")
Back to Journals » International Journal of Nanomedicine » Volume 20
Core-Shell Magnetic Nanocarriers: Fe3O4-Hydroxyapatite/Polysuccinimide Hybrids for Enhanced Oral Bioavailability of Fluorouracil
Authors Zhang W, Wang Q, Zhai F, Fan X, Meng F, Shen G, Zhu Y, Cao J, Yu F
Received 4 December 2024
Accepted for publication 8 March 2025
Published 20 March 2025 Volume 2025:20 Pages 3671—3695
DOI https://doi.org/10.2147/IJN.S507458
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jie Huang
Wenhui Zhang,1 Qiang Wang,2 Fengguo Zhai,1 Xingjun Fan,3 Fanqin Meng,1 Guangzhi Shen,1 Ying Zhu,1 Jingdan Cao,1 Fengbo Yu1
1School of Pharmacy, Mudanjiang Medical University, Mudanjiang, 157011, People’s Republic of China; 2Hongqi People Hospital, Mudanjiang Medical University, Mudanjiang, 157011, People’s Republic of China; 3School of Public Health, Mudanjiang Medical University, Mudanjiang, 157011, People’s Republic of China
Correspondence: Fengbo Yu, School of Pharmacy, Mudanjiang Medical University, Mudanjiang, 157011, People’s Republic of China, Email [email protected]
Objective: This study pioneers a pH-responsive core-shell nanoplatform integrating magnetic Fe3O4-hydroxyapatite (Fe/HAP) with polysuccinimide (PSI) polymer, engineered to enhance tumor-targeted delivery of fluorouracil (5-FU) for liver cancer therapy.
Methods: The individual components—hydroxyapatite (HAP), magnetite (F3O4), iron-doped hydroxyapatite (Fe/HAP), and polysuccinimide (PSI)—were synthesized and systematically characterized through Fourier-transform infrared (FTIR) spectroscopy and scanning electron microscopy (SEM). Through a combination of single-factor experiments and Box-Behnken design (BBD) response surface methodology, the formulation parameters were optimized for two nanoparticle systems: (1) non-magnetic 5-FU-loaded PSI-HAP (designated as 5-FU@DC, where DC denotes “drug carrier”) and (2) magnetic-targeted formulations 5-FU@PSI-Fe/HAP with varying iron content (5-FU@FeDC20, 5-FU@FeDC30, 5-FU@FeDC40). The engineered nanoparticles were thoroughly characterized for their morphological characteristics, hydrodynamic properties (particle size distribution and zeta potential), magnetic responsiveness (vibrating sample magnetometry), and pH-dependent drug release profiles. Nile Red was used to label the drug-loaded nanoparticles, and small animal imaging technology was employed to track their distribution in mice in vivo. Furthermore, in vitro studies examined the effects of these formulations on the proliferation, apoptosis, and migration of Huh-7 liver cancer cells.
Results: The formulations (5-FU@DC and 5-FU@FeDC) were found to form uniform spherical or near-spherical nanoparticles. Vibrating sample magnetometer (VSM) analysis confirmed that the 5-FU@FeDC formulations displayed paramagnetic properties. Zeta potential measurements showed that all prepared systems had negative charges, similar to human biological membranes. All nanoparticles gradually released the drug at pH levels above 5, with the release rate increasing as the pH increased. Compared to the non-magnetic 5-FU@DC formulation, the magnetic 5-FU@FeDC formulations showed significantly longer distribution and retention times in liver tissue and more effectively inhibited the proliferation of Huh-7 cells.
Conclusion: The current study developed a magnetic targeting nano-delivery system using PSI and Fe/HAP as formulation excipients. The system offers uniform particle size, a simple preparation process, and a cost-effective method for targeted drug delivery. It is not only suitable for liver-targeted drug delivery but also applicable for drug delivery to other tissues in the body for anti-tumor drugs.
Keywords: magnetic nanocarriers, Fe3O4-hydroxyapatite core, polysuccinimide coating, fluorouracil, liver cancer
Graphical Abstract:

Introduction
Primary liver cancer (PLC) is considered a major contributor to the global cancer burden, with hepatocellular carcinoma (HCC) being the most prevalent subtype, comprising approximately 90% of PLC cases. As the fourth leading cause of cancer-related mortality worldwide, HCC represents a significant public health challenge. Patients with early-stage HCC are preferred candidates for hepatic resection, transplantation, and radiofrequency. Transarterial chemoembolization (TACE) is the established standard of care for patients with intermediate-stage HCC. In advanced-stage HCC, systemic therapies, such as tyrosine kinase inhibitors and immune checkpoint inhibitors, represent the cornerstone of clinical management.1,2 Surgery is the preferred treatment for HCC, as it offers the only potential for long-term survival and even a cure. However, the majority of liver cancer patients in China are diagnosed at intermediate to advanced stages, leaving them without the option for surgical intervention.3 Fluorouracil (5-FU), the first antimetabolite ever designed and synthesized, remains the most widely used antipyrimidine drug in clinical practice. It exerts its therapeutic effects by inhibiting thymidylate synthase, leading to the conversion of 5-FU into 5-fluoro-2’-deoxyuridine monophosphate. The synthesized metabolite interferes with DNA synthesis, triggering apoptosis and inhibiting the growth of cancer cells.4 It is effective against various cancers, including gastrointestinal tumors, breast cancer, ovarian cancer, cervical cancer, liver cancer, and stomach cancer.5 However, 5-FU is associated with significant adverse effects, particularly the suppression of normal immune function and hematopoiesis, which significantly limits its widespread use and necessitates careful monitoring during treatment.
The introduction of the concept of targeted formulations in 1906 marked the beginning of a new era in drug delivery system research.6 Targeted drug delivery systems offer a more efficient and specific method of delivering drugs to the affected areas compared to traditional dosage forms like solutions, capsules, and tablets. This targeted approach enhances drug bioavailability while reducing the required dosage and minimizing side effects.7 Among these systems, magnetic targeted drug delivery systems (MTDDS) incorporate suitable magnetic components, such as Fe3O4, into the drug delivery system. After administration, an external magnetic field can be applied at specific locations in the body to guide the drug carrier directly to the target tissue, facilitating localized drug release at the site of disease.8,9 Magnetically targeted formulations furnish advantages such as simple preparation processes and good targeting effects,10 rendering them useful as an efficient and rapid local targeted drug delivery system.
Nanocarrier-based drug delivery systems (NDDSs) are composed of nanomaterials with diameters less than 1,000 nm.11 They have broad prospects in biomedical applications due to their efficient drug loading, targeted delivery, and controlled release capabilities.12 The introduction of NDDSs has significantly transformed cancer treatment in clinical practice,13,14 fostering rapid advancements in targeted therapies, combination therapy, and early diagnosis of tumors.15 Because of their specific spatial dimensions, NDDSs show unique surface effects and small-size effects.16,17 Typical nanomaterials have several advantageous features including a high surface-to-volume ratio, strong electrical conductivity, superparamagnetic behavior, spectral shifts, and unique fluorescence properties.18 The high surface-to-volume ratio of nanomaterials enhances their ability to interact with biomolecules or protein groups. This characteristic improves the targeting of NDDSs in tumor treatment, ultimately leading to better therapeutic outcomes while minimizing toxicity to normal cells.11 The targeting mechanisms of NDDSs are broadly classified into four distinct categories: passive targeting, active targeting, physical and chemical targeting. Passive targeting relies on the enhanced permeability and retention (EPR) effect, which allows macromolecular substances—such as liposomes, nanoparticles, and certain macromolecular drugs—to infiltrate tumor tissue more effectively.19 Active targeting, on the other hand, involves modifying the surfaces of NDDSs with specific antigens, lectins, ligands, or other molecules that bind to overexpressed receptors, antigens, and glycoproteins on tumor cells. This modification facilitates the active targeting of tumor cells.20 Currently, passive targeting nanomedicines are clinically more common, while active targeting is more difficult to achieve.21 However, both systems are limited in their accumulation around tumors due to the high interstitial fluid pressure in the tumor microenvironment.22
Research on nano-magnetic delivery systems aims to overcome the challenge passive nano-delivery systems face in accumulating around tumor tissues. Integrating magnetic materials into the delivery system allows anticancer drugs to be concentrated and retained more effectively at the lesion site. Furthermore, the ability of these NDDSs to penetrate tumor tissue enhances the delivery of drugs directly into tumor cells, leading to significant synergistic therapeutic effects while minimizing toxicity. In recent years, numerous studies have reported on the use of nanomagnetic drug delivery systems for the administration of anticancer drugs.23,24 Studies have demonstrated that, for liver cancer treatment drugs, the concentration and retention time of magnetic targeting nano-delivery systems in the target area have significantly improved at the same dosage. Also, the toxicity of these drugs to non-target organs has either not increased or may have even decreased, greatly enhancing both the efficacy and safety of the treatments.25
In this study, 5-FU serves as a model drug, using a biodegradable and biocompatible hydroxyapatite (HAP)26,27 carrier embedded with magnetic Fe3O4 (Fe/HAP) for drug adsorption and release. The carrier is coated with biodegradable polysuccinylamide (PSI)28,29 to encapsulate the drug-loaded Fe/HAP, forming uniformly sized nanoscale magnetic targeted drug delivery systems, 5-FU@PSI-Fe/HAP (5-FU@FeDC, oral administration). This system is specifically designed for liver-targeted delivery of 5-FU. Oral administration helps lower production costs and simplifies formulation, reducing risks associated with injections. Moreover, the drugs can be rapidly concentrated in liver tissue post-administration using an external magnetic field. This targeted approach minimizes potential toxic side effects that might arise from systemic drug circulation with nano-carriers. In this drug delivery system, the PSI coating on Fe/HAP smooths the surface of magnetic HAP particles, preventing size changes due to the aggregation of individual nano-Fe/HAP particles. This enhances drug loading capacity and prolongs and stabilizes drug release. Nano-HAP30 and PSI31 possess inherent anti-proliferative effects on tumor cells, rendering the nano-PSI-Fe/HAP (FeDC) delivery system an effective carrier for anti-tumor drugs. The magnetic targeted drug delivery system 5-FU@PSI-Fe/HAP (5-FU@FeDC) described in this paper offers advantages such as a simple preparation process, low cost, ease of production, and scalability.
Material and Methods
Materials
Fluorouracil (5-FU), Nile Red, and Iron(II) chloride tetrahydrate (FeCl2·4H2O) were purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China); L-Aspartic acid and Iron(III) chloride hexahydrate (FeCl3·6H2O) were procured from Aladdin Industrial Co., Ltd. (Shanghai, China); Fetal bovine serum (FBS) was obtained from Beijing Sino Biological Co., Ltd. (Beijing, China); Dulbecco’s Modified Eagle Medium (DMEM) was provided by Thermo Fisher Scientific (Shanghai, China); Crystal Violet and Hoechst 33342 were purchased from Beyotime Biotechnology Co., Ltd. (Shanghai, China); 4′,6-Diamidino-2-phenylindole (DAPI), Cell Counting Kit-8 (CCK-8), penicillin-streptomycin, and phosphate-buffered saline (PBS) were obtained from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China); Coumarin 6 was purchased from Shanghai Macklin Biochemical Co., Ltd. (Beijing, China); Huh-7 cells was procured from Nanjing Abace Biotechnology Co., Ltd. (Nanjing, China). All other chemicals and solvents used were of analytical or high-performance liquid chromatography (HPLC) grade and were obtained from commercial suppliers.
Methods
Preparation of Excipients
Preparation of HAP
Tween-80 (5 mL) was dissolved in 20 mL butyl acetate and then added to 60 mL of 1 mol/L Na2CO3 solution under constant stirring for 20 min. The emulsion was then slowly dripped into a 0.2 mol/L CaCl2 solution and stirred for 1 h. The resulting precipitate was separated by filtration, rinsed multiple times with deionized water, washed three times with anhydrous ethanol, and dried at 80°C for 12 h to obtain CaCO3 powder. After that, 1 g of the prepared CaCO3 powder was added to 200 mL of deionized water to form a suspension. While stirring, 200 mL of a 0.03 mol/L Na2HPO4 solution was added to the suspension at a rate of 2 mL/min. The reaction was carried out at atmospheric pressure in a 60 °C water bath, with the pH adjusted to 11 using a 20% NaOH solution. After 2 h stirring, the product was separated, and washed with deionized water until neutral, followed by rinses with anhydrous ethanol and dichloromethane. The final product was dried at 80°C for 12 h to yield HAP.
Preparation of Fe3O4
FeCl3•6H2O and FeCl2•4H2O were dissolved in 50 mL of deionized water to achieve concentrations of 0.050 mol/L and 0.028 mol/L, respectively. Ammonia solution (5 mL) was added to this solution, and the mixture was stirred at 20,000 rpm for 20 min, resulting in a black suspension. The nanoparticles were magnetically separated from the supernatant and washed five times with deionized water. After washing, the nanoparticles were collected by magnetic separation and dried at 80 °C for 12 h.
Preparation of Fe/HAP
After preparing CaCO3 following the method described in Preparation of HAP, three CaCO3 suspensions (1.0000 g/200 mL) were prepared. To each suspension, 0.0500 g, 0.0333 g, and 0.0250 g of magnetic Fe3O4 were added, respectively. The mixtures were stirred until homogenous, and the preparation of HAP was continued, as described in Preparation of HAP. After drying, Fe/HAP composites with mass ratios of 20:1, 30:1, and 40:1 (mCaCO3:mFe3O4) were obtained, labeled as Fe/HAP20, Fe/HAP30, and Fe/HAP40 respectively, with Fe/HAP20 containing the lowest CaCO3 and highest Fe3O4 content.
Preparation of PSI
In a 250 mL round-bottom flask, L-aspartic acid (10.0 g) was mixed with phosphoric acid (85%, 6 mL) and deionized water (1 mL). The mixture was heated under reduced pressure using a rotary evaporator at 170°C for 1 hour. After completion of the reaction, the temperature was reduced to 100°C, and 60 mL of N, N-dimethylformamide was added to dissolve the residue. This solution was then slowly added to 500 mL of deionized water under mechanical stirring (700 rpm) and ultrasonic conditions (60°C, 1500 W) at room temperature. The resulting product was collected by filtration, washed with deionized water and dichloromethane, and dried at 80°C for 12 h.
Characterizations
Fourier-transform infrared (FTIR) spectra analysis was performed using a Nicolet iS5 spectrometer (Thermo Fisher Scientific, USA) over a wavenumber range of 400 to 4000 cm−1 with a resolution of 1 cm−1. The morphological characteristics of the samples were evaluated by an FEI Sirion field emission scanning electron microscope (SEM, Thermo Fisher Scientific, USA). Both FTIR spectroscopy and SEM techniques were employed to characterize the structural and morphological properties of HAP, Fe3O4, Fe/HAP, and PSI. Furthermore, the particle size distribution (PSD) of Fe3O4 nanoparticles was determined using a zetasizer zs90 (Malvern Panalytical, USA).
Optimization of the Formulation and Preparation of the 5-FU-Loaded PSI–HAP (5-FU@DC) and PSI-Fe/HAP (5-FU@FeDC) Delivery System
Determination of the Mass Ratio of PSI Coating and HAP or Fe/HAP
The PSI and HAP or Fe/HAP20, Fe/HAP30, and Fe/HAP40 powders were weighed in different mass ratios, and then the PSI powder was added to a dimethylacetamide solution (concentration of 25 mg/mL). The solution was then sonicated to achieve complete dissolution and stored at room temperature. HAP or Fe/HAP was dissolved in ethanol to prepare a suspension (mPSI: mHAP=3:1, 4:1, 5:1, 6:1, 7:1, and 8:1, Fe/HAP ratio was calculated based on the mass of HAP present), followed by sonication (50 W, 35 kHz) to achieve uniformity. At room temperature, the PSI solution is added dropwise into the stirring HAP suspension (1000 rpm). The precipitate was collected by centrifugation (12000 rpm), washed repeatedly with deionized water and ethanol, and dried at 80°C for 12 h. SEM was employed to characterize and determine the mass ratio of PSI coating and HAP or Fe/HAP.
Single-Factor Experiments and BBD Response Surface Optimization Method to Determine the Formulation and Process
The formulations and preparation of 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 were optimized using a single-factor experimental design along with the Box-Behnken design (BBD) response surface optimization method. The optimization was based on entrapment efficiency (EE, %) and drug-loading capacity (DLC, %) as key parameters. The EE and DLC values were then calculated using the following equations:


Where M0 is the total mass of the added drug, M0 is the total mass of the added drug, M1 is the mass of the drug in the carrier, and M2 is the total mass of the blank carrier; 265 nm was selected as the detection wavelength of 5-FU (Supplementary Figure S1); 5-FU standard curve: A=57.75C-0.002362, R2=0.9998. The linearity was good in the concentration range of 0.004 mg/mL~0.02 mg/mL (Supplementary Figure S2), and the accuracy and precision of the method met the requirements.
The effect of various solvents (H2O, 50% Ethanol, 75% Ethanol, and Anhydrous ethanol), and adsorption times (10, 20, 30, and 60 min) on drug loading in HAP, as well as the impact of 5-FU solution concentration (1, 2, and 3 mg/mL) and the 5-FU to HAP mass ratio (1:1, 4:1, 8:1, 12:1, and 16:1, with Fe/HAP calculated based on HAP content) on drug loading in PSI-HAP (DC) preparations, were investigated as process parameters. The main influences were identified by combining the results derived from single-factor experiments. The optimal formulations and processes were determined using the BBD response surface optimization method, with 5-FU concentration, drug-to-HAP or drug-to-Fe/HAP mass ratio, and PSI-to-HAP or PSI-to-Fe/HAP mass ratio as key influencing factors. The 5-FU-containing preparation was prepared as described in Preparation of Excipients. Specifically, 5-FU was added to the HAP or Fe/HAP suspension and sonicated for 1 min followed by the incorporation of PSI solution. The subsequent steps followed the previously described procedure.
Properties of the Determined 5-FU@DC and 5-FU@FeDC Delivery System
Characterizations of the Final Preparations
The structural and morphological characteristics of the drug-loaded systems, including 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40, were thoroughly analyzed using FTIR, SEM, PSD analysis, and zeta potential measurements (Zeta Transfer Standard ZTS1240, Malvern Panalytical, USA). Furthermore, the magnetic properties of 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 were evaluated by vibrating sample magnetometry (VSM) using the Lakeshore 8604 (Lakeshore Cryotronics, Inc, USA) with a magnetic field range of ±1 T.
In vitro Drug Release from Optimally Prepared 5-FU@DC and 5-FU@FeDC
A test preparation of 10 mg is placed in a centrifuge tube and separately immersed in 15 mL of PBS at pH values of 5.4, 6.8, and 7.4. The combined solutions are then incubated in a constant-temperature shaker. Samples are taken at the following time points: 1, 2, 4, 6, 8, 10, 12, 24, 48, 72 and 96 h (n=3). At each time point, 2 mL of the solution is removed and replaced with 2 mL of fresh PBS. After sampling, the solutions are centrifuged and washed, and the release of 5-FU is quantified at 265 nm using a UV-Vis spectrophotometer. Each set of samples is tested in triplicate, and the cumulative release curve of 5-FU is subsequently plotted.
Distribution of 5-FU@DC and 5-FU@FeDC in vivo
The systemic distribution of the compounds was investigated using an animal model. HAP, Fe/HAP20, Fe/HAP30, or Fe/HAP40 were mixed with ethanol along with 5-FU. A 1 mg/mL solution of Nile Red in ethanol was then prepared and combined with the PSI solution. This mixture was subsequently added to the suspension containing HAP or Fe/HAP, and 5-FU. Accordingly, Nile red 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30 and 5-FU@FeDC40 were prepared. Then, 84 female Kunming mice (SPF grade, 4–6 weeks old, weighing 25–30 g) were divided into 5 groups: saline ig group, 5-FU@DC ig group, 5-FU@FeDC20 ig group, 5-FU@FeDC30 ig group, 5-FU@FeDC40 ig group. After administering the treatment, a magnetic field of 5000 Gs was applied above the liver of the mice for 30 min. Before administration, the mice were fasted for 12 h; however, they had unrestricted access to water. Each mouse received an intragastric dose of 30 mg/mL Nile Red (200 mg/kg). Mice were euthanized at various time points: 1, 3, 6, 9, 12, and 24 h post-administration, using cervical dislocation. At each time point, the organs (heart, liver, spleen, lung, and kidney) were collected. The organs were then placed under a live fluorescence imager (NightOWL LB 983 in vivo imaging system) for observation and imaging, using a fixed excitation wavelength of 530 nm and an emission wavelength of 600 nm. This study was approved by the Animal Ethics Committee of Mudanjiang Medical University (IACUC-20231108-120), and all applicable international, national, and institutional guidelines for the care and use of animals were duly followed.
In vitro Pharmacodynamics of 5-FU@DC and 5-FU@FeDC
Cell Culture
Huh-7 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin-streptomycin under standard culture conditions. The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2, with the complete medium replaced every 48 h.
CCK-8 Assay for Evaluating Cell Proliferation Capacity
Cell viability was evaluated using the Cell Counting Kit-8 (CCK-8) assay. Briefly, Huh-7 cells were seeded in 96-well plates at a density of 1 × 104 cells per well and allowed to adhere for 12 h in a humidified incubator at 37°C with 5% CO2. Following cell attachment, the culture medium was removed and replaced with a fresh medium containing different formulations.: 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 at concentrations ranging from 5 to 100 μg/mL (5, 20, 40, 60, 80, and 100 μg/mL). After 24 h of treatment, 10 μL of CCK-8 solution was added to each well containing 100 μL of culture medium, and the plates were incubated for a further 2 h at 37°C. The optical density (OD) of each well was measured at 480 nm using a microplate reader, and cell viability was determined as a percentage based on the manufacturer’s protocol.

Morphological Observation
Huh-7 cells were inoculated and cultured in 6-well plates at 37°C with a 5% CO2 environment for 12 h. Once the cells reached an appropriate confluence, the culture medium was removed, and fresh medium containing 50 μmol/L of 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, or 5-FU@FeDC40 was introduced. A blank control group (medium only) was also established. After 24 h of culture, the cells were washed with PBS buffer and the changes in cell morphology in response to the different treatments were observed under an inverted microscope.
Clone Formation Assay
The colony formation assay was performed to evaluate the inhibitory effects of various formulations on tumor cell proliferation. Huh-7 cells were seeded into 6-well plates at a density of 1,000 cells per well and allowed to adhere for 24 h in a humidified incubator at 37 °C with 5% CO2. Subsequently, the culture medium was replaced with a serum-free medium containing 10 μmol/L of the test formulations: 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40. Cells were maintained under standard culture conditions for 14 days and were monitored daily for growth until colony formation, defined as clusters containing ≥50 cells. Upon reaching this stage, the incubation was terminated. The cells were fixed with 4% paraformaldehyde for 15 min, washed three times with phosphate-buffered saline (PBS), and stained with 0.1% crystal violet for 10 min. After staining, the plates were rinsed with PBS, air-dried, and imaged. The number of colonies was counted, and the colony formation rate was calculated based on the colony count.
Wound Healing Assay
Huh-7 cells in the logarithmic growth phase were seeded into 6-well plates at a density of 5 × 105 cells per well and cultured until reaching approximately 80% confluence. A uniform scratch was introduced into the cell monolayer using a sterile 10 μL pipette tip, and this time point was designated as 0 h. The cells were subsequently treated with serum-free medium containing 50 μmol/L of the test formulations: 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40, with serum-free medium devoid of drugs serving as the negative control. Cell migration was monitored at 12 h and 24 h post-treatment, and images were captured using an inverted phase-contrast microscope. The scratch area was quantified using image analysis software, and the cell migration rate was calculated based on the reduction in scratch width over time.

Apoptosis Assay
To evaluate nuclear apoptosis induction, Huh-7 cells were seeded onto glass coverslips in 24-well plates at a density of 5×104 cells/well and allowed to adhere for 24 h. Cells were exposed to 100 μmol/L concentrations of 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, or 5-FU@FeDC40 for 4 h, with serum-free medium serving as the negative control. Post-treatment, cells were immobilized with 4% paraformaldehyde (15 min, RT), and washed thrice with PBS (pH 7.4). Nuclear chromatin was counterstained with Hoechst 33342 (10 μg/mL, 20 min, 37 °C) to visualize apoptotic features. Following PBS washes, coverslips were mounted with an anti-fade reagent, and the samples were examined using a Leica DMI8 fluorescence microscope with a 20× oil immersion objective (excitation/emission: 350/461 nm).
Cellular Uptake Assay
To evaluate the cellular internalization of 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 in Huh-7 cells, a fluorescence-based cellular uptake assay was performed using coumarin-6 as the fluorescent probe. A 1 mg/mL solution of Coumarin-6 was prepared, and according to the formulation protocol, HAP or Fe/HAP was combined with the drug, and thoroughly mixed to ensure uniform dispersion. The mixture was then supplemented with PSI solution through dropwise addition. After centrifugation, washing, and drying, the fluorescent formulations of Coumarin 6-5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40, which included Coumarin-6-labeled 5-FU@DC and 5-FU@FeDC, were prepared.
Huh-7 cells were seeded in 24-well plates containing cell-climbing slices at a density of 2×105 cells per well and incubated for 12 hours at 37 °C in a 5% CO2 atmosphere. The cells were then treated with 50 μmol/L concentrations of the respective formulations, each containing an equivalent concentration of coumarin-6, for 1, 2, and 4 h. Post-incubation, cells were fixed with 4% paraformaldehyde for 10 minutes, washed three times with PBS buffer, and stained with DAPI for 5 minutes. Finally, after DAPI staining, cells were washed with PBS buffer, treated with an anti-fade reagent, and covered with a coverslip. Fluorescence images of cellular uptake from the different formulations were then captured using a fluorescence microscope.
This optimized methodology facilitated a quantitative comparison of time-dependent internalization kinetics among the four formulations. Dual-channel imaging (blue: DAPI, green: formulations containing coumarin 6, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40) offered high spatial resolution of nanoparticle distribution relative to cell nuclei, providing essential insights into formulation-specific uptake mechanisms and their association with therapeutic potential.
Cytotoxicity Study
The cytotoxic profiles of nanocarriers (DC, FeDC20, FeDC30, FeDC40) without drug loading, were systematically evaluated on Huh-7 hepatocellular carcinoma cells using the standardized Cell Counting Kit-8 (CCK-8) assay protocol. Logarithmic phase Huh-7 liver cancer cells were selected and diluted to a single cell suspension with a concentration of 1×106 cells/mL. The cells were inoculated into a 96-well plate, with each well containing 1×104 cells. After allowing the plate to sit on the workbench for 10 minutes, it was placed in a 37 °C incubator for overnight culture (12 h). Next, 100 μL of DC, FeDC20, FeDC30, and FeDC40, each at mass concentrations of 5, 20, 40, 60, 80, and 100 μg/L, was added to each well. Each group included five replicate wells, and a blank control group (medium only). A solvent blank control group (without cells and drugs) was also established. After a 24-hour incubation period, the drug-containing medium was discarded, and the cells were washed with PBS buffer. Fresh medium was added, followed by the addition of 10 μL of CCK-8 reagent to each well. After incubating for 2 h, the absorbance at 450 nm was measured using a Multiskan FC microplate reader (Molecular Devices, Shanghai, China) to calculate the cell survival rate.
Statistical Analysis
All experimental data were expressed as mean ± standard deviation (SD) with biological replicates (n ≥ 3). Statistical significance was evaluated using GraphPad Prism 10.1 (GraphPad Software, La Jolla, CA, USA). For group comparisons, either a t-test (two groups) or one-way analysis of variance (ANOVA) with post-hoc analysis (multiple groups) was applied based on experimental design. Significance thresholds were defined as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results and Discussion
Structure of HAP, Fe3O4, Fe/HAP, and PSI
The primary characteristic peaks of HAP, shown in Figure 1Aa, align with previously reported literature.32 The absorption peaks at 3554, 3471, 3417, 1637, and 1618 cm−1 correspond to the stretching and bending vibrations of O-H. The asymmetric stretching vibrations of the phosphate group are observed at 1108 and 1043 cm−1, while the double peaks at 614 and 565 cm−1 are associated with the bending vibrations of O-P-O. The FTIR spectrum of the synthesized Fe3O4 nanoparticles (Figure 1Ba) demonstrated characteristic Fe-O vibrational bands at 579 cm−1 and 440 cm−1, corresponding to the octahedral Fe3+-O and tetrahedral Fe2+-O stretching vibrations in the inverse spinel structure of magnetite, respectively, while the weak broad band around 3412 cm⁻1 and 1624 cm⁻1 indicated the presence of trace amounts of surface-adsorbed hydroxyl groups. The FTIR spectra of Fe/HAP20, Fe/HAP30, and Fe/HAP40 composites (Figure 1Ca–Ea) displayed distinct absorption bands associated with both Fe3O4 and HAP, thereby confirming the successful incorporation of magnetite nanoparticles into the HAP matrix. Specifically, the characteristic Fe-O stretching vibrations of Fe3O4 were observed at 570 cm−1 and 430 cm−1, while the phosphate group vibrations of HAP at ~960 cm⁻1 and in the range of 1020–1100 cm⁻1 remained pronounced, indicating that the crystallinity of HAP was preserved after modification. Corresponding SEM analysis further corroborated these findings, revealing that the Fe/HAP composites maintained microstructural characteristics similar to pristine HAP. In Figure 1Fa, the peak at 3438 cm⁻1 corresponded to the stretching vibration of -CO-N-, while the peak at 2925 cm⁻1 was primarily attributed to the stretching vibration of CH2 groups. The peaks at 1717 and 1653 cm−1 represented the carbonyl stretching vibrations of the amide bond present in R-CO-R, while the peaks at 1399 and 1362 cm−1 corresponded to the stretching vibrations of -C-N-. These peaks are characteristic of PSI, confirming the presence of PSI in the composite material.
 |
Figure 1 Characterization of HAP, Fe3O4, Fe/HAP, and PSI ((A) HAP; (B) Fe3O4; (C) Fe/HAP20; (D) Fe/HAP30; (E) Fe/HAP40; (F) PSI; a: FTIR spectra; b: SEM micrograph; c: Particle Size Distribution, PSD). |
SEM analysis revealed that both synthesized HAP and its iron-doped variants (Fe/HAP) demonstrated aggregated morphologies (Figure 1Ab, Cb–Eb). The Fe/HAP composites retained structural characteristics analogous to pure HAP (Figure 1Ab), suggesting that the low-concentration Fe species were uniformly integrated within the HAP matrix, thereby preserving the macrostructure of the original material. This observation was further corroborated by the aggregated morphology of F3O4 particles (Figure 1Bb) and PSI (Figure 1Fb), indicating a tendency for aggregation in the synthesized materials. FTIR and SEM analysis are essential characterization techniques in the preparation of PSI-HAP nanocarriers, providing real-time insights regarding the synthesis of excipients, while maintaining structural integrity, and following morphological changes.
In Figure 1Bc, the average particle size of Fe3O4 nanoparticles is 1129 nm, with a PDI of 0.695. The PDI reflects the particle size distribution, where a value below 0.7 generally aligns with the theoretical model’s range of applicability, indicating a more uniform particle size distribution. A lower PDI value suggests a more homogeneous sample in terms of particle size. However, due to the magnetic properties of the synthesized Fe3O4 nanoparticles, mutual adsorption leads to particle aggregation, resulting in a larger observed particle size and a higher PDI value.
Optimization of the Formulation and Preparation of 5-FU-@DC and 5-FU@FeDC
Mass Ratio of PSI Coating and HAP or Fe/HAP
Figure 2 shows that when the mass ratio of PSI to HAP is around 5:1, and that for PSI to Fe/HAP20, Fe/HAP30, Fe/HAP40 is around 7:1, the addition of PSI disrupts the aggregated state of HAP or Fe/HAP. This process results in the formation of nanoparticles that have relatively uniform and spherical or nearly spherical shapes. Encapsulating Fe3O4 within HAP and coating it with PSI effectively reduces the agglomeration of magnetite nanoparticles. In contrast to HAP alone, the incorporation of Fe3O4 necessitates a higher PSI amount to effectively break the aggregation of Fe/HAP and create dispersed nanoparticles. This may be attributed to the increased attraction between the original HAP particles caused by the presence of the magnetic material. As illustrated in Figure 1, PSI, HAP, and Fe/HAP showed aggregation when isolated. However, within a defined stoichiometric range under aqueous conditions, PSI interacts with HAP or Fe/HAP, leading to the rapid self-assembly of monodisperse, non-aggregated nanoparticles. The observed phenomenon was attributed to the rapid formation of a PSI-derived nanocoating on HAP or Fe/HAP surfaces, which concurrently decreased particle aggregation through electrosteric stabilization and regulated crystallite growth kinetics, facilitating the formation of uniformly dispersed nanoparticles. Previous studies have consistently emphasized the exceptional efficacy of PSI and its derivatives (X-PSI) as coating materials for HAP-based systems. The functional compound X, which may represent targeting moieties (eg, antibodies, peptides), was covalently conjugated to PSI molecules via carbodiimide-mediated amidation, enhancing the system’s functional versatility. However, alternative amino acid-based polymers (eg, polylysine) or chitosan derivatives did not show comparable anti-aggregation effects. This limitation was likely attributed to inadequate interfacial binding energy or mismatched charge distribution, which hindered the effective stabilization of the HAP-based system.
 |
Figure 2 SEM micrographs of DC and FeDC ((A): DC, PSI:HAP=1:5, w/w; (B) FeDC20, PSI:Fe/HAP20=1:7, w/w; (C) FeDC30, PSI: Fe/HAP30=1:7, w/w; (D) FeDC40, PSI: Fe/HAP40=1:7, w/w). |
Results of Single-Factor Experiments
Effect of Solvents on the Drug Loading Capacity of HAP
The results of EE and DLC in Table 1 indicate that while the solubility of 5-FU decreases with reduced solvent polarity, the adsorption capacity of HAP for 5-FU increases. This suggests that non-polar solvents are more effective for 5-FU adsorption by HAP. The incorporation of Fe3O4 enhances the EE, although this improvement is not particularly substantial.
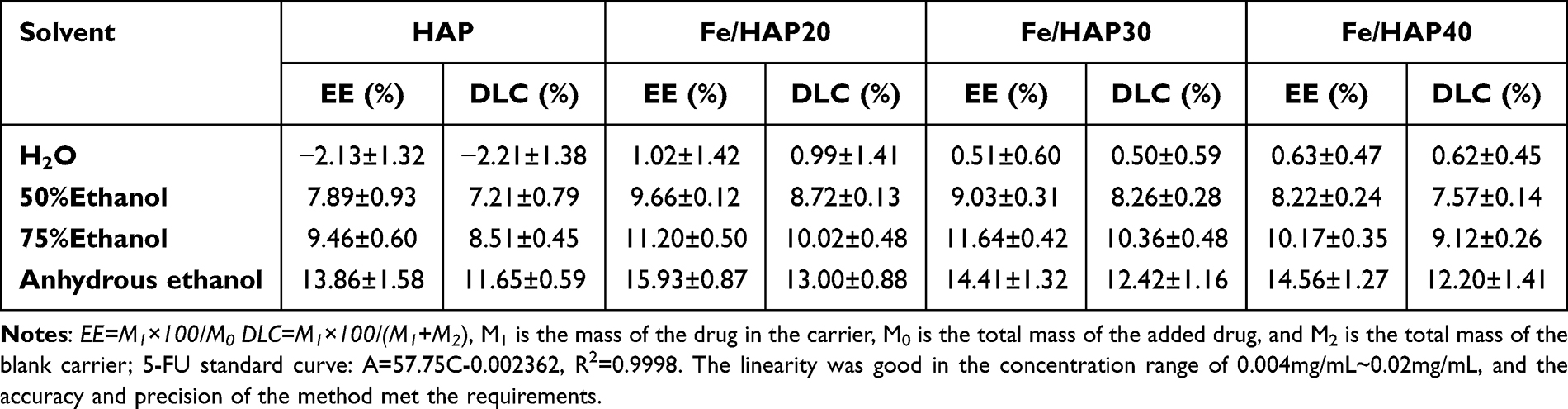 |
Table 1 Effect of Different Solvents on Drug Loading Capacity of HAP (n=3) |
Effect of Adsorption Time on Drug Loading of HAP
As can be seen from Table 2, when the adsorption time reaches 30 min, the adsorption of 5-FU by HAP and Fe/HAP can reach a relative equilibrium and saturation.
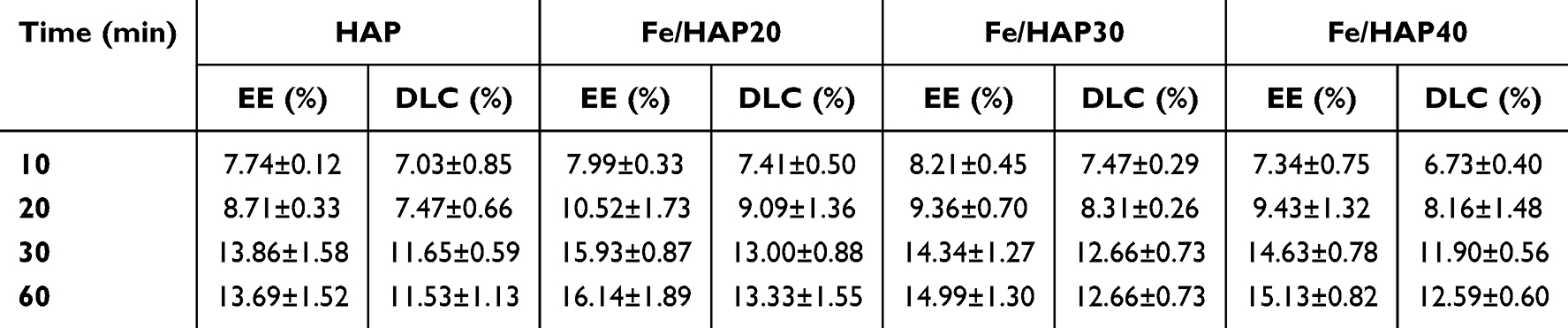 |
Table 2 Effect of Adsorption Time on Drug Loading of HAP (n=3) |
Effect of 5-FU Solution Concentration on Drug Loading of DC and FeDC Preparations
Using anhydrous ethanol as the solvent and an adsorption time of 30 min, the effects of varying concentrations of 5-FU (1 mg/mL, 2 mg/mL, 3 mg/mL) on the encapsulation efficiency and drug loading of DC and FeDC preparations were investigated. The results are presented in Table 3.
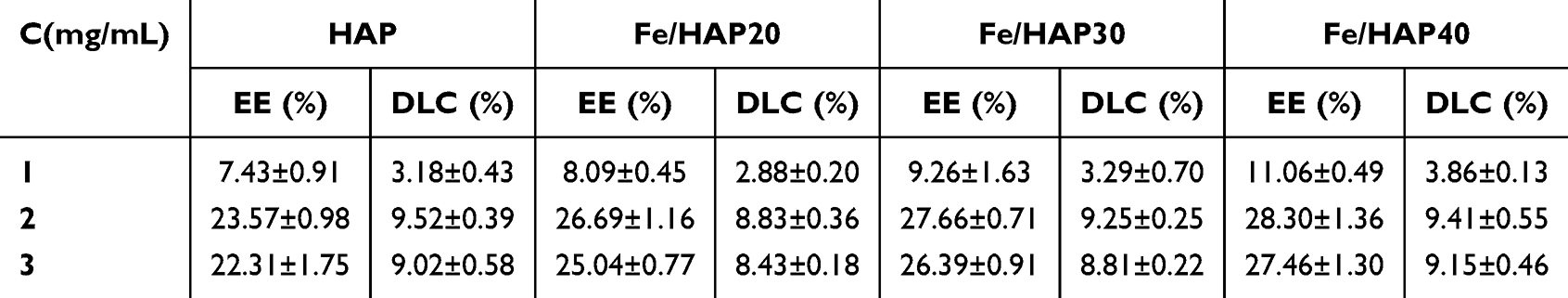 |
Table 3 Effect of 5-FU Solution Concentration on Drug Loading of DC and FeDC Preparations (n=3) |
From Table 3, it can be seen that the concentration of 5-FU has a very significant effect on the encapsulation and drug loading of HAP-adsorbing drugs. When the drug concentration exceeds 2 mg/mL, it significantly enhances the drug encapsulation efficiency. While the inclusion of PSI increases the drug encapsulation rate, the overall drug loading of the preparation slightly decreases compared to HAP alone (Table 1). This reduction is attributed to the increased amount of excipients in the formulation.
Effect of the 5-FU to HAP Mass Ratio on Drug Loading in DC and FeDC Formulations
The solvent used was fixed as anhydrous ethanol, and the drug adsorption time was set at 30 min with a 5-FU concentration of 2 mg/mL. The study investigated the impact of varying drug-to-HAP mass ratios on the entrapment efficiency and drug loading of DC and FeDC preparations, with the results presented in Table 4.
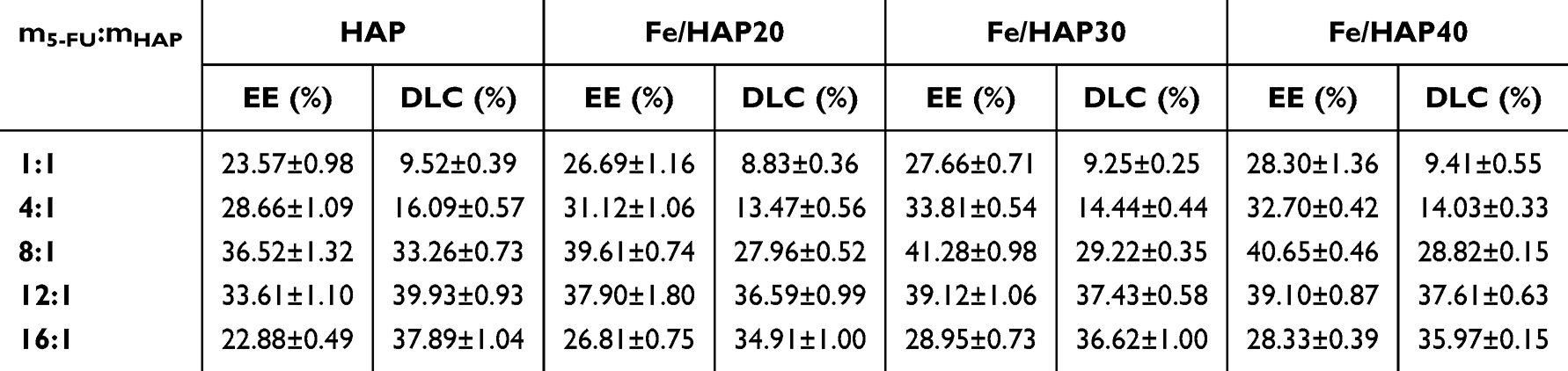 |
Table 4 Effect of the 5-FU to HAP Mass Ratio on Drug Loading in DC and FeDC Formulations (n=3) |
Table 4 demonstrates that the mass ratio of 5-FU to HAP and Fe/HAP has a significant influence on the encapsulation and drug loading of the formulation, particularly enhancing drug loading. The most favorable encapsulation efficiency and drug loading are observed when the mass ratio ranges between 4:1 and 16:1. Furthermore, a comparison of Tables 3 and 4 indicates that the inclusion of Fe3O4 in the PSI-HAP formulations improves drug encapsulation, likely due to the increased amount of PSI added.
Determination of the 5-FU Formulation for DC and FeDC Using the BBD
Based on the single-factor research results, drug concentrations ranging from 1 mg/mL to 3 mg/mL, a drug-to-HAP mass ratio of 4:1 to 16:1, a PSI-to-HAP mass ratio of 4:1 to 6:1, a PSI-to-Fe/HAP40 mass ratio of 6:1 to 8:1, and PSI-to-Fe/HAP20 and Fe/HAP30 mass ratio of 7:1 to 8:1 were identified as the primary influencing factors. The Design-Expert 13.0 software was utilized for experimental design, enabling the measurement of results and the creation of response surface curve graphs (Figure 3), which ultimately guided the determination of the final formulation and process (Table 5).
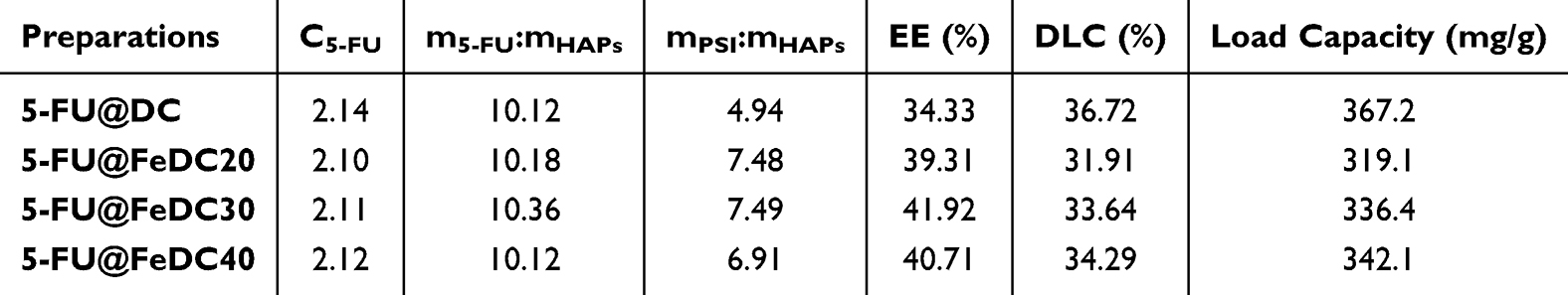 |
Table 5 Optimization Results of Preparation Formula |
 |
Figure 3 3D-response surface plot for optimization of the prepared formulations ((A) 5-FU@DC; (B) 5-FU@FeDC20; (C) 5-FU@FeDC30; (D) 5-FU@FeDC40). |
Figure 3 revealed that key formulation parameters similarly influence system behavior across different pharmaceutical compositions, highlighting underlying mechanistic consistency in their response trends. The experimental results of BBD indicate that the most significant impact on the encapsulation efficiency is the mass ratio of the drug to HAP and Fe/HAP, followed by the drug concentration. The mass ratio of PSI to HAP and Fe/HAP does not significantly affect the encapsulation efficiency. The factors influencing drug loading include the drug concentration, the mass ratio of the drug to HAP and Fe/HAP, and the mass ratio of PSI to HAP and Fe/HAP. Among these, the mass ratio of the drug to HAP and Fe/HAP exerts the greatest effect on drug loading, followed by drug concentration and, finally, the PSI to HAP and Fe/HAP ratio. Overall, incorporating Fe3O4 enhanced the encapsulation efficiency of PSI-HAPs, though increasing the PSI amount slightly decreased final drug loading. However, the drug loading capacity of PSI-HAPs remains above 300 mg/g. Current organic nanomedicines often suffer from low drug loading capacity, with liposomes, for example, frequently unable to exceed 10 mg/g.33 This implies that patients may consume a significant amount of excipients, thereby exacerbating their physical burden.34 Therefore, enhancing drug loading remains a significant challenge in developing anticancer drugs.
In summary, the PSI-HAP nanodelivery system was formulated and synthesized using PSI and HAP as excipients through a straightforward process. The absence of complex instrumentation or harsh reaction conditions simplified manufacturing and facilitated efficient process optimization. During the optimization of process parameters, the adsorption kinetics between HAP and pharmaceutical compounds should be carefully analyzed, as they are considered essential in governing drug loading efficiency and controlled release profiles. Experimental data revealed that a 30-minute contact period was sufficient for HAP to reach saturated drug adsorption. The adsorption capacity of HAP was largely independent of the solubility of the target drugs. Moreover, mechanistic studies indicated that in solid-liquid systems, drug molecules were efficiently captured by HAP’s surface active sites through interfacial mass transfer processes. This multiphase adsorption mechanism substantially enhanced the drug-loading efficiency of HAP. The solvent environment significantly influenced HAP’s adsorption behavior toward various drug molecules, which can limit its loading efficiency and interaction mechanisms. Comprehensive experimental analysis indicates that formulation optimization primarily depends on the precise control of two critical parameters: drug concentration and the drug-to-HAP mass ratio. This streamlined optimization approach minimized developmental complexity while ensuring reproducibility, which is a significant advantage for clinical translation.
Properties of the Final Preparations
Characterization of the Final Preparations
As shown in the Supplementary Figure S3, the infrared characteristic peaks of all formulations closely resemble those of PSI, indicating that the chemical structure of PSI dominates the surface properties of the composite materials. As illustrated in Figure 4A, the final formulations of 5-FU were of relatively uniform nanoparticles. The particle sizes of 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 were 467.1 nm (Figure 4Ab), 467.2 nm (Figure 4Bb), 326.3 nm (Figure 4Cb), and 382 nm (Figure 4Db), respectively. The PDI values were 0.489 (5-FU@DC, Figure 4Ac), 0.369 (5-FU@FeDC20, Figure 4Bc), 0.294 (5-FU@FeDC30, Figure 4Cc), and 0.357 (5-FU@FeDC40, Figure 4Dc), respectively. Compared to Fe3O4 (Figure 1Bc) alone, the particle size and PDI values of the 5-FU@FeDC formulations were significantly reduced, indicating a more uniform particle size distribution. These findings confirm that magnetically induced agglomeration of Fe3O4 nanoparticles was effectively suppressed, resulting in a homogeneous dispersion within the PSI-HAP carrier systems (FeDC20, FeDC30, and FeDC40). Conventional methodologies for achieving nanoparticles—such as high-pressure homogenization, ultrasonication, and sequential filtration—were not required for nanoparticle formation in the PSI-HAP platform. Instead, controlled self-assembly occurred spontaneously when PSI was blended with HAP or Fe/HAP composites at optimized stoichiometric ratios, facilitating uniform nanoparticle dispersion without additional processing steps. Crucially, the particle sizes of all DC and FeDC formulations were approximately 400 nm, a dimension favorable for efficient tumor cell uptake and membrane penetration, thereby increasing intracellular drug delivery.35 The zeta potentials for these formulations were −19.5 mV (5-FU@DC, Figure 4Ac), −22.6 mV (5-FU@FeDC20, Figure 4Bc), −23.2 mV (5-FU@FeDC30, Figure 4Cc), and −23.6 mV (5-FU@FeDC40, Figure 4Dc), respectively, consistent with the negative charge characteristics of human biological membranes. These values indicated that the formulations possess sufficient colloidal stability, which is crucial for preventing aggregation while ensuring prolonged circulation in the bloodstream. A slight increase in zeta potential magnitude with higher iron content (from 5-FU@FeDC20 to 5-FU@FeDC40) indicated that iron incorporation may enhance electrostatic repulsion between particles, thereby contributing to improved nanosystem stability.36 Moreover, the negative surface charge of these formulations is advantageous for minimizing nonspecific interactions with negatively charged cell membranes and serum proteins, potentially reducing premature clearance by the reticuloendothelial system (RES).37 This property is particularly advantageous for targeted drug delivery, as it increases the probability of nanoparticles reaching their intended site of action, consistent with previous studies on negatively charged nanocarriers.38 The consistent zeta potential values across the formulations indicate that 5-FU loading had minimal impact on the surface characteristics of the nanoparticles. This is an important consideration for maintaining the reproducibility and predictability of the drug delivery system.39 Overall, the zeta potential results suggest that these formulations are promising candidates for advanced in vitro and in vivo assessments, highlighting their potential for cancer therapy and broader biomedical applications.40 In addition, the low hemolysis rates observed across all tested concentrations of 5-FU-DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 (Supplementary Figure S4 and Supplementary Table S1) demonstrate the excellent biocompatibility of these formulations.
 |
Figure 4 Characterization of 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 ((A) 5-FU@DC; (B) 5-FU@FeDC20; (C) 5-FU@FeDC30; (D) 5-FU@FeDC40; (a) SEM micrograph; (b) Particle Size Distribution, PSD; (c) Zeta Potential). |
Hysteresis Loop
The saturation magnetization (Ms) values of Fe3O4, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 were 63.98, 0.33, 0.28, and 0.16 emu/g, respectively (Figure 5A), all exhibiting paramagnetic properties. When Fe3O4 was uniformly dispersed in HAP and coated with non-magnetic PSI, the Ms value decreased significantly due to the shielding effect of the HAP and PSI layers. However, in vivo distribution experiments indicated that 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 retain magnetic responsiveness to an external applied magnetic field. The remanent magnetization (Mr) values were 0.51, 0.005, 0.003, and 0.001 emu/g, respectively, and the Hc values were 8.52, 5.50, 3.32, and 3.74 Oe (Figure 5B). Compared to Fe3O4, the Mr and Hc values of 5-FU@FeDC samples were significantly reduced, suggesting superparamagnetic properties likely due to the stabilization by HAP and PSI, which prevents Fe3O4 aggregation caused by magnetic attraction.
 |
Figure 5 Hysteresis curve (Ms, (A)) and coercive forces (Hc, (B)) of the final formulations. |
The superparamagnetic nature of Fe3O4 nanoparticles allows them to be controlled by external magnetic fields, providing opportunities for labeling, separation, and targeted drug delivery to tumor tissues.41 Under an external magnetic field, magnetic drug delivery systems can transport drug-loaded particles directly to the lesion site, showing promise in applications like hyperthermia.42,43 Moreover, due to their ability to significantly reduce transverse relaxation time and their biocompatibility, Fe3O4 nanoparticles can also be applied in MRI imaging.44
In vitro Release Experiment
As shown in Fig. 6, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40, all demonstrate the ability to release 5-FU in environments with pH levels of 5.4, 6.8, and 7.4, following a similar overall release pattern. The observed results confirmed that the incorporation of Fe3O4 nanoparticles had no statistically significant effect on drug release kinetics, consistent with the system’s design, where Fe3O4 served as a magnetically responsive component without interfering with the pH-triggered release mechanism. The formulation shows lower drug release at pH 5.4, characterized by a slower release rate. This sustained and gradual release could enhance drug accumulation in liver cancer cells, potentially improving the therapeutic efficacy of the drug. Consistent with these findings, our prior research45 confirmed that the PSI-HAP delivery system achieves minimal drug leakage under harsh acidic conditions (pH 2.0), ensuring gastric stability during oral administration. Magnetically guided targeting facilitated rapid accumulation at diseased tissues, where pH-sensitive drug release ensured site-specific therapeutic action. The described approach minimized systemic exposure and off-target distribution, significantly reducing drug-associated adverse effects. Furthermore, the developed platform is well-suited for encapsulating labile biomacromolecules including nucleic acids and proteins, protecting them from enzymatic degradation in vivo. Its versatility covers both oral and intestinal administration, extending its potential biomedical applications.
 |
Figure 6 In vitro release curve. |
In vivo Biodistribution
In the control group, fluorescence in the body was mostly metabolized within 6 h, with an evident presence in the liver region at 3 h, although the fluorescence intensity was low (Figure 7). However, all drug administration groups demonstrated that fluorescence largely disappeared within 24 h, indicating a significantly prolonged distribution time and increased fluorescence intensity. In the 5-FU@DC administration group, a small amount of fluorescence was observed in the liver area at 3 h, peaking at 6 h before gradually diminishing at 9 h. By 12 h, only weak fluorescence remained, and it completely disappeared by 24 h. Compared with the 5-FU@DC administration group, the 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 administration groups all showed fluorescence in the liver area at 1 hour, which increased over time, peaked at 3 h, and persisted until 6 h. Fluorescence then began to decrease at 9 h, with a small amount still visible at 12 h, nearly disappearing by 24 h. Throughout all time points, the fluorescence intensity in the liver was greater than that observed in the 5-FU@DC administration group (Figure 7B). In the heart (Figure 7A), spleen (Figure 7C), lungs (Figure 7D), and kidneys (Figure 7E), the three formulations of 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 displayed weak fluorescence at 6 h, which increased at 9 h but remained lower than that of the 5-FU@DC group. By 12 h, there was almost no fluorescence detected, indicating a low distribution of the preparations in these organs.
 |
Figure 7 In vivo biodistribution ((A) Heart; (B) Liver; (C) Spleen; (D) Lung; (E) Kidney). |
Overall, the biodistribution results demonstrated that, compared to the control group, the distribution time of the 5-FU-loaded preparations in various organs was significantly prolonged. While the formulations modified with different proportions of Fe3O4 exhibited improved liver tissue targeting compared to the 5-FU@DC administration group, the strength of this targeting was relatively weak as the amount of Fe3O4 increased, and distribution in the heart, spleen, lungs, and kidneys was reduced.
In vitro Pharmacodynamics of 5-FU@DC and 5-FU@FeDC
CCK-8 Assay Based Evaluation of Cell Proliferation Ability
Figure 8A shows that within the concentration range of 5-FU from 5–100μg/mL, the activity of Huh-7 cells exhibits significant concentration-dependent inhibition by different drugs. 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 all significantly inhibit the Huh-7 cells at all concentrations tested. The half-inhibitory concentration values (IC50) of the formulations 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 after treating Huh-7 cells for 24 h were 63.10, 42.36, 26.85, 25.94, and 25.82 μg/mL, respectively (Figure 8B). Compared with 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 showed significant differences (P<0.0001), while there were no significant differences among 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 (P>0.5). In Huh-7 cells, the 5-FU@FeDC formulations gradually release Fe3O4 alongside 5-FU, resulting in an increased concentration of ferrous ions within the cells. This elevates the intratumoral supply of reactive ferrous ions, which enhances the production of hydroxyl radicals through the Fenton reaction. This process leads to a depletion of glutathione (GSH) due to the overexpression of matrix metalloproteinases in the tumor microenvironment, ultimately promoting ferroptosis in tumor cells.46,47 Moreover, the Fe3O4 in the 5-FU@FeDC formulations can produce a magnetic thermal effect under the influence of the magnetic field, which may also increase the inhibitory effect on tumor cells.48 Therefore, the IC50 values of 5-FU@FeDC are lower, and good anti-tumor effects can be achieved at lower doses.
 |
Figure 8 (A) Proliferation inhibition of drug in Huh-7 cells; (B) The half-inhibitory concentration values (IC50) of the formulations; (C) Cells morphology of drug-treated Huh-7 cells; (D) Blank vector toxicity; (E) Clone formation of Huh-7 cells; (F) Relative clone numbers of Huh-7 cell (**p<0.01, ***p<0.001, ****p<0.0001). |
Morphological Observation
As illustrated in Figure 8C, the control group exhibited vigorous cell growth, characterized by plump cell bodies and well-defined contours, with a clear attachment to the walls. In comparison, cells treated with 5-FU displayed subtle morphological changes, including some cells losing their distinct contours and a slight decrease in cell number, although the overall cellular structure remained relatively intact. However, cells treated with 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 demonstrated significant cell shrinkage, loss of their original morphology, and a substantial reduction in cell number.
Cytotoxicity Study
The cell viability of Huh-7 cells treated with gradient concentrations of blank-DC, blank-FeDC20, blank-FeDC30, and blank-FeDC40 remained above 90% (Figure 8D), indicating that the blank carriers exert negligible effects on cell survival. Therefore, the excipients utilized in the preparation of these nanomaterials demonstrated low toxicity to Huh-7 cells and can be considered biocompatible for Huh-7 hepatocellular carcinoma cells.
Clone Formation Assay
A plate clone assay was conducted to evaluate the effects of different drugs on cell clone formation. The results of the assay (Figure 8E) indicated that in Huh-7 cells, the percentages of clone formation for the 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups were 75.35±1.23%, 41.24±0.97%, 15.05±1.10%, 14.73±0.97%, and 15.19±1.17% of the control group, respectively. In comparison to the treatment with free 5-FU, the 5-FU@DC treatment led to a reduction in the number of cell clones. Moreover, treatment with 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 resulted in an even greater decrease in clone formation, indicating a more pronounced inhibition of Huh-7 cell proliferation. However, no significant differences were observed among the 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups (P > 0.1, Figure 8F).
Wound Healing Assay
The experiment indirectly assesses cell migration ability by comparing the ratios of the scratched areas after treatment with different drugs. The results of the scratch assay (Figure 9A) show that at various time points, the widths of the scratches for each group increased in the following order: blank control group, 5-FU, 5-FU@DC, 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40. The statistical results presented in Figure 9B indicate that the migration rate of the Huh-7 cell control group at 24 h was 59.25±1.20%, suggesting that the wound in this group gradually healed over that time. At both 12 and 24 h, the migration rate of the 5-FU@DC group was significantly lower than that of the 5-FU group (p < 0.001). This demonstrates that the nano-carrier effectively enhanced the poor solubility of 5-FU and, to some extent, inhibited the migration ability of the cells. Furthermore, in Huh-7 cells, the cell migration rates of the 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups at 24 h were all less than 20%, indicating that the cells’ migration ability was greatly restricted. The migration rates of the 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups at both 12 h and 24 h were significantly lower than those of the 5-FU@DC group, while there was no significant difference among the 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups (P >0.1, Figure 9B).
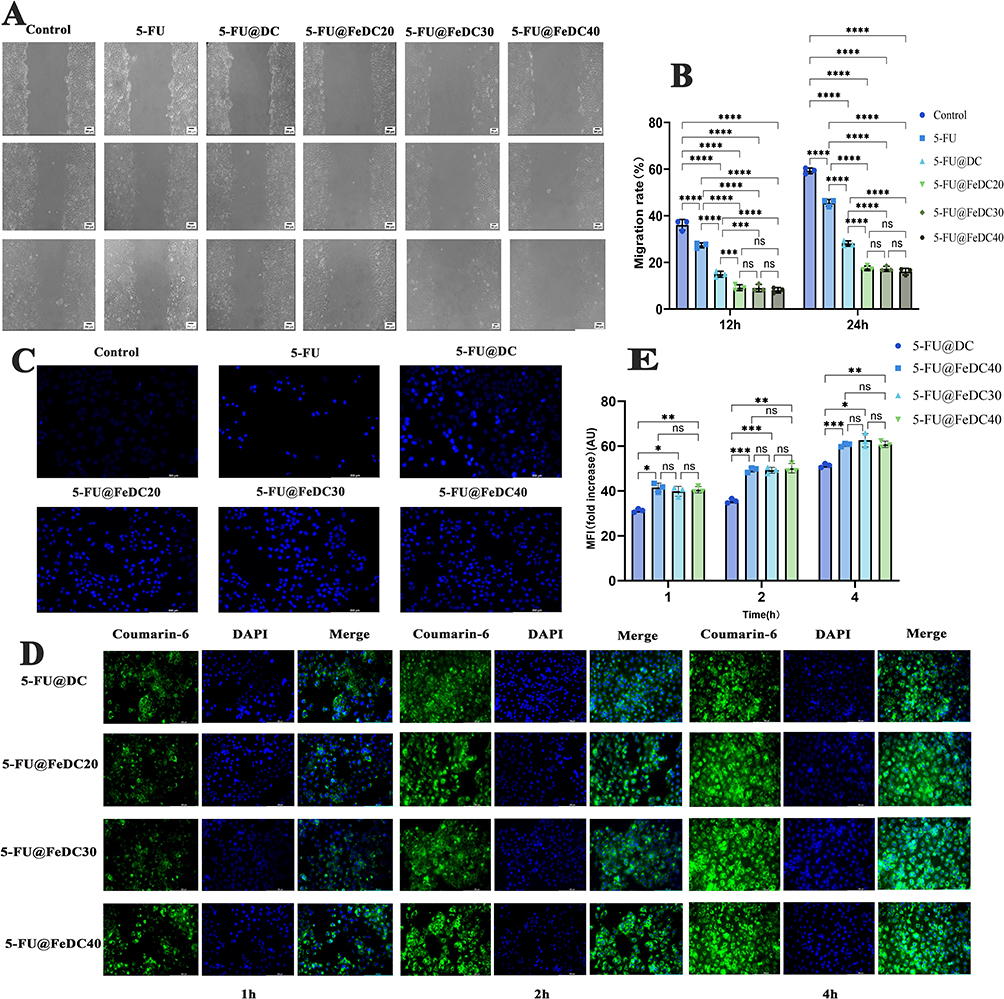 |
Figure 9 (A) Wound healing assay in Huh-7 cells; (B) Migration rate at 12 h and 24 h; (C) Hoechst staining images of Huh-7 cells; (D) Cellular uptake images of Huh-7 cells; (E) Fold increase at 1 h, 2 h and 4 h (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). |
Apoptosis Assay
The apoptosis of Huh-7 cells treated with various nano-drugs was assessed using the Hoechst staining method. As illustrated in Figure 9C, the nuclei of the control group cells appeared intact, with smooth edges and a faint, uniform blue coloration. The 5-FU@DC group showed signs of partial nuclear fragmentation and contraction, exhibiting a brighter blue color. The 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups demonstrated similar and effective pro-apoptotic effects.
Cellular Uptake Assay
To assess the internalization effect of the DC and FeDC in Huh-7 cells, a cell uptake experiment was performed to mimic the formulation’s entry into liver cancer cells at the cancer site. Since 5-FU lacks fluorescent properties, a fluorescent agent, coumarin 6 was incorporated during the formulation preparation, resulting in coumarin 6-labeled nanoparticles that could be tracked visually in subsequent experiments. As illustrated in Figure 9D, the blue represents the cell nuclei stained with DAPI, while the green fluorescence indicates the distribution of nanoparticles within the cells. The green fluorescence observed in each group increased with longer drug incubation times, demonstrating that the uptake of each formulation by the cells is time-dependent, with more nanoparticles entering the cells over time. After 1 hour of incubation, only weak green fluorescence was detected in all groups, indicating the initial entry of nanoparticles. By 2 h, the fluorescence intensity increased compared to 1 hour, and at 4 h, the green fluorescence reached its peak. Compared with the 5-FU@DC group, the 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups showed significantly stronger green fluorescence at 4 h, and the fluorescence was concentrated in the cytoplasm. This indicates that, at this time, the nanoparticles had largely traversed the membrane and entered the interior of the cells.
In Figure 9E, the average fluorescence intensity of the 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups reached approximately 60 AU at the 4-hour mark. In comparison to the 5-FU@FeDC group, the fluorescence intensity of the 5-FU@FeDC formulations at 4 h was significantly higher, with no significant differences among the 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40 groups (P>0.1). This suggests that the 5-FU@FeDC formulations can enter tumor cells in substantial quantities and provide a prolonged sustained-release effect, enhancing its efficacy in inhibiting tumor cell proliferation.
In vitro cellular analysis confirmed that both DC and FeDC nanoparticles demonstrated minimal cytotoxicity while substantially enhancing the therapeutic efficacy of 5-FU against Huh-7 hepatocellular carcinoma cells. These carriers markedly inhibit tumor cell proliferation and migration while promoting apoptosis. FeDC, the magnetically guided variant containing Fe3O4, demonstrated superior antitumor efficacy compared to DC, likely due to the contribution of iron ions in enhancing the anticancer activity of 5-FU. Cellular uptake analysis further confirmed the efficient internalization of both DC and FeDC nanoparticles by Huh-7 cells. Following oral administration, FeDC rapidly localized to the target site under magnetic guidance, ensuring prolonged retention and sustained therapeutic action. FeDC nanoparticles effectively penetrated tumor tissues and were readily internalized by cancer cells. Within the acidic tumor microenvironment, pH-sensitive drug release ensured localized accumulation, maximizing therapeutic impact while minimizing off-target effects.
In summary, this study established novel magnetic drug delivery systems 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40. These systems have particle sizes around 400 nm and feature negative zeta potentials that align with the charge characteristics of human biological membranes. The particles are relatively uniform spheres with good paramagnetic properties and strong drug adsorption capacity. Both 5-FU@DC and 5-FU@FeDC demonstrated effective sustained-release properties. In vivo distribution experiments showed enhanced liver targeting by the 5-FU@FeDC groups, with reduced distribution in other tissues. Among these, the targeting effects of 5-FU@FeDC20 and 5-FU@FeDC30 were similar, while the targeting effect of 5-FU@FeDC40 was comparatively weaker. In vitro, pharmacodynamic results indicated that all three formulations— 5-FU@FeDC20, 5-FU@FeDC30, and 5-FU@FeDC40—showed significant anti-tumor effects on Huh-7 cells, with no significant differences among them. Safety evaluations confirmed that the excipients used in this drug delivery system were safe and non-toxic. A comprehensive assessment of experimental variables identified an optimal Fe3O4-to-HAP mass ratio of approximately 1:30 (w/w) for FeDC synthesis. This was evidenced by the enhanced characteristics of 5-FU@FeDC30, demonstrating reduced particle size and improved uniformity relative to other formulations.
Additionally, according to the IUPAC classification of adsorption-desorption isotherms, the N2 adsorption-desorption isotherms of HAP, Fe/HAPs, DC, and FeDC30 correspond to type IV isotherms (Supplementary Figure S5A). This hysteresis loop suggests that the material has an accumulation of mesopores with an irregular pore structure. The pore size distribution peaks ranged from 2 to 20 nm (Supplementary Figure S5B). Supplementary Table S2 shows that the average pore size of HAP, Fe/HAP20, Fe/HAP30, Fe/HAP40, DC, FeDC30 ranged from 7 to 13 nm, all within the 2–50 nm range, indicating mesopore formation. Compared with HAP and Fe/HAP alone, the specific surface area and pore volume of DC and FeDC30, with an outer PSI coating, were significantly increased. This suggested enhanced adsorption capacity and indicated that the PSI coating disrupted the aggregation of HAP and Fe/HAP, resulting in nanoparticles with smaller particle sizes. The increase in specific surface area was particularly notable for FeDC30. The enhanced specific surface area and pore volume of DC and FeDC30 are particularly advantageous for applications requiring high adsorption capacity, such as drug delivery or catalytic processes. The PSI coating not only stabilizes the nanoparticles but also contributes to their dispersibility, which is critical for achieving uniform performance in practical applications. Furthermore, the mesoporous structure of these materials facilitates efficient mass transfer and accessibility of active sites, which is essential for optimizing their functional performance.
The 5-FU@FeDC discussed in this article represents a novel magnetic sustained-release drug delivery system. This system was easy to prepare and features magnetic Fe/HAP particles as the drug-loading core. PSI was selected as the coating material to diminish stability challenges and prevent uncontrolled drug release caused by Fe/HAP core aggregation. HAP, the primary mineral constituent of human bones and teeth, is distinguished by its endogenous origin and inherent biodegradability.26,27 This biogenic material exhibits exceptional biocompatibility and osteophilic characteristics, aligning with physiological mineralization processes while maintaining low immunogenicity. Its degradation is finely regulated by physiological pH and enzymatic activity, generating calcium-phosphate byproducts that integrate smoothly into native metabolic pathways. The Fe/HAP core retains the characteristic properties of HAP, has strong drug adsorption capabilities, and offers high cell transfection efficiency, making it suitable as a carrier for various anti-tumor drugs, including gene therapies. PSI, a membrane-permeable polymer characterized by low polarity, demonstrates exceptional tumor cell penetration capabilities. Upon internalization, the hydrolysis of imide bonds under acidic intracellular conditions disrupted pH balance, interfering with enzymatic pathways critical for tumor cell survival and apoptosis regulation. The described targeted disruption amplified antitumor efficacy while minimizing off-target effects. The resultant disruption of cellular biochemical equilibrium effectively suppressed tumor proliferation and metastatic progression.31 When mixed with PSI and Fe/HAP in solution, these two components form a non-aggregated, superparamagnetic nanoscale sustained-release drug carrier called PSI-Fe/HAP (FeDC). Guided by an external magnetic field, the PSI-Fe/HAP (FeDC) drug delivery system can swiftly transport drugs to targeted tissues while ensuring long-term retention and sustained release. At the same time, this approach minimizes the potential toxic side effects associated with systemic drug circulation. Moreover, oral administration reduces formulation costs, simplifies process conversion, and avoids the risks associated with injection. The excipients used in this drug delivery system were safe, non-toxic, and cost-effective, and the preparation process was straightforward, facilitating clinical production and scalability.
Further, the PSI-HAP delivery system, constructed using PSI/X-PSI and HAP/Fe-HAP as biocompatible excipients, demonstrates significant translational advantages due to its efficient fabrication process and cost-effective material selection. PSI synthesis relied solely on aspartic acid as the precursor, while HAP was derived from readily available calcium and phosphate salts, ensuring a streamlined fabrication process with minimal impurity incorporation. A key advantage of PSI includes its inherent structural versatility, enabling spontaneous covalent conjugation with amine-containing molecules under catalyst-free conditions, thereby facilitating the incorporation of active targeting functionalities. Crucially, this chemical modification preserves the inherent HAP-binding capacity, a critical feature preliminarily validated by our research group.49 These collective advantages established the PSI-HAP platform as a strong contender for clinical translation. Currently, lipid-based nanoparticles employing passive targeting mechanisms remain the predominant nanocarrier platform in clinical applications. Although advanced delivery systems with active targeting and stimulus-responsive properties enhanced targeting precision, their clinical implementation has been significantly hindered by a range of complex challenges. Such challenges intricate formulation processes, scalability limitations in industrial manufacturing, and potential immunogenicity concerns. However, the PSI-HAP system overcomes key challenges in drug delivery by integrating a straightforward synthesis process with controlled structural modifications while preserving biofunctionality. Unlike conventional nanoparticle platforms facing scalability and immunogenicity concerns, PSI-HAP provides a clinically viable approach with reliable reproducibility. Moreover, its adaptable design facilitates targeted delivery of various anticancer agents, highlighting its potential for application in combination therapies and precision medicine.
Conclusion
This study successfully developed 5-FU@FeDC, a nanomagnetic drug delivery system designed for oral administration. The system integrated pH-responsive release with magnetically guided targeting to facilitate precise 5-FU accumulation in hepatocellular carcinoma tissues, thereby enhancing therapeutic efficacy while reducing systemic exposure. The system offered a cost-effective and scalable manufacturing process, with standardized protocols that support clinical translation. However, thorough pharmacodynamic evaluations and GLP-compliant toxicological assessments are essential to ensure its safety and efficacy for clinical applications.
Acknowledgment
This work was supported by the Program for Young Talents of Basic Research in Universities of Heilongjiang Province [grant number YQJH2024256]. Graphical Abstract is created by Figdraw.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Prim. 2021;7:7.
2. Vogel A, Saborowski A. Current strategies for the treatment of intermediate and advanced hepatocellular carcinoma. Cancer Treat Rev. 2020;82:101946. doi:10.1016/j.ctrv.2019.101946
3. Zhou H, Song T. Conversion therapy and maintenance therapy for primary hepatocellular carcinoma. Biosci Trends. 2021;15(3):155–160. doi:10.5582/bst.2021.01091
4. Grem JL. 5-Fluorouracil: forty-plus and still ticking. A review of its preclinical and clinical development. Invest New Drugs. 2000;18(4):299–313. doi:10.1023/A:1006416410198
5. Heidelberger C, Chaudhuri NK, Danneberg P, et al. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957;179(4561):663–666. doi:10.1038/179663a0
6. Gupta PK. Drug targeting in cancer chemotherapy: a clinical perspective. J Pharmaceut Sci. 1990;79(11):949–962. doi:10.1002/jps.2600791102
7. Zhang P, Ye G, Xie G, et al. Research progress of nanomaterial drug delivery in tumor targeted therapy. Front Bioeng Biotechnol. 2023;11:1240529. doi:10.3389/fbioe.2023.1240529
8. Veiseh O, Gunn JW, Zhang M. Design and fabrication of magnetic nanoparticles for targeted drug delivery and imaging. Adv Drug Delivery Rev. 2010;62(3):284–304. doi:10.1016/j.addr.2009.11.002
9. Xu C, Sun S. New forms of superparamagnetic nanoparticles for biomedical applications. Adv Drug Delivery Rev. 2013;65(5):732–743. doi:10.1016/j.addr.2012.10.008
10. Wang Y, Liu X, Ma S, et al. Progress in cancer therapy with functionalized Fe3O4 nanomaterials. Front Mater Sci. 2023;17(3):230658. doi:10.1007/s11706-023-0658-4
11. Mitchell MJ, Billingsley MM, Haley RM, et al. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
12. Jain P, Kathuria H, Momin M. Clinical therapies and nano drug delivery systems for urinary bladder cancer. Pharmacol Ther. 2021;226:107871. doi:10.1016/j.pharmthera.2021.107871
13. Salvador-Morales C, Grodzinski P. Nanotechnology tools enabling biological discovery. ACS Nano. 2022;16(4):5062–5084. doi:10.1021/acsnano.1c10635
14. Masood F. Polymeric nanoparticles for targeted drug delivery system for cancer therapy. Mater Sci Eng C. 2016;60:569–578. doi:10.1016/j.msec.2015.11.067
15. Zheng N, Wang Q, Zhang S, et al. Recent advances in nanotechnology mediated mitochondria-targeted imaging. J Mat Chem B. 2022;10(37):7450–7459. doi:10.1039/D2TB00935H
16. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update. Bioeng Transl Med. 2019;4(3):e10143. doi:10.1002/btm2.10143
17. Cabral H, Kinoh H, Kataoka K. Tumor-targeted nanomedicine for immunotherapy. Acc Chem Res. 2020;53(12):2765–2776. doi:10.1021/acs.accounts.0c00518
18. Cui G, Su W, Tan M. Formation and biological effects of protein Corona for food‐related nanoparticles. Compr Rev Food Sci Food Saf. 2022;21(2):2002–2031. doi:10.1111/1541-4337.12838
19. Paris A, Tardif N, Galibert MD, et al. AhR and cancer: from gene profiling to targeted therapy. Int J mol Sci. 2021;22(2):752. doi:10.3390/ijms22020752
20. Rozo AJ, Cox MH, Devitt A, et al. Biophysical analysis of lipidic nanoparticles. Methods. 2020;180:45–55. doi:10.1016/j.ymeth.2020.05.001
21. Rosenblum D, Joshi N, Tao W, et al. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi:10.1038/s41467-018-03705-y
22. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583–592. doi:10.1038/nrc1893
23. Wilczewska AZ, Niemirowicz K, Markiewicz KH, et al. Nanoparticles as drug delivery systems. Pharmacol Rep. 2012;64(5):1020–1037. doi:10.1016/S1734-1140(12)70901-5
24. Kumar R, Mondal K, Panda PK, et al. Core–shell nanostructures: perspectives towards drug delivery applications. J Mat Chem B. 2020;8(39):8992–9027. doi:10.1039/D0TB01559H
25. Li J, Li L, Lv Y, et al. The construction of the novel magnetic prodrug Fe3O4@ DOX and its antagonistic effects on hepatocarcinoma with low toxicity. RSC Adv. 2020;10(48):28965–28974. doi:10.1039/D0RA01729A
26. Shao R, Quan R, Zhang L, et al. Porous hydroxyapatite bioceramics in bone tissue engineering: current uses and perspectives. J Ceram Soc Jpn. 2015;123(1433):17–20. doi:10.2109/jcersj2.123.17
27. Köse S, Kankilic B, Gizer M, et al. Stem cell and advanced nano bioceramic interactions. Novel Biomater Regenerat Med. 2018;317–342.
28. Velazco-De-La-Garza J, Avérous L, de Jesús Sosa-Santillán G, et al. Biological properties of novel polysuccinimide derivatives synthesized via quaternary ammonium grafting. Eur Polym J. 2020;131:109705. doi:10.1016/j.eurpolymj.2020.109705
29. Jalalvandi E, Shavandi A. Polysuccinimide and its derivatives: degradable and water soluble polymers. Eur Polym J. 2018;109:43–54. doi:10.1016/j.eurpolymj.2018.08.056
30. Luo Y, Ling Y, Guo W, et al. Docetaxel loaded oleic acid-coated hydroxyapatite nanoparticles enhance the docetaxel-induced apoptosis through activation of caspase-2 in androgen independent prostate cancer cells. J Control Release. 2010;147(2):278–288. doi:10.1016/j.jconrel.2010.07.108
31. Ostrovskaya LA, Korman DB, Varfolomeev SD. Polysuccinimide: experimental antitumor activity. Biophysics. 2015;60(2):298–302. doi:10.1134/S000635091502013X
32. Tank KP, Chudasama KS, Thaker VS, et al. Cobalt-doped nanohydroxyapatite: synthesis, characterization, antimicrobial and hemolytic studies. J Nanopart Res. 2013;15(5):1–11. doi:10.1007/s11051-013-1644-z
33. Pham H, Ramos K, Sua A, et al. Tuning crystal structures of iron-based metal–organic frameworks for drug delivery applications. ACS Omega. 2020;5(7):3418–3427. doi:10.1021/acsomega.9b03696
34. Guo H, Li X, Qu D, et al. Research progress on Fe-based metal-organic frameworks in antitumor drug delivery. Acta Pharma Sin. 2022;1252–1262.
35. Yu W, Liu R, Zhou Y, et al. Size-tunable strategies for a tumor targeted drug delivery system. ACS Cent Sci. 2020;6(2):100–116. doi:10.1021/acscentsci.9b01139
36. Laurent S, Forge D, Port M, et al. Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem Rev. 2008;108(6):2064–2110. doi:10.1021/cr068445e
37. Alexis F, Pridgen E, Molnar LK, et al. Factors affecting the clearance and biodistribution of polymeric nanoparticles. mol Pharmaceut. 2008;5(4):505–515. doi:10.1021/mp800051m
38. Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318.
39. Peer D, Karp JM, Hong S, et al. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotechnol. 2007;2(12):751–760. doi:10.1038/nnano.2007.387
40. Salvati A, Pitek AS, Monopoli MP, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule Corona adsorbs on the surface. Nature Nanotechnol. 2013;8(2):137–143. doi:10.1038/nnano.2012.237
41. Chomoucka J, Drbohlavova J, Huska D, et al. Magnetic nanoparticles and targeted drug delivering. Pharmacol Res. 2010;62(2):144–149. doi:10.1016/j.phrs.2010.01.014
42. Jordan A, Wust P, Fählin H, et al. Inductive heating of ferrimagnetic particles and magnetic fluids: physical evaluation of their potential for hyperthermia. Int j Hyperthermia. 1993;9(1):51–68. doi:10.3109/02656739309061478
43. Guardia P, Di Corato R, Lartigue L, et al. Water-soluble iron oxide nanocubes with high values of specific absorption rate for cancer cell hyperthermia treatment. ACS Nano. 2012;6(4):3080–3091. doi:10.1021/nn2048137
44. Bian R, Wang T, Zhang L, et al. A combination of tri-modal cancer imaging and in vivo drug delivery by metal–organic framework based composite nanoparticles. Biomater Sci. 2015;3(9):1270–1278. doi:10.1039/C5BM00186B
45. Yu F, Wang Q, Liu D, et al. Studies of a novel nano sustained-released drug delivery system with a hydroxyapatite core and polysuccinimide coating structure[J]. Mater Adv. 2024;5(18):7419–7431. doi:10.1039/D4MA00381K
46. Xie S, Sun W, Zhang C, et al. Metabolic control by heat stress determining cell fate to ferroptosis for effective cancer therapy. ACS Nano. 2021;15(4):7179–7194. doi:10.1021/acsnano.1c00380
47. Qin W, Huang J, Yang C, et al. Protease‐Activatable nanozyme with photoacoustic and tumor‐enhanced magnetic resonance imaging for photothermal ferroptosis cancer therapy. Adv Funct Mater. 2023;33(4):2209748. doi:10.1002/adfm.202209748
48. Belyanina I, Kolovskaya O, Zamay S, et al. Targeted magnetic nanotheranostics of cancer. Molecules. 2017;22(6):975. doi:10.3390/molecules22060975
49. Yu F, Wang H, Wang Q, et al. Studies of a novel bone-targeted nano drug delivery system with a HAP core-PSI coating structure for tanshinol injection. J Drug Targeting. 2023;31(7):762–775. doi:10.1080/1061186X.2023.2230528
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.