")
Back to Journals » International Journal of Nanomedicine » Volume 20
Electromechanical Regulation Underlying Protein Nanoparticle-Induced Osmotic Pressure in Neurotoxic Edema
Authors Zheng Z, Nie A, Wu X, Chen S, Zhang L, Yang D, Shi Y, Xiong X, Guo J
Received 27 October 2024
Accepted for publication 28 March 2025
Published 5 April 2025 Volume 2025:20 Pages 4145—4163
DOI https://doi.org/10.2147/IJN.S503181
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Dong Wang
Zihui Zheng,1,2,* Aobo Nie,1,* Xiaojie Wu,1 Shi Chen,1 Lijun Zhang,1 Dongqing Yang,3 Yuqing Shi,1 Xiyu Xiong,1 Jun Guo1,2
1Department of Biochemistry and Molecular Biology, School of Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, 210023, People’s Republic of China; 2State Key Laboratory on Technologies for Chinese Medicine Pharmaceutical Process Control and Intelligent Manufacture, Nanjing University of Chinese Medicine, Nanjing, 210023, People’s Republic of China; 3Department of Public Health, School of Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, 210023, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Guo, School of Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, 210023, People’s Republic of China, Tel +86 13813909055, Email [email protected]
Purpose: Osmotic imbalance is a critical driving force of cerebral edema. Protein nanoparticles (PNs) amplify intracellular osmotic effects by regulating membrane potential and homeostasis of water and multiple ions. This study has investigated how PNs control the neuronal swelling through electromechanical activity.
Methods: The fluorescence resonance energy transfer (FRET)-based Vimentin force probe was used to real-time monitor the osmotic tension in neurons. Patch clamp and the living cell 3D imaging system were applied to explore the relationship between cell electromechanical activity and cell volume in different cytotoxic cell models. Cytoplasmic PN amount measured by the NanoSight instrument, ion contents detected by the freezing point osmometer and ion imaging were performed to investigate the role of PNs in regulating cell swelling.
Results: We observed a close association between neuronal swelling and changes in osmotic tension and membrane potential. The tension effect of biological osmotic pressure (OP) relies on electromechanical cooperation induced by intracellular PN and Ca2+ levels. PNs increment results from cytoplasmic translocation of intracellular various proteins. Alterations in Ca2+ content are involved in the membrane potential transition between depolarization and hyperpolarization in a PN-dependent manner. Chemical signals-mediated sensitization of ion channels has an indispensable effect on PN-induced ion increments. Notably, aquaporin-mediated water influx recovers membrane potential and enhances osmotic tension controlling neuronal swelling.
Conclusion: Our findings indicate that PNs, Ca2+, and water are pivotal in electromechanical cooperation and provide insights into the biological OP mechanisms underlying neurotoxic edema.
Keywords: protein nanoparticle, electromechanical cooperation, osmotic tension, membrane potential, neuronal swelling
Graphical Abstract:

Introduction
Cerebral edema is a secondary symptom of ischemic stroke, leading to intracranial hypertension and cerebral herniation.1 Cytotoxic edema occurs in the early stages of cerebral edema, and it is characterized by excitatory toxicity and intracellular hyperosmosis-induced swelling of neurons and astrocytes.2 Excessive extracellular accumulation of the neurotransmitter glutamate can activate intracellular calcium overload and the abnormal accumulation of intracellular ions, leading to the occurrence of malignant edema. Therefore, osmotic imbalance induces extracellular ions and water flow, as the crucial driving forces of cytotoxic edema.3 Thus, investigating the mechanisms underlying the development of cytotoxic edema could facilitate the timely treatment of cerebral edema.
Our previous studies have shown that protein nanoparticles (PNs) generated in astrocytes and neurons can induce membrane potential changes and upregulate osmosis, which are significant pathogenic factors in cerebral edema.4,5 In neuronal cells, changes in membrane potential require the transmembrane movement of charged ions. The number of ions that flow across the membrane to cause changes in membrane potential is small, so the total intra- and extracellular ion concentrations are not considerably affected.6 Notably, changes in membrane potential can activate various voltage-dependent ion channels, such as the calcium-activated non-selective cation channel SUR1-TRPM4 and chloride channel TMEM16A, facilitating massive ion flux and osmotic regulation.4 Glibenclamide (Gil), an inhibitor of SUR1-TRPM4 channel, reportedly prolongs the closed state of the channel and suppresses neurovascular expression of the channel protein, thereby alleviating cell swelling and cerebral edema.7–9 Similarly, TMEM16A channel inhibitors have been shown to alleviate cerebral infarction and nervous system impairment in ischemic stroke models.10,11 These findings suggest a complex electrophysiological–osmotic cascade regulation in cytotoxic edema.
Cell volume changes depend on imbalanced osmosis-induced water flux. Aquaporins (AQPs) are crucial in facilitating water flux across the membrane. AQP9 overexpression has been reported to accelerate the occurrence of cerebral edema in stroke models, while AQP9 knockdown can reduce the entry of interstitial fluid into cells and delay the formation of neurogenic edema.12 AQP9-positive neurons and astrocytes are widely present in the hypothalamus after brain injury,13,14 suggesting that AQP9 is important in water flux regulation in cytotoxic edema. At present, the regulatory factors of AQP activity remain unclear. Further investigation into the upstream and downstream events of AQP is pivotal for understanding the role of water influx in cell swelling.
Cytotoxic swelling is accompanied by mechanical force transduction.15 The cytoskeleton, with its network structure, generates mechanical stress. Intracellular tension is essentially derived from outward osmotic pressure (OP) and inward cytoskeletal tension, which participate in regulating cell volume by resisting mechanical or OP shocks.16,17 Mechanically sensitive cation channels, such as Piezo1, can sense mechanical force and convert it into electrical or chemical signals within cells. Piezo1 acts as a calcium-permeable ion channel, and its activation is associated with membrane depolarization,18–20 neuronal activity, and cerebral edema.19,21–24 These findings suggest that mechanical force synergistically works with electrochemical signals in regulating cell volume, closely related to transmembrane ion balance and OP.
The amount of intracellular PNs is closely associated with osmosis. PNs can induce ion redistribution on both sides of the cell membrane, rather than simple colloid OP. PNs, sourced from the depolymerization of microfilaments and microtubules, and inflammasomes, can regulate membrane potential and further activate voltage-dependent ion channels.4,25 Therefore, we propose that PNs are key factors in controlling the disequilibrium of electrochemical-osmotic tension in cytotoxic edema.
Intermediate filament (IF), as an important part of the cytoskeleton, plays a role in supporting the cell shape. Transmembrane osmotic difference triggers changes in the IF pulling tension. Herein we used a fluorescence resonance energy transfer (FRET)-IF tension probe, whose tension is positively related to transmembrane OP in living cells.4,5,25–27 Combined with electrophysiological techniques, we observed the occurrence and equilibrium of electrochemistry and osmotic tension, revealing the interaction of PNs, calcium, water, membrane potential, osmosis, and IF tension in the progression of neuronal swelling. Understanding of how electrochemistry-tension disequilibrium regulates water transmembrane flux and biological OP may possibly have applications, in the treatment of ischemic stroke and cerebral edema. Considering the functional redundancy among multiple ion channels, we propose the upstream events of PNs can control membrane potential and the downstream multiple voltage-dependent ion channels. PN is hopefully developed as a new therapeutic strategy for the edema treatment.
Materials and Methods
Cell Culture and Reagents
The human neuroblastoma cell line SH-SY5Y was obtained from the American Type Culture Collection (Manassas, VA, USA). SH-SY5Y cells were cultured in DMEM/F12 media (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin–streptomycin (Gibco) in a 5% CO2 incubator at 37°C. The growth medium was changed every 24 h. All buffer solutions were prewarmed at 37°C before treatment with cells. Cells on coverslips were mounted on a stage equipped with a Bioptechs temperature controller to keep the cells at 37°C.
Taxol (TAX), Sennoside A (Sen A), Nimodipine (Nimo), Cytochalasin D (Cyto D), and nocodazole (Noc) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Heparin, tranilast (Tran), CoCl2, LPS, and 1.2-DCE were purchased from Shanghai Macklin Biochemical Co., Ltd. (Shanghai, China). Glibenclamide (Gil), Niclosamide (Nic), calcium-like peptide-1 (CALP1), and phorbol 12-myristate 13-acetate (PMA) were procured from MedChemExpress (Monmouth Junction, NJ, USA).
Probe Construction and Transfection
Following the principles of fluorescence resonance energy transfer (FRET), we constructed a vimentin–cpstFRET (cpVenus–7aa–cpCerulean) fluorescent tension probe.4,25,26 cpCerulean (cyan) was the donor and cpVenus (yellow) was the acceptor, and they were positioned parallel to each other in the absence of an external force. When stimulated by an external force, the angle of cpstFRET changed, decreasing FRET efficiency. The cells were transfected with the vimentin–cpstFRET plasmid (1 μg) using 3 μL Lipofectamine™ 2000 (Thermo Fisher, US).
cpstFRET Analyses
FRET efficiency was determined by the dipole angle between the donor/enhanced cyan fluorescent protein (eCFP) and the acceptor/enhanced yellow fluorescent protein (eYFP).28 The donor and acceptor were tested using argon lasers at 458 nm and 514 nm, respectively. Images of the donor and acceptor emissions were captured using a Dual View 2 splitter (MAG biosystems at BioVision Technologies, Exton, PA, USA). FRET/acceptor emission ratios were calculated for each pixel in the clearest optical plane for each image field. The cells were applied with a binary mask using ImageJ. A donor mask was generated by applying a threshold to the donor image, and a similar mask was generated for the acceptor channel. CFP/FRET ratios (intensity of the CFP channel divided by that of the FRET channel) were calculated using the equation E = eCFPdonor/eYFPacceptor, negatively correlated with FRET efficiency but positively correlated with force. Pseudocolor was applied using ImageJ to generate the final images. As the strong autofluorescence from serum-containing media, the culture media was replaced with 37 °C prewarmed HEPES buffer, for all FRET and live fluorescence imaging.
siRNA Transfection and Primer Design
Piezo1-siRNA and NC-siRNA were designed and synthesized by GenePharma (Shanghai, China). The sense and antisense sequences of Piezo1-siRNA were 5′-AGAAGAAGAUCGUCAAGUATT-3′ and 5′-UACUUGACGAUCUCUUCUTT-3′, respectively, and those of NC-siRNA were 5′-GUGAGCGUCUAUAUACCAUTT-3′ and 5′-AUGGUAUAUAGACGCUCACTT-3′, respectively.
For siRNA transfection, the cells were mixed in 100:3:1 (volume ratio) OPTI-MEM:plasmid:transfection reagent. After incubation in the dark, 100 μL of the mixture was added to 900 μL serum-free DMEM, and the cells were cultured at 37°C and 5% CO2 for 48 h before subsequent experiments.
Measurement of Cytoplasmic OP and Count Rate of PNs
Cells were cultured in a 100-mm culture dish. When the cultures reached approximately 95% confluence, the cells were washed twice with isotonic HEPES buffer and then treated with specific drugs. Cytoplasmic supernatants were obtained through digestion, centrifugation (14000 g, 4°C), ultrasonication (Sonics & Materials, Newtown, CT, USA), and re-centrifugation. Cytoplasmic OP (representing total ions) was measured with an Osmomat 3000 Freezing Point Osmometer (Gonotec, Berlin, Germany). The count of cytoplasmic nanoparticles was determined using a NanoSight NS300 instrument (Malvern Analytics, Malvern, UK) in the “Protein Size” mode, yielding the kilocycles per second (kcps) value.
Measurement of Intracellular Ca2+/Cl− Levels
Intracellular Ca2+ levels were detected using the calcium fluorescent indicator probe Fluo-4 AM (Beyotime, China). Fluo-4 AM (200 μg) was dissolved in 88.4 μL DMSO, and Pluronic F-127 was added; a working solution of 4 μM was prepared by diluting with HEPES-buffered saline solution. The cells were cultured in the probe working solution and HEPES buffer containing 1% fetal bovine serum. They were then washed with HEPES buffer and incubated, and Ca2+ fluorescence was detected under an inverted fluorescence microscope (Leica Microsystems, Wetzlar, Germany) at excitation and emission wavelengths of 488 nm and 516 nm, respectively.
Intracellular Cl− levels were measured using the N-[ethoxycarbonylmethyl]-6-methoxy-quinolinium bromide (MQAE) probe (Beyotime, China). A working solution was prepared using Kerbs-HEPES buffer, which contained 2.5 mM NaCl, 2.5 mM KCl, 2.7 mM CaCl2, 20 mM HEPES, 1 mM MgCl2, and 16 mM glucose (pH 7.4). The cells were incubated in 100 μL Kerbs-HEPES buffer containing 10 mM MQAE for 40 min and then washed five times. MQAE fluorescence was detected under an inverted fluorescence microscope (Leica Microsystems) at excitation and emission wavelengths of 355 nm and 460 nm, respectively. An increase in intracellular Cl− levels led to a decrease in MQAE fluorescence intensity.
Holographic Label-Free Live Cell Imaging
SH-SY5Y cells were prepared and seeded in a 35-mm confocal culture dish and maintained at 37°C in an environment with 95% O2 and 5% CO2. At approximately 30% density, cell morphology was monitored using a 3D tomographic microscope with a 60× objective lens (Nanolive CXA, Switzerland) and STEVE software (Nanolive CXA).
Electrophysiology
Whole-cell recordings of SH-SY5Y cells were performed in either current- or voltage-clamp mode. The cells were seeded onto polylysine-coated slides. During the experiments, the cells were placed in a bath, and the required solution was perfused. Voltage and current were measured using a MultiClamp 700B amplifier and Digidata 1550B digital converter controlled by pClamp 10.6 (Molecular Devices, Sunnyvale, CT, USA). Glass electrodes (outer diameter 1.5 mm, inner diameter 0.86 mm, World Precision Instruments, USA) were pulled using a puller (P-1000, Narishige) to achieve resistance of 2–5 MΩ when filled with internal solution. Capacitive and series resistance compensations were adjusted before recording. The data sampling rate was set to 2 kHz for cell membrane potential recordings and 10,000 kHz for cell membrane current recordings.
For cell membrane potential recordings, a hypotonic solution was used, which contained (mM) 90 NaCl, 4.8 KCl, 5 NaHCO3, 2.4 MgCl2, 2.5 CaCl2, and 10 HEPES. pH was adjusted to 7.4 with NaOH, and OP was adjusted to 220 mOsm/kg with mannitol. The extracellular isotonic solution contained (mM) 90 NaCl, 4.8 KCl, 5 NaHCO3, 2.4 MgCl2, 2.5 CaCl2, and 10 HEPES. pH was adjusted to 7.4 with NaOH, and OP was adjusted to 300 mOsm/kg with mannitol. The intracellular solution comprised (mM) 139 KCl, 2 MgCl2, Na2ATP, 1 CaCl2, 2 EGTA, and 5 glucose. pH was adjusted to 7.4 with NaOH, and OP was adjusted to 300 mOsm/kg with mannitol.
SUR1-TRPM4 channel current was recorded within a voltage range of −100 to +100 mV. Starting from 0 mV clamping potential, a ramp voltage stimulation scheme of 300 ms was used to obtain a stable current–voltage relationship curve.4 For TMEM16A channel current recordings, a step voltage (20 mV) stimulation protocol was applied from −80 to +80 mV, starting from 0 mV clamping potential and lasting for 1.1 s.4 Finally, for VGCC current recordings, a step voltage (10 mV) stimulation protocol was applied from −70 mV to +10 mV, starting from −90 mV clamping potential and lasting for 300 ms.29
To record SUR1-TRPM4 channel current, SH-SY5Y cells were perfused with an extracellular solution, which comprised (mM) 144 NaCl, 4.3 KCl, 2.5 CaCl2, 1.1 MgCl2, 10 HEPES, and 10 D-glucose. pH was adjusted to 7.4 with NaOH, and OP was adjusted to 300 mOsm/kg with mannitol. The intracellular solution contained (mM) 140 CsCl, 2 MgCl2, 9.8 CaCl2, 10 HEPES, 10 EGTA, and 2 Na2ATP. pH was adjusted to 7.4 with CsOH, and OP was adjusted to 300 mOsm/kg with mannitol. To record TMEM16A channel current, SH-SY5Y cells were perfused with an extracellular solution, which contained (mM) 140 N-methyl-D-glucosamine, 10 tetraethylammonium chloride, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 D-glucose. pH was adjusted to 7.4 with HCl, and OP was adjusted to 300 mOsm/kg with mannitol. The intracellular solution comprised (mM) 140 CsCl, 9.8 CaCl2, 2 MgCl2, 10 EGTA, 10 hEPES, and 2 Na2ATP. pH was adjusted to 7.4 with CsOH, and OP was adjusted to 300 mOsm/kg with mannitol. To record VGCC current, SH-SY5Y cells were perfused with an extracellular solution, which contained (mM) 145 tetraethylammonium chloride, 5 CaCl2, 0.8 MgCl2, 10 HEPES, and 5 glucose. pH was adjusted to 7.4 with CsOH, and OP was adjusted to 300 mOsm/kg with mannitol. The intracellular solution comprised (mM) 135 CsCl, 1 CaCl2, 2 MgCl2, 2 Na2ATP, 11 EGTA, and 10 HEPES. pH was adjusted to 7.4 with CsOH, and OP was adjusted to 300 mOsm/kg with mannitol.
Current density was calculated by dividing the current value obtained by compensating the capacitance and series resistance recorded by the patch clamp amplifier by the cell membrane capacitance.
LC–MS and Proteomic Data Processing
Cultured SH-SY5Y cells were washed twice with isotonic HEPES buffer and then subjected to specific treatments: (1) the control group, (2) co-treatment with microfilaments and microtubules depolymerizers (the CytoD+Noc group) for 30 min, and (3) co-treatments with CytoD–Noc for 30 min, followed by replacement with DMEM/F12 medium and further culturing for 30 min (the CytoD+Noc-removal group). Cytoplasmic supernatants were obtained through digestion, centrifugation (14,000 g, 4°C), ultrasonication (Sonics & Materials), and re-centrifugation.
For filter-aided sample preparation, total protein concentration was determined using a Nanodrop microvolume spectrophotometer. Sample aliquots (100 μg) were used for all subsequent steps. Each sample was mixed with dithiothreitol and alkylated with iodoacetic acid, and then combined with urea. Filter-aided sample preparation filters were activated with NH4HCO3, followed by centrifugation. Peptides were eluted from the membrane, and samples were dried using a vacuum centrifuge.
For MS and data analysis, peptides were resolved on a C18 reverse phase column (Thermo Fisher Scientific EASY-nLC) and analyzed with a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific) using higher-energy collisional dissociation fragmentation. The mass spectrometer was operated in the Top 20 data-dependent mode with automated switching between MS and MS/MS. Raw data were processed using the MaxQuant search engine, mapped to UniProt/SwissProt. Spectra were searched against the UniProt human database. Target decoy analysis was performed by searching a reverse database with an overall false discovery rate of 1% at the protein and peptide levels. Label-free quantification was performed using the LFQ feature included in MaxQuant according to the default parameters.
Finally, the resulting data were subjected to GO and KEGG enrichment analysis. Pathways with a p-value of ≤0.05 were considered significantly enriched and ranked by p-value.
Statistical Analyses
Values represent mean ± standard error of the mean (SEM). Data were analyzed using SPSS v.22.0 (IBM, Armonk, NY, USA). One-way analysis of variance was used for single-factor sample comparisons, and Bonferroni’s post-hoc test was used for comparisons between means. All experiments were repeated using at least three independent biological replicates.
Results
Membrane Potential Changes Regulate Osmotic Tension, Closely Associated with Intracellular PNs and Calcium Signaling
Intracellular PNs can induce membrane potential changes and intracellular hyperosmosis.4,25 To explore the effects of membrane potential on IF tension in neurons and the underlying regulatory mechanisms, we used three voltage-gated channel agonists: Bay K8644 (L-type voltage-gated calcium channel, VGCC), lubiprostone (voltage-gated chloride channel), and Lu AE98134 (voltage-gated sodium channel). These agents individually induce changes in cell membrane potential.30–32 We observed that IF tension increased within 30 min, with Bay K8644 promoting the most significant effect (Figure 1A and B). Besides, Bay K8644 treatment increased intracellular PN production (Figure 1D), cytoplasmic OP (Figure 1E), and intracellular Ca2+/Cl− levels (Figure 1F and G). To further investigate the association between membrane potential and osmotic tension, we induced membrane depolarization by setting the extracellular K+ concentration to 40 mm (under isotonic extracellular conditions) (Figure 1H and J), which increased IF tension (Figure 1I and K) and Ca2+/Cl− levels (Figure 1L and M). These data indicate that membrane potential changes are closely related to osmotic tension and OP regulation.
Figure 1 Continued. Figure 1 Changes in membrane potential induce osmotic tension imbalance, closely associated with PNs and Ca2+. (A) After transfection with vimentin–cpstFRET probe, SH-SY5Y cells were treated with Bay K8644 (1 μM), Lu AE98134 (30 μM), lubiprostone (100 μM), Bay K8644 + Sen A (100 μM) + TAX (10 μM), Bay K8644 + 0 mm Ca2+ (no extracellular Ca2+), Bay K8644 + heparin (0.5 mg/mL), and Bay K8644 + 0 mm Ca2+ + heparin. Representative fluorescent images were captured within 30 min. Scale bar, 10 µm. The representative fluorescent calibration bar was set from 0.1 to 1.5. The dark-blue calibration bar indicates the smallest CFP/FRET ratio (0.10), whereas red indicates the largest CFP/FRET ratio (1.5). (B) Normalized CFP/FRET ratio of vimentin IF tension under different treatments. ****p < 0.0001, Bay K8644 was compared with the control. ***p < 0.001, Lu AE98134 or lubiprostone was individually compared with the Bay K8644 group. (C) Normalized CFP/FRET ratio of vimentin IF tension under different treatments. ***p < 0.001, the additional treatment of Sen A+TAX or 0 mm Ca2++heparin was individually compared with the Bay K8644 group. *p < 0.05, the additional treatment of 0 mm Ca2+ was compared with the Bay K8644+heparin group. (D) Number of cytoplasmic PNs under different treatments. (E) Cytoplasmic OP was detected using a freezing point osmometer. (F) Normalized F30/F0 ratio of intracellular Ca2+ and (G) Cl− fluorescent intensities. The significant differences were statistically analyzed between the Bay K8644 group compared with the control, the additional treatment of Sen A+TAX or 0 mm Ca2++heparin individually compared with the Bay K8644 group. (H) Neuronal cells were treated with an isotonic solution (300 mOsm/kg) containing 40 mm KCl. Representative trace map of membrane potential was recorded using a whole-cell patch clamp. (I) Representative fluorescence images of vimentin IF tension. The calibration bar was set from 0.1 to 1.5. (J) Membrane potential changes after KCl treatment. Each line represents a single neuronal cell. Paired samples were analyzed by t-test. (K) Normalized CFP/FRET ratio of vimentin IF tension. (L) Normalized F30/F0 ratio of intracellular Ca2+ and (M) Cl− fluorescent intensities. Average of ≥3 biologically independent replicates ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.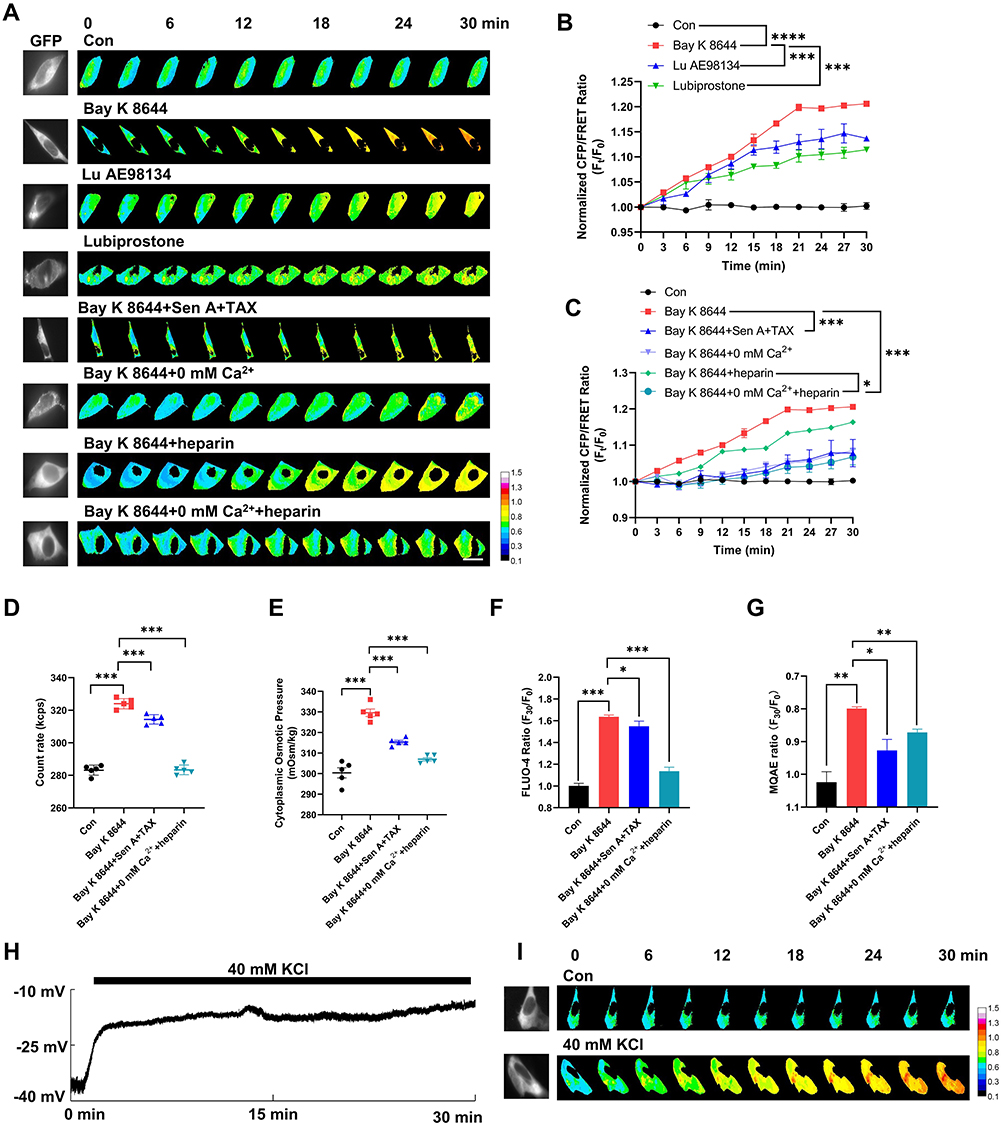

Our previous studies have shown that intracellular PNs, sourced from the depolymerization of microfilaments and microtubules under cytotoxic stimuli,4,25,27 are involved in membrane potential regulation. To assess whether PNs and Ca2+ contribute to higher IF tension and intracellular OP, we co-treated the cells with the microfilaments stabilizer sennoside A (Sen A)33 and microtubules stabilizer taxol (TAX),34 which significantly reversed Bay K8644-induced increases in IF tension, intracellular PN amounts, cytoplasmic OP, and intracellular Ca2+/Cl− levels (Figure 1A and C–G). Similarly, inhibiting intracellular Ca2+ with heparin [an inhibitor of inositol triphosphate receptor (IP3R)]35–37 and the absence of extracellular Ca2+ reversed these effects (Figure 1A and C–G). We thus believe that PNs and calcium signaling play a crucial role in membrane potential-regulated intracellular OP and ion levels.
Extracellular Hypotonicity Induces Membrane Potential Changes Dependent on PN and Calcium Levels
In living cells, IF tension is closely related to changes in transmembrane OP.5,27 To investigate the effects of osmotic tension on membrane potential, we exposed neuronal cells to extracellular hypotonic stress (220 mOsm/kg) to induce cell swelling.38 IF tension increased over time (Figure 2A and B), and the rate of change slowed from the 27th min, plateauing as cell volume increased (Figure 2C and D). This suggests that intracellular mechanical signals stabilized at this point, with water influx nulling. Hypotonic stress also upregulated cytoplasmic OP, amount of intracellular PNs, and Ca2+/Cl− levels (Figure 2E–H). Ion rearrangement between two sides of cell membrane could influence on the membrane potential. We next monitored membrane potential changes in neurons using whole-cell recording in current-clamp mode and found that hypotonic stress induced significant depolarization of membrane potential (Figure 2I), indicating a close correlation between IF tension changes and membrane potential.
 |
Figure 2 Extracellular hypotonicity induces IF tension and membrane potential changes dependent on PNs and Ca2+. (A) Neuronal cells were treated with hypotonic solution (Hypo, 220 mOsm/kg), Hypo + Sen A (100 μM) + TAX (10 μM), Hypo + 0 mm Ca2+, and Hypo + heparin (0.5 mg/mL). Representative fluorescent images of vimentin IF tension were captured within 30 min. Scale bar, 10 µm. The calibration bar was set from 0.1 to 1.5. (B) Normalized CFP/FRET ratio of vimentin IF tension. (C) Ratio of 27 min and 30 min CFP/FRET was analyzed by paired t-test. (D) Normalized ratio (Ft/F0) of cell volume within 30 min. (E) Number of cytoplasmic PNs. (F) Cytoplasmic OP was detected using a freezing point osmometer. (G) Normalized F30/F0 ratio of intracellular Ca2+ and (H) Cl− fluorescent intensities. An increase in intracellular Cl− levels led to a decrease in MQAE fluorescence intensity. The significant differences were statistically analyzed between the Hypo group compared with the control, the additional treatment of Sen A+TAX or 0 mm Ca2+ or heparin or 0 mm Ca2++heparin individually compared with the Hypo group (B, D–H). (I) Representative trace map and (J) variations in membrane potential measured using a whole-cell patch clamp. Average of ≥3 biologically independent replicates ± SEM. The significant differences were statistically analyzed between the additional treatment of Sen A+TAX or 0 mm Ca2+ or heparin or 0 mm Ca2++heparin individually compared with the Hypo group. ns, no significant, *p < 0.05, **p < 0.01, ***p < 0.001. |
Cell swelling can activate mechanically sensitive ion channels, converting mechanical stimulation into electrical or chemical signals.15 Piezo1, a non-selective cation channel, allows Ca2+, K+, and Na+ to pass through,39 generating inward depolarization currents.18–20 To explore the relationship between osmotic tension and electrical activity, we knocked down Piezo1 using siRNA under extracellular hypotonic conditions and assessed membrane potential and osmotic effects. Compared to the si-Con group, the si-Piezo1 group showed recovered hypotonicity-induced IF tension (Figure 3A and B) and membrane depolarization (Figure 3C–F), along with reduced intracellular Ca2+ (Figure 3G) and Cl− (Figure 3H) levels. Piezo1 has been reported to be involved in Ca2+ signaling regulation.40 Our results indicate that Piezo1-mediated transduction of osmotic tension modulates membrane potential and intracellular ion composition.
 |
Figure 3 Piezo1 is associated with IF tension, membrane potential, and intracellular ion level regulation. Neuronal cells were transfected with siRNA-Control (si-Con) and siRNA-Piezo1 (si-Piezo1) under extracellular hypotonic conditions (220 mOsm/kg). (A) Representative fluorescent images (scale bar, 10 µm). The calibration bar was set from 0.1 to 1.5. (B) Normalized CFP/FRET ratio of vimentin IF tension. ***p < 0.001, the si-Con+Hypo group was compared with the si-Con group. *p < 0.05, the si-Piezo1+Hypo group was compared with the si-Con+Hypo group. (C and D) Representative trace map of membrane potential was recorded within 30 min using a whole-cell patch clamp. (E) Statistics of resting membrane potential before treatment. (F) Variations in membrane potential after treatment. (G and H) Normalized F30/F0 ratio of intracellular Ca2+ and Cl− fluorescent intensities. The significant differences were statistically analyzed between the si-Con+Hypo group compared with the si-Con group, the si-Piezo1+Hypo group compared with the si-Con+Hypo group. Average of ≥3 biologically independent replicates ± SEM. ns, no significant, *p < 0.05, **p < 0.01, ***p < 0.001. |
Furthermore, treatments with microfilaments/microtubules stabilizers, absence of extracellular Ca2+, and inhibition of intracellular Ca2+ could recover extracellular hypotonicity-induced IF tension, cell volume, amounts of PNs, cytoplasmic OP, and intracellular ion levels (Figure 2A–H). Regarding membrane potential, PN inhibition recovered the hypotonic-induced depolarization; the absence of both extracellular and intracellular Ca2+ resulted in the transition to the hyperpolarization state (Figure 2I and J). These findings suggest that extracellular hypotonicity induces PN- and calcium-dependent changes in cell membrane potential and intracellular osmosis. In summary, mechanical changes are closely related to electrical and chemical activity in neuronal cells. Their collaboration is key to maintaining biological OP and water flow across the membrane. Electromechanical disequilibrium, rather than sole mechanical change, contributes to the occurrence of neuronal swelling.
Collaboration Between PN-Induced Activation and Chemical Signal-Induced Sensitization of Voltage-Dependent Ion Channels in Increasing PN-OP
To further investigate the effects of PNs on membrane potential and intracellular osmosis, we employed cytochalasin D (CytoD) and nocodazole (Noc) to depolymerize microfilaments and microtubules, respectively.41,42 Our previous research demonstrated that cytoskeletal depolymerization can induce to produce a mass of abnormal PNs in cytoplasm.4,27 Accompanying the increased number of intracellular PNs, we observed an increase in cytoplasmic OP and intracellular Ca2+/Cl− levels (Figure 4A–D). According to the Donnan effect, PN carrying negative charge can adsorb cations and triggered ion rearrangement near the cell membrane. Indeed, these led to membrane depolarization (Figure 4E and F), further confirming the involvement of intracellular PNs in electromechanical cooperation.
 |
Figure 4 PNs contribute to intracellular hyperosmosis, membrane potential changes, and extent of opening of voltage-dependent ion channels. Neuronal cells were treated with 10 μM CytoD and 100 μM Noc. (A) Number of cytoplasmic PNs. (B) Cytoplasmic OP. (C and D) Normalized F30/F0 ratio of intracellular Ca2+ and Cl− fluorescent intensities. (E) Representative trace map of membrane potential was recorded using a whole-cell patch clamp. (F) Membrane potential changes after CytoD+Noc treatment. Each line represents a single neuronal cell; paired samples were analyzed by t-test. (G) Cells were treated with CytoD+Noc and CytoD+Noc+Gil (20 μM). Under the holding potential of 0 mV, cells were subjected to ramp voltage stimulation from −100 mV to +100 mV for 200 ms, and representative current trace of SUR1-TRPM4 channel was recorded. (H) Average current density of SUR1-TRPM4 channel induced at −80 mV. (I) Cells were treated with CytoD+Noc and CytoD+Noc+Nic (1 μM). Under the holding potential of 0 mV, a step voltage stimulation scheme of +20 mV was applied in the voltage range of −80 mV to +80 mV, and representative current trace of TMEM16A channel was recorded. (J) Average current density of TMEM16A channel was calculated at +80 mV. (K) Cells were treated with CytoD+Noc and CytoD+Noc+Nimo (10 μM). At the holding potential of −90 mV, cells were subjected to step voltage stimulation from −70 mV to +10 mV, and representative current trace of VGCCs was recorded. (L) Average current density of VGCCs. The significant differences were statistically analyzed between the Cyto D+Noc group compared with the control, the additional treatment of Gil or Nic or Nimo individually compared with the Cyto D+Noc group (H, J and L). (M and N) Representative fluorescent images (scale bar, 10 µm; the calibration bar was set from 0.1 to 1.5) and normalized CFP/FRET ratio of vimentin IF tension recorded within 30 min. (O) Normalized ratio (Ft/F0) of cell volume within 30 min. Average of ≥3 biologically independent replicates ± SEM. ns, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
We hypothesized that PN-induced membrane depolarization and calcium signaling promote the opening of voltage-dependent ion channels. In this study, we focused on depolarization-dependent and Ca2+-activated ion channels: sulfonylurea receptor 1-transient receptor potential cation channel subfamily M member 4 (SUR1-TRPM4), transmembrane protein 16 A (TMEM16A), and L-type voltage-gated calcium channel (L-VGCC). SUR1-TRPM4 channel is activated by increased intracellular Ca2+ levels and membrane depolarization,43 contributing to edema formation. The opening of TMEM16A channel, a voltage-dependent Ca2+-sensitive chloride channel, exacerbates cerebral infarction.44 Rapid calcium influx during acute cerebral hypoxia causes neuronal damage and death associated with stroke,45 while blocking L-type VGCCs has beneficial effects on brain tissue post-ischemia.46
We investigated the effects of PNs on these ion channels by co-treating neurons with CytoD and Noc. Compared to the control group, CytoD–Noc co-treatment enhanced currents through SUR1-TRPM4 channel, TMEM16A channel, and VGCC, which were suppressed by the inhibitors glibenclamide (Gil), niclosamide (Nic), and nimodipine (Nimo), respectively (Figure 4G–L). The increased number of intracellular PNs, an increase in IF tension and cell volume was observed in the CytoD–Noc group (Figure 4M–O). These results indicate that cytoskeletal depolymerization-induced PNs activate voltage-dependent ion channels. Ion flux through various types of ion channels is responsible for PN-mediated regulation of biological OP and neuronal edema.
The degree of ion channel opening is crucial for regulating intracellular osmosis. Previous reports indicate that calmodulin (CaM) increases Ca2+ sensitivity of SUR1-TRPM4 channel.4,47 Protein kinase C (PKC)-induced phosphorylation also enhances TRPM4 current.47 PKC inhibition prevents ammonia-induced astrocyte swelling,48 PKC activity and expression are upregulated in cytotoxic edema.49 Our prior study showed that CaM and PKC regulate PN-induced ion increments via SUR1-TRPM4 and TMEM16A channel opening.4 In this study, we used CaM and PKC agonists [calcium-like peptide-1 (CALP1) and phorbol 12-myristate 13-acetate (PMA), respectively] in the CytoD–Noc-induced hyperosmotic PN model. Compared to the CytoD+Noc group, a significant increase in channel current was observed upon adding CALP1 or PMA (Figure 5A and B). Correspondingly, IF tension and cell volume showed a significant increase after CaM and PKC agonist treatments (Figure 5C–E).
 |
Figure 5 Sensitization of voltage-dependent ion channels is involved in PN-induced OP. (A) Neuronal cells were treated with CytoD+Noc, CytoD+Noc+CALP1, and CytoD+Noc+PMA. Under the holding potential of 0 mV, cells were subjected to ramp voltage stimulation from −100 mV to +100 mV for 200 ms, and representative current trace of SUR1-TRPM4 channel was recorded. (B) Average current density of SUR1-TRPM4 channel induced at −80 mV. (C and D) Representative fluorescent images and normalized CFP/FRET ratio of vimentin IF tension. Scale bar, 10 µm. The calibration bar was set from 0.1 to 1.5. (E) Normalized ratio (F30/F0) of cell volume. The significant differences were statistically analyzed between the additional treatment of CALP1 or PMA individually compared with the Cyto D+Noc group (B, D and E). (F) Linear relationship between cytoplasmic protein concentration and OP, corresponding to the concentration gradients (CytoD 10 μM + Noc 100 μM, CytoD 20 μM + Noc 200 μM, CytoD 30 μM + Noc 300 μM, and CytoD 40 μM + Noc 400 μM) (G, H and I) Linear relationship between the count rate of cytoplasmic PNs and OP, corresponding to the concentration gradients of CytoD + Noc. Average of ≥3 biologically independent replicates ± SEM. **p < 0.01, ***p < 0.001. |
Furthermore, we found linear positive correlations between cytoplasmic OP and intracellular PN concentrations or amounts (Figure 5F and G). PMA and CALP1 increased the slope of the linear correlation between intracellular PN amounts and cytoplasmic OP (Figure 5H and I). These data indicate that PN-induced hyperosmosis relies on CaM- and PKC-mediated sensitization of nonselective ion channels.
Neurotoxicity Induces Electromechanical Imbalance to Promote Neuronal Edema
Cobalt chloride (CoCl2), a neurotoxin mimicking hypoxia stimulus, induces nervous edema in ischemic stroke.50 Lipopolysaccharide (LPS) activates pro-inflammatory pathways, enhancing transmembrane water permeability and cytotoxic edema.51 Dichloroethane (1,2-DCE) induces cerebral edema through oxidative stress, calcium overload, blood-brain barrier damage, and neurotransmitter changes.52 We established neurotoxic edema models by treating neuronal cells with these neurotoxins to investigate the electromechanical mechanism involving PNs, Ca2+, and water flux.
CoCl2, LPS, and 1,2-DCE induced membrane depolarization and increased IF tension, intracellular PN amounts, cell volume and Ca2+/Cl− levels (Figures 6A, B, G; S1F–H; S2F–H; S3D–F). We investigated the roles of PN inhibition in these neurotoxin-induced electromechanical events. Besides being elicited by microfilaments/microtubules depolymerization, the NLRP3 inflammasome and downstream inflammatory cytokines are major sources of intracellular PNs in cerebral edema.4 Sen A, TAX, and tranilast (Tran, NLRP3 inhibitor)53 were used to stabilize the cytoskeleton and inhibit NLRP3 inflammasome assembly to reduce intracellular PN production. PN inhibition significantly attenuated CoCl2/LPS/1,2-DCE-induced IF tension (Figure 6A) and restored membrane potential (Figure 6C and F), amounts of PNs (Figure 6G), cytoplasmic OP (Figures S1E, S2E and S3C), cell volume (Figures S1F, S2F and S3D), and Ca2+/Cl− levels (Figures S1G and H, S2G and H and S3E and F).
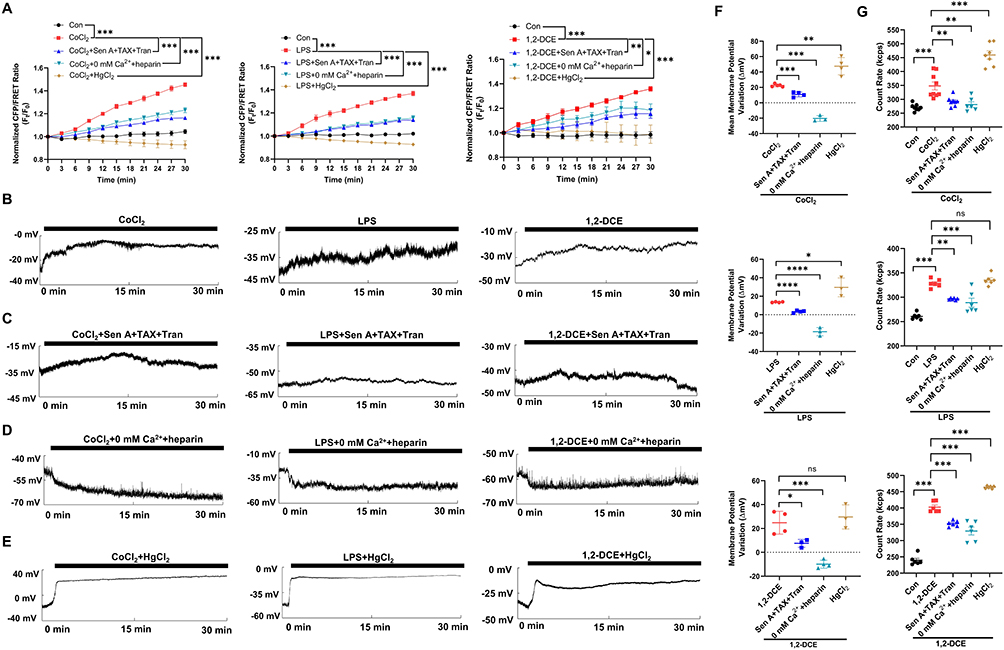 |
Figure 6 Neurotoxins induce electromechanical imbalance dependent on intracellular PNs. Neuronal cells were treated with CoCl2 (200 μM), LPS (1 μg/mL), and 1,2-DCE (1.2 μg/mL), or combined with Sen A + TAX + Tran (20 μM), 0 mm Ca2+ + heparin, and HgCl2 (200 μM). (A) Normalized CFP/FRET ratio of vimentin IF tension. (B–E) Representative trace map of membrane potential was recorded within 30 min under different treatments. (F) Variations in membrane potential. The significant differences were statistically analyzed between the additional treatment of Sen A+TAX+Tran or 0 mm Ca2++heparin or HgCl2 individually compared with the CoCl2 group, the LPS group or 1,2-DCE group. (G) Number of cytoplasmic PNs. The significant differences were statistically analyzed between the individual CoCl2/LPS/1,2-DCE group compared with the control, the additional treatment of Sen A+TAX+Tran or 0 mm Ca2++heparin or HgCl2 individually compared with the CoCl2 group, the LPS group or 1,2-DCE group (A and G). Average of ≥3 biologically independent replicates ± SEM. ns, no significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Combined inhibition of intra- and extracellular Ca2+ significantly reduced CoCl2/LPS/1,2-DCE-induced IF tension (Figure 6A) as well as PN amounts (Figure 6G), cytoplasmic OP, cell volume, and Ca2+/Cl− levels (Figures S1–S3). Interestingly, the depolarization of membrane potential induced by CoCl2/LPS/1,2-DCE was reversed to hyperpolarization (Figure 6D and F). Conversely, carbachol (an agonist of IP3R) induced an increase in Ca2+ levels that reversed NaN3-induced hyperpolarization to depolarization, and PN inhibition also attenuated NaN3-induced membrane potential changes (Figure S4). These findings suggest that Ca2+ is involved in converting membrane potential polarization states.
According to the electrical double-layer theory, high-valence ions (Ca2+) can be absorbed by PNs, displacing low-valence ions (Na+ and K+), thus increasing free monovalent cation levels and intracellular hyperosmosis. Beyond Ca2+, an isotonic solution with low Hg2+ concentrations or high Cd2+ and Mg2+ concentrations was found to increase IF tension and membrane depolarization (Figure S5), similar to the effects of high Ca2+ concentrations on electromechanical changes, suggesting a role of divalent cations in regulating membrane potential and biological OP.
Neuronal edema induced by CoCl2, LPS, and 1,2-DCE is driven by unbalanced water inflow, and AQP channel inhibition seems to reduce cytotoxic edema. Hg2+ (a pan-inhibitor of AQP channels) can reportedly prevent water inflow/outflow.54 Herein we observed that high Hg2+ concentrations (200 μM) abolished the increase in IF tension and cell volume caused by CoCl2, LPS, and 1,2-DCE treatment (Figures 6A, S1F, S2F and S3D). Notably, mercuric chloride (HgCl2) failed to revoke the electrophysiological imbalance caused by CoCl2, LPS, and 1,2-DCE but instead caused stronger membrane potential depolarization (Figure 6E and F), consistent with the results of a previous study.55 In addition, HgCl2 did not decrease intracellular Ca2+/Cl− levels induced by neurotoxins (Figures S1G and H, S2G and H and S3E and F), and even resulted in more intracellular PNs and higher cytoplasmic OP (Figures 6G, S1E, 2E and S3C). These results suggest that AQP-mediated water influx is independent of ionic OP regulation. In summary, electromechanical disequilibrium induced by CoCl2, LPS, and 1,2-DCE facilitates neuronal edema, closely regulated by PNs, Ca2+, and water influx.
AQP-Mediated Water Influx Is Involved in Regulating Membrane Potential and OP-Induced Tension
AQP channels mediate water flux, playing a key role in water homeostasis and edema formation. AQP9 overexpression in cerebral stroke models has been found to aggravate cerebral edema.56 To investigate the impact of AQP9 on IF tension upon exposure to CoCl2, LPS, and 1,2-DCE, green fluorescent protein-tagged vimentin and red fluorescent protein-tagged AQP9 were co-transfected into neuronal cells. AQP9 overexpression aggravated CoCl2/LPS/1,2-DCE-induced increase in IF tension, whereas PN inhibition reversed these effects (Figure 7A and B), suggesting AQP-mediated water influx associated with PN-induced osmosis. Particularly, AQP9 overexpression also facilitated the recovery of CoCl2, LPS, and 1.2-DCE-induced membrane depolarization, eventually achieving a repolarization state (Figure 7C and D). This indicates that water influx through AQP9 channels plays a role in electrical activity regulation, alleviating the depolarization of cell membrane potential and promoting partial recovery of membrane potential.
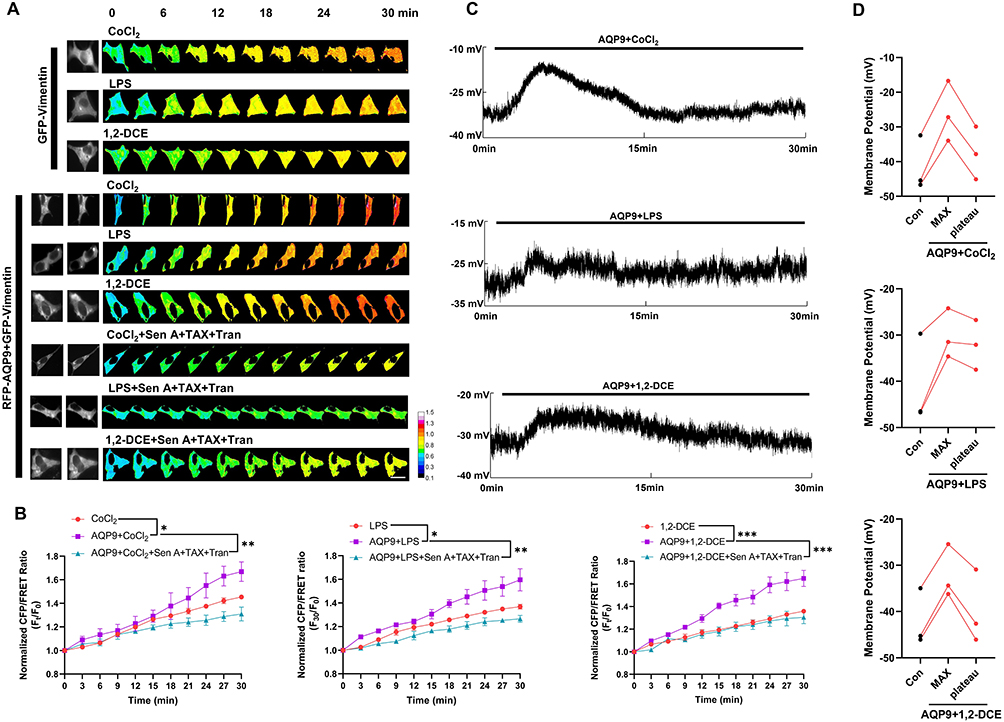 |
Figure 7 AQP is involved in IF tension and membrane potential regulation. Neuronal cells were co-transfected with green fluorescent protein-tagged vimentin probe and red fluorescent protein-tagged AQP9. (A and B) Representative fluorescent images and normalized CFP/FRET ratio of vimentin IF tension under different treatments. Scale bar, 10 µm. The calibration bar was set from 0.1 to 1.5. The significant differences were statistically analyzed between the AQP9+CoCl2/LPS/1,2-DCE group individually compared with the corresponding CoCl2/LPS/1,2-DCE group; the additional treatment of Sen A+TAX+Tran individually compared with the corresponding AQP9+CoCl2/LPS/1,2-DCE group. (C) Representative trace map of cell membrane potential. (D) Cell membrane potential before treatment, max membrane potential after treatment, and the recovered plateau. Each line represents a single neuronal cell. Average of ≥3 biologically independent replicates ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. |
Proteomic Analysis of PN Composition
To explore the generation of PNs in neuronal cells due to cytoskeletal depolymerization, we performed liquid chromatography–mass spectrometry (LC–MS) to identify changes in cytoplasmic PNs in models treated with CytoD and Noc as well as models from which CytoD and Noc were removed. Compared to the control group, the expression of 94 and 78 types of proteins was up- and downregulated, respectively, in the CytoD+Noc group (Figure 8A). Despite these changes, the total amount of PNs increased following CytoD–Noc co-treatment (Figure 4A). This indicates that cytoskeletal depolymerization induces widespread alterations in PN composition.
 |
Figure 8 Proteomic analysis by label-free LC–MS. Identification of different proteins between (1) the control and CytoD+Noc groups and (2) the CytoD–Noc and CytoD–Noc-removal groups. (A and B) Volcano plot of nonredundant proteins. Red dots indicate upregulated proteins, blue dots indicate downregulated proteins, and grey dots indicate proteins with no significant change. (C and D) Classification of identified proteins by subcellular localization. The percentage in parentheses represents the enriched types of proteins. (E and F) GO enrichment analysis categorized by cellular components (CC), biological processes (BP) and molecular functions (MF). Top 20 enriched pathways were ranked by p-value. Pathways with p ≤ 0.05 were considered to be significantly enriched. |
Further, to identify the pathways affected in the CytoD+Noc group, we performed subcellular localization analyses. Categories related to the endoplasmic reticulum, nucleus, mitochondria and vesicles were significantly enriched in the CytoD+Noc group compared to the control group (Figures 8C and S6A), suggesting PN translocation within the cytoplasm. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses revealed enrichment in pathways related to ribosome structure and pathway, vesicle organization and actin binding (Figures 8E and S6). In addition, the different types (40 and 106 types of proteins up- and downregulated) of PNs between the CytoD+Noc and CytoD+Noc-removal groups, mainly originated from the nucleus, mitochondria, vesicles (Figure 8B and D); however, the greatest number of PNs seem to recover in nucleus and vesicles, not endoplasmic reticulum (Figure S6B). Compared with the CytoD+Noc group, GO and KEGG enrichment in the CytoD+Noc-removal group focused on focal adhesion, cell-substrate junction, RNA splicing, cadherin binding, ribosome, etc (Figures 8F; S6D–G). These indicate that some PNs were able to recover when cytoskeletal depolymerization was inhibited. In summary, the production of PNs results from the cytoplasmic translocation of various proteins induced by cytoskeletal depolymerization, and some PNs can reversibly recover through their translocation.
Discussion
Cytotoxic edema is a secondary symptom of ischemic stroke and is challenging to monitor in situ. Osmotic imbalance is widely accepted to be the primary driving force responsible for the swelling of astrocytes and neurons, which depends on ion maldistribution across the cell membrane and the resultant AQP-mediated water influx. Notably, we have found that changes in osmotic tension and electrophysiological activity accompany and mutually participate in neuronal swelling. Electromechanical cooperation, a key regulator of biological OP and water flux, heavily relies on intracellular PN and Ca2+ concentrations. Abnormal increased PNs resulted in the membrane potential changes and Ca2+ contributed to the transition between depolarization and hyperpolarization. Membrane depolarization-induced activation combined with CaM/PKC-induced sensitization of voltage-dependent cation and anion channels control the increase in intracellular ionic OP. AQP-mediated water influx contributes to rebalancing the membrane potential and stabilizing osmotic tension, facilitating neurotoxic edema.
PN-induced OP depends on the activation and sensitization of ion channels. Voltage-dependent ion channels are crucial executors for membrane potential involved in OP regulation. PN-induced membrane potentials activate voltage-dependent non-selective ion channels. Meanwhile, chemical signal-mediated sensitization enhances ion channel function.57–59 Thus, increases in intracellular PNs can induce membrane potential changes and collaborate with chemical signals to facilitate the continuous opening of ion channels, exacerbating the abnormal flux of cations and anions.
PN-induced adsorption of cations is key to regulating membrane potential. PNs carrying a negative charge can absorb cations, leading to a decrease in free cations and a relative increase in anions near the cell membrane,60,61 promoting imbalanced electric potential across the membrane. Notably, Ca2+ plays a crucial role in regulating PN-induced membrane potential. According to the electrical double-layer theory,62,63 high-valence ions, such as Ca2+, Co2+, and Cs2+ (Figure S5), can be absorbed by PNs, replacing low-valence ions (Na+ and K+). Intracellular PN–Ca2+ absorption increases monovalent ions inside the cell membrane, creating a state of negative outside and positive inside, eventually promoting membrane depolarization. Therefore, PNs and Ca2+ play a central role in controlling the electrochemical effects in cytotoxic edema by altering the potential state.
In our previous studies, we focused on intracellular PNs principally derived from cytoskeletal depolymerization and the NLRP3 inflammasome.4,26,27 This proteomic analysis enhances our understanding of PN sources. Cytoskeletal depolymerization seems to promote the cytoplasmic translocation of PNs from the nucleus and mitochondria, particularly most PNs from endoplasmic reticulum. Since the endoplasmic reticulum is responsible for correct protein folding and modification in controlling protein homeostasis, pathological endoplasmic reticulum stress could induce the massive unfolded or misfolded proteins, which transfer and accumulate in the cytoplasm and further amplify the PNs effects in intracellular hyperosmosis. The new finding could provide the strategy to eliminate abnormal protein nanoparticles, such as the ubiquitin proteasome system and autophagy, potentially being the therapeutic strategy in the cerebral edema treatment. In addition, the number of PNs is decreased after the removal of cytoskeleton-depolymerizing stimuli, indicating the flexible and reversible regulation of some PNs in electromechanical cooperation and osmotic homeostasis.
It is widely accepted that intracellular hyperosmosis triggers water influx and subsequent cell swelling, which is highly dependent on AQP function.64,65 Our findings reveal that AQPs are involved in electromechanical activity beyond their role as water channels in cytotoxic edema, providing novel contribution to AQP function in the membrane potential regulation. Water influx facilitates the recovery of membrane potentials, possibly by regulating intracellular PN and Ca2+ concentrations. Furthermore, AQP activation and water influx are independent of ionic OP regulation. The AQP inhibitor HgCl2 can reverse cytotoxicity-induced IF tension and cell volume but cannot recover intracellular ion levels in cytotoxic swelling (Figures S1–S3). Water entering the cell is driven by intracellular hyperosmosis and resisted by IF tension. AQP overexpression further aggravates the increase in IF tension in response to neurotoxic edema. Therefore, the vector sum of outward osmotic tension and inward IF pulling tension determines the volume of water flux. Altogether, OP-tension effects control AQP channel activity, with water influx regulating changes in membrane potential and the extent of opening of voltage-dependent ion channels, resulting in a new OP homeostasis.
Since we have elucidated the mechanism underlying electromechanical cooperation among intracellular PN, calcium and water flux involved in the neurotoxic edema, there are some limitations in this study and the future research direction. Firstly, the number of intracellular PNs cannot be detected in vivo conditions by the existing techniques. In addition, specific classifications of PNs are whether exerting different osmotic effects corresponding to the edema. Our proteomic analysis has found cytoplasmic PNs increment mostly from endoplasmic reticulum; thus, whether these potential unfolded and misfolded proteins are important sources of PNs that cause edema needs to be identified in future research.
Conclusion
In summary, PNs amplify osmotic effects by inducing electromechanical cooperation, which is distinct from ionic OP. Physical OP is determined by the difference in solute quantity across a semipermeable membrane. We propose a new concept of “biological OP” based on membrane potential changes and the resultant selective flow of ions and water across the cell membrane. In the regulation of biological OP, PN–Ca2+ adsorption induces membrane potential changes and collaborates with chemical signals, amplifying the activity of voltage-dependent ion channels. Meanwhile, water influx, as an independent factor, promotes the recovery of membrane potential and OP homeostasis through electromechanical cooperation in living cells. We believe these findings will provide a new perspective on cytomechanics, interpreting the occurrence and development of cytotoxic edema. As PN is a central link involved in controlling neurotoxic edema, how to effectively remove abnormal PNs will be a key problem to solve in the drug development for alleviating cerebral edema. The ubiquitin proteasome system-mediated degradation of misfolding proteins and autophagy-mediated elimination of inflammation-related proteins involved in the PNs-induced osmosis and edema will be further studied in the future direction.
Abbreviations
1,2-DCE, dichloroethane; AQP, aquaporins; Cyto D, CaM, calmodulin; CALP1, calcium-like peptide-1; CoCl2, cobalt chloride; Cytochalasin D; FRET, fluorescence resonance energy transfer; eCFP, enhanced cyan fluorescent protein; eYFP, enhanced yellow fluorescent protein; Gil, glibenclamide; GO, Gene ontology; HgCl2, mercuric chloride; IF, intermediate filament; IP3R, inositol triphosphate; KEGG, Kyoto Encyclopedia of Genes and Genomes; LC–MS, liquid chromatography–mass spectrometry; LPS, lipopolysaccharide; L-VGCC, L-type voltage-gated calcium channel; MQAE, N-[ethoxycarbonylmethyl]-6-methoxy-quinolinium bromide; Nic, Niclosamide; Nimo, nimodipine; Noc, nocodazole; OP, osmotic pressure; PMA, phorbol 12-myristate 13-acetate; PKC, Protein kinase C; PN, protein nanoparticle; Sen A, sennoside A; SEM, standard error of the mean; SUR1-TRPM4, sulfonylurea receptor 1-transient receptor potential cation channel subfamily M member 4; TAX, taxol; Tran, tranilast; TMEM16A, transmembrane protein 16 A.
Acknowledgments
This investigation was supported by the grants from National Natural Science Foundation of China (No. 82073826 and 82273908), and the Innovation Projects of State Key Laboratory on Technologies for Chinese Medicine Pharmaceutical Process Control and Intelligent Manufacture (No. NZYSKL240203).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Schleicher RL, Vorasayan P, Mccabe ME, et al. Analysis of brain edema in RHAPSODY. International Journal of Stroke. 2024;19(1):68–75. doi:10.1177/17474930231187268
2. Halstead MR, Geocadin RG. The Medical Management of Cerebral Edema: past, Present, and Future Therapies. Neurotherapeutics. 2019;16(4):1133–1148. doi:10.1007/s13311-019-00779-4
3. Hladky SB, Barrand MA. Alterations in brain fluid physiology during the early stages of development of ischaemic oedema. Fluids Barriers CNS. 2024;21(1):51. doi:10.1186/s12987-024-00534-8
4. Zheng Z, Qiu Z, Xiong X, et al. Co-activation of NMDAR and mGluRs controls protein nanoparticle-induced osmotic pressure in neurotoxic edema. Biomedicine & Pharmacotherapy. 2023;169:115917. doi:10.1016/j.biopha.2023.115917
5. Zheng Z, Wang Y, Li M, et al. Albumins as Extracellular Protein Nanoparticles Collaborate with Plasma Ions to Control Biological Osmotic Pressure. International Journal of Nanomedicine. 2022;17:4743–4756. doi:10.2147/IJN.S383530
6. Delpire E, Gagnon KB. Water Homeostasis and Cell Volume Maintenance and Regulation. Current Topics in Membranes. 2018;81:3–52.
7. Simard JM, Woo SK, Bhatta S, et al. Drugs acting on SUR1 to treat CNS ischemia and trauma. Current Opinion in Pharmacology. 2008;8(1):42–49. doi:10.1016/j.coph.2007.10.004
8. Simard JM, Chen M, Tarasov KV, et al. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nature Medicine. 2006;12(4):433–440. doi:10.1038/nm1390
9. Simard JM, Geng Z, Woo SK, et al. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism. 2009;29(2):317–330. doi:10.1038/jcbfm.2008.120
10. Centeio R, Ousingsawat J, Schreiber R, et al. Ca2+ Dependence of Volume-Regulated VRAC/LRRC8 and TMEM16A Cl− Channels. Frontiers in Cell and Developmental Biology. 2020;8:596879. doi:10.3389/fcell.2020.596879
11. Liu PY, Zhang Z, Liu Y, et al. TMEM16A Inhibition Preserves Blood-Brain Barrier Integrity After Ischemic Stroke. Frontiers in Cellular Neuroscience. 2019;13:360. doi:10.3389/fncel.2019.00360
12. Wang BF, Cui ZW, Zhong ZH, et al. Curcumin attenuates brain edema in mice with intracerebral hemorrhage through inhibition of AQP4 and AQP9 expression. Acta Pharmacol Sin. 2015;36(8):939–948. doi:10.1038/aps.2015.47
13. Liu H, Yang M, Qiu GP, et al. Aquaporin 9 in rat brain after severe traumatic brain injury. Arq Neuropsiquiatr. 2012;70(3):214–220. doi:10.1590/S0004-282X2012000300012
14. Badaut J. Aquaglyceroporin 9 in brain pathologies. Neuroscience. 2010;168(4):1047–1057. doi:10.1016/j.neuroscience.2009.10.030
15. Kefauver JM, Ward AB, Patapoutian A. Discoveries in structure and physiology of mechanically activated ion channels. Nature. 2020;587(7835):567–576. doi:10.1038/s41586-020-2933-1
16. Chen T, Guo Y, Shan J, et al. Vector Analysis of Cytoskeletal Structural Tension and the Mechanisms that Underpin Spectrin-Related Forces in Pyroptosis. Antioxidants & Redox Signaling. 2019;30(12):1503–1520.
17. Bober BG, Love JM, Horton SM, et al. Actin-myosin network influences morphological response of neuronal cells to altered osmolarity. Cytoskeleton. 2015;72(4):193–206. doi:10.1002/cm.21219
18. Li J, Hou B, Tumova S, et al. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515(7526):279–282. doi:10.1038/nature13701
19. Coste B, Mathur J, Schmidt M, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330(6000):55–60. doi:10.1126/science.1193270
20. Romac JM, Shahid RA, Swain SM, et al. Piezo1 is a mechanically activated ion channel and mediates pressure induced pancreatitis. Nature Communications. 2018;9(1):1715. doi:10.1038/s41467-018-04194-9
21. Douguet D, Honoré E. Mammalian Mechanoelectrical Transduction: structure and Function of Force-Gated Ion Channels. Cell. 2019;179(2):340–354. doi:10.1016/j.cell.2019.08.049
22. Zhu J, Xian Q, Hou X, et al. The mechanosensitive ion channel Piezo1 contributes to ultrasound neuromodulation. Proceedings of the National Academy of Sciences of the United States of America. 2023;120(18):e2300291120. doi:10.1073/pnas.2300291120
23. Qu S, Hu T, Qiu O, et al. Effect of Piezo1 Overexpression on Peritumoral Brain Edema in Glioblastomas. AJNR American Journal of Neuroradiology. 2020;41(8):1423–1429. doi:10.3174/ajnr.A6638
24. Wang YY, Zhang H, Ma T, et al. Piezo1 mediates neuron oxygen-glucose deprivation/reoxygenation injury via Ca2+calpain signaling. Biochemical and Biophysical Research Communications. 2019;513(1):147–153. doi:10.1016/j.bbrc.2019.03.163
25. Qian Z, Wang Q, Qiu Z, et al. Protein nanoparticle-induced osmotic pressure gradients modify pulmonary edema through hyperpermeability in acute respiratory distress syndrome. Journal of Nanobiotechnology. 2022;20(1):314. doi:10.1186/s12951-022-01519-1
26. Zheng Z, Wang T, Chen J, et al. Inflammasome-Induced Osmotic Pressure and the Mechanical Mechanisms Underlying Astrocytic Swelling and Membrane Blebbing in Pyroptosis. Frontiers in Immunology. 2021;12:688674. doi:10.3389/fimmu.2021.688674
27. Zhang J, Wang Y, Zheng Z, et al. Intracellular ion and protein nanoparticle-induced osmotic pressure modify astrocyte swelling and brain edema in response to glutamate stimuli. Redox Biology. 2019;21:101112. doi:10.1016/j.redox.2019.101112
28. Guo J, Wang Y, Sachs F, et al. Actin stress in cell reprogramming. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(49):E5252–61. doi:10.1073/pnas.1411683111
29. Geng X, Shi H, Ye F, et al. Matrine inhibits itching by lowering the activity of calcium channel. Scientific Reports. 2018;8(1):11328. doi:10.1038/s41598-018-28661-x
30. Long MA, Jin DZ, Fee MS. Support for a synaptic chain model of neuronal sequence generation. Nature. 2010;468(7322):7322):394–9. doi:10.1038/nature09514
31. Cuppoletti J, Blikslager AT, Chakrabarti J, et al. Contrasting effects of linaclotide and lubiprostone on restitution of epithelial cell barrier properties and cellular homeostasis after exposure to cell stressors. BMC Pharmacology. 2012;12(1):3. doi:10.1186/1471-2210-12-3
32. Von schoubye NL, Frederiksen K, Kristiansen U, et al. The sodium channel activator Lu AE98134 normalizes the altered firing properties of fast spiking interneurons in Dlx5/6(±) mice. Neuroscience Letters. 2018;662:29–35. doi:10.1016/j.neulet.2017.10.004
33. Madineni A, Alhadidi Q, Shah ZA. Cofilin Inhibition Restores Neuronal Cell Death in Oxygen-Glucose Deprivation Model of Ischemia. Molecular Neurobiology. 2016;53(2):867–878. doi:10.1007/s12035-014-9056-3
34. Yang CH, Horwitz SB. Taxol(®): the First Microtubule Stabilizing Agent. International Journal of Molecular Sciences. 2017;18(8):1733. doi:10.3390/ijms18081733
35. Zhao H, Tong G, Liu J, et al. IP3R and RyR channels are involved in traffic-related PM(2.5)-induced disorders of calcium homeostasis. Toxicology and Industrial Health. 2019;35(5):339–348. doi:10.1177/0748233719843763
36. Kaplin AI, Snyder SH, Linden DJ. Reduced nicotinamide adenine dinucleotide-selective stimulation of inositol 1,4,5-trisphosphate receptors mediates hypoxic mobilization of calcium. The Journal of Neuroscience. 1996;16(6):2002–2011. doi:10.1523/JNEUROSCI.16-06-02002.1996
37. Zhang XB, Spergel DJ. Kisspeptin inhibits high-voltage activated Ca2+ channels in GnRH neurons via multiple Ca2+ influx and release pathways. Neuroendocrinology. 2012;96(1):68–80. doi:10.1159/000335985
38. Kang C, Xie L, Gunasekar SK, et al. SWELL1 is a glucose sensor regulating β-cell excitability and systemic glycaemia. Nature Communications. 2018;9(1):367. doi:10.1038/s41467-017-02664-0
39. Gnanasambandam R, Bae C, Gottlieb PA, et al. Ionic Selectivity and Permeation Properties of Human PIEZO1 Channels. PLoS One. 2015;10(5):e0125503.
40. Ye Y, Barghouth M, Dou H, et al. A critical role of the mechanosensor PIEZO1 in glucose-induced insulin secretion in pancreatic β-cells. Nature Communications. 2022;13(1):4237. doi:10.1038/s41467-022-31103-y
41. Shoji K, Ohashi K, Sampei K, et al. Cytochalasin D acts as an inhibitor of the actin-cofilin interaction. Biochemical and Biophysical Research Communications. 2012;424(1):52–57. doi:10.1016/j.bbrc.2012.06.063
42. Scott SJ, Suvarna KS, Pp D. Synchronization of human retinal pigment epithelial-1 cells in mitosis. Journal of Cell Science. 2020;133(18):jcs247940. doi:10.1242/jcs.247940
43. Launay P, Fleig A, Perraud AL, et al. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109(3):397–407. doi:10.1016/S0092-8674(02)00719-5
44. Al-hosni R, Ilkan Z, Agostinelli E, et al. The pharmacology of the TMEM16A channel: therapeutic opportunities. Trends in Pharmacological Sciences. 2022;43(9):712–725. doi:10.1016/j.tips.2022.06.006
45. Rowland MJ, Ezra M, Winkler A, et al. Calcium channel blockade with nimodipine reverses MRI evidence of cerebral oedema following acute hypoxia. Journal of Cerebral Blood Flow and Metabolism. 2019;39(2):285–301. doi:10.1177/0271678X17726624
46. PisanI A, Calabresi P, Tozzi A, et al. L-type Ca2+ channel blockers attenuate electrical changes and Ca2+ rise induced by oxygen/glucose deprivation in cortical neurons. Stroke. 1998;29(1):196–201. doi:10.1161/01.str.29.1.196
47. Woo SK, Kwon MS, Ivanov A, et al. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. The Journal of Biological Chemistry. 2013;288(5):3655–3667. doi:10.1074/jbc.M112.428219
48. Jia G, Wang R, Yue Y, et al. Activation of Protein Kinase Cδ Contributes to the Induction of Src/EGF Receptor/ERK Signaling in Ammonia-treated Astrocytes. Journal of Molecular Neuroscience: MN. 2020;70(7):1110–1119. doi:10.1007/s12031-020-01517-8
49. Yeung PK, Shen J, Chung SS, et al. Targeted over-expression of endothelin-1 in astrocytes leads to more severe brain damage and vasospasm after subarachnoid hemorrhage. BMC Neuroscience. 2013;14(1):131. doi:10.1186/1471-2202-14-131
50. Kim HB, Yoo JY, Yoo SY, et al. Neuregulin-1 inhibits CoCl(2)-induced upregulation of excitatory amino acid carrier 1 expression and oxidative stress in SH-SY5Y cells and the hippocampus of mice. Molecular Brain. 2020;13(1):153. doi:10.1186/s13041-020-00686-2
51. Song TT, Bi YH, Gao YQ, et al. Systemic pro-inflammatory response facilitates the development of cerebral edema during short hypoxia. Journal of Neuroinflammation. 2016;13(1):63. doi:10.1186/s12974-016-0528-4
52. Xiang Y, Zhang X, Tian Z, et al. Molecular mechanisms of 1,2-dichloroethane-induced neurotoxicity. Toxicological Research. 2023;39(4):565–574. doi:10.1007/s43188-023-00197-x
53. Huang Y, Jiang H, Chen Y, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Molecular Medicine. 2018;10(4):e8689. doi:10.15252/emmm.201708689
54. Callies C, Cooper TG, Yeung CH. Channels for water efflux and influx involved in volume regulation of murine spermatozoa. Reproduction. 2008;136(4):401–410. doi:10.1530/REP-08-0149
55. Liang GH, Järlebark L, Ulfendahl M, et al. Mercury (Hg2+) suppression of potassium currents of outer hair cells. Neurotoxicology and Teratology. 2003;25(3):349–359. doi:10.1016/s0892-0362(03)00008-4
56. Dibas A, Yang MH, Bobich J, et al. Stress-induced changes in neuronal Aquaporin-9 (AQP9) in a retinal ganglion cell-line. Pharmacol Res. 2007;55(5):378–384. doi:10.1016/j.phrs.2007.01.021
57. Vydra Bousova K, Zouharova M, Jiraskova K, et al. Interaction of Calmodulin with TRPM: an Initiator of Channel Modulation. International Journal of Molecular Sciences. 2023;24(20):15162. doi:10.3390/ijms242015162
58. Nilius B, Prenen J, Tang J, et al. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. The Journal of Biological Chemistry. 2005;280(8):6423–6433. doi:10.1074/jbc.M411089200
59. Dutta AK, Khimji AK, Liu S, et al. PKCα regulates TMEM16A-mediated Cl⁻ secretion in human biliary cells. American Journal of Physiology Gastrointestinal and Liver Physiology. 2016;310(1):G34–42. doi:10.1152/ajpgi.00146.2015
60. Lang GE, Stewart PS, Vella D, et al. Is the Donnan effect sufficient to explain swelling in brain tissue slices? Journal of the Royal Society Interface. 2014;11(96):20140123. doi:10.1098/rsif.2014.0123
61. Yu Z, Moomaw JF, Thyagarajapuram NR, et al. A mechanistic model to account for the Donnan and volume exclusion effects in ultrafiltration/diafiltration process of protein formulations. Biotechnology Progress. 2021;37(2):e3106. doi:10.1002/btpr.3106
62. López-garcía JJ, Horno J, Grosse C. Influence of the dielectrophoretic force in mixed electrical double layers. Journal of Colloid and Interface Science. 2013;405:336–343. doi:10.1016/j.jcis.2013.05.036
63. Bohinc K, Bossa GV, May S. Incorporation of ion and solvent structure into mean-field modeling of the electric double layer. Advances in Colloid and Interface Science. 2017;249:220–233. doi:10.1016/j.cis.2017.05.001
64. Previch LE, Ma L, Wright JC, et al. Progress in AQP Research and New Developments in Therapeutic Approaches to Ischemic and Hemorrhagic Stroke. International Journal of Molecular Sciences. 2016;17(7):1146. doi:10.3390/ijms17071146
65. Vella J, Zammit C, Di Giovanni G, et al. The central role of aquaporins in the pathophysiology of ischemic stroke. Frontiers in Cellular Neuroscience. 2015;9:108. doi:10.3389/fncel.2015.00108
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.