")
Back to Journals » Journal of Inflammation Research » Volume 17
Dissecting Innate and Adaptive Immunity in Inflammatory Bowel Disease: Immune Compartmentalization, Microbiota Crosstalk, and Emerging Therapies
Authors Yue N, Hu P, Tian C, Kong C, Zhao H, Zhang Y , Yao J, Wei Y, Li D , Wang L
Received 18 August 2024
Accepted for publication 12 November 2024
Published 29 November 2024 Volume 2024:17 Pages 9987—10014
DOI https://doi.org/10.2147/JIR.S492079
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Ningning Yue,1,* Peng Hu,2,* Chengmei Tian,3 Chen Kong,1 Hailan Zhao,1 Yuan Zhang,4 Jun Yao,1 Yuqi Wei,5 Defeng Li,1 Lisheng Wang1
1Department of Gastroenterology, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, People’s Republic of China; 2School of Health Science and Engineering, University of Shanghai for Science and Technology, Shanghai, People’s Republic of China; 3Department of Emergency, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, People’s Republic of China; 4Department of Medical Administration, Huizhou Institute of Occupational Diseases Control and Prevention, Huizhou, People’s Republic of China; 5Department of Rehabilitation, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Defeng Li; Lisheng Wang, Department of Gastroenterology, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University; the First Affiliated Hospital, Southern University of Science and Technology), No.1017, Dongmen North Road, Luohu District, Shenzhen, 518020, People’s Republic of China, Tel +86 755 25533018, Email [email protected]; [email protected]
Abstract: The intestinal immune system is the largest immune organ in the human body. Excessive immune response to intestinal cavity induced by harmful stimuli including pathogens, foreign substances and food antigens is an important cause of inflammatory diseases such as celiac disease and inflammatory bowel disease (IBD). Although great progress has been made in the treatment of IBD by some immune-related biotherapeutic products, yet a considerable proportion of IBD patients remain unresponsive or immune tolerant to immunotherapeutic strategy. Therefore, it is necessary to further understand the mechanism of immune cell populations involved in enteritis, including dendritic cells, macrophages and natural lymphocytes, in the steady-state immune tolerance of IBD, in order to find effective IBD therapy. In this review, we discussed the important role of innate and adaptive immunity in the development of IBD. And the relationship between intestinal immune system disorders and microflora crosstalk were also presented. We also focus on the new findings in the field of T cell immunity, which might identify novel cytokines, chemokines or anti-cytokine antibodies as new approaches for the treatment of IBD.
Keywords: ulcerative colitis, Crohn’s disease, inflammatory bowel disease, immunology, therapy
Graphical Abstract:

Introduction
Inflammatory bowel disease (IBD), encompassing Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic relapsing inflammatory disorder of the gastrointestinal (GI) tract. The condition is characterized pathologically by intestinal inflammation and epithelial damage.1 Notably, the prevalence of IBD exceeds 0.3% in Western nations and is progressively rising in newly industrialized countries with their increasingly Westernized societies.2 UC primarily involves the rectum, affecting part of the colon or the entire colon in an inverted continuous pattern. In contrast, CD typically affects the ileum and colon, with intermittent involvement of other GI tract regions. The predominant clinical symptoms for IBD include recurrent abdominal pain, diarrhea, and rectal bleeding. In certain cases, extraintestinal manifestations such as uveitis, erythema nodosum, and primary sclerosing cholangitis can also occur in conjunction with the aforementioned symptoms.3,4 IBD necessitates lifelong management to prevent disease deterioration, along with close collaboration between general practitioners and specialists. Currently, the medical interventions utilized for IBD patients primarily consist of 5-aminosalicylates, glucocorticoids, immunosuppressive agents, and biologics.5 Although favorable therapeutic effects of these drugs are observed in certain patients, long-term outcomes for many remain suboptimal. Additionally, they are often associated with significant adverse effects, including increased risk of infection, drug tolerance, and other immune-related complications.6
The precise mechanisms underlying the pathogenesis of IBD remain elusive; however, an increasing body of evidence suggests that IBD is the result of an inappropriate immune response to intestinal microorganisms in genetically predisposed hosts. However, the intricate mechanisms underlying gene-environment-immune interactions remain underexplored, particularly in relation to accurately predicting and preventing individualized IBD, identifying at-risk individuals, and developing more targeted therapies. The GI mucosa is constantly exposed to a vast array of microbial and food antigens. Intestinal commensal bacteria fulfill numerous vital physiological functions in the host, including digestion and metabolism of non-absorbable food components, vitamin production, regulation of the epithelial barrier, and development of the intestinal immune system.7 However, the intestinal lumen also provides the access to pathogenic microorganisms responsible for tissue damage. The intestinal immune system thus undertakes a very delicate immunomodulatory role of maintaining the ability to mount a rapid and effective immune response against invading pathogens while tolerating commensal microorganisms and harmless dietary components. A disruption in this dynamic balance predisposes the host to developing IBD. Research indicates that both innate and adaptive immunity play an important role in the pathophysiogenesis of IBD.8 Intestinal innate immunity, a non-specific immune response, constitutes the first line of protection against pathogenic microorganisms. It is primarily comprised of intestinal epithelial cells (IECs), Paneth cells, macrophages, dendritic cells (DCs), and innate lymphoid cells (ILCs), which respond initially and rapidly to microorganisms by recognizing pattern recognition receptors (PRRs).9 In contrast to innate immunity, intestinal adaptive immunity is highly specific and immune-memorizing, in which T cells are the primary participants.10 In general, CD is mediated by T helper (Th) 1 cells whereas UC is mediated by Th2 cells.11 However, the precise mechanisms by which interactions between the innate and adaptive immune systems trigger and regulate inflammatory responses in different IBD subtypes, such as CD and UC, remain poorly understood. Moreover, the molecular pathways driving immune dysregulation and the transition from protective immunity to pathological states that cause tissue damage during disease progression continue to pose significant challenges in research. All in all, the etiology of IBD is complex, and the exact functions of the intestinal immune system in this disease context remain unclear. This uncertainty contributes to the lack of response to immunotherapeutic strategies in a significant proportion of IBD patients and highlights gaps in our understanding of disease pathology.12 Therefore, a thorough investigation of the interactions between intestinal immune cells at various stages of IBD, along with strategies to restore the microbial-immune balance, will be crucial for future research and the development of innovative therapies.
In this review, we detail the pivotal roles of intestinal innate and adaptive immunity in the pathogenesis of IBD and their interactions with gut microbes. Furthermore, we highlight recent advancements in immunomodulators, which have undoubtedly opened new possibilities for the treatment of IBD.
Intestinal Immune Compartmentalization and Immune Cell Composition
The intestine represents the largest and most complicated immune organ in the body and is constantly exposed to foreign antigens and potential immune stimuli (Figure 1). The gut is divided into multiple anatomically and functionally diverse sections, each exposed to different environmental stresses.13 Continuous crosstalk between the immune system and outside environmental signals is essential for maintaining local tissue homeostasis. However, any changes to these signals can disturb the makeup and function of the immune cells, thereby increasing the susceptibility to IBD. Therefore, understanding the constituents and roles of the innate and adaptive immune responses in distinct regions of the GI tract, including the proximal small intestine, distal small intestine, and large intestine, will contribute to unraveling the pathogenesis of IBD and developing therapeutic strategies tailored to different intestinal locales.
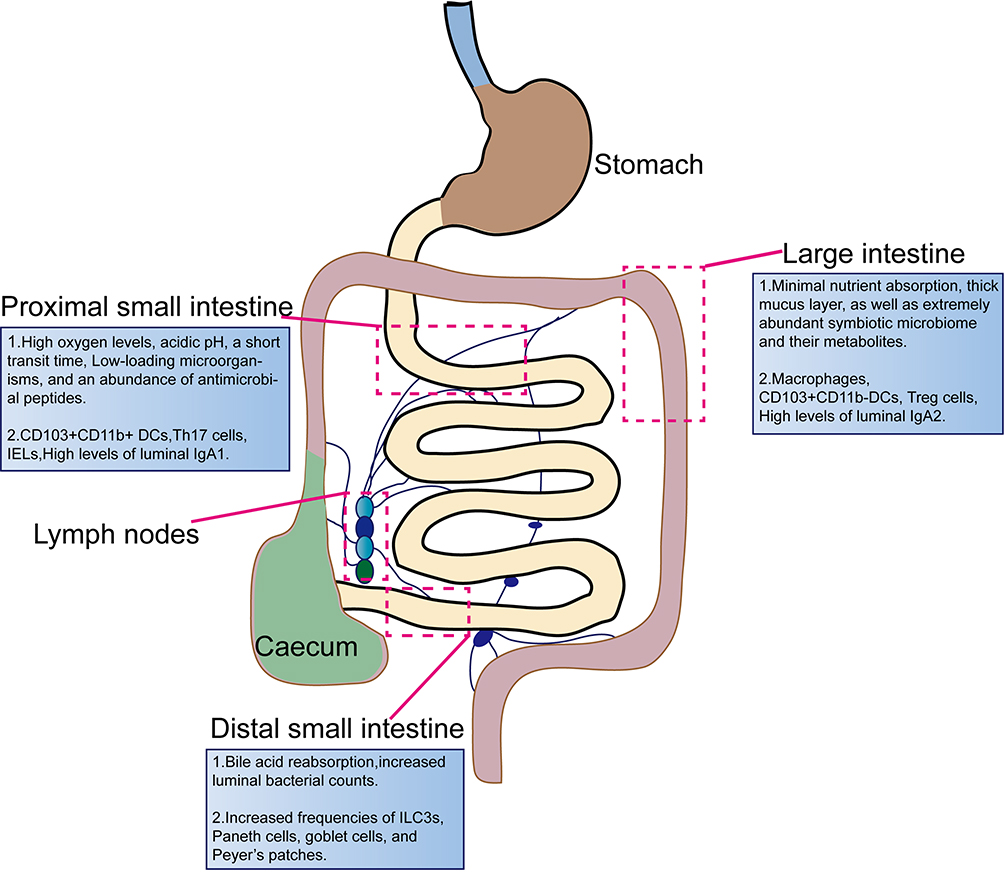 |
Figure 1 The compartment of intestinal immune system. Five major immune zones exist in the gastrointestinal (GI) tract: the proximal small intestine consists of the duodenum and jejunum, and the distal small intestine of the ileum, the large intestine contained colonic tissue, as well as the mesenteric and intestinal draining lymph nodes, and intestinal-associated lymphoid nodes. The proximal small intestine, features long, thin villi and serves as the primary site for food digestion and absorption. Key immune cells present in this area include CD103+CD11b+ dendritic cells (DCs), Th17 cells, intraepithelial lymphocytes (IELs), and elevated levels of luminal IgA1.The distal small intestine, villi become shorter and flatter, while the mucus layer thickens. Although nutrient absorption is reduced in this region, it plays a vital role in bile acid reabsorption. In response to increased bacterial presence, levels of innate lymphoid cells type 3 (ILC3s), Paneth cells, goblet cells, and Peyer’s patches rise. Compared to the small intestine, the large intestine has limited nutrient absorption capacity, a thicker mucosal layer, and a more diverse bacterial flora. It contains a higher abundance of goblet cells, rare Paneth cells, and increased numbers of immune cells such as macrophages, CD103+CD11b− DCs, and regulatory T (Treg) cells, along with elevated levels of luminal IgA2. |
Proximal Small Intestine
The proximal small intestine, divided into the duodenum and jejunum, exhibits distinctive structural and functional features. Long and dense villi protrude into the lumen, significantly increasing the epithelial surface area for efficient digestion and absorption. The length of the villi gradually decreases from the duodenum to the ileum, reflecting the varying absorptive ability along the length of the small intestine. The epithelial surface of the small intestine is merely covered by a relatively loose mucosal layer, facilitating the isolation of intestinal microorganisms while allowing unimpeded nutrient absorption. Paneth cells are unique to the crypt of the small intestine, whose distribution gradually increases from the duodenum to the ileum, aligning with the changing microbial landscape along the small intestine.14 Paneth cells secrete antimicrobial peptides that exert antimicrobial effects in response to interleukin-22 (IL-22) and pattern recognition receptors, such as toll-like receptor (TLR) and nucleotide-binding oligomerization domain 2 (NOD2). Additionally, they produce pro-epidermal growth factor (pro-EGF), WNT3, and Notch ligand 3, which are essential for maintaining normal crypt stem cell activity. Consequently, alterations in Paneth cell function are closely associated with the pathology of CD. Therefore, strategies aimed at promoting the differentiation of intestinal epithelial cells into Paneth cells are crucial for the treatment of CD.15 The proximal small intestine possesses distinct physiological characteristics, including high oxygen levels, acidic pH, a short transit time, and an abundance of antimicrobial peptides. Consequently, the microbial load is significantly reduced in this region (105 bacteria per mL in the proximal small intestine compared to up to 1012 bacteria per mL in the ileum and large intestine), which is consistent with the distribution of macrophages along the intestine. CD103+CD11b+ DCs predominantly reside in the proximal small intestine and are rarely observed in the colon (Figure 1). CD103+CD11b+ DCs in the lamina propria (LP) of the small intestine are mature pro-inflammatory cells that rely on interferon regulatory factor 4 (IRF4) to facilitate the differentiation of Th17 cells and the production of IL-17. In contrast, DCs located within the epithelium exhibit a tolerogenic phenotype. Food-derived retinoic acid (ATRA) enhances actinomyosin contractility, promoting the migration of LP DCs toward the epithelium. Once there, these DCs, influenced by ATRA and mucins, adopt a tolerogenic phenotype, playing a critical role in inducing tolerance to dietary antigens.16,17 Therefore, strategies that induce the migration of LP DCs to intraepithelial DCs may offer therapeutic potential for CD. Additionally, in humans and mice, epithelial lymphocyte counts are highest in the proximal small intestine and decrease progressively along the length of small intestine. This phenomenon appears to be associated with higher concentrations of intestinal CCL25, CCR9, AHR ligands, G protein-coupled receptors 18 (GPR18), and retinoic acid (RA) in the small intestine.18–22 In the LP, CCR9 induces the localization of activated CD4+ and CD8+ T cells to the small intestine.23 Moreover, the LP CD4+ T cells exhibit high diversity and have the ability to differentiate into various subpopulations, including effector T cells (Th1, Th2, and Th17 cells) and regulatory T (Treg) cells. However, the distribution and function of these subpopulations differ noticeably across the length of the gut. The abundance of Foxp3+ Treg cells in the LP increases progressively from the small intestine to the colon, whereas Th17 cells in the LP are increased in the small intestine, correlating with its unique intestinal environment (Figure 1). Interestingly, it has been hypothesized that preferential access of adherent microorganisms to the small intestinal epithelium may underlie the increased population of Th17 cells in this gut region.24 This immune adaptation underscores the close relationship between microbial load and immune cell function across different gut regions. In contrast, the frequencies of Th1 and Th2 cells do not show significant variations throughout the length of the intestine. Additionally, tissue-resident memory (TRM) T cells are essential for local immune responses, demonstrating functional specialization through metabolic reprogramming to respond rapidly to their microenvironment. In the proximal small intestine, TRM cells facilitate immune surveillance due to a high antigenic load, while in the colon, they exhibit enhanced anti-inflammatory properties that help balance the dense commensal microbiota. Single-cell RNA sequencing analyses have revealed that CD4+CD103+ TRM cells display a distinct inflammatory profile in CD patients,25 whereas CD8+ TRM cells aggregate and shift toward an inflammatory state in the colonic mucosa of UC patients.26 Altogether, the immune system of the proximal small intestine undergoes adaptations to meet the functional requirements of nutrient absorption and pathogen encounters. These adaptations constitute the basis of the unique immune environment within this specific intestinal region.
Distal Small Intestine
The distal small intestine, specifically the ileum, exhibits certain anatomical and functional characteristics that distinguish it from the proximal small intestine. The villi in the ileum are shorter and wider, and the mucus layer is thicker. Although the contribution of the distal small intestine to nutrient absorption is comparatively less, it plays a vital role in bile salt and vitamin B12 absorption. There is also an increase in the luminal bacterial population (107 bacteria per mL) that requires an enhancement in the epithelial barrier, such as an increase in the number of Paneth cells producing antimicrobial peptides and mucus-producing goblet cells.13 Liu et al demonstrates that a Western diet increases the abundance of intestinal Clostridium difficile, mediates the conversion of primary to secondary bile acids in the ileum, and activates the farnesoid X receptor (FXR) in Paneth cells and myeloid cells. Myeloid cells respond to FXR activation to enhance the production of type I interferon (IFN), which sequentially influences intestinal epithelial cells, leading to the development of abnormal Paneth cells. Notably, discontinuation of the Western diet may allow for potential reversibility of these Paneth cell defects, highlighting important implications for the treatment of IBD.27 In addition, in the ileum, increased density of Peyer’s patches may enhance antigen uptake by microfolded cells (M cells) (Figure 1).28 An intriguing study suggests that TRPV1+ sensory neurons, which innervate the intestine, can respond to Salmonella enterica serovar Typhimurium (STm) through calcitonin gene-related peptide (CGRP). This response modulates the density of M cells in ileum Peyer’s patch follicle-associated epithelia (FAE), thereby limiting invasion by STm. These findings highlight the key role of interactions between the nerves, microbes and immunity in maintaining the dynamic homeostasis of the gut’s complex microenvironment and offer new insights for IBD treatment.29 The distribution and composition of macrophages in the distal small intestine appear to be similar to that in the proximal small intestine. However, CD103+CD11b−DCs counts are slightly increased in the ileum compared to the duodenum. Microorganisms colonizing the ileum also contribute to shaping the ileal immune landscape. For example, segmental filamentous bacteria (SFB) preferentially colonize the ileum, inducing an increase in Th17 cells in the ileum and thereby draining lymph nodes.30 Conversely, there is no apparent difference in the abundance of Foxp3+ Treg cells in the ileum compared to the proximal small intestine. In conclusive, the immune compartmentalization of the distal and proximal small intestine is not wholly distinct.
Large Intestine
The large intestine consists mainly of the cecum, colon, rectum, as well as the anus, and is characterized by a flat epithelial surface lacking villi, rendering it minimally involved in digestion. Rather, it serves as a prominent reservoir for a vast number of commensal bacteria with up to 1012 bacteria per mL (Figure 1). To detect and react to this microbial burden, TLR4, TLR2, and TLR5 are robustly expressed coupled with an increase in the count of mucus-secreting goblet cells.13,31 The colon is covered with a double mucus layer, featuring a loose external layer and a denser inner layer, restricting direct contact between the bacteria and the intestinal epithelium. Parikh et al utilizing single-cell RNA sequencing technology has shown that healthy human colonic epithelial cells, specifically BEST4/OTOP2 cells, selectively express uroguanylin. This peptide is crucial for maintaining luminal homeostasis in a pH-dependent manner through the guanylate cyclase 2C receptor signaling. Notably, the number of uroguanylin-producing cells is significantly reduced in IBD patients, suggesting a novel pathogenic mechanism. These findings provide a theoretical foundation for the development of therapies based on uroguanylin mimetics for the treatment of IBD.32 In mice, the abundance of macrophages is greatest in the colon and least in the proximal small intestine.33 However, the abundance of macrophages appear to be similar between distinct regions of the human intestine.34 Colonic and small intestinal macrophages both highly express MHCII, F4/80, CD64, CD163, and CX3C chemokine receptor1(CX3CR1).35–37 However, colonic macrophages also exhibit elevated expression of CD40, CD209, CCL2, CCR5, and formyl-peptide receptors (FPRs).38,39 Furthermore, macrophages in the colonic region are closer to the epithelial surface than in the small intestine, enabling a more swift response to pathogenic invasion and underscoring the significance of the microbiota and their metabolites for both the development and the function of colonic macrophages (Figure 1).40 CD103+CD11b−DCs represent the predominant CD103+DC subpopulation within the colon, exhibiting functional specialization that induces interferon (IFN)-γ secretion from lymphocytes in the LP and colon epithelium, and triggering an early and reversible early anti-inflammatory response in the intestinal epithelium.33,41 The Foxp3+ Treg cell population in the LP of the intestine increases progressively in abundance from the small intestine to the colon, adapting to the environment of the intestine. In germ-free or antibiotic-treated mice, the number of Foxp3+ Treg cells is decreased in the colon but not in the small intestine, suggesting that the majority of Foxp3+ Treg cells in the colon develop in response to the microbiota.42 A recent research indicates that the promoter of polysaccharide A (PSA) synthesis in Bacteroides fragilis is more frequently “on” in healthy individuals, while it is often “off” in patients with IBD. This shift is associated with elevated levels of phages and acts through Treg cells. Importantly, this phase change can be reversed as inflammation subsides. This suggests that phages influence bacterial function and, consequently, the host’s immune response and disease progression through DNA inversion states. These findings provide new insights into the role of the gut microbiota in IBD and may lead to innovative strategies for its diagnosis and treatment.43 Moreover, in the mouse colon, the distribution of Th17 cells exhibits a negative correlation with the distribution of Treg cells, whereas in humans, the abundance of Th17 cells is increased in the large intestine.33 Furthermore, both humans and mice harbor the majority of their intestinal plasma cells (PCs) in the LP, with the highest density of PCs observed in the proximal small intestine and distal large intestine, approximately 75–80% and 90%, respectively. Throughout the intestine are IgA-producing PCs that exhibit specificity towards the microbiota and autoantigens. Humans generate both IgA1 and IgA2, with IgA1-producing PCs dominating in the proximal small intestine and IgA2-producing PCs dominating in the colon.44 Notably, under conditions of bacterial overgrowth, the typical prevalence of IgA1 in the small intestine can shift towards IgA2.45 Concordantly, IgA2 possesses a stiff structure, being more resistant to bacterial proteases, thereby indicating its adaptation for bacterial-rich colonic environments.46 In conclusion, the intricate interactions between immune cells and gut microbes in large intestine are crucial for maintaining local homeostasis and responding to inflammation. These findings not only enhance our understanding of the pathological mechanisms underlying IBD but also open new avenues for developing targeted therapeutic strategies.
Implications of Intestinal Immune Compartmentalization
Regional immune specialization facilitates the localization of inflammatory diseases within the gut. Considerable data suggest that dysfunction of Paneth cells increases the susceptibility to CD.47–50 Furthermore, the aggregation of cells encoding the IL-23 receptor gene, such as DCs, macrophages, and Th17 cells, constitutes another risk factor for CD.51 Specific intestinal microorganisms are associated with various pathological sites of CD. For example, adherent invasive Escherichia coli (AIEC), predominantly present in the small intestine, are frequently observed in the ileal mucosa in CD patients.52 In addition, specific knockdown of IL-10 receptor signaling in macrophages results in colitis, but not small bowel disease.53 Moreover, CD11c+ cell activation-dependent tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6)-deficient mice exhibit decreased counts of Foxp3+ Treg cells and loss of immune tolerance specifically in the small intestine, but not in the colon.54 Contrarily, mice with CD11c+ cells lacking αVβ8 integrin fail to activate transforming growth factor β (TGF-β), leading to reduced Treg cell numbers and inflammation in the colon, but not in the small intestine.55 Understanding intestinal regional immune specialization highlights the potential of immune strategies in the treatment of IBD based on specific intestinal regions.
Intestinal Immune-Microecology Interactions in IBD
The intestine harbors trillions of microorganisms and serves as the primary site of host immunity and interactions with both commensal and pathogenic microbes. Over thousands of years of evolution, the intestinal immune system and gut microorganisms have established a symbiotic partnership. In healthy individuals, the gut microbiota is essential for regulating the growth and function of both innate and adaptive immune cells, while the immune system modulates the composition and abundance of the gut microbiota. However, disruption of this interdependent relationship due to gut dysbiosis may underlie the pathophysiology of IBD.
Colonization of the intestinal flora is essential for the proper development of the intestinal immune system, as evidenced by immune defects observed in germ-free animals.7 Microbial metabolites exert profound regulatory effects on immune cell development and differentiation. For example, short-chain fatty acids generated by Clostridium clusters VI, XIVa, and XVIII promote the differentiation of Foxp3+ Treg cells via activating TGF-β signaling in the epithelium.56 Butyrate, by inhibiting histone deacetylase (HDAC), suppresses pro-inflammatory cytokine expression in macrophages, hinders DCs development and maturation, and maintains low responsiveness to commensal bacteria.57,58 Another microbial metabolite, acetate, activates mTOR signaling to drive the differentiation of Th1, Th17, and Treg cells through acetylation of p70 S6 kinase (S6K) and phosphorylation of ribosomal protein S6 (rS6).59 Additionally, in the small intestine, pyruvate and lactate activate GPR31 signaling to induce dendritic protrusion of CX3CR1+ phagocytes.60
Moreover, bacterial components can modulate the host immune response by engaging PRRs like TLRs and NOD-like receptors (NLRs). For instance, polysaccharide A (PSA) from Bacteroides fragilis induces IL-10 production in Foxp3+ Treg cells by acting directly on TLR2.61 Furthermore, bacteria themselves play a pivotal role in the development and maturation of the intestinal immune system. For example, administration of Lactobacillus rhamnosus CNCM I-3690 in mice enhances colonic barrier function, as evidenced by increased mucus production and restoration of goblet cell populations.62 The probiotic Clostridium butyricum stimulates the production of IL-10 by intestinal macrophages via TLR2/MyD88 signaling, thereby exerting inhibitory effects on dextran sodium sulfate (DSS)-induced colitis.63 Additionally, in the small intestine, specific antigens derived from SFB are recognized by MHCII-expressing CD11c+DCs, inducing the development and differentiation of Th17 cells, subsequently promoting IL-17 expression.64 This observation underscores the intricate interplay between bacterial antigens and the adaptive immune system.
In turn, gut immune cells exert direct and indirect effects on the microflora. For example, IL-22 suppresses the expansion of SFB, thereby suppressing the differentiation of Th17 cells and preventing Th17-mediated bowel inflammation.64 Deficiency in IL-22 has been associated with alterations in the composition of the colonic microbiota, including a decreased abundance of Lactobacillus and increased levels of other genera, resulting in a heightened susceptibility to DSS-induced colitis.60 Furthermore, the major histocompatibility complex class I-like molecule, Cd1d, expressed by DCs and IECs, enhances the colonization of gut symbionts by controlling Paneth cell function.65 Secretory immunoglobulin A (SIgA) assumes a critical role in maintaining intestinal microbiota homeostasis. A deficiency in IgA can lead to abnormal expansion of SFB in the intestine, triggering a potent immune response in the intestinal mucosa.66
Collectively, the gut microbiota plays a pivotal position in regulating host immune defenses against bacterial components and metabolites. Conversely, the gut immune system exerts precise regulation over microbial composition, diversity, as well as transport. Microecological and immune dysfunctions constitute the underlying pathogenesis of bowel inflammation. Hence, maintaining a stable balance between the intestinal flora and the immune system is essential for overall health.
Innate Immune in IBD
The intestinal innate immune system, as the primary defensive barrier of the body, recognizes and initiates the inflammatory responses against microorganisms (Figure 2). It is comprised of the intestinal epithelial barrier, innate immune cells, and innate immune molecules, collectively responsible for this crucial process. These innate immune cells recognize foreign bacteria through intra- and extracellular PRRs, such as TLRs and NLRs, that activate multiple signaling pathways. This activation culminates in the recruitment of inflammatory cells and the generation of pro-inflammatory cytokines, chemokines, as well as antimicrobial peptides. A growing body of evidence emphasizes the significance of innate immune disorders in conjunction with IBD.
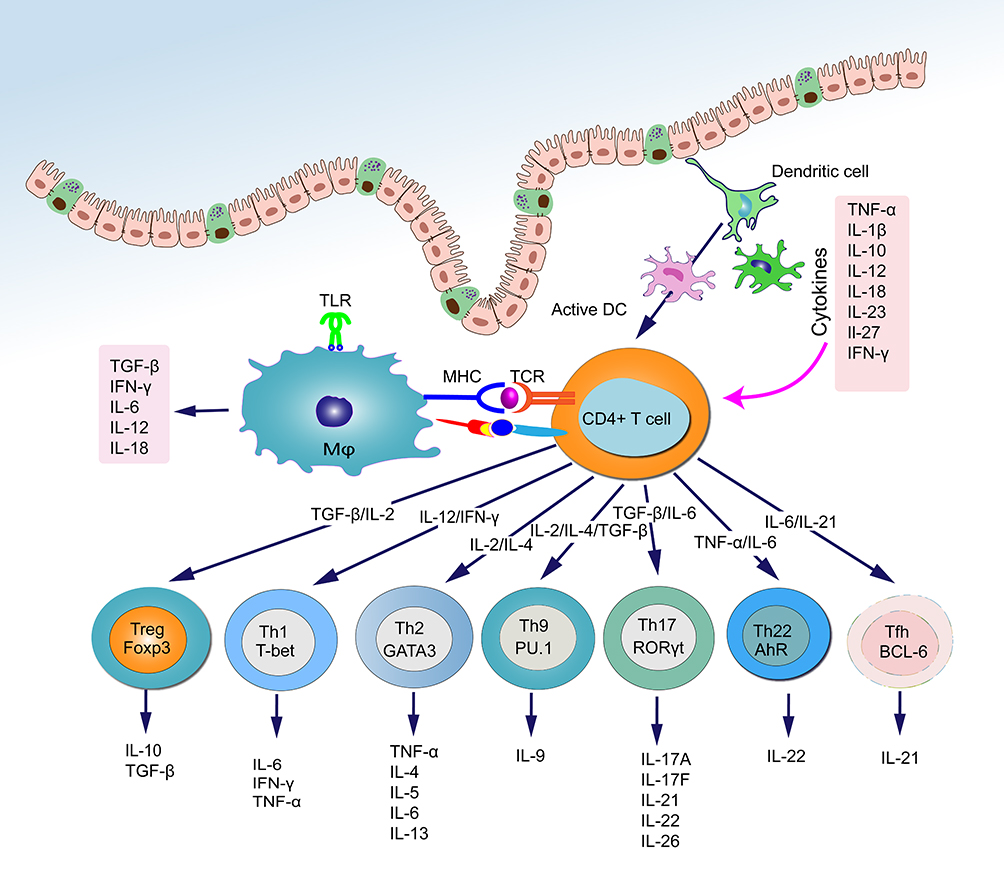 |
Figure 2 Schematic representation of the interaction of intestinal immune system and cytokines. The recognition and uptake of antigens by macrophages and DCs trigger the differentiation of naïve CD4+ T cells into various effector T cell subtypes, including Treg, Th1, Th2, Th17, Th9, Th22, and T follicular helper (Tfh) cells. These effector T cells play a crucial role in maintaining intestinal immune homeostasis and defending against pathogen invasion by secreting specific cytokines. In turn, cytokines released by activated effector T cells can influence the differentiation direction of naïve CD4+ T cells and regulate the interconversion and function of different T cell subtypes. |
Intestinal Epithelial Barrier
The intestinal epithelial barrier assumes a critical function in maintaining a precise equilibrium between gut luminal contents and mucosal immune responses. The intestinal epithelium acts as a physical barricade to prevent the passage of bacteria and other antigens derived from the GI tract into the bloodstream. In IBD patients, increased intestinal permeability is frequently observed, and these abnormalities may arise from defects in epithelial barrier function.67 Additional defense against bacterial intrusion exists in the form of specialized epithelial cells, comprised of goblet cells and Paneth cells.
Goblet cells contribute to the protective mucus layer covering the gut epithelium. This mucosal layer primarily consists of mucin-2 (MUC2), calcium-activated chloride channel regulator 1 (CLCA1), and Fcγ binding protein (FCGBP), which are essential for mucosal defense and repair.68–70 Goblet cell exhaustion is frequently reported as a common characteristic of colitis. For instance, MUC2-deficient mice were found to develop spontaneous colitis.71 Likewise, patients with active UC exhibit a reduced number of goblet cells and a weakening of core mucus structural components.68 Impaired intercrypt goblet cells and defective intercrypt mucus have been observed in UC patients, facilitating the onset and progression of this disease.72 Other than mucin production and secretion, goblet cells also deliver luminal antigens via goblet cell-associated antigen passages (GAPs). This process contributes to maintaining preexisting Treg cells within the LP, imparting tolerance properties to LP DCs and promoting IL-10 production by LP macrophages, thus establishing a bridge to the immune system.73,74 Consequently, impaired delivery of intestinal luminal antigens by goblet cells results in increased susceptibility to colitis.75,76
Paneth cells, another highly specialized epithelial cell population, perform a crucial function in maintaining intestinal homeostasis and modulating the microbiota within the small intestinal. Paneth cells are positioned at the base of small intestinal crypts and secrete antimicrobial peptides (such as lysozyme, defensins, and regenerating islet-derived protein IIIγ (REGIIIγ)) and inflammatory mediators in response to sensing pathogen-associated pattern molecules (PAMPs) through TLR and NOD2 receptor. This ensures the protection and regulation of the homeostasis between commensal bacteria and the host mucosa.77,78 IBD patients frequently exhibit aberrant Paneth cell function, which disrupts the microecological balance and impairs mucosal repair.79
Several critical susceptibility genes and pathogenic pathways impacting Paneth cell function have been studied include microbial signaling (NOD2), autophagy (ATG16L1), and endoplasmic reticulum (ER) stress.79,80 Notably, NOD2 is highly expressed in ileal Paneth cells and closely associated with α-defensin production. Ileal CD patients carrying NOD2 risk variants exhibit reduced levels of human enteric alpha-defensins (HD5 and HD6) secreted by Paneth cells, although not necessarily associated with the degree of inflammation in CD patients.81 Intriguingly, NOD2 (-/-) mice, when challenged with Helicobacter hepaticus, developed granulomatous inflammation in the ileum, which is a typical characteristic of CD.82 Furthermore, NOD2 is involved in the sorting of Paneth cell-derived AMP, and deficiency in NOD2 directs AMP to lysosomes instead of staying in dense core vesicles (DCVs).83 Thus, in NOD2-deficient patients, CD can be initiated by the lack of Paneth cell-derived AMP. ATG16L1 plays a protective role in CD by maintaining autophagy and responding to environmental stimuli in Paneth cells. Defective ATG16L1 impairs autophagy and alters the proteomic abundance profile in Paneth cells, restricting Paneth cell defensin and antimicrobial peptide cytosolic secretion.84 Moreover, ATG16L1-deficient Paneth cells acquire unexpected functions, including involvement in peroxisome proliferator-activated receptor (PPAR) signaling, lipid metabolism, and acute phase reactants responses.84 Thus, CD may be triggered by the dysfunction of ATG16L1-deficient Paneth cells. Furthermore, variants in ER stress-related genes such as X-box binding protein-1(XBP1) and anterior gradient-2(AGR2) in Paneth cells are linked to an increased susceptibility for CD.85,86 Deletion of XBP-1 in mouse IECs, along with subsequent ER stress, results in Paneth cell damage and spontaneous colitis.85 Similarly, Agr2 (-/-) mice exhibit disrupted Paneth cell and goblet cell functions, increasing ER stress and leading to severe terminal ileitis and colitis.86 However, NOD2, ATG16L1, and XBP1 are not exclusive to Paneth cells, as their functions in other cell groups may also increase risk for IBD. Thus, Paneth cell dysfunction alone appears to be insufficient in provoking intestinal inflammation, and may require additional inflammatory stimulation or injury.
Macrophages and DCs
Macrophages and DCs are responsible for initiating innate immunity, being closely associated with the onset and development of IBD (Figure 2). In healthy individuals, intestinal LP macrophages are characterized by a lack of CD14 and exhibit a predominantly hyporeactive phenotype towards inflammatory stimuli. These macrophages secrete anti-inflammatory molecules like IL-10, contributing to Treg cell differentiation and suppressing Th1 and Th17 cell responses. Simultaneously, they retain their phagocytic and bactericidal functions, thus serving as key contributors to intestinal immune homeostasis.87 However, an abundance of CD14+ macrophages has been found in patients with CD that generate substantial amounts of pro-inflammatory cytokines, including TNF-α, IL-23, and IL-6.88 These CD14+ macrophages also stimulate the secretion of IFN-γ by IL-23- and TNF-α-dependent LP monocytes (LPMC), thereby further initiating aberrant macrophage differentiation with high IL-23 production capacity.89 In addition, normal intestinal macrophages lack the expression of triggering receptors expressed on myeloid cells 1 (TREM-1), which promotes the secretion of pro-inflammatory factors. However, an increased number of TREM-1+ macrophages is observed in IBD patients and correlates with disease activity.90 Consequently, targeting the TREM-1 signaling pathway holds promise as a potential therapeutic approach for IBD. Moreover, all macrophages in the intestinal LP appear to express CX3CR1 receptors. CX3CR1+ macrophages derived from Ly6Chi precursors are recruited to the inflamed intestine via CCR2. In an inflammatory environment, the differentiation of Ly6Chi monocytes is blocked at an immature stage, rendering them highly responsive to TLR signaling. As a result, these immature macrophages release high levels of pro-inflammatory mediators such as TNF-α, IL-1β, IL-6, as well as IL-12, while reducing the levels of IL-10. This skewed cytokine profile promotes the immune response mediated by Th1 and Th17 cells.36,91,92 In contrast, mature CX3CR1hi macrophages in the LP retain a hyporeactive and anti-inflammatory character.93 Studies on CX3CR1 (-/-) mice have shown a decreased abundance of LP macrophages and an increased translocation of commensal bacteria to the mesenteric lymph nodes. These alterations contribute to the exacerbation of DSS-induced colitis. Notably, disease severity can be ameliorated by the transfer of CX3CR1+macrophages.94
In contrast to macrophages, DCs serve as specialized antigen-presenting cells (APCs) that constitute an interface between innate and adaptive immunity. They play a critical role in initiating both protective pro-inflammatory and tolerogenic immune responses. DCs possess distinctive characteristics, including a unique stellate morphology, high MHC II expression, and the ability to capture antigens and migrate to draining lymph nodes. In these lymph nodes, DCs trigger naive T cells and guide their differentiation into distinct phenotypes, including inflammatory (Th1, Th2, and Th17) or Treg cells.95 The intestinal tissue-resident DCs are sentinels for intestinal mucosal immunity and facilitate the sampling of bacteria by myeloid differentiation primary response gene 88 (MyD88)-dependent signaling via TLRs and CX3CR1. This process represents the initial step in the initiation of adaptive immunity.96 In healthy individuals, LP CD103+CX3CR1−DCs promote the differentiation of Foxp3+ Treg cells and IgA secretion, relying on RA and TGF-β to induce immune tolerance.97,98 Importantly, the number of DCs is elevated in the inflamed mucosa of patients with IBD, while the CD103+ subpopulation is specifically reduced.99 Furthermore, duodenal LP PD-L1+ DCs and colonic LP XCR1+ DCs serve as distinct tolerogenic subpopulations that are critical for maintaining intestinal homeostasis. Mice lacking PD-L1+ and XCR1+ DCs result in elevated levels of Th1/Th17 cells and decreased Treg cells, thereby creating a pro-inflammatory intestinal environment and exacerbating disease severity in colitis models.100 Conversely, in an inflammatory milieu, Ly6Chi monocytes heavily infiltrate the colon and differentiate into pro-inflammatory CD103 − CX3CR1+ DCs, producing high levels of IL-12, IL-23, inducible nitric oxide synthase (iNOS) and TNF.101 Moreover, intestinal DCs from IBD patients demonstrate increased expression of several PRRs, as TLR2 and TLR4, enhancing their capacity to recognize microbial antigens and exacerbating the immune response.102 In summary, just as macrophages and DCs contribute to maintaining intestinal tolerance, aberrant function of these cells may represent a contributing factor in the pathogenesis of IBD.
Innate Lymphoid Cells (ILCs)
ILCs constitute a crucial part of the intestinal innate immune system, contributing to the maintenance of intestinal mucosal homeostasis and the development of IBD. Originating from a common lymphoid progenitor cell population, ILCs represent the innate counterpart of T lymphocytes. However, they lack the diverse antigen receptors expressed by T and B cells.103,104 Primarily residing in the LP of the intestine, ILCs maintain healthy immune responses to commensals and pathogens, while also participating in organ development, tissue protection and regeneration, and preserving mucosal homeostasis.104,105 Based on their developmental pathways, expression of transcription factors, and cytokine secretion profiles, ILCs can be subdivided into three subsets: group 1 ILCs, group 2 ILCs, and group 3 ILCs.
Group 1 ILCs include ILC1s and natural killer (NK) cells, whose functions correspond to Th1 cells and CD8+T cells, respectively. The growth and function of these cells are dependent on the T box transcription factor T-bet. They respond to IL-12, IL-15, and IL-18, producing IFN-γ and TNF. Consequently, they contribute to immune responses against intracellular pathogens and tumors.106,107 Group 2 ILCs, represented by ILC2s, functionally resemble Th2 cells. Their development is reliant on GATA3 and vincristine-related orphan nuclear receptor (ROR) alpha, which are stimulated by IL-25, IL-33, and thymic stromal lymphopoietin (TSLP). ILC2s secrete IL-6 and Th2 cell-related cytokines such as IL-4, IL-5, IL-9, IL-13, and granulocyte macrophage colony-stimulating factor (GM-CSF). They perform a pivotal task in combating helminth infection and regulating tissue repair.108–110 Group 3 ILCs, including NCR− ILC3s and NCR+ ILC3s (NKp44, NKp46), are comparable to Th17 and Th22 cells in terms of their function. The development and maturation of these ILCs depend on the regulation of RORγt and AHR, and the secretion of IL-17, IL-22, and TNF-α in response to IL-1 and IL-25 stimulation. They participate in the innate immune response against extracellular bacteria and in the control of intestinal commensals.111
Various subtypes of ILCs exist in the human intestinal mucosa and are engaged in maintaining gut homeostasis. However, the dysregulation and imbalance of these cells can facilitate the onset of IBD. The balance between ILC1s and ILC3s has been identified as particularly significant in the context of IBD. Under IL-12 stimulation, ILC3s acquire the ability to secrete IFN-γ and exhibit ILC1-like characteristics through upregulation of T-bet expression and downregulation of RORγt expression.112 According to Bernink et al, CD patients exhibit an elevated abundance of ILC1s and a decreased number of ILC3s in intestinal tissues, which contribute to intestinal inflammation through IFN-γ secretion.113 Thus, dysregulation of ILC subgroup homeostasis due to intercellular transformation contributes to the pathogenesis of IBD. Nevertheless, not all ILC3 subsets are decreased in IBD. ILCs isolated from inflamed colon tissue of CD patients exhibit increased gene expression of key ILC3-associated cytokines such as IL-17A and IL-22, cytokine receptors (such as IL-23R), and transcription factors (such as RORγt and AHR).114 IL-22 secreted by ILC3s promotes the generation of antimicrobial peptides and mucin by epithelial cells, serving as protection against invading pathogens.115 Furthermore, IBD patients exhibit upregulated expression of AHR, which is crucial for the function of group 3 ILCs. A study in AHR (-/-) mice demonstrates a decrease in ILC3s and an increase in Th17 cells, suggesting a potential role for ILC3s in negatively regulating Th17 cells and inhibiting T cell-mediated intestinal inflammation.116 However, ILC3s stimulated with IL-23 produce IL-17 and IFN-γ, which induce the development of intestinal inflammation.117,118 This evidence suggests that ILC3s can either inhibit or induce intestinal inflammatory responses under different circumstances. Thus, the role of ILC3s in IBD is complicated and necessitates further investigation and clarification.
In addition, NK cells in group 1 ILCs not only produce IFN-γ to encourage the recruitment of additional NK cells from peripheral blood but also highly express perforin to directly kill pathogens, which contributes to the induction of the immune response. Notably, NK cells can exacerbate intestinal inflammation by upregulating NK cell group 2D (NKG2D) expression, promoting ER stress in IECs.119 Takayama et al discovered that the equilibrium between NKp44+ and NKp46+ NK cells was disrupted in the bowel mucosa of CD patients. NKp46+ NK cells may mediate the pathogenesis of CD by activating intestinal inflammatory macrophages through IFN-γ.120 Moreover, LPNK cells undergo metabolic reprogramming during IBD. In the setting of acute colitis, LPNK cells are more cytotoxic to target cells compared to in healthy individuals through an upregulation in the expression of glycolysis and oxidative phosphorylation-related genes, potentially as a response to impending microbial infection.121
The precise role of group 2 ILCs in IBD pathogenesis remains poorly understood, as they are virtually undetectable in the healthy adult intestine.122 Forkel et al reported an increase in group 1 ILCs in CD patients, whereas group 2 ILCs were elevated in UC patients.123 In the oxazolone-induced colitis mouse model, an increase in IL-13+ ILC2s suggested they play a proinflammatory role in UC.124 Furthermore, under inflammatory conditions, group 2 ILCs are plastic and can convert to group 1 ILCs or group 3 ILCs. For example, IL-13+ ILC2s have been detected in CD patients and can obtain the ability to generate IFN-γ via IL-12/IL-12R signaling, associated with increased T-bet expression.125 Similarly, AHR activation inhibits ILC2 function while enhancing ILC3 function, and alterations in AHR expression-mediated ILC2s/ILC3s conversion influence the intestinal immune response in IBD.126 However, ILC2s may also exert a protective role in IBD. Notably, ILC2-derived IL-13 promotes the differentiation of intestinal stem cells into goblet and Turf cells, facilitating the restoration of damaged intestinal mucosa.127 Moreover, in an acute colitis mouse model, IL-33-activated ILC2s exert protective effects and reduce disease severity by secreting the growth factor amphiregulin (AREG), possibly through upregulation of tight junction protein-1 and MUC-2 expression.110 Overall, the role of group 2 ILCs in the human gut remains elusive, and further investigations are warranted to uncover the mechanisms underlying immune imbalance in IBD.
Unusual T Cells (γδT Cells)
The T cell receptor (TCR) on the surface of γδT cells is a heterodimer comprised of γ and δ chains (TCRγδ), enabling direct recognition and binding of antigen molecules without requiring MHC molecules and APCs. γδT cells exert their biological effects mainly by lysing target cells and secreting various cytokines (IL-17, TNF-α, and IFN-γ) and chemokines. Additionally, they can serve as APCs and present antigens to αβT cells. In healthy individuals, γδT cells in the gut constitute a small subset of IL-17-producing T lymphocytes. However, during intestinal microbial infections, IL-17-producing γδT cells expand their population to enhance host defense against infections.128 Furthermore, in the context of IBD, tissue-resident γδT cells exhibit increased IL-17 production, contributing to the regulation of host pathogen defense and intestinal inflammation. Notably, this function can be inhibited by short-chain fatty acids derived from intestinal microbes.129 Moreover, γδT cells display an anti-inflammatory effect by secreting apoptosis inhibitory factor 5 (API5), promoting the viability of ATG16L1 gene-deficient Paneth cells.130 Studying the role of γδT cells in IBD has received limited attention, and further exploration is required to elucidate their specific functions.
Adaptive Immune System in IBD
Mounting research supports the contribution of adaptive immunity in the etiology of IBD. Unlike innate immunity, adaptive immunity is an acquired, highly specific, and long-lasting immunity with the capacity for immune memory (Figure 2). Recognition of intestinal microbial antigens by APCs (DCs and macrophages) initiates a complex interplay of pro-inflammatory as well as anti-inflammatory signals. Disruption of the delicate balance between these signals in IBD results in the migration of leukocytes to the intestinal mucosa, triggering aberrant activation of T cells and perpetuating inflammation through excessive release of cytokines and chemokines.131,132 Typically, activated naive CD4+ T cells can differentiate into diverse effector T cell subpopulations (Th1, Th2, Th9, Th17, Th22, and T follicular helper (Tfh) cells) or Treg cells (Figure 2).133 Notably, the T cell populations driving immune responses in CD and UC appear to be distinct, which may account for the differences in the phenotypes seen in clinical practice and the variable responses to new targeted therapies.6,9 Therefore, gaining a comprehensive understanding of the mechanisms underlying adaptive immunity in CD and UC will assist in the development and targeted selection of therapeutic agents.
T Cell Immune Responses in CD
CD is typically considered to be associated with Th1 and Th17 cells, whereas Th2 and CD1d-restricted NKT cells (IL-13) are more commonly associated with UC.134 Activation of Th1 cells is primarily induced by IFN-γ, leading to the activation of signal transducer and activator of transcription 1(STAT1) and upregulation of T-bet expression. This, in turn, up-regulates IL-12R and activates STAT4 and NF-κB signaling pathways in the presence of IL-12, promoting the production of IFN-γ, IL-12, and TNF-α. These events facilitate macrophage activation and restrict epithelial cell proliferation.135,136 Furthermore, Th1 cell responses can be intensified by IL-15 and IL-18. Th1 cells are essential for combating intracellular pathogens. Under the influence of low TGF-β concentrations, naive CD4+ T cells can polarize into Th17 cells when exposed to IL-6 or IL-1β through STAT3 or mTOR signaling. This differentiation leads to the secretion of IL-17A, IL-17F, IL-21, and IL-22, which play a role in the recruitment and activation of neutrophils, thereby promoting inflammatory responses.137,138 Notably, IL-21 and IL-23 promote the proliferation and functional maintenance of Th17 cells.139 Th17 cells functionally contribute to the removal of extracellular bacteria and fungi. Treg cells, in a homeostatic state, regulate the activity of Th1 and Th17 cells and prevent uncontrolled inflammation. However, in CD, disruption of the epithelial barrier by intestinal microbial dysbiosis triggers a pro-inflammatory immune response in DCs and M cells. This dysbiosis leads to a replacement of the regulatory Treg cells with the inflammatory Th1 and Th17 cells. Studies have revealed significantly higher proportions of Th17 and Th1 cells in patients with active CD compared to those in remission.140 In addition, CD patients exhibited a stronger immune response from Th1 and Th17 cells compared to UC and normal controls, resulting in increased generation of IFN-γ and IL-17, subsequent death of IECs, and recruitment of inflammatory cells.141
IL-12 and IL-23 are heterodimeric cytokines sharing the p40 subunit while paired with the p35 and p19 subunits, respectively, and are considered to serve a critical role in T cell-mediated inflammatory responses.142 IL-23 has been recognized as a key factor in the pathogenesis of CD. CD-associated Th17 cells expressing an upregulated IL-23R, in the presence of IL-23, activate multiple pathways via the Janus kinase (JAK) and STAT pathway, such as P38-MAPK, PI3K-Akt and NF-κB signaling pathway. Consequently, several effector cytokines, including IL17A, IL17F, and IL22, are released in CD.143 Furthermore, the amplification of apoptosis-resistant TNFR2+IL23R+ T cells in response to IL-23 results in heightened expression of IFN-γ, T-bet, IL-17A, and RORγt, all associated with resistance to anti-TNF treatment in CD and further supporting a role for IL-23 in CD pathogenesis.144 Additionally, IL-17 expression is enhanced in the mucosa and serum of IBD patients, particularly in the setting of CD compared to UC.145 However, blocking IL-17A has been shown to be ineffective in the treatment of CD and may even exacerbate the disease.146 These findings suggest that IL-17A may also exert a protective role in maintaining barrier integrity and Treg cell function.147
In contrast to effector T cells, Treg cells differentiate in response to IL-2 and high levels of TGF-β. These cytokines induce the upregulation of Foxp3 expression via the JAK-1-STAT5 or JAK-3-PI3K signaling pathways, promoting IL-10 and TGF-β secretion. This regulatory response contributes to the control of inflammatory processes.56 Indeed, in patients with Foxp3 mutations, Treg cells are absent in number or function, which tends to cause more severe intestinal inflammation.148 Furthermore, a lower abundance of Treg cells in the small intestine of CD patients is associated with reduced IL-2 production by ILC3, which is influenced by gut microbes.149 Conversely, adult and pediatric CD patients demonstrate increased numbers of Treg cells in both inflamed and non-inflamed tissues, which implies that IBD pathogenesis may be attributed to a deficiency in Treg cell function rather than an insufficient number of Treg cells.150 Similarly, despite high levels of TGF-β in CD patients, the ability of Treg cells to suppress the local inflammatory response is compromised, probably due to the suppression of TGF-β signaling in the inflammatory environment.151 Impaired trafficking of Treg cells also contributes to the emergence of IBD. For example, CD patients exhibit lower expression levels of integrin α4β7 on Treg cells compared to control patients, hampering Treg cell movement and decreasing the ability to suppress immune responses. This idea is reinforced by the fact that elevating the levels of integrin α4β7 restores the normal function of Treg cells.152 However, the specific differences in Treg cell number and functional changes between UC and CD remain unclear.
T Cell Immune Responses in UC
Compared to healthy individuals, UC patients exhibit increased secretion of IL-5 and IL-13 and decreased production of IL-4. Thus, UC is considered to be an atypical Th2 cell-mediated disease (Figure 3).11,153 Activation of Th2 cells is primarily initiated by IL-4, which results in the activation of STAT6 and upregulation of the transcription factor GATA-3. This cascade induces the release of IL-4, IL-5, IL-9, IL-13, IL-21, coupled with IL-25, known for their roles in antiparasitic responses and allergic reactions.154 Interestingly, intestinal IL-33 has been shown to enhance GATA-3 expression, implying that the elevated IL-33 levels may be the reason for the increased Th2 cell counts in UC patients. Intriguingly, intestinal biopsies from active UC patients demonstrate higher expression levels of IL-33 compared to those from inactive UC patients and healthy subjects, further demonstrating the association between Th2 cell responses and UC.155 Moreover, it has been demonstrated that the majority of IL-13 in UC is derived from CD1d-restricted non-classical NKT cells.153,156 IL-13 can compromise epithelial barrier function by influencing epithelial tight junctions, apoptosis, and repair processes.153 Evidence from a mouse study demonstrates the protective effect of specific antibodies targeting IL-13 in an oxazolone-induced colitis model.156 However, specific inhibition of IL-13 signaling by IL-13 inhibitors in UC patients has proven insufficient to shield them from the illness, which poses a challenge to the therapeutic targeting of IL-13.157 Therefore, the precise role of IL-13 in the pathogenesis of UC necessitates further elucidation. Furthermore, IL-17-producing Th17 cells, IL-9-producing Th9 cells, and CD-mediating Th1 cells frequently appear in the LP mucosa of UC patients and are engaged in the pathological process of UC (Figure 3) (see 5.1 for Th1 and Th17 cell features).158
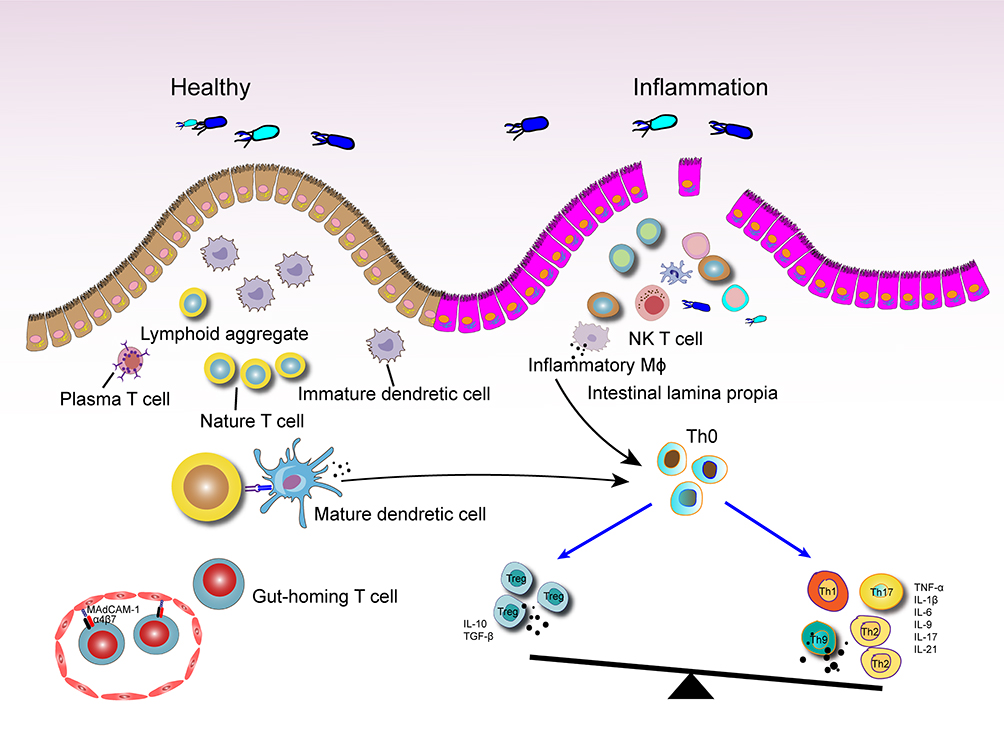 |
Figure 3 Potential modalities of the role of excessive T-cell immune responses in UC. Damage to the intestinal barrier and increased intestinal permeability facilitate bacterial invasion. When macrophages and DCs recognize bacterial antigens, their functional state shifts from a tolerant to an activated phenotype, resulting in the production of pro-inflammatory factors. Following antigen processing, these immune cells present the antigens to naïve CD4+ T cells, disrupting the balance between Treg cells and effector T cells in the inflammatory environment. Circulating T cells expressing integrin-α4β7 migrate to areas of intestinal inflammation by binding to mucosal addressin cell adhesion molecule 1(MAdCAM-1) on intestinal vascular endothelial cell. |
Th1 cells and related cytokines have been demonstrated to be crucial in the early stages of UC, with the disease shifting from a Th1 cell-driven immune response to a Th2 cell-driven immune response as the disease progresses.159 Among these cytokines, IL-9 plays a significant role in the evolution of UC. Elevated levels of IL-9 have been discovered in mice with oxazolone-induced colitis. Functionally, IL-9 impairs intestinal barrier function and hinders the healing of mucosal injury in vivo. In turn, deficiency in IL-9 and transcription factor PU.1 (a key transcription factor for Th9 cell development) inhibits acute and chronic colitis in mouse models.160 A prominent feature of UC is the formation of crypt abscesses, resulting from the accumulation of neutrophils and lymphocytes in the crypt epithelium. Evidence suggests that IL-9 and IL-17 play a crucial role in crypt abscess formation in UC by inducing the release of IL-8, recruiting neutrophils, and secreting matrix metalloproteinases.161,162 Moreover, enhanced generation of IL-23 and IL-17 has been noted in colitis mouse models and UC patients, highlighting the involvement of the IL-23/IL-17 axis in the pathogenesis of UC.163,164 Initially, the IL-23 inhibitors were considered effective only in CD; however, a recent phase III clinical trial has demonstrated the anti-inflammatory effects of ustekinumab in UC patients.165 Th17 cells can also exert protective effects by producing anti-inflammatory cytokines, such as IL-22, which influences antimicrobial peptide production, mucus secretion, and epithelial cell differentiation and proliferation.166 Studies in UC patients and colitis mice have shown that IL-22 also promotes the growth of intestinal commensal bacteria and maintains intestinal microecological homeostasis through mediating host glycosylation or AHR signaling.167,168 Therefore, IL-22 amplification holds potential therapeutic implications for UC. However, several studies have shown that Th22 cells, a crucial source of IL-22 in the intestine, are significantly decreased in UC patients but not in CD patients.169 Moreover, Th17 cells have demonstrated plasticity, being able to convert to IFN-γ-producing Th1 or Th17/Th1 cells in response to IL-12 or IL-23 signaling, which contributes to the pathogenesis of colitis.170 Therefore, strategies that prevent the transition of Th17 cell precursors to Th1-like cells or inhibit the activity of Th1 or Th1-like cells deserve attention as potential interventions. Furthermore, the disruption of the Th17/Treg cell balance not only contributes to CD but is also implicated in the development of UC. Therefore, restoring the balance of Th17/Treg cells could have beneficial therapeutic effects on UC.
Besides CD4+ T cells, CD8+ T cells also play a role in the pathogenesis of IBD. Colonic tissue-resident memory CD8+ T cells possess the ability to rapidly respond to repetitive antigen exposure and may be involved in tissue damage in IBD. Nevertheless, the cellular phenotype, transcriptional regulation, and effector functions of CD8+ T cells, as well as their specific influence on IBD pathogenesis remain elusive. The constitution of CD8+ T cells in the colon is extremely heterogeneous, encompassing both effector and post-effector differentiated CD8+ T cells. CD8+ effector T cells related with UC are implicated in tissue destruction and TNF-α production, whereas CD8+ post-effector T cells expressing IL-26 possess innate characteristics that drive immune regulation and alleviate excessive inflammation.171 Interestingly, UC patients exhibit an increase in IL-26-producing CD8+ T cells compared to healthy individuals. However, in UC, these cells are persistently activated and display a “depleted” signature. To explore any functional implications, researchers employed a humanized IL-26 transgenic mice model and discovered that IL-26 inhibited the induction of pro-inflammatory cytokines(such as TNF-α) and chemokines (such as CXCL9 and CXCL10), which play a potentially protective role during the acute phase of UC.172 Thus, the imbalance between CD8+ post-effector T cells expressing IL-26 and CD8+ effector T cells may encourage tissue damage and participate in the etiology of UC.
B Cell Immune Responses in CD and UC
In addition to T cells, B cells have also emerged as active participants in the pathogenesis of CD. Normally, humoral immunity contributes to maintaining the balance between the intestinal microorganisms and the host immune system. B cells control the microbiota and prevent bacterial assault on epithelial cells by secreting immunoglobulin A (IgA) into the lumen. Perturbations in immunoglobulin subclasses observed in CD and UC indicate abnormalities in humoral immunity.173 Defective humoral immunity can result in inflammation in the small intestine by disrupting the balance of bile acid.174 Interestingly, CD patients exhibit persistently aberrant B cell responses, accompanied by increased molecular maturation of IgA and IgG, which relates to the formation of granuloma tissue.175 Furthermore, CD patients tend to generate antibodies against CD-associated bacterial antigens (such as bacterial flagellin (CBir1) and Escherichia coli outer membrane protein C). Although the pathogenic role of these antibodies in CD remains to be established, these observations convey a valuable message regarding humoral immune studies in IBD: the more antibacterial antibodies that are present, the higher their titers and the more severe the clinical process.176
However, B cells have not been adequately studied in UC. On the one hand, intestinal CD11b+ B cells are important immunosuppressive cells, crucial for maintaining intestinal homeostasis by secreting IgA and thereby ameliorating colitis.177 On the other hand, UC patients display an increased presence of naive B cells and IgG PCs. However, the diversity and maturation of the B cells appear to be diminished in the setting of UC.178,179 IgG rich PCs can enhance the immune response by recruiting inflammatory monocytes and Th17 cells via FcγR-dependent mechanisms.180 Moreover, a significantly decreased abundance of CD24hiCD38hiand CD5+ regulatory B cells (Bregs) has been observed in peripheral blood and intestinal tissues of UC patients, along with lower serum IL-10 levels. The abundance of Bregs and the levels of IL-10 exhibit a negative correlation with Mayo clinical scores in UC patients.181 Collectively, evidence from these studies points to a potential role of dysregulated B cell responses in the development of UC.
Updated Immunomodulator Approaches in IBD
Considering the overwhelming evidence that immune activation and inflammation are crucial for the pathogenesis of IBD, it is not astonishing that biologics and small molecule agents targeting the immune system have achieved breakthroughs in the clinical management of IBD. Table 1 summarizes clinical trials in IBD involving novel immune-therapeutics that have been completed or are ongoing. Currently, the most successful biologic agents for the treatment of IBD have turned out to be antibodies targeting TNF-α, of which infliximab (IFX), adalimumab (ADL), golimumab (GOLI), and certolizumab pegol (CZP) have received approval form the US Food and Drug Administration (FDA). The independent efficacy of these anti-TNF agents precisely reflects the pleiotropic role of TNF-α in the pathogenesis of IBD. Although anti-TNF therapy is quite potent, it is frequently limited by an initial non-response or loss of therapeutic effect over time. The non-response to anti-TNF-α treatment in IBD may be attributed to factors such as immunogenicity and proteolytic degradation of biological agents, as well as a unique network of interactions between immune cells.182–184 In light of these limitations, the evaluation of switching to a second anti-TNF-α drug or a novel drug with a distinct mechanism of action has been considered. The success of such a conversion depends on several factors, including the reasons for discontinuing the previous drug and the order of drug administration.185,186
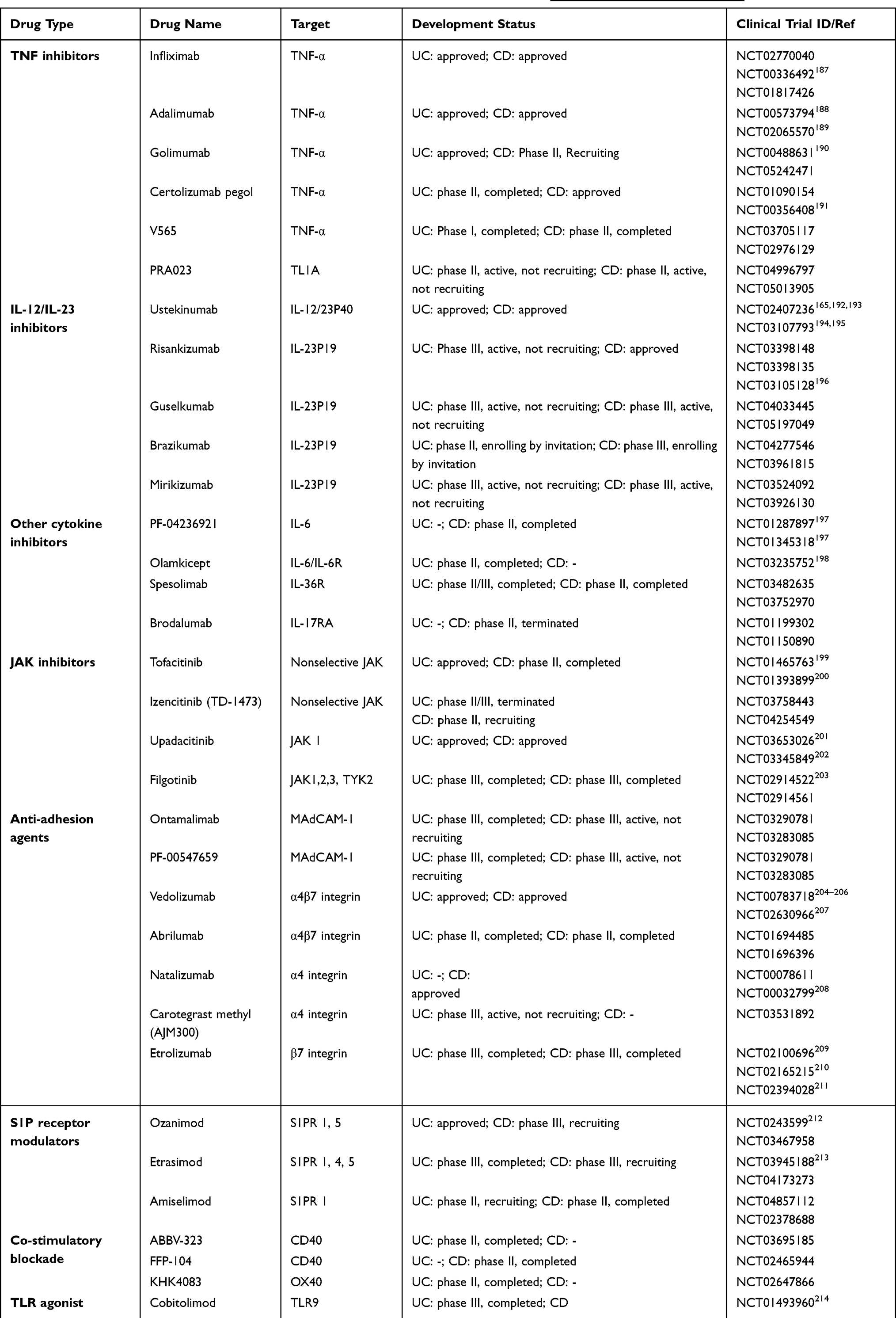 |
Table 1 Clinical Trials in IBD Involving Biological and Small Molecule Therapies (https://www.clinicaltrials.gov/) |
Agents targeting IL-12/IL-23 pathways have reported promising efficacy in the treatment of IBD. This includes IL-12/IL-23 p40 inhibitors (ustekinumab and briakinumab) and IL-23 p19 inhibitors (risankizumab, guselkumab, brazikumab, and mirikizumab). Currently, ustekinumab is the only approved drug in this class for both CD and UC treatment. Data from the phase III UNITI and UNIFI trials have shown that the long-term efficacy and safety of ustekinumab induction (130 mg) and maintenance (90 mg) was superior to placebo for moderate to severe CD and UC patients.165,215 Moreover, phase III ADVANCE and MOTIVATE trials have demonstrated the effectiveness and tolerability of risankizumab (600mg and 1200mg) in inducing remission in patients with moderate to severe CD.196 The phase III FORTIFY trial has further supported the maintenance of clinical remission for 1 year in patients with moderate to severe CD using risankizumab (180mg and 360mg).216 Thus, risankizumab has become the first approved inhibitor that specifically targets IL-23 p19 for the therapy of adult patients with moderate to severe CD based on the results from these two clinical trials. This represents a significant milestone in CD therapy. However, it is important to note that the consistency of such encouraging results for all drugs in this class relies on the outcomes of ongoing clinical trials.
The comparative efficacy of selective IL-23 inhibitors versus IL-12/IL-23 inhibitors was initially investigated in phase III UltIMMa-1 and UltIMMa-2 clinical trials involving psoriasis patients, wherein risankizumab demonstrated superior efficacy to ustekinumab.217 A similar phase III trial is currently ongoing to compare the efficacy of these two drugs in adult subjects with moderate to severe CD (NCT04524611). Encouraged by the favorable efficacy of antibodies targeting IL-23 p40 and p19, research efforts have turned towards strategies that target the IL-23/IL-17 axis within IBD. Activation of the IL-17R signaling pathway, which modulates the expression of α4β7 integrin on T cells and enhances intestinal homing and colonic inflammation, has been noted in IBD patients who did not respond to anti-TNF or anti-α4β7 therapy, thus highlighting IL-17R as a promising therapeutic target.218 Moreover, IL-17A inhibitors (secukinumab and ixekizumab) and IL-17A receptor inhibitors (brodalumab) have exhibited therapeutic benefits for psoriasis.219 However, their efficacy in the treatment of IBD has yielded conflicting results, with some studies reporting worsened colitis accompanied by compromised barrier function, potentially attributable to the protective effect of IL-17A on intestinal epithelial barrier integrity.220,221
However, the intricate cytokine network in IBD poses challenges when targeting a single cytokine, as it may lead to compensatory pro-inflammatory cytokine pathways. Therefore, alternative strategies that focus on cytokine signal transduction pathways have been explored to achieve more reliable and complete remission. One such pathway is the JAK-STAT signaling pathway, which is activated by cytokines and exerts a significant influence on IBD pathogenesis. Tofacitinib, a first-generation oral pan-JAK inhibitor, gained FDA authorization in 2018 for the clinical treatment of UC patients based on 3 phase III clinical trials (OCTAVE 1 and 2 induction trials and OCTAVE maintenance trial). The outcomes of these trials showed that tofacitinib induced and maintained clinical remission for 52 weeks in UC patients who were unresponsive to conventional or anti-TNF-α therapy.199 However, the clinical use of first-generation JAK inhibitors was limited due to their non-selectivity, which resulted in serious toxic side effects such as tumors, infections, and cardiovascular diseases, despite achieving good efficacy.199 To address these concerns, second-generation JAK inhibitors, such as upadacitinib (a selective JAK1 inhibitor), have been developed to more precisely regulate cytokine signal transduction in the JAK-STAT pathway, effectively resolving the safety issues in the first-generation JAK inhibitors.201 Upadacitinib received its first approval in China in February 2023 as a therapeutic option for adult patients diagnosed with moderately to severely active UC who had an insufficient response, intolerance to, or contraindication to one or more TNF-α inhibitors. Additionally, other potential and selective JAK inhibitors (filgotinib, ivarmacitinib, and izencitinib) are being investigated for improved safety and efficacy.
Another therapeutic approach in IBD involves homing of lymphocytes to the gut through inhibition of the interaction between the α4β7 integrin and adhesion molecule-1 (MAdCAM1) expressed by intestinal endothelial cells. Anti-integrins or anti-MAdCAM1 have been shown to be promising as IBD therapy. Natalizumab, a monoclonal antibody to α4 integrin, is the first anti-integrin antibody sanctioned by the FDA for the treatment of CD patients. Although it is effective in inducing and sustaining clinical remission in CD patients, natalizumab use was largely discontinued due to cases of progressive multifocal leukoencephalopathy, a consequence of natalizumab inhibiting lymphocyte homing to the brain via the α4β1 integrin and resulting in JC virus reactivation.222 Subsequently, vedolizumab (an anti-α4β7 integrin antibody) was developed to selectively inhibit lymphocyte migration into the inflamed intestine and demonstrated favorable therapeutic effects in UC and CD patients. Therefore, vedolizumab was approved by the FDA in 2014 as a treatment option for patients with moderately to severely active UC and CD who had previously experienced treatment failure with conventional therapy or anti-TNF-α agents. Significantly, a recent phase IV clinical trial reported vedolizumab was superior to placebo in realizing clinical remission of UC patients with chronic pouchitis after ileal pouch-anal anastomosis (IPAA).223 In addition, etrolizumab (anti-β7 integrin), carotegrast methyl (anti-α4 integrin), and ontamalimab (anti-MAdCAM-1) have shown therapeutic potential in IBD patients, and clinical trials to assess their efficacy and safety are still ongoing.224–226 The development of several sphingosine-1-phosphate (S1P) modulators for the treatment of IBD has also yielded encouraging results. Ozanimod, a selective agent targeting S1P receptor subtypes 1 and 5, effectively inhibits lymphocytes migration to sites of intestinal inflammation. The phase III True North trial revealed a significantly higher clinical response rate for ozanimod compared to placebo during induction and maintenance therapy.212 Consequently, in May 2021, the FDA granted approval for the use of ozanimod as an induction and maintenance therapy for patients with moderately to severely active UC. Additionally, other S1P modulators, including etrasimod and amiselimod, have been developed and are undergoing clinical trials with initial results showing promise.213,227
It is worth noting that most of the clinical trials primarily focused on clinical remission rather than immunological endpoints. Incorporating immunological endpoints into the design of future clinical studies is crucial, as it may prompt a shift towards combination therapies targeting multiple processes. Other potential therapeutic strategies and their combinations should be considered in the future, such as Treg cell therapy, the transfer of tolerogenic DCs, induction of regulatory immune responses, modulation of ILCs and B cells, and co-stimulatory blockade among others.178,228–233 Additionally, novel therapeutic approaches aimed at modulating immune pathways in IBD are being rapidly developed and translated into clinical applications. These include bacteriophage therapy, engineered microbiota, intestinal organoid technology, and nanovesicle therapy (Figure 4).43,234–236 Patient enrollment criteria and recruitment timing play pivotal roles in treatment outcomes since the inflammatory response evolves over time. Specifically, if dysregulated immune responses begin early in the disease process, investigators may not be able to improve the disease by giving immunomodulatory interventions after clinical diagnosis. In addition, notable differences in mucosal cellular immune responses between CD and UC have been identified.237 The diverse responses to highly selective agents such as cytokine inhibitors, JAK inhibitors, and lymphocyte homing blockade underscore the complexity of IBD immunopathogenesis, which is a regulated by a multilayered interplay of microbial and genetic factors.238 Leveraging the rich data from preclinical and clinical trials to study the precise intrinsic phenotypes and biomarkers of inflammation holds promise for stratifying and individualizing patients. This endeavor promotes the appropriate prescription of biological and small molecule therapies, thereby providing improved treatment options for future clinical trials.
 |
Figure 4 Novel therapies to modulate immunological pathways for IBD. |
Concluding Remarks
A wealth of evidence derived from animal models and clinical trials emphasizes the pivotal roles of innate and adaptive immune responses in the development and maintenance of inflammatory pathologies in IBD. However, the immunological mechanisms involved in the pathogenesis of IBD remain enigmatic. The precise balance between intestinal immune cell types and mechanisms of action changes significantly along the length of the intestine, reflecting differences in intestinal lumen contents, function, and anatomy. Consequently, the unique challenges posed by the gut necessitate constant immune system adaptations to maintain an appropriate immune landscape. In IBD, immune cells interact and are highly coordinated, forming a complex network that drives a chronic inflammatory response which result in persistent damage to the intestinal mucosa and immune dysregulation. Furthermore, the dialogue between the gut microbiota and its metabolites and the host is crucial for maintaining the immune landscape of the gut, which may explain why therapeutic interventions efficacious in genetically analogous autoimmune conditions (eg, secukinumab in psoriasis) yield opposite results in IBD.220 Furthermore, although T cells are key drivers of the intestinal inflammatory response, the precise contribution of distinct T cell subsets in orchestrating the intricate inflammatory mechanisms underlying the disparate clinical phenotypes, CD and UC, remains incompletely understood. Hence, identifying reliable markers that distinguish immune mechanisms driving UC and CD could significantly enhance our understanding of the distinct immune cell types and functional contributions involved in different stages of IBD.
As the clinical implementation of diverse immunotherapies gains momentum, several pressing challenges remain. First, a major issue with biologic and small molecule agents is the poor response or loss of response, which reflects the fact that the immune mechanisms of IBD are more complex than previously recognized. Therefore, the development of therapeutic agents should take into account factors such as the specific location and nature of inflammation, plasticity of immune cells, and cytokine production patterns and their mechanisms of action. Another challenge is to develop novel detection technologies, such as single-cell sequencing, to predict disease trajectories in order to unravel the critical stages of the immune dysregulation. To this end, comprehensive genomic, transcriptomic, and immunomic analyses are imperative, necessitating the collection of immune cells and tissues from individuals at genetic and environmental risk for IBD. This approach will facilitate the search for meaningful changes in responses to immune challenges rather than just comparing differences in immune cell types and functions between healthy and diseased cases. Finally, the discovery of accurate early diagnostic tools or biomarkers holds significant potential for providing effective treatments aimed at preventing, delaying, or halting the disease progression. A deeper unraveling of the intricate immunopathogenic mechanisms in IBD, coupled with the development of accurate diagnostic modalities, promises to foster tremendous therapeutic advances. Armed with this knowledge, clinicians will be empowered to intervene effectively in the future, employing redesigned or new immunomodulatory and anti-inflammatory approaches to mitigate disease progression.
Abbreviations
IBD, Inflammatory bowel disease; GI, Gastrointestinal; CD, Crohn’s disease; UC, Ulcerative colitis; IECs, Intestinal epithelial cells; DCs, Dendritic cells; ILCs, Innate lymphoid cells; PRRs, Pattern recognition receptors; Th, T helper; IL, Interleukin; LP, Lamina propria; Treg, Regulatory T; TRM, Tissue-resident memory; SFB, Segmental filamentous bacteria; TLR, Toll-like receptor; IFN, Interferon; PCs, Plasma cells; NOD2, Nucleotide-binding oligomerization domain 2; ATG16L1, Autophagy related protein 16 like protein 1; TNF, Tumor necrosis factor; TGF-β, Transforming growth factor β; NLRs, NOD-like receptors; MUC2, Mucin-2; ER, Endoplasmic reticulum; TREM-1, Triggering receptor expressed on myeloid cells 1; APCs, Antigen presenting cells; NK, Natural killer; TCR, T cell receptor; Tfh, T follicular helper; JAK, Janus kinase; STAT, Signal transducer and activator of transcription; FDA, Food and Drug Administration; MAdCAM1, Adhesion molecule-1; S1P, Sphingosine-1-phosphate.
Funding
This work was supported by Science and Technology Innovation Committee of Shenzhen (JCYJ20210324113802006, JCYJ2022053015180024, and JCYJ20210324113613035).
Disclosure
The authors declare no competing conflict of interest in this work.
References
1. Hodson R. Inflammatory bowel disease. Nature. 2016;540(7634):S97–S97. doi:10.1038/540S97a
2. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet Lond Engl. 2017;390(10114):2769–2778. doi:10.1016/S0140-6736(17)32448-0
3. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361(21):2066–2078. doi:10.1056/NEJMra0804647
4. Rogler G, Singh A, Kavanaugh A, Rubin DT. Extraintestinal manifestations of inflammatory bowel disease: current concepts, treatment, and implications for disease management. Gastroenterology. 2021;161(4):1118–1132. doi:10.1053/j.gastro.2021.07.042
5. Na SY, Moon W. Perspectives on current and novel treatments for inflammatory bowel disease. Gut Liver. 2019;13(6):604–616. doi:10.5009/gnl19019
6. Baumgart DC, Le Berre C. Newer biologic and small-molecule therapies for inflammatory bowel disease. J Med. 2021;385(14):1302–1315. doi:10.1056/NEJMra1907607
7. Pickard JM, Zeng MY, Caruso R, Núñez G. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017;279(1):70–89. doi:10.1111/imr.12567
8. Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13(1):3–10. doi:10.1016/j.autrev.2013.06.004
9. Ramos GP, Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. 2019;94(1):155–165. doi:10.1016/j.mayocp.2018.09.013
10. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med. 2020;383(27):2652–2664. doi:10.1056/NEJMra2002697
11. Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol Baltim Md. 1996;157(3):1261–1270.
12. Friedrich M, Pohin M, Jackson MA, et al. IL-1-driven stromal-neutrophil interactions define a subset of patients with inflammatory bowel disease that does not respond to therapies. Nat Med. 2021;27(11):1970–1981. doi:10.1038/s41591-021-01520-5
13. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14(10):667–685. doi:10.1038/nri3738
14. Lueschow SR, McElroy SJ. The paneth cell: the curator and defender of the immature small intestine. Front Immunol. 2020;11:587. doi:10.3389/fimmu.2020.00587
15. Ragab M, Schlichting H, Hicken M, et al. Azathioprine promotes intestinal epithelial cell differentiation into paneth cells and alleviates ileal crohn’s disease severity. Sci Rep. 2024;14(1):12879. doi:10.1038/s41598-024-63730-4
16. Rivera CA, Randrian V, Richer W, et al. Epithelial colonization by gut dendritic cells promotes their functional diversification. Immunity. 2022;55(1):129–144.e8. doi:10.1016/j.immuni.2021.11.008
17. Persson EK, Uronen-Hansson H, Semmrich M, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity. 2013;38(5):958–969. doi:10.1016/j.immuni.2013.03.009
18. Rivera-Nieves J, Ho J, Bamias G, et al. Antibody blockade of CCL25/CCR9 ameliorates early but not late chronic murine ileitis. Gastroenterology. 2006;131(5):1518–1529. doi:10.1053/j.gastro.2006.08.031
19. Kunkel EJ, Campbell JJ, Haraldsen G, et al. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J Exp Med. 2000;192(5):761–768. doi:10.1084/jem.192.5.761
20. Li Y, Innocentin S, Withers DR, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147(3):629–640. doi:10.1016/j.cell.2011.09.025
21. Wang X, Sumida H, Cyster JG. GPR18 is required for a normal CD8αα intestinal intraepithelial lymphocyte compartment. J Exp Med. 2014;211(12):2351–2359. doi:10.1084/jem.20140646
22. Reis BS, van Konijnenburg DP H, Grivennikov SI, Mucida D. Transcription factor T-bet regulates intraepithelial lymphocyte functional maturation. Immunity. 2014;41(2):244–256. doi:10.1016/j.immuni.2014.06.017
23. Wermers JD, McNamee EN, Wurbel MA, Jedlicka P, Rivera-Nieves J. The chemokine receptor CCR9 is required for the T-cell-mediated regulation of chronic ileitis in mice. Gastroenterology. 2011;140(5):1526–1535.e3. doi:10.1053/j.gastro.2011.01.044
24. Ivanov II, de Llanos Frutos R, Manel N, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4(4):337–349. doi:10.1016/j.chom.2008.09.009
25. Yokoi T, Murakami M, Kihara T, et al. Identification of a unique subset of tissue-resident memory CD4+ T cells in crohn’s disease. Proc Natl Acad Sci U S A. 2023;120(1):e2204269120. doi:10.1073/pnas.2204269120
26. Boland BS, He Z, Tsai MS, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol. 2020;5(50):eabb4432. doi:10.1126/sciimmunol.abb4432
27. Liu TC, Kern JT, Jain U, et al. Western diet induces paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe. 2021;29(6):988–1001.e6. doi:10.1016/j.chom.2021.04.004
28. Kobayashi N, Takahashi D, Takano S, Kimura S, Hase K. The roles of peyer’s patches and microfold cells in the gut immune system: relevance to autoimmune diseases. Front Immunol. 2019;10:2345. doi:10.3389/fimmu.2019.02345
29. Lai NY, Musser MA, Pinho-Ribeiro FA, et al. Gut-innervating nociceptor neurons regulate peyer’s patch microfold cells and SFB levels to mediate salmonella host defense. Cell. 2020;180(1):33–49.e22. doi:10.1016/j.cell.2019.11.014
30. Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–498. doi:10.1016/j.cell.2009.09.033
31. Price AE, Shamardani K, Lugo KA, et al. A map of toll-like receptor expression in the intestinal epithelium reveals distinct spatial, cell type-specific, and temporal patterns. Immunity. 2018;49(3):560–575.e6. doi:10.1016/j.immuni.2018.07.016
32. Parikh K, Antanaviciute A, Fawkner-Corbett D, et al. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature. 2019;567(7746):49–55. doi:10.1038/s41586-019-0992-y
33. Denning TL, Norris BA, Medina-Contreras O, et al. Functional specializations of intestinal dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J Immunol Baltim Md. 2011;187(2):733–747. doi:10.4049/jimmunol.1002701
34. Nagashima R, Maeda K, Imai Y, Takahashi T. Lamina propria macrophages in the human gastrointestinal mucosa: their distribution, immunohistological phenotype, and function. J Histochem Cytochem off J Histochem Soc. 1996;44(7):721–731. doi:10.1177/44.7.8675993
35. Bain CC, Origin SA. Differentiation, and function of intestinal macrophages. Front Immunol. 2018;9:2733. doi:10.3389/fimmu.2018.02733
36. Bain CC, Scott CL, Uronen-Hansson H, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6(3):498–510. doi:10.1038/mi.2012.89
37. Tamoutounour S, Henri S, Lelouard H, et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol. 2012;42(12):3150–3166. doi:10.1002/eji.201242847
38. Mahida YR, Patel S, Gionchetti P, Vaux D, Jewell DP. Macrophage subpopulations in lamina propria of normal and inflamed colon and terminal ileum. Gut. 1989;30(6):826–834. doi:10.1136/gut.30.6.826
39. Gross-Vered M, Trzebanski S, Shemer A, et al. Defining murine monocyte differentiation into colonic and ileal macrophages. eLife. 2020;9:e49998. doi:10.7554/eLife.49998
40. Kang B, Alvarado LJ, Kim T, et al. Commensal microbiota drive the functional diversification of colon macrophages. Mucosal Immunol. 2020;13(2):216–229. doi:10.1038/s41385-019-0228-3
41. Muzaki ARBM, Tetlak P, Sheng J, et al. Intestinal CD103(+)CD11b(-) dendritic cells restrain colitis via IFN-γ-induced anti-inflammatory response in epithelial cells. Mucosal Immunol. 2016;9(2):336–351. doi:10.1038/mi.2015.64
42. Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:6015):337–341. doi:10.1126/science.1198469
43. Carasso S, Zaatry R, Hajjo H, et al. Inflammation and bacteriophages affect DNA inversion states and functionality of the gut microbiota. Cell Host Microbe. 2024;32(3):322–334.e9. doi:10.1016/j.chom.2024.02.003
44. Sterlin D, Fadlallah J, Adams O, et al. Human IgA binds a diverse array of commensal bacteria. J Exp Med. 2020;217(3):e20181635. doi:10.1084/jem.20181635
45. Kett K, Baklien K, Bakken A, Kral JG, Fausa O, Brandtzaeg P. Intestinal B-cell isotype response in relation to local bacterial load: evidence for immunoglobulin A subclass adaptation. Gastroenterology. 1995;109(3):819–825. doi:10.1016/0016-5085(95)90389-5
46. Steffen U, Koeleman CA, Sokolova MV, et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat Commun. 2020;11(1):120. doi:10.1038/s41467-019-13992-8
47. Patman G. Crohn’s disease. TCF1 regulates paneth cell α-defensins. Nat Rev Gastroenterol Hepatol. 2014;11(9):517. doi:10.1038/nrgastro.2014.128
48. Shanahan MT, Carroll IM, Grossniklaus E, et al. Mouse paneth cell antimicrobial function is independent of Nod2. Gut. 2014;63(6):903–910. doi:10.1136/gutjnl-2012-304190
49. Klionsky DJ. Crohn’s disease, autophagy, and the paneth cell. N Engl J Med. 2009;360(17):1785–1786. doi:10.1056/NEJMcibr0810347
50. Liu B, Gulati AS, Cantillana V, et al. Irgm1-deficient mice exhibit paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2013;305(8):G573–584. doi:10.1152/ajpgi.00071.2013
51. Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62(10):1505–1510. doi:10.1136/gutjnl-2012-303954
52. Nguyen HTT, Dalmasso G, Müller S, Carrière J, Seibold F, Darfeuille-Michaud A. Crohn’s disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology. 2014;146(2):508–519. doi:10.1053/j.gastro.2013.10.021
53. Zigmond E, Bernshtein B, Friedlander G, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40(5):720–733. doi:10.1016/j.immuni.2014.03.012
54. Han D, Walsh MC, Cejas PJ, et al. Dendritic cell expression of the signaling molecule TRAF6 is critical for gut microbiota-dependent immune tolerance. Immunity. 2013;38(6):1211–1222. doi:10.1016/j.immuni.2013.05.012
55. Païdassi H, Acharya M, Zhang A, et al. Preferential expression of integrin αvβ8 promotes generation of regulatory T cells by mouse CD103+ dendritic cells. Gastroenterology. 2011;141(5):1813–1820. doi:10.1053/j.gastro.2011.06.076
56. Tanoue T, Atarashi K, Honda K. Development and maintenance of intestinal regulatory T cells. Nat Rev Immunol. 2016;16(5):295–309. doi:10.1038/nri.2016.36
57. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111(6):2247–2252. doi:10.1073/pnas.1322269111
58. Singh N, Thangaraju M, Prasad PD, et al. Blockade of dendritic cell development by bacterial fermentation products butyrate and propionate through a transporter (Slc5a8)-dependent inhibition of histone deacetylases. J Biol Chem. 2010;285(36):27601–27608. doi:10.1074/jbc.M110.102947
59. Park J, Kim M, Kang SG, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015;8(1):80–93. doi:10.1038/mi.2014.44
60. Morita N, Umemoto E, Fujita S, et al. GPR31-dependent dendrite protrusion of intestinal CX3CR1+ cells by bacterial metabolites. Nature. 2019;566(7742):110–114. doi:10.1038/s41586-019-0884-1
61. Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker HC, Kasper DL. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe. 2014;15(4):413–423. doi:10.1016/j.chom.2014.03.006
62. Martín R, Chamignon C, Mhedbi-Hajri N, et al. The potential probiotic Lactobacillus rhamnosus CNCM I-3690 strain protects the intestinal barrier by stimulating both mucus production and cytoprotective response. Sci Rep. 2019;9(1):5398. doi:10.1038/s41598-019-41738-5
63. Hayashi A, Sato T, Kamada N, et al. A single strain of Clostridium butyricum induces intestinal IL-10-producing macrophages to suppress acute experimental colitis in mice. Cell Host Microbe. 2013;13(6):711–722. doi:10.1016/j.chom.2013.05.013
64. Goto Y, Panea C, Nakato G, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40(4):594–607. doi:10.1016/j.immuni.2014.03.005
65. Nieuwenhuis EES, Matsumoto T, Lindenbergh D, et al. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J Clin Invest. 2009;119(5):1241–1250. doi:10.1172/JCI36509
66. Suzuki K, Meek B, Doi Y, et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci U S A. 2004;101(7):1981–1986. doi:10.1073/pnas.0307317101
67. Michielan A, D’Incà R. Intestinal permeability in inflammatory bowel disease: pathogenesis, clinical evaluation, and therapy of leaky gut. Mediators Inflamm. 2015;628157. doi:10.1155/2015/628157
68. van der Post S, Jabbar KS, Birchenough G, et al. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut. 2019;68(12):2142–2151. doi:10.1136/gutjnl-2018-317571
69. Nyström EEL, Birchenough GMH, van der Post S, et al. Calcium-activated Chloride Channel Regulator 1 (CLCA1) controls mucus expansion in colon by proteolytic activity. EBioMedicine. 2018;33:134–143. doi:10.1016/j.ebiom.2018.05.031
70. Ehrencrona E, van der Post S, Gallego P, et al. The IgGFc-binding protein FCGBP is secreted with all GDPH sequences cleaved but maintained by interfragment disulfide bonds. J Biol Chem. 2021;297(1):100871. doi:10.1016/j.jbc.2021.100871
71. der Sluis MV, De Koning BAE, Bruijn ACJMD, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131(1):117–129. doi:10.1053/j.gastro.2006.04.020
72. Nyström EEL, Martinez-Abad B, Arike L, et al. An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function. Science. 2021;372:eabb1590. doi:10.1126/science.abb1590
73. McDole JR, Wheeler LW, McDonald KG, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature. 2012;483(7389):345–349. doi:10.1038/nature10863
74. Kulkarni DH, Gustafsson JK, Knoop KA, et al. Goblet cell associated antigen passages support the induction and maintenance of oral tolerance. Mucosal Immunol. 2020;13(2):271–282. doi:10.1038/s41385-019-0240-7
75. Knoop KA, McDonald KG, Coughlin PE, et al. Synchronization of mothers and offspring promotes tolerance and limits allergy. JCI Insight. 2020;5(15):e137943. doi:10.1172/jci.insight.137943
76. Knoop KA, Gustafsson JK, McDonald KG, et al. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci Immunol. 2017;2(18):eaao1314. doi:10.1126/sciimmunol.aao1314
77. Gubatan J, Holman DR, Puntasecca CJ, Polevoi D, Rubin SJ, Rogalla S. Antimicrobial peptides and the gut microbiome in inflammatory bowel disease. World J Gastroenterol. 2021;27(43):7402–7422. doi:10.3748/wjg.v27.i43.7402
78. Salzman NH, Bevins CL. Dysbiosis--a consequence of paneth cell dysfunction. Semin Immunol. 2013;25(5):334–341. doi:10.1016/j.smim.2013.09.006
79. Wang SL, Shao BZ, Zhao SB, et al. Impact of paneth cell autophagy on inflammatory bowel disease. Front Immunol. 2018;9:693. doi:10.3389/fimmu.2018.00693
80. Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020;16(1):38–51. doi:10.1080/15548627.2019.1635384
81. Wehkamp J, Salzman NH, Porter E, et al. Reduced paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A. 2005;102(50):18129–18134. doi:10.1073/pnas.0505256102
82. Biswas A, Liu YJ, Hao L, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A. 2010;107(33):14739–14744. doi:10.1073/pnas.1003363107
83. Wang H, Zhang X, Zuo Z, et al. Rip2 is required for Nod2-mediated lysozyme sorting in paneth cells. J Immunol Baltim Md. 2017;198(9):3729–3736. doi:10.4049/jimmunol.1601583
84. Jones EJ, Matthews ZJ, Gul L, et al. Integrative analysis of Paneth cell proteomic and transcriptomic data from intestinal organoids reveals functional processes dependent on autophagy. Dis Model Mech. 2019;12(3):dmm037069. doi:10.1242/dmm.037069
85. Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134(5):743–756. doi:10.1016/j.cell.2008.07.021
86. Zhao F, Edwards R, Dizon D, et al. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2-/- mice. Dev Biol. 2010;338(2):270–279. doi:10.1016/j.ydbio.2009.12.008
87. Delfini M, Stakenborg N, Viola MF, Boeckxstaens G. Macrophages in the gut: masters in multitasking. Immunity. 2022;55(9):1530–1548. doi:10.1016/j.immuni.2022.08.005
88. Smith AM, Rahman FZ, Hayee B, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206(9):1883–1897. doi:10.1084/jem.20091233
89. Kamada N, Hisamatsu T, Okamoto S, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. 2008;118(6):2269–2280. doi:10.1172/JCI34610
90. Caër C, Gorreja F, Forsskåhl SK, et al. TREM-1+ macrophages define a pathogenic cell subset in the intestine of crohn’s disease patients. J Crohns Colitis. 2021;15(8):1346–1361. doi:10.1093/ecco-jcc/jjab022
91. Bain CC, Bravo-Blas A, Scott CL, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929–937. doi:10.1038/ni.2967
92. Bernardo D, Marin AC, Fernández-Tomé S, et al. Human intestinal pro-inflammatory CD11chighCCR2+CX3CR1+ macrophages, but not their tolerogenic CD11c-CCR2-CX3CR1- counterparts, are expanded in inflammatory bowel disease. Mucosal Immunol. 2018;11(4):1114–1126. doi:10.1038/s41385-018-0030-7
93. Weber B, Saurer L, Schenk M, Dickgreber N, Mueller C. CX3CR1 defines functionally distinct intestinal mononuclear phagocyte subsets which maintain their respective functions during homeostatic and inflammatory conditions. Eur J Immunol. 2011;41(3):773–779. doi:10.1002/eji.201040965
94. Medina-Contreras O, Geem D, Laur O, et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest. 2011;121(12):4787–4795. doi:10.1172/JCI59150
95. Cerovic V, Bain CC, Mowat AM, Milling SWF. Intestinal macrophages and dendritic cells: what’s the difference? Trends Immunol. 2014;35(6):270–277. doi:10.1016/j.it.2014.04.003
96. Arques JL, Hautefort I, Ivory K, et al. Salmonella induces flagellin- and MyD88-dependent migration of bacteria-capturing dendritic cells into the gut lumen. Gastroenterology. 2009;137(2):579–587,587.e1–2. doi:10.1053/j.gastro.2009.04.010
97. Sun CM, Hall JA, Blank RB, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204(8):1775–1785. doi:10.1084/jem.20070602
98. Chieppa M, Rescigno M, Huang AYC, Germain RN. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203(13):2841–2852. doi:10.1084/jem.20061884
99. Magnusson MK, Brynjólfsson SF, Dige A, et al. Macrophage and dendritic cell subsets in IBD: ALDH+ cells are reduced in colon tissue of patients with ulcerative colitis regardless of inflammation. Mucosal Immunol. 2016;9(1):171–182. doi:10.1038/mi.2015.48
100. Moreira TG, Mangani D, Cox LM, et al. PD-L1+ and XCR1+ dendritic cells are region-specific regulators of gut homeostasis. Nat Commun. 2021;12(1):4907. doi:10.1038/s41467-021-25115-3
101. Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209(1):139–155. doi:10.1084/jem.20101387
102. Silva MA, Quera R, Valenzuela J, Salim SY, Söderholm JD, Perdue MH. Dendritic cells and toll-like receptors 2 and 4 in the ileum of Crohn’s disease patients. Dig Dis Sci. 2008;53(7):1917–1928. doi:10.1007/s10620-007-0105-x
103. Hepworth MR, Fung TC, Masur SH, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science. 2015;348(6238):1031–1035. doi:10.1126/science.aaa4812
104. Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517(7534):293–301. doi:10.1038/nature14189
105. Kim CH, Hashimoto-Hill S, Kim M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol. 2016;37(1):68–79. doi:10.1016/j.it.2015.11.003
106. Eberl G, Colonna M, Di Santo JP, McKenzie ANJ. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348(6237):aaa6566. doi:10.1126/science.aaa6566
107. Kim J, Ryu S, Kim HY. Innate lymphoid cells in tissue homeostasis and disease pathogenesis. Mol Cells. 2021;44(5):301–309. doi:10.14348/molcells.2021.0053
108. Wong SH, Walker JA, Jolin HE, et al. Transcription factor RORα is critical for nuocyte development. Nat Immunol. 2012;13(3):229–236. doi:10.1038/ni.2208
109. Li J, Glover SC. Innate lymphoid cells in inflammatory bowel disease. Arch Immunol Ther Exp. 2018;66(6):415–421. doi:10.1007/s00005-018-0519-5
110. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A. 2015;112(34):10762–10767. doi:10.1073/pnas.1509070112
111. Wu Y, Shen J. Innate lymphoid cells in crohn’s disease. Front Immunol. 2020;11:554880. doi:10.3389/fimmu.2020.554880
112. Vonarbourg C, Mortha A, Bui VL, et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt(+) innate lymphocytes. Immunity. 2010;33(5):736–751. doi:10.1016/j.immuni.2010.10.017
113. Bernink JH, Peters CP, Munneke M, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14(3):221–229. doi:10.1038/ni.2534
114. Geremia A, Arancibia-Cárcamo CV, Fleming MPP, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. 2011;208(6):1127–1133. doi:10.1084/jem.20101712
115. Leung JM, Loke P. A role for IL-22 in the relationship between intestinal helminths, gut microbiota and mucosal immunity. Int J Parasitol. 2013;43(3–4):253–257. doi:10.1016/j.ijpara.2012.10.015
116. Qiu J, Guo X, Chen ZME, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39(2):386–399. doi:10.1016/j.immuni.2013.08.002
117. Buonocore S, Ahern PP, Uhlig HH, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464(7293):1371–1375. doi:10.1038/nature08949
118. Chen L, He Z, Slinger E, et al. IL-23 activates innate lymphoid cells to promote neonatal intestinal pathology. Mucosal Immunol. 2015;8(2):390–402. doi:10.1038/mi.2014.77
119. Hosomi S, Grootjans J, Tschurtschenthaler M, et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J Exp Med. 2017;214(10):2985–2997. doi:10.1084/jem.20162041
120. Takayama T, Kamada N, Chinen H, et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology. 2010;139(3):882–892,892.e1–3. doi:10.1053/j.gastro.2010.05.040
121. Zhou H, Xie X, Jiang B, Ke C. NKp46+ lamina propria natural killer cells undergo metabolic reprogramming in a mouse experimental colitis model. Inflamm Res off J Eur Histamine Res Soc Al. 2020;69(4):401–414. doi:10.1007/s00011-020-01324-2
122. Ochel A, Tiegs G, Neumann K. Type 2 innate lymphoid cells in liver and gut: from current knowledge to future perspectives. Int J Mol Sci. 2019;20(8):1896. doi:10.3390/ijms20081896
123. Forkel M, van Tol S, Höög C, Michaëlsson J, Almer S, Mjösberg J. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established crohn’s disease and ulcerative colitis. J Crohn's Colitis. 2019;13(1):67–78. doi:10.1093/ecco-jcc/jjy119
124. Camelo A, Barlow JL, Drynan LF, et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J Gastroenterol. 2012;47(11):1198–1211. doi:10.1007/s00535-012-0591-2
125. Lim AI, Menegatti S, Bustamante J, et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med. 2016;213(4):569–583. doi:10.1084/jem.20151750
126. Li S, Bostick JW, Ye J, et al. Aryl hydrocarbon receptor signaling cell intrinsically inhibits intestinal group 2 innate lymphoid cell function. Immunity. 2018;49(5):915–928.e5. doi:10.1016/j.immuni.2018.09.015
127. Xiong X, Cheng Z, Wu F, et al. Berberine in the treatment of ulcerative colitis: a possible pathway through Tuft cells. Biomed Pharma Biomed Pharm. 2021;134:111129. doi:10.1016/j.biopha.2020.111129
128. Chen YS, Chen IB, Pham G, et al. IL-17-producing γδ T cells protect against Clostridium difficile infection. J Clin Invest. 2020;130(5):2377–2390. doi:10.1172/JCI127242
129. Dupraz L, Magniez A, Rolhion N, et al. Gut microbiota-derived short-chain fatty acids regulate IL-17 production by mouse and human intestinal γδ T cells. Cell Rep. 2021;36(1):109332. doi:10.1016/j.celrep.2021.109332
130. Matsuzawa-Ishimoto Y, Yao X, Koide A, et al. The γδ IEL effector API5 masks genetic susceptibility to paneth cell death. Nature. 2022;610(7932):547–554. doi:10.1038/s41586-022-05259-y
131. Di Sabatino A, Lenti MV, Giuffrida P, Vanoli A, Corazza GR. New insights into immune mechanisms underlying autoimmune diseases of the gastrointestinal tract. Autoimmun Rev. 2015;14(12):1161–1169. doi:10.1016/j.autrev.2015.08.004
132. Giuffrida P, Corazza GR, Di Sabatino A. Old and new lymphocyte players in inflammatory bowel disease. Dig Dis Sci. 2018;63(2):277–288. doi:10.1007/s10620-017-4892-4
133. DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol. 2016;16(3):149–163. doi:10.1038/nri.2015.18
134. de Souza HSP, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13(1):13–27. doi:10.1038/nrgastro.2015.186
135. Ruterbusch M, Pruner KB, Shehata L, Pepper M. In vivo CD4+ T cell differentiation and function: revisiting the Th1/Th2 paradigm. Annu Rev Immunol. 2020;38:705–725. doi:10.1146/annurev-immunol-103019-085803
136. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3(2):133–146. doi:10.1038/nri1001
137. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. 2018;87:1–15. doi:10.1016/j.jaut.2017.12.007
138. Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol. 2019;41(3):283–297. doi:10.1007/s00281-019-00733-8
139. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi:10.1146/annurev.immunol.021908.132710
140. Cui X, Jiao C, Wang D, et al. Elevated levels of IL-27 are associated with disease activity in patients with crohn’s disease. Mediators Inflamm. 2021;2021:5527627. doi:10.1155/2021/5527627
141. Sakuraba A, Sato T, Kamada N, Kitazume M, Sugita A, Hibi T. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn’s disease. Gastroenterology. 2009;137(5):1736–1745. doi:10.1053/j.gastro.2009.07.049
142. Teng MWL, Bowman EP, McElwee JJ, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015;21(7):719–729. doi:10.1038/nm.3895
143. Schmitt H, Neurath MF, Atreya R. Role of the IL23/IL17 pathway in crohn’s disease. Front Immunol. 2021;12:622934. doi:10.3389/fimmu.2021.622934
144. Schmitt H, Billmeier U, Dieterich W, et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in crohn’s disease. Gut. 2019;68(5):814–828. doi:10.1136/gutjnl-2017-315671
145. Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52(1):65–70. doi:10.1136/gut.52.1.65
146. Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61(12):1693–1700. doi:10.1136/gutjnl-2011-301668
147. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol. 2013;8:477–512. doi:10.1146/annurev-pathol-011110-130318
148. Bacchetta R, Passerini L, Gambineri E, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. 2006;116(6):1713–1722. doi:10.1172/JCI25112
149. Zhou L, Chu C, Teng F, et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature. 2019;568(7752):405–409. doi:10.1038/s41586-019-1082-x
150. Reikvam DH, Perminow G, Lyckander LG, et al. Increase of regulatory T cells in ileal mucosa of untreated pediatric crohn’s disease patients. Scand J Gastroenterol. 2011;46(5):550–560. doi:10.3109/00365521.2011.551887
151. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J Clin Invest. 2001;108(4):601–609. doi:10.1172/JCI12821
152. Goldberg R, Scotta C, Cooper D, et al. Correction of defective T-regulatory cells from patients with crohn’s disease by ex vivo ligation of retinoic acid receptor-α. Gastroenterology. 2019;156(6):1775–1787. doi:10.1053/j.gastro.2019.01.025
153. Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129(2):550–564. doi:10.1016/j.gastro.2005.05.002
154. Kmieć Z, Cyman M, Ślebioda TJ. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Adv Med Sci. 2017;62(1):1–16. doi:10.1016/j.advms.2016.09.001
155. Seidelin JB, Coskun M, Kvist PH, Holm TL, Holgersen K, Nielsen OH. IL-33 promotes GATA-3 polarization of gut-derived T cells in experimental and ulcerative colitis. J Gastroenterol. 2015;50(2):180–190. doi:10.1007/s00535-014-0982-7
156. Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17(5):629–638. doi:10.1016/s1074-7613(02)00453-3
157. Danese S, Rudziński J, Brandt W, et al. Tralokinumab for moderate-to-severe UC: a randomised, double-blind, placebo-controlled, phase IIa study. Gut. 2015;64(2):243–249. doi:10.1136/gutjnl-2014-308004
158. Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. 2019;20(8):970–979. doi:10.1038/s41590-019-0415-0
159. Mavroudis G, Magnusson MK, Isaksson S, et al. Mucosal and systemic immune profiles differ during early and late phases of the disease in patients with active ulcerative colitis. J Crohn's Colitis. 2019;13(11):1450–1458. doi:10.1093/ecco-jcc/jjz072
160. Gerlach K, Hwang Y, Nikolaev A, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol. 2014;15(7):676–686. doi:10.1038/ni.2920
161. Baraldo S, Faffe DS, Moore PE, et al. Interleukin-9 influences chemokine release in airway smooth muscle: role of ERK. Am J Physiol Lung Cell Mol Physiol. 2003;284(6):L1093–L1102. doi:10.1152/ajplung.00300.2002
162. Pearl DS, Shah K, Whittaker MA, et al. Cytokine mucosal expression in ulcerative colitis, the relationship between cytokine release and disease activity. J Crohn's Colitis. 2013;7(6):481–489. doi:10.1016/j.crohns.2012.07.022
163. Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116(5):1310–1316. doi:10.1172/JCI21404
164. Ohman L, Dahlén R, Isaksson S, et al. Serum IL-17A in newly diagnosed treatment-naive patients with ulcerative colitis reflects clinical disease severity and predicts the course of disease. Inflamm Bowel Dis. 2013;19(11):2433–2439. doi:10.1097/MIB.0b013e3182a563cb
165. Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381(13):1201–1214. doi:10.1056/NEJMoa1900750
166. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12(5):383–390. doi:10.1038/ni.2025
167. Nagao-Kitamoto H, Leslie JL, Kitamoto S, et al. Interleukin-22-mediated host glycosylation prevents Clostridioides difficile infection by modulating the metabolic activity of the gut microbiota. Nat Med. 2020;26(4):608–617. doi:10.1038/s41591-020-0764-0
168. Mar JS, Ota N, Pokorzynski ND, et al. IL-22 alters gut microbiota composition and function to increase aryl hydrocarbon receptor activity in mice and humans. Microbiome. 2023;11(1):47. doi:10.1186/s40168-023-01486-1
169. Leung JM, Davenport M, Wolff MJ, et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol. 2014;7(1):124–133. doi:10.1038/mi.2013.31
170. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci U S A. 2015;112(22):7061–7066. doi:10.1073/pnas.1415675112
171. Ray K. Deciphering the role of CD8+ T cells in IBD: from single-cell analysis to biomarkers. Nat Rev Gastroenterol Hepatol. 2020;17(10):595. doi:10.1038/s41575-020-00362-9
172. Corridoni D, Antanaviciute A, Gupta T, et al. Single-cell atlas of colonic CD8+ T cells in ulcerative colitis. Nat Med. 2020;26(9):1480–1490. doi:10.1038/s41591-020-1003-4
173. Scott MG, Nahm MH, Macke K, Nash GS, Bertovich MJ, MacDermott RP. Spontaneous secretion of IgG subclasses by intestinal mononuclear cells: differences between ulcerative colitis, Crohn’s disease, and controls. Clin Exp Immunol. 1986;66(1):209–215.
174. Mohammed AD, Mohammed Z, Roland MM, et al. Defective humoral immunity disrupts bile acid homeostasis which promotes inflammatory disease of the small bowel. Nat Commun. 2022;13(1):525. doi:10.1038/s41467-022-28126-w
175. Timmermans WMC, van Laar JAM, van der Houwen TB, et al. B-cell dysregulation in crohn’s disease is partially restored with infliximab therapy. PLoS One. 2016;11(7):e0160103. doi:10.1371/journal.pone.0160103
176. Mow WS, Vasiliauskas EA, Lin YC, et al. Association of antibody responses to microbial antigens and complications of small bowel crohn’s disease. Gastroenterology. 2004;126(2):414–424. doi:10.1053/j.gastro.2003.11.015
177. Fu Y, Wang Z, Yu B, et al. Intestinal CD11b+ B cells ameliorate colitis by secreting immunoglobulin A. Front Immunol. 2021;12:697725. doi:10.3389/fimmu.2021.697725
178. Spencer J, Bemark M. Human intestinal B cells in inflammatory diseases. Nat Rev Gastroenterol Hepatol. 2023;20(4):254–265. doi:10.1038/s41575-023-00755-6
179. Uzzan M, Martin JC, Mesin L, et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat Med. 2022;28(4):766–779. doi:10.1038/s41591-022-01680-y
180. Castro-Dopico T, Dennison TW, Ferdinand JR, et al. Anti-commensal IgG drives intestinal inflammation and type 17 immunity in ulcerative colitis. Immunity. 2019;50(4):1099–1114.e10. doi:10.1016/j.immuni.2019.02.006
181. Wang X, Zhu Y, Zhang M, Wang H, Jiang Y, Gao P. Ulcerative colitis is characterized by a decrease in regulatory B cells. J Crohn's Colitis. 2016;10(10):1212–1223. doi:10.1093/ecco-jcc/jjw074
182. Atreya R, Neurath MF. Mechanisms of molecular resistance and predictors of response to biological therapy in inflammatory bowel disease. Lancet Gastroenterol Hepatol. 2018;3(11):790–802. doi:10.1016/S2468-1253(18)30265-6
183. Biancheri P, Brezski RJ, Di Sabatino A, et al. Proteolytic cleavage and loss of function of biologic agents that neutralize tumor necrosis factor in the mucosa of patients with inflammatory bowel disease. Gastroenterology. 2015;149(6):1564–1574.e3. doi:10.1053/j.gastro.2015.07.002
184. Martin JC, Chang C, Boschetti G, et al. Single-cell analysis of crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell. 2019;178(6):1493–1508.e20. doi:10.1016/j.cell.2019.08.008
185. Gisbert JP, Marín AC, McNicholl AG, Chaparro M. Systematic review with meta-analysis: the efficacy of a second anti-TNF in patients with inflammatory bowel disease whose previous anti-TNF treatment has failed. Aliment Pharmacol Ther. 2015;41(7):613–623. doi:10.1111/apt.13083
186. Viola A, Pugliese D, Renna S, et al. Outcome in ulcerative colitis after switch from Adalimumab/golimumab to infliximab: a multicenter retrospective study. Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver. 2019;51(4):510–515. doi:10.1016/j.dld.2018.10.013
187. Singh S, Proudfoot JA, Dulai PS, et al. No benefit of concomitant 5-aminosalicylates in patients with ulcerative colitis escalated to biologic therapy: pooled analysis of individual participant data from clinical trials. Am J Gastroenterol. 2018;113(8):1197–1205. doi:10.1038/s41395-018-0144-2
188. Ryan C, Sobell JM, Leonardi CL, et al. Safety of adalimumab dosed every week and every other week: focus on patients with hidradenitis suppurativa or psoriasis. Am J Clin Dermatol. 2018;19(3):437–447. doi:10.1007/s40257-017-0341-6
189. D’Haens GR, Sandborn WJ, Loftus EV, et al. Higher vs standard adalimumab induction dosing regimens and two maintenance strategies: randomized SERENE CD trial results. Gastroenterology. 2022;162(7):1876–1890. doi:10.1053/j.gastro.2022.01.044
190. Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):96–109.e1. doi:10.1053/j.gastro.2013.06.010
191. Loftus EV, Colombel JF, Schreiber S, et al. Safety of long-term treatment with certolizumab pegol in patients with crohn’s disease, based on a pooled analysis of data from clinical trials. Clin Gastroenterol Hepatol. 2016;14(12):1753–1762. doi:10.1016/j.cgh.2016.07.019
192. Li K, Marano C, Zhang H, et al. Relationship between combined histologic and endoscopic endpoints and efficacy of ustekinumab treatment in patients with ulcerative colitis. Gastroenterology. 2020;159(6):2052–2064. doi:10.1053/j.gastro.2020.08.037
193. Danese S, Sands BE, Abreu MT, et al. Early symptomatic improvement after ustekinumab therapy in patients with ulcerative colitis: 16-week data from the UNIFI trial. Clin Gastroenterol Hepatol. 2022;20(12):2858–2867.e5. doi:10.1016/j.cgh.2022.02.050
194. Danese S, Vermeire S, D’Haens G, et al. Treat to target versus standard of care for patients with crohn’s disease treated with ustekinumab (STARDUST): an open-label, multicentre, randomised Phase 3b trial. Lancet Gastroenterol Hepatol. 2022;7(4):294–306. doi:10.1016/S2468-1253(21)00474-X
195. Kucharzik T, Wilkens R, D’Agostino MA, et al. Early ultrasound response and progressive transmural remission after treatment with ustekinumab in crohn’s disease. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc. 2023;21(1):153–163.e12. doi:10.1016/j.cgh.2022.05.055
196. D’Haens G, Panaccione R, Baert F, et al. Risankizumab as induction therapy for crohn’s disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet Lond Engl. 2022;399(10340):2015–2030. doi:10.1016/S0140-6736(22)00467-6
197. Danese S, Vermeire S, Hellstern P, et al. Randomised trial and open-label extension study of an anti-interleukin-6 antibody in crohn’s disease (ANDANTE I and II). Gut. 2019;68(1):40–48. doi:10.1136/gutjnl-2017-314562
198. Zhang S, Chen B, Wang B, et al. Effect of induction therapy with olamkicept vs placebo on clinical response in patients with active ulcerative colitis: a randomized clinical trial. JAMA. 2023;329(9):725–734. doi:10.1001/jama.2023.1084
199. Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723–1736. doi:10.1056/NEJMoa1606910
200. Panés J, Sandborn WJ, Schreiber S, et al. Tofacitinib for induction and maintenance therapy of crohn’s disease: results of two phase IIb randomised placebo-controlled trials. Gut. 2017;66(6):1049–1059. doi:10.1136/gutjnl-2016-312735
201. Danese S, Vermeire S, Zhou W, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet Lond Engl. 2022;399(10341):2113–2128. doi:10.1016/S0140-6736(22)00581-5
202. Loftus EV, Panés J, Lacerda AP, et al. Upadacitinib induction and maintenance therapy for crohn’s disease. N Engl J Med. 2023;388(21):1966–1980. doi:10.1056/NEJMoa2212728
203. Feagan BG, Danese S, Loftus EV, et al. Filgotinib as induction and maintenance therapy for ulcerative colitis (SELECTION): a phase 2b/3 double-blind, randomised, placebo-controlled trial. Lancet Lond Engl. 2021;397(10292):2372–2384. doi:10.1016/S0140-6736(21)00666-8
204. Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699–710. doi:10.1056/NEJMoa1215734
205. Colombel JF, Sands BE, Rutgeerts P, et al. The safety of vedolizumab for ulcerative colitis and crohn’s disease. Gut. 2017;66(5):839–851. doi:10.1136/gutjnl-2015-311079
206. Arijs I, De Hertogh G, Lemmens B, et al. Effect of vedolizumab (anti-α4β7-integrin) therapy on histological healing and mucosal gene expression in patients with UC. Gut. 2018;67(1):43–52. doi:10.1136/gutjnl-2016-312293
207. Schwartz DA, Peyrin-Biroulet L, Lasch K, Adsul S, Danese S. Efficacy and safety of 2 vedolizumab intravenous regimens for perianal fistulizing crohn’s disease: ENTERPRISE Study. Clin Gastroenterol Hepatol. 2022;20(5):1059–1067.e9. doi:10.1016/j.cgh.2021.09.028
208. Sandborn WJ, Colombel JF, Enns R, et al. Natalizumab induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2015;372(21):2074. doi:10.1056/NEJMx140055
209. Peyrin-Biroulet L, Hart A, Bossuyt P, et al. Etrolizumab as induction and maintenance therapy for ulcerative colitis in patients previously treated with tumour necrosis factor inhibitors (HICKORY): a phase 3, randomised, controlled trial. Lancet Gastroenterol Hepatol. 2022;7(2):128–140. doi:10.1016/S2468-1253(21)00298-3
210. Vermeire S, Lakatos PL, Ritter T, et al. Etrolizumab for maintenance therapy in patients with moderately to severely active ulcerative colitis (LAUREL): a randomised, placebo-controlled, double-blind, phase 3 study. Lancet Gastroenterol Hepatol. 2022;7(1):28–37. doi:10.1016/S2468-1253(21)00295-8
211. Sandborn WJ, Panés J, Danese S, et al. Etrolizumab as induction and maintenance therapy in patients with moderately to severely active Crohn’s disease (BERGAMOT): a randomised, placebo-controlled, double-blind, phase 3 trial. Lancet Gastroenterol Hepatol. 2023;8(1):43–55. doi:10.1016/S2468-1253(22)00303-X
212. Sandborn WJ, Feagan BG, D’Haens G, et al. Ozanimod as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2021;385(14):1280–1291. doi:10.1056/NEJMoa2033617
213. Sandborn WJ, Vermeire S, Peyrin-Biroulet L, et al. Etrasimod as induction and maintenance therapy for ulcerative colitis (ELEVATE): two randomised, double-blind, placebo-controlled, phase 3 studies. Lancet Lond Engl. 2023;401(10383):1159–1171. doi:10.1016/S0140-6736(23)00061-2
214. Atreya R, Reinisch W, Peyrin-Biroulet L, et al. Clinical efficacy of the Toll-like receptor 9 agonist cobitolimod using patient-reported-outcomes defined clinical endpoints in patients with ulcerative colitis. Dig Liver Dis. 2018;50(10):1019–1029. doi:10.1016/j.dld.2018.06.010
215. Feagan BG, Sandborn WJ, Gasink C, et al. Ustekinumab as induction and maintenance therapy for crohn’s disease. N Engl J Med. 2016;375(20):1946–1960. doi:10.1056/NEJMoa1602773
216. Ferrante M, Panaccione R, Baert F, et al. Risankizumab as maintenance therapy for moderately to severely active crohn’s disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet Lond Engl. 2022;399(10340):2031–2046. doi:10.1016/S0140-6736(22)00466-4
217. Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet Lond Engl. 2018;392(10148):650–661. doi:10.1016/S0140-6736(18)31713-6
218. Belarif L, Danger R, Kermarrec L, et al. IL-7 receptor influences anti-TNF responsiveness and T cell gut homing in inflammatory bowel disease. J Clin Invest. 2019;129(5):1910–1925. doi:10.1172/JCI121668
219. Ghoreschi K, Balato A, Enerbäck C, Sabat R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet Lond Engl. 2021;397(10275):754–766. doi:10.1016/S0140-6736(21)00184-7
220. Fauny M, Moulin D, D’Amico F, et al. Paradoxical gastrointestinal effects of interleukin-17 blockers. Ann Rheum Dis. 2020;79(9):1132–1138. doi:10.1136/annrheumdis-2020-217927
221. Maxwell JR, Zhang Y, Brown WA, et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity. 2015;43(4):739–750. doi:10.1016/j.immuni.2015.08.019
222. Van Assche G, Van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for crohn’s disease. N Engl J Med. 2005;353(4):362–368. doi:10.1056/NEJMoa051586
223. Travis S, Silverberg MS, Danese S, et al. Vedolizumab for the treatment of chronic pouchitis. N Engl J Med. 2023;388(13):1191–1200. doi:10.1056/NEJMoa2208450
224. Danese S, Colombel JF, Lukas M, et al. Etrolizumab versus infliximab for the treatment of moderately to severely active ulcerative colitis (GARDENIA): a randomised, double-blind, double-dummy, phase 3 study. Lancet Gastroenterol Hepatol. 2022;7(2):118–127. doi:10.1016/S2468-1253(21)00294-6
225. Matsuoka K, Watanabe M, Ohmori T, et al. AJM300 (carotegrast methyl), an oral antagonist of α4-integrin, as induction therapy for patients with moderately active ulcerative colitis: a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Gastroenterol Hepatol. 2022;7(7):648–657. doi:10.1016/S2468-1253(22)00022-X
226. Reinisch W, Sandborn WJ, Danese S, et al. Long-term safety and efficacy of the anti-MAdCAM-1 monoclonal antibody ontamalimab [SHP647] for the treatment of ulcerative colitis: the open-label study TURANDOT II. J Crohn's Colitis. 2021;15(6):938–949. doi:10.1093/ecco-jcc/jjab023
227. D’Haens G, Danese S, Davies M, Watanabe M, Hibi T. A phase II, multicentre, randomised, double-blind, placebo-controlled study to evaluate safety, tolerability, and efficacy of amiselimod in patients with moderate to severe active crohn’s disease. J Crohn's Colitis. 2022;16(5):746–756. doi:10.1093/ecco-jcc/jjab201
228. Cook L, Stahl M, Han X, et al. Suppressive and gut-reparative functions of human type 1 T regulatory cells. Gastroenterology. 2019;157(6):1584–1598. doi:10.1053/j.gastro.2019.09.002
229. Cifuentes-Rius A, Desai A, Yuen D, Johnston APR, Voelcker NH. Inducing immune tolerance with dendritic cell-targeting nanomedicines. Nat Nanotechnol. 2021;16(1):37–46. doi:10.1038/s41565-020-00810-2
230. Li MC, He SH. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J Gastroenterol. 2004;10(5):620–625. doi:10.3748/wjg.v10.i5.620
231. Cassinotti A, Passamonti F, Segato S. Cell therapy in inflammatory bowel disease. Pharmacol Res. 2021;163:105247. doi:10.1016/j.phrs.2020.105247
232. Saez A, Gomez-Bris R, Herrero-Fernandez B, Mingorance C, Rius C, Gonzalez-Granado JM. Innate lymphoid cells in intestinal homeostasis and inflammatory bowel disease. Int J Mol Sci. 2021;22(14):7618. doi:10.3390/ijms22147618
233. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov. 2020;19(12):860–883. doi:10.1038/s41573-020-0081-9
234. Guo P, Wang W, Xiang Q, et al. Engineered probiotic ameliorates ulcerative colitis by restoring gut microbiota and redox homeostasis. Cell Host Microbe. 2024;32(9):1502–1518.e9. doi:10.1016/j.chom.2024.07.028
235. Hammerhøj A, Chakravarti D, Sato T, Jensen KB, Nielsen OH. Organoids as regenerative medicine for inflammatory bowel disease. iScience. 2024;27(6):110118. doi:10.1016/j.isci.2024.110118
236. Li DF, Yang MF, Xu J, et al. Extracellular vesicles: the next generation theranostic nanomedicine for inflammatory bowel disease. Int J Nanomed. 2022;17:3893–3911. doi:10.2147/IJN.S370784
237. Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi:10.1146/annurev.immunol.20.100301.064816
238. Lee JWJ, Plichta D, Hogstrom L, et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe. 2021;29(8):1294–1304.e4. doi:10.1016/j.chom.2021.06.019
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Stem Cell Therapy in Inflammatory Bowel Disease: A Review of Achievements and Challenges

Tian CM, Zhang Y, Yang MF, Xu HM, Zhu MZ, Yao J, Wang LS, Liang YJ, Li DF
Journal of Inflammation Research 2023, 16:2089-2119
Published Date: 16 May 2023
A Multicentre Study of the Clinical and Epidemiological Profile of Inflammatory Bowel Disease in Northeast Brazil
Brito CAA, Celani LMS, Araújo MVTD, Lucena MT, Vasconcelos GBS, Lima GAS, Nóbrega FJF, Diniz GTN, Lucena-Silva N, Toneto GT, Falcão JVDC, Barbosa PM, Oliveira PRFD, Dantas LSX, Fernandes LKC, Araújo SAD, Martinelli VF
Clinical and Experimental Gastroenterology 2023, 16:87-99
Published Date: 21 June 2023
Cytomegalovirus Pneumonia in Inflammatory Bowel Disease: Literature Review and Clinical Recommendations
Ren K, Yong C, Wang Y, Wei H, Zhao K, He B, Cui M, Chen Y, Wang J
Infection and Drug Resistance 2023, 16:6195-6208
Published Date: 13 September 2023
Microbial Disruptions in Inflammatory Bowel Disease: A Comparative Analysis
Ma J, Wang K, Wang J, Zeng Q, Liu K, Zheng S, Chen Y, Yao J
International Journal of General Medicine 2024, 17:1355-1367
Published Date: 6 April 2024
Obstacles to Medication Adherence for Patients with Inflammatory Bowel Disease: A Qualitative Study in East China
Xu F, Xing J, Fan M, Zhu Z, Chen Y, Hu W, Zhou Y
Patient Preference and Adherence 2024, 18:2481-2494
Published Date: 8 December 2024