")
Back to Journals » International Journal of Nanomedicine » Volume 19
Development of Dual Diagnostic-Therapeutic Nanoformulation Effective Against Pancreatic Cancer in Animal Model
Authors Huang Y, Wang Y, Zheng T, Nie S, Wang Y, Shen H, Mo F
Received 10 April 2024
Accepted for publication 23 August 2024
Published 6 September 2024 Volume 2024:19 Pages 9121—9143
DOI https://doi.org/10.2147/IJN.S464788
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Farooq A. Shiekh
Yanan Huang,1,* Yunfeng Wang,2,* Tianyu Zheng,1,* Shuang Nie,1 Yanli Wang,3 Hui Shen,1 Fengfeng Mo1
1Department of Naval Nutrition and Food Hygiene, Faculty of Navy Medicine, Naval Medical University, Shanghai, People’s Republic of China; 2Department of Gastroenterology, Changhai Hospital, Shanghai, People’s Republic of China; 3International Joint Research Center of Human-Machine Intelligent Collaborative for Tumor Precision Diagnosis and Treatment of Hainan Province, Hainan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanli Wang; Fengfeng Mo, Email [email protected]; [email protected]
Purpose: Erythrocytes and fibroblasts in the pancreatic cancer tumor microenvironment promote tumor cell growth and invasion by providing nutrients and promoting immunosuppression. Additionally, they form a barrier against the penetration of chemotherapeutic drugs. Therefore, the search for diversified tumor-targeting materials plays an essential role in solving the above problems.
Methods: Physicochemical characterization of Graphene fluorescent nanoparticles (GFNPs) and nanomedicines were analyzed by transmission electron microscopy (TEM), elemental analyzers and ultraviolet fluorescence (UV/FL) spectrophotometer. Localization of GFNPs in cell and tissue sections imaged with laser confocal microscope, fluorescence scanner and small animal in vivo imager. Qualitative detection and quantitative detection of GFNPs and GFNPs-GEM were performed using High performance liquid chromatography (HPLC).
Results: Based on the 3 nm average dimensions, GFNPs penetrate vascular endothelial cells and smooth muscle cells, achieve up to label 30% tumor cells and 60% cancer-associated fibroblasts (CAFs) cells, and accurately label mature red blood cells in the tumor microenvironment. In orthotopic transplanted pancreatic cancer models, the fluorescence intensity of GFNPs in tumors showed a positive correlation with the cycle size of tumor development. The differential spatial distribution of GFNPs in three typical clinical pancreatic cancer samples demonstrated their diagnostic potential. To mediate the excellent targeting properties of GFNPs, we synthesized a series of nanomedicines using popular chemotherapeutic drugs, in which complex of GFNPs and gemcitabine (GFNPs-GEM) possessed stability in vivo and exhibited effective reduction of tumor volume and fewer side effects.
Conclusion: GFNPs with multiple targeting tumor microenvironments in pancreatic cancer possess diagnostic efficiency and therapeutic potential.
Keywords: graphene fluorescent nanoparticles, cancer-associated fibroblasts, pancreatic cancer cells, dual-targeting, chemo-target therapy
Graphical Abstract:

Introduction
Pancreatic ductal adenocarcinoma (PDAC) has the highest mortality rate of all major cancers, with a 5-year mortality rate higher than 92%, and most patients succumb to their disease within the first year.1,2 It is the third leading cause of cancer deaths in both the US and Canada according to the National Cancer Institute and the Canadian Cancer Society, respectively.3 In China, the incidence and mortality rate of pancreatic cancer is also increasing year by year. At present, surgical resection followed by adjuvant chemotherapy is the most favorable therapy for early-stage pancreatic cancer.4 Nevertheless, PDAC is locally invasive and highly metastatic, rendering most patients not suitable for surgery. Alternatively, radiotherapy (RT) has limitations in safely targeting pancreatic cancer.5
The tumor epithelium of Pancreatic ductal adenocarcinoma (PDAC) is present in dense mesenchyme and is thought to be a key mediator of disease progression through direct action on cancer cells and indirect action on the tumor immune microenvironment.6 Moreover, the tumor microenvironment (TME), which contains a heterogeneous mix of CAFs, immune cells, macrophages, vasculatures, and the extracellular matrix (ECM), inhibits the spread of chemotherapeutic drugs to tumor connective tissue hyperplasia, and inflammation is the two main features of PDAC.7–9 Connective tissue is composed of ECM, CAFs, infiltrating immune cells, and endothelial cells, along with the ECM, vascular system, and CAFs, which act as biophysical barriers to chemotherapy and actively promote tumor progression and metastasis.10 CAFs are mainly formed by intrinsic fibroblasts or stellate cells in tissues stimulated by growth factors. Epithelial cells, endothelial cells, bone marrow mesenchymal stem cells, and other cells in tumor tissues can also differentiate into CAFs. Extracellular matrix proteins produced by CAFs in the tumor stroma impede the effective delivery of chemotherapeutic agents,11,12 resulting in poor responses in patients with pancreatic cancer.13
Due to recent advances in nanotechnology, the application of nanoparticles in cancer therapy has become a leading area in cancer research.12,14,15 Nanotherapeutics can achieve better therapeutic effects on tumors than free drugs based on the enhanced permeability and retention (EPR) effect of tumors.16 However, the benefits of anti-tumor nanotherapeutics such as Doxil and Abraxane, two nanotherapeutics approved by the Food and Drug Administration (FDA) for use in solid tumors, are still far from satisfactory.17 As an extension, active targeting drug delivery systems, which are modified with targeting moieties with special affinity to the corresponding antigens overexpressed in tumor tissues, have been explored to achieve additional anti-tumor efficacy compared to those based on the EPR effect alone.18,19 Therefore, suitable antigens overexpressed by both cells and the corresponding targeting moieties can be selected for active drug delivery systems that simultaneously target both myeloma cells and CAFs to achieve a strong therapeutic effect for PDAC treatment.20 In tumors, cancer cell surface antigens have homologous or heterologous adhesion properties.21 These properties are attributed to plasma membrane proteins on the surface of cancer cells, including N-calmodulin, galactoglucan-3, and epithelial cell adhesion molecules,22 and the surface-encapsulated cancer cell membrane (CCM) of NPs competes for homotypic cancer cell surface antigens, acquiring immune escape and homology-targeting capabilities that can be used for highly specific cancer targeting and effective cancer Treatment. Studies have found that cancer cell membranes and fibroblast membrane-camouflaged nanoparticles can target cancer cells and fibroblasts, respectively, via homologous targeting.23,24 Encouraged by this, the fusion of cancer cell and fibroblast membranes provides a potent dual-targeting capability. Compared with conventional complex synthetic nanoparticles (NPs), low-cost yield-synthesized harmless fluorescent NPs are well suited for achieving dual-targeting effects and have the advantages of rapid metabolism and tumor-homing capabilities.
Despite the importance of CAFs in creating an optimal niche for extensive tumor cell growth, most studies have focused on tumor cells.13,25 Therefore, to effectively use nanoparticles for therapeutic purposes, it is important to elucidate the extent of nanoparticle uptake and retention in tumor cells and CAFs.26,27 To make significant progress in this field, we need to consider the tumor microenvironment, especially CAFs that promote tumor growth. Researchers have demonstrated the potential of targeting both tumor cells and CAFs to reap the full benefits of cancer nanomedicine.26,28,29 We previously validated the recognition of graphene quantum dot nanomaterials in hepatocellular carcinoma tissue, paracellular tissue, and normal tissue based on the difference in permeability between tumor cell membranes and normal cell membranes.30,31 Unlike the TME of liver cancer, the tumor tissue stroma of PDAC is highly fibrotic and is a typical model used to study the simultaneous dual targeting of GFNPs to tumor cells and CAFs.32 The expression of surface markers of CAFs varies by subtype. Alpha smooth muscle Actin (αSMA) is a marker of activated fibroblasts and is often used as a major indicator of the efficacy of targeted CAFs therapy. In this study, we further investigated the mechanism of single-cell recognition of GFNPs in the PDAC tissue microenvironment, which could abundantly accumulate in tumors and were highly colocalized with CAFs and pancreatic cancer cells, localized in the nuclei of both SMA-CAFs and adjacent tumor cells. The fluorescence signal intensity was proportional to the tumor development cycle and tumor size. Considering that the significant side effects of GEM limit its application prospects, it is a classical treatment for pancreatic cancer.33 We used GFNPs to modify GEM into GFNPs-GEM to improve the efficiency of pancreatic cancer treatment and reduce side effects. Our study also highlighted the potential use of the dual-targeting performance of GFNPs in the TME to improve drug efficacy and reduce drug toxicity.
Materials and Methods
Synthesis of Graphene Fluorescent Nanoparticles
GFNPs were obtained by hydrothermal reaction as follows: pyrene (0.5 g, 98% purity) was added to 25 mL HNO3 and reacted at 80°C for 24 h. At the end of the reaction, the solution was cooled and filtered through 150 mL of deionized water, and the excess acid was filtered through a 0.22 µm membrane. The residue was transferred to Na2SO3 solution (50 mL, 0.5 M), and the solution was stirred for 0.5 h. The solution was transferred to a reaction vessel and allowed to react in a vacuum drying chamber at 200°C for 12 h. The solution was cooled and filtered through a 0.22 µm membrane to retain the liquid. Vacuum drying at 70 °C was used in subsequent experiments.
Synthesis of Graphene Fluorescent Nanoparticles and Chemotherapeutic Drug
The GFNPs-GEM solution was diluted with physiological saline at a concentration of 1:5 and reacted at 140 rpm for 12 h on a shaker at 37°C. For bufalin, trametinib and sorafenib (MCE, China), we added 0.1 mL of each drug, dissolved in DMSO (10 mg mL−1), dropwise (20 µL per 15 sec) to a 0.6 mL aqueous solution containing GFNPs (1 mg mL−1) under slight vortex. The solution was centrifuged twice (20,000 × g, 30 min) and the pellet was resuspended in 1 mL of sterile PBS. Pellets that were difficult to resuspend were ultrasonicated for 5 s with a 1/8 inch probe tip (Sonics & Materials) at 40% intensity. The nanoparticles were lyophilized in 5% saline/sucrose solution.
Characterization Analysis of Graphene Fluorescent Nanoparticles and Chemotherapeutic Drug
Morphological and particle size observations were performed using transmission electron microscopy (TEM) (Japan 200CX). Absorption and fluorescence spectra were recorded using a spectrophotometer (Hitachi 3100, Japan) and fluorescence spectrophotometer (Hitachi 7000, Japan), respectively. Fourier-transform infrared (FT-IR) spectra of the dried samples were recorded using a Bio-Rad FTIR spectrometer FTS165. Elemental content analysis (C, H, N, and S) was performed using an organic element analyzer (Vario Micro cube, Germany). The quantum yields (QYs) of GFNPs aqueous solutions were determined by comparing the integrated photoluminescence (PL) intensities (excited at 360 nm) and the absorbency values (at 360 nm) using quinine sulfate (quantum yields (QYs): 0.55) in sulfuric acid (0.1 mol L−1, η = 1.33) as a reference. The molecular weight of GFNP was determined using matrix-assisted laser desorption tandem time-of-flight mass spectrometry (MALDI-TOF) 7090 (Shimadzu, Japan). Qualitative detection and quantitative detection of GFNPs and GFNPs-GEM were performed using HPLC (Agilent, USA). To simulate GEM release in the tumor microenvironment, GFNPs-GEM was dialyzed at 37 degrees in a 500Kd dialysis bag contained PBS (pH 6.5) for 30 minutes.
Cell Culture
The murine pancreatic cancer cell line Pan02 and human pancreatic cell line SW1990 were purchased from the Cell Resource Center, IBMS, CAMS/PUMC (Beijing, China). Cell lines were authenticated by short tandem repeat (STR)profiling and verified to be mycoplasma-negative. All cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO, USA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37 °C and 5% CO2.
Human Samples
Fresh pancreatic cancer samples were also obtained from Changhai Hospital, Shanghai, China (n = 6, 83.3% males and 16.7% females, age distribution: 40–75 years old) to evaluate the correlation between tumor tissue and GFNPs signals by in vivo imaging system (IVIS, PerkinElmer, USA) analysis. All procedures were conducted with the approval of the Ethical Committee of Changhai Hospital (CHEC2022-018) and in accordance with the relevant regulations and guidelines. Patient consent was obtained before the start of the study.
Animal Models
All mice were housed in specific pathogen-free facilities under a 12 h light/dark cycle and controlled temperature (20–25°C) with standard rodent chow (Shanghai Jihui Laboratory Animal Breeding Co., Shanghai, China) and water provided ad libitum. All mice used in the experiments were male and approximately 6 weeks old. The health status of the mice was checked following the protocols, and The University Committee on Use and Care of Animals of Naval Medical University approved all the animal protocols used in this study. For in Vivo studies, SW1990, Pan02, and tD Tomato-Bxpc-3 cells were injected subcutaneously or orthotopically into the left thighs of BALB/c, C57BL/6, or nude mice (Laboratory Animal Resources, Chinese Academy of Sciences, Shanghai, China). The number of mice per group for each experiment is detailed in the figure legends. Tumor size (length X width2 X 0.5) was measured twice a week after injection. The mice were euthanized at approximately 3–4 weeks and the tumors were harvested for weight measurement and tissue analyses.
Orthotopic Transplanted Pancreatic Cancer Models in Nude Mice with Different Cycles
Two million pancreatic cancer cells (SW1990 cells) were injected orthotopically into the pancreas of nude mice to establish xenograft tumor models with cycles of 0, 7, 14, 21, and 28 days. After completion of the modeling, the mice were injected with a fluorescent nanoprobe into the tail vein. Thirty minutes later, the mice were euthanized, the tumor tissues were removed, and the changes in fluorescent signals within the tumor during the four weeks of modeling were analyzed qualitatively and quantitatively using a small animal live imager, with mice of sham group as a blank control. The pancreatic tumor sections were panoramic imaged by fluorescence scanner (3DHISTECH, Hungary), browsed by Caseviewer Software and analyzed by Indica labs (USA). Local imaging of tissue and tumor cells observed by Confocal laser scanning microscope (FV1000, Olympus, Japan) and inverted fluorescent microscope (Leica, German).
Subcutaneous Tumor and Orthotopic Pancreatic Cancer Models in Nude Mice with Different Amounts of Tumor-Forming Cells
Orthotopic pancreatic cancer models: two, four and six million pancreatic cancer cells (SW1990 cells) were injected orthotopically into the pancreas of nude mice to establish a xenograft tumor model with a cycle of 14 days. After completion of the modeling, the tail vein was injected with a nano fluorescent probe, and 30 min later, the mice were euthanized, and the tumor tissues were removed. The changes in fluorescent signals within the tumors of different groups were analyzed qualitatively and quantitatively using a small-animal live imager. All mice were tumor bearing and injected with GFNPs. Five parallel samples in each group.
Subcutaneous tumor models: three, six and nine million pancreatic cancer cells (SW1990 cells) were injected subcutaneously into nude mice to establish a heterogeneous subcutaneous tumor model with a tumor formation cycle of 14 days. After completion of the modeling, the mice were injected with a fluorescent nanoprobe in the tail vein, and 30 min later, the mice were euthanized and the tumor tissues were removed. The changes in fluorescence signals within the tumors of different groups were analyzed qualitatively and quantitatively using a small animal live imager, with five parallel samples from each group.
Subcutaneous tumor model after labeling: to-Tomato red fluorescent protein gene was transfected into human pancreatic cancer cells (BXPC-3) using lentiviral transfection technology to construct fluorescent luminescent labeled cells; then different doses of fluorescent cells (16, 32, 48 and 64 million) were used to establish a subcutaneous tumor model of pancreatic cancer, the tumor formation cycle was 30 days, and the tumor volume was measured every ten days. After 30 days of modeling, the mice were injected with 100 mg/kg of fluorescent nanoprobe in the tail vein, and 30 minutes later, the tumors were removed and the tumors were observed to coincide with the two fluorescence channels (474 nm excitation/560 nm excitation) using a small animal live imager, and the fluorescence signal values of the different tumors under the two channels were counted. Tumors were weighed and recorded, frozen sections were prepared, nuclei were stained with TOPRO3, frozen sections were scanned in the full area using a fluorescence scanner, and the positivity of tumor cells, the proportion of positive tumor cells labeled with nanoprobes, and the colocalization of the two fluorescence channels at the cellular level were analyzed using the Halo technique.
Animal Models of Pancreatic Metastasis in Mice with Spleen Injection
C57BL/6 mice were weighed and subsequently divided into two groups, the sham-operated group and the model group, with five animals in each group. Mice were anesthetized by intraperitoneal injection of sodium pentobarbital and the skin of the surgical field was disinfected with 75% alcohol. The left upper abdomen of each mouse was incised obliquely for approximately 1.0 cm, and the spleen was gently pulled out from the abdominal cavity using tissue forceps. After the injection, the spleen was gently returned to its original position. After the injection in each group, the needle eye was compressed with a 75% alcohol cotton ball for 2–3 minutes to stop bleeding and kill any possible extravasated cancer cells to prevent intra-abdominal implantation and metastasis, and suture the wound. After awakening from anesthesia, the mice were maintained at the SPF level and observed daily. Mice were injected with 100 mg/kg GFNPs via the tail vein after 21 days and euthanized within 30 min. The heart, liver, spleen, lungs, kidneys, and pancreas were separated. Specimens from each group were fixed in formalin and cut into frozen sections for immunohistochemistry (IHC) and immunofluorescence (IF).
Different Types of Clinically Isolated Pancreatic Cancer Samples
Six samples each of PDAC, neuroendocrine tumor (NET), and adenosquamous carcinoma (ASC) tissues smaller than 2 cm were collected. Fresh tissues were incubated in 500 µg/mL of GFNPs solution at 4°C for 30 min as soon as possible after surgery, and the relationship between the tumor and fluorescence signal intensity was analyzed by in vivo imaging to compare the fluorescence signals in the three types of small pancreatic cancer tissues, while orthotopic imaging of frozen tissue sections was used for determination. Cellular labeling of GFNPs was determined by orthotopic imaging of the frozen tissue sections.
Immunofluorescence Staining
Frozen section fixation Frozen sections were rewarmed from the refrigerator, dried, fixed in paraformaldehyde for 60 min, and washed three times in PBS (pH7.4) on a decolorization shaker for 5 min each time after the paraformaldehyde was completely dry. Antigen repair Tissue sections were placed in a repair cassette filled with citric acid antigen repair buffer (pH 6.0) and subjected to antigen repair in a microwave oven. Heat was applied for 5 min, ceased for 5 min, and then decreased for 10 min. During this process, the buffer should be prevented from evaporating excessively and the slides should not dry out. After natural cooling, the slide was placed in PBS (pH 7.4) and washed 3 times for 5 min each by shaking on a decolorization shaker. (The repair solution and intensity of repair are determined by the tissue) Drawing circles: after the slides have been slightly shaken dry, draw circles around the tissue with a histochemical pen (to prevent antibody run-off). Closure: Bovine serum albumin (BSA) dropwise to the circle and incubated for 30 minutes in the dark. The primary antibody was added by gently shaking off the blocking solution, adding a drop of PBS to the section. Incubating overnight at 4°C in a wet box protected from light (moderate water was added to the wet box to prevent evaporation of the antibody) and the slides were washed three times in PBS (pH 7.4) on a decolorization shaker for 5 min each. The sections were shaken dry, covered with secondary antibodies of the corresponding species in a circle, incubated for 50 min at room temperature, and protected from light. Blocking: The slides were washed three times for 5 min each on a decolorization shaker in PBS (pH 7.4), shaken, dried, and blocked with an anti-fluorescence quenching blocker. Microscopic examination and photography: Sections were observed under a fluorescence microscope and images were taken. (UV excitation wavelength 330–380 nm, emission wavelength 420 nm, blue light; FITC excitation wavelength 465–495 nm, emission wavelength 515–555 nm, green light; CY3 excitation wavelength 510–560 nm, emission wavelength 590 nm, red light; CY5 excitation wavelength 608–648 nm, emission wavelength 672–712 nm, pink light).
In vivo Treatments
For GEM and GFNPs-GEM treatments, mice were treated with GEM (10 mg/kg body weight), GFNPs-GEM (10 mg/kg body weight), or the solvent was tail intravenously injected once every three days for 3 weeks beginning on day 10 after subcutaneous wild-type SW1990 cell inoculation. Before treatment initiation, mice were randomized into treatment or control groups with similar average tumor volumes. Daily weighing and recording of body weight and tumor volume in mice.
Statistical Analysis
Statistical analysis was performed using the GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA). Data points are presented as mean ± SD. All experiments were repeated at least three times. After confirming that the values followed a normal distribution, a two-tailed Student’s t-test was applied to determine the significance of the differences between two groups of independent samples. Pearson’s correlation analysis was performed to determine the correlations between the two variables. The survival rate was analyzed using the Kaplan-Meier method. Statistical significance was set at p < 0.05. All p values are indicated in the figure (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant).
Fluorescence colocalization analysis: Tissue sections are mounted on the scanner and scanned as they are moved gradually under the lens of the scanner, while imaging is performed to scan all the tissue information on the sections and form a folder containing all the tissue information. The folder can be opened with CaseViewer 2.2 software and magnified from 1–400x for observation. Using the Halo v3.0.3 11.3 14 analysis software with the Indica Labs - HighPlex FL v3.1.0 module, the number of positive cells, total cells, and fluorescent pattern dual-labeled cells in the target area of each section are quantified, and the positive rate (%) is calculated. Display diagram of the application of the analysis software were listed in supplementary materials.
Results
Characterization and Safety Evaluation of Graphene Fluorescent Nanoparticles
The morphology and particle size distribution of GFNPs with negative charge are shown in Figure 1A and B. GFNPs exhibits excellent monodisperse properties, whose particle size distribute mainly in the range of 2–4 nm. The functionalized groups of the GFNPs were characterized using FT-IR spectroscopy (Figure 1C). Functionalized SO3− group is observed at 1034 cm−1, and the characteristic peak at 613 cm−1 corresponds to the C-S bond. O-H was observed at 3400 cm−1 and C-OH was observed at 1100 cm−1. These results indicated that the GFNPs were co-functionalized with OH and SO3−. Percentages of the chemical elements are shown in Table 1, where C, H, S, and N were 10.28%, 1.25%, 15%, and 2.11%, respectively. The plasmon resonance peaks of the GFNPs were at approximately 370 and 406 nm, respectively (Figure 1D). GFNPs have strong absorption bands that extend to ~360 nm, bright emission wavelength at 510 nm excited between 372 nm and 488 nm, a wide photoluminescence excitation (PLE) spectrum with a quantum yield of up to 41.79%, and exhibit excitation-dependent emission behavior (Figure 1E). The aqueous solution emitted bright blue fluorescence when irradiated with ultraviolet (UV) light. MALDI analysis indicated that the molecular weight was approximately 286.27(Figure 1F). After intravenous tail injection, GFNPs showed no significant difference in organ HE (hematoxylin-eosin) staining compared to the control (Figure S1), which safely metabolized from the tumor tissue and other organs from 0.5 h to 2 h (Figure S2).
 |
Table 1 Chemical Element Content Ratio |
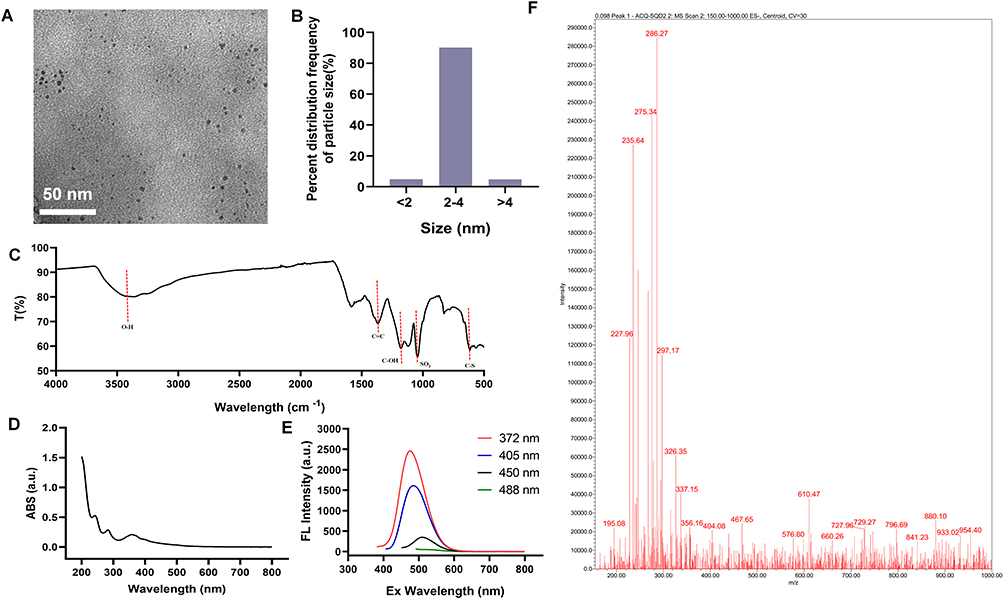 |
Figure 1 GFNPs physical chemistry characterization. (A) TEM image. Scale bar =100 nm. (B) Size distribution. (C) FT-IR spectrogram. (D) Absorption spectrum. (E) Fluorescence excitation emission spectrum. (F) Molecular weight profile. |
Biodistribution of Graphene Fluorescent Nanoparticles in Different Size Orthotopic Pancreatic Tumor Tissues
GFNPs showed the strongest signal in the tumor tissue of the orthotopic pancreatic cancer tumor model constructed from SW1990 cells (Figure 2A), followed by the liver and kidney tissues. GFNPs marked tumor neovascularization of 1-week tumor model in Figure 2B. Perfect colocalization of GFNPs and nuclei inside frozen sections of 2-weeks pancreatic cancer tissues is shown in Figure 2C. The fluorescence signal became weaker from inside to outside in the 4-week tumor, whose surface area was significantly larger than that of the 1- and 2-week tumors (Figure 2D). There was no significant difference in the fluorescence signal of most tissues except in orthotopic pancreatic cancer tumor to the total fluorescence intensity of the 4-week tumor mouse was significantly higher than that of the 1-week tumor mouse, and the distribution ratio of GFNPs in the liver and kidney decreased and pancreatic tumor tissue increased, respectively (Figure 2E and F). After deducting the autofluorescence interference of the tissues, the GFNPs signals of the heart, liver, spleen, lung, kidney, and brain in the tumor-bearing mice were the same as those in the mice of sham group. The GFNPs signals of continuously proliferating tumors of different sizes were significantly larger than those of pancreatic tissues in sham group (Figure 2G). The GFNPs fluorescence area was similar to the M-cherry-marked SW1990 cell signal and completely overlapped the M-cherry fluorescent signal in the SW1990 cell spheroid (Figures S3 and 4).
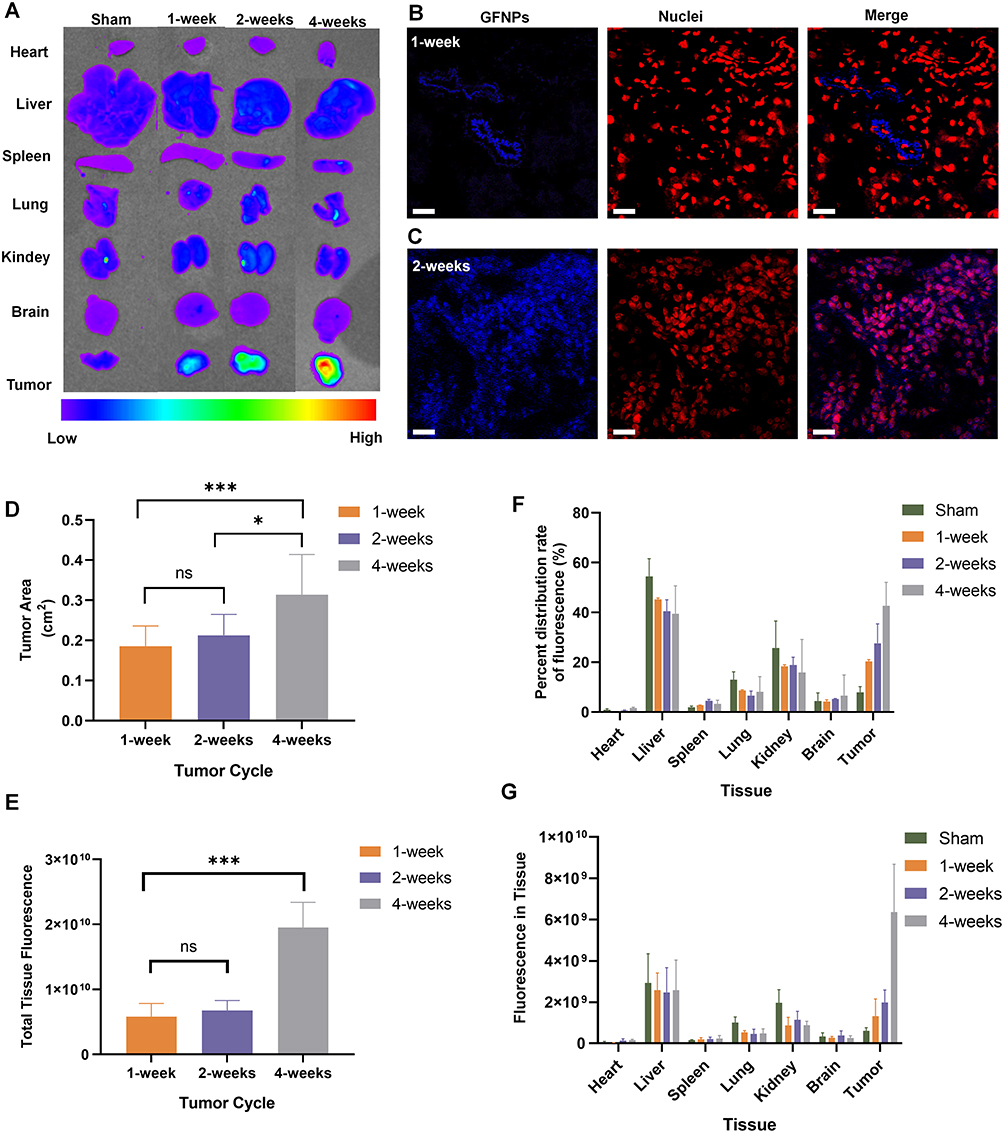 |
Figure 2 GFNPs distribution on murine orthotopic pancreatic tumor model at different stages of development. (A) In vivo imaging results of various organs of nude mice (Ex474 nm/Em525 nm) after tail vein injection of 100 mg/kg GFNPs for 30 min in orthotopic pancreatic cancer tumor models at different stages of development. (B and C) The plot of laser confocal imaging results of GFNPs after making 20 µm frozen sections of 1-week and 2-weeks pancreatic cancer tumor tissue, with nuclei stained with TOPRO3, scale bar = 20 µm. (D)Statistical results of the surface area of pancreatic cancer tumor tissue at 1, 2 and 4 weeks of growth. n=8. (E) Total fluorescence intensity of organs of nude mice with different tumor development cycles after tail vein injection of 100 mg/kg GFNPs for 30 min. n=8. (F)Percent distribution rate of fluorescence in the organs of nude mice with different tumor development cycles after tail vein injection of 100 mg/kg GFNPs for 30 min. n=8. (G)Fluorescence semi-quantitative statistics of GFNPs in various organs of nude mice with different tumor development cycles after tail vein injection of 100 mg/kg GFNPs for 30 min. n=8. Notes: Data was exhibited as the Mean ± SEM * p < 0.05, *** p < 0.001. |
Antibodies like ki67 and SMA are usually used to rapidly analyze proliferating tumor cells and CAFs formed by pancreatic stellate cell activation in the TME. Immunostaining of frozen tumor tissue sections provided a clear view of each cell type. Ki67 positive areas of IHC staining became increasingly larger, indicating tumor cell growth and invasion (Figure 3A). Pancreatic cancer tissues contain abundant neovascularization, and we demonstrated that GFNPs enter the tumor microenvironment by crossing vimentin-labeled vascular endothelial cells (Figure 3B). CAFs closely surrounded the tumor cells in 2-week tumor tissues. Figure 3C shows that GFNPs were located in the nuclei of tumor cells immediately adjacent to CAFs, showing colocalization with the tumor cell nuclear protein Ki67. In addition, CAFs nuclei near Ki67-positive tumor cells also exhibited GFNPs signals.
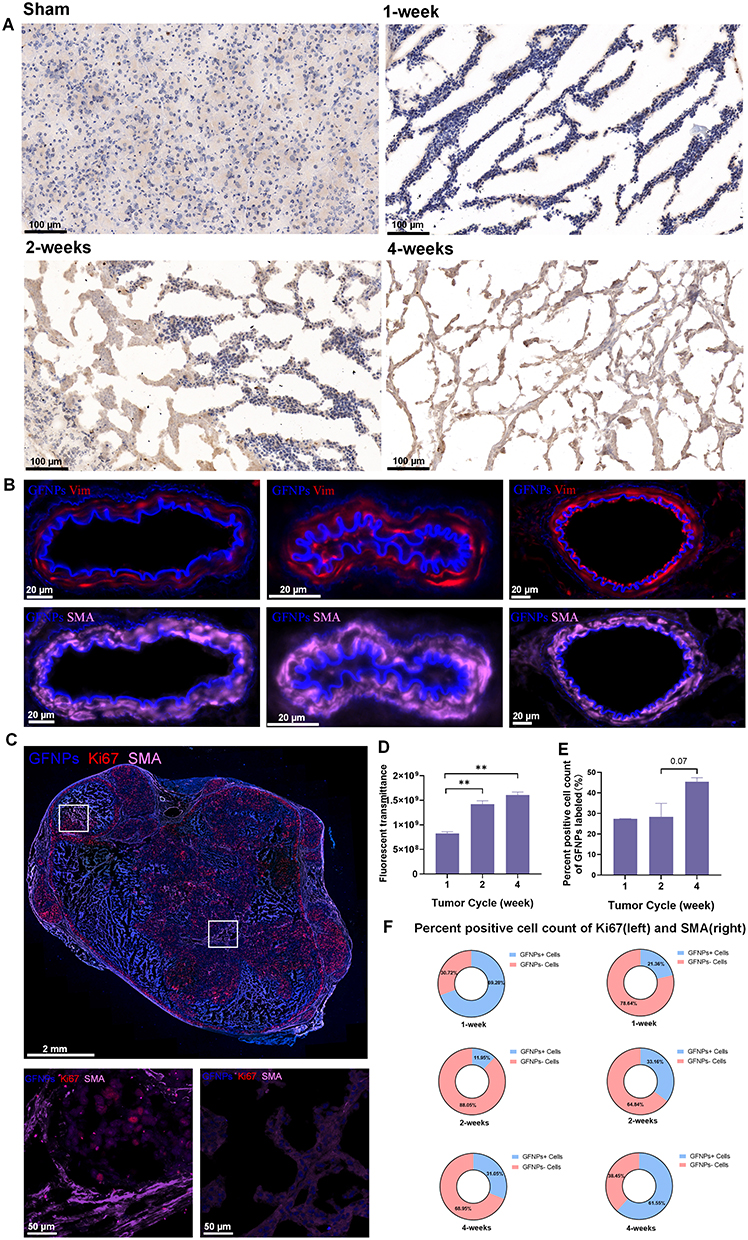 |
Figure 3 Analysis of the fluorescence diagnostic effect of GFNPs on murine orthotopic pancreatic tumor tissue at different stages of development. (A) Immunohistochemical tracings of Ki67 in pancreas tissue of sham group and orthotopic transplanted pancreatic tumor tissue at 1, 2 and 4 weeks. scale bar = 100 µm. (B) Immunofluorescence staining of vascular structure in TME. Blue-GFNPs; Pink-SMA; Red-Vimentin. scale bar = 20 µm. (C) Immunofluorescence staining scanning of orthotopic transplanted pancreatic tumor tissue (up) and partial detail view of the white box (bottom). scale bar = 2 mm (up), scale bar = 50 µm (bottom), Pink-SMA; Blue-GFNPs; Red-Ki67. (D) Fluorescent transmittance in orthotopic transplanted pancreatic tumor tissue at 1, 2 and 4 weeks. (E) Percent positive cell count of GFNPs labeled in orthotopic transplanted pancreatic tumor tissue sections at 1, 2 and 4 weeks. (F) Percent positive cell count of Ki67 (left) and SMA (right) in different pancreatic tumor tissue. Notes: Datas were exhibited as the Mean ± SEM. ** p < 0.01. |
IHC-positive regions almost overlapped with IF in the whole tissue, based on the morphology of adjacent tissue sections. IHC and IF staining were combined to assess the colocalization of Ki67-positive tumor cells and SMA-positive CAFs with GFNPs in 1-, 2 and 4 -week tumor tissue, respectively (Figure S5). In addition to qualitative analysis, we also quantified the fluorescence intensity in 1-, 2 and 4 -week tumor tissue using the Halo technique. We analyzed the percentage of positive cells (GFNPs+/PI+) inside the 1, 2 and 4 -week tumor tissue according to their fluorescence differences. GFNPs labeled approximately 45% of nuclei in the 4 -week tumor tissue, which was higher than that in the 1- and 2-week tumor tissues (Figure 3D and E). Halo software analyzed colocalization between kinds of cells in TME and cells stained by GFNPs (Figure S6). Highest percentage of Ki67-positive cells co-localized with GFNPs in 1-week tumor tissue, and earlier stages of tumorigenesis, the better marking effect. Proportion of GFNPs labeling CAFs positively correlated with tumor progression (Figure 3F).
Orthotopic pancreatic cancer tumor development involves more than just proliferation of tumor cells. We used different doses of tumor cells combined with orthotopic and subcutaneous tumor models to observe GFNPs signals in tumors with different cell volumes and the same proliferation cycle. The fluorescence intensity of GFNPs in the tumor decreased as the number of tumor-forming cells increased (Figure 4A and B). Although the number of planted SW1990 cells was multiplied, there was no difference in tumor volume and weight between the groups after two weeks (Figure 4C and D). However, these phenomena were not observed in the subcutaneous tumor model formed by SW1990 cells, where the fluorescence signal of the subcutaneous tumor increased as the number of planted SW1990 cells increased (Figure 4E and F), and the subcutaneous tumor volume and weight increased incrementally (Figure 4G and H). To further investigate the relationship between the initial tumor cell dose and the targeting signal of GFNPs, we employed a third tumor model using a fluorescently labeled pancreatic cancer cell line (tD-Tomato-BXPC-3) to establish a subcutaneous tumor model in nude mice. The in vivo fluorescence of tD-Tomato-BXPC-3 cells is shown in Figure 4I, and the fluorescence of GFNPs in subcutaneous tumors formed by tD-Tomato-BXPC-3 is shown in Figure 4J. Similar to the trend for subcutaneous tumors formed by SW1990 cells, both the volume and weight of the subcutaneous tumor formed by tD-Tomato-BXPC-3 cells gradually increased (Figure 4K and L). We found that the red fluorescence intensity of the subcutaneous tissue itself increased with the initial cell volume by analyzing the fluorescent signal of tD-Tomato-BXPC-3 in vivo (Figure 4M). However, the fluorescence intensity of GFNPs in subcutaneous tumors showed no upward or downward trend, with the strongest labeling signal of GFNPs in subcutaneous tumors formed by 32 million tD-Tomato-BXPC-3 cells (Figure 4N). We analyzed the results of Ki67 IHC and HE staining of adjacent subcutaneous tumor tissue slides, and different magnification images are shown in Figure 4O and P. Enlarged marked box in Figure 4O shows that the GFNP signal of GFNPs could not overlap with TOPRO3 stained cell nuclei in the tissue area without tD-Tomato-BXPC-3 cells (Figure 4P).
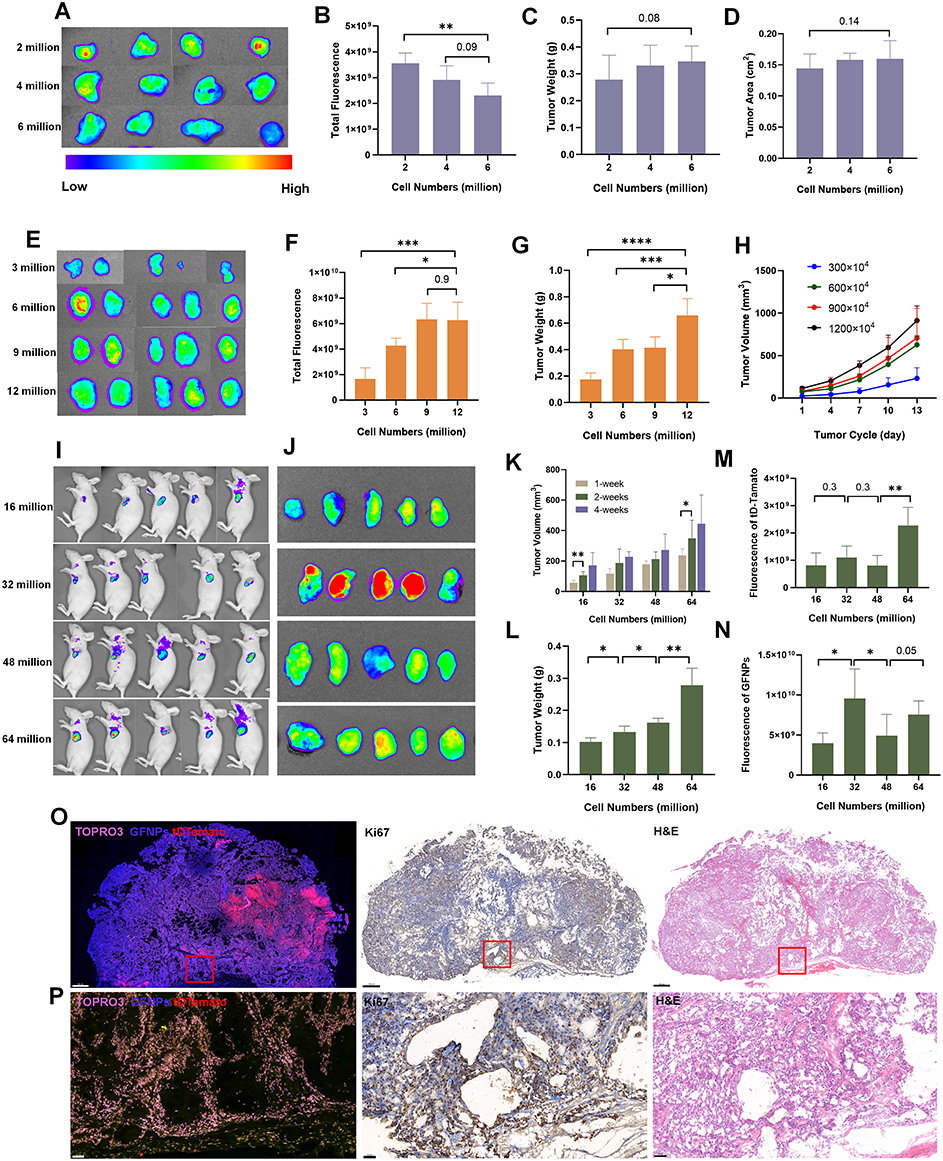 |
Figure 4 Fluorescence analysis of GFNPs on pancreatic cancer tissues with different numbers of tumor-forming cells. (A) Fluorescence imaging of tissue in vitro of murine orthotopic pancreatic cancer tissues (Ex474 nm/Em525 nm). n=4 (B–D) Fluorescence intensity, tumor weight and tumor surface area of murine orthotopic pancreatic cancer tissues formed by different numbers of SW1990 cells (Ex474 nm/Em525 nm). n=4 (E) Fluorescence imaging of tissue in vitro of murine subcutaneous pancreatic cancer tissues. n=5 (F–H) Fluorescence intensity, tumor weight and tumor volume of subcutaneous pancreatic tumor tissue formed by different numbers of SW1990 cells (Ex474 nm/Em525 nm). n=5 (I and J) Fluorescence imaging of tissue in vivo and in vitro of murine subcutaneous pancreatic cancer tissues formed by different numbers of fluorescently labeled Bxpc-3 tD Tomato tumor cells. n=5 (K and L) Tumor weight and tumor volume of subcutaneous pancreatic tumor tissue formed by different numbers of Bxpc-3 tD-Tomato tumor cells. n=5 (M and N) Fluorescence intensity of tD-Tomato channel (Ex 550 nm/Em 590 nm) and GFNPs channel (Ex 474 nm/Em 525 nm) of pancreatic cancer subcutaneous tumors tissue. n=5 (O and P) View of Ki67 stained by IF, IHC and HE stain between adjacent subcutaneous tumor tissue section formed by BXPC-3-tD Tomato tumor cells, panoramic (top) and local (bottom) views of stain results. Top: scale bar = 500 µm, bottom: scale bar = 50 µm. Blue-GFNPs; Pink-TOPRO3; Red-tD Tomato. Notes: Datas were exhibited as the Mean ± SEM. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. |
Fluorescence Labeling Effect of Graphene Fluorescent Nanoparticles on Liver Metastasis of Pancreatic Cancer
Pan02 cells underwent hematogenous transfer to the liver after injection into the spleen. As shown in Figure 5A (left), a large white tumor nodule was evident in the livers of the mice (red circle), and we successfully established a pancreatic tumor metastasis model. In vivo imaging of the liver and pancreas in Figure 5A (right) shows that the fluorescence of the white nodules in the liver was significantly higher than that of the non-white nodules. The increased fluorescence of the lung and pancreatic tissues indicated that the tumor cells also invaded the adjacent lung and pancreatic tissues (Figure 5B), with the most pronounced change in fluorescence in the liver (Figure 5C). We investigated the spatial location of GFNPs in relation to tumor cells and CAFs in pancreatic tumor metastasis. IF signals of Ki67 almost completely overlapped with propidium iodide (PI), GFNPs showed local signal overlap with the tumor nucleus antibody protein Ki67, and CAFs were also distributed in the vicinity of the double-positive region for GFNPs and Ki67 (Figure 5D). Ki67-positive Pan02 cells also exist in cancerous lung tissues. Panoramic IF imaging of cancerous lung tissue revealed that a large number of strongly Ki67-positive tumor cells were surrounded by SMA-positive CAFs, with CAFs surrounding the cancerous cells in a circle. Dense tumor cells were distributed both inside and outside the circle. GFNPs were mainly located in the cytoplasm of tumor cells and CAFs in the TME (Figure 5E).
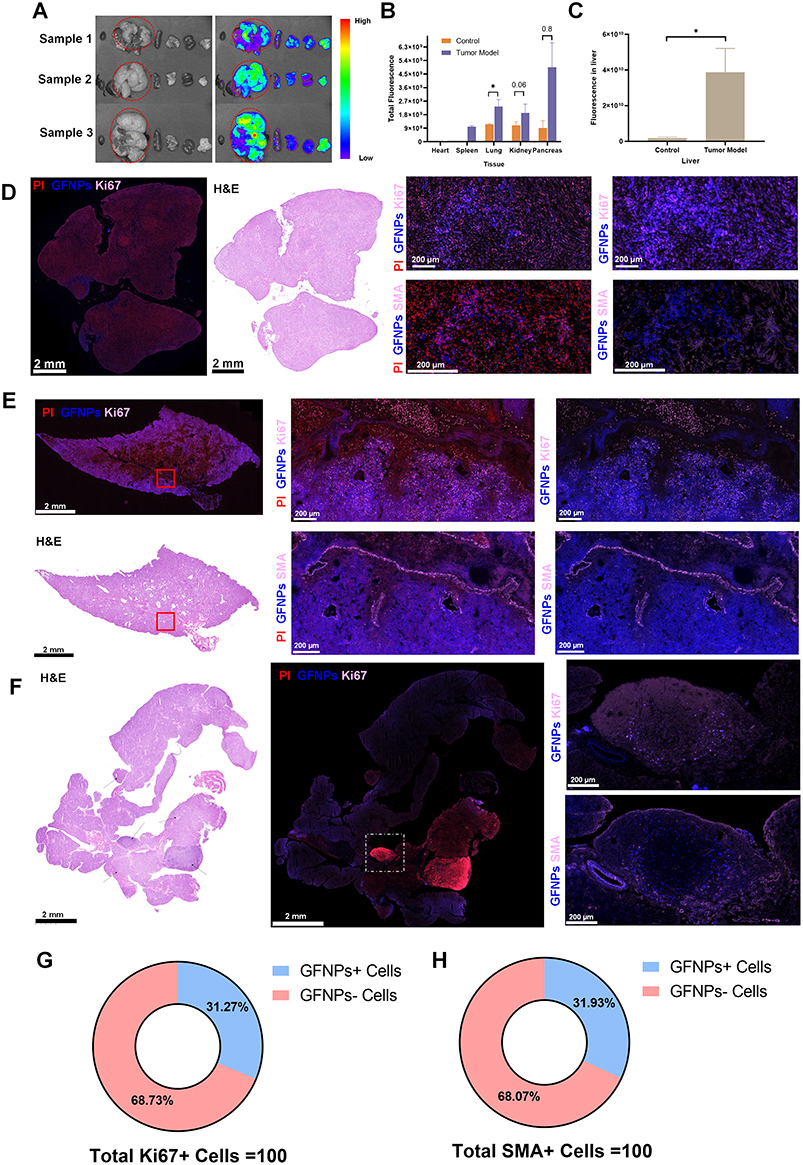 |
Figure 5 Localization analysis of GFNPs in pancreatic cancer metastasize to liver cancer murine model. (A) Photographs and fluorescence imaging of tissue in vitro of three models of pancreatic cancer metastasize to liver cancer murine derived from Pan02 cells (Ex474 nm/Em525 nm). (B and C) Fluorescence intensity of the heart, liver, spleen, lung, kidney and pancreas between sham group and tumor model group. n=5. (D)HE and immunofluorescence staining results of adjacent tissue section derived from liver cancer tissue. Blue-GFNPs; red-PI; pink-Ki67. Scanning scale bar=2 mm. Detailed scale bar=200 µm. (E) Immunofluorescence and HE staining results of adjacent locations scan of a lung tissue section from Pan02 cell-derived pancreatic metastases tumor model. Blue-GFNPs; red-PI; pink-SMA. Scanning scale bar=2 mm. Detailed scale bar=200 µm. (F) Immunofluorescence and HE staining results of adjacent locations scan of a pancreatic tissue section from Pan02 cell-derived pancreatic metastases tumor model. Blue-GFNPs; red-PI; pink-Ki67.Scanning scale bar=2 mm. Partial scale bar=200 µm. (G) Percent positive cell count of Ki67 at pancreatic tissue. (H) Percent positive cell count of SMA+ cells at pancreatic tissue. Notes: Datas were exhibited as the Mean ± SEM. * p < 0.05. |
We observed an increased fluorescent signal in the pancreatic tissue of liver metastasis tumor-bearing mice compared to blank controls, and by HE staining of the whole pancreatic tissue, we found multiple tumor cell clusters in the pancreatic tissue (Figure 5F, black arrow). Similarly, PI signals in the pancreatic tissue were significantly higher than those in the other areas (Figure 5F, right). One of the cancerous pancreatic tissues in Figure 5A was magnified, and GFNPs showed local signal overlap with Ki67-positive Pan02 cells and SMA-positive CAFs exit in the vicinity of the double-positive area for GFNPs and Ki67.The proportion of both CAFs and tumor cells in the TME was elevated after carcinogenesis of the pancreatic tissue. GFNPs could recognize over 30% of tumor cells and CAFs (Figure 5G and H).
Labeling Effects and Mechanisms of Graphene Fluorescent Nanoparticles in Clinical Samples of Different types of Pancreatic Cancer
To investigate the labeling effect of GFNPs on clinical pancreatic cancer samples, three typical tumors, pancreatic neuroendocrine tumor (NET), adenosquamous carcinoma (ASC), and pancreatic ductal adenocarcinoma (PDAC), were selected as subjects. Patient and pathological information of the clinical tumor samples are shown in Table S1 (Ethical Number: CHEC2022-018). We compared the fluorescence intensity of GFNPs in each clinical tumor tissue after incubation with GFNPs. The signal intensities of GFNPs in the three types of tumor tissues were NET, ASC, and PDAC in descending order (Figure 6A). We performed HE, IHC, and IF staining, and analyzed the three pancreatic tumor tissues using serial sectioning techniques. Because of the low vascular density and high tumor cell density of ASC, GFNPs labeled less than 1% of the ASC tumor cells (Figure 6B) and 25% of the CAFs (Figure 6C). Pathological results from patients with ASC showed a positive Ki67 rate of 70% and positive SMA expression in the mesenchyme. Unlike the abundant vascular structures of PDAC, adenosquamous carcinoma tissues had fewer vascular structures, and the nuclei of tumor cells showed marked heterogeneity, being significantly larger than those of other cells in the TME. GFNPs were mostly distributed in the cytoplasm of tumor cells, with stronger fluorescent signals in the cytoplasm around ki67-positive tumor cells (Figure 6D), and did not have an obvious spatial co-location with SMA-positive CAFs close to Ki67-positive tumor cells in the tumor stroma (Figure 6E). Strong Ki67 and SMA expression was observed in IHC and IF staining of adjacent ASC tissue slides (Figure 6F and G). IHC staining helped us to recognize cell morphology of the ASC microenvironment more clearly. Unlike ASC tissue, there were indeed few positive areas for Ki67 in IHC and IF staining of PDAC tissue. In PDAC tissues, GFNPs overlapped with some Ki67-positive tumor cells and were extensively located in the cytoplasm of cells adjacent to tumor cells (Figure 7A). Combined with the detailed plot of adjacent tissue section in Figure 7B, CAFs closely surround Ki67-positive tumor cells in PDAC tissue, and GFNPs are distributed across the dense fibroblast layer in the immediate vicinity of the tumor cells. IHC results further verified IF results (Figure 7C and D). GFNPs identified ~35% Ki67-positive tumor cells and~30% SMA-positive CAFs in PDAC (Figure 7E and F). IHC pathology results from patients with NET showed a Ki67 positivity rate of 3% and a diagnosis of NET-G2. Combined with the IF staining results of Ki67 and SMA in NET tissue, it is clear that GFNPs are predominantly distributed in the cytoplasm of all cells (Figure 8A and B). GFNPs fluorescent signals and Ki67-positive islet B cells completely overlapped with surrounding tumor cells (Figure 8A). In the same mechanism as previously described of entry into mouse tumor TME, GFNPs act on vascular endothelial cells and vascular smooth muscle cells in the NET (Figure 8C and D), and then enter into TME. Similarly, GFNPs recognized more than 30% of tumor cells and CAFs of NET sample (Figure 8E and F).
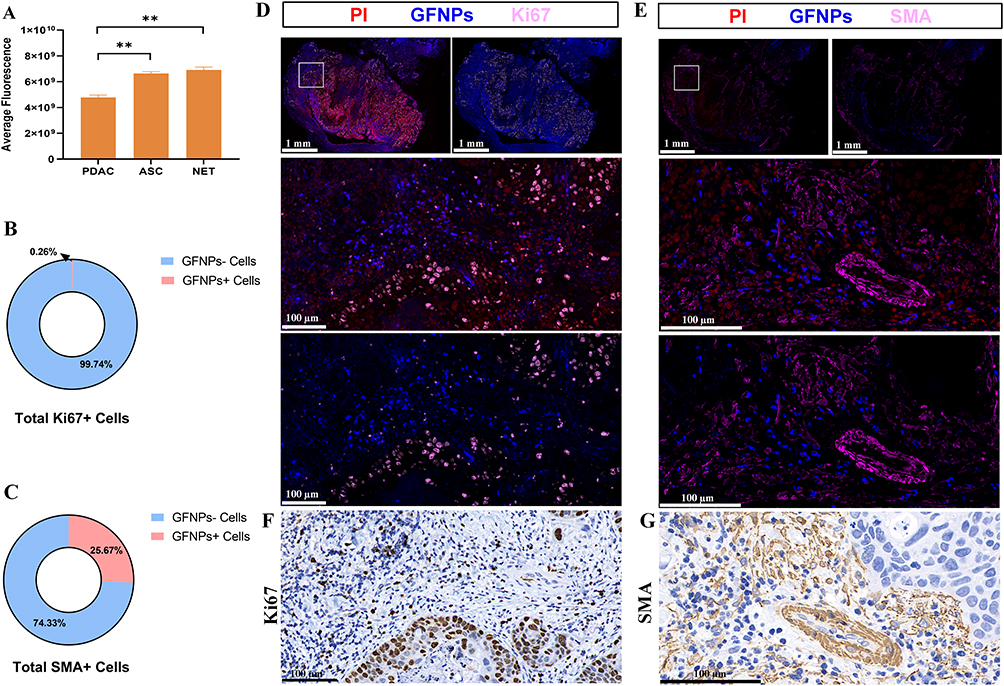 |
Figure 6 Qualitative and semi-quantitative analysis of GFNPs in ASC patient sample. (A) Fluorescence intensity of GFNPs in tumor tissues of different types of human pancreatic cancer samples. (B) Percent positive cell count of Ki67 at ASC patient sample. (C) Percent positive cell count of SMA at ASC patient sample. (D) Panoramic and detailed view of immunofluorescence of ASC patient sample. Scanning scale bar=1 mm. Partial scale bar=100 µm. Blue-GFNPs; red-PI; pink-Ki67. (E) Panoramic and detailed view of immunofluorescence of ASC patient sample. Scanning scale bar=1 mm. Partial scale bar=100 µm. Blue-GFNPs; red-PI; pink-SMA. (F) Detailed view of immunohistochemistry of Ki67 at adjacent locations of ASC patient sample. Scale bar=100 µm. (G) Detailed view of immunohistochemistry of SMA at adjacent locations of ASC patient sample. Scale bar=100 µm. Notes: Data was exhibited as the Mean ± SEM. ** p < 0.01. |
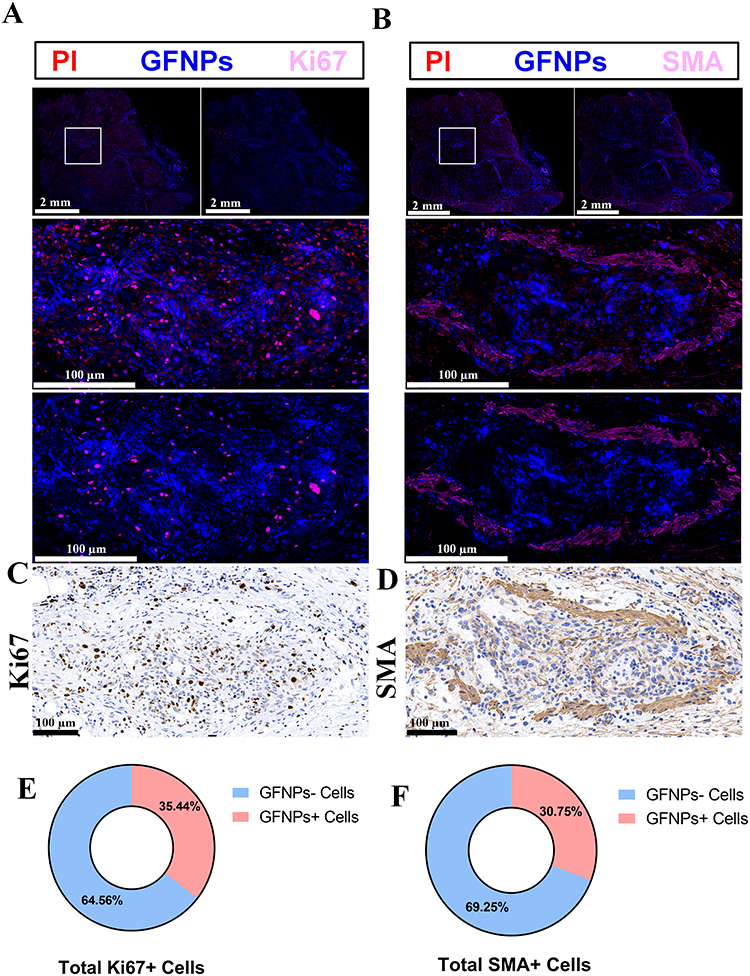 |
Figure 7 Qualitative and semi-quantitative analysis of GFNPs in PDAC patient sample. (A) Panoramic and detailed view of immunofluorescence of PDAC patient sample. Scanning scale bar=2 mm. Partial scale bar=100 µm. Blue-GFNPs; red-PI; pink-Ki67. (B) Panoramic and detailed view of immunofluorescence of PDAC patient sample. Scanning scale bar=2 mm. Partial scale bar=100 µm. Blue-GFNPs; red-PI; pink-SMA. (C) Detailed view of immunohistochemistry of Ki67 at adjacent locations of PDAC patient sample. Scale bar=100 µm. (D) Detailed view of immunohistochemistry of SMA at adjacent locations of PDAC patient sample. Scale bar=100 µm. (E) Percent positive cell count of Ki67 at PDAC patient sample. (F) Percent positive cell count of SMA at PDAC patient sample. Notes: Datas were exhibited as the Mean ± SEM. |
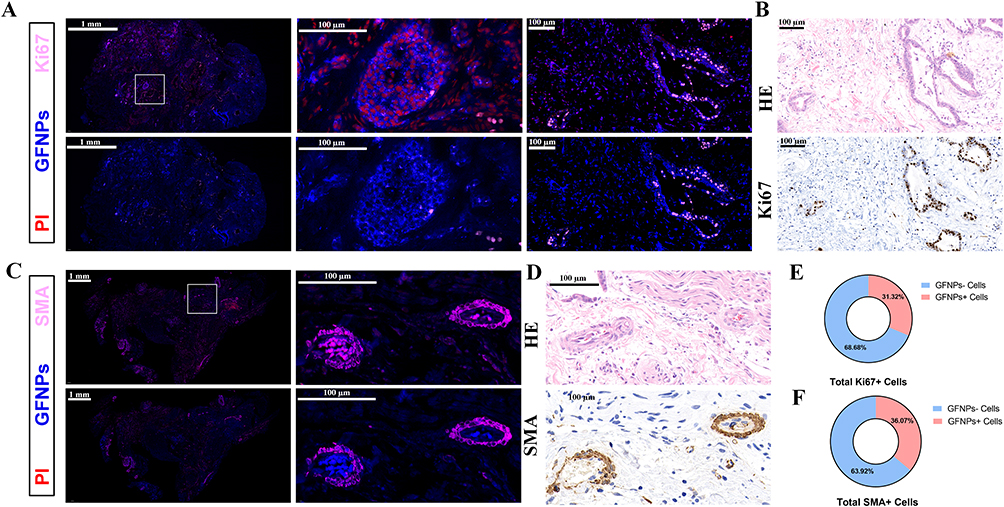 |
Figure 8 Qualitative and semi-quantitative analysis of GFNPs in NET patient sample. (A) Panoramic and detailed view of immunofluorescence of NET patient sample. Scanning scale bar=1 mm. Partial scale bar=100 µm. Blue-GFNPs; red-PI; pink-Ki67. (B) Detailed view of HE (up) and immunohistochemistry (down) of Ki67 at adjacent locations of NET patient sample. Scale bar=100 µm. (C) Panoramic and detailed view of immunofluorescence of NET patient sample. Scanning scale bar=1 mm. Partial scale bar=100 µm. Blue-GFNPs; red-PI; pink-SMA. (D) Detailed view of HE (up) and immunohistochemistry (down) of SMA at adjacent locations of NET patient sample. Scale bar=100 µm. (E) Percent positive cell count of Ki67 at PDAC patient sample. (F) Percent positive cell count of SMA at PDAC patient sample. Note: Datas were exhibited as the Mean ± SEM. |
Synthesis and Tumor Chemo-Target Therapy of Graphene Fluorescent Nanoparticles and Gemcitabine Based on Diversified Targeting Effects
We selected four typical anti-tumor drugs that were modified by GFNPs to obtain nanodrugs. Transmission electron micrographs showed that they all had better dispersion and more homogeneous particle sizes, mainly in the range of 30–50 nm (Figure 9A and B). The UV spectra of the nanomaterials are shown in Figure 9C. These four nanodrugs could enter the tumor cells (Figure 9D). In particular, the nanomedicine consisting of gemcitabine adhered to the tumor cell membrane at 6 h and entered the tumor cell lysosomes at 24 h, which was consistent with the localization characteristics of the nanomaterials after entering the cells (Figure 9E). We simulate GFNPs-GEM access to tumor tissue in vivo by dialysis experiments. We collected the substances for HPLC detection after 30 minutes dialysis in PBS (pH 6.5) on account of molecular weights of GFNPs and GEM were both within 500 Kd, and found that these two substances still existed after dialysis (Figure 9F). Uncombined GFNPs and GEM were dispersed outside the dialysis bag. Next, we used GFNPs-GEM in treating mouse tumor. Animal experiments showed that GFNPs-GEM was highly effective in the treatment of pancreatic cancer, significantly improving the efficacy of GEM alone in less than two weeks of treatment, and improved the survival rate of mice compared to GEM alone, but the difference was not significant (Figure 9G and H).
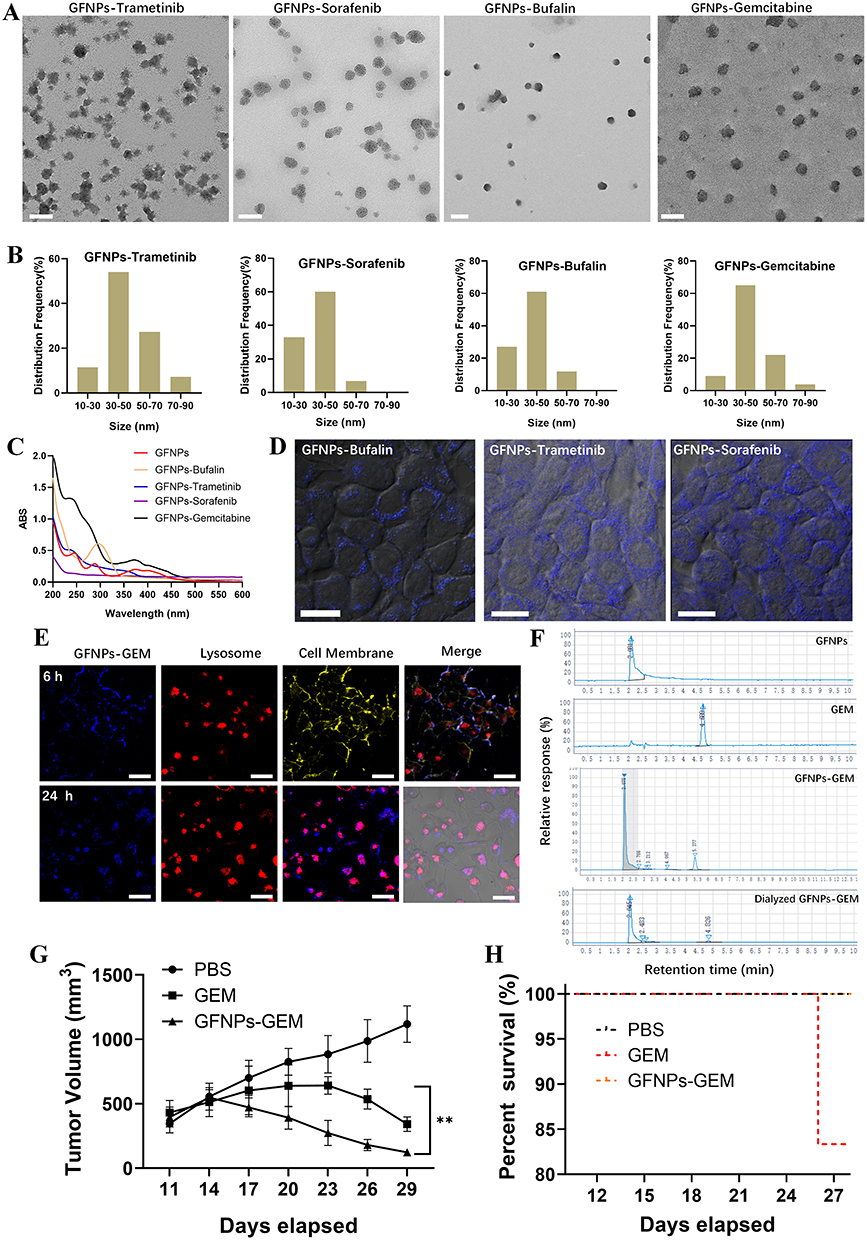 |
Figure 9 Synthesis and tumor chemo-target therapy nanomedicines. (A) TEM of four nanomedicines. Scale bar =100 nm. (B) Histogram of particle size statistics of four nanomedicines. (C) Ultraviolet absorption spectrum of four nanomedicines. (D and E) Laser confocal results of cellular co-localization of four nanomedicines. Scale bar=20 µm. (F) High performance liquid chromatography of GFNPs, GEM, GFNPs-GEM and dialyzed GFNPs-GEM with 500kd. (G) Mice subcutaneous tumor volume during GFNPs-GEM nanomedicine treatment. n=5. (H) Survival curve during GFNPs-GEM nanomedicine treatment. Notes: Data was exhibited as the Mean ± SEM. ** p < 0.01. |
Discussion
Difference of GFNPs fluorescence localization between 2D cells and 3D cell spheroid existed in vitro. In fact, GFNPs cannot entry into 2D cells normally because of negative charge and the background fluorescence intensity is higher than in the cells. GFNPs could entry into partial cell of 3D cell spheroid and the background fluorescence intensity was close to that of cells (Figure S7). Phenomenon in pancreatic tumor tissue is consistent with our previous results in primary liver cancer.30 Small water-soluble nanoparticles with proper surface charge, such as GFNPs, may be targeted to reticuloendothelial cell cells in the liver, spleen, and other organs, and are finally excreted by the kidney.34 All primary tumors originate from tumor cells that act on surrounding normal cells, in which tumor cells can alter the gene expression of normal cells in the same environment.35 The fluorescence signal of GFNPs inside the tumor tissue changed from one point to the surrounding area, similar to the invasion pattern of tumor cells. Tumor volume can act as a surrogate for tumor progression in models with the same number of inoculated tumor cells. The fluorescence signal intensity of GFNPs was proportional to the tumor proliferation cycle (Figure S8). Herein, we hypothesized that such changes in GFNPs fluorescence signals are related to certain cells in the TME of pancreatic cancer, which affect tumor proliferation. Single-cell suspension of PDAC tumors from untreated patients were processed for scRNA-seq to identify the cell populations present in the tissue, which including cancer cells, fibroblasts, ductal cells, etc.36 It has been reported to increase malignancy through interactions with the abundant stroma. CAFs commonly identified by the expression of SMA are a major source of Col1 fibers in tumors and promote tumor progression.37 These are major contributors to PDAC progression through pro-tumor signaling and the generation of fibrosis, accounting for a large proportion of PDAC stroma.38 The amount of SMA-positive area decreased significantly as the tumor grade increased from Grade1 to Grade3.39 Although the concept of differentiation of mouse orthotopic pancreatic cancer has not yet been proposed, we found that the spatial relationship between SMA-positive CAFs in the tumor stroma and tumor cells is related to the cycle of tumor proliferation with IF quantification, which revealed that the proportion of SMA-positive CAFs was reduced by half between 2-weeks of 4-weeks tumor. Studies on 4T1 tumors showed that more than 85% of NPs were delivered to the SMA+ stroma.40 GFNPs were localized in the nuclei of both SMA-expressing fibroblasts and adjacent tumor cells, demonstrating outstanding dual-targeting ability, which can simultaneously target cancer cells and CAFs without complex targeting ligand modifications.41 The therapeutic bottleneck in PDAC arises from the dense stroma. Smaller NPs are better candidates for future therapeutics because of their deeper tumor penetration.42,43 Nanoparticles can be efficiently and rapidly internalized by macrophages and vascular smooth muscle cells via i.v. injection,44 then enter into tumors through transendothelial pathways, which are the dominant mechanisms of nanoparticle extravasation into tumors.45 PDAC includes abundant ducts and tumor neovascularization, and GFNPs possess this size advantage and the ability to specifically mark the medial side of the catheter lumen, showing an adjacent position to vimentin and an out-adjacent position to SMA. In comparison with other NPs showed only a few localized in the vicinity of tumor vessels, GFNPs are not only abundantly deposited nearby tumor vessels but also penetrate tumor tissues to reach those regions far away from tumor vessels.
Current studies on the localization and quantification of individual tumor cells have focused on circulating tumor cells, and researchers have proposed tumor treatments based on the capture of tumor cells.46,47 Tumor cells are labeled in advance with bioluminescence and fluorescence when studying the interaction between tumor cells and drugs; however, such treatments have many problems. Wang reported the design and synthesis of a trifunctional probe for observing and counting cancer cells using both fluorescence imaging and inductively coupled plasma mass spectrometry (ICP-MS); however, the components were also very complex.48 We expected to find a relationship between the initial tumor cell number and the TME fluorescence signal after internalization of GFNPs. Based on simple synthetic GFNPs and excellent performance in actively targeting tumor cells, we found that the size and weight of tumors formed by different numbers of tumor cells did not differ significantly. Therefore, in this case, we cannot directly determine the malignancy of the tumor, but the fluorescent signal of the GFNPs showed a significant decrease in the orthotopic pancreatic model. It is the tumorigenesis stage rather than the tumor volume that affects the targeting efficiency of nanoparticles. Li found most tumor-targeted nanoparticles were concentrated in tumor-associated macrophages in tumor blood vessels, the density of which decreases with tumor progression. These results revealed that the tumor-targeting efficiency of tumor-targeted nanoparticles decreases with tumor progression.49 We hypothesized that there is a correlation between the initial tumor number and tumor-associated macrophages in the tumor vasculature. The targeting efficiency of GFNPs negatively correlated with time after tumor inoculation in orthotopic pancreatic tumors. However, we did not reach the same conclusion in the subcutaneous pancreatic tumor model. IHC studies using an antibody against proliferating cell nuclear antigen revealed that small subcutaneous tumors contained a larger fraction of proliferating cells than large tumors.50 Tumors growing in the liver had significantly lower vessel densities, especially in the center, coincident with central necrosis, than subcutaneous tumors. However, the macromolecular vascular permeability in orthotopic liver tumors is significantly higher than that in subcutaneous tumors.51 In contrast, orthotopic tumors metastasize more readily than subcutaneous tumors because of the differences in the types and ratios of various cell types between the orthotopic pancreatic model and subcutaneous tumors, particularly CAFs. IHC of SMA in tumor sections and immunoblot analysis of SMA in tumor extracts from orthotopic and subcutaneous tumors revealed that tumors identified a higher number of SMA-CAFs in orthotopic tumors.37 The fluorescence intensity of GFNPs in subcutaneous tumors formed by tD-Tomato-BXPC-3 cells did not show a single linear variation, rather, the strongest fluorescence signal was observed in subcutaneous tumors formed by specific cell counts. GFNPs co-localized with the nuclei of tumor cells in areas where tD-tomato signals were presented.
In addition to the primary focus, tumor cells also metastasize to the other organs along the transvascular or lymphatic route. Nearly all deaths caused by solid cancers occur as a result of metastasis and the formation of secondary tumors in distant organs such as the lungs, liver, brain and bone.52 Liver metastases are commonly detected in a range of malignancies, including colorectal, pancreatic, melanoma, lung, and breast cancer. Interactions between tumor cells and the tumor microenvironment play an important role in engraftment, survival, and progression of metastases. Various cells are implicated in promoting and sustaining metastases in the liver.53,54 Tumor foci in the liver have a tortuous vascular architecture, heterogeneous blood flow, significantly lower vascular density, and significantly higher vascular permeability than normal liver tissues.51 We also observed that the fluorescence of GFNPs was significantly higher in the tumor nodules of the liver than in the liver tissue without tumor nodules, and the increased fluorescence in the lung and pancreatic tissues indicated that the tumor cells also invaded adjacent lung and pancreatic tissues. CDK2 overexpression facilitates lymph node metastasis. The overexpression of cyclin E and CDK2 may promote early cancer progression. Increased cyclin E protein levels are related to elevated CDK2 levels, which is further linked to higher Ki67 levels.53 In cancerous liver sections, the fluorescence signals of the tumor nucleus antibody protein Ki67 and the nuclear PI almost completely overlapped. The GFNPs and the tumor nucleus antibody protein Ki67 showed localized signal overlap, and the SMA-CAFs were distributed in the vicinity of the double-positive areas of GFNPs and Ki67. The same phenomenon has also been observed in invaded pancreatic tissues. However, GFNPs did not colocalize with metastatic pancreatic SMA-CAFs, in contrast to the highly metastasized cancerous liver.55
GFNPs fluorescently label clinical pancreatic cancer tumors with varying degrees of malignancy and intensities.56,57 PDAC is an invasive epithelioid tumor, occurring in 80–90% of pancreatic cancers, usually accompanied by a ductal lumen, in which the tumor cells are often cubic and a marked pro-fibro-mesenchymal reaction is typical.58,59 As a highly fibrotic pancreatic tumor, GFNPs can label CAFs in the PDAC mesenchyme and proportion of tumor cells.60 NETs are a type of pancreatic tumor that is predominantly characterized by neuroendocrine differentiation. These are less common, accounting for 1–2% of all pancreatic tumors, and are also called islet cell tumors. They are well-differentiated, with well-defined single nodules showing dense fibrosis.61,62 RBC can absorb DNA-bearing tumorigenic mutations from cancer cell lines, but require cell-to-cell contact. Researchers have confirmed mature RBC acquired DNA from lung cancer cell lines in vitro and contained long DNA fragments with special characteristic63 Based on the specific binding of GFNPs to DNA histones, GFNPs can then label mature erythrocytes in the NET tumor microenvironment. GFNPs demonstrate very good fluorescent labeling of cells in the tumor microenvironment of NET, which can identify nearly all types of cells in the TME and co-localize fluorescently with molecules secreted by pancreatic islet β cells.64 ASC is an epithelial malignancy of the pancreas that accounts for approximately 2% of all pancreatic malignancies with ductal and squamous epithelial differentiation.65 ASC tumors often contain necrotic islets, which is a known clinical feature of ASC. This could have been fluorescently labeled specifically by GFNPs. In addition, it exhibited a strong fluorescent signal inside cells surrounding Ki67-positive tumor cells, but the fluorescent signal did not overlap with CAFs in the stroma. In summary, GFNPs have the strongest fluorescent signal for NET-labeling but are less specific, with better specificity for fluorescent labeling of the other two malignancies.
Trametinib, sorafenib and bufalin are all clinical oncology drugs with bad target therapy effect. Given its excellent stability and fluorescent targeted labeling of tumor tissue, we explored the possibility of binding GFNPs to chemical agents. We have confirmed that GFNPs could combined with three kinds of drugs through self-assembly to form nanomedicine below 40nm particle size. Nanomaterials between 20 nm and 100nm can penetrate deep into tumors and are not easily cleared during blood circulation.66 Gemcitabine is the gold-standard drug for treating pancreatic cancer. However, its anticancer efficacy is severely limited by instability and poor cellular uptake.67 To enhance the clinical efficacy of GEM, we constructed a novel nanodrug delivery system based on the dual-targeting performance of GFNPs against pancreatic cancer tumor cells and CAFs. GFNP-GEM nanoformulation exhibited uniform particle size about ~30 nm. With the features of a small size and stable formulation, the obtained GEM nanoformulation could effectively accumulate at the tumor site and rapidly uptake into cells. Dialysis products of simulating acidic TME confirmed the stability of GFNPs-GEM before released into the TME. GFNPs-GEM nanoformulation displayed a more potent anticancer activity than free GEM in vivo. Moreover, the nanodrug displayed significantly reduced adverse effects and satisfactory biocompatibility. Benefiting from the advantageous features of the dual-targeting performance of GFNPs against tumor cells and CAFs and nanotechnology-based drug delivery, this GEM nanosystem constitutes a promising therapeutic candidate for pancreatic cancer treatment. Our study also highlighted the potential use of the dual-targeting performance of GFNPs in the TME to improve drug efficacy and reduce drug toxicity.
Conclusion
We confirmed the target-specificity of GFNPs in tumor tissues, specifically, the longer the tumor progressed, the higher the targeting efficiency of GFNPs, and the stronger the fluorescence exhibited by the tumor tissue. Targeting effect of GFNPs in tumor tissues was significantly linearly related to the tumor progression time and tumor size. Furthermore, GFNPs could recognize metastatic lesions in mice. All these targeting effects are related to the specific labeling of GFNPs with tumor cells and CAFs, which are the two most abundant cell types in the pancreatic cancer tumor microenvironment. The evidence we have provided indicates that GFNPs have the potential for clinical diagnosis, and we have also conducted some related research on the emerging in-surgery rapid pathological diagnosis technology. Results showed that GFNPs can achieve rapid diagnosis of three typical clinical pancreatic tumors. Although the specific targeting mechanisms cannot match exactly with what was discovered in the mouse experiments, which also provide basis for future mechanistic studies of pancreatic cancer in clinical settings. Currently, many environmentally responsive nanoparticles and multi-level nanoparticles can deliver drugs to tumor sites, but their synthesis process is complex. Unlike water-soluble nanomaterials with simpler synthesis processes, GFNPs can not only effectively diagnose, but also combined clinical chemotherapy drugs in various ways. Compared to traditional nanomaterials delivery systems, the nanomedicines formed by GFNPs are smaller in size, which make it easier and more efficient to deliver chemotherapy drugs to highly fibrotic pancreatic tumor tissues. We believe that there is still a greater scope for exploration on the application and mechanism of GFNPs in pancreatic cancer therapy.
Abbreviations
IF, Immunofluorescence; IHC, Immunohistochemistry; HE, Hematoxylin-eosin; CAFs, Cancer-associated fibroblasts; GFNPs, Graphene fluorescent nanoparticles; SMA, Alpha-smooth muscle actin; Vim, Vimentin; PI, Propidium Iodide; TME, Tumor microenvironment; GEM, Gemcitabine; PDAC, Pancreatic ductal adenocarcinoma; pNET, Pancreatic neuroendocrine tumor; ASC, Adenosquamous carcinoma; RT, Radiotherapy; ECM, Extracellular matrix; EPR, Enhanced permeability and retention; FDA, Food and Drug Administration; NPs, Nanoparticles; IVIS, In vivo imaging system; SPF, Specified Pathogen Free; ICP-MS, Inductively coupled plasma mass spectrometry; TEM, Transmission electron microscopy; FTIR, Fourier transform infrared spectroscopy; QYs, quantum yields; PL, photoluminescence; PLE, Photoluminescence excitation; MALDI-TOF, matrix-assisted laser desorption tandem time-of-flight mass spectrometry. RBC, Red Blood Cells. HPLC, High performance liquid chromatography.
Data Sharing Statement
All data and materials are available.
Ethics Approval and Consent to Participate
All experimental procedures were carried out according to the ARRIVE Guidelines and the UK Animals (Scientific Procedures) Act, 1986, and associated with the National Research Council’s Guide for the Care and Use of Laboratory Animals. In addition, we tested the diagnostic effect of GFNPs in human pancreatic cancer samples, following the principles of the Code of Ethics of the World Medical Association (Helsinki Declaration) for experiments involving humans, and the study was approved by the ethics committee of Changhai Hospital (CHEC2022-018).
Consent for Publication
All authors consent for publication.
Acknowledgments
The authors thank Shuang Nie and Tianyu Zheng for their assistance with this study. This paper has been uploaded to ResearchSquare as a preprint: https://www.researchsquare.com/article/rs-2445059/v1.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Sci-Tech Innovation 2030 Brain Science and Brain-Like Intelligence Technology Project [grant number 2022ZD0208100] and the Naval Medical University Youth Start-up Fund [grant number 2022QN016].
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported in this study.
References
1. Gentiluomo M, Canzian F, Nicolini A, Gemignani F, Landi S. Germline genetic variability in pancreatic cancer risk and prognosis. Semin Cancer Biol. 2022;79. doi:10.1016/j.semcancer.2020.08.003
2. Park W, Chawla A, O’Reilly EM. O’Reilly E M.Pancreatic Cancer: a Review. JAMA. 2021;326(9):851–862. doi:10.1001/jama.2021.13027
3. Oba A, Croce C, Hosokawa P, et al. Prognosis based definition of resectability in pancreatic cancer a road map to new guidelines. Ann Surg. 2022;275(1):175–181. doi:10.1097/sla.0000000000003859
4. Mokdad AA, Minter RM, Zhu H, et al. Neoadjuvant therapy followed by resection versus upfront resection for resectable pancreatic cancer: a propensity score matched analysis. J Clin Oncol. 2017;35(5):515–522. doi:10.1200/jco.2016.68.5081
5. Khorana AA, Tullio K, Elson P, et al. Time to initial cancer treatment in the United States and association with survival over time: an observational study. PLoS One. 2019;14(3). doi:10.1371/journal.pone.0213209
6. Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17(8):487–505. doi:10.1038/s41575-020-0300-1
7. Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–860. doi:10.1038/nm.4123
8. Wang RK, Chen JR, Wang W, et al. CD40L-armed oncolytic herpes simplex virus suppresses pancreatic ductal adenocarcinoma by facilitating the tumor microenvironment favorable to cytotoxic T cell response in the syngeneic mouse model. J Immunother Cancer. 2022;10(1):e003809. doi:10.1136/jitc-2021-003809
9. Encarnacion-Rosado J, Kimmelman AC. Kimmelman A C.Harnessing metabolic dependencies in pancreatic cancers. Nat Rev Gastroenterol Hepatol. 2021;18(7):482–492. doi:10.1038/s41575-021-00431-7
10. Li YX, Zhu XX, Wu X, et al. ACLP promotes activation of cancer-associated fibroblasts and tumor metastasis via ACLP-PPAR gamma-ACLP feedback loop in pancreatic cancer. Cancer Lett. 2022;544:215802. doi:10.1016/j.canlet.2022.215802
11. Indolfi L, Ligorio M, Ting DT, et al. A tunable delivery platform to provide local chemotherapy for pancreatic ductal adenocarcinoma. Biomaterials. 2016;93:71–82. doi:10.1016/j.biomaterials.2016.03.044
12. Wu LM, Zhang F, Chen XN, et al. Self-assembled gemcitabine prodrug nanoparticles show enhanced efficacy against patient-derived pancreatic ductal adenocarcinoma. ACS Appl Mater Interfaces. 2020;12(3):3327–3340. doi:10.1021/acsami.9b16209
13. Lu JQ, Liu XS, Liao YP, et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat Commu. 2017;8:1.
14. Tong QS, Miao WM, Huang H, et al. ATumor-penetrating nanomedicine improves the chemoimmunotherapy of pancreatic cancer. Small. 2021;17(29):e2101208. doi:10.1002/smll.202101208
15. Ji Y, Liu XS, Li J, et al. Use of ratiometrically designed nanocarrier targeting CDK4/6 and autophagy pathways for effective pancreatic cancer treatment. Nat Commu. 2020;11(1). doi:10.1038/s41467-020-17996-7
16. Dong XY, Chu DF, Wang Z. Leukocyte-mediated delivery of nanotherapeutics in inflammatory and tumor sites. Theranostics. 2017;7(3):751–763. doi:10.7150/thno.18069
17. Shan XZ, Li SM, Sun BJ, et al. Ferroptosis-driven nanotherapeutics for cancer treatment. J Control Release. 2020;319:322–332. doi:10.1016/j.jconrel.2020.01.008
18. Alhussan A, Palmerley N, Smazynski J, et al. Potential of gold nanoparticles in current radiotherapy using a co-culture model of cancer cells and cancer associated fibroblasts. Cancers. 2022;14(15):3586. doi:10.3390/cancers14153586
19. Liu JY, Ren LW, Li S, et al. The biology, function, and applications of exosomes in cancer. Acta Pharm Sin B. 2021;11(9):2783–2797. doi:10.1016/j.apsb.2021.01.001
20. Zhao J, Wang HM, Hsiao CH, et al. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials. 2018:159(215–228. doi:10.1016/j.biomaterials.2018.01.014
21. Peng W, Zhang Y, Zhu R, Mechref Y. Mechref Y.Comparative membrane proteomics analyses of breast cancer cell lines to understand the molecular mechanism of breast cancer brain metastasis. Electrophoresis. 2017;38(17):2124–2134. doi:10.1002/elps.201700027
22. Zhang D, Ye Z, Wei L, Luo H, Xiao L. Cell membrane-coated porphyrin Metal–organic frameworks for cancer cell targeting and O 2 -evolving photodynamic therapy. ACS Appl Mater Interfaces. 2019;11(43):39594–39602. doi:10.1021/acsami.9b14084
23. Harris JC, Scully MA, Day ES. Cancer cell membrane-coated nanoparticles for cancer management. Cancers. 2019;11(12):1836. doi:10.3390/cancers11121836
24. Li J, Zhen X, Lyu Y, Jiang Y, Huang J, Pu K. Cell membrane coated semiconducting polymer nanoparticles for enhanced multimodal cancer phototheranostics. ACS Nano. 2018;12(8):8520–8530. doi:10.1021/acsnano.8b04066
25. Zhang XH, Detering L, Sultan D, et al. CC Chemokine receptor 2-targeting copper nanoparticles for positron emission tomography-guided delivery of gemcitabine for pancreatic ductal adenocarcinoma. ACS Nano. 2021;15(1):1186–1198. doi:10.1021/acsnano.0c08185
26. Feng JX, Xu MJ, Wang JH, et al. Sequential delivery of nanoformulated alpha-mangostin and triptolide overcomes permeation obstacles and improves therapeutic effects in pancreatic cancer. Biomaterials. 2020;241:119907. doi:10.1016/j.biomaterials.2020.119907
27. Mardhian DF, Storm G, Bansal R, Prakash J. Nano-targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo. J Control Release. 2018;290(1–10):1–10. doi:10.1016/j.jconrel.2018.09.031
28. Zhao T, Zhang R, He Q, Zhou H, Song X. Partial ligand shielding nanoparticles improve pancreatic ductal adenocarcinoma treatment via a multifunctional paradigm for tumor stroma reprogramming. Acta Biomater. 2022;145:122–134. doi:10.1016/j.actbio.2022.03.050
29. Yu QW, Qiu Y, Li JP, et al. Targeting cancer-associated fibroblasts by dual-responsive lipid-albumin nanoparticles to enhance drug perfusion for pancreatic tumor therapy. J Control Release. 2020;321:564–575. doi:10.1016/j.jconrel.2020.02.040
30. Lei ZD, Ding L, Yao CJ, et al. A highly efficient tumor-targeting nanoprobe with a novel cell membrane permeability mechanism. Adv Mater. 2019;31(12). doi:10.1002/adma.201807456
31. He JY, Li CC, Ding L, et al. Tumor targeting strategies of smart fluorescent nanoparticles and their applications in cancer diagnosis and treatment. Adv Mater. 2019;31(40). doi:10.1002/adma.201902409
32. Zhang T, Ren Y, Yang P, Wang J, Zhou H. Zhou H.Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022;13(10). doi:10.1038/s41419-022-05351-1
33. Zhao W, Yang S, Li C, et al. Amphiphilic dendritic nanomicelle-mediated delivery of gemcitabine for enhancing the specificity and effectiveness. Int J Nanomed. 2022;17:3239–3249. doi:10.2147/ijn.s371775
34. Riviere JE. Riviere J.Pharmacokinetics of nanomaterials: an overview of carbon nanotubes, fullerenes and quantum dots. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1(1):26–34. doi:10.1002/wnan.24
35. Seidel S, Garvalov B. Acker T.Isolation and culture of primary glioblastoma cells from human tumor specimens. Methods Mol Biol. 2015;1235:263–275. doi:10.1007/978-1-4939-1785-3_19
36. Moncada R, Barkley D, Wagner F, et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol. 2020;38(3):333–342. doi:10.1038/s41587-019-0392-8
37. Penet M, Kakkad S, Pathak A, et al. Structure and function of a prostate cancer dissemination-permissive extracellular matrix. Clin Cancer Res. 2017;23(9):2245–2254. doi:10.1158/1078-0432.ccr-16-1516
38. Sharbeen G, McCarroll J, Akerman A, et al. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma determine response to SLC7A11 inhibition. Cancer Res. 2021;81(13):3461–3479. doi:10.1158/0008-5472.can-20-2496
39. Shinkawa T, Ohuchida K, Mochida Y, Sakihama K, Iwamoto C. Abe T, et al.Subtypes in pancreatic ductal adenocarcinoma based on niche factor dependency show distinct drug treatment responses. J Exp Clin Cancer Res. 2022;41(1):89. doi:10.1186/s13046-022-02301-9
40. Murakami M, Ernsting M, Undzys E, Holwell N, Foltz W, Li S-D. Docetaxel conjugate nanoparticles that target α-smooth muscle actin-expressing stromal cells suppress breast cancer metastasis. Cancer Res. 2013;73(15):4862–4871. doi:10.1158/0008-5472.can-13-0062
41. Zang S, Huang K, Li J, et al. Metabolic reprogramming by dual-targeting biomimetic nanoparticles for enhanced tumor chemo-immunotherapy. Acta Biomater. 2022;148:181–193. doi:10.1016/j.actbio.2022.05.045
42. Yang C, Bromma K, Chithrani D. Peptide mediated in vivo tumor targeting of nanoparticles through optimization in single and multilayer in vitro cell models. Cancers. 2018;10(3):84. doi:10.3390/cancers10030084
43. Yohan D, Cruje C, Lu X, Chithrani DB. Size-dependent gold nanoparticle interaction at nano-micro interface using both monolayer and multilayer (Tissue-Like) cell models. Nanomicro Lett. 2016;8(1):44–53. doi:10.1007/s40820-015-0060-6
44. Wang Y, Li L, Zhao W, Dou Y, An H. Targeted therapy of atherosclerosis by a broad-spectrum reactive oxygen species scavenging nanoparticle with intrinsic anti-inflammatory activity. ACS Nano. 2018;12(9):8943–8960. doi:10.1021/acsnano.8b02037
45. Sindhwani S, Syed AM, Ngai J, et al. The entry of nanoparticles into solid tumours. Nature Mater. 2020;19(5):566–575. doi:10.1038/s41563-019-0566-2
46. Song Y, Shi Y, Huang M, et al. Bioinspired engineering of a multivalent aptamer-functionalized nanointerface to enhance the capture and release of circulating tumor cells. Angew Chem Int Ed Engl. 2019;58(8):2236–2240. doi:10.1002/anie.201809337
47. Kedarisetti P, Bouvet V, Shi W, et al. Enrichment and ratiometric detection of circulating tumor cells using PSMA- and folate receptor-targeted magnetic and surface-enhanced Raman scattering nanoparticles. Biomed Opt Express. 2020;11(11):6211–6230. doi:10.1364/boe.410527
48. Zhang Z, Luo Q, Yan X, et al. Integrin-targeted trifunctional probe for cancer cells: a “seeing and counting” approach. Anal Chem. 2012;84(21):8946–8951. doi:10.1021/ac302029w
49. Zhang J, Zhang W, Yang M, et al. Passive cancer targeting with a viral nanoparticle depends on the stage of tumorigenesis. Nanoscale. 2021;13(26):11334–11342. doi:10.1039/d1nr01619a
50. Yoon S, Fidler I, Beltran P, Bucana C, Wang Y, Fan D. Fan D.Intratumoral heterogeneity for and epigenetic modulation of mdr-1 expression in murine melanoma. Melanoma Res. 1997;7(4):275–287. doi:10.1097/00008390-199708000-00002
51. Fukumura D, Yuan F, Monsky W, Chen Y, Jain RK. Jain R.Effect of host microenvironment on the microcirculation of human colon adenocarcinoma. Am J Pathol. 1997;151(3):679–688.
52. Eckhardt B, Francis P, Parker B, Anderson RL. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat Rev Drug Discov. 2012;11(6):479–497. doi:10.1038/nrd2372
53. Tsilimigras D, Brodt P, Clavien P, et al. Liver metastases. Nat Rev Dis Primers. 2021;7(1):27. doi:10.1038/s41572-021-00261-6
54. Lee J, Stone M, Porrett P, et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature. 2019;567(7747):249–252. doi:10.1038/s41586-019-1004-y
55. Cheng J, Gong Z. Zhu B.Correlation analysis of alpha -SMA~+ CAFs with clinic pathological characteristics and tumor-infiltrated immune cells in patients with lung adenocarcinoma. Immunol J. 2015;31(3):230–233.
56. Bhayana R, Baliyan V, Kordbacheh H, Kambadakone A. Hepatobiliary phase enhancement of liver metastases on gadoxetic acid MRI: assessment of frequency and patterns. Eur Radiol. 2021;31(3):1359–1366. doi:10.1007/s00330-020-07228-3
57. Gupta P, Rana P, Marodia Y, et al. Contrast-enhanced ultrasound of solid pancreatic head lesions: a prospective study. Eur Radiol. 2022;32(10):6668–6677. doi:10.1007/s00330-022-08854-9
58. Ligorio M, Sil S, Malagon-Lopez J, et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell. 2019;178(1):160–175. doi:10.1016/j.cell.2019.05.012
59. Carstens JL, de Sampaio PC, Yang DL, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. 2017;8(27):15095. doi:10.1038/ncomms15095
60. Takesue S, Ohuchida K, Shinkawa T, et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer-associated fibroblasts. Int J Oncol. 2020;56(2):596–605. doi:10.3892/ijo.2019.4951
61. Xue Y, Reid M, Pehlivanoglu B, et al. Morphologic variants of pancreatic neuroendocrine tumors: clinicopathologic analysis and prognostic stratification. Endocr Pathol. 2020;31(3):239–253. doi:10.1007/s12022-020-09628-z
62. Bulloni M, Sandrini G, Stacchiotti I, et al. Automated analysis of proliferating cells spatial organisation predicts prognosis in lung neuroendocrine neoplasms. Cancers. 2021;13(19):4875. doi:10.3390/cancers13194875
63. Liang N, Jiao Z, Zhang C, et al. Mature red blood cells contain long DNA fragments and could acquire DNA from lung cancer tissue. Adv. Sci. 2023;10(7):1. doi:10.1002/advs.202206361
64. Chu X, Gao X, Jansson L, Quach M, Skogseid B, Barbu A. Multiple microvascular alterations in pancreatic islets and neuroendocrine tumors of a Men1 mouse model. Am J Pathol. 2013;182(6):2355–2367. doi:10.1016/j.ajpath.2013.02.023
65. Närhi K, Nagaraj A, Parri E, et al. Spatial aspects of oncogenic signalling determine the response to combination therapy in slice explants from Kras-driven lung tumours. J Pathol. 2018;245(1):101–113. doi:10.1002/path.5059
66. Sun Q, Zhou Z, Qiu N, Shen Y. Rational design of cancer nanomedicine: nanoproperty integration and synchronization. Adv Mater. 2017;29(14). doi:10.1002/adma.201606628
67. Han H, Hou Y, Chen X, et al. Metformin-induced stromal depletion to enhance the penetration of gemcitabine-loaded magnetic nanoparticles for pancreatic cancer targeted therapy. J Am Chem Soc. 2020;142(10):4944–4954. doi:10.1021/jacs.0c00650
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.