")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 17
Employing Hexahydroquinolines as PfCDPK4 Inhibitors to Combat Malaria Transmission: An Advanced Computational Approach
Authors Oduselu GO, Elebiju OF , Ogunnupebi TA, Akash S, Ajani OO, Adebiyi E
Received 2 July 2024
Accepted for publication 18 September 2024
Published 23 September 2024 Volume 2024:17 Pages 83—105
DOI https://doi.org/10.2147/AABC.S476404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Maria Miteva
Gbolahan O Oduselu,1 Oluwadunni F Elebiju,1,2 Temitope A Ogunnupebi,1,2 Shopnil Akash,3 Olayinka O Ajani,1,2 Ezekiel Adebiyi1,4,5
1Covenant University Bioinformatics Research (CUBRe), Covenant University, Ota, OG, Nigeria; 2Department of Chemistry, Covenant University, Ota, OG, Nigeria; 3Department of Pharmacy, Daffodil International University, Dhaka, Bangladesh; 4African Center of Excellence in Bioinformatics & Data Intensive Science, Makerere University, Kampala, Uganda; 5Division of Applied Bioinformatics, German Cancer Research Center (DKFZ), Heidelberg, Germany
Correspondence: Ezekiel Adebiyi, Email [email protected]
Background: Existing antimalarial drugs primarily target blood-stage parasites, but there is a need for transmission-blocking drugs to combat malaria effectively. Plasmodium falciparum Calcium-dependent Protein Kinase 4 (CDPK4) is a promising target for such drugs. This study employed advanced in silico analyses of hexahydroquinolines (HHQ) derivatives to identify PfCDPK4 inhibitors capable of disrupting malaria transmission. Structure-based virtual screening (SBVS) was employed to discover HHQ derivatives with the highest binding affinities against the 3D structure of PfCDPK4 (PDB 1D: 4QOX).
Methods: Interaction analysis of protein-ligand complexes utilized Discovery Studio Client, while druglikeness and ADMET properties were assessed using SwissADME and pkCSM web servers, respectively. Quantum mechanical calculations of the top hits were conducted using density functional theory (DFT), and GROMACS was employed to perform the molecular dynamics (MD) simulations. Binding free energy was predicted using the MMPBSA.py tool from the AMBER package.
Results: SBVS identified ten best hits possessing docking scores within the range of − 11.2 kcal/mol and − 10.6 kcal/mol, surpassing the known inhibitor, BKI-1294 (− 9.9 kcal/mol). Among these, 4-[4-(Furan-2-carbonyl)piperazin-1-yl]-1-(naphthalen-2-ylmethyl)-2-oxo-4a,5,6,7,8,8a-hexahydroquinoline-3-carbonitrile (PubChem ID: 145784778) exhibited the highest binding affinity (− 11.2 kcal/mol) against PfCDPK4.
Conclusion: Comparative analysis of this compound with BKI-1294 using advanced computational approaches demonstrated competitive potential. These findings suggest the potential of 4-[4-(Furan-2-carbonyl)piperazin-1-yl]-1-(naphthalen-2-ylmethyl)-2-oxo-4a,5,6,7,8,8a-hexahydroquinoline-3-carbonitrile as a promising PfCDPK4 inhibitor for disrupting malaria transmission. However, further experimental studies are warranted to validate its efficacy and safety profile.
Keywords: antimalarial drugs, drug design, gametocidal activity, malaria, molecular dynamics
Graphical Abstract:

Introduction
Malaria, a disease caused by Plasmodium parasites, continues to be a major global health concern, especially in regions where the disease is prevalent.1 The process of developing effective antimalarial drugs is both time-consuming and costly, often spanning up to 15 years before reaching commercial availability.2 To address this issue, Medicines for Malaria Venture (MMV) has established clear guidelines through target product profiles (TPPs) and target candidate profiles (TCPs) for new malaria treatments. TCP5 focuses on compound candidates that block human-to-mosquito parasite transmission.3 Current antimalarial medications are tailored to specific stages of the parasite’s life cycle, primarily targeting blood-stage parasites to treat symptomatic malaria. To achieve the elimination and eventual eradication of malaria, there is a critical need to transition the focus from primarily curative treatments to strategies aimed at preventing disease transmission.3 While current antimalarial drugs primarily target symptomatic blood-stage parasites, there is a growing recognition that interrupting disease transmission is crucial for malaria control and eventual eradication.4 Presently, the existing drugs used to treat malaria primarily focus on eliminating asexual blood stages. However, malaria transmission rates remain high due to the continued infectiousness of circulating gametocytes to mosquitoes weeks after treatment. This persistent transmission impedes effective disease control and eradication efforts.5 Preventing transmission of malaria, whether through blocking the human-mosquito cycle or targeting distinct processes, can reduce the risk of reinfection and effectively limit the spread of drug resistance.2
The Calcium-dependent Protein Kinase 4 (CDPK4) enzyme in Plasmodium falciparum presents a promising target for the development of antimalarial drugs that block transmission.5 Unlike human kinases, CDPK4 has a small gatekeeper residue, serine, which offers unique possibilities for selectively inhibiting the parasite’s viability and transmission.3 Among the CDPK family, PfCDPK4 stands out as a critical kinase for the sexual blood stage of Plasmodium falciparum, but not the asexual blood stage, making it an ideal target for transmission-blocking drugs.6 Inhibition of PfCDPK4 disrupts Plasmodium microgamete exflagellation, thus preventing malaria transmission. The unique structural characteristics of Plasmodium kinases, coupled with the need for targeted transmission-blocking drugs, make PfCDPK4 an attractive target for antimalarial drug development, offering more opportunities for medicinal chemists to combat the persistent malaria menace.
Currently, the only drug recommended by the World Health Organization (WHO) as a malaria transmission-blocking drug is primaquine.7 However, the use of this drug is limited due to potential side effects such as gastrointestinal intolerance and hemolysis in patients with glucose-6-phosphate dehydrogenase deficiency.8 Efforts to develop inhibitors against transmission-blocking PfCDPK4 have yielded promising results. Early examples, such as BKI-1, have demonstrated potent inhibition of PfCDPK4. A significant challenge, though, is ensuring these inhibitors remain effective in the bloodstream long enough to clear gametocytes after administration.6 Various PfCDPK4 inhibitors, including pyrrolopyrimidines bumped kinase inhibitors (BKI),9 phenothiazines,3 imidazopyrazines,10 and 5-aminopyrazole-4-carboxamide derivatives5 have been developed. Yet, none have progressed to advanced pre-clinical studies, primarily due to the requirement for long-term bioavailability to remain active against gametocytes.11 PfCDPK4 inhibitors, when ingested by mosquitoes during a blood meal containing malaria gametocytes, inhibit CDPK4 activity, thereby preventing the exflagellation of male gametocytes.12 This necessitates the development of inhibitors that are non-toxic, long-lasting, and effective at doses that allow for extended human treatment exposure.
Hexahydroquinolines (HHQ), identified through extensive screening of the Novartis-GNF Malaria Box, have emerged as a class of heterocyclic compounds with notable transmission-blocking potential against early- and late-stage Pf gametocytes, prompting the need for pharmacokinetic and pharmacodynamic optimization.13 HHQ compounds have a unique structure consisting of a fused heterocyclic structure, which includes a dihydropyridine (DHP) ring containing nitrogen and a cyclohexane ring.14 HHQs are known for their wide spectrum of pharmacological activities including anticancer, antioxidant, antibacterial, antifungal, antitubercular and antimalarial activities.14,15 Furthermore, advancements in computational approaches and target-based drug discovery have facilitated the identification of lead compounds and guided structure-activity relationship studies. Target-based drug discovery for malaria is not a novel concept, and numerous antimalarial drug candidates identified through this approach have advanced to human trials. This includes molecules designed to target specific enzymes such as dihydroorotate dehydrogenase (DHODH) and dihydrofolate reductase (DHFR).4 Given the high attrition rates and costs associated with drug discovery, selecting the most promising targets for drug candidate development is paramount.16 This study delves into advanced in silico analyses of HHQ derivatives, aiming to identify PfCDPK4 inhibitors capable of disrupting the transmission of the malaria parasite from humans to mosquitoes. This research contributes significantly to the overarching goal of malaria elimination.
Materials and Methods
Ligand Library Preparation
The PubChem database (https://pubchem.ncbi.nlm.nih.gov/) was explored and the chemical structures of 16,525 hexahydroquinoline derivatives were obtained. These structures were then filtered using Lipinski’s rule of five (RO5), which includes the following criteria: clog P ≤ 5, molecular weight ≤ 500 g/mol, hydrogen bond acceptors ≤ 10, rotatable bonds ≤ 10, hydrogen bond donors ≤ 5, and topological polar surface area ≤ 140 Å. This strategy was employed because unfavorable physicochemical properties can individually or collectively hinder metabolic stability, absorption (such as solubility and permeability), and ultimately oral bioavailability.17 After filtering, a total of 11,579 hexaquinoline derivatives that passed the rule were downloaded in their.sdf formats. The structures were imported into the OpenBabel interface of PyRx software18 for energy minimization and conversion to their corresponding pdbqt formats. All the compounds were minimized, the energy minimization parameters used are as follow: uff forcefield, conjugate gradients optimization algorithm and a total number of 200 steps. The prepared ligands were taken further for the structure-based virtual screening. In addition to the hexaquinoline derivatives screened, we also included BKI-1 (a reported potent inhibitor of PfCDPK4),9 1294 (a N-methylpiperidine analog of BKI-1),19 DXR (the co-crystallized ligand of the PfCDPK4 target) and primaquine.6
Protein Preparation
A BLAST analysis using NCBI-BLASTp (https://blast.ncbi.nlm.nih.gov/Blast) was employed to determine the selectivity of the PfCDPK4 and ensure it is non-human homologs.20 The UniProtKB accession ID for PfCDPK4 (Q8IBS5) was employed in the blast analysis against the human calcium/calmodulin-dependent protein kinases found in the NCBI database. Using a standard expectation value (E value) of 0.005 and a percentage identity at 35%, PfCDPK4 did not show significant similarity with the human host kinases. The recent availability of crystal structure of targets PfCDPK4 (PDB: 4QOX and 4RGJ) has provided valuable insights into its binding pocket, facilitating the design of selective inhibitors.6 However, 4RGJ does not have a co-crystallized ligand, which makes identification of binding pocket a bit difficult to access. Hence, the crystal structure of PfCDPK4 with PBD:4QOX was downloaded from RCSB PDB.21 The crystal structure was obtained using X-ray crystallography at a resolution of 2.75 Å. The 3D protein structure also possesses a co-crystallized ligand, 3-(3-bromobenzyl)-1-tert-butyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (DXR), in its binding site. The crystal structure of PfCDPK4 is characterized by a very small gatekeeper residue, serine which exposes an enlarged hydrophobic pocket in the ATP-binding site that is not present in human protein kinases.3,22 This hydrophobic pocket can accommodate a large aromatic group that can function as the target inhibitor. The downloaded 4QOX structure was prepared using UCSF Chimera software23 by removing the native co-ligand, water molecules, and other non-amino acid residues. Also, the Dock Prep option of the Chimera software was employed to add hydrogen atoms and Gasteiger charges using the AMBER ff14SB standard residues. The protein structure was further minimized using the following energy minimization parameters: Steepest descent steps: 300, Steepest descent step size: The prepared and minimized protein structure was taken further as the target for the structure-based virtual screening.
Structure-Based Virtual Screening (SBVS)
Virtual screening is one of the commonly used computer-aided drug design (CADD) techniques employed in the identification of lead compounds with good binding affinities to a protein of interest by screening a large chemical database.24 Virtual screening approaches are usually structure-based, or ligand-based. Structure-based virtual screening (SBVS) employs the three-dimensional structure of the target protein to conduct molecular docking studies against compounds within chemical libraries.25 Before the virtual screening, the docking protocol was validated by re-docking the co-crystallized ligand, DXR, into the binding pocket of the active site of 4QOX structure using Autodock vina.26,27 The docked coligand-protein complex was aligned to that of the native coligand-protein complex to obtain the root-mean-square distance (RMSD) value. RMSD values are reliable measures of variability when assessing highly similar proteins, such as different conformations of the same protein. RMSD data derived from structural comparisons of different-sized protein pairs cannot be directly compared because the RMSD value is inherently influenced by the number of atoms involved in the structural alignment.28 The RMSD is zero when structures are identical, and its magnitude increases as the dissimilarity between the two structures increases. The SBVS of the ligand library against the 4QOX crystal structure was also conducted using AutoDock vina. The grid box for the virtual screening was set to cover the binding pocket of the co-crystallized ligand using grid center points set to x=29.735, y=−13.440, z=−0.777, and the grid box size set to x=34, y=34, z=34. After the screening, the compounds were ranked in order of their binding affinities to identify the best hits.
Interaction Analysis and Qualitative Structural Assessment of the Four Best Hits in the Docking Model
The visualisation and interaction analysis of the best hits were performed using Discovery Studio 2021 Client.29 This also revealed the relationships between the chemical structures of the compound and the amino acid residues at the binding site of the 4QOX protein target. This was carried out to examine the compound features and functional groups responsible for the different binding interactions observed in the post-screening analysis. The position and characteristics feature of the functional group determine the type of interaction the compounds exhibited with the amino acid residues at the active site of the target.30 These interactions can introduce a synergistic effect (positive interactions that increase activity) or an antagonistic effect (an interaction that reduces activity) of compounds.31
Determination of Druglikeness and ADMET
The drug-likeness properties of the HHQs were determined using the RO5. The ligand library was initially filtered using RO5 before downloading from PubChem Database, however, this was further confirmed using SwissADME.32 In the drug discovery and development phase, it is evident that considering the pharmacokinetic (PK) characteristics, particularly ADMET (absorption, distribution, metabolism, excretion, and toxicity) is crucial. Therefore, the pkCSM web server was used to screen ADMET properties of the hexahydroquinoline derivatives under investigation.33 These screenings involved evaluating calculated physicochemical and ADMET-related parameters. The SMILES strings of the ligands served as input for the analysis on both platforms. Undesirable pharmacokinetic features in potential drug candidates significantly contribute to the high failure rate in clinical trials.34 Therefore, predicting the pharmacokinetic properties of compounds can provide insights into their safety and toxicity profiles, ultimately reducing the rate of drug attrition.35 Consequently, it is imperative to conduct experimental pre-clinical assessments to confirm or validate the predicted pharmacokinetic characteristics and drug-likeness of the compounds.36
Density Functional Theory (Quantum Mechanics)
Density functional theory (DFT) was used to perform quantum mechanical calculations on of the four best hits from the virtual screening. The geometry optimization of the compounds was achieved using the DFT-B3LYP/6-311G(d,p) level of theory on GaussView 6.0.16 and Gaussian 09 software package programme.37 The 6–311G (d,p) was employed as the basic set due to the standard theory level for some of the major elements found in organic compounds (C, H, N, S and F). Also, the DFT method, Becke, 3-parameter, Lee-Yang-Parr (B3LYP) was employed to interpret the Lowest Unoccupied Molecular Orbital (LUMO) analysis in the optimized structures.37 Both HOMO and LUMO make up the Frontier Molecular Orbital (FMO) and they describe the energies occupied by electrons and the unoccupied energy levels, respectively. The calculation of HOMO-LUMO energy levels and several other critical factors influencing conductivity activity, such as the Ionization potential (I), electron affinity (A), energy band gap (∆Egap), hardness (η), softness (S) and electronegativity (χ), chemical potential (μ), electrophilicity index (ω) were also carried out using Equations 1–6.36 The ∆Egap indicates the stability and reaction of molecules and describes the difference between the HOMO and LUMO. The molecular characteristics of hardness and softness elucidate how electronic transitions and dipole induction occur within the atoms of a compound, whereas electronegativity delineates both the inclination and degree of bond polarization. Electrophilicity describes the strength of electron attraction.38








Molecular Dynamics Simulation
Molecular dynamics (MD) simulations have gained significant attention in the field of drug development research for their ability to provide insights into the dynamic behaviour and stability of protein-ligand complexes.39 This analysis holds significant importance in unravelling the structure-function relationship of the target, as well as the fundamental dynamics of the protein-ligand interactions, which serves as a crucial guide in the journey of drug discovery and design.40 To investigate the potential interactions of the best hit (compound 01) with the Plasmodium falciparum calcium-dependent protein kinase 4 (CDPK4) enzyme (PDB ID 4QOX), we initiated a 200-ns all-atom MD simulation. This was also compared with the MD simulation of the BKI-1294-4QOX complex, due to the reported PfCDPK4 inhibitory potential of BKI-1294, being a bumped kinase inhibitor (BKI).19 These simulations were performed using the CHARMM36 force field41 and the GROMACS version 2024 software package.42 To acquire a molecular topology file suitable for the CHARMM36 force field, we employed the SwissParam online platform43 and also utilized an explicit water model. To neutralize the system’s charge, sodium ions (Na+) and chloride ions (Cl–) were added accordingly. For compound 01–4QOX complex, 47 Na+ ions and 41 Cl– ions were introduced to replace 88 solute molecules in the system while for compound BKI-1294-4OQX complex, 44 Na+ ions and 41 Cl– ions were introduced to replace 85 solute molecules in the system.
The MD simulations included structure minimization using the CHARMM36 force field, and equilibration at 310 K for 100 seconds in the NPT ensemble. Hydrogen atoms were constrained using the Lincs technique and van der Waals forces were calculated with a cutoff value using a switching method of 12–14. Long-range electrostatic interactions were computed using the particle mesh Ewald (PME) approach.44 The system size adjustment for the barostat was targeted at 1 bar, and the temperature was kept constant at 310 K throughout the simulations. A manufacturing run of 100,000,000 and an integration time step of 2 fs were also used, resulting in a 200 ns total simulation time. After the MD simulations, VMD was used to analyze and visualize the trajectory data.45 Bio3D on the Galaxy Europe platform,46 and XMGRACE (version 5.1.19) were also used. A number of properties were assessed to gain insights into the behaviour of the complexes, such as hydrogen bonds (H-bonds), solvent-accessible surface area (SASA), radius of gyration (Rg), root mean square deviation (RMSD), and root mean square fluctuation (RMSF).
Binding Free Energy Calculation
To calculate the free interaction energy between molecules and the protein target in this study, MM-PBSA approach was used. The binding free energy (ΔG) was calculated using Equation 9, and the MMPBSA.py tool from the AMBER package was utilized:47

In this context, Gcomplex refers to the free energy related to the creation of a complex, Greceptor represents the free energy of the receptor, and Gligand indicates the free energy of the ligand.
Result and Discussion
Structure-Based Virtual Screening (SBVS)
The re-docking of the co-crystallized ligand, DXR, into the binding pocket of the active site of the 4QOX structure, revealed the validity of the docking protocol employed for the SBVS. The redocking provided an acceptable RMSD value of 0.04 Å when aligned with the native complex (Figure 1). After the virtual screening of the different 11,579 hexaquinoline derivatives in the binding pocket of the protein target, the ten best hits (Figure 2) were identified using a python script that ranked the compounds according to their binding affinities. The lower the value of the binding affinity, the better the binding.48 Generally, the binding energy of molecules higher than −6.0 kcal/mol is usually considered as potential drug candidates.49 The binding affinities of the ten best hits from the SBVS revealed that all these compounds had better binding affinities than the co-crystallized ligand DXR, BKI-1, compound BKI-1294 and primaquine (Table 1).
 |
Table 1 Binding Affinity Against Targeted Receptor |
 |
Figure 1 Validation of the docking protocol (A) the conformation of the native DXR (red), and docked DXR (yellow) in the 4QOX protein target (B) 2D interaction of the native DXR-4QOX complex, and (C) 2D interaction of the docked DXR-4QOX. |
 |
Figure 2 Structural representation of the best hits from the SBVS, and the reference compounds. |
Interaction Analysis and Qualitative Structural Assessment of the Best Four Hits in the Docking Model
The 2D interactions within the binding pocket of the protein target (4QOX) were scrutinized for the top four hits (Figure 3).
 |
Figure 3 Interaction Analysis and Qualitative structural assessment of the four best hits in the docking model (A) Compound 01 (B) Compound 02 (C) Compound 03 (D) Compound 04. |
Druglikeness and Predicted TPSA Analysis
The drug-likeness properties of the ten best HHQ hits from the SBVS were assessed using the SwissADME web server, and it was observed that none of the compounds violated the Lipinski rule of five (Table 2).
 |
Table 2 Data on Druglikeness of the Ten Best Hits |
Pharmacokinetic Properties Investigation
The summary of the predictive ADMET profiling as obtained from the pkCSM web server is reported in Table 3.
 |
Table 3 ADMET Data of the Ten Best Hits |
Energetic Parameters and Quantum Chemical Descriptors
The energetic parameters and quantum chemical descriptors of the four top hits were analyzed using the DFT-B3LYP/6-311G(d,p) basis set in the Gaussian 09 software package and GaussView 6.0.16.50 To assess the predictive chemical and biological properties of the best hits, various parameters such as ionization potential, electron affinity, energy gap, chemical hardness/softness, electronegativity, chemical potential, and electrophilicity index were examined (Table 4).
 |
Table 4 Energetic Parameters and Quantum Chemical Descriptors of the Four Best Hits |
Frontier Molecular Orbitals (FMOs)
The reactivity of compounds, as determined by quantum mechanics, is governed by the frontier molecular orbitals (FMOs), consisting of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).51 The HOMO identifies the specific region within compounds capable of electron donation, while the LUMO identifies the specific atoms within the compounds capable of accepting electrons (Figure 4).
 |
Figure 4 Highest occupied orbitals (HOMO) and Lowest unoccupied orbitals (LUMO) of the four best hits. |
Molecular Electrostatic Potential (MEP) Surface Result Analysis
MEP are usually employed to predict the electrophilic and nucleophilic sites on a compound for reaction as well as hydrogen bonding interactions.52 This analysis is used to predict the impact sensitivity of CHNO energetic compound and it is commonly carried out on molecular van der Waals (vdW) surface (Figure 5).53
 |
Figure 5 Molecular Electrostatic Potential diagram as calculated at the B3LYP/6-311G(d,p) level (A) Compound 01 (B) Compound 02 (C) Compound 03 (D) Compound 04. |
Molecular Dynamics (MD) Simulation Analysis
Root‑mean‑square Deviation (RMSD) Analysis
Analyzing RMSD is crucial for evaluating the structural stability of proteins or protein-ligand complexes over a defined time frame.54 The RMSD analysis was conducted for the compound 01–4QOX and BKI-1294-4QOX complexes over a simulation period of 200 ns, as illustrated in Figure 6.
 |
Figure 6 RMSD analysis of the c-alpha of the protein-ligand complexes during the molecular dynamics simulations for the time scale of 200 ns. |
Root‑mean‑square Fluctuation (RMSF)
The Root Mean Square Fluctuation (RMSF) analysis stands as a critical parameter in the identification of both rigid and flexible regions within the protein structure. Serving as a standard measure of particle deviation from its original position, RMSF aids in evaluating the flexibility of the protein’s backbone elements and the ligand.55 In this study, RMSF was conducted for the entire protein structure combined with the ligands (compound 01 and BKI-1294), providing insight into the mean displacement of each atom (Figure 7).
 |
Figure 7 RMSF plot of compound 01–4QOX (black) and BKI-1294-4QX (red) complexes. |
Radius of Gyration Analysis
The radius of gyration (Rg) estimates the mass of atoms relative to that of the center of complex’s mass. The observations of Rg suggest the impact of ligand movements within the binding site on the compactness of the protein structure.56 Generally, protein structures that are loosely packed exhibit higher Rg values, whereas tightly packed protein structures have lower Rg values.54 The Rg plots for the protein 4QOX, Compound 01–4QOX complex, and BKI-1294-4QOX complex are depicted in Figure 8.
 |
Figure 8 The radius of gyration (Rg) graph of the protein (4QOX) and complexes with ligands (compound 01 and BKI-1294). |
Solvent Accessible Surface Area (SASA) Analysis
The SASA analysis is a crucial parameter for assessing unfolding and characterizing the hydrogen bond network between amino acid residues and surrounding water molecules.54 When a compound binds to a protein, it can induce conformational changes that affect SASA. Typically, the introduction of a ligand leads to an expansion of the protein structure and an increase in SASA, indicating increase in exposure of the protein surface to solvent.57 The SASA plots for the protein 4QOX, Compound 01–4QOX complex, and BKI-1294-4QOX complex are depicted in Figure 9.
 |
Figure 9 Solvent Accessible Surface Analysis (SASA) graph of the protein (4QOX) and complexes with ligands (compound 01 and BKI-1294). |
Hydrogen Bond Analysis
The analysis of intermolecular hydrogen bonding is crucial for understanding the binding affinity of protein-ligand complexes.56 A higher number of hydrogen bonds typically indicates stronger binding between the amino acid residues of the protein and the ligand. The total number of hydrogen bonds formed in the Compound 01–4QOX complex and the BKI-1294-4QOX complex is illustrated in Figure 10.
 |
Figure 10 Number of hydrogen bonds observed for the Compound 01–4QOX complex (black) and the BKI-1294 complex (red) throughout the MD simulation of 200 ns. |
Binding Free Energy Calculation
To study the molecular binding interactions in protein-ligand complexes, we calculated the binding free energy using the MM-PBSA method.This technique takes into account many interactions, including bonded forces (such as van der Waals force) and non-bonded forces (such as electrostatic forces. The complexes binding free energies were calculated with the Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) method using the final 10 nanoseconds of the trajectory. Based on the binding affinity scores, we used the MMGBSA approach to calculate the free energy binding (∆G_bind) of Compound 01 and BKI-1294 in the protein target (4QOX). When the values of ∆G_bind are lower, it indicates a greater protein-ligand binding affinities. Table 5 and Figure 11 demonstrate the correlation between the molecular docking outcomes and the calculated free binding energies outcomes for the Compound 01 and BKI-1294.
 |
Table 5 Results of Binding Energy of the Ligands |
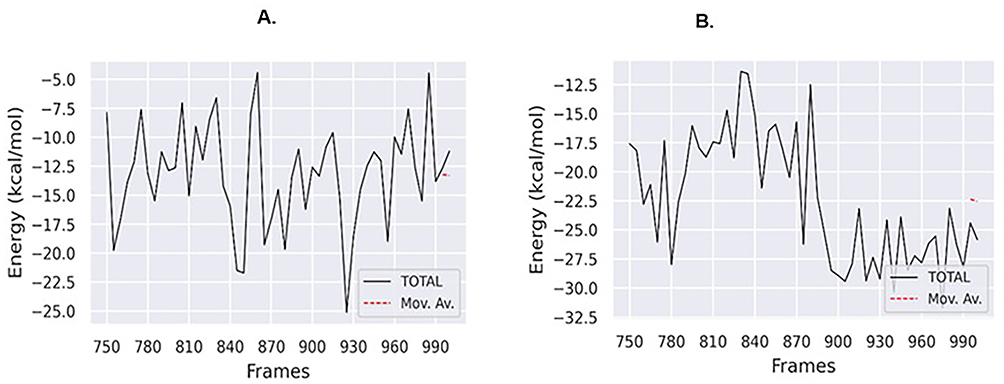 |
Figure 11 Binding free energy plots for (A) compound 01–4QOX and (B) BKI-1294-4QX. |
Discussion
Ojo et al reported compound BKI-1 as a lead in PfCDPK4 inhibition with an IC50 value of 4 nM and a PfNF54 exflagellation EC50 value of 35 nM,9 while an iterative modification of the BKI-1 resulted in compound BKI-1294, which possessed a better absorption, exposure, safety profile, and efficacy in transmission blocking.19 Compound 01 (PubChem ID: 145784778) was the most effective compound in silico with a binding affinity of −11.2 kcal/mol against the Plasmodium falciparum calcium-dependent protein kinase 4 (CDPK4) enzyme (PDB ID 4QOX). Comparison of the binding affinities of compounds BKI-1 and BKI-1294 with that of the HHQ best hits from the SBVS revealed that they had lesser binding affinities, which means they are possible candidates for PfCDPK4 inhibition. Since a number of the HHQ derivatives displayed a robust binding affinity with the target, they warrant further exploration through wet-lab synthesis and subsequent evaluation for their potential as drug candidates in practical or biological applications. However, in this study, we focused on an advanced computational approach to examine the potency of the best hits from the SBVS.
The interaction analysis aimed to elucidate the specific functional groups and heterocycles associated with the HHQ, shedding light on their contributions to the observed binding affinities (Figure 3). Aher and Roy (2016) explored the structural requirements for PfCDPK-4 inhibitors using various in silico approaches.58 Their study revealed that most PfCDPK-4 inhibitors bind to the natural pocket of ATP through hydrogen bonding interactions (Asp148, Tyr150, Lys78, Glu197) and hydrophobic interactions (Leu76, Val84, Ala97, Ile98, Leu133, Leu145, Val146, Val149, Tyr150, Ile214, Phe216, Leu218, Met131, Leu200). Comparing these findings to our study, certain similarities emerged. In our docking studies, Compound 1 was found to interact with amino acid residues of 4QOX. Specifically, it formed conventional hydrogen bonds with Lys231 and carbon hydrogen bonds with Lys78 and Glu154 (Figure 3A). Also, the naphthyl group on N-1 of the HHQ compound 1 interacted via pi-sigma, pi-alkyl and pi-sulfur with Met131, Ile214, Ala97, Leu200, and Val84. These observations further reveal that interactions at certain amino acids contribute to the inhibitory efficiency of the ligands. For compound 2, one oxygen atom of the nitrophenyl group linked directly to N-1 of the HHQ interacted with amino acid residue Lys231 through hydrogen bonding. Other interactions were observed from the phenyl group linked at position C-7, including pi-alkyl interaction with Val84 and Ile214, and pi-sulfur interaction with Met131, among others. Carbazole linked at position C-4 of the HHQ moiety in compound 3 interacted with Leu200, Ala97, Ile214 and Val84 through pi-alkyl bonding. It also interacted via hydrogen bonding with Asp148. The amido linker connecting the pyridine to HHQ at position C-3 interacted with Leu76 and Gly153 through hydrogen bonding. Additionally, the HHQ moiety of compound 4 interacted via pi-alkyl bonding with Leu76. The thiazole linked directly to the N-1 of HHQ interacted via hydrogen bonding with Tyr150, and the dimethylphenyl linked to thiazole interacted with Val84, Ala97, Met131, Leu200, and Ile214 through pi-alkyl and alkyl bonding.
The drug-likeness properties of the ten best HHQ hits from the SBVS possessed molecular weight ≤ 500 g/mol, hydrogen bond acceptors ≤ 10, hydrogen bond donors ≤ 5, and topological polar surface area ≤ 140 Å. This suggests that all the ten best hits are likely to be orally active drug candidates in humans.59 Furthermore, previously proposed inhibitors for PfCDPK4 such as the BKI-1 etc., face challenges in advancing to advanced pre-clinical studies, primarily due to the requirement for prolonged exposure and sufficient concentration in the bloodstream until gametocytes are cleared post-administration.6,58 Therefore, the design and study of new PfCDPK4 inhibitors are contingent on their ability to maintain favourable oral bioavailability.10 Two main approaches—preclinical animal models and computational methods—have been employed in oral bioavailability predictions.60 Among the computational methods, the calculation of topological polar surface area (TPSA) stands out. This method utilizes functional group contributions derived from a vast database of structures, offering a rapid means of virtual bioavailability screening for a large molecule collection.61,62 A TPSA value of ≤140 Å indicates a higher likelihood of good oral bioavailability.63 It is crucial to acknowledge that TPSA is just one of the factors influencing bioavailability, and the overall pharmacokinetic profile involves a complex interplay of various molecular properties. Experimental validation is necessary to confirm bioavailability predictions. The TPSA prediction using the SwissADME web server revealed that the ten best hits from the SBVS exhibited values ranging from 58.20 to 128.83 Å, with a predicted oral bioavailability of 0.55 (55%), comparable to BKI-1 and BKI-1249. Also, compound 01 which had the best binding affinity from the docking studies had a similar TPSA value of 80.79 Å2 to that of BKI-1294 with a TPSA value of 79.54 Å2. All the best hits possess a TPSA value of ≤140 Å, suggesting their likelihood of good oral bioavailability.
The pharmacokinetic properties investigation revealed the ADMET profiling of the ten best hits (Table 3). The assessment of absorption properties included examination of water solubility (Log S), Caco-2 Permeability x 10−6, and Human Intestinal Absorption (%). Distribution properties were evaluated based on VDss (human) and BBB permeability. Metabolism properties were assessed using CYP450 1A2 Inhibitor and CYP450 2D6 Substrate. Excretion properties were determined by analyzing Total Clearance (mL/min/kg) and Renal OCT2 substrate. Lastly, toxicity properties were examined, including max. tolerated dose (Human) (log mg/kg/day), AMES Toxicity, and Hepatotoxicity. The water solubility suggests the solubility of a compound in water at 25°C. The water solubility report showed that compound 09 had a high solubility value of −2.892 log mol/L whereas the other derivatives had lesser solubility tendencies. Caco-2 is a human colorectal adenocarcinoma cell that is utilized as an in vitro model to predict how medicines are absorbed in the human gut.64 High Caco-2 permeability is characterized by predicted values > 8×10−6 cm/s. However, it was discovered that the Caco-2 permeability of the best hits ranged between (0.372–1.798) x 10−6 cm/s. Human intestinal absorption measures the proportion of compound that is absorbed through the human small intestine. A compound with an absorbance of less than 30% is considered to be poorly absorbed. The % intestinal absorption of all the compounds ranges from 81% to 100%, suggesting that all the compounds have high tendencies of absorption. Compounds 02 and 07 were predicted to possess human intestinal absorption of 100%. The distribution property, such as Blood-Brain Barrier (BBB) permeability, serves as an indicator of a drug’s capability to penetrate the brain. This parameter holds significant importance in mitigating potential side effects and toxicity associated with drugs that exert their pharmacological activity within the brain.65 For a given compound, a BBB value > 0.3 is considered to readily cross the blood-brain barrier while a BBB value < −1 is considered to be poorly distributed to the brain. Compound 03 possesses a value of 0.323, hence, it will readily cross the BBB while compounds 06 and 08 with values of −1.014 and −1.178 respectively, will be poorly distributed to the brain. Another distribution parameter considered is the steady state volume of distribution (VDss), which refers to the theoretical volume that the total dose of a drug would need to be uniformly distributed to give the same concentration as in blood plasma. A higher value of VDss (logVDss > 0.45) signifies the tendency of a drug to be distributed in the tissue rather than the plasma. VDss is considered low if log VDss < −0.15. The VDss of the compounds ranged between −0.492 and 0.781.
Most of the compounds were predicted not to inhibit the cytochrome P450 enzyme, except for compounds 04 and 09 which tend to inhibit CYP450 1A2 and CYP450 2D6, respectively. Cytochrome P450 enzyme is an important detoxification enzyme in the body, mainly in the liver. Inhibition of this enzyme can affect drug metabolism, hence, it is ideal that the compounds do not inhibit the cytochrome P450 enzyme. Since the liver and kidneys are the primary routes of excretion, the molecules size is significant. Drug-like substances with molecular weight (MW) <300 is eliminated via bile, while those with MW > 500 is eliminated through urine. Substance with MW in between these ranges are eliminated by both route.36 The excretion level is determined by total clearance rate and Organic Cation Transporter 2 (OCT2). The determination of drug clearance involves assessing the proportionality constant CL tot, which predominantly encompasses hepatic clearance (metabolism in the liver and biliary clearance) and renal clearance (excretion via the kidneys). This parameter is closely linked to bioavailability and plays a crucial role in establishing dosing rates necessary to attain steady-state concentrations.66 Compounds 01 and 09 had that highest total clearance values with 0.836 and 0.859 mg/kg/day, respectively. The OCT2 is a renal uptake transporter that plays an important role in renal clearance of drugs. Except for compound 09, all the best hits are not renal OCT2 substrates. Toxicity is an important parameter in determining whether to proceed with an active molecule. Seven of the compounds exhibited tendency for a degree of hepatoxicity, while three of the compounds were determined to exhibit tendency for AMES toxicity.
The HOMO and LUMO energies of the four best hits were determined by applying Koopman’s approximation to convert from atomic units (a.u) to electron volts (eV), yielding the LUMO and HOMO energies (ELUMO and EHOMO) for each compound. The energy gaps of all compounds ranged from 2.1152 eV to 4.0314 eV. Compounds 01, 02, and 03 exhibited low energy band values, suggesting good reactivity, as lower energy gap values indicate increased reactivity. It is noteworthy that compounds with higher softness levels degrade more quickly than those with lower softness levels, whereas hardness is a fundamental property indicating durability. Higher values of chemical hardness signify stable compounds with low reactivity, while higher softness values denote less stable compounds with higher reactivity.67 The order of softness was determined as 02>03>01>04, while the order of hardness was 04>01>03>02, with hardness being inversely related to softness. Compound 04 displayed the highest ionization potential, while compound 02 exhibited the highest electron affinity. The electronegativity values of compounds 01, 02, 03, and 04 were 3.4555, 4.9883, 3.1517, and 4.6770 eV, respectively, indicating their electron attraction capacities. All of the top hits had negative values of chemical potential (μ), indicating exceptional stability and the possibility of forming stable complexes with the 4QOX target. A chemical species’ propensity to receive an electron is indicated by its electrophilic tendency, which is measured by electrophilicity (ω). Compound 02 demonstrated a strong electrophilic tendency with a value of 11.764 eV. All top hits exhibited electrophilicity values higher than 1.5 eV, indicating their strong electrophilic nature.47 In conclusion, the quantum chemical descriptors provided insight into the potential of the top hits as 4QOX targets.
The HOMOs are predominantly localized over the naphthyl group of compound 1, which could be responsible for the concentration of interactions (pi-alkyl, pi-sulfur, pi-sigma) observed between the compound and the 4QOX target, as depicted in Figure 3A. Meanwhile, the LUMO of compound 1 is primarily localized over the piperazinyl group, the nitrogen atom of the cyano group, and the nitrogen atom of HHQ. For compound 2, both HOMO and LUMO are predominantly distributed over the entire molecule, except around the phenyl group on the C-7 of HHQ. Similarly, in compound 3, both HOMO and LUMO are observed mainly at the carbazolyl ring, the pyridinyl group, and the amido group, as these are the electron-donating and accepting groups attached to the HHQ. For instance, the amido group is a strong activating group with a high tendency to donate electron density to the attached ring.68 Likewise, in compound 4, the HOMO and LUMO are observed mainly around the HHQ ring, thiazole, and dimethylphenyl groups of the compound.
The MEPs for the optimized geometries of the best hits were calculated using GaussView program and their MEP maps at B3LYP/6-311G(d,p) level were plotted, as shown in Figure 5. The negative electrostatic potential is represented by the red color and this is related to electrophilic reactivity, whereas the positive electrostatic potential is indicated by the blue color and this is related to nucleophilic reactivity. Electrostatic potential increases in the order red<orange<yellow<green<blue. As observed from Figure 5, the red colour region or electrophilic reactivity of the compounds are mainly localized on the oxygen atoms, with portions of the blue region or the nucleophilic reactivity.
In this study, the RMSD analysis was conducted for the compound 01–4QOX and BKI-1294-4QOX complexes over a simulation period of 200 ns, as illustrated in Figure 6. For the compound 01–4QOX complex, the RMSD analysis indicated that the protein remained stable with an average value of 0.2 nm throughout the MD simulation. However, the RMSD of compound 01 within the complex exhibited stability around 0.3 nm after an initial increase from 0 ns until 58 ns. Subsequently, there was another increase until approximately 0.8 nm, maintaining this stability until the end of the 200 ns simulation. In contrast, for the BKI-1294-4QOX complex, both the BKI-1294 ligand and the protein displayed an average RMSD value of 0.25 nm and 0.5 nm, respectively, throughout the 200 ns MD simulation. Notably, the RMSD of the BKI-1294-4QOX complex remained relatively constant during the analysis, suggesting stability of the complex. It was also observed that both complexes (compound 01–4QOX and BKI-1294-4QX) have slightly similar RMSF patterns with majority of the atoms lower than 0.2 nm.
Furthermore, the average Rg value for the protein 4QOX is 2.49 nm, while for the Compound 01–4QOX complex and BKI-1294-4QOX complex, it increases to 2.59 nm and 2.61 nm, respectively. This indicates that the protein structure of 4QOX is tightly packed, and upon introduction of the ligands (Compound 01 and BKI-1294), the Rg values increase, reflecting a more loosely packed complex. These changes may be attributed to conformational shifts resulting in alterations to the secondary protein structure during molecular dynamics simulations.54 Also, the introduction of the ligands (Compound 01 and BKI-1294) in the protein binding site led to a notable increase in the solvent accessible surface area, with SASA values increasing from an average value of 230 nm2 for the protein structure alone, to 320 nm2 and 315 nm2 for Compound 01–4QOX complex, and BKI-1294-4QOX complex, respectively. This also further confirmed that the introduction of the ligands led to the expansion of the protein structure, with both ligands used have a similar effect on the SASA value. Interestingly, the BKI-1294-4QOX complex exhibited the highest number of hydrogen bonds, with predominantly 3 to 4 bonds throughout the 200 ns simulation. In contrast, the Compound 01–4QOX complex predominantly maintained one hydrogen bond during the simulation, with occasional instances of two hydrogen bonds being observed. This observation aligns with the specific conventional hydrogen bond detected in our molecular docking studies between Lys231 and the oxygen of the furuyl group on Compound 01, as shown in Figure 3. The MM-PBSA research demonstrated that the binding energy of BKI-1294 complex is strong and has increased stability, same with the Compound 01–4QOX complex.
Conclusion
The pursuit of antimalarial drug candidates targeting transmission-blocking, beyond symptomatic blood-stage parasites, highlights the need for novel approaches in malaria treatment. Hexahydroquinolines (HHQ), identified through extensive screening of the Novartis-GNF Malaria Box, have emerged as promising candidates with significant transmission-blocking potential against early- and late-stage Pf gametocytes. In this study, we conducted advanced in silico analyses of HHQ derivatives obtained from the PubChem database, aiming to identify PfCDPK4 inhibitors capable of disrupting malaria transmission from humans to mosquitoes. Our computational approach encompassed structure-based virtual screening (SBVS), druglikeness assessments, predicted topological polar surface area (TPSA), ADMET studies, pharmacokinetic properties evaluation, density functional theory (DFT), molecular electrostatic potential (MEP) analysis, and molecular dynamics (MD) simulations. The SBVS identified ten top hits exhibiting binding affinities ranging from −11.2 kcal/mol to −10.6 kcal/mol. Notably, the compound 4-[4-(Furan-2-carbonyl)piperazin-1-yl]-1-(naphthalen-2-ylmethyl)-2-oxo-4a,5,6,7,8,8a-hexahydroquinoline-3-carbonitrile (PubChem ID: 145784778) demonstrated the highest binding affinity (−11.2 kcal/mol) against PfCDPK4 (PDB ID: 4QOX). We compared this top hit to BKI-1294, a known inhibitor, to assess its competitive potential. Compound 01, exhibiting the best binding affinity from docking studies, shared a similar topological polar surface area (TPSA) value (80.79 Å2) with BKI-1294 (79.54 Å2). MD simulation analyses further supported the suitability of Compound 01 as a potential PfCDPK4 inhibitor, demonstrating stability within the complex. However, it is essential to note that these in silico findings provide theoretical insights and require validation through experimental biological screening, including in vitro, in vivo, pre-clinical, and clinical trials. Such studies are crucial to confirm the efficacy and safety profile of Compound 01 as a promising antimalarial drug candidate with transmission-blocking properties.
Acknowledgment
The authors appreciate the Covenant University Bioinformatics Research (CUBRe) management for making available the necessary tools and infrastructure important for the successful execution of the project.
Funding
This work was funded by the West Africa Sustainable Leadership and Innovation Training in Bioinformatics Research (WASLITBRe) through grant number: 5U2RTW010679.
Disclosure
All authors report no conflicts of interest in this work.
References
1. World Health Organization. World Malaria Report 2022. World Health Organization.; 2022.
2. Umumararungu T, Nkuranga JB, Habarurema G, et al. Recent developments in antimalarial drug discovery. Bioorg Med Chem. 2023:88–89. doi:10.1016/j.bmc.2023.117339
3. Moolman C, van der Sluis R, Beteck RM, Legoabe LJ. An update on development of small-molecule plasmodial kinase inhibitors. Molecules. 2020;25(21):1–45. doi:10.3390/molecules25215182
4. Yang T, Ottilie S, Istvan ES, et al. Trends in MalDA, accelerating malaria drug discovery. Trends Parasitol. 2021;37(6):493–507. doi:10.1016/j.pt.2021.01.009
5. Huang W, Hulverson MA, Zhang Z, et al. 5-Aminopyrazole-4-carboxamide analogues are selective inhibitors of Plasmodium falciparum microgametocyte exflagellation and potential malaria transmission blocking agents. Bioorganic Med Chem Lett. 2016;26(22):5487–5491. doi:10.1016/j.bmcl.2016.10.014
6. Ong HW, Adderley J, Tobin AB, Drewry DH, Doerig C. Parasite and host kinases as targets for antimalarials. Expert Opin Ther Targets. 2023;27(2):151–170. doi:10.1080/14728222.2023.2185511
7. Wadi I, Singh P, Nath M, Anvikar AR, Sinha A. Malaria transmission-blocking drugs: implications and future perspectives. Future Med Chem. 2020;12(11):1071–1101. doi:10.4155/fmc-2020-0026
8. Yu S, Wang J, Luo X, et al. Transmission-blocking strategies against malaria parasites during their mosquito stages. Front Cell Infect Microbiol. 2022:12(February):1–14. doi:10.3389/fcimb.2022.820650
9. Ojo KK, Pfander C, Mueller NR, et al. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J Clin Invest. 2012;122(6):2301–2305. doi:10.1172/JCI61822
10. Vidadala RSR, Ojo KK, Johnson SM, et al. Development of potent and selective Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4) inhibitors that block the transmission of malaria to mosquitoes. Eur J Med Chem. 2014;74:562–573. doi:10.1016/j.ejmech.2013.12.048
11. Mustière R, Vanelle P, Primas N. Plasmodial Kinase Inhibitors Targeting Malaria: recent Developments. Molecules. 2020;25(24). doi:10.3390/MOLECULES25245949
12. Van Voorhis WC, Doggett JS, Parsons M, et al. Extended-spectrum antiprotozoal bumped kinase inhibitors: a review. Exp Parasitol. 2017;180:71–83. doi:10.1016/j.exppara.2017.01.001
13. Vanaerschot M, Lucantoni L, Li T, et al. Hexahydroquinolines are antimalarial candidates with potent blood-stage and transmission-blocking activity. Nat Microbiol. 2017;2(10):1403–1414. doi:10.1038/s41564-017-0007-4
14. Ranjbar S, Edraki N, Firuzi O, Khoshneviszadeh M, Miri R. 5-Oxo-hexahydroquinoline: an attractive scaffold with diverse biological activities. Mol Divers. 2019;23(2):471–508. doi:10.1007/s11030-018-9886-4
15. Çetin G, Çetin B, Çolak B, et al. A new perspective for biological activities of novel hexahydroquinoline derivatives. J Res Pharm. 2022;26(1):219–230. doi:10.29228/JRP.120
16. Forte B, Ottilie S, Plater A, et al. Prioritization of Molecular Targets for Antimalarial Drug Discovery. ACS Infect Dis. 2021;7(10):2764–2776. doi:10.1021/acsinfecdis.1c00322
17. Zeng S, Ye Y, Xia H, et al. Current advances and development strategies of orally bioavailable PROTACs. Eur J Med Chem. 2023:
18. Dallakyan S, Olson AJ. Small molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263(January):1–11. doi:10.1007/978-1-4939-2269-7
19. Ojo KK, Eastman RT, Vidadala R, et al. A specific inhibitor of pfcdpk4 blocks malaria transmission: chemical-genetic validation. J Infect Dis. 2014;209(2):275–284. doi:10.1093/infdis/jit522
20. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi:10.1016/S0022-2836(05)80360-2
21. Burley SK, Berman HM, Bhikadiya C, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019;47(D1):D464–D474. doi:10.1093/nar/gky1004
22. Huang W, Ojo KK, Zhang Z, et al. SAR Studies of 5-Aminopyrazole-4-carboxamide Analogues as Potent and Selective Inhibitors of Toxoplasma gondii CDPK1. ACS Med Chem Lett. 2015;6(12):1184–1189. doi:10.1021/acsmedchemlett.5b00319
23. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
24. Salmaso V, Moro S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: an overview. Front Pharmacol. 2018;9(AUG):1–16. doi:10.3389/fphar.2018.00923
25. Malathi K, Ramaiah S. Bioinformatics approaches for new drug discovery: a review. Biotechnol Genet Eng Rev. 2018;34(2):243–260. doi:10.1080/02648725.2018.1502984
26. Trott O, Olson AJ. AutoDock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009;31(2):455–461. doi:10.1002/jcc
27. Joshi T, Joshi T, Sharma P, et al. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur Rev Med Pharmacol Sci. 2020;24(8):4529–4536. doi:10.26355/eurrev_202004_21036
28. Carugo O, Pongor S. A normalized root-mean-spuare distance for comparing protein three-dimensional structures. Protein Sci. 2008;10(7):1470–1473. doi:10.1110/ps.690101
29. Sravani M, Kumaran A, Tulshiram Dhamdhere A, Senthil Kumar N. Computational molecular docking analysis and visualisation of anthocyanins for anticancer activity. Int J Res Appl Sci Biotechnol. 2021;8(1):154–161. doi:10.31033/ijrasb.8.1.18
30. Oduselu GO, Afolabi R, Ademuwagun I. Structure-based pharmacophore modeling, virtual screening, and molecular dynamics simulation studies for identification of Plasmodium falciparum 5-aminolevulinate synthase inhibitors. Front Med. 2023;9(1022429):1–15. doi:10.3389/fmed.2022.1022429
31. Sharma J, Bhardwaj VK, Das P, Purohit R. Identification of naturally originated molecules as γ-aminobutyric acid receptor antagonist. J Biomol Struct Dyn. 2020;1(1):1. doi:10.1080/07391102.2020.1720818
32. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(March):1–13. doi:10.1038/srep42717
33. Azzam KAL. SwissADME and pkCSM webservers predictors: an integrated online platform for accurate and comprehensive predictions for in silico ADME/T properties of artemisinin and its derivatives. Complex Use Miner Resour. 2023;325(2):14–21. doi:10.31643/2023/6445.13
34. Agamah FE, Mazandu GK, Hassan R, et al. Computational/in silico methods in drug target and lead prediction. Brief Bioinform. 2020;21(5):1663–1675. doi:10.1093/bib/bbz103
35. Oduselu GO, Aderohunmu DV, Ajani OO, Elebiju OF, Ogunnupebi TA, Adebiyi E. Synthesis, in silico and in vitro antimicrobial efficacy of substituted arylidene-based quinazolin-4(3H)-one motifs. Front Chem. 2023;11(1264824):1–18. doi:10.3389/fchem.2023.1264824
36. Akash S, Bayıl I, Hossain S, et al. Novel computational and drug design strategies for inhibition of human papillomavirus ‑ associated cervical cancer and DNA polymerase theta receptor by Apigenin derivatives. Sci Rep. 2023:1–22. doi:10.1038/s41598-023-43175-x
37. Silvarajoo S, Osman UM, Kamarudin KH, et al. Dataset of theoretical Molecular Electrostatic Potential (MEP), Highest Occupied Molecular Orbital-Lowest Unoccupied Molecular Orbital (HOMO-LUMO) band gap and experimental cole-cole plot of 4-(ortho-, meta- and para-fluorophenyl)thiosemicarbazide isomers. Data Br. 2020;32:106299. doi:10.1016/j.dib.2020.106299
38. Benjamin I, Udoikono AD, Louis H, et al. Antimalarial potential of naphthalene-sulfonic acid derivatives: molecular electronic properties, vibrational assignments, and in-silico molecular docking studies. J Mol Struct. 2022;1264:133298. doi:10.1016/j.molstruc.2022.133298
39. Hasan AH, Murugesan S, Amran SI, et al. Novel thiophene Chalcones-Coumarin as acetylcholinesterase inhibitors: design, synthesis, biological evaluation, molecular docking, ADMET prediction and molecular dynamics simulation. Bioorg Chem. 2022;119(July 2021):105572. doi:10.1016/j.bioorg.2021.105572
40. Liu X, Shi D, Zhou S, Liu H, Liu H, Yao X. Molecular dynamics simulations and novel drug discovery. Expert Opin Drug Discov. 2018;13(1):23–37. doi:10.1080/17460441.2018.1403419
41. Sahu S, Ghosh SK, Kalita J, Dutta M, Bhat HR. Design, synthesis and antimalarial screening of some hybrid 4-aminoquinoline-triazine derivatives against pf-DHFR-TS. Exp Parasitol. 2016;163:38–45. doi:10.1016/j.exppara.2016.01.010
42. Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi:10.1093/bioinformatics/btt055
43. Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem. 2011;32(11):2359–2368. doi:10.1002/jcc
44. Safarizadeh H, Garkani-Nejad Z. Molecular docking, molecular dynamics simulations and QSAR studies on some of 2-arylethenylquinoline derivatives for inhibition of Alzheimer’s amyloid-beta aggregation: insight into mechanism of interactions and parameters for design of new inhibitors. J Mol Graph Model. 2019;87:129–143. doi:10.1016/j.jmgm.2018.11.019
45. Aksimentiev A, Arkhipov A, Birnbaum R, et al. Using VMD; 2019. Available from: http://www.ks.uiuc.edu/Training/Tutorials/.
46. Grant BJ, Rodrigues APC, Elsawy KM, Mccammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22(21):2695–2696. doi:10.1093/bioinformatics/btl461
47. Hamza S, Tayab A, Uzairu A, et al. Molecular docking studies of some benzoxazole and benzothiazole derivatives as VEGFR-2 target inhibitors: in silico design, MD simulation, pharmacokinetics and DFT studies. Intelligent Pharmacy. 2024;2(2):232–250. doi:10.1016/j.ipha.2023.11.010
48. Akinnusi PA, Olubode SO, Adebesin AO, et al. Structure-based scoring of anthocyanins and molecular modeling of PfLDH, PfDHODH, and PfDHFR reveal novel potential P. falciparum inhibitors. Informatics Med Unlocked. 2023;38(March):101206. doi:10.1016/j.imu.2023.101206
49. Nath A, Kumer A, Zaben F, Khan MW. Investigating the binding affinity, molecular dynamics, and ADMET properties of 2,3-dihydrobenzofuran derivatives as an inhibitor of fungi, bacteria, and virus protein. Beni-Suef Univ J Basic Appl Sci. 2021;10(1). doi:10.1186/s43088-021-00117-8
50. Ayar A, Aksahin M, Mesci S, Yazgan B, Gül M, Yıldırım T. Antioxidant, cytotoxic activity and pharmacokinetic studies by SwissAdme, Molinspiration, Osiris and DFT of PhTAD-substituted dihydropyrrole derivatives. Curr Comput Aided Drug Des. 2021;17:1–12. doi:10.2174/1573409917666210223105722
51. Olawale F, Olofinsan K, Iwaloye O, Chukwuemeka PO, Elekofehinti OO. Screening of compounds from Nigerian antidiabetic plants as protein tyrosine phosphatase 1B inhibitor. Comput Toxicol. 2022;21(October 2021):100200. doi:10.1016/j.comtox.2021.100200
52. Rajalakshmi K, Gunasekaran S, Kumaresan S. Density functional theory, comparative vibrational spectroscopic studies, highest occupied molecular orbital and lowest unoccupied molecular orbital analysis of Linezolid. Indian J Phys. 2015;89(6):525–538. doi:10.1007/s12648-014-0618-z
53. Cao W, Ding Z, Hang X, et al. Theoretical study of a series of 4,4′-azo-1: h −1,2,4-triazol-5-one based nitrogen-rich salts as potential energetic compounds. RSC Adv. 2018;8(42):23805–23816. doi:10.1039/c7ra13424j
54. Surti M, Patel M, Adnan M, et al. Ilimaquinone (marine sponge metabolite) as a novel inhibitor of SARS-CoV-2 key target proteins in comparison with suggested COVID-19 drugs: designing, docking and molecular dynamics simulation study. RSC Adv. 2020;10(62):37707–37720. doi:10.1039/d0ra06379g
55. Verma S, Debnath U, Agarwal P, Srivastava K, Prabhakar YS. In silico exploration for new antimalarials: arylsulfonyloxy acetimidamides as prospective agents. J Chem Inf Model. 2015;55(8):1708–1719. doi:10.1021/acs.jcim.5b00392
56. Thillainayagam M, Malathi K, Ramaiah S. In-Silico molecular docking and simulation studies on novel chalcone and flavone hybrid derivatives with 1, 2, 3-triazole linkage as vital inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biomol Struct Dyn. 2017;36(15):3993–4009. doi:10.1080/07391102.2017.1404935
57. Rahman MM, Saha T, Islam KJ, et al. Virtual screening, molecular dynamics and structure–activity relationship studies to identify potent approved drugs for Covid-19 treatment. J Biomol Struct Dyn. 2020;1(1):1–11. doi:10.1080/07391102.2020.1794974
58. Aher RB, Roy K. Exploring structural requirements for the inhibition of: plasmodium falciparum calcium-dependent protein kinase-4 (Pf CDPK-4) using multiple in silico approaches. RSC Adv. 2016;6(57):51957–51982. doi:10.1039/c6ra05692j
59. Fu M, Ge M, Yang W, et al. Discovery of a potent and selective cell division cycle 7 inhibitor from 6-(3-fluoropyridin-4-yl)thieno[3,2-d]pyrimidin-4(3H)-one derivatives as an orally active antitumor agent. Acta Pharm Sin B. 2024;14(2):893–896. doi:10.1016/j.apsb.2023.11.026
60. Beheshti S, Shayanfar A. Prediction of the oral bioavailability correlation between humans and preclinical animals. Eur J Drug Metab Pharmacokinet. 2020;45(6):771–783. doi:10.1007/s13318-020-00636-2
61. Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43(20):3714–3717. doi:10.1021/jm000942e
62. Prasanna S, Doerksen R. Topological polar surface area: a useful descriptor in 2D-QSAR. Curr Med Chem. 2008;16(1):21–41. doi:10.2174/092986709787002817
63. Xie YZ, Peng CW, Su ZQ, et al. A practical strategy for exploring the pharmacological mechanism of luteolin against COVID-19/asthma comorbidity: findings of system pharmacology and bioinformatics analysis. Front Immunol. 2022:12(January):1–18. doi:10.3389/fimmu.2021.769011
64. Ren S, Lien EJ. Caco-2 cell permeability vs human gastrointestinal absorption: QSPR analysis. Prog Drug Res. 2000;54:1–23. doi:10.1007/978-3-0348-8391-7_1
65. Sobańska AW, Wanat K, Brzezińska E. Prediction of the blood-brain barrier permeability using RP-18 thin layer chromatography. Open Chem. 2019;17(1):43–56. doi:10.1515/chem-2019-0005
66. Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–4072. doi:10.1021/acs.jmedchem.5b00104
67. Kaddouri Y, Abrigach F, Yousfi EB, El Kodadi M, Touzani R. New thiazole, pyridine and pyrazole derivatives as antioxidant candidates: synthesis, DFT calculations and molecular docking study. Heliyon. 2020;6(1). doi:10.1016/j.heliyon.2020.e03185
68. Truce WE, Tichenor GJW. Effect of activating group on trans stereoselectivity of thiolate additions to activated acetylenes. J Org Chem. 1972;37(15):2391–2396. doi:10.1021/jo00980a007
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.