")
Back to Journals » Cancer Management and Research » Volume 17
Effects of Tp53 Gene Mutations on the Survival of Non-Small Cell Lung Cancer (NSCLC); A Short Review
Authors Zhang C, Yang C, Shi Q
Received 6 September 2024
Accepted for publication 14 December 2024
Published 15 January 2025 Volume 2025:17 Pages 65—82
DOI https://doi.org/10.2147/CMAR.S495006
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lu-Zhe Sun
Chi Zhang,1,2,* Chao Yang,3,* Qingming Shi1,2
1Department of Oncology, Anhui Chest Hospital, Hefei, 230022, People’s Republic of China; 2Anhui Medical University Clinical College of Chest, Hefei, 230022, People’s Republic of China; 3Department of Urology, Anhui Provincial Children’s Hospital, Hefei, 230022, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qingming Shi, Email [email protected], [email protected]
Abstract: Non-small cell lung cancer (NSCLC) is the leading cause of cancer-related deaths worldwide. Mutations within the TP53 gene represent critical molecular events in NSCLC, contributing to the tumorigenesis in the pulmonary epithelial tissues. TP53 is a widely researched prognostic indicator in NSCLC, and pathological investigations have revealed a weak to mild negative predictive effect for TP53. Mutated p53 protein may have some pro-oncogenic impact, and the variations may change tumor inhibitors into oncogenes. The diverse mutational spectrum of TP53 in NSCLC with different mutations is linked to varied treatment responses. In contrast, first-line chemotherapeutics to this progress are limited, however, randomized trials with new chemotherapeutics have shown significant survival benefits. This review highlighted the critical influence of TP53 gene mutations on pathological-sensitivity and overall survival outcomes in NSCLC. Further research is needed to explore TP53 mutation-specific pathways and their effects on NSCLC progression and treatment effectiveness.
Keywords: non-small cell lung cancer, TP53 gene, treatment, survival analysis, mutations
Graphical Abstract:

Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide, accounting for over a million new cases each year. Most of such cases are ascribable to external factors like smoking.1,2 Changes or mutations within the TP53 gene represent a critical molecular event in lung cancer, significantly contributing to the tumorigenesis in the epithelial tissues of the pulmonary. Lung malignancies among humans are categorized into 2 principal forms of lung cancer: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) comprising multiple subtypes. Among these, adenocarcinoma (AC) is the utmost prevalent sub-type of NSCLC, with its occurrence growing globally. Adenocarcinoma (AC) is the peak prevailing form of lung carcinoma among the individuals who have never smoked.1,3,4 In 1991, Dr. Curt Harris and his collaborators established a TP53 mutations database to assist the reclamation and investigation of identified mutations. The up-to-date description comprises more than 80401 alterations. In this issue, more than 6870 variants are recorded. The most extensively studied genetic alterations in pulmonary tumors are TP53-tumor suppressor gene (MIM#191170) happen in just around 50–55% of NSCLC and over 69% of SCLC.4,5
Studies have revealed that TP53 mutated bands in lung malignancy exhibit hotspots which are seldom perceived in former kinds of humanoid tumors, signifying the presence of carcinogenic mutations. Yet tobacco smoke susceptibility is the most eminent and extensively premeditated mutagen, compared to other factors inferred with work-related and ecological influences contributing to lung cancer.6–8
The progression of cancer can be elucidated through a Darwinian framework, wherein cells undergo selection based on their adaptability to an altered environment. Survival advantage is conferred upon cells exhibiting superior fitness, achieved through sequential accrual of genetic modifications in genes governing cell proliferation, lifespan, stress response, interactions with neighboring cells, and genetic integrity. Among the multifarious genetic-pathways implicated in cancer advancement, the TP53-gene (OMIM #191170) emerges as a pivotal nexus, transcending the tissue and cellular origins of tumors. Perturbation of the TP53-pathway is a hallmark of the common aggressive cancers, commonly mediated by allele loss, point mutations (predominantly missense), and protein inactivation via sequestration by viral antigens or cellular proteins.9–11 The International Agency for Research on Cancer meticulously curates a comprehensive repository of such mutations.12,13 We go over the salient features of the complex interplay between TP53 and lung cancer development in this review.
TP53, situated on chromosome 17p13, comprises 11 exons spanning 20 kilobases, encoding a predominantly nuclear phosphoprotein with a molecular weight of 53 kDa. This gene belongs to a family of highly conserved genes, including at least two other members, TP63 and TP73.13–16 However, TP53 distinguishes itself through its unique tumor-suppressive function. TP53-deficient mice exhibit normal development but display a heightened susceptibility to early-onset tumors, unlike TP73- or TP63-deficient counterparts, which manifest intricate developmental anomalies and physiological aberrations but do not demonstrate increased cancer incidence.4 The pivotal role of TP53 in cancer defense is underscored by Li-Fraumeni Syndrome (LFS), an inherited syndrome predisposing individuals to diverse cancers, often associated with the hereditary transmission of a mutant TP53 allele.16–19 The p53 protein conforms to the classic structure of a sequence-specific transcription factor, featuring an acidic N-terminus housing transactivation domain, a hydrophobic central core binding to specific DNA sequences (consensus repeats of RRRCA/TA/TGYYY), and a basic C-terminus encompassing oligomerization and regulatory domains. Predominantly, identified mutations aggregate within the DNA-binding domain, underscoring its pivotal biochemical role in p53’s function as a tumor suppressor. Almost all tissues express the constitutively repressed p53 protein. Different signal classes can de-repress p53 and induce accumulation of post-translational modifications.19–21 These signals include genotoxic chemicals (damaging DNA), constitutively activated growth signaling cascades (oncogenic stress), and other stressors like ribonucleotide deficiency or hypoxia. Consequently, p53 is situated at the intersection of numerous, distinct stress-response pathways.22 Once activated, p53 induces a range of coordinated antiproliferative effects, including those on angiogenesis, apoptosis, DNA repair, and differentiation, as well as the cell cycle.21 Transcriptional regulation (either activation or repression) and direct binding to other proteins to modify their activity are the mechanisms by which p53 controls these processes.23,24 An incomplete list of effectors engaged in signaling pathways downstream of p53 is given in Figure 1. Depending on the type and strength of the inducing signal, the tissue under consideration, and the degree of cell differentiation, individual cells may have distinct combinations of effectors triggered by p53.25–27 Additionally, it is important to consider the concurrent activation of other pathways that influence the regulation of cell growth and survival.
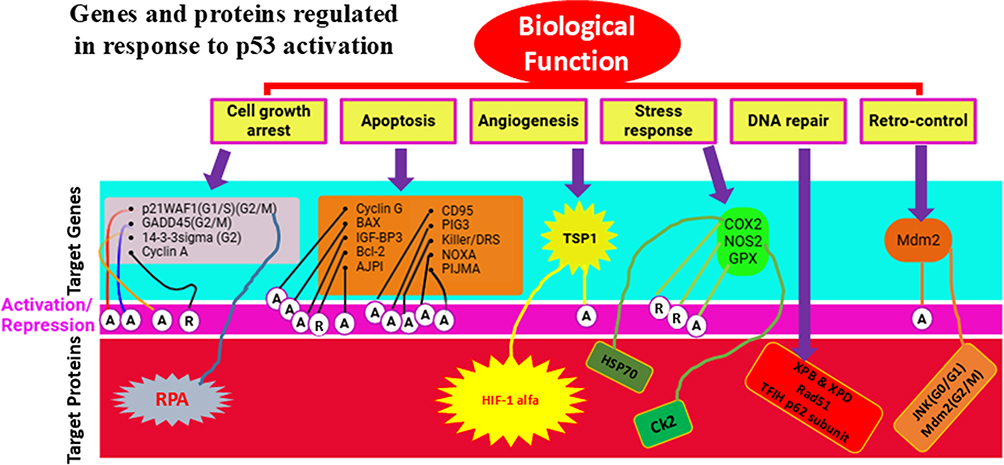 |
Figure 1 Genes and proteins are regulated in response to p53 activation. A: activation; R: repression; G0, G1, S, G2, M are the cell cycle phases. Data from et al.27 |
TP53 Genetic Alterations
Assortment of TP53 Mutations in Malignancy
TP53, which is distinct from utmost other tumor suppressor genes in that more than 75% of known mutations are missense, resulting in substitution of a solitary amino acid (AA). TP53 exhibits unique mutation patterns compared to other tumor suppressor genes eg APC, RB, and BRCA1 (Breast cancer type 1) are frequently altered by deletions, frameshift alterations, and nonsense mutations disrupting protein sequence and function.2,12 Mutations in TP53 are dispersed across all coding-exons, with a substantial prevalence in exons 4–9, which encode the protein’s DNA-binding domain.26–28 Exon 2 (1st coding-exon, programming extreme N-terminus) and exon 11 (the closing exon, representing the extreme C-terminus) account for barely 1.5k of 15k alterations discovered to IARC TP53-database, (R5 version). This is especially interesting because these sections have important post-translational modification sites and regulatory domains, which are essential for controlling the protein’s function. While the C-terminus is involved in controlling DNA-binding activity, the N-terminus comprises the binding-site for MDM2, the primary-regulator of p53-protein stability.29–31 Additionally, both domains have locations for post-translational changes, such as sumoylation (at lysine 386), acetylation (at lysines 371, 373, and 382), and phosphorylation (at serines 6, 9, 15, 20, 376, 378, and 392). It is noteworthy that in human malignancies, none of these residues had mutations.
A significant number of mutations occur in p53’s DNA-binding domain, which has a complicated structure consisting of 2 β-sheets stacked in a toasty-like manner and joined by loops and helices.32–34 The binding of a zinc atom stabilizes these loops, which is critical to the domain’s structural stability. Notably, mutations do not occur uniformly across all residues in this region. Approximately 30% of the mutations occur at six “hotspot” codons identified in nearly all cancer types (codons 175, 245, 248, 249, 273, 282). Located at the protein’s DNA-binding interface, these residues are essential for both protein structure (codons 175, 249, 282) and protein–DNA interactions (codons 245, 248, 273). Nevertheless, inactivating mutations can also be found at a number of additional locations, such as residues that are involved in zinc binding (codons 176, 179, 238, 242). These changes together account for about 5% of all mutations.35–37 There are two widely accepted theories that explain the observed differences in the frequency of mutations between residues. First, the suggestion is for targeted mutagenesis.38–41 Out of the six “hotspot” mutations found, five of them are located at cytosines that are part of di-pyrimidine repeats (CpG sites). The human genome has many methylation sites, and one typical mechanism for these mutations is the spontaneous deamination of methylated cytosine-to-thymine.42–45
Furthermore, a number of codons are recognized locations for DNA damage brought on by external carcinogens.45–47 For example, aflatoxin-B1, a cancer-causing mycotoxin, causes G to T transversing at codon 249, whereas tobacco smoke components cause G to T transversions at codons 157, 158, 248, and 273. As a result, connections between different mutation types and cancer risk factors can be found by comparing the mutation patterns of various tumors.
Functional selection is the subject of the second explanation. For DNA-binding capabilities, just about all commonly altered-codons are crucial.48–51 It has been suggested that the position of the mutation within the protein may affect how many wild-type p53 functions are disrupted. In fact, the data points to the fact that not every mutation has the same functional consequences. The identical mutation at codon 273 and the Arginine-Histidine mutation at codon 175 show higher in-vitro effects.52–54 It is up to dispute whether these variations are related to the clinical features of malignancies. Recent research, however, indicates that the position of the mutation might affect how the body reacts to cytotoxic treatment.
Why p53 is Frequently Altered in Humanoid Malignancies
There is no clear motive to regard the TP53 gene as being located in a hypermutable area of the humanoid genome. Thus, the increased incidence of alterations observed in malignancy is most probably the result of an assortment process: cells with an intact, functional TP53-pathways are incapable to advance to a full neo-plastic phenotype, necessitating the commotion of this route for cancer to grow fast. When malignant cells hold normal TP53-alleles, the protein’s expressions or regulations are commonly influenced via additional mechanisms, such as, amplification and/or overexpression of the p53 regulatory protein MDM2).27,55,56 According to the theory put forth above, p53 function activation and regulation happen as a typical defensive mechanism against neoplastic alteration ensuing the following mentioned mechanisms.
Furthermore, the data points to the possibility that p53 protein might be rendered inactive by means additionally than direct mutations, which thwarts it from folding into proper wild-type conformation needed for DNA binding. The renowned but excessively restrictive description of p53 as a “guardian of the genome” against DNA damage originated from its identification of this DNA-damage pathway that was the first p53-regulatory pathway to be identified.57
It is hypothesized that all cancer cells probably display some level of p53 malfunction, either because of direct gene alterations or indirect changes impacting other p53 pathway components. The second pathway uses the p14arf protein (ARF-tumor suppressor) as its key effector and functions, at least in part, independently of accumulated DNA damage. The INK4a/CDKN2A gene, this furthermore codes for the p16 cyclin kinase inhibitor, also yields an alternate product called P14arf.58,59 These two proteins have different reading frames, but their exon 2 DNA sequences are similar, and different promoters’ control how these proteins are expressed. By binding to mdm2, p14arf inhibits mdm2’s capacity to repress p53. This may be achieved by isolating mdm2 from p53 and locating it in the nucleolus, which is home to p14arf. Growth-promoting (GP) and carcinogenic stimuli, like transfection of adenoviral protein (AP) E1A, which repossesses and suppresses the retinoblastoma protein, or an active Ras gene, activates the action of p14arf. It is yet unknown how p14arf recognizes these cancer-causing signals; however, it’s suggested that DAP-kinase, a protein-kinase that is frequently methylated-inactivated in several cancers, may be regarded in this mechanism.59–62 Dysregulation of pathways such as β-catenin/c-Myc pathway or the Ras/Rb/E2F pathway can activate p14arf during neoplastic alteration, resulting in growth inhibition that is dependent on p53. Thus, the only cells that can develop into complete neoplasia are those that are able to avoid this p53-dependent repression.27,62
There is less knowledge about the 3rd path, which arbitrates non-genotoxic signs. The pathways for p53 activations are given in the Figure 2. It has been demonstrated that hypoxia causes p53 levels to rise over time in a way that is independent of mdm2 activity and different from DNA-damage signaling. The specifics of this mechanism’s operation are still unknown, though.61–64 These three mechanisms are frequently implicated in the pathogenesis of most cancer types. DNA impairment arises as a result of disclosure to environmental mutagens, as well as via endogenic mechanisms like polymerase-errors or deficiencies in DNA restoration. Activation of growth-signaling pathways serves as a ubiquitous feature across nearly all primary-cancers. Hypoxia represents a common occurrence at numerous stages of solid tumor progression.57,64–67 Consequently, throughout the initiation and progression of cancer, diverse assortment of stresses congregate on p53 via these all three mechanisms, operating either independently or in tandem.
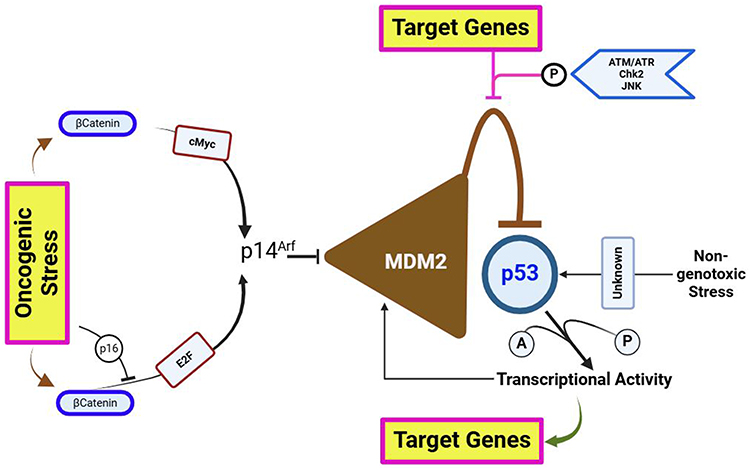 |
Figure 2 The three distinct and convergent pathways of p53 activations. These pathways act as channels for various types of selection pressure during neoplastic transformation. |
TP53 Variations in Lung Malignancy
Alterations in TP53 in lung cancer, including frequent damage of heterozygosity (LOH) observed in lung cancer cell lines and tumor samples at TP53 gene locus on chromosome 17p13, indicate the likely involvement of this gene in lung cancer pathogenesis. Genetic abnormalities of TP53 in lung cancers have been linked with low survival prognosis and augmented cellular resistance to treatment.68–71 The highest frequency of TP53 alterations is observed in specimens of small-cell lung carcinoma (SCLC). Conversely, TP53 mutations are most prevalent in squamous cell carcinomas and less frequent in adenocarcinomas among NSCLC tumor samples.68,69 Somatic-mutation and augmented expression of TP53 are commonly detected in approximately 23% and 65% of NSCLC, both with and without allele loss at 17p13, primarily located inside the DNA-binding sphere of TP53. Since coding alterations of TP53 typically happen, initially, in lung cancer and may be necessary for sustaining malignant phenotype, acquired TP53 alterations persist throughout cancer progression and metastatic dissemination.70–73 Studies by Chang et al reported a 23.2% occurrence of TP53 mutations in initial tumors and a 21.4% incidence in metastatic lymph nodes among patients with NSCLC who experienced invasive resection, with a 92.9% concordance in TP53-gene status between primary-tumors growths and metastatic lymph nodes.74–76 This finding suggests that TP53 alterations often follow lymph nodes metastasis, with utmost mutations occurring before metastasis initiation and persisting via subsequent stages of cancer growth development, indicating no choice in contrast to TP53 alterations during metastasis.
TP53 Mutations
Prognosis of NSCLC Patients in Wild-Type TP53 and Mutant TP53
TP53 is the utmost and widely researched prognostic indicator in NSCLC, and investigations have revealed a weak to mild -ve predictive effect for TP53. Some studies demonstrated that abnormal TP53 is a prognostic aspect in NCSCL at any stage and histology, including squamous cells carcinoma (SCC) and adenocarcinoma.77–80 Furthermore, no significant variations were identified, comparing various antibodies employed in the immunohistochemical (IHC) experiments.44 The DNA mutations and protein expression were separately analyzed by Mitsudomi et al in lung cancer, and the concordance between these two assays up to 70% abnormal TP53 was generally more frequent in SCC, but their importance as a predictive was important in adenocarcinomas. Shortly, existence of an abnormal TP53 may indicate an illness with clear symptoms and, thus, a possibly shorter lifespan.81,82 Adjuvant treatment for Cancer and Leukemia Group B (CALGB) patients with tumors ≥4 cm significantly affected both overall survival (OS) and disease-free survival (DFS) in Phase III studies. OS and DFS were linked to the IHC staining for TP53, BCL-2, blood group antigen A, and mucin and mucin expression was 45% and TP53 was 47%, respectively.83,84 Univariate examination for DFS revealed significant impact for inclusion of the COX model and correlation between mucin and TP53. In another advanced NSCLC study, TP53 expression of proteins was assessed by IHC in tissue specimens, where the prevalence of TP53 protein upregulation was 52% (132 patients). Untreated TP53-positive patients had significantly shorter overall survival than patients with TP53-negative tumors (P = 0.03; HR 1.89; 95% CI, 1.07–3.34). However, these TP53-positive patients also had a significantly greater survival benefit from adjuvant chemotherapy (P = 0.02; HR 0.54) than patients with TP53-negative tumors (P = 0.26; interaction P = 0.02; HR 1.40), indicating that TP53 protein overexpression is a significant marker of poor prognosis as well as a significant predictive marker for a differentially greater benefit from adjuvant chemotherapy in completely resected NSCLC patients.84 Individuals with advanced NSCLC who had undergone neoadjuvant chemo were examined for TP53 genotype of tumors.85 The existence of mutant TP53 genotype was significantly associated with resistance to chemotherapy. The mutant TP53 genotype identified non-responders with a sensitivity of 94%. Different evidence suggests that TP53 changes are correlated with a survival benefit. Because of this, most individuals have poor prognosis; nevertheless, there is no evidence to definitively prove that TP53 status plays a part in the treatment of individual NSCLC patients.86–89
TP53 Mutational Spectra Between Never-Smokers and Smokers
As NSCLC is categorized into numerous subtypes and their prevalence has altered during the previous decades. Tobacco use is the most significant risk factor for lung cancer, and the risk of lung cancer rises with the amount of cigarettes smoked and the smoking period.90,91 It’s responsible for >90% of lung cancers in males and up to 80% in females in Europe and the US. The TP53 alterations in female nonsmokers with AC were mostly transient (up to 83%). However, among smokers, TP53 alterations were mostly transversions (up to 60%) and deletions (20%). According to current studies, smoking promotes lung cancer by a direct mutagenesis impact on DNA as seen in >50% cases, thus TP53 gene must be recognized as one of the most common targets of tobacco smoking-related DNA alterations. Individuals via tobacco-related cancer had a greater incidence of up to 71% of mutations in TP53 than people who had never smoked (up to 47%) (Figure 3 and Table 1). Multiple investigations have previously shown hotspots on TP53 gene, with G: C to T: A (G to T) alterations being a typical outcome in tobacco-related lung cancers. Ninety percent of the guanines are experiencing these transversions incidents resulting in non-transcribed DNA.31,38,92,93 The mutational-hotspots inside Tp53 gene, for instance, codon 157, have been noticed for tobacco-associated lung cancer; however, similar alterations are seldom detected in other malignancies. The prevalence of G to T alterations in nonsmokers is much less than in smokers. For smokers and non-smokers, respectively, the percentage of G to T alterations to G: C to A: T (G to A) transversions was 1.0 and 0.34. The spectrum of G to T transversions is apparently produced by polycyclic-aromatic hydrocarbons (PAH) found in tobacco smoking. One of the most thoroughly studied members of the PAH class is benzo[α]pyrene, and its main metabolite is benzo[α]pyrene diol epoxide (BPDE), one of the most potent carcinogens present in tobacco smoke in large quantities. The BPDE-DNA adduct pattern in TP53 gene in BEC (bronchial-epithelial cells) corresponds to G to T alterational-hotspots at codons (C) 157, 248, and 273 in smoking-related lung malignancies. G to T transversions are characteristic for massive adduct-producing mutagens, which include the PAH class, and BPDE adducts at these C (codons).45,94
 |
Figure 3 Mutations of TP53 existence/occurrence with smoking duration. The percentages of TP53 mutations and wild-type are denoted in the Figure: Mutated (M) and Wild-type (WT). |
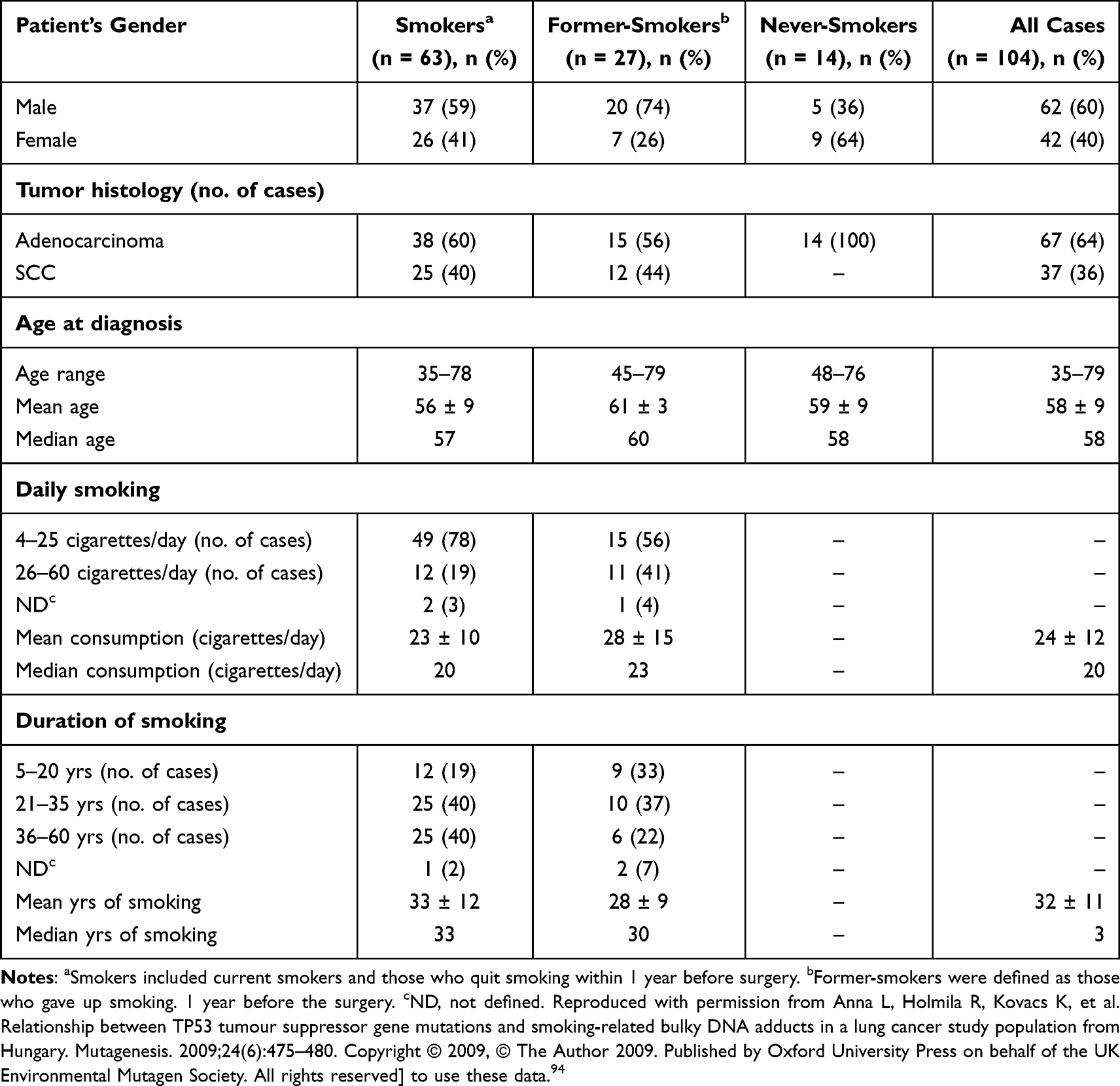 |
Table 1 Details and Characteristics of Smoking for Lung Cancer.94 |
Alternatively, individuals with AC who were not smokers are more probable to have alterations in the epidermal growth factor receptor (EGFR) tyrosine kinase and respond well to its blockers than individuals with tobacco-related lung cancers. It was observed that TP53 alterations are strongly associated with smokers, but EGFR alterations are statistically much more prevalent in females and non-smokers in AC. Moreover, the incidence of alterations in K-RAS and TP53 varies between individuals with lung cancer who were not smokers and those with tobacco-related lung cancer. TP53 mutations represent a clinically relevant mechanism of resistance to EGFR-TKIs, regardless of their generation.90 Smokers have the majority of TP53 changes, indicating that tobacco exposure plays a role in TP53 mutations. Females and non-smokers are more likely to have EGFR mutations, suggesting that other variables including genetic or environmental predispositions may be involved. The efficiency of EGFR-targeted treatments may be diminished by mutations that confer resistance. Aggressive tumor behavior and a worse overall survival rate may be associated with combined EGFR/TP53 mutations. Knowledge of the interaction between EGFR and TP53 may guide combination therapy approaches, such as concurrent EGFR inhibition and p53 reactivation.90,94 Researchers can get new understanding of the intricate connections between TP53 changes and EGFR mutations in AC by investigating these regions, which can ultimately help develop more potent treatment plans.
TP53 Mutations in Cancer Progression: “Late” or “Early” Event?
In contrast, TP53 alterations appear to develop at any early stage in various forms of cancers that are directly induced by external factors inducing cancers. This is true for smoker’s lung cancers, non-melanoma skin tumors following UV exposure, and the majority of esophageal tumors. In such tumors, TP53 alterations are frequently evident in dysplastic and hyperplastic lesions.83–85 It is important to use caution when interpreting the role of TP53 alterations in the timings of cancer progression. Indeed, many investigations are retrospective, using pathological archives, and a greater prevalence of TP53 alterations was associated with histological staging. In comparison, there are only a few prospective studies in which the same lesions are biopsied many times to track the buildup of genetic-lesions. One of such investigation has demonstrated that TP53 mutations may develop at the metaplastic stage.77,79,81,82 Figure 4 offers a model that demonstrates how TP53 alteration might arise as an initial or final incident in cancer development. In accordance with our premise that assortment pressure is the factor for the development of an alteration, we propose that the site of the variation depends on the biological mechanism of stimulating p53 protein activity. In tissues extensively subjected to carcinogens (like lung tissues in smokers), the p53 protein is activated in healthy cells as a natural reaction to DNA damages.83–85 This initiation causes damaged cells to be suppressed, and individual cells with variant TP53 would escape and go on to expand clonally. In contrast, in tissues where contaminants do not directly trigger the growth of cancer, other genetic modifications (because of inherited abnormalities or poor DNA repairmen’s) happen 1st and cause the establishment of an initial lesion w/o TP53 alteration. Though, these genetic modifications will cause oncogenic stress, which stimulates the p53-protein via the p14arf pathway.75–82 Therefore, the existence of wild-type (WT) p53 is a restricting event for the evolution of such lesions, and only such cells that have missing TP53 activity will be capable to advance to invasive malignancy. The model in Figure 4 only displays two stages of collection for TP53 alterations, but it’s evident that this approach may be extended to clarify the incidence of TP53 alterations at nearly every step of cancer progression. Further variables, including hypoxia, telomere erosion, or loss of adhesion qualities, may potentially add to the selection forces operating on the p53 checkpoint.70,74,75
 |
Figure 4 TP53 mutations as an “early” or “late” event in carcinogenesis. |
Mutant p53 Protein: More Than Just “Loss of Function”
One of the utmost startling pathological findings about modifications to TP53 is that many do not result in protein loss. On the contrary, most cancerous cells acquire altered p53 protein and even maintain this high amount of protein in metastatic regions. These elevated planes are attributable to the reality that altered p53 is more persistent than WT, resulting in protein aggregation.68–71 Therefore, cancer cells act as if they are selectively keeping the altered p53 protein. This feature cannot be explained just on the assumption that the mutation promotes the deactivation of protein activity (loss of function, LOF). It was proposed that mutated p53 protein may have some pro-oncogenic impact, and that variation was really changing this tumor inhibitor into some sort of oncogene.58,63,64,66
There are at least two mechanisms by which mutant p53 might induce cancer. The 1st is a dominant negative effect (DNE). This impact correlates to the altered protein’s ability to combine with the product of remaining WT allele and deactivate its activity. Therefore, DNE completely disables p53 protein activity, even if the wild-type protein is still produced in cells. The 2nd mechanism is called gain-of-function (GOF), and this does not need the existence of a WT allele. Indeed, altered p53 accumulates in cells which lost the WT allele, indicating that the mutant protein promotes itself.60–62 The mechanisms of this GOF are currently unclear. Mutant p53 has been demonstrated to increase transcription of genes including the multidrug resistance gene MDR1, but w/o directly interacting in the region of the promoter. In recent times, it has been revealed that altered p53 may form highly stable complexes with products of another member of TP53 gene family, p73 and p63, which inhibit their trans-activation potential. These proteins perform many functions in variation and development, and they may also operate as an inhibitor when p53 is deactivated.55,56 Therefore, the sequestration of these proteins by altered p53 may confer a further survival benefit, clarifying why mutant p53 protein is selectively maintained in cancerous cells. Figure 5 demonstrates in what way LOF, DNE, and GOF all exhibit the phenotypic consequences of altered p53. For all known hotspot mutations, the initial consequence is LOF. Though, the degree of LOF varies, and certain mutant proteins may maintain some transcriptional activity for subsets of target-genes. DNE is an extra feature of some altered p53 proteins. To enable mutant p53 to interact with WT, the protein’s C-terminal oligomerisation region must be intact.53,56 The properties of GOF alterations are poorly understood. On the molecular level, criteria for GOF are not exactly as those for DNE, subsequently binding of altered p53 to p63 and p73 does not need the oligomerization domain but appears to include a component of the core, DNA-binding domain. Moreover, it is possible that altered p53 will attach to previously unknown protein or DNA targets.52
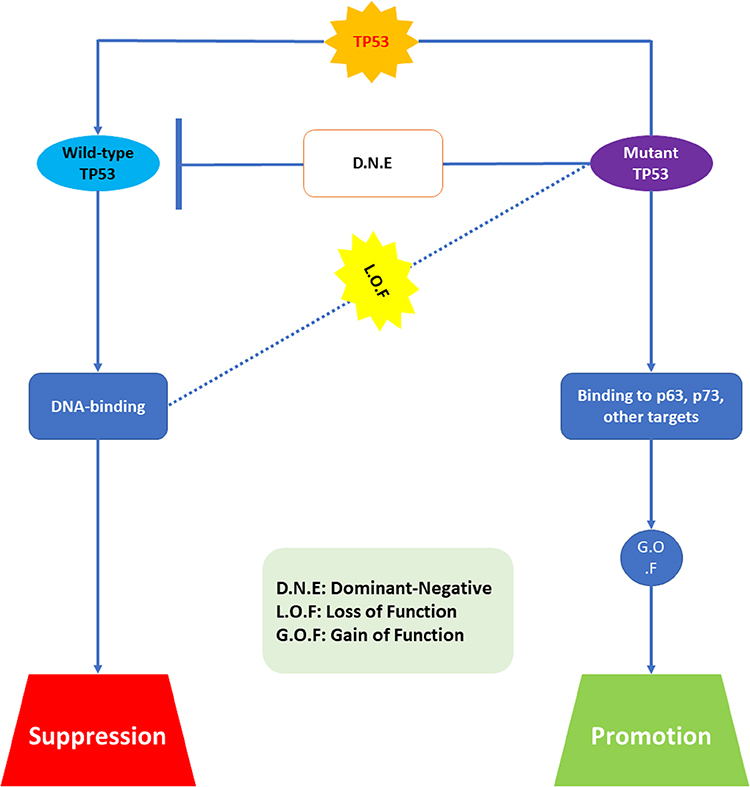 |
Figure 5 Biological properties of mutant p53. Abbreviations: DNE, dominant-negative; LOF, loss of function; GOF, gain of function. |
Therapeutic Strategies for NSCLC with TP53 Mutation
TP53 mutations play a crucial role in the tumorigenesis of lung epithelial cells, occurring in approximately 50% of NSCLC cases. Given that TP53 mutations are present in up to 50% of NSCLC cases, with their incidence increasing annually, targeting these mutations to eliminate their oncogenic effects is vital for the successful treatment of NSCLC. Multiple investigations show that TP53 alterations give chemo-resistance (CR) to lung cancers cells in vitro and in vivo.47,48,50 Evaluating TP53 status importance in deciding on chemo/radiation treatment. For example, it is widely recognized that tumors with the altered TP53 are utmost susceptible to ionizing radiation compared to those with WT TP53. The widespread deactivation of TP53 in human malignancies implies that the rebuilding of the TP53-mediated pathway in cancerous cells may be an appealing tumor cells-specific technique for treating cancers.48,49 Virus-based treatment techniques for mutant TP53-carrying tumors are among the most sophisticated methods to therapeutically target the TP53 pathway in cancer. WT TP53 restoration in altered TP53-carrying cancers is possible by gene therapy, which involves introducing an entire cDNA copy grounded on an adenovirus (Adp53). Because of the common alteration in TP53 in lung-cancers, concurrent therapy with various types of chemotherapy and TP53 gene substitution techniques have been investigated in both clinical and preclinical environments. In pre-clinical tests with lung-cancer cell lines expressing mutant TP53, the TP53-replacement method enhanced chemotherapy and radiation outcomes. TP53 gene-therapy also investigated in clinical-trials in lung-cancer patients, and clinical investigations have really shown a favorable impact of the combination of Adp53-gene therapy with chemo-therapeutic medicines and radiation.21,27,28,32,33
In a Phase-I clinical study, patients with NSCLC whose malignancies had progressed despite traditional therapies were given the Adp53 gene directly into their tumors without any additional treatment.95 In this study, 89% of the patients were assessed with a decrease in tumor size. In another experiment, Adp53 was provided via bronchoscopic intra-tumoral injection in individuals with advanced endo-bronchial NSCLC, in which (50%) patients demonstrated considerable reduction in airway barrier. Individuals with advanced NSCLC and atypical TP53 activity got intra-tumoral Adp53 injections along with intravenous cisplatin, and 71% patients showed greatest clinical reaction of steady illness; up to 8% attained a partial response; the remaining maximum individuals showed advanced disease.12,14,15,20–22,95 As previously stated, reconstituting the disordered TP53 tumor suppressor pathways, such as introducing Adp53 in tumor cells, is one of the most favorable approaches for enhanced therapy. Recently, numerous techniques for identifying small compounds that hit altered TP53 have been used ie RITA.95 RITA reactivates TP53 and causes apoptosis via disrupting its connection with HDM-2. It was postulated as a critical chemical to hit wild-type TP53 malignancies, which may show tolerance to medications that restore mutant-TP53 activity, like PRIMA-1 (p53 reactivation and production of large programmed cell death). According to Zhao et al demonstrated that RITA-induced reaction is reliant on the existence of mutant-TP53 and is maintained by restoration of apoptosis.77 They found that RITA is a potential lead in the creation of anticancer medicines which again activate the tumor suppressive activity of TP53, regardless of whether they show mutant or WT TP53.77,96 PRIMA-1 binds to mutant TP53 core and restores its WT conformation, causing apoptosis of tumor cells. Moreover, PRIMA-1MET/APR-246, a methylated structural analog of the compound PRIMA-1 may act synergistically with cisplatin or other frequently prescribed antitumor drugs to activate tumors cell apoptosis. Another research found that PRIMA-1 did not cause apoptosis but did significantly impair cell viability in NSCLC cell lines expressing various TP53-proteins: A549 (p53wt), LX1 (p53R273H), and SKMes1 (p53R280K). With adriamycin, PRIMA-1 increases the adriamycin-induced cell death in A549 and LX1.1,2 Adp53 gene treatment, along with small molecules, like PRIMA-1 and PRIMA-1MET, which may repair the transcriptional activity of mutant-TP53, or RITA, which intervenes with MDM2-directed TP53 erosion, have been verified in preclinical settings, and some are presently in clinical trials (Figure 6). Furthermore, innovative TP53-based treatment methods for NSCLC can be coupled with traditional and molecular-targeting cancer therapies, such as EGFR-TKI. The investigation of somatic genetics of the TP53-pathway in tumors has been verified to be valuable in the search of targeted therapeutics, and TP53 mutational status could serve as a prognostic indicator in some malignancies.6–9
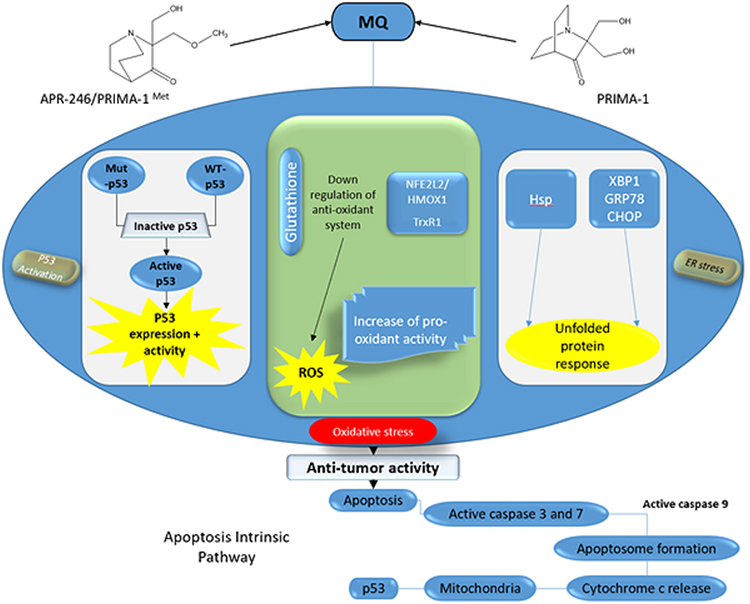 |
Figure 6 Therapeutic Strategies for NSCLC with TP53 Mutation. |
The primary challenge moving forward is to develop comprehensive approaches that analyze the complex interactions between p53 and other cellular factors. This requires expanding our understanding of p53’s role beyond individual signaling pathways to encompass entire networks of interactions.11,12
Non-small cell lung cancer (NSCLC) poses a significant-rising occurrence of this ailment among younger individuals in recent years. NSCLC was diagnosed at an advanced or metastatic stage in 79.7% of the cases. Median overall survival stood at 8 ± 0.72 months. Univariate analysis identified tumor stage, CRP levels, and patients’ general condition, assessed by the World Health Organization’s Performance Status (PS) score at admission, as significant survival influencers. Multivariate analysis pinpointed elevated CRP levels and a PS index of ≥2 as predictors of unfavorable prognosis. Despite therapeutic advancements, the prognosis for young NSCLC patients remains bleak. Timely diagnosis and intervention are crucial for enhancing survival prospects.24
Therapeutic Associations of TP53
Disruption of p53 in Human Cancer Cells Alters the Responses to Therapeutic Agents
The information presented here has significant implications for understanding and evaluating the use of therapeutic drugs in cancer treatment. It confirms previous research indicating that p53 mutations contribute to treatment resistance.27 However, this study expands on previous findings by demonstrating that while antimetabolite and DNA-damaging drugs also operate in a p53-dependent manner, their therapeutic effects differ significantly. Specifically, colorectal cancer cells with disrupted p53 exhibit increased susceptibility to radiation and adriamycin-induced apoptosis but decreased susceptibility to 5-fluorouracil (5-FU)-induced apoptosis.28,33 Interestingly, these observations align with results obtained from normal mouse intestinal cells, where p53 deficiency enhances the resistance to 5-FU. Furthermore, the cytotoxicity induced by 5-FU in normal mouse colonic epithelial cells is associated with RNA metabolism rather than DNA metabolism, consistent with observations in the human cells examined. These findings suggest that disruptions in RNA metabolism may underlie at least some of the major effects of 5-FU.33,61,62 The contrasting responses of p53-deficient cells to 5-FU compared to DNA-damaging agents, along with the inhibitory effect of uridine on apoptosis, indicate that the current model may be insufficient. This suggests that agents targeting RNA metabolism, in addition to those targeting DNA metabolism, may be worth exploring in cancer treatment. It is important to note that extrapolating these findings from a single-cell line to the heterogeneous nature of naturally occurring cancers would be premature. The observed cellular responses in vitro were more pronounced than those observed in vivo, highlighting the complexity of translating laboratory findings to clinical settings. Despite these limitations, further elucidation of the molecular mechanisms underlying 5-FU activation of p53, and induction of apoptosis will provide valuable insights into the action of this critical chemotherapeutic drug.63,65,70,71,74,77,78
Furthermore, after 5-FU treatment, conventional colony formation assays did not reveal any effects of p53 disruption. However, p53 disruption significantly influenced the extent of 5-FU-induced apoptosis in tissue culture and the responses of tumor xenografts to 5-FU in vivo. These findings highlight the challenges of extrapolating in vitro results to in vivo contexts and suggest that apoptosis may serve as a more reliable indicator of drug response in vivo compared to colony formation in certain scenarios. Our results hold relevance for colorectal cancer therapy, given that 5-FU is the primary drug utilized in this context. It’s crucial to emphasize that our findings do not suggest ineffectiveness of 5-FU in cancers with p53 mutations. Tumors with p53 mutations are less prone to respond to 5-FU compared to tumors without such mutations. Drug responses, including those to 5-FU, are multifaceted and are unlikely to be solely attributed to a single genetic alteration. The strength of our approach lies in its capability to identify specific genetic modifications and assess their impact on drug sensitivity. This methodology could potentially serve as a model for understanding the role of other genetic alterations in response to established or novel medications.32,45,66,72,97
p53 Alterations and Survival in Stage-I
According to Ahrendt et al, p53 gene mutations were observed in 55% (n = 188) of cancers. Statistical analysis revealed that p53 gene mutations were significantly linked with reduced OS (P < 0.05). Four years OS was significantly less in patients with p53 variations compared to those with WT p53. Additionally, the age of patient and pathological tumor-node-metastasis (TNM) stage was prognostic of OS which was significantly reduced in patients with metastases in N2 lymph nodes (P < 0.001).45,65,98
In this study, p53 alteration status and pathological stage were comparatively non-significant. In contrast, p53 alteration status did not correlate with worse outcomes in patients with stage II or IIIA NSCLC (P > 0.05) except stage I. Thus, p53 alterations were independently prognostic of reduced survival only in patients with pathological stage I NSCLC. Patients whose tumors harbored truncating, structural, or DNA contact p53 mutations had significantly worse survival compared to those with wild-type p53 (P = 0.01).14,22,99
The p53 mutations were linked with significantly less survival than WT p53. In stage I NSCLC individuals, those with alterations predicted to affect p53 function or structure mutations had significantly worse four-year overall actuarial survival compared to those with wild-type p53 (P = 0.001). P53 gene alteration is a significant predictor of poor outcomes in patients with NSCLC, with the greater risk of deaths being most pronounced in those with stage-I disease. Furthermore, the clinical behavior of NSCLC tumors harboring a p53 mutation varies depending on the type and location of the mutation and is also associated with smoking and other carcinogens.9,10,12
Numerous studies have examined the prognostic role of p53 alterations in NSCLC. Molina-Vila et al concluded that nondisruptive TP53 mutations are an independent prognostic factor for shorter survival in advanced NSCLC. Clinical trials are warranted to determine whether patients with these types of mutations could benefit from drugs that reactivate mutant p53.14,15,18,20,23–25
Overall Survival (OS) and TP53 Mutations
In a cohort study conducted by Molina-Vila et al, participants with TP53 mutations had an average overall survival (OS) of 17.5 months compared to 22.9 months for patients with WT TP53.35 Recognizing that different types of TP53 mutations have varying impacts on protein functionality, the study classified TP53 alterations into disruptive and nondisruptive categories. It was found that patients with advanced NSCLC with these two types of TP53 mutations represent distinct prognostic groups.35,100–103, 104
In this investigation, patients with nondisruptive mutations exhibited a median overall survival (OS) of 13.3 months, contrasting with 24.6 months observed in patients with wild-type TP53 or harboring disruptive mutations (P < 0.001). Notably, the correlation between nondisruptive mutations and shorter survival persisted even when patients were stratified based on EGFR status.5,25,32,42,46,92,101 Epidermal growth factor receptor (EGFR) mutations are detected in 15% of NSCLC patients in the United States, with a notably higher prevalence among Asian populations. These mutations are more commonly observed in female patients and individuals who do not smoke. Targeting EGFR primarily involves two main strategies: tyrosine kinase inhibitors (TKIs) and monoclonal antibodies.103
Within the EGFR wild-type (EGFR-WT) cohort, patients with non-disruptive TP53 alterations exhibited an average OS of 8.5 months, contrasting with 15.6 months observed in other patients (P = 0.003) represented in Figure 7. Among the EGFR-mutated (EGFR-mut) individuals, those with TP53 nondisruptive mutations demonstrated an OS of 17.8 months, compared to 28.4 months in other patients (P = 0.04).6,90,92,97 Multivariate analysis revealed that the presence of a nondisruptive TP53 mutation emerged as the sole independent prognostic factor for OS.35
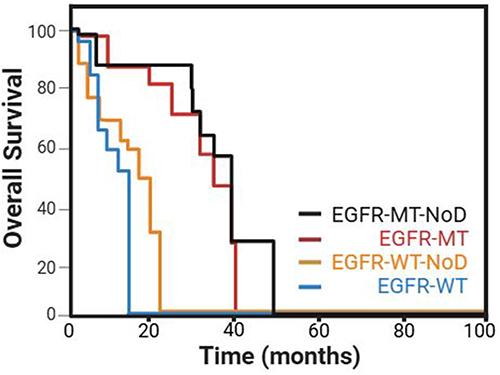 |
Figure 7 Kaplan–Meier plots of OS among EGFR wild-type (EGFR-WT) or EGFR mutated (EGFR-MT) cohort, patients with non-disruptive (NoD) TP53 alterations. Data from et al.90 |
Conclusion and Future Challenges
In conclusion, this review highlights the critical influence of TP53 gene mutations on treatment sensitivity and overall survival outcomes in non-small cell lung cancer (NSCLC). The diverse mutational spectrum of TP53 in NSCLC presents a complex challenge, with different mutations linked to varied treatment responses and prognostic outcomes. Targeted therapies and precision medicine strategies emerge as promising approaches to improve clinical results, emphasizing the need for continued research into TP53 mutation-specific pathways and their effects on NSCLC progression and treatment effectiveness. By enhancing our understanding of TP53 mutations in NSCLC, we can drive the development of more effective therapeutic interventions and improve survival rates for patients with this aggressive cancer. Advancements in the NSCLC treatment landscape have led to better median overall survival (OS) and 1-year survival rates. Despite these advancements, recent reviews indicate that the contribution of improved first-line chemotherapy to this progress is limited. While pivotal randomized trials with new chemotherapy agents have shown statistically significant survival benefits, subsequent trials comparing these regimens with other platinum doublets have not consistently replicated these superior outcomes. This inconsistency underscores the challenges in achieving sustained improvements in survival benefits with third-generation chemotherapy agents in NSCLC.
Acknowledgments
This work was supported by the Anhui Provincial Key Clinical Speciality Construction Project in 2020 ((2020) No. 243).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was studied the Anhui Provincial Key Clinical Speciality Construction Project in 2020 ((2020) No. 243).
Disclosure
All the authors declare that they have no conflict of interest.
References
1. Sun H, Ren P, Chen Y, et al. Optimal therapy for concomitant EGFR and TP53 mutated non-small cell lung cancer: a real-world study. BMC Cancer. 2023;23(1):198. doi:10.1186/s12885-023-10637-4
2. Machado-Rugolo J, Baldavira CM, Prieto TG, et al. Concomitant TP53 mutation in early-stage resected EGFR-mutated non-small cell lung cancer: a narrative approach in a genetically admixed Brazilian cohort. Braz J Med Biol Res. 2023;56:e12488. doi:10.1590/1414-431X2023e12488
3. Yang Y, Huang J, Wang T, et al. Decoding the evolutionary response to ensartinib in patients with ALK-positive NSCLC by dynamic circulating tumor DNA sequencing. J Thorac Oncol. 2021;16(5):827–839. PMID: 33588113. doi:10.1016/j.jtho.2021.01.1615
4. Christopoulos P, Kirchner M, Bozorgmehr F, et al. Identification of a highly lethal V3+ TP53+ subset in ALK+ lung adenocarcinoma. Int J Cancer. 2019;144(1):190–199. PMID: 30255938. doi:10.1002/ijc.31893
5. Cao J, Gu J, Liang Y, Wang B. Evaluate the prognosis of MYC/TP53 comutation in Chinese patients with EGFR-positive advanced NSCLC using next-generation sequencing: a retrospective study. Technol Cancer Res Treat. 2022;21:15330338221138213. doi:10.1177/15330338221138213
6. Liu DH, Zhao ZR, Lin YB, et al. Prognostic effect of TP53 and PKD co-mutations in patients with resected epidermal growth factor receptor-mutated lung adenocarcinoma. Ann Surg Oncol. 2019;26(6):1934–1941. doi:10.1245/s10434-019-07254-6
7. Tamiya A, Koh Y, Isa SI, et al. Impact of somatic mutations on prognosis in resected non-small-cell lung cancer: The Japan Molecular Epidemiology for lung cancer study. Cancer Med. 2020;9(7):2343–2351. doi:10.1002/cam4.2897
8. Mäki-Nevala S, Sarhadi VK, Rönty M, et al. Hot spot mutations in Finnish non-small cell lung cancers. Lung Cancer. 2016;99:102–110. doi:10.1016/j.lungcan.2016.06.024
9. Glaser M, Rasokat A, Prang D, et al. Clinicopathologic and molecular characteristics of small-scale ROS1-mutant non-small cell lung cancer (NSCLC) patients. Lung Cancer. 2023;184:107344. doi:10.1016/j.lungcan.2023.107344
10. Meucci S, Keilholz U, Heim D, Klauschen F, Cacciatore S. Somatic genome alterations in relation to age in lung adenocarcinoma. Int J Cancer. 2019;145(8):2091–2099. doi:10.1002/ijc.32265
11. Sakai T, Matsumoto S, Ueda Y, et al. Clinicogenomic features and targetable mutations in NSCLCs harboring BRAF non-V600E mutations: a multi-institutional genomic screening study (LC-SCRUM-Asia). J Thorac Oncol. 2023;18(11):1538–1549. doi:10.1016/j.jtho.2023.07.024
12. Hu X, Xu H, Xue Q, Wen R, Jiao W, Tian K. The role of ERBB4 mutations in the prognosis of advanced non-small cell lung cancer treated with immune checkpoint inhibitors. Mol Med. 2021;27(1):126. doi:10.1186/s10020-021-00387-z
13. Li XM, Li WF, Lin JT, et al. Predictive and prognostic potential of TP53 in patients with advanced non-small-cell lung cancer treated with EGFR-TKI: analysis of a phase III randomized clinical trial (CTONG 0901). Clin Lung Cancer. 2021;22(2):100–109.e3. doi:10.1016/j.cllc.2020.11.001
14. Si J, Hao Y, Wei J, et al. Clinical outcomes of immune checkpoint inhibitors to treat non-small cell lung cancer patients harboring epidermal growth factor receptor mutations. BMC Pulm Med. 2023;23(1):158. doi:10.1186/s12890-023-02466-9
15. Xie X, Qiu G, Chen Z, et al. Characteristics and prognosis of EGFR mutations in small cell lung cancer patients in the NGS era. Clin Transl Oncol. 2024;26(2):434–445. doi:10.1007/s12094-023-03263-w
16. de Anta JM, Jassem E, Rosell R, et al. TP53 mutational pattern in Spanish and Polish non-small cell lung cancer patients: null mutations are associated with poor prognosis. Oncogene. 1997;15(24):2951–2958. doi:10.1038/sj.onc.1201475
17. Wei W, Shi F, Xu Y, et al. The enrichment of Fanconi anemia/homologous recombination pathway aberrations in ATM/ATR-mutated NSCLC was accompanied by unique molecular features and poor prognosis. J Transl Med. 2023;21(1):874. doi:10.1186/s12967-023-04634-1
18. Jao K, Tomasini P, Kamel-Reid S, et al. The prognostic effect of single and multiple cancer-related somatic mutations in resected non-small-cell lung cancer. Lung Cancer. 2018;123:22–29. doi:10.1016/j.lungcan.2018.06.023
19. Cavagna RO, Pinto IA, Escremim de Paula F, et al. Disruptive and truncating TP53 mutations are associated with African-ancestry and worse prognosis in Brazilian patients with lung adenocarcinoma. Pathobiology. 2023;90(5):344–355. doi:10.1159/000530587
20. Qian H, Hou C, Zhang Y, et al. Effects of concurrent TP53 mutations on the efficacy and prognosis of targeted therapy for advanced EGFR mutant lung adenocarcinoma. Cancer Genet. 2023;278–279:62–70. doi:10.1016/j.cancergen.2023.08.006
21. Zhao Z, Wan J, Guo M, et al. Expression and prognostic significance of m6A-related genes in TP53-mutant non-small-cell lung cancer. J Clin Lab Anal. 2022;36(1):e24118. doi:10.1002/jcla.24118
22. Moes-Sosnowska J, Szpechcinski A, Chorostowska-Wynimko J. Clinical significance of TP53 alterations in advanced NSCLC patients treated with EGFR, ALK and ROS1 tyrosine kinase inhibitors: an update. Tumour Biol. 2024;46(s1):S309–S325. doi:10.3233/TUB-230034
23. Shajani-Yi Z, de Abreu FB, Peterson JD, Tsongalis GJ. Frequency of somatic TP53 mutations in combination with known pathogenic mutations in colon adenocarcinoma, non-small cell lung carcinoma, and gliomas as identified by next-generation sequencing. Neoplasia. 2018;20(3):256–262. doi:10.1016/j.neo.2017.12.005
24. Proulx-Rocray F, Routy B, Nassabein R, et al. The prognostic impact of KRAS, TP53, STK11 and KEAP1 mutations and their influence on the NLR in NSCLC patients treated with immunotherapy. Cancer Treat Res Commun. 2023;37:100767. doi:10.1016/j.ctarc.2023.100767
25. Nishio M, Paz-Ares L, Reck M, et al. RELAY, Ramucirumab Plus Erlotinib (RAM+ERL) in untreated metastatic EGFR-mutant NSCLC (EGFR+ NSCLC): association between TP53 status and clinical outcome. Clin Lung Cancer. 2023;24(5):415–428. doi:10.1016/j.cllc.2023.02.010
26. Barta JA, McMahon SB. Lung-enriched mutations in the p53 tumor suppressor: a paradigm for tissue-specific gain of oncogenic function. Mol Cancer Res. 2019;17(1):3–9. doi:10.1158/1541-7786.MCR-18-0357
27. Guimaraes D, Hainaut P. TP53: a key gene in human cancer. Biochimie. 2002;84(1):83–93. doi:10.1016/S0300-9084(01)01356-6
28. Luo J, Mei Z, Lin S, Xing X, Qian X, Lin H. Integrative pan-cancer analysis reveals the importance of PAQR family in lung cancer. J Cancer Res Clin Oncol. 2023;149(12):10149–10160. doi:10.1007/s00432-023-04922-9
29. La Fleur L, Falk-Sörqvist E, Smeds P, et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer. 2019;130:50–58. doi:10.1016/j.lungcan.2019.01.003
30. Ma X, Rousseau V, Sun H, et al. Significance of TP53 mutations as predictive markers of adjuvant cisplatin-based chemotherapy in completely resected non-small-cell lung cancer. Mol Oncol. 2014;8(3):555–564. doi:10.1016/j.molonc.2013.12.015
31. Ma X, Le Teuff G, Lacas B, et al. Prognostic and predictive effect of TP53 mutations in patients with non-small cell lung cancer from adjuvant cisplatin-based therapy randomized trials: a LACE-bio pooled analysis. J Thorac Oncol. 2016;11(6):850–861. doi:10.1016/j.jtho.2016.02.002
32. Fu J, Tong Y, Xu Z, et al. Impact of TP53 mutations on EGFR-tyrosine kinase inhibitor efficacy and potential treatment strategy. Clin Lung Cancer. 2023;24(1):29–39. doi:10.1016/j.cllc.2022.08.007
33. Gibelin C, Couraud S. Somatic alterations in lung cancer: do environmental factors matter? Lung Cancer. 2016;100:45–52. doi:10.1016/j.lungcan.2016.07.015
34. Schwaederlé M, Lazar V, Validire P, et al. VEGF-A expression correlates with TP53 mutations in non-small cell lung cancer: implications for antiangiogenesis therapy. Cancer Res. 2015;75(7):1187–1190. doi:10.1158/0008-5472.CAN-14-2305
35. Molina-Vila MA, Bertran-Alamillo J, Gascó A, et al. Nondisruptive p53 mutations are associated with shorter survival in patients with advanced non-small cell lung cancer. Clin Cancer Res. 2014;20(17):4647–4659. doi:10.1158/1078-0432.CCR-13-2391
36. Aisner DL, Sholl LM, Berry LD, et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations-the lung cancer mutation consortium (LCMC2). Clin Cancer Res. 2018;24(5):1038–1047. doi:10.1158/1078-0432.CCR-17-2289
37. Zhang C, Wang K, Lin J, Wang H. Non-small-cell lung cancer patients harboring TP53/KRAS co-mutation could benefit from a PD-L1 inhibitor. Future Oncol. 2022;18(27):3031–3041. doi:10.2217/fon-2022-0295
38. Mathiot L, Nigen B, Goronflot T, et al. Prognostic impact of TP53 mutations in metastatic nonsquamous non-small-cell lung cancer. Clin Lung Cancer. 2024;25(3):244–253.e2. doi:10.1016/j.cllc.2023.12.004
39. Lee SY, Jeon HS, Hwangbo Y, et al. The influence of TP53 mutations on the prognosis of patients with early stage non-small cell lung cancer may depend on the intratumor heterogeneity of the mutations. Mol Carcinog. 2015;54(2):93–101. doi:10.1002/mc.22077
40. Woo HG, Wang XW, Budhu A, et al. Association of TP53 mutations with stem cell-like gene expression and survival of patients with hepatocellular carcinoma. Gastroenterology. 2011;140(3):1063–1070. doi:10.1053/j.gastro.2010.11.034
41. Blasi M, Kuon J, Lüders H, et al. First-line immunotherapy for lung cancer with MET exon 14 skipping and the relevance of TP53 mutations. Eur J Cancer. 2024;199:113556. Erratum in: Eur J Cancer. 2024 Jul;205:114130. doi: 10.1016/j.ejca.2024.114130. doi:10.1016/j.ejca.2024.113556
42. Qin K, Hou H, Liang Y, Zhang X. Prognostic value of TP53 concurrent mutations for EGFR- TKIs and ALK-TKIs based targeted therapy in advanced non-small cell lung cancer: a meta-analysis. BMC Cancer. 2020;20(1):328. doi:10.1186/s12885-020-06805-5
43. De B, Farooqi AS, Mitchell KG, et al. Benchmarking outcomes for molecularly characterized synchronous oligometastatic non-small-cell lung cancer reveals EGFR mutations to be associated with longer overall survival. JCO Precis Oncol. 2023;7:e2200540. doi:10.1200/PO.22.00540
44. Li VD, Li KH, Li JT. TP53 mutations as potential prognostic markers for specific cancers: analysis of data from The Cancer Genome Atlas and the International Agency for Research on Cancer TP53 database. J Cancer Res Clin Oncol. 2019;145(3):625–636. doi:10.1007/s00432-018-2817-z
45. Nelson HH, Wilkojmen M, Marsit CJ, Kelsey KT. TP53 mutation, allelism and survival in non-small cell lung cancer. Carcinogenesis. 2005;26(10):1770–1773. doi:10.1093/carcin/bgi125.
46. Pezzuto F, Hofman V, Bontoux C, et al. The significance of co-mutations in EGFR-mutated non-small cell lung cancer: optimizing the efficacy of targeted therapies? Lung Cancer. 2023;181:107249. doi:10.1016/j.lungcan.2023.107249
47. Liu J, Gao J. Efficacy of immunotherapy as second-line or later-line therapy and prognostic significance of KRAS or TP53 mutations in advanced non-small cell lung cancer patients. Eur J Cancer Prev. 2023;32(6):590–599. doi:10.1097/CEJ.0000000000000799
48. Feng H, Xu H, Shi X, et al. TP53 exon 5 mutation indicates poor progression-free survival for patients with stage IV NSCLC. Front Biosci. 2023;28(7):147. doi:10.31083/j.fbl2807147
49. Omasini P, Mascaux C, Jao K, et al. Effect of coexisting KRAS and TP53 mutations in patients treated with chemotherapy for non-small-cell lung cancer. Clin Lung Cancer. 2019;20(3):e338–e345. doi:10.1016/j.cllc.2018.12.009
50. Wang H, Shan Q, Guo J, et al. PDL1 high expression without TP53, KEAP1 and EPHA5 mutations could better predict survival for patients with NSCLC receiving atezolizumab. Lung Cancer. 2021;151:76–83. doi:10.1016/j.lungcan.2020.11.006
51. Netter J, Lehmann-Che J, Lambert J, et al. Functional TP53 mutations have no impact on response to cytotoxic agents in metastatic colon cancer. Bull Cancer. 2015;102(2):117–125. doi:10.1016/j.bulcan.2014.12.010
52. Christopoulos P, Kluck K, Kirchner M, et al. The impact of TP53 co-mutations and immunologic microenvironment on outcome of lung cancer with EGFR exon 20 insertions. Eur J Cancer. 2022;170:106–118. doi:10.1016/j.ejca.2022.04.020
53. Zhang L, Zhang T, Shang B, Li Y, Cao Z, Wang H. Prognostic effect of coexisting TP53 and ZFHX3 mutations in non-small cell lung cancer patients treated with immune checkpoint inhibitors. Scand J Immunol. 2021;94(3):e13087. doi:10.1111/sji.13087
54. Scalera S, Mazzotta M, Cappuzzo F, Ciliberto G, Maugeri-Saccà M. KEAP1 and TP53 mutations in lung cancer: more is better. reply to: “Survival analysis of TP53 co-mutations should be interpreted more cautiously”. J Thorac Oncol. 2022;17(3):e40–e41. doi:10.1016/j.jtho.2021.12.001
55. Li H, Yang L, Wang Y, et al. Integrative analysis of TP53 mutations in lung adenocarcinoma for immunotherapies and prognosis. BMC Bioinf. 2023;24(1):155. doi:10.1186/s12859-023-05268-2
56. Zhu M, Kim J, Deng Q, et al. Loss of p53 and mutational heterogeneity drives immune resistance in an autochthonous mouse lung cancer model with high tumor mutational burden. Cancer Cell. 2023;41(10):1731–1748.e8. doi:10.1016/j.ccell.2023.09.006
57. Lane DP. Cancer. P53, guardian of the genome. Nature. 1992;358:15–16. doi:10.1038/358015a0
58. Bueno R, Stawiski EW, Goldstein LD, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48(4):407–416. doi:10.1038/ng.3520
59. Honda T, Seto K, Endo S, et al. The possibility of mutations of RAS signaling genes and/or TP53 in combination as a negative prognostic impact on pathological stage I non-small cell lung cancer. Cancer Med. 2023;12(19):19406–19413. doi:10.1002/cam4.6535
60. Song P, Zhang F, Li Y, et al. Concomitant TP53 mutations with response to crizotinib treatment in patients with ALK-rearranged non-small-cell lung cancer. Cancer Med. 2019;8(4):1551–1557. doi:10.1002/cam4.2043
61. Saleh MM, Scheffler M, Merkelbach-Bruse S, Wolf J, Buettner R. Reply to: “Survival analysis of TP53 co-mutations should be interpreted more cautiously”. J Thorac Oncol. 2022;17(6):e57–e59. doi:10.1016/j.jtho.2022.03.004
62. Pellinen T, Paavolainen L, Martín-Bernabé A, et al. Fibroblast subsets in non-small cell lung cancer: associations with survival, mutations, and immune features. J Natl Cancer Inst. 2023;115(1):71–82. doi:10.1093/jnci/djac178
63. Bearz A, Martini JF, Jassem J, et al. Efficacy of lorlatinib in treatment-naive patients with ALK-positive advanced NSCLC in relation to EML4: ALK variant type and ALK with or without TP53 mutations. J Thorac Oncol. 2023;18(11):1581–1593. doi:10.1016/j.jtho.2023.07.023
64. Offin M, Chan JM, Tenet M, et al. Concurrent RB1 and TP53 alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J Thorac Oncol. 2019;14(10):1784–1793. doi:10.1016/j.jtho.2019.06.002
65. Corsini EM, Mitchell KG, Mehran RJ, et al. Colorectal cancer mutations are associated with survival and recurrence after pulmonary metastasectomy. J Surg Oncol. 2019;120(4):729–735. doi:10.1002/jso.25630
66. Jiang L, Huang J, Higgs BW, et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS Genet. 2016;12(4):e1005895. doi:10.1371/journal.pgen.1005895
67. Kim JY, Jung J, Kim KM, Lee J, Im YH. TP53 mutations predict poor response to immunotherapy in patients with metastatic solid tumors. Cancer Med. 2023;12(11):12438–12451. doi:10.1002/cam4.5953
68. Assoun S, Theou-Anton N, Nguenang M, et al. Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer. 2019;132:65–71. doi:10.1016/j.lungcan.2019.04.005
69. Gao W, Jin J, Yin J, et al. KRAS and TP53 mutations in bronchoscopy samples from former lung cancer patients. Mol Carcinog. 2017;56(2):381–388. doi:10.1002/mc.22501
70. Chen J, Yang H, Teo ASM, et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat Genet. 2020;52(2):177–186. doi:10.1038/s41588-019-0569-6
71. Hoyos D, Zappasodi R, Schulze I, et al. Fundamental immune-oncogenicity trade-offs define driver mutation fitness. Nature. 2022;606(7912):172–179. Erratum in: Nature. 2022 Jun;606(7914):E5. doi: 10.1038/s41586-022-04879-8. doi:10.1038/s41586-022-04696-z
72. Wu Z, Su J, Li FL, et al. YAP silencing by RB1 mutation is essential for small-cell lung cancer metastasis. Nat Commun. 2023;14(1):5916. doi:10.1038/s41467-023-41585-z
73. Marcoux N, Gettinger SN, O’Kane G, et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J Clin Oncol. 2019;37(4):278–285. doi:10.1200/JCO.18.01585
74. Chang F, Syrjänen S, Tervahauta A, et al. Tumourigenesis associated with the p53 tumour suppressor gene. Br J Cancer. 1993;68:653–661. doi:10.1038/bjc.1993.404
75. Chang F, Syrjänen S, Syrjänen K. Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncol. 1995;13(4):1009–1022. doi:10.1200/JCO.1995.13.4.1009
76. Chang F, Syrjänen S, Kurvinen K, Syrjänen K. The p53 tumor suppressor gene as a common cellular target in human carcinogenesis. Am J Gastroenterol. 1993;88(2):174–186.
77. Zhao L, Qu X, Wu Z, Li Y, Zhang X, Guo W. TP53 somatic mutations are associated with poor survival in non-small cell lung cancer patients who undergo immunotherapy. Aging. 2020;12(14):14556–14568. doi:10.18632/aging.103502
78. Frankell AM, Dietzen M, Al Bakir M, et al. The evolution of lung cancer and impact of subclonal selection in TRACERx. Nature. 2023;616(7957):525–533. Erratum in: Nature. 2024 Jul;631(8022):E15. doi: 10.1038/s41586-024-07738-w. doi:10.1038/s41586-023-05783-5
79. Vokes NI, Chambers E, Nguyen T, et al. Concurrent TP53 mutations facilitate resistance evolution in EGFR-mutant lung adenocarcinoma. J Thorac Oncol. 2022;17(6):779–792. doi:10.1016/j.jtho.2022.02.011
80. West HJ, McCleland M, Cappuzzo F, et al. Clinical efficacy of atezolizumab plus bevacizumab and chemotherapy in KRAS-mutated non-small cell lung cancer with STK11, KEAP1, or TP53 comutations: subgroup results from the phase III IMpower150 trial. J Immunother Cancer. 2022;10(2):e003027. doi:10.1136/jitc-2021-003027
81. Mitsudomi T, Hamajima N, Ogawa M, Takahashi T. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: a meta-analysis. Clin Cancer Res. 2000;6(10):4055–4063.
82. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376(22):2109–2121. doi:10.1056/NEJMoa1616288
83. George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. doi:10.1038/nature14664
84. Tsao MS, Aviel-Ronen S, Ding K, et al. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. J Clin Oncol. 2007;25(33):5240–5247. doi:10.1200/JCO.2007.12.6953
85. Custodio AB, González-Larriba JL, Bobokova J, et al. Prognostic and predictive markers of benefit from adjuvant chemotherapy in early-stage non-small cell lung cancer. J Thorac Oncol. 2009;4(7):891–910. doi:10.1097/JTO.0b013e3181a4b8fb
86. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev. 2016;25(1):16–27. doi:10.1158/1055-9965
87. Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol. 1979;31(2):472–483. doi:10.1128/JVI.31.2.472-483.1979
88. Linzer DI, Levine AJ. Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17(1):43–52. doi:10.1016/0092-8674(79)90293-9
89. Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278(5701):261–263. doi:10.1038/278261a0
90. Ferrara MG, Belluomini L, Smimmo A, et al. Meta-analysis of the prognostic impact of TP53 co-mutations in EGFR-mutant advanced non-small-cell lung cancer treated with tyrosine kinase inhibitors. Crit Rev Oncol Hematol. 2023;184:103929. doi:10.1016/j.critrevonc.2023.103929
91. Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19(9):495–509. doi:10.1038/s41568-019-0179-8
92. Canale M, Andrikou K, Priano I, et al. The role of TP53 mutations in EGFR-mutated non-small-cell lung cancer: clinical significance and implications for therapy. Cancers. 2022;14(5):1143. doi:10.3390/cancers14051143
93. Deben C, Deschoolmeester V, Lardon F, Rolfo C, Pauwels P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: evaluating their prognostic and predictive value. Crit Rev Oncol Hematol. 2016;99:63–73. doi:10.1016/j.critrevonc.2015.11.019
94. Anna L, Holmila R, Kovacs K, et al. Relationship between TP53 tumour suppressor gene mutations and smoking-related bulky DNA adducts in a lung cancer study population from Hungary. Mutagenesis. 2009;24(6):475–480. doi:10.1093/mutage/gep031
95. Swisher SG, Roth JA, Nemunaitis J, et al. Adenovirus-mediated p53 gene transfer in advanced non-small-cell lung cancer. J Natl Cancer Inst. 1999;91(9):763–771. doi:10.1093/jnci/91.9.763
96. Stockhammer P, Grant M, Wurtz A, et al. Co-occurring alterations in multiple tumor suppressor genes are associated with worse outcomes in patients with EGFR-mutant lung cancer. J Thorac Oncol. 2024;19(2):240–251. doi:10.1016/j.jtho.2023.10.001
97. Mogi A, Kuwano H. TP53 mutations in nonsmall cell lung cancer. J Biomed Biotechnol. 2011;2011:583929. doi:10.1155/2011/583929
98. Plaja A, Moran T, Carcereny E, et al. Small-cell lung cancer long-term survivor patients: how to find a needle in a haystack? Int J Mol Sci. 2021;22(24):13508. doi:10.3390/ijms222413508
99. Cao H, Ma Z, Huang Q, et al. Clinicopathologic features, concurrent genomic alterations, and clinical outcomes of patients with KRAS G12D mutations in resected lung adenocarcinoma. Eur J Cancer. 2024;202:113985. doi:10.1016/j.ejca.2024.113985
100. Ønnesen EMT, Stougaard M, Meldgaard P, Lade-Keller J. Prognostic value of KRAS mutations, TP53 mutations and PD-L1 expression among lung adenocarcinomas treated with immunotherapy. J Clin Pathol. 2023;77(1):54–60. doi:10.1136/jcp-2022-208574
101. Uras IZ, Moll HP, Casanova E. Targeting KRAS mutant non-small-cell lung cancer: past, present and future. Int J Mol Sci. 2020;21(12):4325. doi:10.3390/ijms21124325
102. Liu S, Yu J, Zhang H, Liu J. TP53 co-mutations in advanced EGFR-mutated non-small cell lung cancer: prognosis and therapeutic strategy for cancer therapy. Front Oncol. 2022;12:860563. doi:10.3389/fonc.2022.860563
103. Duma N, Santana-Davila R, Molina JR. Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc. 2019;94(8):1623–1640. doi:10.1016/j.mayocp.2019.01.013
104. Soussi T. The TP53 Website-UMD TP53 mutation database. TP53 mutation database [Online]. Available from: https://p53.fr/.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.