")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Design and Evaluation of Andrographolide Analogues as SARS-CoV-2 Main Protease Inhibitors: Molecular Modeling and in vitro Studies
Authors Suriya U, Intamalee P , Saeeng R, Wilasluck P, Deetanya P, Wangkanont K, Kanjanasirirat P , Wongwitayasombat C, Nutho B
Received 24 December 2024
Accepted for publication 5 May 2025
Published 15 May 2025 Volume 2025:19 Pages 3907—3924
DOI https://doi.org/10.2147/DDDT.S514193
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Utid Suriya,1 Pansachon Intamalee,2,3 Rungnapha Saeeng,2,3 Patcharin Wilasluck,4,5 Peerapon Deetanya,4,5 Kittikhun Wangkanont,4,5 Phongthon Kanjanasirirat,6 Chanikarn Wongwitayasombat,7 Bodee Nutho8
1Department of Biochemistry, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand; 2Department of Chemistry and Center for Innovation in Chemistry, Faculty of Science, Burapha University, Chonburi, 20131, Thailand; 3The Research Unit in Synthetic Compounds and Synthetic Analogues from Natural Product for Drug Discovery (RSND), Burapha University, Chonburi, 20131, Thailand; 4Center of Excellence for Molecular Biology and Genomics of Shrimp, Department of Biochemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand; 5Center of Excellence in Molecular Crop, Department of Biochemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand; 6Department of Pathobiology, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand; 7Biomedical Science Program, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand; 8Department of Pharmacology, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand
Correspondence: Bodee Nutho, Email [email protected]
Background: The COVID-19 pandemic, caused by SARS-CoV-2, highlights the urgent need for novel antiviral agents targeting key viral proteins. The main protease (Mpro) is a crucial enzyme for viral replication, making it an attractive drug target. Andrographolide, a natural compound with known antiviral properties, serves as a promising scaffold for inhibitor development.
Objective: This study aimed to design, synthesize, and evaluate C-12 dithiocarbamate andrographolide analogues as potential SARS-CoV-2 Mpro inhibitors using computational and experimental approaches.
Methods: A structure-based drug design approach was employed to design andrographolide derivatives. Molecular dynamics simulations were conducted to assess binding interactions and stability. The hit compound was synthesized and evaluated using an enzyme inhibition assay against SARS-CoV-2 Mpro. Cytotoxicity was assessed in HepG2, HaCaT, and HEK293T cells to determine safety profiles.
Results: Among the designed compounds, compound 1, incorporating a 2,4,5-trifluorobenzene moiety, exhibited the strongest binding affinity and stable interactions with key Mpro residues (H41, M49 and M165). Enzyme inhibition assay confirmed ~70% inhibition at 100 μM, with moderate to low cytotoxicity (CC50 values comparable to andrographolide).
Conclusion: Compound 1 represents a promising non-covalent SARS-CoV-2 Mpro inhibitor. Further structural optimization is necessary to enhance potency, selectivity, and safety for therapeutic applications.
Keywords: COVID-19, SARS-CoV-2 main protease, andrographolide analogues, MD simulations, enzyme-based assay
Introduction
The coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has had a profound global impact, resulting in over seven million deaths worldwide (https://covid19.who.int/, accessed on March 11, 2025). While immunity can be achieved through natural infection or vaccination—both of which help reduce mortality risks—the emergence of new variants presents significant challenges. These variants often exhibit increased transmissibility, infectivity, and the potential to evade existing immunity.1,2 Consequently, there is an urgent need to discover and develop effective antiviral agents to slow disease progression and complement ongoing vaccine development efforts.
SARS-CoV-2 is a positive-sense, single-stranded RNA virus belonging to the Coronaviridae family and the Betacoronavirus genus. Its genome encodes nine accessory proteins and four structural proteins: the spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins. In addition, the RNA genome of SARS-CoV-2 includes sixteen non-structural proteins (NSP1 to NSP16) expressed as part of the replicase polyproteins 1a and 1ab (pp1a and pp1ab).3 Key non-structural proteins—such as the main protease (Mpro, also known as 3-chymotrypsin-like protease or 3CLpro), papain-like protease (PLpro), helicase (NSP13), and RNA-dependent RNA polymerase (RdRp)—are potential targets for COVID-19 therapies.4 Specifically, Mpro plays a crucial role in viral replication by cleaving pp1a and pp1ab at multiple sites to produce essential mature NSP required for forming the RNA replicase-transcriptase complex.5 Furthermore, the absence of closely related homologs in humans enhances Mpro’s attractiveness as a target for COVID-19 treatment, minimizing the risk of off-target effects.6
Structurally, SARS-CoV-2 Mpro is composed of three distinct domains: domain I (residues 8–101), domain II (residues 102–184), and domain III (residues 201–303), with domains II and III linked by a long loop.6 These domains form a homodimer, a configuration that exhibits significantly higher hydrolytic activity than the monomeric form.7 A key factor in the formation of this dimeric structure is the salt bridge interaction between Arg4 of one monomer and Glu290 of the other.8 Additionally, the catalytic dyad, consisting of His41 and Cys145 located in the respective domains I and II, acts as a nucleophile and a general acid/base, playing a crucial role in the cleavage of the viral polyprotein.7
In response to the COVID-19 pandemic, substantial efforts have been made to develop drugs specifically targeting SARS-CoV-2 Mpro. Among these, nirmatrelvir, a potent covalent inhibitor of Mpro, combined with ritonavir under the brand name Paxlovid, has been approved in several countries for COVID-19 treatment.9 Clinical studies show that nirmatrelvir can reduce hospitalization risk by 89% in unvaccinated COVID-19 patients.10 Another effective Mpro inhibitor, ensitrelvir (S-217622, marketed as Xocova), is a non-covalent, non-peptidomimetic drug that has shown clinical benefits in reducing COVID-19 severity and has been approved for use in Japan.11 In addition, leritrelvir12 and simnotrelvir13 have received conditional approval in China. While the success of nirmatrelvir and ensitrelvir demonstrates the effectiveness of targeting Mpro in combating SARS-CoV-2, concerns remain about the potential emergence of drug resistance due to mutations in the viral genome. Mutations such as S144A, E166V, and A173V in SARS-CoV-2 Mpro variants have been shown to reduce the potency of both nirmatrelvir and ensitrelvir.14 Therefore, developing new antivirals that can effectively inhibit SARS-CoV-2 Mpro is a pressing need.
Natural products offer a promising candidate for drug discovery due to their structural diversity and biological activity.15 The semisynthetic modification of natural products allows for the introduction of various functional groups onto their core structures, enhancing their pharmacological properties.16 Andrographolide, the primary active compound in Andrographis paniculata, is known for its broad-spectrum antiviral activities, including against SARS-CoV-2.17 In our previous in vitro study on semisynthetic derivatives of andrographolide, we observed promising outcomes. Compounds 3k, 3l, 3m, and 3t demonstrated more than 50% inhibitory activity against SARS-CoV-2 Mpro at a concentration of 10 μM.18 Furthermore, molecular dynamics (MD) simulations combined with binding free energy calculations showed that the (S)-enantiomer of compound 3l exhibited the strongest binding affinity to Mpro. This suggests potential for further structural optimization to enhance binding efficiency and bioactivity. Specifically, the γ-crotonolactone, benzyl, and p-fluorobenzyl groups in (S)-3l were well-positioned within the S1, S2, and S4 pockets of Mpro, respectively, which may contribute to its potent inhibitory activity.
Building on these promising results, we employed a structure-based drug design approach to develop new andrographolide analogues using compound (S)-3l as the template. Drawing inspiration from ensitrelvir—the first oral non-covalent SARS-CoV-2 Mpro inhibitor featuring a 2,4,5-trifluorobenzene pharmacophore—we designed novel analogues by incorporating a 2,4,5-trifluorobenzene group at the P2 position (Figure 1). This moiety is known to occupy the hydrophobic S2 pocket of SARS-CoV-2 Mpro, mimicking the P2 position of ensitrelvir and potentially enhancing binding affinity.19 To further explore the effects on protein–ligand interactions, we also varied the chemical substituents at the P2 position. Therefore, the aim of this study is to design and evaluate C-12 dithiocarbamate andrographolide derivatives as potential inhibitors of SARS-CoV-2 Mpro through a combined in silico and in vitro approach. We hypothesize that the introduction of a dithiocarbamate and a 2,4,5-trifluorobenzene moiety will enhance binding affinity and inhibitory activity against SARS-CoV-2 Mpro by stabilizing key interactions within the active site. The findings provide a basis for developing potent andrographolide-based analogues, contributing to ongoing antiviral drug discovery efforts against COVID-19.
 |
Figure 1 Rational design of novel andrographolide analogues based on ensitrelvir framework. The atomic labels of the core structure of andrographolide are also provided. |
Materials and Methods
Computational Details
Preparation of Protein and Ligand
The previously docked structure of the andrographolide analogue (S)-3l in complex with SARS-CoV-2 Mpro 18 served as the template for our structure-based drug design approach. To create models of the newly designed andrographolide analogues (1–6) bound to protomer A (chain A) of SARS-CoV-2 Mpro, we performed structural modifications on the p-fluorobenzene moiety of the original (S)-3l molecule. These modifications transformed (S)-3l into the corresponding analogues 1–6 (Figure 2), utilizing the small molecules tool available in Discovery Studio Visualizer (BIOVIA, San Diego, CA, USA). For comparison, the structure of ensitrelvir bound to SARS-CoV-2 Mpro was retrieved from the Protein Data Bank (PDB ID: 7VU6). The protonation states of ionizable amino acids (Asp, Glu, Lys, Arg, and His) were considered at pH 7.4 and set based on our previous study.18,20 The chemical structures of 1–6 were constructed using the Gaussview 5.0 program and were structurally optimized at the B3LYP/6-31G(d) level of theory using the Gaussian 16 program.21 Next, the electrostatic potential (ESP) charges of each ligand were calculated through single-point calculations at the HF/6-31G(d) level of theory. The antechamber and parmchk2 modules implemented in AMBER22 were used to generate the restrained ESP (RESP) charges and the missing molecular parameters for the ligands, respectively. The AMBER ff19SB force field 22 and the generalized AMBER force field 2 (GAFF2)23 were adopted to treat the bonded and nonbonded parameters for the protein and ligand, respectively. Missing hydrogen atoms were added to the protein–ligand complex using the LEaP module of AMBER22. Each system was subsequently solvated with the OPC explicit solvation model,24 ensuring a minimum buffer thickness of 10 Å, and was electrostatically neutralized with sodium (Na+) counterions. The newly added hydrogen atoms in the protein–ligand complex and solvent molecules underwent energy minimization through 1000 steps of steepest descent (SD), followed by 2000 steps of conjugate gradient (CG) methods. Next, the protein and ligand were energy-minimized using the same approach, with 1000 steps of SD and 2000 steps of CG, while the solvent molecules were kept fixed. Finally, the entire system underwent complete energy minimization, employing 1000 iterations for the SD method and 2000 iterations for the CG method.
 |
Figure 2 Chemical structures of designed andrographolide analogues 1–6. |
Molecular Dynamics Simulations
To comprehensively assess the stability and dynamic behavior of the protein–ligand complexes in an aqueous environment, MD simulations were conducted using the SANDER and PMEMD modules of the AMBER22 software package.25 The modeled system was executed under periodic boundary conditions with the isothermal–isobaric (NPT) scheme, as detailed in our previous study.18,26 Long-range electrostatic interactions were addressed using the particle-mesh Ewald summation method,27 while a 10-Å cutoff distance was established for nonbonded interactions. The SHAKE algorithm28 was employed to maintain all hydrogen atom bonds at a constant length, allowing for a 2-fs time step of integration. To control the pressure and temperature of each system, the Berendsen barostat,29 with a pressure-relaxation time of 1 ps, and the Langevin thermostat,30 with a damping frequency of 2 ps⁻¹, were applied. The simulated system was gradually thermalized from 10 to 300 K over 200 ps using the canonical ensemble (NVT), with positional restraints of 30.0 kcal/mol·Å² on the Cα atoms of the protein. Following this, the complex underwent NPT equilibration for 1300 ps, which included four steps of restrained MD simulations. During these steps, the restraints on the atoms of the protein active site were progressively decreased from 30 to 20, 10, and finally 5 kcal/mol·Å², followed by an additional 500 ps without any restraints. Subsequently, the entire system was subjected to NPT ensemble MD simulations at 300 K and 1 atm until a total simulation time of 500 ns was reached. MD trajectories were collected every 10 ps for analysis. The post-dynamic trajectories were examined for their structural features using the CPPTRAJ module31 of AMBER22. Per-residue decomposition free energy calculations were performed using the MMPBSA.py module32 to further quantify the contributions of individual residues to ligand binding, allowing for a more detailed understanding of the key interactions driving inhibitory activity. 3D visualizations were generated using the UCSF Chimera33 and ChimeraX34 programs.
Experimental Details
General Information for the Synthesis of Compound 1
All chemicals were purchased from commercial suppliers and used without further purification. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Avance 400 MHz spectrometer using chloroform-d (CDCl3) as the solvent. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane (TMS, δ 0.00) or CDCl3 (δ 7.26) as internal standards. Carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker Avance 100 MHz spectrometer using CDCl3 as the solvent. Chemical shifts (δ) are reported in ppm relative to CDCl3 (δ 77.0) as the internal standard. High-resolution mass spectrometry (HRMS) data were obtained at Naresuan University, Thailand. Analytical thin-layer chromatography (TLC) was performed on precoated silica gel 60F-254 plates (E. Merck, Darmstadt, Germany). Column chromatography used silica gel 60 PF254 (E. Merck, Darmstadt, Germany). Melting points were determined using a Griffin melting point apparatus and are reported uncorrected.
Preparation of 14-Acetyl-3, 19-Isopropylidene Andrographolide as a Precursor for the Synthesis of Compound 1
The synthetic pathway for producing 14-acetyl-3,19-isopropylidene andrographolide, which serves as a precursor for the synthesis of compound 1 is shown in Scheme 1.
 |
Scheme 1 The procedure for the synthesis of 14-acetyl-3,19-isopropylidene andrographolide. |
Step 1: Protection of Andrographolide with 2,2-Dimethoxypropane
Andrographolide (2.0016 g, 5.71 mmol) was dissolved in acetone (60 mL). 2,2-Dimethoxypropane (3.5 mL) and PPTS (0.7177 g) were added, and the reaction mixture was stirred at room temperature for 2 hours. The progress of the reaction was monitored by TLC using 70% ethyl acetate (EtOAc)/hexane as the eluent. Upon completion, the reaction was quenched by adding saturated sodium hydrogen carbonate (NaHCO3) solution until the pH of the aqueous layer was neutral. The mixture was extracted with EtOAc (x3) and the combined organic layers were washed with saturated sodium chloride (NaCl) solution, dried over anhydrous sodium sulfate (Na2SO4), and concentrated under reduced pressure to afford the crude product. The crude product was purified by column chromatography using 50% EtOAc/hexane as the eluent.35 The 3,19-isopropylidene andrographolide was obtained as a white solid in 91% yields (2.03 g) (Rf = 0.78) and was further performed acetylation reaction.
3,19-isopropylideneandrographolide; 1H NMR (400 MHz, CDCl3): δ6.96 (t, J = 7.0 hz, 1H), 5.03 (brt, 1H), 4.91 (s, 1H), 4.62 (s, 1H), 4.45 (dd, J = 10.0, 6.0 hz, 1H), 4.27 (dd, J = 10.0, 1.5 hz, 1H), 3.96 (d, J = 11.5 hz, 1H), 3.49 (dd, J = 9.0, 4.0 hz, 1H), 3.18 (d, J = 11.5 hz, 1H), 2.60–2.54 (m, 2H), 2.42 (dt, J = 12.5, 3.0 hz, 2H), 2.05–1.92 (m, 1H), 1.85–1.78 (m, 1H), 1.78–1.70 (m, 2H), 1.41 (s, 3H), 1.36 (s, 3H), 1.34–1.22 (m, 4H), 1.20 (s, 3H), 0.96 (s, 3H).
Step 2: Acetylation
The 3,19-isopropylidene andrographolide (0.5007 g, 1.28 mmol) was dissolved in EtOAc (3.5 mL) in a round-bottom flask. Triethylamine (0.8047 mL, 5.77 mmol) in EtOAc (4.0 mL) was added, followed by dropwise addition of acetyl chloride (320 μL, 4.49 mmol). The reaction mixture was stirred at room temperature for 30 minutes. The progress of the reaction was monitored by TLC using 50% EtOAc/hexane as the eluent. Upon completion, the reaction was quenched by the addition of saturated NaHCO3 solution until the aqueous layer reached neutral pH. The mixture was extracted with EtOAc (x3) and the combined organic layers were washed with saturated NaCl solution, dried over Na2SO4, and concentrated under reduced pressure to afford the crude product. The crude product was purified by column chromatography using 20% EtOAc/hexane as the eluent. Each collected fraction was analyzed by TLC. The desired fractions were combined and concentrated under reduced pressure, then further dried under vacuum to obtain product as yellow liquid in 89% yields (0.49 g) (Rf=0.92). 14-acetyl-3,19-isopropylidene andrographolide; 1H NMR (400 MHz, CDCl3): δ 7.03 (td, J = 6.9, 1.7 hz, 1H), 5.93 (d, J = 6.1 hz, 1H), 4.89 (s, 1H), 4.55 (dd, J = 11.3, 6.1 hz, 1H), 4.54 (s, 1H), 4.24 (dd, J = 11.2, 2.0 hz, 1H), 3.95 (d, J = 11.6 hz, 1H), 3.50 (dd, J = 8.6, 4.0 hz, 1H), 3.18 (d, J = 11.6 hz, 1H), 2.47–2.37 (m, 3H), 2.12 (s, 3H), 2.05–1.93 (m, 2H), 1.88–1.68 (m, 3H), 1.40 (s, 3H), 1.36 (s, 3H), 1.32–1.22 (m, 4H), 1.19 (s, 3H), 0.94 (s, 3H).
Synthesis and Characterization of Andrographolide Analogue (Compound 1)
The synthetic pathway for the production of compound 1 is outlined in Scheme 2 as follows:
 |
Scheme 2 The procedure for the synthesis of compound 1. |
Step 1: Dithiocarbamate Generation
A solution of 1-(2,4,5-trifluorobenzyl)piperazine (370 mg, 1.61 mmol) in CH3CN (3 mL) was prepared at room temperature. Carbon disulfide (CS2) was then added at 3 °C. The mixture was stirred at room temperature until complete conversion was confirmed by TLC.36
Reaction with Andrographolide Derivative
To the reaction mixture, 14-acetyl-3,19-isopropylidene andrographolide (695 mg, 1.61 mmol) was added and stirred at room temperature. After confirming complete conversion by TLC, the reaction mixture was cooled, and excess silica gel was added, followed by stirring overnight.
Isolation of Product
The reaction mixture was filtered, washed with EtOAc, and the organic solvent was removed under reduced pressure to yield the compound 12-((1-(2,4,5-trifluorobenzyl)piperazine-4-carbonothioyl)thio)-3,19-isopropylidene-14-deoxyandrographolide as a pale brown solid in 80% yield (873 mg, 1.28 mmol). This compound was then used in the second step to synthesize compound 1.
Precursor of Compound 1
12-((1-(2,4,5-trifluorobenzyl)piperazine-4-carbonothioyl)thio)-3,19-isopropylidene-14-deoxyandrographolide: Pale brown solid (873 mg, 80%); 1H NMR (400 MHz, CDCl3): δ 7.42 (t, J = 1.4 hz, 1 h), 7.25–7.19 (m, 1H), 6.95–6.88 (m, 1H), 5.21 (dd, J = 12.3, 4.2 hz, 1H), 4.98 (s, 1H), 4.80 (dd, J = 4.1, 1.5 hz, 2H), 4.75 (s, 1H), 4.30 (brs, 2H), 3.94 (d, J = 11.6 hz, 1H), 3.92 (brs, 2H), 3.54 (s, 2H), 3.44 (dd, J = 9.2, 4.0 hz, 1H), 3.15 (d, J = 11.6 hz, 1H), 2.53 (s, 4H), 2.41 (dt, J = 12.7, 2.4 hz, 1H), 1.98–1.93 (m, 2H), 1.92–1.68 (m, 6H), 1.40 (s, 3H), 1.35 (s, 3H), 1.31–1.21 (m, 3H), 1.18 (s, 3H), 0.89 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 195.83, 172.63, 157.76, 155.32, 148.62, 147.72, 146.65, 134.63, 132.02, 119.49, 108.96, 99.29, 70.41, 64.14, 54.51, 53.98, 52.97, 52.21, 46.40, 39.02, 38.70, 38.40, 38.12, 34.54, 27.79, 25.69, 25.49, 23.79, 16.53; HRMS (ESI) m/z calcd for C35H45F3N2O4S2 [M+H]+ 679.2851, found 679.2854.
Step 2
A solution of 12-((1-(2,4,5-trifluorobenzyl)piperazine-4-carbonothioyl)thio)-3,19-isopropylidene-14-deoxyandrographolide (347 mg, 0.51 mmol) in 2 M HCl (1 mL) was stirred for 15 minutes. After complete conversion was confirmed by TLC, the mixture was filtered, washed with EtOAc, and the organic solvent was removed under reduced pressure. Compound 1 (12-((1-(2,4,5-trifluorobenzyl)piperazine-4-carbonothioyl)thio)-3,19-hydroxy-14-deoxyan drographolide) was obtained as a white solid in 67% yield (219 mg, 0.34 mmol).
Compound 1
(12-((1-(2,4,5-trifluorobenzyl)piperazine-4-carbonothioyl)thio)-3,19-hydroxy-14-deo xyandrographolide: White solid (219 mg, 67%); 1H NMR (400 MHz, CDCl3): δ 7.54–7.39 (m, 2H), 7.06 (s, 1H), 5.10 (d, J = 9.6 hz, 1H), 4.96 (s, 1H), 4.84 (s, 2H), 4.69 (s, 1H), 4.32–4.17 (m, 5H), 3.44–3.29 (m, 4H), 2.56–2.06 (m, 7H), 1.74–1.40 (m, 6H), 1.25 (s, 3H), 1.05–0.82 (m, 3H), 0.64 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 196.96, 172.44, 158.19, 148.70, 148.44, 145.95, 131.27, 122.53, 122.35, 108.41, 106.41, 80.30, 70.27, 64.13, 64.04, 55.27, 54.10, 53.32, 46.87, 39.10, 38.85, 36.59, 26.66, 26.46, 23.93, 22.27, 15.50; HRMS (ESI) m/z calcd for C32H41F3N2O4S2 [M+H]+ 639.2538, found 639.2539.
SARS-CoV-2 Mpro in vitro Assay
The assays for protease activity and inhibition were conducted following the methods outlined in previous studies.20,37,38 In brief, enzyme kinetics were carried out using SARS-CoV-2 Mpro at a concentration of 0.2 µM and the fluorogenic peptide substrate E(EDANS)TSAVLQSGFRK(DABCYL) (Biomatik), with excitation and emission wavelengths set at 340 and 490 nm, respectively. During the initial screening for inhibitory activity, enzymatic activity was assessed both with and without compound concentrations of 100 μM. To determine the half-maximal inhibitory concentration (IC50), the initial rate of substrate cleavage (25 μM) was measured at various concentrations of the selected compounds. The IC50 values were analyzed using GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA, USA). The Cheng-Prusoff equation39 was applied to calculate the inhibitory constant (Ki) using a previously reported Km value of 51 μM.37
Cytotoxicity
HepG2 (ATCC HB-8065), HEK293T (ATCC CRL-3216), and HaCaT (Catalog No. T0020001) cell lines, obtained from the American Type Culture Collection (ATCC, USA) and AddexBio (San Diego, USA), respectively, were seeded in 96-well clear plates (Corning, Acton, MA, USA) at densities of 1×104, 1.5×104, and 1×104 cells per well. The cells were cultured for 24 hours at 37°C with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Detroit, MI, USA) supplemented with 10% fetal bovine serum (FBS, Merck, Schuchardt, Darmstadt, Germany) and 1% penicillin-streptomycin (P/S, Gibco). For HEK293T cells, 2 mM L-glutamine (Hyclone, GE Healthcare, USA) was also added. After incubation, cells were treated with compound 1 at concentrations of 100, 50, 25, 12.5, 6.25, 3.13, and 1.56 μM for 24 hours under the same conditions. Ensitrelvir (Sigma-Aldrich, St. Louis, MO, USA) served as the positive control. Following treatment, the medium was replaced with serum-free medium containing MTT reagent [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Abcam, Cambridge, UK) at a final concentration of 0.5 mg/mL. The cells were incubated for an additional 3 hours at 37°C with 5% CO2. The MTT-containing medium was then discarded, and DMSO (Merck, Schuchardt, Darmstadt, Germany) was added to dissolve the formazan crystals. Absorbance was measured at 570 nm using a microplate reader (Biometrics Technologies, Wilmington, DE, USA). CC50 values were calculated using GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA, USA).
Results and Discussion
This study employed a combined in silico and in vitro approach to design and evaluate C-12 dithiocarbamate andrographolide analogues as potential SARS-CoV-2 Mpro inhibitors. Computational analysis guided the selection of structural modifications that enhance ligand binding affinity and stability within the Mpro active site. Among the tested compounds, compound 1 emerged as the most promising candidate, demonstrating notable inhibitory activity against SARS-CoV-2 Mpro while maintaining a favorable cytotoxicity profile. These findings highlight the potential of andrographolide derivatives for antiviral development and underscore the need for further studies to optimize their potency and safety for clinical applications, as discussed in the following sections.
Molecular Dynamics of Designed Compounds Bound to SARS-CoV-2 Mpro
Structural Stability and Compactness of Protein–Ligand Complex
The stability of the protein–ligand complexes was assessed using root-mean-square deviation (RMSD), which is commonly employed to evaluate the dynamic stability of modeled complexes in an aqueous environment.40–44 As shown in Figure 3A, all simulated systems exhibited similar RMSD trends, characterized by increased fluctuations during the initial 100 ns, followed by an equilibration phase between 400 and 500 ns. Note that all modeled complexes displayed slightly lower RMSD values during the simulation time compared to ensitrelvir, suggesting that these complexes could form stable and equilibrate more rapidly in the aqueous environment.
 |
Figure 3 Analysis of structural dynamics. (A) Time evolution of the backbone root-mean-square deviation (RMSD) of the amino acids within 5 Å of the ligand. (B) The calculated radius of gyration (Rg) of the designed compounds and ensitrelvir in complex with SARS-CoV-2 Mpro. |
The compactness of the protein structure in complex with the designed compounds was further assessed by calculating the radius of gyration (Rg) of the protein Cα atoms, as depicted in Figure 3B. The average Rg values ranged from 25.33 to 25.45 Å, indicating minimal variation across all modeled complexes. This consistency indicates that the protein maintained a tightly packed structure throughout the simulations. Additionally, the Rg values are in agreement with previous MD studies on andrographolide analogues18 and nirmatrelvir,45 further supporting the stability of these complexes. Based on these findings, the structural coordinates from the final 50 ns (450–500 ns) of MD simulations were selected for subsequent analyses.
Atomic Contacts and Intermolecular Hydrogen Bonds of Protein–Ligand Complex
To briefly investigate the binding capability of the designed compounds toward SARS-CoV-2 Mpro, the number of atomic contacts (# contacts) between each compound and the active site residues was calculated using a 3.5 Å cutoff. Any atoms within this distance were counted as contacts. As shown in Figure 4, the # contacts for these complexes remained relatively stable throughout the simulation period. During the final 50 ns, the # contacts for compounds 1–6 ranged from 3 to 20, with ensitrelvir exhibiting the highest # contacts at 27. Notably, compound 1 exhibited similar # contacts (20 ± 5 atoms) to ensitrelvir (27 ± 6 atoms), suggesting that compound 1 may bind more favorably to the active site residues of SARS-CoV-2 Mpro compared to the other designed compounds. Additionally, the # contacts of compound 1 are comparable to other reported Mpro inhibitors, such as lapatinib46 and fragmented-lapatinib aminoquinazoline analogues,47 further supporting its potential as a promising inhibitor.
 |
Figure 4 Number of atomic contacts (# contacts) between the ligand and the active site residues plotted over the entire 500-ns simulation period. The average # contacts from the final 50 ns is represented as mean ± SD. |
Next, we performed an analysis of hydrogen bond (H-bond) formation to quantitatively evaluate the intermolecular interactions between the active site amino acids and the designed compounds. The analysis was based on two geometric criteria: (i) an acceptor-donor distance ≤ 3.5 Å and (ii) an acceptor-H-donor angle ≥ 120°. As illustrated in Figure 5, the designed andrographolide analogues demonstrated limited H-bonding with the active site residues of SARS-CoV-2 Mpro, forming only 1 to 3 H-bonds. In comparison, ensitrelvir established 5 to 6 H-bonds. These findings could provide valuable insights into the mechanisms of compound recognition and the energetic contributions to ligand binding, which are discussed further in the following section.
 |
Figure 5 Time evolution of the number of intermolecular hydrogen bonds (H-bonds) between the surrounding amino acids and the ligand. |
Water Accessibility at the SARS-CoV-2 Mpro Active Site
To assess water accessibility at the SARS-CoV-2 Mpro active site, solvent-accessible surface areas (SASAs) were calculated for amino acid residues within 5 Å of each ligand. Only protomer A contained the ligand, while protomer B represented the apo form (Figure 6A). The time evolution of SASA for each system is shown in Figure 6B. The results show that the average SASAs for the apo form of compounds 1–6 ranged from approximately 890 to 1200 Å2, while ensitrelvir showed an average SASA of 1086 ± 101 Å2. Upon ligand binding in the active site, the average SASAs for all simulated models decreased by approximately 110 to 414 Å2. This observation is consistent with previous MD studies on lopinavir/ritonavir,20 peptidomimetic inhibitors (including compounds N3, 11a, 13b, and 14b),48 and halogenated baicalein,49 all of which reported significant reductions in SASA during the binding process. Among the designed compounds, 1, 2, and 5 showed slightly higher SASA values than ensitrelvir but remained lower than the other analogues. These findings indicate that the designed compounds are well-embedded within the SARS-CoV-2 Mpro binding pocket.
 |
Figure 6 (A) Representation of the SARS-CoV-2 Mpro homodimer, showing protomer A (light blue) with the ligand bound and protomer B (light pink) in the unbound state. (B) Calculated solvent-accessible surface area (SASA) of the six modeled complexes during 500-ns MD simulation. Amino acids within a 5-Å radius of the ligand were selected for SASA calculations. The average SASA values (mean ± SD) for protomer A and protomer B, derived from the last 50 ns of individual MD simulations, are depicted in light blue and pink, respectively. |
End-State Binding Free Energy Calculations
To evaluate the binding strength of the designed compounds against SARS-CoV-2 Mpro in a dynamic system, 500 snapshots from the last 50 ns of MD simulations were analyzed to calculate ∆Gbind using the solvated interaction energy (SIE) method. The SIE method is an end-point, physics-based scoring function that directly calibrates solvation terms for binding energy calculations in protein–ligand complexes.50,51 Compared to the commonly used MM/PB(GB)SA method, SIE provides a faster and more efficient estimation of binding free energies by eliminating the need for computationally intensive entropy calculations.
∆Gbind is a widely used thermodynamic parameter for evaluating protein–ligand binding affinity, making it a valuable criterion for identifying promising drug candidates.52,53 As shown in Table 1, the ΔGbind values for the designed compounds ranged from −9.69 to −6.73 kcal/mol, with ensitrelvir exhibiting a ΔGbind of −9.92 kcal/mol. Notably, the predicted ΔGbind for ensitrelvir closely aligned with its experimentally derived value of −10.37 kcal/mol, calculated from its IC50 (0.049 ± 0.001 µM54) using the formula 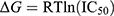 . This agreement could validate the predictive method’s reliability and support the accuracy of the results. In addition, van der Waals interactions (ΔEvdW) and molecular surface-based energies (ΔGcavity) played a key role in ligand binding, significantly contributing to the overall binding affinity. Although coulombic interactions (ΔEcoul) were favorable, their contribution was often offset by unfavorable reaction field energies (ΔGRF), reducing their overall impact on binding energy. For further investigation, compound 1, which exhibited the lowest ΔGbind, along with the highest # contacts (see Figure 4) among the designed compounds, was selected for experimental evaluation of its biological activity against SARS-CoV-2 Mpro, as detailed in the following section. Besides, the calculated energy terms in Table 1 highlight the critical role of specific noncovalent interactions in drug recognition. The designed compounds showed significantly greater contributions from van der Waals interactions compared to electrostatic interactions (8- to 22-fold lower), consistent with previous studies emphasizing the hydrophobic nature of the SARS-CoV-2 Mpro binding pocket.20,48,49 In addition, we can draw conclusions about the impact of P2 substituents on binding affinity against SARS-CoV-2 Mpro. Among the designed compounds (1–6), those containing isobutyl (2) and cyclohexyl (3) moieties exhibited weaker binding affinity to Mpro, suggesting that aliphatic and bulkier aliphatic groups may not favorably interact with the S2 pocket of Mpro. In contrast, the introduction of benzyl and fluorobenzyl moieties enhanced binding affinity, following the trend: 2,4,5-trifluorobenzyl (1) > benzyl (4) > p-fluorobenzyl (6) > m-fluorobenzyl (5). These findings highlight the critical role of benzyl and fluorobenzyl substitutions in enhancing the binding affinity of andrographolide analogues toward SARS-CoV-2 Mpro.
. This agreement could validate the predictive method’s reliability and support the accuracy of the results. In addition, van der Waals interactions (ΔEvdW) and molecular surface-based energies (ΔGcavity) played a key role in ligand binding, significantly contributing to the overall binding affinity. Although coulombic interactions (ΔEcoul) were favorable, their contribution was often offset by unfavorable reaction field energies (ΔGRF), reducing their overall impact on binding energy. For further investigation, compound 1, which exhibited the lowest ΔGbind, along with the highest # contacts (see Figure 4) among the designed compounds, was selected for experimental evaluation of its biological activity against SARS-CoV-2 Mpro, as detailed in the following section. Besides, the calculated energy terms in Table 1 highlight the critical role of specific noncovalent interactions in drug recognition. The designed compounds showed significantly greater contributions from van der Waals interactions compared to electrostatic interactions (8- to 22-fold lower), consistent with previous studies emphasizing the hydrophobic nature of the SARS-CoV-2 Mpro binding pocket.20,48,49 In addition, we can draw conclusions about the impact of P2 substituents on binding affinity against SARS-CoV-2 Mpro. Among the designed compounds (1–6), those containing isobutyl (2) and cyclohexyl (3) moieties exhibited weaker binding affinity to Mpro, suggesting that aliphatic and bulkier aliphatic groups may not favorably interact with the S2 pocket of Mpro. In contrast, the introduction of benzyl and fluorobenzyl moieties enhanced binding affinity, following the trend: 2,4,5-trifluorobenzyl (1) > benzyl (4) > p-fluorobenzyl (6) > m-fluorobenzyl (5). These findings highlight the critical role of benzyl and fluorobenzyl substitutions in enhancing the binding affinity of andrographolide analogues toward SARS-CoV-2 Mpro.
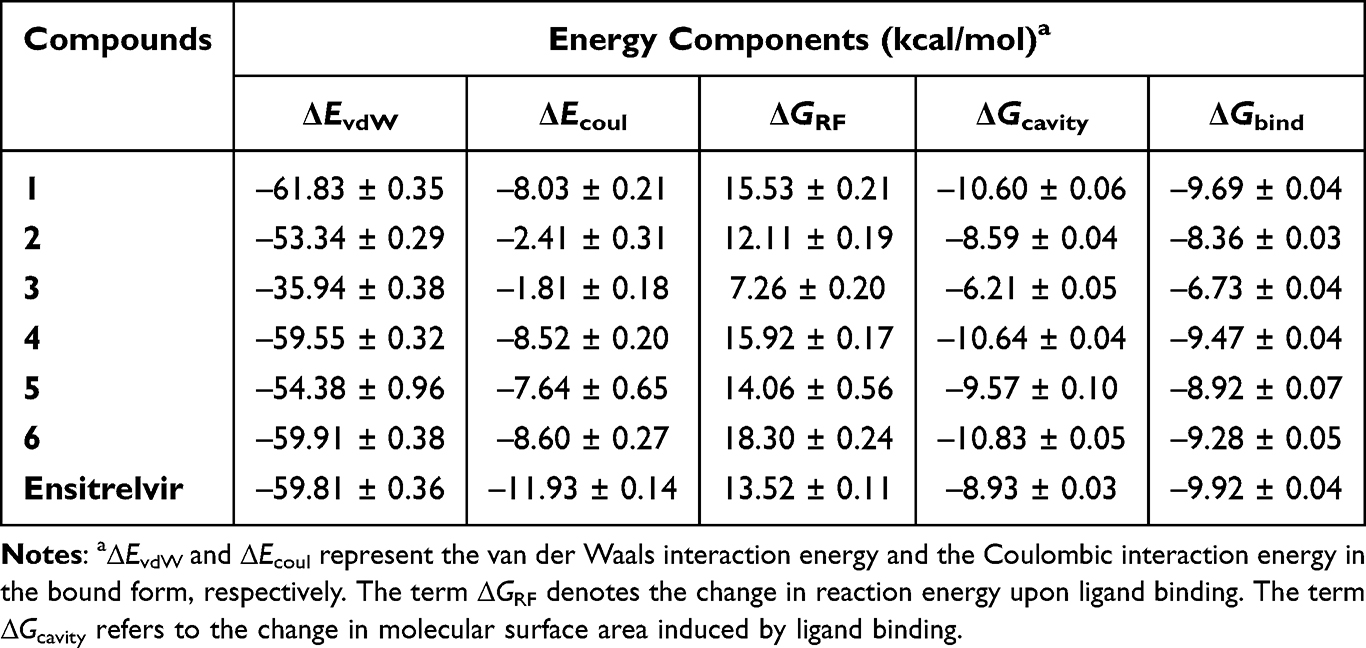 |
Table 1 ΔGbind Values and Their Energetic Components (in kcal/mol) for the Complexes of the Designed Compounds and Ensitrelvir with SARS-CoV-2 Mpro Estimated Using the End-State SIE Method |
It should be noted that a few reports have investigated andrographolide derivatives as potential COVID-19 therapeutics, exploring different mechanisms of action. While our study focuses on direct Mpro inhibition, other research highlights alternative antiviral pathways. For instance, Schulte et al 2022,55 demonstrated that andrographolide derivatives can modulate the KEAP1/NRF2 signaling pathway, enhancing cellular defense mechanisms and thereby reducing viral replication. Meanwhile, Thomas et al 2024,56 employed a cheminformatics-driven approach to identify andrographolide derivatives as dual inhibitors of SARS-CoV-2 methyltransferases (nsp14 and nsp16). These enzymes play a key role in viral RNA capping, a process essential for evading host immune recognition.
In vitro Evaluation of Anti-SARS-CoV-2 Mpro Activity and Predicted Key Binding Residues
The inhibitory activity of compound 1 against SARS-CoV-2 Mpro was evaluated using an Mpro inhibition assay with a fluorogenic substrate. Initially, the relative inhibition at a concentration of 100 μM was measured for both andrographolide and compound 1. Compound 1 demonstrated approximately 70% inhibition of Mpro activity (Figure 7A), which is more potent than the parent andrographolide and previously reported inhibitors, such as caffeic acid ester derivatives (which showed 58.0–68.8% relative inhibition at 100 μM).48 This finding suggests that the designed compound exhibits enhanced binding affinity and inhibitory activity against SARS-CoV-2 Mpro. Subsequently, a dose-response curve was generated to determine the IC50 value of compound 1. As depicted in Figure 7B, the IC50 value of compound 1 was calculated to be 72.0 ± 3.9 μM (Ki ~ 48.3 μM), placing it within a similar micromolar range as known potent non-covalent inhibitors, including X77 and ML188,57 as well as semi-synthetic compounds such as baicalein and brominated baicalein (IC50 values of 63 ± 1 and 56 ± 1 μM, respectively).49 These results highlight the potential of compound 1 as a promising scaffold for further development as a SARS-CoV-2 Mpro inhibitor.


To better understand the mechanism of inhibitory action, the amino acids acting as hot-spot residues were predicted using the MM/GBSA method. Residues with a free energy decomposition 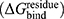 value of ≤ −1.0 kcal/mol were identified as key binding amino acids. As illustrated in Figure 7C, six amino acids—H41, T45, M49, M165, D187, and R188—were considered critical contributors to the binding of compound 1. Among these, M49 and M165 were the most influential, consistent with previously reported key residues involved in the binding of 12-dithiocarbamate-14-deoxyandrographolide analogues,18 brominated baicalein,49 and a newly designed compound MI-23.58 These findings indicate that compound 1 establishes strong intermolecular interactions with the catalytic residue H41, as well as with the substrate-binding residues, which are crucial for Mpro inhibition.59
value of ≤ −1.0 kcal/mol were identified as key binding amino acids. As illustrated in Figure 7C, six amino acids—H41, T45, M49, M165, D187, and R188—were considered critical contributors to the binding of compound 1. Among these, M49 and M165 were the most influential, consistent with previously reported key residues involved in the binding of 12-dithiocarbamate-14-deoxyandrographolide analogues,18 brominated baicalein,49 and a newly designed compound MI-23.58 These findings indicate that compound 1 establishes strong intermolecular interactions with the catalytic residue H41, as well as with the substrate-binding residues, which are crucial for Mpro inhibition.59
Furthermore, the inhibitory mechanism of compound 1 can be attributed to its ability to form several non-covalent interactions within the active site of SARS-CoV-2 Mpro. The 2,4,5-trifluorobenzene moiety in compound 1 enhances hydrophobic interactions, particularly with M49 and M165, which are crucial for ligand stabilization. Furthermore, H-bonding interactions involving H41 further strengthen binding affinity and enzyme inhibition. These key interactions align with those observed in other non-covalent Mpro inhibitors, such as ensitrelvir,19 α-mangostin derivatives,60 and ebsulfur derivatives.61 Given that compound 1 exhibits comparable binding affinity to known inhibitors, it is likely that its mechanism of action involves interference with the substrate-binding region, thereby preventing the proteolytic processing of viral polyproteins.
A structural comparison between compound 1 and ensitrelvir reveals both similarities and differences in their binding modes and inhibitory activities. Ensitrelvir is a non-covalent SARS-CoV-2 Mpro inhibitor that incorporates a 2,4,5-trifluorobenzyl moiety, the same as in compound 1, which enhances hydrophobic interactions within the S2 pocket of Mpro. Both compounds also interact with Met49, a key residue involved in ligand stabilization. However, ensitrelvir exhibits significantly greater potency, with an IC50 of 0.013 μM, compared to compound 1 (IC50 = 72.0 μM). This difference is likely due to the optimized P1 and P1’ substitutions of ensitrelvir, which include a 1-methyl-1H-1,2,4-triazole moiety in the S1 pocket, enhancing H-bonding with H163, and a 6-chloro-2-methyl-2H-indazole moiety in the P1’ position, which contributes to additional hydrophobic interactions.19 While compound 1 exhibits promising inhibition of SARS-CoV-2 Mpro, further structural optimization, such as modifying the P1 moiety to strengthen interactions with H163 and optimizing a P1’ moiety that forms stronger hydrophobic contacts with surrounding residues could help bridge the potency gap between compound 1 and clinically approved inhibitors.
Cytotoxic Effects on Representative Cell Lines
Understanding the cytotoxicity profile of the identified hit compound is important for assessing its therapeutic potential during drug development. In this study, HaCaT cells (human keratinocyte cells), HEK293T cells (human embryonic kidney cells), and HepG2 cells (human liver cancer cells) were used to represent three major organ systems. As shown in Table 2 and Figures S1–S3 in the supplementary data, compound 1 demonstrated moderate to low cytotoxicity, with CC50 values of 13.24 ± 2.25 µM in HaCaT cells, 41.02 ± 2.62 µM in HEK293T cells, and 42.26 ± 1.16 µM in HepG2 cells. While compound 1 exhibited higher toxicity than ensitrelvir, its cytotoxicity levels were comparable to those of its parent compound, andrographolide, across all tested cell lines. These results indicate that compound 1 enhances binding to SARS-CoV-2 Mpro without significantly compromising the viability of liver and kidney cells, which are essential for drug metabolism and excretion. However, the increased sensitivity of HaCaT cells to compound 1 (CC50 = 13.24 ± 2.25 µM) may limit its potential use in dermatological applications. Compared to other Mpro inhibitors, compound 1 exhibits a toxicity profile similar to several non-covalent Mpro inhibitors, including α-mangostin analogues,60 sulfonamide chalcones,62 and flavonoid derivatives.63 While its cytotoxicity profile appears favorable, further selectivity studies against human proteases and long-term toxicity evaluations in preclinical models are essential to confirm its safety for therapeutic development.
 |
Table 2 Cytotoxic Effects of Compound 1 Indicated by CC50 Values (µM) in Three Different Cell Lines: HaCaT Cells (Human Keratinocyte Cells), HEK293T Cells (Human Embryonic Kidney Cells), and HepG2 Cells (Human Liver Cancer Cells) |
However, this study has certain limitations that should be mentioned. While the in vitro enzyme inhibition assay provides valuable insights into the inhibitory activity of compound 1 against SARS-CoV-2 Mpro, it does not fully capture its antiviral efficacy in a cellular environment. Another significant limitation is the lack of cytotoxicity testing in lung cell lines, which are more physiologically relevant for SARS-CoV-2 infection. Although cytotoxicity assessments in HaCaT, HEK293T, and HepG2 cells provide initial safety insights, they may not accurately reflect the effects of the compound in lung tissues, the primary site of SARS-CoV-2 infection. Future studies should incorporate lung-derived epithelial cells to provide a more relevant and comprehensive assessment of cytotoxicity. Moreover, the absence of in vivo validation limits the ability to assess the pharmacokinetic and toxicity profiles, which are critical for therapeutic development. To address these limitations, further investigations, including cell-based antiviral assays and animal model studies, are necessary to confirm its clinical applicability and therapeutic potential.
Conclusions and Future Perspectives
This study highlights the successful design and evaluation of C-12 dithiocarbamate andrographolide analogues as SARS-CoV-2 Mpro inhibitors, supporting the development of novel antiviral therapeutics. Using a structure-based drug design approach, six analogues (1–6) were developed and analyzed through MD simulations, which revealed their stable binding within the Mpro active site. Among them, compound 1, incorporating a 2,4,5-trifluorobenzene moiety, exhibited the most favorable binding energy (ΔGbind = −9.69 kcal/mol) and formed strong interactions with key active site residues (H41, M49, and M165). Experimental validation further supported the computational findings, as compound 1 demonstrated ~70% inhibition of Mpro at 100 μM with an IC50 value of 72 μM. Cytotoxicity assessments indicated moderate to low toxicity, comparable to andrographolide, suggesting a favorable safety profile in vitro. These findings underscore the potential of andrographolide derivatives as promising scaffolds for SARS-CoV-2 antiviral drug development. To enhance its therapeutic potential, future studies should focus on optimizing compound 1 by improving its potency, selectivity, and pharmacokinetic properties through structural modifications and analogue development. In addition, further in vitro and in vivo evaluations are necessary, such as testing against clinically relevant Mpro mutants, conducting antiviral assays in SARS-CoV-2-infected cells, and assessing pharmacokinetic and toxicity profiles in animal models. These investigations will provide critical insights into its therapeutic feasibility and potential for clinical translation.
Beyond its relevance to SARS-CoV-2, the findings of this study have broader implications for antiviral drug discovery. The structure-based optimization of andrographolide derivatives could serve as a basis for developing inhibitors against other coronaviruses, such as SARS-CoV-1 and Middle East Respiratory Syndrome Coronavirus (MERS-CoV), which share structural and functional similarities in their Mpro.64,65 Additionally, the computational drug design strategies utilized in this study may also be applicable to other viruses that depend on cysteine proteases for their life cycle, such as human rhinovirus, enterovirus 71 and picornavirus.66–68 Given the ongoing risk of emerging coronaviruses, the identification of natural product-derived inhibitors as broad-spectrum antiviral agents presents a promising strategy for pandemic preparedness. Future research could extend these findings by evaluating optimized compound 1 and its analogues against a range of viral proteases to assess their potential for broader antiviral applications. This study contributes to the ongoing efforts to develop effective antiviral therapeutics against COVID-19 and emerging coronaviruses, reinforcing the significance of natural product-derived inhibitors in antiviral drug discovery campaigns.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Acknowledgments
We would like to thank the Faculty of Science, Mahidol University, for research facilities and computational resources and the Department of Chemistry, Faculty of Science, Mahidol University, for access to the Gaussian 16 software. We also thank the Excellent Center for Drug Discovery (ECDD), Faculty of Science, Mahidol University, for providing the HaCaT and HEK293T cell lines.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research project has been funded by Mahidol University (Fundamental Fund: fiscal year 2024 by National Science Research and Innovation Fund (NSRF)) (Grant No. FF-059/2567) to B.N., and the Agricultural Research Development Agency (ARDA) grant #PRP6605030320 to R.S. Funding for the protease activity assay is supported by the National Research Council of Thailand (NRCT) (Grant No. N34A670082) to K.W..
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Uriu K, Ito J, Kosugi Y, et al. Transmissibility, infectivity, and immune evasion of the SARS-CoV-2 BA.2.86 variant. Lancet Infect Dis. 2023;23(11):e460–1. doi:10.1016/S1473-3099(23)00575-3
2. Kaku Y, Okumura K, Padilla-Blanco M, et al. Virological characteristics of the SARS-CoV-2 JN.1 variant. Lancet Infect Dis. 2024;24(2):e82. doi:10.1016/S1473-3099(23)00813-7
3. Wong L-YR, Perlman S. Immune dysregulation and immunopathology induced by SARS-CoV-2 and related coronaviruses—are we our own worst enemy? Nat Rev Immunol. 2022;22(1):47–56. doi:10.1038/s41577-021-00656-2
4. Li G, Hilgenfeld R, Whitley R, De Clercq E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat Rev Drug Discov. 2023;22(6):449–475. doi:10.1038/s41573-023-00672-y
5. Zhao Y, Zhu Y, Liu X, et al. Structural basis for replicase polyprotein cleavage and substrate specificity of main protease from SARS-CoV-2. Proc Natl Acad Sci U S A. 2022;119(16):e2200366119.
6. Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi:10.1038/s41586-020-2223-y
7. Hu Q, Xiong Y, Zhu GH, et al. The SARS-CoV-2 main protease (Mpro): structure, function, and emerging therapies for COVID-19. MedComm. 2022;3(3):e145. doi:10.1002/mco2.151
8. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi:10.1126/science.abb3405
9. Westberg M, Su Y, Zou X, et al. An orally bioavailable SARS-CoV-2 main protease inhibitor exhibits improved affinity and reduced sensitivity to mutations. Sci Transl Med. 2024;16(738):eabq7360. doi:10.1126/scitranslmed.adi0979
10. Hammond J, Leister-Tebbe H, Gardner A, et al. Oral Nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N Engl J Med. 2022;386(15):1397–1408. doi:10.1056/NEJMoa2118542
11. Mukae H, Yotsuyanagi H, Ohmagari N, et al. A randomized phase 2/3 study of Ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in Japanese patients with mild-to-moderate COVID-19 or asymptomatic SARS-CoV-2 infection: results of the phase 2a part. Antimicrob Agents Chemother. 2022;66(10):e0069722. doi:10.1128/aac.00697-22
12. Zhan Y, Lin Z, Liang J, et al. Leritrelvir for the treatment of mild or moderate COVID-19 without co-administered ritonavir: a multicentre randomised, double-blind, placebo-controlled Phase 3 trial. eClinicalMedicine. 2024;67:102359. doi:10.1016/j.eclinm.2023.102359
13. Cao B, Wang Y, Lu H, et al. Oral Simnotrelvir for adult patients with mild-to-moderate Covid-19. N Engl J Med. 2024;390(3):230–241. doi:10.1056/NEJMoa2301425
14. Iketani S, Mohri H, Culbertson B, et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature. 2023;613(7944):558–64.
15. Atanasov AG, Zotchev SB, Dirsch VM, Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20(3):200–216. doi:10.1038/s41573-020-00114-z
16. Majhi S, Das D. Chemical derivatization of natural products: semisynthesis and pharmacological aspects—a decade update. Tetrahedron. 2021;78:131801. doi:10.1016/j.tet.2020.131801
17. Sa-Ngiamsuntorn K, Suksatu A, Pewkliang Y, et al. Anti-SARS-CoV-2 activity of andrographis paniculata extract and its major component andrographolide in human lung epithelial cells and cytotoxicity evaluation in major organ cell representatives. J Nat Prod. 2021;84(4):1261–1270. doi:10.1021/acs.jnatprod.0c01324
18. Nutho B, Wilasluck P, Deetanya P, et al. Discovery of C-12 dithiocarbamate andrographolide analogues as inhibitors of SARS-CoV-2 main protease: in vitro and in silico studies. Comput Struct Biotechnol J. 2022;20:2784–2797. doi:10.1016/j.csbj.2022.05.053
19. Unoh Y, Uehara S, Nakahara K, et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J Med Chem. 2022;65(9):6499–6512. doi:10.1021/acs.jmedchem.2c00117
20. Nutho B, Mahalapbutr P, Hengphasatporn K, et al. Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry. 2020;59(18):1769–1779. doi:10.1021/acs.biochem.0c00160
21. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 16 Rev. C.01. Wallingford (CT): Gaussian, Inc.; 2016.
22. Tian C, Kasavajhala K, Belfon KAA, et al. ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J Chem Theory Comput. 2020;16(1):528–552. doi:10.1021/acs.jctc.9b00591
23. Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi:10.1002/jcc.20035
24. Izadi S, Anandakrishnan R, Onufriev AV. Building water models: a different approach. J Phys Chem Lett. 2014;5(21):3863–3871. doi:10.1021/jz501780a
25. Case DA, Cheatham TE, Darden T, et al. The amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. doi:10.1002/jcc.20290
26. Nutho B, Tungmunnithum D. Anti-aging potential of the two major flavonoids occurring in Asian water lily using in vitro and in silico molecular modeling assessments. Antioxidants. 2024;13(5):601. doi:10.3390/antiox13050601
27. Darden T, York D, Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. doi:10.1063/1.464397
28. Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–341. doi:10.1016/0021-9991(77)90098-5
29. Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. doi:10.1063/1.448118
30. Uberuaga BP, Anghel M, Voter AF. Synchronization of trajectories in canonical molecular-dynamics simulations: observation, explanation, and exploitation. J Chem Phys. 2004;120(14):6363–6374. doi:10.1063/1.1667473
31. Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–3095. doi:10.1021/ct400341p
32. Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8(9):3314–3321. doi:10.1021/ct300418h
33. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
34. Meng EC, Goddard TD, Pettersen EF, et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 2023;32(11):e4710. doi:10.1002/pro.4792
35. Sirion U, Kasemsook S, Suksen K, Piyachaturawat P, Suksamrarn A, Saeeng R. New substituted C-19-andrographolide analogues with potent cytotoxic activities. Bioorg Med Chem Lett. 2012;22(1):49–52. doi:10.1016/j.bmcl.2011.11.085
36. Arsakhant P, Sirion U, Chairoungdua A, et al. Design and synthesis of C-12 dithiocarbamate andrographolide analogues as an anticancer agent. Bioorg Med Chem Lett. 2020;30(14):127252. doi:10.1016/j.bmcl.2020.127263
37. Deetanya P, Hengphasatporn K, Wilasluck P, Shigeta Y, Rungrotmongkol T, Wangkanont K. Interaction of 8-anilinonaphthalene-1-sulfonate with SARS-CoV-2 main protease and its application as a fluorescent probe for inhibitor identification. Comput Struct Biotechnol J. 2021;19:3364–3371. doi:10.1016/j.csbj.2021.05.053
38. Wansri R, Lin ACK, Pengon J, et al. Semi-synthesis of N-aryl amide analogs of piperine from piper nigrum and evaluation of their antitrypanosomal, antimalarial, and anti-SARS-CoV-2 main protease activities. Molecules. 2022;27(9):2841. doi:10.3390/molecules27092841
39. Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. doi:10.1016/0006-2952(73)90196-2
40. Suriya U, Srikuea R, Chokpanuwat T, et al. A diarylheptanoid derivative mediates glycogen synthase kinase 3β to promote the porcine muscle satellite cell proliferation: implications for cultured meat production. Biochem Biophys Res Commun. 2024;736:150850. doi:10.1016/j.bbrc.2024.150850
41. Suriya U, Mahalapbutr P, Wimonsong W, Yotphan S, Choowongkomon K, Rungrotmongkol T. Quinoxalinones as a novel inhibitor scaffold for EGFR (L858R/T790M/C797S) tyrosine kinase: molecular docking, biological evaluations, and computational insights. Molecules. 2022;27(24):8901. doi:10.3390/molecules27248901
42. Suriya U, Mahalapbutr P, Geronikaki A, et al. Discovery of furopyridine-based compounds as novel inhibitors of Janus kinase 2: in silico and in vitro studies. Int J Biol Macromol. 2024;260:129308. doi:10.1016/j.ijbiomac.2024.129308
43. Nukulkit S, Nalinratana N, Aree T, et al. Maeruines A−E, elusive indole alkaloids from stems of Maerua siamensis and their inhibitory effects on cyclooxygenases and HT-29 colorectal cancer cell proliferation. Phytochemistry. 2025;229:114291. doi:10.1016/j.phytochem.2024.114291
44. Mahalapbutr P, Sabuakham S, Nasoontorn S, Rungrotmongkol T, Silsirivanit A, Suriya U. Discovery of amphotericin B, an antifungal drug as tyrosinase inhibitor with potent anti-melanogenic activity. Int J Biol Macromol. 2023;246:125587. doi:10.1016/j.ijbiomac.2023.125587
45. Rungruangmaitree R, Phoochaijaroen S, Chimprasit A, Saparpakorn P, Pootanakit K, Tanramluk D. Structural analysis of the coronavirus main protease for the design of pan-variant inhibitors. Sci Rep. 2023;13(1):7055. doi:10.1038/s41598-023-34305-6
46. Sanachai K, Somboon T, Wilasluck P, et al. Identification of repurposing therapeutics toward SARS-CoV-2 main protease by virtual screening. PLoS One. 2022;17(6):e0269563. doi:10.1371/journal.pone.0269563
47. Adediji A, Sroithongmoon A, Suroengrit A, et al. Design, synthesis, and antiviral activity of fragmented-lapatinib aminoquinazoline analogs towards SARS-CoV-2 inhibition. Eur J Med Chem. 2025;286:117303. doi:10.1016/j.ejmech.2025.117303
48. Pojtanadithee P, Isswanich K, Buaban K, et al. A combination of structure-based virtual screening and experimental strategies to identify the potency of caffeic acid ester derivatives as SARS-CoV-2 3CLpro inhibitor from an in-house database. Biophys Chem. 2024;304:107125. doi:10.1016/j.bpc.2023.107125
49. Hengphasatporn K, Wilasluck P, Deetanya P, et al. Halogenated baicalein as a promising antiviral agent toward SARS-CoV-2 main protease. J Chem Inf Model. 2022;62(6):1498–1509. doi:10.1021/acs.jcim.1c01304
50. Sulea T, Cui Q, Purisima EO. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 2. benchmark in the CSAR-2010 scoring exercise. J Chem Inf Model. 2011;51(9):2066–2081. doi:10.1021/ci2000242
51. Sulea T, Purisima EO. The solvated interaction energy method for scoring binding affinities. Methods Mol Biol. 2012;819:295–303.
52. Verma K, Mahalapbutr P, Suriya U, et al. In silico screening of DNA gyrase B potent flavonoids for the treatment of clostridium difficile infection from phytoHub database. Braz Arch Biol Technol. 2021;64:e21200402.
53. Thirunavukkarasu MK, Suriya U, Rungrotmongkol T, Karuppasamy R. In silico screening of available drugs targeting non-small cell lung cancer targets: a drug repurposing approach. Pharmaceutics. 2021;14(1):59. doi:10.3390/pharmaceutics14010059
54. Lin M, Zeng X, Duan Y, et al. Molecular mechanism of ensitrelvir inhibiting SARS-CoV-2 main protease and its variants. Commun Biol. 2023;6(1):694. doi:10.1038/s42003-023-05071-y
55. Schulte B, König M, Escher BI, et al. Andrographolide derivatives target the KEAP1/NRF2 axis and possess potent anti-SARS-CoV-2 activity. ChemMedChem. 2022;17(5):e202100732. doi:10.1002/cmdc.202100732
56. Thomas J, Ghosh A, Ranjan S, Satija J. Cheminformatics approach to identify andrographolide derivatives as dual inhibitors of methyltransferases (nsp14 and nsp16) of SARS-CoV-2. Sci Rep. 2024;14(1):9801. doi:10.1038/s41598-024-58532-7
57. Luttens A, Gullberg H, Abdurakhmanov E, et al. Ultralarge virtual screening identifies SARS-CoV-2 main protease inhibitors with broad-spectrum activity against coronaviruses. J Am Chem Soc. 2022;144(7):2905–2920. doi:10.1021/jacs.1c08402
58. Qiao J, Li Y-S, Zeng R, et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371(6536):1374–1378. doi:10.1126/science.abf1611
59. Song L, Gao S, Ye B, et al. Medicinal chemistry strategies towards the development of non-covalent SARS-CoV-2 Mpro inhibitors. Acta Pharm Sin B. 2024;14(1):87–109. doi:10.1016/j.apsb.2023.08.004
60. Pyae NY, Maiuthed A, Phongsopitanun W, et al. N-containing α-mangostin analogs via smiles rearrangement as promising cytotoxic, antitrypanosomal, and SARS-CoV-2 main protease inhibitory agents. Molecules. 2023;28(3):1104. doi:10.3390/molecules28031104
61. Sinsulpsiri S, Nishii Y, Xu-Xu QF, et al. Unveiling the antiviral inhibitory activity of ebselen and ebsulfur derivatives on SARS-CoV-2 using machine learning-based QSAR, LB-PaCS-MD, and experimental assay. Sci Rep. 2025;15(1):6956. doi:10.1038/s41598-025-91235-1
62. Pojtanadithee P, Hengphasatporn K, Suroengrit A, et al. Identification of promising sulfonamide chalcones as inhibitors of SARS-CoV-2 3CLpro through structure-based virtual screening and experimental approaches. J Chem Inf Model. 2023;63(16):5244–5258. doi:10.1021/acs.jcim.3c00663
63. Khamto N, Utama K, Boontawee P, et al. Inhibitory activity of flavonoid scaffolds on SARS-CoV-2 3CLpro: insights from computational and experimental investigations. J Chem Inf Model. 2024;64(3):874–891. doi:10.1021/acs.jcim.3c01477
64. Li J, Lin C, Zhou X, et al. Structural basis of the main proteases of coronavirus bound to drug candidate PF-07321332. J Virol. 2022;96(8):e02013–21. doi:10.1128/jvi.02013-21
65. Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281(18):4085–4096. doi:10.1111/febs.12936
66. Wen W, Qi Z, Wang J. The function and mechanism of Enterovirus 71 (EV71) 3C protease. Curr Microbiol. 2020;77(9):1968–1975. doi:10.1007/s00284-020-02082-4
67. Wang QM, Shu-Hui C. Human rhinovirus 3C protease as a potential target for the development of antiviral agents. Curr Protein Pept Sci. 2007;8(1):19–27. doi:10.2174/138920307779941523
68. Sun D, Chen S, Cheng A, Wang M. Roles of the picornaviral 3C proteinase in the viral life cycle and host cells. Viruses. 2016;8(3):82. doi:10.3390/v8030082
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.