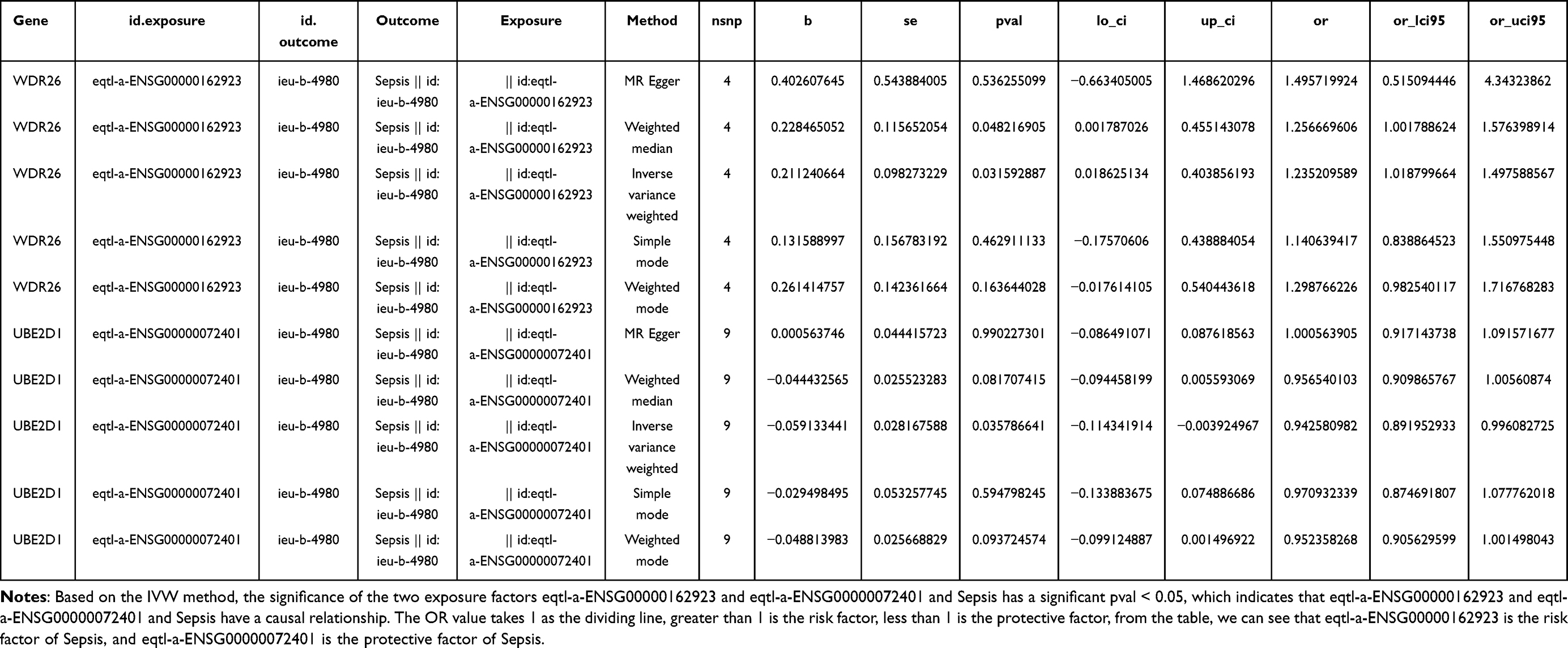 |
Table 1 MR Analysis Results |
 |
Table 2 The Heterogeneity Test |
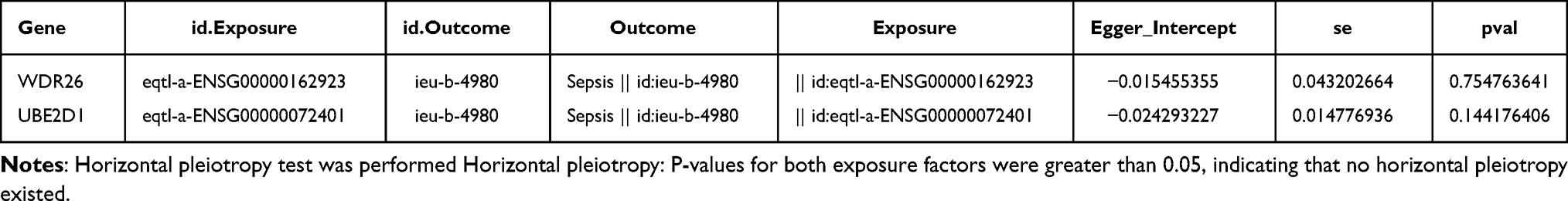 |
Table 3 The Horizontal Pleiotropy Test |
 |
Figure 3 WDR26 was a risk factor and UBE2D1 was a protective factor. Notes: (A) Scatterplot of Mendelian randomization analysis, where a positive slope of the line indicates a risk factor and a negative slope of the line indicates a safety factor. (B) Forest plot of Mendelian randomization analysis, eqtl-a-ENSG00000162923 is to the right of the parameter line for risk factors and eqtl-a-ENSG00000072401 is to the left of the parameter line for protective factors. (C) Funnel plot of Mendelian randomization analysis, 2 genes are almost symmetrical based on IVW method. (D) Mendelian randomization analysis of the line Leave-one-out analysis is shown. |
WDR26 and UBE2D1 had the Strongest Interaction with RANBP10
In terms of the likelihood for the biomarkers to distinguish sepsis from normal samples, the ROC curve illustrated that WDR26 and UBE2D1 provide precise distinction between sepsis and normal samples (AUC>0.7) in the training and validation sets (Figure 4A and B). Subsequently, neural network model was further constructed (Figure 4C). Confusion matrix and ROC curve outcomes indicated that the ANN model held a strong diagnostic power (Figure 4D and E). Possible functions and pathways of the biomarkers were further elaborated. The enrichment analysis results of GSEA pointed out that UBE2D1 was significantly enriched in 54 pathways such as ‘ubiquitin-mediated protein hydrolysis’ (Figure 4F). WDR26 was significantly enriched in 40 pathways like ‘ribosome’ (P<0.05) (Figure 4G). Eventually, the GeneMANIA network revealed that 20 genes, such as GID8, were interconnected with WDR26 and UBE2D1, with the strongest interaction being with the RANBP10. These genes may predict signalling pathways such as ubiquitin-like protein binding enzyme activity (Figure 4H).
 |
Figure 4 WDR26 and UBE2D1 had the strongest interaction with RANBP10. Notes: (A) ROC curve of the biomarker in the GSE28750 dataset. (B) ROC curve of biomarker in GSE95233 dataset. (C) Artificial neural network diagnostic model. (D and E) Evaluation of the artificial neural network diagnostic model (validation set). (F) Enrichment analysis of UBE2D1. (G) Enrichment analysis of WDR26. (H) Genemina. |
Amounts of 9 Immune Cells Were Significant Discrepancies Between Sepsis and Normal
Immune cell infiltration analysis offers valuable guidance in predicting disease course and response to treatment. The relative ratio of 22 immune cell types was visualised in Figure 5A, and among 9 immune cells such as B cells memory and Macrophages M0, there were significant discrepancies between sepsis and normal group (P<0.05) (Figure 5B). Furthermore, the correlation between different immune cells exhibited that T cells CD8 were negatively correlated with Macrophages M0, Macrophages M1, Neutrophils and Plasma cells (P<0.05). It was positively correlated with B cells memory and NK cells resting (P<0.05) (Figure 5C). Figure 5D illustrated that WDR26 was negatively associated with Plasma cells (P<0.01), nevertheless, it was positively correlated with T cells CD4 naive and T cells CD8 (P<0.05). UBE2D1 was positively correlated with T cells CD4 naive (P<0.05). Afterwards, ceRNA network facilitated the investigation of gene functions and their regulation mechanism at a broader level. The network displayed that UBE2D1 interacted with miRNAs including hsa-miR-106b-5p and lncRNAs such as AGAP11, whereas WDR26 interacted with miRNAs like hsa-miR-30E-5p and lncRNAs such as IER3-AS1 (Figure 5E). Regarding the expression of biomarkers, it was apparent that the expression levels of UBE2D1 and WDR26 were significantly higher in sepsis than the normal group (P<0.05) (Figure 5F–G).
 |
Figure 5 Amounts of 9 immune cells were significant discrepancies between sepsis and normal. Notes: (A) Immune cell percentage stacked plot. (B) Immune cell percentage box plot. (C) Differential immune cell correlation analysis. (D) Correlation analysis of biomarkers with differential immune cells. (E) Diagram of ceRNA regulatory network. (F) Expression analysis of candidate biomarkers in the training set. (G) Expression analysis of candidate biomarkers in the validation set.*: P < 0.05, **: P < 0.01, ***: P < 0.001, ****P < 0.0001. |
The Expression of UBE2D1 Was Considerably Higher in the Model Group
It was apparent that the control mice had no distinct changes in their lungs, while the model mice had lung damage (Figure 6A). Western blotting and qRT-PCR were conducted to detect the protein and mRNA expression of the biomarkers in the model and control groups. The results demonstrated that the expression of UBE2D1 was considerably higher in the model group than in the control, whereas the expression of WDR26 was remarkably lower in the model group (P < 0.05) (Figure 6B–D).
 |
Figure 6 The expression of UBE2D1/WDR26 in the model group. Notes: (A) Lung tissues of model and control mice. (B and C) Detection of biomarker proteins in the model and control groups by Western blotting. (D) Detection of biomarker mRNA expression in the model and control groups by qRT-PCR. *: P < 0.05, **: P < 0.01, ***: P < 0.001. |
Discussion
Sepsis results from a dysregulated host response to infection. Timely recognition and diagnosis of sepsis are consistently associated with better prognosis. Numerous bioinformatics studies have explored sepsis-associated signaling pathways and molecular dysregulation mechanisms, identifying several biomarkers. However, investigations into the mechanisms and biomarkers related to ubiquitination during sepsis remain limited.
In this study, we first screened sepsis-related DEGs in the GSE28750 dataset and constructed a WGCNA co-expression network. Subsequently, DEGs, module genes, and UbRGs were intersected to identify differential UbRGs for further analysis. Spearman correlation, KEGG enrichment, and GO enrichment analyses were performed to explore their potential roles in sepsis. Notably, few GO terms or KEGG pathways associated with ubiquitination were highlighted, including GO regulation of protein catabolic processes, KEGG pathways in “ubiquitin-mediated proteolysis”, “autophagy”, and “protein processing in the endoplasmic reticulum”. Next, we integrated 90 eQTL data of differential UbRGs obtained from the IEU OpenGWAS database into the MR analysis, identifying two sepsis ubiquitination-related biomarkers, WDR26 and UBE2D1. Subsequently, we performed ROC curve, ANN, and expression analyses of these biomarkers using the training set GSE28750 and validation set GSE95233 and revealed that WDR26 and UBE2D1 were highly expressed in sepsis patients than that of healthy controls (Human samples in the database). Furthermore, these biomarkers exhibited strong discriminative and diagnostic performance in distinguishing sepsis samples from control samples.
UBE2D1 (ubiquitin-conjugating enzyme E2D1) is a member of the E2 ubiquitin-conjugating enzyme family that plays a critical role in the degradation of dysfunctional or aged proteins. Proteins with E2 ubiquitinase activity have been reported to modulate LPS-induced macrophage polarization toward an M2 phenotype by regulating the molecular switch that governs the LPS-induced endocytosis of TLR4 and the subsequent transition from MyD88 to TRIF-dependent signaling24 Furthermore, Wang25 identified UBE2D1 as a potential biomarker for diabetes-associated sepsis through bioinformatic analysis, and animal experiments indicated that UBE2D1 expression was significantly upregulated, consistent with our experimental findings. These studies suggest that UBE2D1 plays a critical role in the regulation of sepsis-related signaling pathways.
WDR26 is a relatively novel human gene identified through bioinformatics during the screening of human cardiac developmental candidate genes. Studies have demonstrated that deletion of the WDR26 gene and overexpression or underexpression of the protein, regulates apoptosis, autophagy, metastasis, and other biological process. WDR26 also plays a crucial roles in signal transduction and transcriptional regulation.26–28 For instance, Liu29 demonstrated that WDR26 overexpression remarkably activates the Nrf2/HO-1 axis, where Nrf2 serves is a key transcriptional regulator of antioxidant and cytoprotective pathways. Similarly, Feng26 reported that WDR26 promotes hypoxia-induced autophagy in hypoxic H9c2 cells, and further studies revealed that WDR26 enhances the mitochondrial membrane potential, that subsequently increases Parkin translocation to the mitochondria and increases the ubiquitylation of mitochondrial proteins during hypoxia in H9c2 cells. The regulation of autophagy and mitochondrial function is believed to play an important role in sepsis, and by regulating both of them, the damage of sepsis to human organs can be alleviated to a certain extent30 The above study shows that WDR26 can affect autophagy and mitochondrial function, which suggests that WDR26 can play a role in sepsis through autophagy and other pathways. Our study links WDR26 to sepsis, which to some extent bridges the gap between WDR26 and sepsis in this direction and also provides new perspectives on the treatment of sepsis.
In the current study, we also performed GSEA enrichment analysis of UBE2D1 and WDR26, in which UBE2D1 was enriched to 54 pathways and WDR26 was enriched to 40 pathways, both are associated with the ‘ubiquitin-mediated protein hydrolysis’ pathway. Ubiquitin-mediated protein degradation is one of the important protein degradation mechanisms in cells and is widely involved in a variety of cell biological processes, including cell cycle regulation, signaling, DNA repair, immune response and quality control. The main steps of the ubiquitin-proteasome system protein degradation pathway can be divided into the following four steps: the carboxyl group of the glycine end of ubiquitin is attached to the sulfhydryl group of ubiquitin-activating enzyme E1; E1 delivers activated ubiquitin to the ubiquitin-conjugating enzyme E2 through the process of cross esterification; ubiquitin-conjugating enzyme E3 attaches ubiquitin bound to E2 to target proteins and releases E2, forming a specific ubiquitinated proteins; the ubiquitinated proteins are recognized and bound by specific proteasomes, and eventually proteolysis is catalyzed by proteases into short peptides or amino acids. Through ubiquitin-mediated protein degradation, cells can regulate the expression level and functional status of different proteins in response to different biological processes. Several E3 ubiquitin ligases have been identified to play important roles in the development of sepsis.31
Innate and adaptive immune systems play a pivotal in the progression of sepsis. In this study, nine immune cell types were identified to be of significant value in patients with sepsis using the CIBERSORT algorithm. During the early stages of sepsis, neutrophil and monocyte macrophage levels are elevated. This phenomenon is believed to be primarily attributed to delayed neutrophil and monocyte-macrophage apoptosis and the release of immature neutrophils. Although their numbers do not decline, significant functional changes occur within these immune cell types. As sepsis progresses, excessive apoptosis of neutrophils and monocyte-macrophages leads to immunosuppression, increasing the risk of secondary infections or death32 Lymphocyte apoptosis is a key factor in the development of the immunosuppressed phase of sepsis33 B cells play a crucial immunomodulatory role in the circulation by presenting antigens to T cells and differentiating into plasma cells that produce tissue-specific antibodies. B-cell dysfunction, a hallmark of sepsis, impairs in the ability of B cells to produce antibodies and to efficiently clear pathogens. A study reported that B-cell-deficient mice exhibit a diminished inflammatory response to bacterial sepsis34 T cells are essential for maintaining immune function, promoting health, and preventing disease. They primarily differentiate into CD4+ and CD8+ T cell subsets. Sepsis remarkably reduces CD4 T cells numbers, and most patients recover pre-sepsis levels of CD4 T cells within a month, a failure to restore adequate numbers of immunoreactive CD4 T cells is associated with a poor prognosis, particularly in older adults35 CD8+ T cells are the primary responders to viral infection, and effector CD8+ T cells secrete pro-inflammatory cytokines, such as IFN-γ and TNF, to inhibit viral replication. They also express various chemokines to recruit other inflammatory cells to sites of infection36 We further analyzed the association between the ubiquitination-related genes WDR26, UBE2D1 and key immune cell populations. WDR26 exhibited a significant negative correlation with plasma cells and a significant positive correlation with naive T cells CD4+ T cells and CD8+ T cells, and UBE2D1 demonstrated a significant positive correlation with naive CD4+ T cells. These findings suggest that they regulate the onset and progression of sepsis by modulating sepsis immune mechanisms.
To validate the expression of these two key genes in sepsis, we constructed a mouse model of sepsis. Analysis of genes expression levels in this model revealed downregulation of WDR26 and upregulation of UBE2D1 compared to that of the NC group. However, this finding is inconsistent with the bioinformatics analysis, that was performed by comparing biomarkers expression in sepsis and control samples from both the training and validation sets. The analysis revealed that the expression levels of both WDR26 and UBE2D1 were upregulated in the blood samples of patients with sepsis. A potential explanation for this may be the notable differences between human and mice transcriptomes, despite their genetic similarities.37
Our study has several strengths. First, this is the first study to explore the causal effect of ubiquitination-related genes on sepsis risk using MR analysis, the results of MR analysis may be more plausible than those of traditional observational studies because they minimize bias from confounding factors and reverse causation. These identified causal relationships provide candidate target genes for future mechanistic studies. However, this study has certain limitations that warrant consideration. Although we validated the association between WDR26, UBE2D1 and sepsis using RT-qPCR and Western blotting, experimental and clinical validation in a larger sample sizes is needed to increase the robustness and reliability of our conclusions. In addition, during the validation process, we found that the bioinformatics analysis showed that the expression levels of UBE2D1 and WDR26 were significantly higher in sepsis than in the normal group. Based on the results of this stable study, UBE2D1 and WDR26 can be considered in the future as potential assays to further investigate their role in the course of sepsis treatment, and prognostic assessment. However, the results of animal experiments differed from the bioinformatics results. We conclude that although animal models are able to mimic some of the features of sepsis, they cannot fully reproduce the complexity of human sepsis. In addition, physiological differences between humans and animals may lead to different gene expression patterns. The results suggest that we should include more clinical samples for experimental studies in the future. On this basis, we need to explore more deeply how UBE2D1 and WDR26 specifically affect sepsis, and further elucidation of their mechanisms of action will help us to promote their application in the clinic more quickly! Additionally, direct mechanistic studies supporting our results are lacking. To elucidate the relationship between ubiquitination genes and sepsis, further studies are needed to investigate their effects on the immune system, pathogens, and disease-susceptibility pathways.
Conclusion
In this study, we performed bioinformatics analyses and validation experiments to identify WDR26 and UBE2D1 as potential biomarkers for sepsis. These findings offer valuable insights and directions for the development of targeted therapeutic strategies for sepsis. However, further validation through comprehensive experimental and clinical studies are required.
Ethics Approval and Consent to Participate
The study was approved by the Ethics Committee of The People’s Hospital of the Ningxia Hui Autonomous Region (Yinchuan, China; accession No. [2019] Luncheon Review [Scientific] No. (053)). The experimental samples in this study were derived from animal model tissues and did not require the provision of patient informed consent. Animal experiments were approved by the Experimental Animal Management and Use Committee of Ningxia Medical University (Yinchuan, China, No. IACUC-NYLAC-2020-188) and were conducted in accordance with the Guidelines for the Use of Experimental Animals of Ningxia Medical University. The study endeavored to reduce the number of animals used and animal suffering.
Funding
This study was supported by the National Natural Science Foundation of China (82260370, 81960360); Ningxia Natural Science Foundation (2023AAC03457).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rudd KE, Johnson SC, Agesa KM. et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the global burden of disease study. Lancet. 2020;395(10219):200–211. doi:10.1016/S0140-6736(19)32989-7
2. Li Q, Kaur A, Okada K, McKenney RJ, Engebrecht J. Differential requirement for BRCA1-BARD1 E3 ubiquitin ligase activity in DNA damage repair and meiosis in the Caenorhabditis elegans germ line. PLoS Genet. 2023;19(1):e1010457. doi:10.1371/journal.pgen.1010457
3. Swatek KN, Komander D. Ubiquitin modifications. Cell Res Apr. 2016;26(4):399–422.
4. Dang F, Nie L, Wei W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021;28(2):427–438. doi:10.1038/s41418-020-00648-0
5. Han S, Wang R, Zhang Y, et al. The role of ubiquitination and deubiquitination in tumor invasion and metastasis. Int J Biol Sci. 2022;18(6):2292–2303. doi:10.7150/ijbs.69411
6. Li F, Li Y, Liang H, et al. HECTD3 mediates TRAF3 polyubiquitination and type I interferon induction during bacterial infection. J Clin Invest. 2018;128(9):4148–4162. doi:10.1172/JCI120406
7. Qian Y, Wang Z, Lin H, et al. TRIM47 is a novel endothelial activation factor that aggravates lipopolysaccharide-induced acute lung injury in mice via K63-linked ubiquitination of TRAF2. Signal Transduct Target Ther. 2022;7(1):148. doi:10.1038/s41392-022-00953-9
8. Xiong X, Lu L, Wang Z, et al. Irisin attenuates sepsis-induced cardiac dysfunction by attenuating inflammation-induced pyroptosis through a mitochondrial ubiquitin ligase-dependent mechanism. Biomed Pharmacother. 2022;152:113199. doi:10.1016/j.biopha.2022.113199
9. Walley KR, Boyd JH, Kong HJ, Russell JA. low low-density lipoprotein levels are associated with, but do not causally contribute to, increased mortality in sepsis. Crit Care Med. 2019;47(3):463–466. doi:10.1097/ccm.0000000000003551
10. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
11. Wodrich MD, Sawatlon B, Busch M, Corminboeuf C. The genesis of molecular volcano plots. Acc Chem Res. 2021;54(5):1107–1117. doi:10.1021/acs.accounts.0c00857
12. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. doi:10.1093/bioinformatics/btw313
13. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559. doi:10.1186/1471-2105-9-559
14. Wang L, Wang D, Yang L, et al. Cuproptosis related genes associated with Jab1 shapes tumor microenvironment and pharmacological profile in nasopharyngeal carcinoma. Front Immunol. 2022;13:989286. doi:10.3389/fimmu.2022.989286
15. Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. doi:10.1016/j.xinn.2021.100141
16. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
17. Hemani G, Zheng J, Elsworth B, et al. The MR-base platform supports systematic causal inference across the human phenome. Elife. 2018;7. doi:10.7554/eLife.34408
18. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-egger method. Eur J Epidemiol. 2017;32(5):377–389. doi:10.1007/s10654-017-0255-x
19. Bae SC, Lee YH. Alcohol intake and risk of rheumatoid arthritis: a Mendelian randomization study. Z Rheumatol. 2019;78(8):791–796. Alkoholkonsum und Risiko der rheumatoiden Arthritis: eine Mendel-Randomisierungsstudie. doi:10.1007/s00393-018-0537-z
20. Wang X, Zhao D, Cheng L, Gao J, Li J, Geng C. Mendelian randomization explores the causal relationships between obesity, diabetes, inflammation and nonalcoholic fatty liver disease. Medicine. 2023;102(38):e34638. doi:10.1097/md.0000000000034638
21. Hu J, Song J, Chen Z, et al. Reverse causal relationship between periodontitis and shortened telomere length: bidirectional two-sample Mendelian random analysis. Front Immunol. 2022;13:1057602. doi:10.3389/fimmu.2022.1057602
22. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi:10.1186/1471-2105-12-77
23. Beck MW. NeuralNetTools: visualization and analysis tools for neural networks. J Stat Softw. 2018;85(11):1–20. doi:10.18637/jss.v085.i11
24. Arora H, Wilcox SM, Johnson LA, et al. The ATP-binding cassette gene ABCF1 functions as an E2 ubiquitin-conjugating enzyme controlling macrophage polarization to dampen lethal septic shock. Immunity. 2019;50(2):418–431.e6. doi:10.1016/j.immuni.2019.01.014
25. Wang X, Wang LT, Yu B. UBE2D1 and COX7C as potential biomarkers of diabetes-related sepsis. Biomed Res Int. 2022;2022(1):9463717. doi:10.1155/2022/9463717
26. Feng Y, Zhao J, Hou H, et al. WDR26 promotes mitophagy of cardiomyocytes induced by hypoxia through Parkin translocation. Acta Biochim Biophys Sin. 2016;48(12):1075–1084. doi:10.1093/abbs/gmw104
27. Zhen R, Moo C, Zhao Z, et al. Wdr26 regulates nuclear condensation in developing erythroblasts. Blood. 2020;135(3):208–219. doi:10.1182/blood.2019002165
28. Ye Y, Tang X, Sun Z, Chen S. Upregulated WDR26 serves as a scaffold to coordinate PI3K/ AKT pathway-driven breast cancer cell growth, migration, and invasion. Oncotarget. 2016;7(14):17854–17869. doi:10.18632/oncotarget.7439
29. Liu Y, Zhao Y, Li K, Miao S, Xu Y, Zhao J. WD-40 repeat protein 26 protects against oxidative stress-induced injury in astrocytes via Nrf2/HO-1 pathways. Mol Biol Rep. 2022;49(2):1045–1056. doi:10.1007/s11033-021-06925-6
30. Yin X, Xin H, Mao S, Wu G, Guo L. The role of autophagy in sepsis: protection and injury to organs. Front Physiol. 2019;10:1071. doi:10.3389/fphys.2019.01071
31. Shao S, Zhou D, Feng J, et al. Regulation of inflammation and immunity in sepsis by E3 ligases. Front Endocrinol. 2023;14:1124334. doi:10.3389/fendo.2023.1124334
32. Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274(1):330–353. doi:10.1111/imr.12499
33. Cao C, Yu M, Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. 2019;10(10):782. doi:10.1038/s41419-019-2015-1
34. Kelly-Scumpia KM, Scumpia PO, Weinstein JS, et al. B cells enhance early innate immune responses during bacterial sepsis. J Exp Med. 2011;208(8):1673–1682. doi:10.1084/jem.20101715
35. Martin MD, Badovinac VP, Griffith TS. CD4 T cell responses and the sepsis-induced immunoparalysis state. Front Immunol. 2020;11:1364. doi:10.3389/fimmu.2020.01364
36. Sun L, Su Y, Jiao A, Wang X, Zhang B. T cells in health and disease. Signal Transduct Target Ther. 2023;8(1):235. doi:10.1038/s41392-023-01471-y
37. Lin S, Lin Y, Nery JR, et al. Comparison of the transcriptional landscapes between human and mouse tissues. Proc Natl Acad Sci USA. 2014;111(48):17224–17229. doi:10.1073/pnas.1413624111
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.