")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Cordycepin Ameliorates Renal Interstitial Fibrosis by Inhibiting Drp1-Mediated Mitochondrial Fission
Authors Sun Y , Jin S, Chen J, Zhang J, Lu Y, Gu Q, Yan Z, Chen W, Chen A, Fang Y, Geng W, Xu X, Song N
Received 30 September 2024
Accepted for publication 13 February 2025
Published 24 February 2025 Volume 2025:19 Pages 1271—1287
DOI https://doi.org/10.2147/DDDT.S498525
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Yingxue Sun,1,* Shi Jin,1,* Jun Chen,2,* Jian Zhang,1 Yufei Lu,1 Qiuyu Gu,1 Zhixin Yan,1 Weize Chen,1 Annan Chen,1 Yi Fang,1 Wenye Geng,3 Xialian Xu,1 Nana Song1
1Department of Nephrology, Zhongshan Hospital, Fudan University; Shanghai Medical Center of Kidney; Shanghai Institute of Kidney and Dialysis; Shanghai Key Laboratory of Kidney and Blood Purification; Hemodialysis Quality Control Center of Shanghai, Shanghai, 200032, People’s Republic of China; 2Department of Pathology, Changzheng Hospital, Naval Military Medical University, Shanghai, 200003, People’s Republic of China; 3Scientific Research Department of Shanghai Medical College, Fudan Zhangjiang Institute, Fudan University, Shanghai, 201203, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xialian Xu; Nana Song, Division of Nephrology, Zhongshan Hospital, Fudan University, Shanghai, People’s Republic of China, Tel +86-021-64041990-2138, Fax +86-021-64038038, Email [email protected]; [email protected]
Objective: This study aimed to investigate the mechanisms and specific targets of cordycepin in the treatment of renal fibrosis using a unilateral ischemia-reperfusion (UIR) model.
Methods: A UIR mouse model was established, followed by intraperitoneal injections of cordycepin and Mdivi-1. Masson’s trichrome staining and PAS staining were used to identify renal tubulointerstitial fibrosis and assess the degree of renal injury. Fibrosis markers and mitochondrial dynamics-related proteins were evaluated using Western blotting, while differential gene expression and pathway enrichment were analyzed by RNA-seq. Molecular docking, molecular dynamics simulations and surface plasmon resonance were conducted to validate the specific binding sites of cordycepin on the target protein Drp1. Immunofluorescence and in vitro experiments further elucidated the therapeutic mechanism of cordycepin.
Results: In vivo experiments showed that intraperitoneal injection of cordycepin significantly reduced renal inflammation and fibrosis, lowered serum creatinine levels, and decreased collagen deposition. Transcriptome analysis revealed that cordycepin treatment downregulated the mitochondrial fission pathway and upregulated the mitochondrial fusion pathway. Western blotting showed reduced levels of fibrosis markers α-SMA and FN, as well as downregulation of Drp1, MFF, and Fis1, and upregulation of OPA1 and Mfn2. In vitro, cordycepin inhibited TGF-β-induced injury in NRK-52E cells, reducing Drp1 expression and IL-6 secretion. Crosstalk experiments confirmed that decreased IL-6 levels were crucial for cordycepin anti-fibrotic effects by suppressing fibroblast activation.
Conclusion: Cordycepin ameliorates renal fibrosis by targeting Drp1 to inhibit mitochondrial fission in injured renal tubular epithelial cells, reducing IL-6 secretion and inhibiting fibroblast activation.
Keywords: cordycepin, Drp1, renal fibrosis, mitochondrial fission, UIR, IL-6
Introduction
According to estimates by the World Health Organization (WHO), kidney-related diseases are projected to cause up to 10 million deaths worldwide.1 Chronic kidney disease (CKD) has diverse effects, impairing various systemic functions through complex pathophysiological mechanisms, leading to a range of serious health complications.2,3 Renal interstitial fibrosis is a key turning point in CKD, indicating the shift from early to advanced stages.4 Renal interstitial fibrosis significantly reduces renal function and can lead to end-stage renal disease (ESRD), which requires dialysis or transplantation.5 Consequently, therapeutic strategies focused on targeting fibrosis have become a crucial research priority aimed at slowing the progression of CKD.6
The kidney is one of the most energy-demanding organ with high basal metabolic rates and abundant mitochondria.7 Mitochondrial dysfunction is considered a trigger for fibroblast activation and fibrosis in various organs, such as pulmonary fibrosis and ocular fibrosis.8,9 In the kidneys, abundant energy is needed to drive active transport to reabsorb ions in proximal tubules. Thus, proximal tubules contain more mitochondria than any other renal structures. Maintaining mitochondrial homeostasis is critical to maintain proper proximal tubule function.7 Disruption of mitochondrial homeostasis is associated with both acute kidney injury (AKI) and CKD and may serve as an early indicator of renal fibrosis.10 Excessive mitochondrial fission, along with reductions in mitochondrial DNA (mtDNA), impaired oxidative phosphorylation, and increased reactive oxygen species (ROS) generation, contributes to AKI-to-CKD transition.11 Mitochondrial dynamin-related protein 1 (Drp1) is the key factor in mitochondrial fission. Inhibition of Drp1 reduces oxidative stress and promotes recovery from renal injury induced by cisplatin or ischemia.12 However, the underlying mechanisms by which mitochondrial fission in the proximal tubule leads to renal fibrosis are unclear.
Injured epithelial cells play a crucial role in fibrosis, as they release various cytokines that activate fibroblasts, thereby promoting fibrosis.13 Among these cytokines, IL-6 has been shown to play a central role in renal fibrosis. IL-6 can bind to the membrane-bound IL-6R (IL-6 receptor) or to soluble IL-6R to activate the classical signaling and trans-signaling pathways. Both modes of signaling converge on glycoprotein 130 (gp130) to activate the intracellular Janus kinase (JAK) and signal transducer and activator of transcription (STAT) pathway.14 Circulating sIL-6R levels are elevated in CKD and associated with high serum fibroblast growth factor 23 levels.15 Inhibiting IL-6 trans-signaling can attenuate renal fibrosis by reducing fibroblast accumulation.16 It was reported that mtROS-mitochondrial fission/fusion axis trigger cytokine homeostasis imbalance in hepatocytes.17 Excessive mitochondrial fission and oxidative stress exacerbates inflammation by promoting release of cytokines such as NF-κB, IL-1β and IL-6.18 Inhibition of mitochondria-derived ROS alleviates IL-6/JAK2/STAT3-mediated inflammatory responses in human aortic smooth muscle cells.19 Thus, we hypothesized that mitochondrial fission drives secretion of IL-6 from epithelial cells to activate fibroblasts and promote renal fibrosis.
Cordyceps sinensis (CS) is highly esteemed in traditional Chinese medicine for its medicinal value. In 1951, German scientist Cunningham identified cordycepin as the primary active component of Cordyceps sinensis. Cordycepin exhibits a range of biological activities, including anti-inflammatory, anti-tumor, antioxidant, anti-aging and immunomodulatory effects. Studies indicate that cordycepin has therapeutic potential for treating fibrosis in various tissues and organs.20,21 Multiple studies have demonstrated that cordycepin can mitigate the severity of CKD and impede the progression of renal failure.22 Additionally, it has been found to improve renal function, reduce urinary protein, serum urea nitrogen and creatinine.23 Currently, the precise mechanisms underlying the bioactivities of cordycepin are under investigation. It was reported that cordycepin can reverse Drp1-mediated aberrant mitochondrial fission to exert neuroprotective effects in Parkinson’s disease.24 Consequently, our study aimed to investigate whether cordycepin could mitigate the progression of renal fibrosis by inhibiting Drp1-mediated mitochondrial division and explore its potential as a therapeutic target.
Materials and Methods
Chemicals and Reagents
Cordycepin was provided by Shanghai Guobao Enterprise Development Center (Shanghai, China). Media and additives for culture of NRK-49F cells were purchased from Gibco-Invitrogen Corporation (Grand Island, NY, USA). Recombinant IL-6 and IL-6R protein were purchased from Proteintech (Wuhan, China). Other reagents and sources were listed below: Abcam (Cambridge, UK), anti-rabbit α-SMA monoclonal antibody, anti-rabbit fibronectin monoclonal antibody, anti-rabbit Drp1 monoclonal antibody; Proteintech (Wuhan, China), anti-rabbit MFF monoclonal antibody, anti-rabbit Fis1 antibody, anti-mouse GAPDH antibody; Abclonal (Wuhan, China), anti-rabbit OPA1 monoclonal antibody and anti-rabbit Mfn2 monoclonal antibody.
Animal Experiments
Male BALB/c mice aged 8 weeks (weight 22–25g) were used in this study. All animals were bred in Zhongshan Hospital affiliated to Fudan University. Thirty-six male BALB/c mice were randomly assigned to six groups: Sham group (n=6), UIR+NS group (n=6), UIR+C-L group (n=6), UIR+C-M group (n=6), UIR+C-H group (n=6) and UIR+Mdivi-1 group (n=6). Mice in both the UIR+NS group and cordycepin treatment group were anesthetized via intraperitoneal injection of 1% phenobarbitone (50 mg/kg body weight). The procedure involved making an incision along the abdominal midline, locating the left kidney pedicle, and clamping the renal vessel for 30 minutes. During this process, temperature was maintained at 37°C. Subsequently, the clamp was removed to restore perfusion. The sham group received a normal diet. UIR+C-L, UIR+C-M, and UIR+C-H groups respectively received intraperitoneal injections of cordycepin for 14 consecutive days beginning from 7 days after UIR surgery (5 mg/(kg·d), 10 mg/(kg·d), 20 mg/(kg·d)). UIR+NS group received equal volumes of normal saline via intraperitoneal injection. Additionally, UIR+Mdivi-1 group was intraperitoneally injected with Mdivi-1 (50 mg/(kg·d)) on the 7th day post-surgery, serving as a positive control. After 21 days, the mice were euthanized for kidney extraction.
Histopathology
Kidney tissues were fixed in 4% paraformaldehyde, paraffin-embedded, and cut into 5-μm sections. For pathologic staining, paraffin sections were stained with a Masson’s trichrome staining kit (Solarbio, Beijing, China). Renal fibrosis scores were determined using a scoring system and evaluated under light microscopy by a pathologist blinded to the origin of the preparations. ImageJ was used to quantify the degree of fibrosis by adjusting the color setting threshold. The image analysis program was configured to detect areas of blue-stained collagen within each renal transverse section stained by Masson’s trichrome. The area of renal parenchyma was calculated by reverse selection of background. The fractional collagen area represents the proportion of collagen area compared to the total renal parenchyma. Paraffin-embedded 5-μm kidney sections were stained with periodic acid-Schiff (PAS). Histological injury scores were evaluated by light microscopy in a blinded manner. Severity was determined in a semi-quantitative manner by the percentage of tubules manifesting epithelial necrosis, loss of the brush border, cast formation, and tubular dilation. A five-point scale was used, as follows: 0, no injury; 1, less than 25%; 2, 25–50%; 3, 50–75%; and 4, more than 75%.
Immunohistochemistry
For immunohistochemistry, 5-μm kidney sections were incubated with primary antibody α-SMA (working dilution 1:200). Renal sections of formalin-fixed, paraffin-embedded samples were stained. The results of each group were quantified by counting the positive area of staining for the fibrocyte activation marker α-SMA in each of five high-magnification fields (magnification×200). The degree of fibrosis in each group was quantitatively evaluated by the percentage of positive area.
Cell Culture and Treatment
NRK-52E (Cell Bank, Chinese Academy of Sciences) and NRK-49F cells (Zhong Qiao Xin Zhou, China) were cultured in DMEM/F12 high-glucose medium supplemented with 10% FBS, 4500 mg/L D-glucose, 110 mg/L Sodium Pyruvate, 100 U/mL penicillin, and 100 mg/mL streptomycin. The cells were grown in a 5% CO2 atmosphere at 37°C until cell fusion. When the cells reach 80% confluence, the medium was replaced with serum-free medium, and treatment was applied for 48 hours. In the TGF-β-induced NRK-52E model, TGF-β was used at a concentration of 10 ng/mL, cordycepin (C-L at 10 μmol/L, C-M at 20 μmol/L, C-H or Cord at 40 μmol/L), and Mdivi-1 at 10 μmol/L.
Western Blotting Analysis
The affected kidney was removed from −80°C and ground into homogenate to extract protein, or renal tubular epithelial cell lysate was utilized for protein extraction, followed by Western blotting experiments. The protein samples were loaded and separated on sodium dodecyl sulfate-polyacrylamide gel. The gel was then transferred to the PVDF membrane and blocked with 5% non-fat milk. The PVDF membrane was incubated with primary antibody against GAPDH (1:10,000), α-SMA (1:2000), FN (1:1000), Drp1 (1:2000), MFF (1:2000), Fis1 (1:2000), OPA1 (1:2000) and Mfn2 (1:1000) overnight at 4°C, followed by incubation with a secondary antibody conjugated with horseradish peroxidase (HRP). Proteins were visualized using chemiluminescent ECL, and the final result was normalized to GAPDH expression.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from cells or kidney tissue samples with TRIzol reagent (Invitrogen, USA). The purity of RNA was assessed by measuring the absorbance at 260 nm and 280 nm, with an A260/A280 ratio within the range of 1.8 to 2.0. The first-strand cDNA was synthesized by reverse transcription using oligo(dT) and Superscript II according to the synthesis kit manufacturer (TAKARA, Japan). Quantitative PCR was conducted by SYBR-green PCR. The relative expression level of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA was used as the internal reference for each gene evaluation. The primer sequences are provided in Table S1.
Immunofluorescence Staining
We conducted immunofluorescence co-staining of paraffin sections. Briefly, after being blocked by 1%BSA, the slices were incubated with the mixed primary antibodies. To assess the extent of interstitial fibrosis, paraffin sections were incubated with anti-rabbit α-SMA antibodies (1:200) or anti-mouse E-cadherin (1:100). After washing with phosphate-buffered saline (PBS), the immunoreaction was detected by Donkey Anti-Rabbit IgG with Alexa Fluor 488-conjugated secondary antibodies (1:100; Thermo Fisher Scientific) and Alexa Fluor 594 coupled Donkey anti-mouse IgG (1:100; Absin). Images were taken under a confocal microscope for observation and acquisition.
Transmission Electron Microscopy (TEM)
Fresh renal cortex and medullary junction tissues were immediately placed in EP tubes containing pre-prepared fixative for TEM and stored at 4°C. Then, the samples underwent dehydration at room temperature, followed by resin penetration, embedding and polymerization. The resin blocks were cut into ultrathin sections, 60–80 nm in thickness, and the tissues were fished out onto the 150 meshes copper grids with formvar film. During the dyeing process, the copper grids were immersed in a 2% uranium acetate-saturated alcohol solution for 8 minutes, followed by three rinses with 70% alcohol. Subsequently, the grids were stained for 8 minutes with a 2.6% lead citrate solution and then washed three times with ultra-pure water. The copper grids were then allowed to dry at room temperature overnight. Transmission electron microscopy was employed to observe and collect images for subsequent analysis.
Cell Counting Kit-8 (CCK-8) Assay
Renal tubular epithelial cells were seeded at a density of 5000 cells per well in 96-well plates. Following treatment, cell proliferation was detected by incubating with CCK-8 reagent. Absorbance was quantified at 450 nm using a microplate reader.
Mitochondrial Membrane Potential Assay (JC-1 Staining)
Mitochondrial membrane potential (MMP) was measured using the mitochondrial membrane potential assay kit with JC-1 (Beyotime, Nanjing, China), according to the manufacturer’s instructions. Specifically, cells were harvested and washed twice with phosphate-buffered saline (PBS). Subsequently, the cells were incubated with JC-1 probe at a 1:200 dilution at room temperature for 20 minutes. When the MMP was high, JC-1 accumulated in its polymerized form, producing red fluorescence. In contrast, a decrease in membrane potential led to JC-1 remaining in its monomeric form, resulting in an increase in green fluorescence. MMP was then measured using flow cytometry or microplate reader.
ROS Detection
ROS detection was performed using a ROS assay kit purchased from Beyotime Biotechnology (Nanjing, China). DCFH-DA was diluted to a working concentration of 10 μM using serum-free medium at a ratio of 1:1000. After removing the cell culture medium, the cells were washed with PBS and then incubated with 10 μM DCFH-DA at 37°C for 20 minutes. Fluorescence was measured using a microplate reader with an excitation wavelength of 488 nm and an emission wavelength of 525 nm.
Mitochondrial Morphology
Live renal tubular epithelial cells were stained with 100 nM MitoTracker Red (Invitrogen, California, USA) probe at 37°C for 30 min. After staining, the staining solution was replaced and the cells were washed with serum-free fresh medium. At the same time, Drp1 co-staining was performed to observe.
Transcriptomic Analysis
We randomly selected kidney tissues from three mice in each group. Total RNA was extracted using standard protocols, and rigorous quality control measures were implemented. Initially, RNA degradation was assessed via 1% agarose gel electrophoresis. Subsequently, RNA concentration was precisely measured using the Qubit 2.0 Fluorometer, and RNA integrity was evaluated with the Qsep nucleic acid detector (BiOptic Inc). Upon passing quality control, mRNA was enriched with Oligo (dT)-coated magnetic beads and subsequently fragmented into short fragments by the addition of fragmentation buffer. The first strand of cDNA was synthesized utilizing six-base random primers, employing the short fragments as templates. The second cDNA strand was synthesized by adding buffer, dNTPs, RNase H, and DNA Polymerase I. The complete sequencing library was then constructed through PCR amplification, followed by high-throughput sequencing via the Illumina platform. Following quality control, comparative analysis, and in-depth transcriptome analysis of the sequencing data, we quantified gene expression levels using a standardized method, counts per million (CPM) reads, to assess differential gene expression. To assess variations in gene expression across different conditions, differential expression analysis was conducted using the Wald test implemented in the DESeq2 package in R. Differentially expressed genes were identified based on a threshold of |log2FoldChange| > 0.43 and P < 0.05. Furthermore, enrichment analysis was primarily conducted using hypergeometric tests or Fisher’s exact tests to evaluate the enrichment of differentially expressed genes within specific pathways.
Molecular Docking and Molecular Dynamics Simulation
The 2D structure of cordycepin was obtained from the PubChem database (http://pubchem.ncbi.nlm.nih.gov/) and subsequently imported into ChemOffice 20.0 to generate the 3D structure, which was saved as a mol2 file. The PDB format file of the target protein, Drp1, was downloaded from the RCSB PDB database (http://www.rcsb.org/). Using PyMOL 2.6.0, Drp1 was prepared by removing water molecules and phosphate groups, and then saved in PDB format. Molecular Operating Environment (MOE) 2019 software was employed to minimize the energy of cordycepin, prepare the target protein, and identify potential active sites. Molecular docking was then performed in MOE 2019, with 50 docking runs set for the computation.
To further investigate the binding interactions between cordycepin and Drp1, MD simulations were carried out using the docking complex as the initial state. MD simulations were performed with Amber 24 (San Francisco, CA, USA) using the ff19SB force field for system parameterization. The system was solvated using the TIP3P water model and neutralized with counter ions. Once the system energy was minimized, the temperature was gradually increased from 0 K to 310.15 K over 500 ps. Restraints were applied during the heating process under the NVT ensemble, followed by system equilibration at 310.15 K. Finally, a 100 ns molecular dynamics simulation was conducted under the NPT ensemble, maintaining periodic boundary conditions throughout. Covalent bonds involving hydrogen atoms were constrained using the SHAKE algorithm. Analysis of the MD results, including RMSD, RMSF, radius of gyration, solvent-accessible surface area (SASA), and hydrogen bonding, was conducted using AmberTools 23.
Surface Plasmon Resonance (SPR) Experiment
The experiments were conducted at 25 °C using a BIAcore 1K system equipped with CM5 sensor chips, with data processed using BIAcore 1K Evaluation software (Cytiva) in accordance with the manufacturer’s guidelines. Briefly, a CM5 sensor chip was prepared by activating it with a solution containing 200 μM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 50 μM N-hydroxysuccinimide (NHS) at a flow rate of 10 μL/min for 420 seconds. The protein sample, mixed with 180 μL of 10 mm sodium acetate (pH 5.0), was immobilized on the activated surface at the same flow rate for 420 seconds, repeated over two runs. Following immobilization, the surface was blocked with 1 M ethanolamine (10 μL/min for 420 seconds). A neighboring channel, serving as a reference, underwent the same activation and blocking steps but used PBS (pH 5.0) during immobilization. Both channels were subsequently equilibrated with PBS. Molecule stock solutions were serially diluted in PBS and injected at a flow rate of 10 μL/min for 150 seconds per run. Regeneration was achieved after each flow using 10 mm glycine-HCl (pH 2.0) for 5 minutes at 10 μL/min. Binding data from the sample were corrected by subtracting those from the reference and analyzed using Biacore Insight (v. 2.0, Cytiva). Association and dissociation constants were determined by globally fitting the data to a 1:1 Langmuir binding model using BIAcore 1K Evaluation software (Cytiva, Marlborough, MA, USA).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 9.0.0 for Windows (GraphPad software, San Diego, CA). All results were presented as the mean ± SEM of n trials. Two-tailed unpaired t-test was used for comparison between two groups. Comparisons among multiple groups were performed using one-way analysis of variance (ANOVA). The Tukey’s test was employed for post hoc pairwise comparisons between groups. Differences between groups were considered statistically significant when P < 0.05.
Results
Cordycepin Significantly Mitigated Renal Fibrosis and Renal Injury in UIR Mice
We established a renal fibrosis model with UIR and evaluated the therapeutic effects of cordycepin (Figure 1A Flow Chart). The results showed that UIR-injured mice exhibited emaciation, reduced activity and thinner fur. The kidneys showed significant morphological changes, including renal atrophy, yellowish-white surfaces and diffuse fine particles. Kidneys with cordycepin treatment displayed varying degrees of improvement (Figure 1B). Masson staining was employed to evaluate the extent of collagen deposition (Figure 1C), and semi-quantitative scoring was utilized to determine the percentage of renal fibrosis areas. The results indicated that fibrosis was significantly diminished with the treatment with cordycepin. The therapeutic effect of cordycepin is dose-dependent. Furthermore, PAS staining results (Figure 1C) revealed that the UIR+C-H group exhibited the most pronounced therapeutic effect, with a significantly lower tubular injury score. Immunohistochemical analysis of α-SMA demonstrated that cordycepin significantly inhibited UIR-induced α-SMA (P<0.0001) levels. We evaluated the renal index (absolute kidney mass/body mass) and serum creatinine levels (Figure 1D). The findings indicated an elevation in serum creatinine levels in the UIR+NS group, whereas cordycepin treatment demonstrated a dose-dependent reduction in both the renal index and serum creatinine levels. Western blotting analysis (Figure 1E) revealed that the expression levels of α-SMA (P =0.0101) and Fibronectin (P=0.0014) were elevated in the UIR+NS group. The expression levels of α-SMA (P<0.05) and Fibronectin (P<0.05) were significantly reduced in groups treated with different doses of cordycepin. This finding was corroborated by immunofluorescence co-staining (Figure 1F), which revealed a decrease in α-SMA expression and an increase in E-cadherin expression in the cordycepin groups. These results robustly confirmed that cordycepin significantly mitigated renal fibrosis and kidney injury in UIR mice.

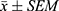
Transcriptome Analysis of Kidney
Principal component analysis (PCA) and Spearman correlation-based clustering revealed significant transcriptomic differences between the three groups, with high intra-group correlation (Figure 2A). Bioinformatics analysis of two key comparisons, UIR+NS vs Sham and UIR+C-H vs UIR+NS, identified 10,784 and 11,551 differentially expressed genes, respectively (Figure 2B). Hierarchical clustering and heatmap visualization (Figure 2C) showed distinct gene expression patterns. Cordycepin treatment downregulated fibrosis-related genes, including FN1, as well as mitochondrial fission genes such as Drp1 (encoded by DNM1L), while promoting mitochondrial fusion genes such as OPA1 (Figure 2D). The heatmap (Figure 2E) clearly displayed that cordycepin effectively inhibited IL-6/STAT3 inflammatory signaling genes. Gene Ontology (GO) analysis (Figure 2F) showed that the UIR+C-H group was significantly enriched in pathways related to mitochondrial fission and mitochondrial fusion. Other enriched pathways closely associated with mitochondrial fission included oxidative stress (ROS), which is often an indication of excessive mitochondrial fission, and the decrease in MMP, which typically suggests early mitochondrial fission. KEGG analysis (Figure 2G) highlighted significant enrichment in the TGF-β signaling pathway and mitophagy. These findings suggested that cordycepin regulated mitochondrial dynamics, contributing to its therapeutic effects in renal fibrosis.
 |
Figure 2 Transcriptome Analysis of Kidney. (A) PCA-plot of gene expression changes. The dots indicate samples. (B) Bar plot of DEGs in groups. (C) Clustered heatmap of groups. (D) Box plots of selected genes of interest. (E) DEGs expression (normalized counts) of fibrosis markers, inflammation factors and mitochondrial dynamics genes. (F) GO analysis of DEGs (enriched for pathways involved in mitochondrial processes). (G) KEGG analysis of DEGs. |
Cordycepin Inhibited Drp1-Mediated Aberrant Mitochondrial Fission in Injured Renal Tubular Epithelial Cells Both in Vivo and in Vitro
Transmission Electron Microscopy (Figure 3A) revealed that mitochondria in the renal tubular epithelial cells of the sham group were predominantly intact, displaying elongated, rod-like structures with clear internal cristae. In contrast, the UIR+NS group displayed numerous edematous, round, and small mitochondria, with shortened cristae and a juvenile morphology, indicating an increase in abnormal mitochondrial fission. Treatment with cordycepin and Mdivi-1 (a selective inhibitor of the mitochondrial fission protein Drp1) significantly improved these mitochondrial and cellular abnormalities, restoring elliptical mitochondria with intact membranes, abundant matrix, and no noticeable collagen deposition. Western blotting (Figure 3B) showed that cordycepin and Mdivi-1 significantly reduced the expression of mitochondrial fission proteins Drp1, MFF, and Fis1, while promoting the fusion proteins OPA1 and Mfn2, which had been reduced in the UIR+NS group. Cordycepin demonstrated a more pronounced effect on improving mitochondrial dynamics compared to Mdivi-1. In vitro experiments using NRK-52E cells treated with TGF-β showed an increase in the expression of Drp1, MFF, and Fis1, along with a decrease in OPA1 and Mfn2 levels (Figure 3C). However, treatment with cordycepin and Mdivi-1 effectively restored a balance between mitochondrial fission and fusion proteins. PCR analysis (Figure 3D) further corroborated these observations, confirming the regulatory effects of both treatments. ROS assays (Figure 3E) demonstrated that both cordycepin and Mdivi-1 effectively reduced oxidative stress. Co-staining with Mitotracker Red and Drp1 immunofluorescence (Figure 3F) indicated that mitochondria in the TGF-β-treated group exhibited fragmentation or punctate morphology, with increased Drp1 fluorescence. Treatment with cordycepin and Mdivi-1 effectively reversed the extent of mitochondrial fragmentation in NRK-52E cells. MMP serves as a crucial indicator of mitochondrial function. Fluorescence and flow cytometry results (Figure 3G and H) revealed that TGF-β induced aberrant mitochondrial division in NRK-52E, leading to a significant reduction in MMP. However, cordycepin and Mdivi-1 restored MMP, further suggesting their potential role in ameliorating mitochondrial function by inhibiting aberrant mitochondrial fission.

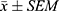
Cordycepin Reduced IL-6 secretion From Tubular Epithelial Cells by Inhibiting Drp1-Mediated Aberrant Mitochondrial Fission
We performed immunofluorescence co-staining for IL-6 and LTL (Figure 4A). The results showed a significant increase in IL-6 secretion from injured renal tubular epithelial cells in the UIR+NS group. In contrast, the UIR+C-H and UIR+Mdivi-1 groups exhibited a marked reduction in IL-6 levels in renal tubular epithelial cells. Western blotting analysis (Figure 4B) further revealed that IL-6 protein levels were significantly upregulated in the UIR+NS group, while both the UIR+C-H and UIR+Mdivi-1 groups showed a substantial downregulation of IL-6 expression. Additionally, ELISA results (Figure 4C) indicated that serum IL-6 levels were markedly elevated in the UIR+NS group, whereas the UIR+C-H and UIR+Mdivi-1 groups exhibited significantly lower serum IL-6 levels. Mdivi-1 treatment also reduced levels of renal fibrosis proteins (Figure 4D). In vitro experiments confirmed that TGF-β treatment induced a significant increase in IL-6 levels in NRK-52E cells (Figure 4E and F) and HK-2 cells (Figure S1A), and IL-6 secretion in the cell supernatant was also substantially elevated (Figure 4G and Figure S1B). However, treatment with cordycepin (Cord) and Mdivi-1 significantly suppressed IL-6 expression and secretion in renal tubular epithelial cells. These findings strongly supported the conclusion that cordycepin reduced IL-6 secretion in epithelial cells by inhibiting Drp1-mediated abnormal mitochondrial fission.

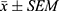
Cordycepin Inhibited Fibroblast Infiltration and Activation by Reducing IL-6 secretion
Fibroblast activation and proliferation play a central role in the pathogenesis of renal fibrosis. We performed immunofluorescence co-staining for IL-6 and α-SMA (Figure 5A). The results revealed a significant increase in IL-6 secretion from injured renal tubular epithelial cells, accompanied by an accumulation of α-SMA-positive fibroblasts in the adjacent interstitial tissue. In contrast, treatment with cordycepin or Mdivi-1 reduced IL-6 secretion from tubular epithelial cells and decreased the number of α-SMA-positive fibroblasts in the surrounding stroma. These findings suggest that IL-6 secreted by injured tubular cells plays a crucial role in fibroblast activation. To further validate this observation, we conducted a crosstalk experiment using conditioned media from treated renal tubular epithelial cells (PTECs) to stimulate fibroblasts. Western blotting and PCR analysis (Figure 5B and C) showed that conditioned media from TGF-β-treated PTECs promoted fibroblast activation, as indicated by increased expression of α-SMA and FN. However, conditioned media from PTECs treated with TGF-β and either cordycepin or Mdivi-1 significantly reduced the expression of both α-SMA and FN. Additionally, CCK-8 assay results (Figure 5D) demonstrated that conditioned media from TGF-β-treated PTECs also stimulated fibroblast proliferation, while the presence of cordycepin inhibited both fibroblast proliferation and activation.

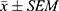
Molecular Dynamics and SPR Analysis of Cordycepin Binding to Drp1
To further elucidate the potential mechanism by which cordycepin affects Drp1, we performed molecular dynamics simulations of the cordycepin-Drp1 complex. Specific residues involved in various interactions between Drp1 and cordycepin were illustrated in Figure 6A–C. The binding results indicated that cordycepin could enter the binding region of the Drp1 target protein, exhibiting two classical binding modes. Free energy calculations revealed a binding affinity of approximately −16.0 kcal/mol, suggesting a strong interaction between cordycepin and Drp1. RMSD analysis showed that the dynamics simulations reached a steady state, confirming the stable binding between the two molecules. Additionally, the SASA results further indicated a close interaction between the ligand and the protein. We performed SPR experiments (Figure 6D–F), and the results clearly confirmed the direct binding between cordycepin and Drp1. The kinetic model showed that the dissociation constant (KD) was 2.75 μM, indicating that cordycepin could effectively bind to Drp1. The dissociation constant and corresponding rate constants were listed in Figure 6E. The affinity model also yielded a good KD of 6.87 μM (Figure 6F). This level of affinity provided a solid foundation for further studies on the mechanism of action of cordycepin and its potential for drug development.
 |
Figure 6 (A) Molecular dynamics simulation of Cordycepin with Drp1; (B and C): RMSD and SASA analyzation of MD simulation; (D-F): SPR analysis of cordycepin binding to Drp1; (G) Molecular mechanism of inhibiting renal fibrosis by inhibiting mitochondrial division with Cordycepin. |
Discussion
Chronic kidney disease (CKD) affects approximately 10–14% of the global population, with its prevalence rising annually.2 Renal fibrosis is a key pathological process in the progression of CKD. As the disease advances, fibrosis worsens, leading to irreversible impairment of kidney function. Therefore, effective interventions targeting renal fibrosis are crucial not only to slow the progression of CKD but also to improve patient outcomes and quality of life.
Cordycepin, known as 3′-deoxyadenosine, is a compound structurally distinct from adenosine due to the absence of a 3′-hydroxyl group. This unique structural feature grants cordycepin a wide range of pharmacological effects. Research indicates that cordycepin can effectively treat renal ischemia-reperfusion injury by modulating inflammation, regulating apoptosis, and mitigating oxidative stress.23 Cordycepin has been demonstrated to regulate the expression of the miR193b-5p/MCL-1 axis to treat diabetic nephropathy.25 Additionally, cordycepin inhibits albumin-induced epithelial-to-mesenchymal transition in renal tubular cells by attenuating the production of reactive oxygen species.26 Furthermore, cordycepin exhibits anti-hyperuricemia effects by inhibiting URAT1.27 These multifaceted mechanisms highlight the significant therapeutic potential of cordycepin in the treatment of various kidney diseases, establishing a robust foundation for future clinical applications. Additionally, it was reported that cordycepin could inhibit the activation of renal myofibroblast to alleviate renal fibrosis.28 However, the underlying mechanisms still need to be thoroughly elucidated. Thus, to deepen the understanding of the underlying mechanisms involved anti-fibrotic effect of cordycepin, we performed bulk RNA sequencing with or without cordycepin administration following UIR. KEGG enrichment analysis revealed that mitochondrial fission and IL-6/STAT3 inflammatory signaling related genes were reduced by cordycepin, which indicated that cordycepin may reduce renal fibrosis by targeting mitochondrial and inflammatory reactivation.
The normal functioning of renal tubular epithelial cells relies on mitochondrial homeostasis to provide sufficient ATP.29 Disruption of mitochondrial homeostasis reduces ATP availability and impairs tubular cell function, playing a pivotal role in the pathophysiology of renal fibrosis.30 Additionally, excessive ROS generated by damaged mitochondria triggers inflammation and fibrosis. Mitochondrial dynamics is essential for mitochondrial component integration, energy transfer, and cellular signaling. An imbalance in mitochondrial fission and fusion leads to tubular cell stress, further exacerbating kidney injury. Mitochondria operate as a dynamic network to maintain their function through continuous cycles of fission and fusion.31 Dynamin-related protein 1 (Drp1), a GTPase in the dynamin family, promotes mitochondrial fission by interacting with the mitochondrial membrane.32 During the progress of fission, Drp1 is recruited to the mitochondrial membrane by mitochondrial dynamics proteins of 49 kDa and 51 kDa (Mid49 and Mid51), as well as the fission factor (MFF). Together, they form a ring-like structure with Fis1 to drive mitochondrial separation.33,34 Studies have shown that in various pathological conditions, the expression levels of Drp1 and Fis1 are elevated in damaged renal tubular epithelial cells, while the expression of mitochondrial fusion proteins Mfn1, Mfn2, and OPA1 are significantly reduced.35 Inhibiting Drp1 has been shown to improve ischemic kidney injury by preventing mitochondrial fission.36 In studies related to neuroinflammation, cordycepin has shown to impede Drp1-mediated abnormal mitochondrial fission.24 This suggests that the influence of cordycepin on mitochondrial dynamics plays a crucial role in its therapeutic effects. In the present study, the interaction between cordycepin and Drp1 was examined by molecular docking and dynamics simulations, showing a significant binding affinity. This suggested that Drp1 may serve as a critical target for the anti-fibrotic effects of cordycepin in renal tissues. We conducted a comprehensive investigation into the therapeutic effects of cordycepin on mitochondrial function. Our results demonstrated that cordycepin inhibited Drp1-mediated mitochondrial fission in renal tubular epithelial cells, reduced oxidative stress, increased MMP, and improved mitochondrial function.
Mitochondrial damage plays a critical role in cellular immune responses, with previous studies demonstrating that excessive mitochondrial fission can significantly promote the expression and secretion of IL-6.37 This phenomenon is particularly important in the progression of renal fibrosis.38 IL-6 exacerbates fibrosis by driving fibroblast proliferation, differentiation, migration, and the synthesis of extracellular matrix (ECM) components.16,39 In our experiments, we further observed that mitochondrial dysfunction, mediated by Drp1-dependent fission, enhanced IL-6 secretion, leading to injury in renal tubular epithelial cells and activation of adjacent fibroblasts. Following renal tubular injury, we observed a significant upregulation of IL-6, accompanied by a marked infiltration of fibroblasts surrounding IL-6-positive cells. This suggested that IL-6 acted as a critical signaling mediator between injured tubular cells and fibroblasts. By inhibiting Drp1-mediated mitochondrial fission in UIR mice using cordycepin or Mdivi-1, we found a significant reduction in IL-6 expression, highlighting the essential role of Drp1-mediated fission in promoting IL-6 secretion. This finding is not only relevant to the kidney but may also apply to other organs, where mitochondrial damage and excessive IL-6 secretion are similarly linked to the progression of inflammation and fibrosis. Unveiling this mechanism provides new insights for future therapeutic strategies targeting mitochondrial dynamics and IL-6 signaling pathways, especially in fibrotic diseases affecting the kidney and other organs.
These findings underscore the growing interest in mitochondrial-targeted therapeutic strategies, which hold significant potential for mitigating renal fibrosis.40 Targeting mitochondrial dynamics and function may offer novel therapeutic approaches for treating fibrosis.
Conclusion
The proposed mechanism of this study is summarized in the schematic diagram in Figure 6G. Following UIR, ischemic injury occurs in renal tubular epithelial cells, leading to mitochondrial dysfunction. This dysfunction is characterized by an imbalance in mitochondrial dynamics, including increased fission and decreased fusion. It also involves reduced MMP and elevated levels of mitochondrial oxidative stress. Consequently, the IL-6 inflammatory signaling pathway is activated, which in turn promotes the proliferation and activation of surrounding fibroblasts, contributing to the progression of fibrosis. Our study is the first to demonstrate that cordycepin mitigates renal fibrosis by inhibiting Drp1-mediated mitochondrial fission in injured renal tubular epithelial cells, reducing oxidative stress, and decreasing IL-6 secretion, ultimately attenuating fibroblast activation. This discovery identifies a novel therapeutic target for clinical application in the treatment of CKD-related fibrosis, offering new strategies for its management.
Ethics Approval and Informed Consent
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Fudan University under approval number 2023-109, and all procedures were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All possible efforts were made to minimize animal suffering.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the National Natural Science Foundation of China (82070710 and 82200792), Shanghai Science and Technology Innovation Action Plan (21S219029001 and 22410714200-International Science and Technology Cooperation Projects), Shanghai Key Laboratory of Kidney and Blood Purification, Shanghai Science and Technology Commission (20DZ2271600), Shanghai Municipal Hospital Frontier Technology Project supported by Shanghai Shen Kang Hospital Development Center (SHDC2202230), Natural Science Foundation of Fujian Province (2022J011419), and Shanghai Municipal Key Clinical Specialty (shslczdzk02501).
Disclosure
The authors declare that they have no competing interests.
References
1. Foreman KJ, Marquez N, Dolgert A, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet. 2018;392(10159):2052–2090. doi:10.1016/S0140-6736(18)31694-5
2. Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. 2014;124(6):2299–2306. doi:10.1172/JCI72267
3. Djudjaj S, Boor P. Cellular and molecular mechanisms of kidney fibrosis. Mol Aspects Med. 2019;65:16–36. doi:10.1016/j.mam.2018.06.002
4. Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet. 2017;389(10075):1238–1252. doi:10.1016/S0140-6736(16)32064-5
5. Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol. 2018;80:309–326. doi:10.1146/annurev-physiol-022516-034227
6. Peek JL, Wilson MH. Cell and gene therapy for kidney disease. Nat Red Nephrol. 2023;19(7):451–462. doi:10.1038/s41581-023-00702-3
7. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. 2017;13(10):629–646. doi:10.1038/nrneph.2017.107
8. Bueno M, Calyeca J, Rojas M, Mora AL. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020;33:101509. doi:10.1016/j.redox.2020.101509
9. Liu J, Pan Z, Tong B, et al. Artesunate protects against ocular fibrosis by suppressing fibroblast activation and inducing mitochondria-dependent ferroptosis. FASEB J. 2023;37(6):e22954. doi:10.1096/fj.202201867R
10. Zhang L, Miao M, Xu X, et al. From physiology to pathology: the role of mitochondria in acute kidney injuries and chronic kidney diseases. Kidney Dis. 2023;9(5):342–357. doi:10.1159/000530485
11. Jiang M, Bai M, Lei J, et al. Mitochondrial dysfunction and the AKI-to-CKD transition. Am J Physiol Renal Physiol. 2020;319(6):F1105–F1116. doi:10.1152/ajprenal.00285.2020
12. Doke T, Susztak K. The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development. Trends Cell Biol. 2022;32(10):841–853. doi:10.1016/j.tcb.2022.03.012
13. Yamashita N, Kramann R. Mechanisms of kidney fibrosis and routes towards therapy. Trends Endocrinol Metab. 2024;35(1):31–48. doi:10.1016/j.tem.2023.09.001
14. Ridker PM, Rane M. Interleukin-6 signaling and Anti-Interleukin-6 therapeutics in cardiovascular disease. Circ Res. 2021;128(11):1728–1746. doi:10.1161/CIRCRESAHA.121.319077
15. Karina D-B, Alia H, Ronen L, et al. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. 2018;94(2):315–325. doi:10.1016/j.kint.2018.02.026
16. Chen W, Yuan H, Cao W, et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics. 2019;9(14):3980–3991. doi:10.7150/thno.32352
17. Shi B, Liu Q, Xu C, Zhang Z, Cai J. Chlorantraniliprole induces mitophagy, ferroptosis, and cytokine homeostasis imbalance in grass carp (Ctenopharyngodon idella) hepatocytes via the mtROS-mitochondrial fission/fusion axis. Pestic Biochem Physiol. 2024;200:105830. doi:10.1016/j.pestbp.2024.105830
18. Zhang J, Li W-J, Chen S-Q, et al. Mutual promotion of mitochondrial fission and oxidative stress contributes to mitochondrial-DNA-mediated inflammation and epithelial-mesenchymal transition in paraquat-induced pulmonary fibrosis. World J Emerg Med. 2023;14(3):209–216. doi:10.5847/wjem.j.1920-8642.2023.057
19. Hsu C-Y, Vo TT, Lee C-W, et al. Carbon monoxide releasing molecule-2 attenuates angiotensin II-induced IL-6/Jak2/Stat3-associated inflammation by inhibiting NADPH oxidase- and mitochondria-derived ROS in human aortic smooth muscle cells. Biochem Pharmacol. 2022;198:114978. doi:10.1016/j.bcp.2022.114978
20. Lan T, Yu Y, Zhang J, et al. Cordycepin ameliorates nonalcoholic steatohepatitis by activation of the AMP-activated protein kinase signaling pathway. Hepatology. 2021;74(2):686–703. doi:10.1002/hep.31749
21. Wang H, Duan M, Xu M, et al. Cordycepin ameliorates cardiac hypertrophy via activating the AMPKα pathway. J Cell Mol Med. 2019;23(8):5715–5727. doi:10.1111/jcmm.14485
22. Sun T, Dong W, Jiang G, et al. Cordyceps militaris improves chronic kidney disease by affecting TLR4/NF-κB redox signaling pathway. Oxid Med Cell Longev;2019. 7850863. doi:10.1155/2019/7850863
23. Han F, Dou M, Wang Y, et al. Cordycepin protects renal ischemia/reperfusion injury through regulating inflammation, apoptosis, and oxidative stress. Acta Biochim Biophys Sin. 2020;52(2):125–132. doi:10.1093/abbs/gmz145
24. Zhang X-L, Huang W-X, Tang P-C, et al. Anti-inflammatory and neuroprotective effects of natural cordycepin in rotenone-induced PD models through inhibiting Drp1-mediated mitochondrial fission. Neurotoxicology. 2021;84:1–13. doi:10.1016/j.neuro.2021.02.002
25. Zheng R, Zhang W, Song J, Zhong Y, Zhu R. Cordycepin from Cordyceps militaris ameliorates diabetic nephropathy via the miR193b-5p/MCL-1 axis. Chin Med. 2023;18(1):134. doi:10.1186/s13020-023-00842-5
26. Xiao L, Ge Y, Sun L, et al. Cordycepin inhibits albumin-induced epithelial-mesenchymal transition of renal tubular epithelial cells by reducing reactive oxygen species production. Free Radic Res. 2012;46(2):174–183. doi:10.3109/10715762.2011.647688
27. Yong T, Chen S, Xie Y, et al. Cordycepin, a characteristic bioactive constituent in cordyceps militaris, ameliorates hyperuricemia through URAT1 in hyperuricemic mice. Front Microbiol. 2018;9:58. doi:10.3389/fmicb.2018.00058
28. Li L, He D, Yang J, Wang X. Cordycepin inhibits renal interstitial myofibroblast activation probably by inducing hepatocyte growth factor expression. J Pharmacol Sci. 2011;117(4):286–294. doi:10.1254/jphs.11127FP
29. Zhang J, Wei Q, Wu S-K, et al. Inhibition of Drp1-mediated mitochondrial fission improves contrast-induced acute kidney injury by targeting the mROS-TXNIP-NLRP3 inflammasome axis. Int Immunopharmacol. 2024;133:112001. doi:10.1016/j.intimp.2024.112001
30. Brage PC, Alves MG, Rodrigues AS, Oliveira PF. Mitochondrial pathophysiology on chronic kidney disease. Int J mol Sci. 2022;23(3):1776. doi:10.3390/ijms23031776
31. Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–531. doi:10.1146/annurev-physiol-021115-105011
32. Liu A, Hatch AL, Higgs HN. Effects of phosphorylation on Drp1 activation by its receptors, actin, and cardiolipin. mol Biol Cell. 2024;35(2):1–15. doi:10.1091/mbc.E23-11-0427
33. Qin L, Xi S. The role of mitochondrial fission proteins in mitochondrial dynamics in kidney disease. Int J mol Sci. 2022;23(23):14725. doi:10.3390/ijms232314725
34. Ren J, Liu J, Zhang J, et al. Dynamin-related protein 1 binding partners MiD49 and MiD51 increased mitochondrial fission in vitro and atherosclerosis in high-fat-diet-fed ApoE-/- mice. Int J mol Sci. 2024;25(1):244. doi:10.3390/ijms25010244
35. Jia X, Zhu L, Zhu Q, Zhang J. The role of mitochondrial dysfunction in kidney injury and disease. Autoimmun Rev. 2024;23(6):103576. doi:10.1016/j.autrev.2024.103576
36. Wang Y, Liu Q, Cai J, et al. Emodin prevents renal ischemia-reperfusion injury via suppression of CAMKII/DRP1-mediated mitochondrial fission. Eur J Pharmacol. 2022;916:174603. doi:10.1016/j.ejphar.2021.174603
37. Nechemia-Arbely Y, Barkan D, Pizov G, et al. IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol. 2008;19(6):1106–1115. doi:10.1681/ASN.2007070744
38. Li Y, Zhao J, Yin Y, et al. The role of IL-6 in fibrotic diseases: molecular and cellular mechanisms. Int J Biol Sci. 2022;18(14):5405–5414. doi:10.7150/ijbs.75876
39. Zhang Y, Qin X, Yang Y, et al. Ginkgo biloba extract attenuates cisplatin-induced renal interstitial fibrosis by inhibiting the activation of renal fibroblasts through down-regulating the HIF-1α/STAT3/IL-6 pathway in renal tubular epithelial cells. Phytomedicine. 2023;115:154809. doi:10.1016/j.phymed.2023.154809
40. Zhao X, Li Y, Yu J, et al. Role of mitochondria in pathogenesis and therapy of renal fibrosis. Metabolism. 2024;155:155913. doi:10.1016/j.metabol.2024.155913
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.