")
Back to Journals » International Journal of Nanomedicine » Volume 19
Enhanced in vivo Stability and Antitumor Efficacy of PEGylated Liposomes of Paclitaxel Palmitate Prodrug
Authors Wu X, Wang X, Zhang H, Chen H, He H, Lu Y , Tai Z , Chen J, Wu W
Received 8 September 2024
Accepted for publication 4 November 2024
Published 9 November 2024 Volume 2024:19 Pages 11539—11560
DOI https://doi.org/10.2147/IJN.S488369
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Xin Wu,1– 3,* Xinyu Wang,3,* Haiyan Zhang,3,* Hang Chen,3,* Haisheng He,2 Yi Lu,1,2 Zongguang Tai,1 Jianming Chen,3 Wei Wu1,2
1Shanghai Skin Disease Hospital, School of Medicine, Tongji University, Shanghai, 200443, People’s Republic of China; 2School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery, Ministry of Education, Shanghai, 201203, People’s Republic of China; 3Shanghai Wei Er Lab, Shanghai, 201707, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Wu, School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery, Ministry of Education, Shanghai, 201203, People’s Republic of China, Tel/Fax +86-21-64175590, Email [email protected] Zongguang Tai, Shanghai Skin Disease Hospital, School of Medicine, Tongji University, Shanghai, 200443, People’s Republic of China, Tel/Fax +86-36803155, Email [email protected]
Purpose: The clinical use of paclitaxel (PTX) in cancer treatment is limited by its poor water solubility, significant toxicity, and adverse effects. This study aimed to propose a straightforward and efficient approach to enhance PTX loading and stability, thereby offering insights for targeted therapy against tumors.
Patients and Methods: We synthesized a paclitaxel palmitate (PTX-PA) prodrug by conjugating palmitic acid (PA) to PTX and encapsulating it into liposomal vehicles using a nano delivery system. Subsequently, we investigated the in vitro and in vivo performance as well as the underlying mechanisms of PTX-PA liposomes (PTX-PA-L).
Results: PTX had a remarkable antitumor effect in vivo and significantly decreased the myelosuppressive toxicity of PTX. Moreover, the introduction of PA increased the lipid solubility of PTX, forming a phospholipid bilayer as a membrane stabilizer, prolonging the circulation time of the drug and indirectly increasing the accumulation of liposomes at the tumor site. Our in vivo imaging experiments demonstrated that PTX-PA-L labeled with DiR has greater stability in vivo than blank liposomes and that PTX-PA-L can target drugs to the tumor site and efficiently release PTX to exert antitumor effects. In a mouse model, the concentration of PTX at the tumor site in the PTX-PA-L group was approximately twofold greater than that of Taxol. However, in a nude mouse model, the concentration of PTX at the tumor site in the PTX-PA-L group was only approximately 0.8-fold greater than that of Taxol. Furthermore, the originally observed favorable pharmacodynamics in normal mice were reversed following immunosuppression. This may be caused by differences in esterase distribution and immunity.
Conclusion: This prodrug technology combined with liposomes is a simple and effective therapeutic strategy with promising developmental prospects in tumor-targeted therapy owing to its ability to convert PTX into a long-circulating nano drug with low toxicity, high pharmacodynamics, and good stability in vivo.
Keywords: paclitaxel palmitate liposome, prodrug, membrane stabilizer, esterase metabolism, Immunity
Introduction
PTX, an active ingredient extracted from the bark of Taxus brevis,1 has been approved for the treatment of ovarian cancer, lung cancer, breast cancer, colon cancer and lymphoma because of its ability to promote tubulin polymerization, induce tumor cell mitosis, block mitosis in the G2 and M phases for a long time, and promote tumor cell division and death.2,3 However, the clinical application of PTX is limited by its poor water solubility and unsatisfactory pharmacodynamics.4,5 The clinically available formulation of paclitaxel (Taxol®) consists of polyoxyethylene castor oil (Cremophor EL, CrEL) and absolute ethanol as a mixed solvent (50:50, v/v) to increase the solubility of the drug.6 However, numerous studies have shown that CrEL has toxic and adverse effects, including neutropenia, peripheral sensory neuropathy, hyperlipidemia, neurotoxicity, and hypersensitivity reactions.7–9 To avoid the use of CrEL, several novel delivery systems have been developed either to solubilize PTX or to deliver it to target places or both. Compared with Taxol®, a PTX albumin nanosuspension (Abraxane®) utilizes albumin in place of CrEL as the carrier system and thus has been proven to be safer and more tolerable, with an increase in efficacy of approximately 50%.10–12 However, the use of albumin may elicit immunological responses, and there is no significant difference in clinical efficacy between Abraxane® and Taxol® in the treatment of some specific cancers, such as breast cancer and non-small cell lung cancer.13,14 Alternatively, a PTX polymer micelle injection named Genexol-PM® was developed by replacing CrEL with a copolymer, methoxy polyethylene glycol-co-poly (lactic acid).15,16 However, polymeric micelle formulations suffer from poor circulation stability in vivo, unsatisfactory biocompatibility, and low responsiveness in the cellular microenvironment.17,18
Liposomes are alternative carrier systems for the solubilization and delivery of poorly soluble drugs. Owing to the incomparable biocompatibility and safety of liposomal constituents, ie, endogenous phospholipids and cholesterols, liposomes remain the first choice in the development of carrier drug delivery systems.19 To solubilize PTX and adapt it for intravenous delivery, PTX liposomes have been successfully developed without the use of CrEL. Nevertheless, the liposomal PTX formulation suffers from a very low loading capacity (approximately 3 mg/mL) and poor physical stability,20 which is typical of the enlarged size and formation of aggregates following the reconstitution of freeze-dried formulations.21 Our pilot study also indicated that PTX liposomes have poor stability during transport in vivo and are prone to premature leakage.22–24 Various methods are currently available to improve the stability of liposomes, such as enhancing the rigidity of liposome membranes25,26 or embedding rigid nanomaterials into liposomal structures.27 In contrast to methods that rigidify liposomes, modifying the drug molecules themselves to endow them with a high affinity for lipid bilayers is a more efficient strategy for enhancing drug loading and preventing premature leakage.
In our previous study, PTX-lipid conjugates were synthesized and encapsulated in phospholipid/cholesterol liposomes. The combination of long-chain saturated fatty acids with PTX substantially elevated the lipid solubility of PTX, thus increasing its loading efficiency.28 More importantly, the integration of lipophilic PTX prodrugs stabilized the liposomes and thereby prolonged the circulation time in vivo (Scheme 1). By overcoming the problems of low loading and instability of traditional PTX liposomes, PTX prodrug-loaded liposomes demonstrate enhanced anticancer efficacy. A lipid chain length dependency was observed in terms of loading efficiency, stabilization, and antitumor efficacy, with PTX palmitate (PTX-PA) having the longest chain length.28
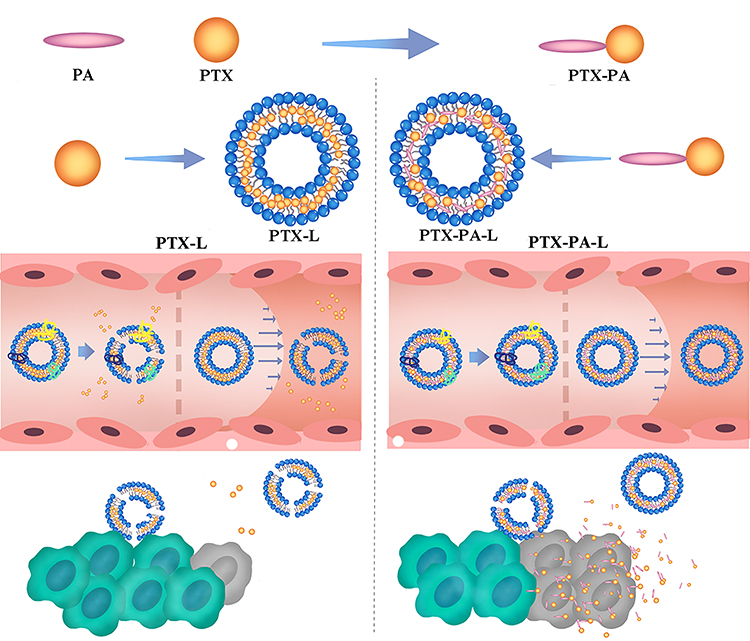 |
Scheme 1 Synthesis and preparation of paclitaxel palmitate liposomes. Paclitaxel palmitate, a prodrug and liposomal membrane stabilizer, can form stable prodrug nanoliposomes (termed PTX-PA-L). Compared with the simple liposome of paclitaxel (called PTX-L), PTX-PA-L is more stable in the blood, and the drug is not easy to leak. The role of PTX-PA-L in preventing PTX leakage caused by plasma proteins could lead to more drugs being delivered to the tumor site. |
The current study aimed to further explore both the in vitro and in vivo performance and underlying mechanisms of PTX-PA liposomes. In vivo pharmacodynamics were evaluated in either a normal or a nude mouse model or a normal mouse model grafted with a 4T1 immunodeficient breast tumor or a Lewis lung tumor. Subsequently, we explored the long-term circulation, biodistribution, tumor targetability and in vivo liposomal stability, as well as the function and number of macrophages and immunity-related factors (Scheme 2). Finally, the safety of PTX-PA liposomes was evaluated. The results demonstrated that prodrug technology combined with liposomes was a simple and effective strategy for enhancing PTX loading and in vivo stability and thus has promising potential for tumor-targeted therapy.
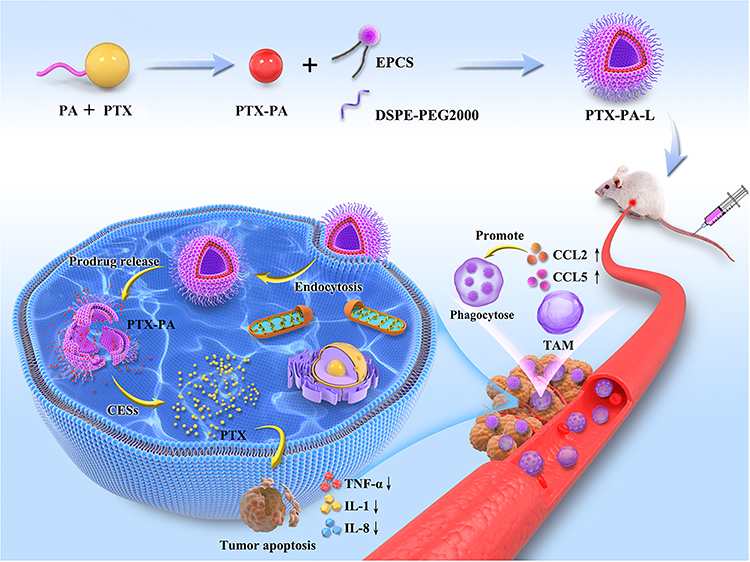 |
Scheme 2 Schematic illustration of targeted anticancer prodrug nanoliposomes that induce apoptosis and improve antitumor immunity. PTX and PA were obtained by the prodrug technique to obtain PTX-PA, which was then coassembled with liposomes to obtain PTX-PA-L. After intravenous injection, PTX-PA-L can maintain a long cycle in vivo and ensure preferential accumulation at the tumor site while entering the body, being engulfed by macrophages, becoming activated and migrating to tumor tissue by regulating the expression levels of immune factors such as CCL2, CCL5, TNF-α, IL-8, and IL-1, among others, exerting its outstanding antitumor effect. |
Materials and Methods
Materials
PTX (China Resources Meilian Pharmaceutical Co., Ltd., Chongqing, China); PA (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China); egg yolk lecithin PC-98T (AVT (Shanghai) Pharmaceutical Technology Co., Ltd., Shanghai, China); hydrogenated soybean phosphatidylcholine (HSPC; Lipoid GmbH, Germany); endothelial progenitor cells (EPCS; Lipoid GmbH, Germany); cholesterol (AVT (Shanghai) Pharmaceutical Technology Co., Ltd. Shanghai, China); 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000; Lipoid GmbH, Germany); 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC), 4-dimethylaminopyridine (DMAP, ethyl acetate, petroleum ether, and dichloromethane (Shanghai Pharmaceutical Chemical Reagent Co., Ltd., Shanghai, China); GF 254 silica gel plate for thin layer analysis (Ocean Chemical, Qingdao, China); isopropanol (chromatographically pure), methanol (HPLC grade), and acetonitrile (HPLC grade) (Thermo Fisher (China) Co., Ltd., Shanghai, China). All other chemical reagents were of analytical or chromatographic grade. Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and phosphate-buffered saline (PBS) were purchased from Gibco Life Technologies (Grand Island, NY, USA). The lipophilic near-infrared dye DiR was acquired from Maokang Biotechnology (Shanghai, China).
Taxol for injection was purchased from Yangzijiang Pharmaceutical Group Co., Ltd. For use, it was combined with normal saline (NS), prepared on the same day, and used within 1 h of preparation.
Animals
Female ICR mice (aged 4 weeks, 18–22 g), female BALB/c nude mice (aged 4–6 weeks, 16–20 g), and male and female SD rats (aged 5–6 weeks, 160–200 g) (Zhejiang Weitong Lihua Experimental Animal Technology Co., LTD, Zhejiang, China, SCXK (Zhe) 2019–0001) were raised at 25±2°C with a relative humidity of 47.5±2.5%. This study has been reviewed and approved by the Experimental Animal Ethics Committee of Shanghai Skin Disease Hospital (SYXK (Hu) 2020–0002). All animal experiments were conducted in accordance with the ethical standards described in the Guide for the Care and Use of Laboratory Animals issued by the NIH.
Cell Culture
Mouse 4T1 cells and Lewis cells (Stem Cell Bank of the Chinese Academy of Sciences, Beijing, China) were monolayer cultured adherently in an incubator containing 10% FBS (Gibco, USA) in 37.5% CO2. Mouse primary macrophages and T-cell immunodeficiency (TID) model mouse primary macrophages were extracted from the abdominal cavity and legs of ICR mice and TID model mice.
Synthesis of a PTX-PA Conjugate
PTX-PA was obtained by esterification of PTX with PA.29,30 The synthetic route is shown in Supplementary Figure 1A. Briefly, 1 mmol of PTX was added to the reaction vessel, followed by the addition of an appropriate amount of anhydrous dichloromethane, the acid-binding agent DMAP (1.2 mmol), EDC (1.2 mmol) and PA (1.2 mmol), and the mixture was stirred overnight in an ice bath under the protection of N2 until the reaction was complete. The reaction process was monitored by thin-layer chromatography. The reaction solution was washed twice with citric acid aqueous solution and twice with saturated brine, and the organic phase was rotary evaporated to remove dichloromethane to obtain the white solid target product PTX-PA, whose purity was determined by HPLC. The crude reaction product was separated and purified by column chromatography to obtain PTX-PA. In addition, the structure of the prodrug was verified by NMR hydrogen, carbon, and ESI mass spectrometry.
Preparation of PTX-PA-Loaded Nanoliposomes
PTX-PA liposomes (PTX-PA-L) were prepared by using the thin film hydration method.31 Briefly, 20 mg of PTX-PA, 400 mg of PC98-T, and 30 mg of DSPE-PEG2000 were precisely weighed into a 500 mL round-bottomed flask and fully dissolved by the addition of 5 mL of dichloromethane. Dichloromethane was subsequently removed at 45°C to form a uniform film on the inner wall of the flask. After the addition of 10 mL of redistilled water, the mixture was preheated to the same temperature in a round-bottomed flask, shaken, and hydrated to obtain crude PTX-PA-L, which was further downsized by probe ultrasonication (Ningbo Xinzhi Biotechnology Co., Ltd., JY92-IIN, Zhejiang, China) for 90s and filtered through a 0.22-μm microporous membrane to obtain PTX-PA-L.31
Characterization of PTX-PA-L
After staining with phosphotungstic acid, the morphology of PTX-PA-L was observed via high-resolution transmission electron microscopy (TEM; Japan Electronics Co., Ltd., JEM-1400, Japan). Briefly, after 50-fold dilution with PBS (pH 7.4), 2–3 drops of PTX-PA-L were coated on the surface of a copper mesh covered with carbon film and negatively stained with 1% phosphotungstic acid for 3 min. The samples were allowed to dry naturally, and the morphology was observed and recorded.31 After 10-fold dilution with distilled water, the particle size, polydispersity index (PDI), and zeta potential of PTX-PA-L were measured with a Malvern particle size analyzer (Malvern Panalytical, NANO-S, Britain).
The encapsulation efficiency (EE%) and drug loading (DL%) of PTX-PA-L were determined by an ultrafiltration-centrifugation method. Two hundred microliters of the liposome dispersion were accurately measured in an ultrafiltration tube (Millipore, MW cutoff 30 kDa, USA) and centrifuged at 4000 r/min for 10 min (Hunan Kecheng Instrument Equipment Co., Ltd., H3-20KR, Hunan, China). The filtrate was collected to determine the content of the unencapsulated free drug (Wfree) by HPLC. Another 500 μL of PTX-PA-L was completely destroyed by methanol to determine the total content (Wtotal) of PTX-PA. The lyophilized product of PTX-PA-L (1 mL) was weighed (WT). The EE% and DL% were calculated according to the following formulas:
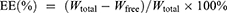
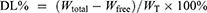
Additionally, any changes in the particle size, PDI, or drug content were detected following storage at 25°C for two weeks or dilution with 0.9% NaCl solution to evaluate the storage or antidilution stability, each experiment was done in triplicate in parallel.
In vitro Drug Release
The release kinetics of PTX-PA-L were quantitatively detected by drug release experiments. PTX-PA-L was diluted 2 times for use. In addition, 3 parts of PBS (containing 30% ethanol, pH 5.5) solution was added to the dissolving cup, and the stirring speed of the dissolution tester (Shenzhen Huasong Analytical Instrument Co., Ltd., DS-808, Shenzhen, China) was set at 100 rpm, and the solution temperature was 37±1°C. One milliliter of diluted liposome solution was added to PBS, and the mixture was stirred. Quantitative samples were collected at each predetermined time point, and an equal amount of fresh release medium was added. After filtration with a 0.22 μm membrane filter, the supernatant was collected by ultrafiltration and centrifugation. The drug concentration was determined by HPLC, and the cumulative release of PTX-PA was calculated.
In vivo Antitumor Effect on Xenograft Tumor Models
Thirty ICR mice and BALB / c nude mice were taken to vaccinate 4T1 cells in the right axilla to establish the mouse and nude mouse breast cancer model.32,33 When the tumor volume reached 100 mm3, the mice and nude mice were randomly divided into 3 groups of 10 mice each for intravenous injection (i.v.). The three groups were injected with NS, Taxol®(15 mg/kg PTX equivalent) or PTX-PA-L (19.18 mg/kg, 15 mg/kg PTX equivalent) every 2 days for a total of 3 times. The tumor volume (TV) and body weight of the tumor-bearing animals were monitored during the treatment period. On the third day after the end of treatment, the tumors were dissected and weighed. The drug efficacy of each treatment group was evaluated by calculating the tumor inhibition rate (TIR). Tumor volume was calculated according to equation (1) (a × b2)/2, where “a” and “b” represent the long and short diameters of the tumor, respectively. The calculation formula for the TIR was (2) [Wcontrol − Wtreat]/Wcontrol × 100%, where Wcontrol and Wtreat represents the average weight of tumors in the control group and the treatment group.32
In vivo Antitumor Study in a TID Model
A mouse model of TID was established by intraperitoneal injection of the immunosuppressive agent cyclosporin A (CsA) into ICR mice aged 4–5 weeks. CsA was dissolved in DMSO and then diluted to 0.2 mg/mL CsA in normal saline containing 20% sulfobutyl ether-β-cyclodextrin. Briefly, CsA solution was injected intraperitoneally at a dose of 25 mg/kg/d once every other day for a total of three doses. The activities of the mice, such as loss of appetite, unresponsiveness, loss of hair luster and partial hair loss, were regularly observed. A certain number of representative mice were selected for spleen inspection, and the percentages of peripheral CD3+, CD4+ and CD8+ T-cell subsets were determined to assess whether the model was successfully established. In this study, 4T1 cells and Lewis cells were subcutaneously seeded in the right axilla of ICR mice and TID model mice, with 30 animals per model group for a total of four groups.
When the tumor volume reached 100 mm3, mice from each model group were again randomly divided into 3 subgroups of 10 mice each for i.v. The three subgroups were injected with NS, Taxol (15 mg/kg PTX equivalent) or PTX-PA-L (19.18 mg/kg, 15 mg/kg PTX equivalent) every 2 days for a total of 3 times. Changes in tumor growth and body weight of the tumor-bearing mice were monitored during the treatment period. On the third day after the end of treatment, the tumor tissues and main organs were dissected, and the TIR (%) was calculated utilizing the same methods as those used for the evaluation of antitumor efficacy. Representative tumor samples were collected from each group for tissue analysis. For biological analysis, tumor tissues were stained with hematoxylin and eosin (H&E) and subjected to immunofluorescence analysis with a Ki67 detection kit.34
In vivo Pharmacokinetics and Tissue Distribution
Pharmacokinetics of different forms of PTX-PA, including all forms PTX-PA (T-PTX-PA), liposome-loaded PTX-PA (L-PTX-PA), and free PTX-PA in plasma, were studied in healthy SD rats. Six rats, half male and half female, were intravenously infused with PTX-PA-L (10 mg/kg, 7.8 mg/kg PTX equivalent) (5 minutes per rat, rats were fasted for 12h before administration and drinking water ad libitum). Plasma T-PTX-PA and L-PTX-PA were measured at 0, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 72, 120, and 168 h post-administration.
The pharmacokinetics were also studied in 4T1 cell-xenografted ICR mice and BALB/c nude mice models as described above. When the tumor volume reached approximately 500 mm3, each group of mice and nude mice was randomly divided into two subgroups (30 animals in each subgroup) and injected i.v with Taxol® (15 mg/kg PTX equivalent) or PTX-PA-L (19.18 mg/kg, 15 mg/kg PTX equivalent), mice were fasted for 12h before administration and drinking water ad libitum.35 After administration, blood from mouse eyeballs was extracted and placed into anticoagulant tubes at 0.1, 0.5, 1, 5, 12, 24, 72, 120, 168, and 336 h, and plasma was collected by centrifugation. Then, the mice were sacrificed, and the heart, liver, spleen, lung, kidney, and tumor tissues were dissected, individually weighed and ground into tissue homogenates.36,37 After that, the internal standard (IS) carbamazepine (10 ng/mL) was added to both the homogenate and plasma samples. After vortexing, the homogenates were extracted with acetonitrile. Following centrifugation at 13,000 rpm for 10 min, 150 μL of the supernatant was collected and mixed with 300 μL of 30% acetonitrile.38,39 Finally, 5 μL of the sample was injected into the HPLC‒MS/MS system for detection.40 The combination of full scan and QED scan was used to monitor PTX, PTX-PA and carbamazepine in MRM mode with m/z 308.1 → 876.32, 546.3 → 1114.5, and 237.1 → 194.0, respectively. The run time was 7 min, and the data were collected in positive polarity mode.
In vivo and ex vivo Imaging
In vivo and ex vivo imaging was performed on 4T1 xenografted mice and nude mouse tumor models. DiR was dissolved in DMSO to prepare a 1 mm stock solution of DiR, which was then diluted to 1 μm with normal saline to prepare free DiR for later use. To prepare DiR (cellular membrane dark red fluorescent probe)-labeled PTX-PA-L (DiR@PTX-PA-L), DIR was added to the oil phase during liposome preparation, and the amount of other raw materials and preparation process were consistent with those used for PTX-PA-L. Then, DiR was loaded into the phospholipid bilayer, and the liposomes were completely labeled. Mice and nude mice were randomly divided into three groups (n = 30) and injected i.v. with free DiR-labeled blank liposomes (DiR@Blank Lip) or DiR@PTX-PA-L (19.18 mg/kg, 15 mg/kg PTX equivalent). Immediately after the animals were anesthetized with isoflurane at selected time points, fluorescence images of the whole body were captured at excitation/emission wavelengths of 748/780 nm using an IVIS Spectrum in vivo imaging system (PerkinElmer, USA). Subsequently, the corresponding mice and nude mice were sacrificed to collect the main organs and tumor tissues, and the fluorescence intensity of each organ and tumor tissue was quantitatively analyzed.
In vitro Uptake of PTX-PA-L by Macrophages
Macrophages were extracted from the peritoneal cavity of ICR mice and TID model mice. Cells were seeded on 6-well plates, and 3 complex wells for each group were incubated for 2 hours in an incubator with 5% CO2 saturation at 37°C. DiR@PTX-PA-L was prepared by loading lipophilic DiR into prodrug liposomes using DiR as a cell membrane fluorescent probe. After the supernatant was discarded and the cells were washed with PBS three times, DiR@PTX-PA-L was added to the media, and the cells were collected at 0.5, 1, and 24 h and analyzed by flow cytometry utilizing FlowJo software for data analysis. In the same way, the cells were inoculated into 6-well plates for 24 h and incubated with PTX-PA-L loaded with DiR at 37°C for 2 h. Nuclear restaining was performed with 4ʹ,6-diamidino-2-phenylindole (DAPI), the cells were collected and fixed with 4% paraformaldehyde solution, and then the uptake of PTX-PA-L by the macrophages was observed using confocal laser scanning microscopy (Nikon, Japan).41 To quantify the uptake of PTX-PA-L by macrophages derived from ICR mice and TID mice, HPLC-MS/MS (Thermo Fisher, USA) was used for precise quantification of differences in drug uptake by macrophages from the two sources.
The contents of PTX and PTX-PA in the macrophages of the mice and nude mice were detected by HPLC-MS/MS (Thermo Fisher, USA). After washing with PBS, PTX-PA-L (100 ng/mL−1 PTX equivalent) was added to the culture dishes of the two types of macrophages and cocultured for 6, 8 or 10 h. After treatment, live cells were collected, counted, resuspended in 250 μL of ultrapure water, and lysed by repeated freeze‒thaw cycles. The cell lysate was further processed, and then 10 μL of IS carbamazepine (10 ng/mL) was added to the cell lysate dispersion. After mixing, the solution was extracted with acetonitrile and centrifuged at 13000 rpm for 10 min. The intracellular drug content was detected by injecting 5 μL of the supernatant into the HPLC‒MS‒MS system.
In vivo Uptake of PTX-PA-L by Tumor-Associated Macrophages (TAMs)
The in vivo uptake of PTX-PA-L by TAMs in mice and nude mice was investigated (n = 3). DiR@PTX-PA-L was injected i.v. at a dose of 19.18 mg/kg (15 mg/kg PTX equivalent). Then, the tumor-bearing mice and nude mice were sacrificed, and the tumors were dissected, snap-frozen, and sectioned. Tumor sections were incubated overnight with rabbit anti-mouse F4/80 antibody (Abcam, Cambridge, MA), stained with fluorescein isothiocyanate (FITC)-conjugated secondary antibody, counterstained with DAPI for nuclei, and visualized under a fluorescence microscope. The mean fluorescence intensity of colocalization of F4/80 and DiR in tumor sections was semiquantitatively calculated using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA).
Immunohistochemistry
To investigate whether the content of TAMs in tumor tissues leads to differences in antitumor efficacy, we further evaluated the expression of a characteristic TAM marker (CD68+) in tumor-bearing mice and nude mice treated with prodrug liposomes by immunohistochemistry (IHC). After antigen retrieval and inactivation, the tumor sections were incubated in normal goat serum, stained with 3,3ʹ-diaminobenzidine (DAB)/H2O2 solution, counterstained with hematoxylin and observed. Positively stained areas were measured and quantified using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD, USA). The density of TAM-positive cells is expressed as the number of cells per square millimeter (mm2).
Expression Levels of Immune Factors
The levels of chemokines (CCL2 and CCL5) and tumor factors (TNF-α, IL-1, and IL-8) in the serum of normal mice, TID model mice were detected using ELISA kits according to the manufacturer’s instructions. Sera were extracted from the blood samples of mice and nude mice by centrifugation at 400 × g for 20 min, and the corresponding ELISA kit (Nanjing Sunberga Biotechnology Co., Ltd., Nanjing, China) was used for detection and analysis (n = 3).
Biosafety Assessment
To further evaluate the safety of PTX-PA-L, a maximum tolerated dose (MTD) study was conducted by means of dose escalation (n = 6, 3 females, and 3 males). In addition, to evaluate the long-term therapeutic effect of PTX-PA-L on tumor-bearing mice, ICR mice and Balb/c nude mice were vaccinated with 4T1 cells. After grouping, the therapeutic dose of the drug was given three consecutive times, and the animals were sacrificed after natural death or less than 60 days (animals was considered dead when tumor diameter was >20mm). The survival of each tumor-bearing mice was recorded and analyzed by Kaplan-Meier method. After the experiments, the main organs of representative mice were dissected and weighed.42
Statistical Analysis
The data are expressed as the means ± standard deviations. All analyses were performed using IBM SPSS Statistics for Windows, version 19 (IBM Corp., Armonk, N.Y., USA), and t tests and one-way ANOVA were used for comparisons between two or more groups. *P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences.
Results and Discussion
Preparation and Characterization of PTX-PA-L
The synthesis of PTX-PA was successful, as verified by mass spectrometry and hydrogen NMR spectroscopy, with a yield of 70% (Supplementary Figure 1B–D). PTX-PA-L was prepared by a thin-film hydration method. Dynamic light scattering revealed that PTX-PA-L was a uniform particle with a mean size of 78.0 ± 2.9 nm and a PDI of 0.176 ± 0. 02 and a zeta potential of –18.1 ± 0.21 mV (Figure 1A and B). TEM revealed the typical spherical shape of the liposomes (Figure 1C). In addition, the prodrug PTX-PA showed excellent encapsulation efficiency in the lipid bilayer, with an EE of 98.28 ± 0.69% (Supplementary Table 1) and a corresponding drug loading capacity of 7.24 ± 0.053 (Supplementary Table 2).
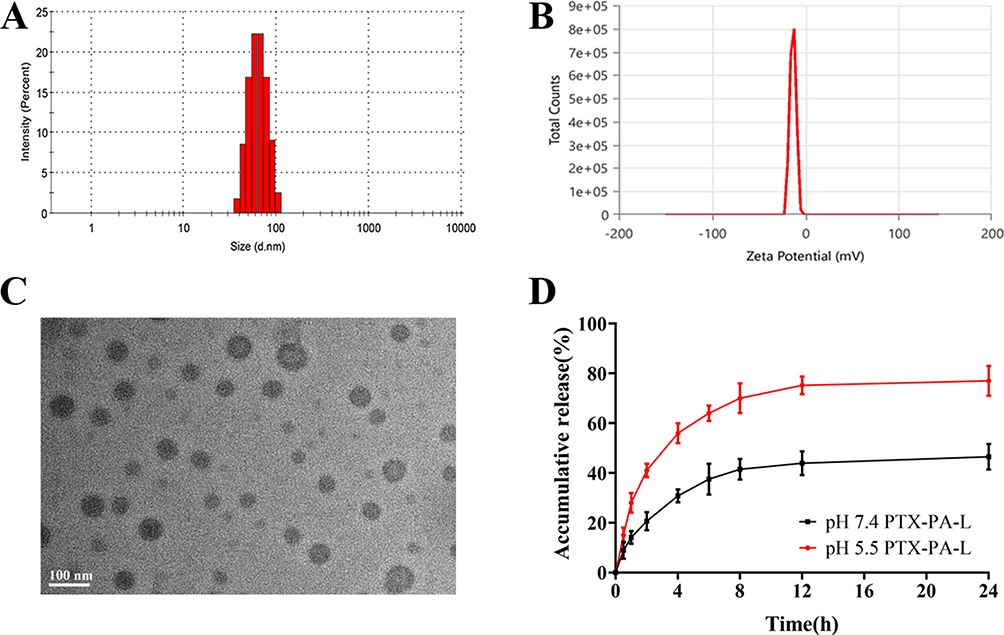 |
Figure 1 (A) Particle size of PTX-PA-L. (B) Zeta potential of PTX-PA-L. (C) TEM image of PTX-PA-L. (D) PTX-PA release profiles from PTX-PA-L in PBS (containing 30% ethanol). The scale bar is 100 nm. The data are shown as the means ± SDs (n = 3). |
PTX-PA-L showed good stability without any signs of aggregation or precipitation after storage at 25°C for at least two weeks or after dilution with 0.9% NaCl solution followed by incubation for 48 h (Figure 2A–C). There was also no significant change in the PDI. The results showed that PTX-PA-L had good stability in vitro, and PTX-PA had good compatibility with the phospholipid bilayer, showing good membrane stability.43,44 The release kinetics of PTX-PA-L were tested in a mixed medium of PBS containing 30% ethanol at pH 5.5 and pH 7.4. Approximately 80% of PTX-PA-L was released in an acidic environment (pH 5.5) within 24 h, and approximately 40% was released in a neutral environment (Figure 1D), indicating that PTX-PA-L has good stability in a neutral environment, which will increase the integrity of liposomes in blood and release more of the active drug PTX when it is transported to more acidic tumor sites.
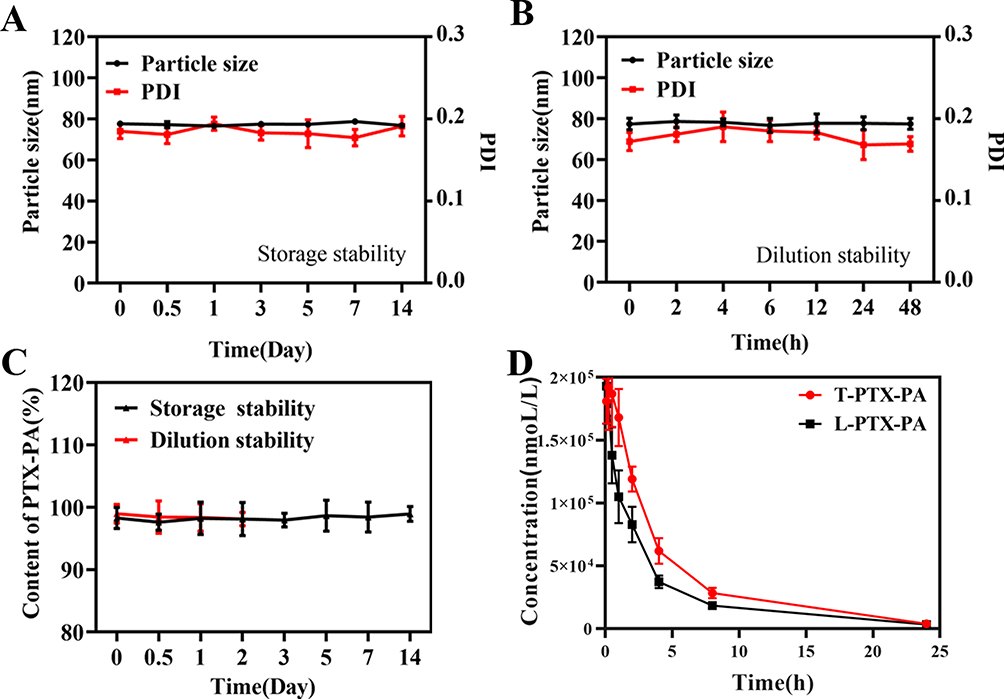 |
Figure 2 (A) Particle size and PDI of PTX-PA-L under 25°C storage conditions (n = 3). (B) Particle size and PDI after 0.9% NaCl dilution of PTX-PA-L (n = 3). (C) The change in the drug content of PTX-PA-L with 25°C storage and 0.9% NaCl dilution (n = 3). (D) Blood drug concentration-time curve of SD rats within 24 h after a single intravenous infusion of 10 mg/kg PTX-PA-L (n = 6). |
In vivo Stability of PTX-PA-L
The in vivo stability of PTX-PA-L was evaluated in SD rats. After intravenous infusion at a dose of 10 mg/kg (7.8 mg/kg PTX equivalent), the plasma L-PTX-PA AUC0–168 h level accounted for 69.72% of the T-PTX-PA. In the first 4 h after administration, more than 95% of the PTX-PA was present in an encapsulated form. This can be clearly seen from the plasma profiles of T-PTX-PA and L-PTX-PA within 24 h. After entering the body, PTX-PA-L was mainly distributed in the form of L-PTX-PA (Figure 2D). After the synthesis of PTX-PA, PTX can increase the stability of PTX-PA-L in plasma by improving lipid solubility, increasing the circulation time of liposomes, and increasing the amount of drug entering the tumor site in the form of liposomes.
Efficacy in a Xenograft Mouse Tumor Model
The in vivo antitumor efficacy of PTX-PA-L was evaluated by establishing a tumor model in which mice were subcutaneously transplanted with 4T1 cells (Figure 3A). The animals were treated with Taxol® (15 mg/kg PTX equivalent) and PTX-PA-L (19.18 mg/kg, 15 mg/kg PTX equivalent). Compared with Taxol®, PTX-PA-L significantly inhibited tumor growth in the 4T1 mouse model (Figure 3B–E). Notably, tumor regression was observed in the mice in the PTX-PA-L group from day 6 of treatment, and the tumors were completely cured in 3 of the 10 mice at the end of the treatment. PTX-PA has no pharmacological activity and needs to be metabolized into PTX to exert its curative effect. Therefore, we conducted a pharmacokinetic study to detect the content of PTX in tumor tissue by using HPLC-MS/MS.45
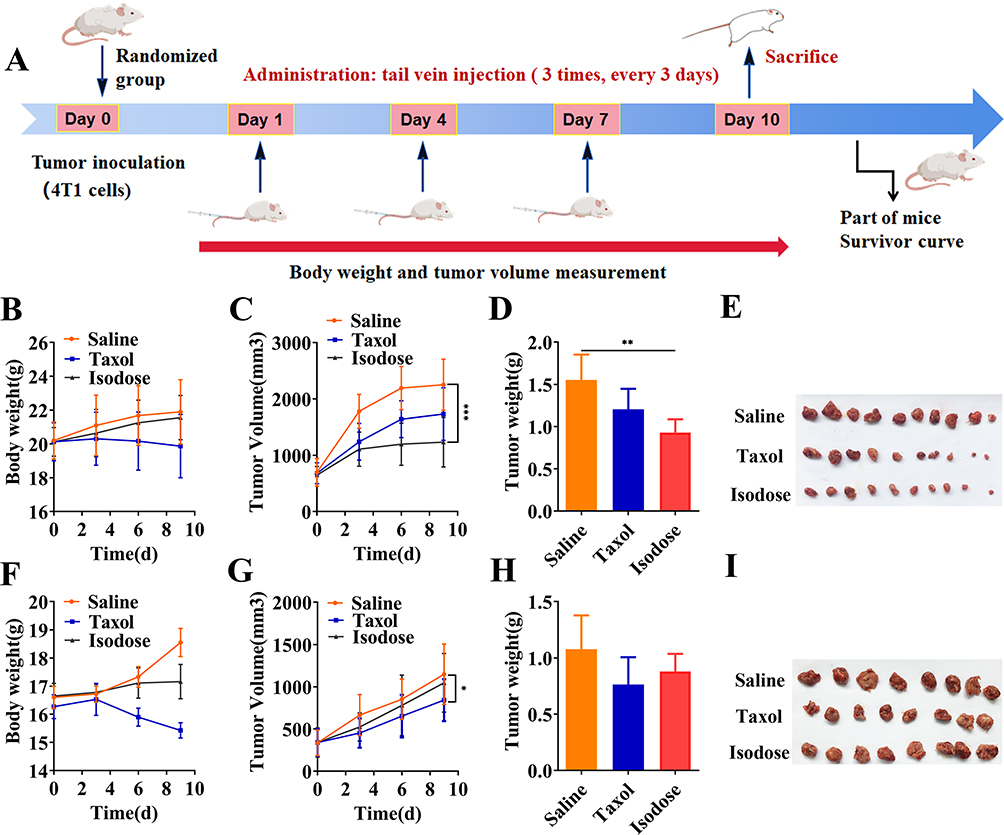 |
Figure 3 Therapeutic efficacy in vivo. Isodose refers to PTX-PA-L (Taxol-equivalent dose). (A) Schematic representation of the animal experiments and treatment process. (B) Weight curves of 4T1 tumor-bearing mice during the experiment. (C) Tumor growth curves of 4T1 tumor-bearing mice during the experiment. (D) Weights of tumors removed from 4T1 tumor-bearing mice subjected to the indicated treatments. (E) Representative images of solid tumors harvested from 4T1 tumor-bearing mice on day 10. (F) 4T1 tumor-bearing nude mouse weight curves during the experiment. (G) Growth curves of 4T1 tumor-bearing nude mice during the experiment. (H) Weights of tumors removed from 4T1 tumor-bearing nude mice for the indicated treatments. (I) Representative images of 4T1 tumor-bearing nude mouse solid tumors harvested on day 10. The data are presented as the mean ± SD (n = 10).*P < 0.05; **P < 0.01; ***P < 0.001. |
The results showed that when the drug was injected directly into the mice, the area under the curve (AUC) of plasma PTX in the PTX-PA-L group was 17 times greater than that in the Taxol group (706.53 vs 41.56 μg‧h/mL) (Table 1 and Figure 4A). Following i.v. administration, only a small portion of PTX actually reached the tumor site in the Taxol group, and more PTX was found to be distributed to other tissues and organs (Figure 4A). The AUC of PTX in tumors in the PTX-PA-L group was three times greater than that in the Taxol® group (44.81 vs 14.80 μg‧h/mL) (Table 1 and Figure 4D). Over time, PTX-PA-L gradually became enriched at the tumor site and released the prodrug PTX-PA, and PTX-PA was effectively converted into the active parent drug PTX in the tumor tissue (Figure 4C). PTX could still be detected 14 days after administration at the tumor site (Figure 5A–C), which is consistent with the significant efficacy of PTX-PA-L in a mouse tumor model. It was reported that the modification of PTX-PA-L surfaces by PEGylation could prolong the retention half-life of liposomes in blood circulation.46 Through the high permeability and retention (EPR) effect of solid tumors, they can effectively accumulate in the tumor site after entering the body.47,48 In addition, the rigidity of the liposome membrane was enhanced by the prodrug PTX-PA, which could effectively reduce the influence of plasma protein and blood flow shear force during the circulation process, thus preventing drug leakage and increasing the drug concentration at the tumor site. In summary, PTX-PA-L exhibited the advantages of long circulation, high enrichment in tumor tissues and efficient conversion into PTX to exert its therapeutic activity.
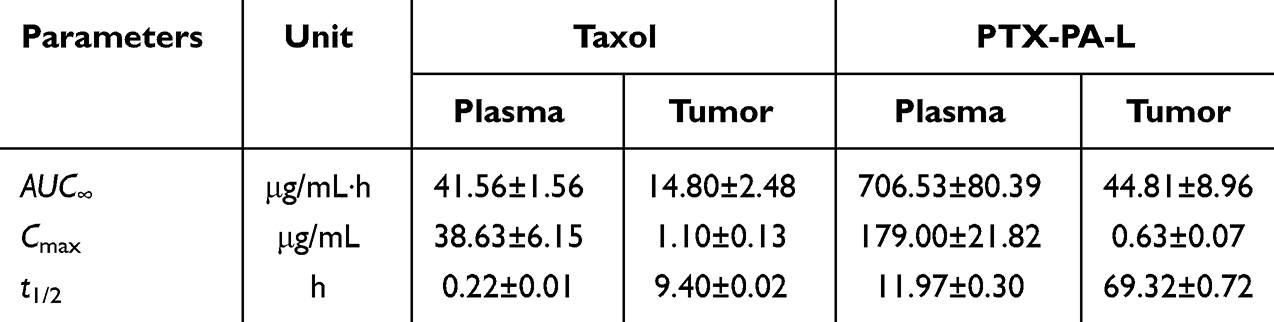 |
Table 1 Pharmacokinetic Parameters of PTX in Mouse Plasma and Tumors After Treatment with Taxol and PTX-PA-L |
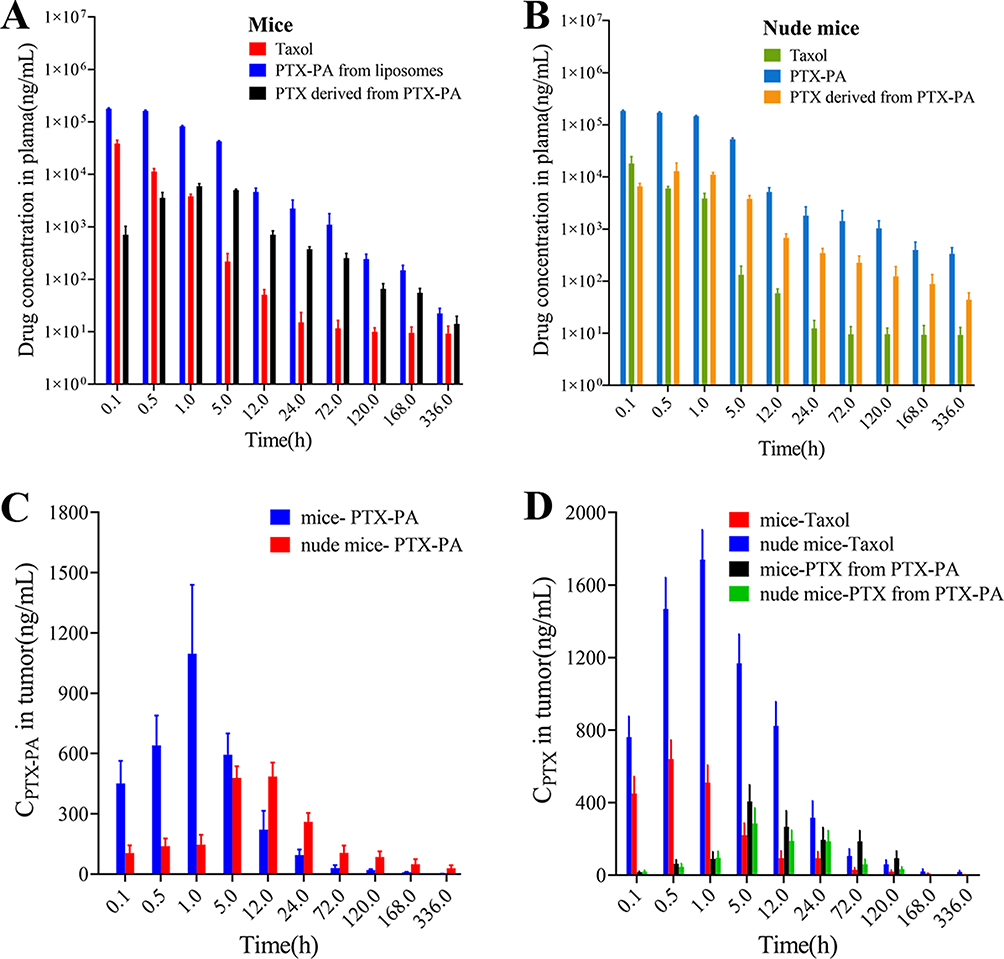 |
Figure 4 (A) Concentrations of PTX and PTX-PA in the plasma of mice treated with i.v. Taxol and i.v. PTX-PA-L. (B) Concentrations of PTX and PTX-PA in the plasma of nude mice treated with i.v. Taxol and i.v. PTX-PA-L. (C) Concentrations of PTX-PA in the tumors of mice and nude mice treated with i.v. PTX-PA-L. (D) Concentrations of PTX in the tumors of mice and nude mice treated with i.v. Taxol and i.v. PTX-PA-L.The data are presented as the mean ± SD (n = 3). |
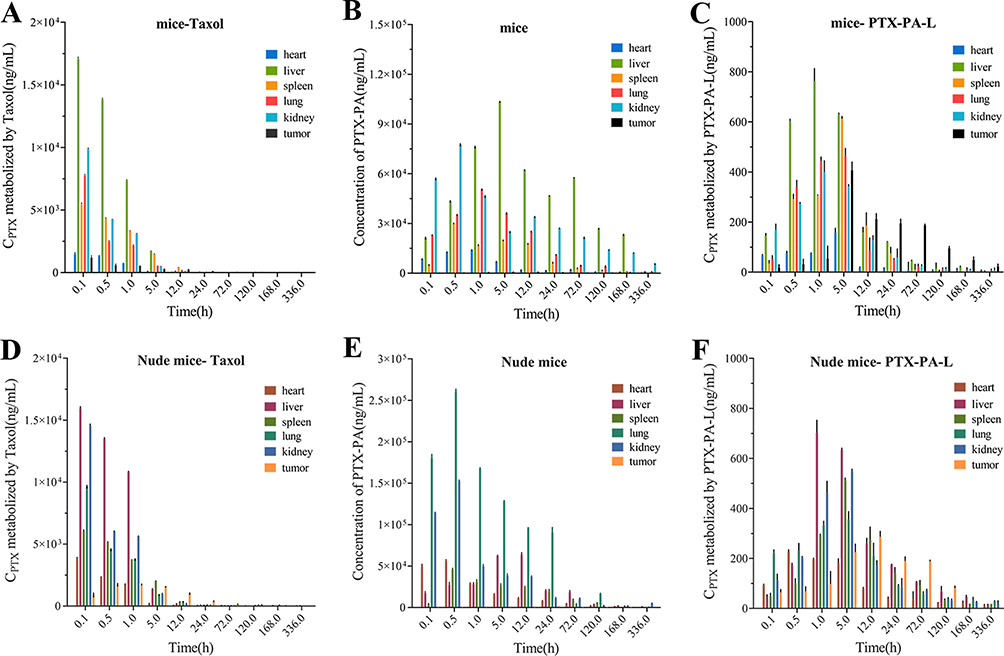 |
Figure 5 (A) Concentrations of PTX in the tissue of mice treated with i.v. Taxol. (B) Concentrations of PTX-PA in the tissue of mice injected with i.v. PTX-PA-L. (C) Concentrations of PTX in the tissue of mice injected with i.v. PTX-PA-L. (D) Concentrations of PTX in the tissue of nude mice injected with i.v. Taxol. (E) Concentrations of PTX-PA in the tissue of nude mice injected with i.v. PTX-PA-L. (F) Concentrations of PTX in the tissue of nude mice injected with i.v. PTX-PA-L. The lower limit of detection was 5 ng/mL. The data are presented as the mean ± SD (n = 3). |
Efficacy in a Xenografted Nude Mouse Tumor Model
To further explore the in vivo antitumor efficacy of PTX-PA-L, tumor models were successfully established by injecting 4T1 cells into nude mice subcutaneously. Surprisingly, the tumor inhibitory effect of PTX-PA-L (19. 18 mg/kg) was significantly inferior to that of Taxol® (15 mg/kg) (Figure 3F–I). To explore the underlying mechanisms, in vivo pharmacokinetics and tissue distribution were evaluated in a 4T1 breast cancer nude mouse tumor model (Figure 4B and D 5D–F). The AUC of plasma PTX in the PTX-PA-L group was approximately 29 times greater than that in the Taxol® group (1095.13 vs 37.62 μg‧h/mL), but the AUC of PTX at the tumor site in the PTX-PA-L group was only 0.8 times greater than that in the Taxol® group (41.08 vs 51.35 μg‧h/mL) (Table 2), indicating that the PTX concentration metabolized at the tumor site in nude mice was lower in the PTX-PA-L group.
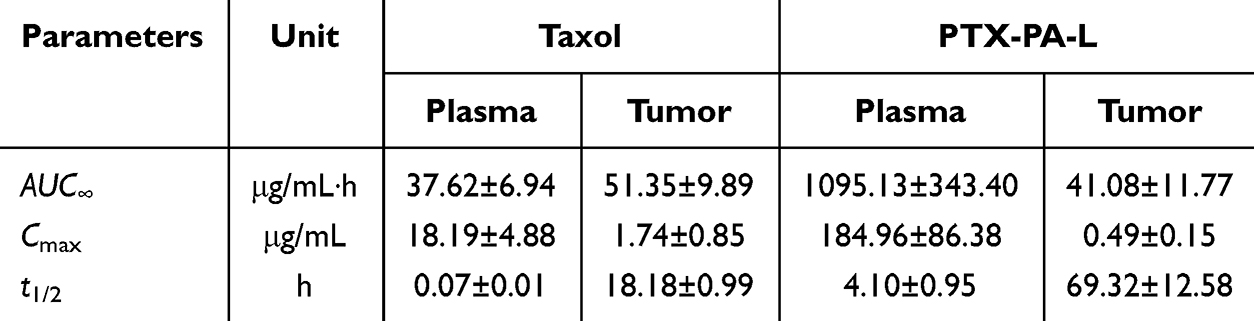 |
Table 2 Pharmacokinetic Parameters of PTX in the Plasma and Tumors of Nude Mice Treated with Taxol and PTX-PA-L |
In addition, we compared the pharmacokinetic parameters of PTX-PA in plasma and tumors of mice and nude mice, and found that the area under the drug-time curve of PTX-PA in mice plasma was smaller than that in nude mice plasma (740.43 vs 1047.52 μg‧h/mL) (Table 3). Surprisingly, the area under the drug-time curve of PTX-PA in the tumor site of the mice was greater than that in the tumor site of the nude mice (44.81 vs 33.36 μg‧h/mL) (Table 3), which may be related to the limited distribution and concentration of metabolic esterase in the nude mice. Esterase is involved in drug activation and is closely related to drug metabolism, playing an important role in the body. Moreover, esterase metabolism is affected not only by the drug structure but also by the metabolic pathway and time. However, differences in metabolic esterase species also affect metabolic processes. Due to the significant differences in plasma esterase activity and expression levels between mice and nude mice, the degree of ester prodrug hydrolysis is also different in the two animal models. Compared with those in nude mice, the plasma esterase activity and expression level in mice were greater, so the more active drug PTX could be hydrolyzed in vivo, thus exerting a stronger antitumor effect.
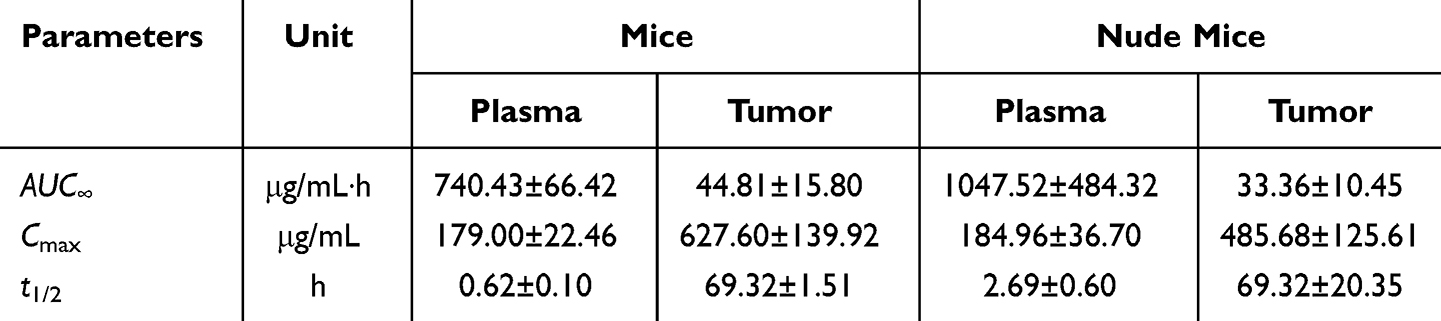 |
Table 3 Pharmacokinetic Parameters of PTX-PA in Mouse and Nude Mouse Plasma and Tumors |
In vivo Tumor Targeting of PTX-PA-L
In vivo fluorescence imaging after labeling the liposomes with DiR was carried out to further verify the in vivo stability of the prodrug liposomes.49,50 The distribution of DiR@PTX-PA-L and DiR@Blank Lip, with free DiR as a control, after i.v. injection was monitored using an in vivo imaging system. Compared with free DiR, DiR@Blank Lip and DiR@PTX-PA-L had significantly greater circulation stability and better tumor targeting efficiency (Figure 6A).
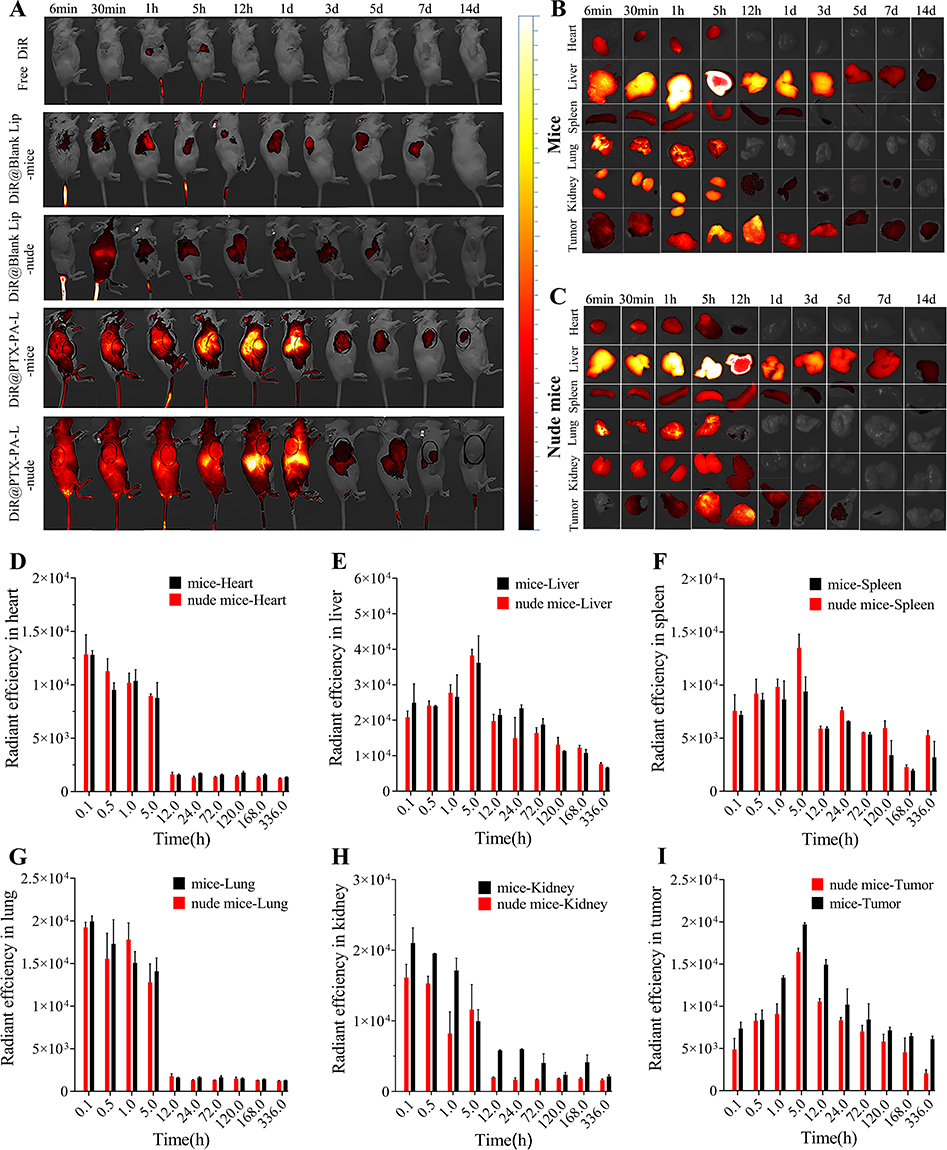 |
Figure 6 In vitro imaging and distribution of PTX-PA-L-formulated DiR in 4T1 tumor-bearing mice and nude mice. (A) In vivo fluorescence imaging of 4T1 tumor-bearing mice and nude mice at 0.1 h, 0.5 h, 1 h, 5 h, 12 h, 24 h, 72 h, 120 h, 168 h and 336 h after IV injection of free DiR, DiR-loaded blank liposomes, or DiR-loaded PTX-PA-L via the tail vein. DiR dissolved in DMSO and DiR-loaded blank liposomes were included as a reference. The black dotted circles indicate tumor regions. (B) In vitro imaging of 4T1 tumor-bearing mice at 6 min, 30 min, 1 h, 5 h, 12 h, 1 d, 3 d, 5 d, 7 d and 14 d after IV injection of DiR-loaded PTX-PA-L via the tail vein. (C) In vitro imaging of 4T1 tumor-bearing nude mice at 6 min, 30 min, 1 h, 5 h, 12 h, 1 d, 3 d, 5 d, 7 d and 14 d after IV injection of DiR-loaded PTX-PA-L via the tail vein. (D) Quantitative evaluation of the fluorescence intensity of the excised heart at different time points postadministration. (E) Quantitative evaluation of the fluorescence intensity of the excised liver at various time points postadministration. (F) Quantitative evaluation of the fluorescence intensity of the excised spleen at different time points postadministration. (G) Quantitative evaluation of the fluorescence intensity of the excised lung at various time points postadministration. (H) Quantitative evaluation of the fluorescence intensity of the excised kidney at various time points postadministration. (I) Quantitative evaluation of the fluorescence intensity of the excised tumor at various time points postadministration. The data are presented as the means ± SDs (n = 3). |
Interestingly, the fluorescence intensity of PTX-PA-L was greater than that of the blank liposomes. We speculate that the long hydrophobic chains of PTX-PA may play an important role in maintaining the structure and stability of the liposomal membrane by interspersing in the phospholipid bilayer. The entry of DiR@Blank Lip into the body induced partial rupture of the liposomal phospholipid membrane to release DiR, and the fluorescence of DiR was quenched in a water environment, attenuating the fluorescence signal. In conclusion, the introduction of PA enhanced the lipid solubility of PTX, and it acted as a membrane stabilizer to increase the stability of PTX-PA-L.
Tumor targeting capability was evaluated following i.v. administration of the liposomes. The free DiR dye control did not exhibit any fluorescence in the dissected tumors. Accumulation of fluorescence was detected in the tumors for both the DiR@Blank Lip and DiR@PTX-PA-L groups. Differently, in mice, the fluorescence of DiR@PTX-PA-L in vivo increased in a time-dependent manner, peaking at approximately 5 h, and a certain amount of fluorescence could still be detected even at 336 h after administration. In nude mice, after injection of DiR@PTX-PA-L, the localization of the fluorescence signal at the tumor site was not obvious, and the signal gradually weakened over time, and the fluorescence at the tumor site basically disappeared after 168h. To accurately verify the targeting of DiR @ PTX-PA-L at tumor sites in mice and nude mice, in vivo imaging and fluorescence quantification were performed on major organs of mice and nude mice at each time point (Figure 6B–I). The results were similar to the live imaging results, in which the mice showed strong fluorescence signal at all time points. Especially at 336h, the fluorescence intensity of the mouse tumor site was kept at a high level, about 3 times that of the mouse tumor site in nude mice. The above results showed that PTX-PA-L has better drug transport capacity and tumor targeting in mice.
Artificial Immunosuppression Induced in the Xenograft Mouse Tumor Model
By intraperitoneal injection of the immunosuppressant cyclosporin A (CsA), normal mice were immunosuppressed to establish a model similar to that of nude mice (Supplementary Figures 2 and 3). Two tumor models were established by inoculating 4T1 breast cancer cells and Lewis lung cancer cells subcutaneously into normal mice and immunosuppressed mice, respectively, to verify the reasons for the poor efficacy of PTX-PA-L in nude mice. Compared with that of the immunosuppressed mice receiving Taxol® (15 mg/kg), the tumor suppressor effect of PTX-PA-L (19.18 mg/kg) was significantly lower than that of Taxol® (Figure 7), which is consistent with the efficacy results in the nude mouse tumor model. The above results indicated that the efficacy of PTX-PA-L was different between normal tumor-bearing mice and immunodeficient tumor-bearing mice, which further demonstrated that the anti-tumor effect of PTX-PA-L may be related to the immune system.
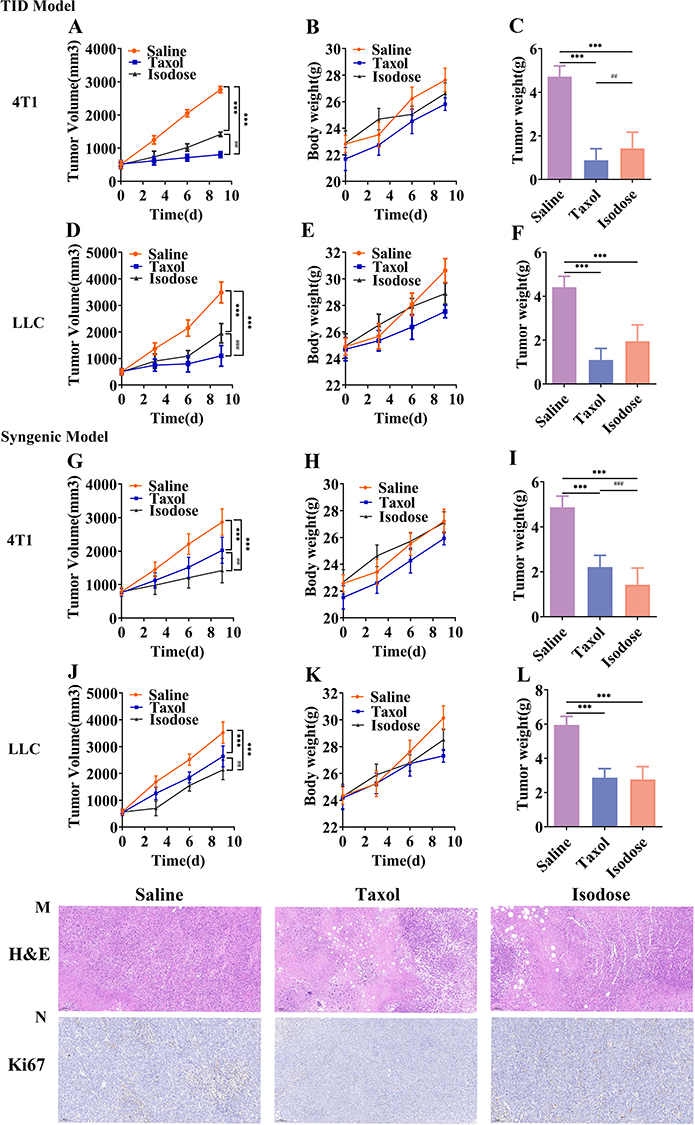 |
Figure 7 In vivo antitumor efficacy of PTX-PA-L in a TID model. Isodose refers to PTX-PA-L (Taxol-equivalent dose). (A) Tumor growth curves of 4T1 TID model tumor-bearing mice during the experiment. (B) 4T1 TID model tumor-bearing mouse weight curves during the experiment. (C) Weights of tumors removed from 4T1 TID model tumor-bearing mice subjected to the indicated treatments. (D) Growth curves of LLC TID model tumor-bearing mice during the experiment. (E) Growth curves of LLC TID model tumor-bearing mice during the experiment. (F) Weights of tumors removed from LLC TID model tumor-bearing mice subjected to the indicated treatments. (G) Growth curves of 4T1 tumor-bearing mice during the experiment. (H) 4T1 tumor-bearing mouse weight curves during the experiment. (I) Weights of tumors removed from 4T1 tumor-bearing mice subjected to the indicated treatments. (J) Growth curves of LLC tumor-bearing mice during the experiment. (K) Growth curves of LLC tumor-bearing nude mice during the experiment. (L) Weights of tumors removed from LLC tumor-bearing mice subjected to the indicated treatments. Representative tumors were excised from mice in each group and subjected to histological analysis 10 days after the first injection of various drugs. (M) Images of H&E staining. (N) IHC staining (Ki67-positive cells are stained in brown) are presented for tumors. Scale bars: 10 μm and 20 μm. The data are presented as the mean ± SD (n = 10). ***P <0.001 compared with the saline group. ##P <0.01 and ###P < 0.001 compared with the Taxol group. Abbreviations: TID, T-cell immunodeficiency; IHC, immunohistochemistry. |
In addition, the detection of related immune factors showed that the expression levels of CCL2, CCL5 and TNF-α in the PTX-PA-L group were significantly greater than those in the Taxol group before immunosuppression (p<0.01) (Figure 8A and B), but they were all reduced after immunosuppression, demonstrating that immune system deficiency reduced the chemotactic function of some immune factors.51 Since CCL2 and CCL5 promote tumor progression through the recruitment and activation of various immune cells,52,53 their high expression in cancer cells can inhibit tumor progression in mice. Before immunosuppression, immune factors, which have normal recruitment and chemotactic effects on PTX-PA-L and have a good inhibitory effect on tumor growth, are normally secreted. Insufficient chemotactic function led to insufficient inhibition of tumor growth, and PTX-PA-L was less effective than Taxol. In conclusion, the pharmacodynamic differences between mouse and nude mouse tumor models were strongly correlated with the expression levels of immune factors. In addition, a reduction in tumor factor levels can effectively reflect tumor suppressor effects. As the main immune cells in tumors, TAMs can produce a series of tumor factors, such as IL-1 and IL-8, which are crucial for maintaining immune tolerance and tumor growth.53,54 We detected the levels of the two tumor factors by ELISA and found that the expression levels of IL-1 and IL-8 in the PTX-PA-L group were significantly lower than those in the Taxol group before immunosuppression (p< 0. 01) (Figure 8A and B), and the tumors grew well at this time. However, the opposite effects were observed after immunosuppression. These results showed that the expression levels of IL-1 and IL-8 were negatively correlated with the antitumor efficacy of PTX-PA-L in vivo.51 The above results show that prodrug liposomes in normal mice can achieve overall regulation through the levels of immune factors and tumor factors secreted by immune cells and TAMs, respectively, to achieve good antitumor efficacy.55
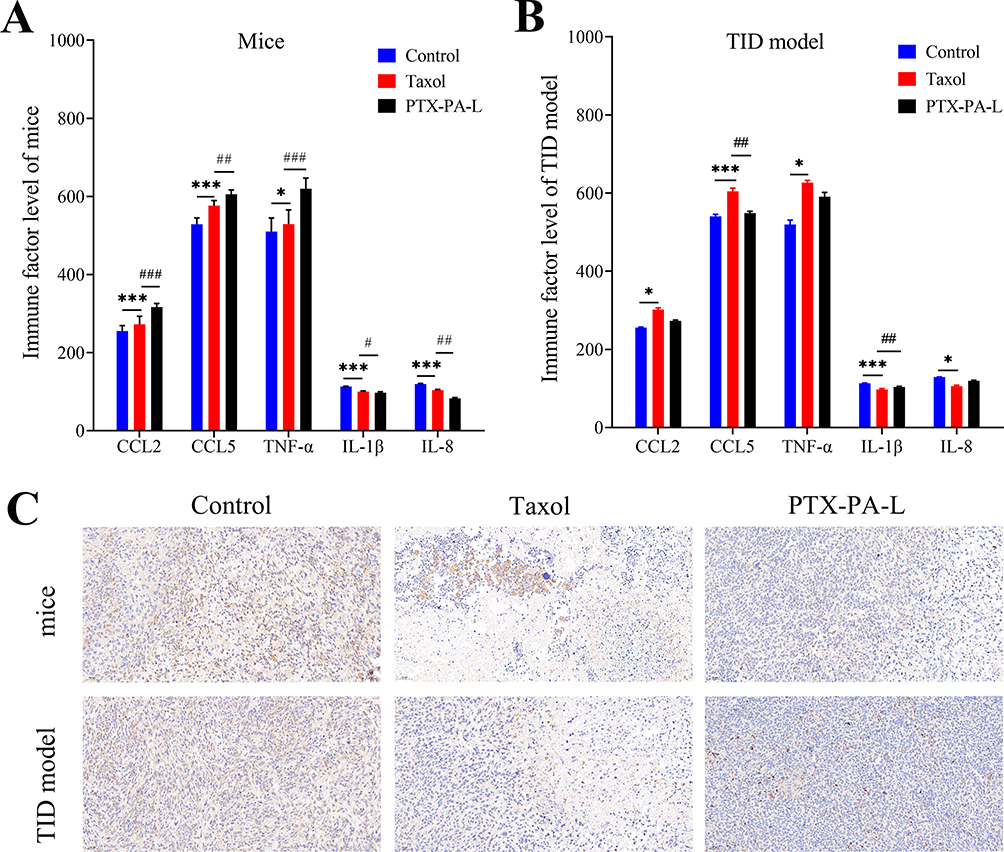 |
Figure 8 (A) Quantification of CCL2, CCL5, TNF-α, IL-1β and IL-8 levels in mice; (B) quantification of CCL2, CCL5, TNF-α, IL-1β and IL-8 levels in the TID model group; (C) Immunohistochemical staining of different experimental groups within the tumor stroma (**P < 0.01). The data are presented as the mean ± SD, n = 3. *P < 0.05 and ***P < 0.001 compared with the control group. #P <0.05, ##P <0.01 and ###P < 0.001 compared with the Taxol group. Abbreviation: TID, T-cell immunodeficiency. |
Macrophages play an important role in the immune system and play key roles in the phagocytosis and transport of liposomal drugs in the body. We cocultured macrophages with DiR@PTX-PA-L and analyzed the phagocytosis of DiR@PTX-PA-L by the macrophages by flow cytometry and laser confocal microscopy before immunosuppression. The results and confocal microscopy images of normal mouse macrophages revealed only weak fluorescence outside the macrophages (Figure 9A, B, D and E), indicating that more DiR@PTX-PA-L entered the cells, while the intracellular fluorescence intensity was obvious, indicating that normal mouse macrophages had good uptake of PTX-PA-L. To quantify the drug uptake more accurately in macrophages, we detected the phagocytic efficiency of PTX-PA-L by macrophages by HPLC-MS/MS. The results showed that PTX-PA-L had high phagocytic efficiency (Figure 9F and G). After immunosuppression, there was strong fluorescence outside the macrophages but only weak fluorescence in the cells. The intracellular drug concentration also decreased, indicating that the uptake of PTX-PA-L by macrophages significantly decreased after immunosuppression.56 Moreover, PTX-PA-L can be efficiently transported to tumor tissue in vivo before immunosuppression (Figure 9C). In summary, preimmunosuppressive macrophages have a stronger ability to phagocytose PTX-PA-L and can deliver it to the tumor site so that the prodrug liposomes can enter the body and reach the tumor microenvironment (TME) smoothly.57–59
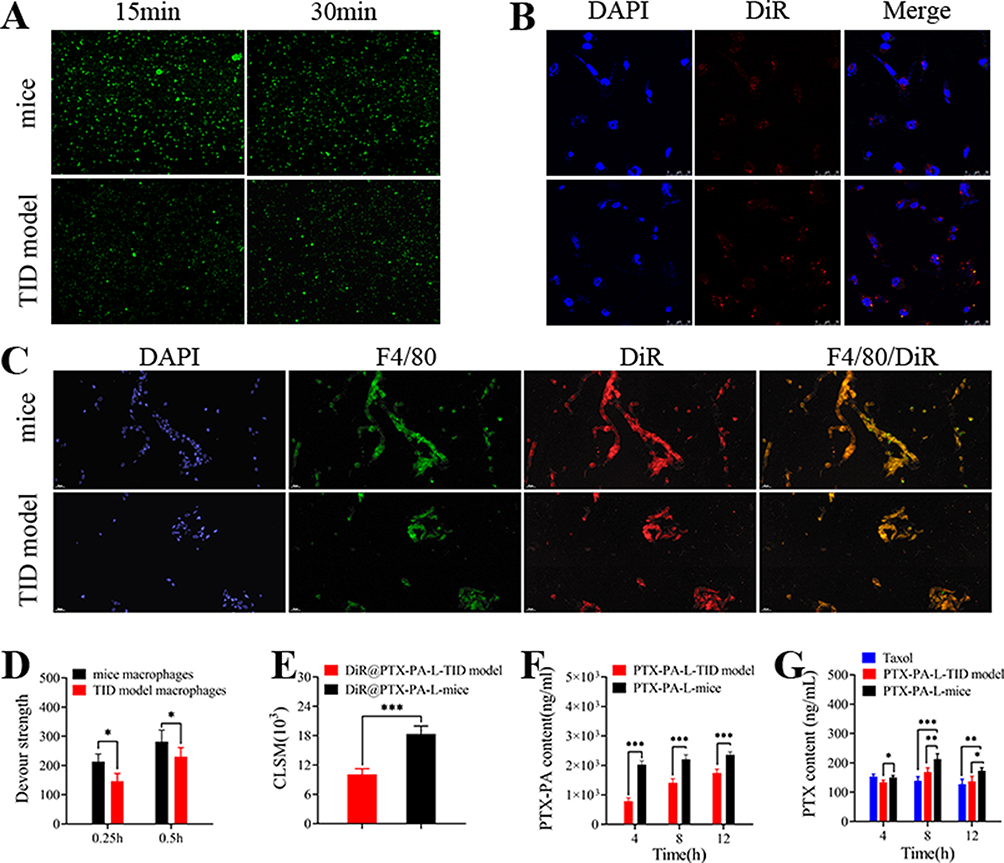 |
Figure 9 Note: Study of mouse and TID model mouse macrophage cellular uptake in vitro. (A) PTX-PA-L uptake by mouse and TID model mouse macrophages was measured by FCM. (B) PTX-PA-L uptake by mouse and TID mouse macrophages was photographed via CLSM. Blue (DAPI) represents the nucleus; red (PTX-PA-L) represents drugs. The scale bar is 10 µm. (C) Fluorescence microscopy images of tumors obtained from 4T1 tumor-bearing mice and 4T1 TID model tumor-bearing mice treated with DiR-loaded PTX-PA-L. (D) Mean fluorescence intensity of mouse and TID model mouse macrophage uptake assessed by FCM. (E) Mean fluorescence intensity of macrophages taken up by mice and TID model mice, as assessed by CLSM. (F) Measurement of the intracellular PTX-PA content at different time points in PTX-PA-L-treated mouse and TID model mouse macrophages by HPLC-MS/MS. (G) Measurement of the intracellular PTX content at different time points in macrophages from Taxol- and PTX-PA-L-treated mice and TID model mice by HPLC-MS/MS. In vivo tumor targeting and TAM uptake of DiR-loaded PTX-PA-L nanoformulations in 4T1 tumor-bearing mice and 4T1 TID model tumor-bearing mice. Scale bar, 100 µm. Each value represents the mean ± SD. (n = 3, *P < 0.05, **P < 0.01, ***P < 0.001). Abbreviations: TID, T-cell immunodeficiency; FCM, flow cytometry; CLSM, confocal laser scanning microscopy; DAPI, 4ʹ,6-diamidino-2-phenylindole; HPLC-MS/MS, High performance liquid chromatography-tandem mass spectrometry. |
The degree of depletion of TAMs in the TME is directly correlated with the antitumor effect of the drug. The in vivo TAMs of mice before and after immunosuppression were tracked by fluorescence to understand the depletion of TAMs in the respective tumors.60 The results showed that before immunosuppression, large numbers of TAMs were depleted in the tumor slices of the mice, and the number of CD68+ cells was significantly reduced, indicating that the efficacy of PTX-PA-L was better than that of Taxol before immunosuppression (Figure 8C). Since PTX-PA-L can selectively produce cytotoxicity to macrophages, induced macrophage depletion can inhibit tumor growth. After immunosuppression, the depletion of macrophages induced by PTX-PA-L to inhibit tumor growth was weakened due to the reduced cytotoxicity of PTX-PA-L to macrophages when PTX-PA-L was less effective than Taxol. In summary, the normal immune system has a better-promoting effect on the depletion of TAMs, achieves the best destruction effect on TAMs, and achieves a better tumor growth inhibition effect.61
In recent years, increasing evidence has shown that the body’s innate and acquired immune responses play important roles in conventional tumor treatment, but the screening and evaluation of anticancer drugs have always been based on models of immunodeficient mice transplanted with human tumors. Little attention has been given to the role of immunity in tumor therapy,62 especially given to the new antitumor dosage forms currently used in clinical practice. Since the immune system cannot be avoided when drugs enter the body, we need to fully consider the relationships and interactions between drugs or preparations, tumors and the immune system in clinical research.63 In clinical practice, the screening and evaluation of drugs have always been carried out by constructing tumor models in immunodeficient animals,64 which may weaken or even negate the impact of the body’s immune system on tumor treatment. Mouse tumor models and more related tumor models should also be considered.
In vivo Biosafety Evaluation of PTX-PA-L
Toxicity was evaluated in healthy mice via tail vein injection of PTX-PA-L. The MTD in the PTX-PA-L group was much greater than that in the Taxol® group (Figure 10A). The safety experiments showed that the increase in MTD proved that PTX-PA-L had good biological safety. Long-term safety results showed that organ weight did not decrease significantly after three consecutive treatments (Figure 10B), and that the survival of mice and nude mice was significantly prolonged (Figure 10C and D), indicating that animals treated with PTX-PA-L had better resistance and safety than those treated with Taxol®. When PTX-PA-L enters the body, the drug is released slowly due to its long circulation time, and a high drug concentration can be maintained at the tumor site for a long time, thereby preventing the toxic and adverse effects commonly observed with some conventional antitumor drug pulse therapies. Despite the high conversion rate of PTX-PA-L to PTX, PTX-PA-L has lower toxicity and greater safety. In addition, because the prodrug itself has the characteristics of low activity and high safety, the liposome carrier has the safety characteristics of high bioavailability, nontoxicity after injection into the body, and no immune response.65 These remarkable advantages offer great prospects for the translation of lipid prodrug technology.
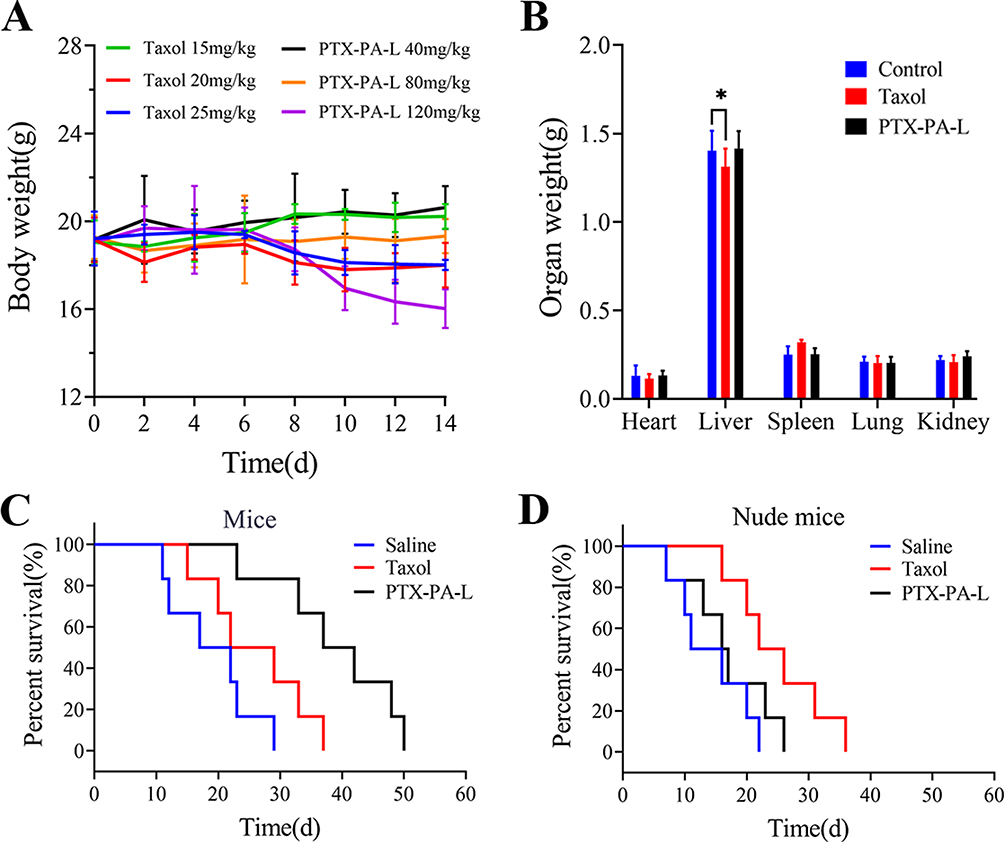 |
Figure 10 In vivo toxicity of PTX-PA-L compared with that of Taxol. Isodose refers to PTX-PA-L (Taxol-equivalent dose). (A) Body weight change of ICR mice after treatment with different doses of Taxol and PTX-PA-L (n = 6). (B) Weights of the heart, liver, spleen, lungs, and kidneys (n = 3). (C) The survival rate of mice following different treatments (n = 6). (D) The survival rate of nude mice following different treatments (n = 6). *P < 0.05 compared with the control group. |
Conclusion
The rational combination of prodrug technology with liposomes is a simple and effective therapeutic strategy for treating malignant tumors. The lipid-soluble prodrug PTX-PA encapsulated in liposomes acts as a membrane stabilizer and is interspersed in the phospholipid bilayer, which significantly improves the stability of the prodrug liposomes and prolongs the half-life in vivo. PTX can be transformed into a nano drug with low toxicity, high efficiency, and good in vivo tolerance, exhibiting broad development prospects in tumor-targeted therapy. On the one hand, the pharmacodynamic reversal in the mouse CDX and nude mouse CDX models may be due to differences in the metabolism of prodrug liposomes in vivo due to the physiological defects of the two organisms. In addition, it is speculated that a deficiency in immune function will hinder the normal function of the body’s immune system, which will limit the effects of related immune factors and the number and function of immune cells, which will reduce the ability of prodrug liposomes to be transported to tumor lesions in vivo. In addition to good stability, high safety and long circulation, the dosing period of the preparation can be adjusted, and the dosage can be increased to compensate for the difference in drug efficacy that may be caused by physiological defects.
Data Sharing Statement
Data will be available upon reasonable request.
Acknowledgments
This study was financially supported by the Shanghai Oriental Talent Youth Project (CYQN2023017), the Shanghai QingPu District Industry-University-Research Cooperative Development Funding Project (2024-07, China), and the High-level Talents of Fujian University of Chinese Medicine (X2019006-Talents, China).
Author Contributions
All authors reviewed and agreed to the version of the manuscript. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest.
References
1. Wang C, Wang Y, Wang Y, et al. Characterization, pharmacokinetics and disposition of novel nanoscale preparations of paclitaxel. Int J Pharm. 2011;414(1–2):251–259. doi:10.1016/j.ijpharm.2011.05.014
2. Chakravarthi SS, Robinson DH. Enhanced cellular association of paclitaxel delivered in chitosan-PLGA particles. Int J Pharm. 2011;409(1–2):111–120. doi:10.1016/j.ijpharm.2011.02.034
3. Akhlaghi SP, Saremi S, Ostad SN, et al. Discriminated effects of thiolated chitosan-coated pMMA paclitaxel-loaded nanoparticles on different normal and cancer cell lines. Nanomedicine. 2010;6(5):689–697. doi:10.1016/j.nano.2010.01.011
4. Seligson AL, Terry RC, Bressi JC, et al. A new prodrug of paclitaxel: synthesis of Protaxel. Anticancer Drugs. 2001;12(4):305–313. doi:10.1097/00001813-200104000-00002
5. Ellis M. Taxane-based chemotherapy for node-positive breast cancer--take-home lessons. N Engl J Med. 2010;362(22):2122–2124. doi:10.1056/NEJMe1003854
6. Kim SC, Kim DW, Shim YH, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72(1–3):191–202. doi:10.1016/S0168-3659(01)00275-9
7. Ma P, Rahima BS, Feng L, et al. 2ʹ-Behenoyl-paclitaxel conjugate containing lipid nanoparticles for the treatment of metastatic breast cancer. Cancer Lett. 2013;334(2):253–262. doi:10.1016/j.canlet.2012.08.009
8. Sledge GW, Neuberg D, Bernardo P, et al. Phase III trial of doxorubicin, paclitaxel, and the combination of doxorubicin and paclitaxel as front-line chemotherapy for metastatic breast cancer: an intergroup trial (E1193). J Clin Oncol. 2003;21(4):588–592. doi:10.1200/JCO.2003.08.013
9. Joshi N, Shanmugam T, Kaviratna A, et al. Proapoptotic lipid nanovesicles: synergism with paclitaxel in human lung adenocarcinoma A549 cells. J Control Release. 2011;156(3):413–420. doi:10.1016/j.jconrel.2011.07.025
10. Ding S, Xiong J, Lei D, et al. Recombinant nanocomposites by the clinical drugs of Abraxane(®) and Herceptin(®) as sequentially dual-targeting therapeutics for breast cancer. J Cancer. 2018;9(3):502–511. doi:10.7150/jca.22163
11. Yuan H, Guo H, Luan X, et al. Albumin nanoparticle of paclitaxel (Abraxane) decreases while taxol increases breast cancer stem cells in treatment of triple negative breast cancer. Mol Pharm. 2020;17(7):2275–2286. doi:10.1021/acs.molpharmaceut.9b01221
12. Hama M, Ishima Y, Chuang V, et al. Evidence for delivery of abraxane via a denatured-albumin transport system. ACS Appl Mater Interfaces. 2021;13(17):19736–19744. doi:10.1021/acsami.1c03065
13. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi:10.1056/NEJMoa1304369
14. Liu J, Bai Y, Li Y, Li X, Luo K. Reprogramming the immunosuppressive tumor microenvironment through nanomedicine: an immunometabolism perspective. EBioMedicine. 2024;107:105301. doi:10.1016/j.ebiom.2024.105301
15. Y KT, Kim DW, Chung JY, et al. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10(11):3708–3716. doi:10.1158/1078-0432.CCR-03-0655
16. Mu W, Chu Q, Liu Y, et al. A review on nano-based drug delivery system for cancer chemoimmunotherapy. Nanomicro Lett. 2020;12(1):142. doi:10.1007/s40820-020-00482-6
17. Hutchinson AD, Hosking JR, Kichenadasse G, et al. Objective and subjective cognitive impairment following chemotherapy for cancer: a systematic review. Cancer Treat Rev. 2012;38(7):926–934. doi:10.1016/j.ctrv.2012.05.002
18. Tam YT, Shin DH, Chen KE, et al. Poly(ethylene glycol)-block-poly(d,l-lactic acid) micelles containing oligo(lactic acid)(8)-paclitaxel prodrug: in vivo conversion and antitumor efficacy. J Control Release. 2019;298:186–193. doi:10.1016/j.jconrel.2019.02.017
19. Shah S, Dhawan V, Holm R, et al. Liposomes: advancements and innovation in the manufacturing process. Adv Drug Deliv Rev. 2020;154–155:102–122. doi:10.1016/j.addr.2020.07.002
20. Li L, Zhan Q, Yi K, et al. Engineering Lipusu with lysophosphatidylcholine for improved tumor cellular uptake and anticancer efficacy. J Mater Chem B. 2022;10(11):1833–1842. doi:10.1039/D1TB02823E
21. Zhang J, Pan Y, Shi Q, et al. Paclitaxel liposome for injection (Lipusu) plus cisplatin versus gemcitabine plus cisplatin in the first-line treatment of locally advanced or metastatic lung squamous cell carcinoma: a multicenter, randomized, open-label, parallel controlled clinical study. Cancer Commun. 2022;42(1):3–16.
22. Berry G, Billingham M, Alderman E, et al. The use of cardiac biopsy to demonstrate reduced cardiotoxicity in AIDS Kaposi’s sarcoma patients treated with pegylated liposomal doxorubicin. Ann Oncol. 1998;9(7):711–716. doi:10.1023/A:1008216430806
23. Yin Y, Wu X, Yang Z, et al. The potential efficacy of R8-modified paclitaxel-loaded liposomes on pulmonary arterial hypertension. Pharm Res. 2013;30(8):2050–2062. doi:10.1007/s11095-013-1058-8
24. Bhardwaj U, Burgess DJ. Physicochemical properties of extruded and non-extruded liposomes containing the hydrophobic drug dexamethasone. Int J Pharm. 2010;388(1–2):181–189. doi:10.1016/j.ijpharm.2010.01.003
25. Song J, Waugh RE. Bending rigidity of SOPC membranes containing cholesterol. Biophys J. 1993;64(6):1967–1970. doi:10.1016/S0006-3495(93)81566-2
26. Najafinobar N, Mellander LJ, Kurczy ME, et al. Cholesterol alters the dynamics of release in protein independent cell models for exocytosis. Sci Rep. 2016;6:33702. doi:10.1038/srep33702
27. Chen ZJ, Yang SC, Liu XL, et al. Nanobowl-supported liposomes improve drug loading and delivery. Nano Lett. 2020;20(6):4177–4187. doi:10.1021/acs.nanolett.0c00495
28. Xin W, Xinmei C, Xinyu W, et al. Paclitaxel-lipid prodrug liposomes for improved drug delivery and breast carcinoma therapy. Chin Chem Lett. 2024;35(2):108756. doi:10.1016/j.cclet.2023.108756
29. Zhou Y, Miao Y, Huang Q, et al. A redox-responsive self-assembling COA-4-arm PEG prodrug nanosystem for dual drug delivery suppresses cancer metastasis and drug resistance by downregulating hsp90 expression. Acta Pharm Sin B. 2023;13(7):3153–3167. doi:10.1016/j.apsb.2022.11.024
30. Sun X, Zhao R, Zhao E, et al. Targeting CD44-positive ovarian cancers via engineered paclitaxel prodrug nanoparticles for enhanced chemotherapeutic efficacy. Biomed Pharmacother. 2022;154:113655. doi:10.1016/j.biopha.2022.113655
31. Patil YP, Jadhav S. Novel methods for liposome preparation. Chem Phys Lipids. 2014;177:8–18. doi:10.1016/j.chemphyslip.2013.10.011
32. Kinoshita R, Ishima Y, Chuang V, et al. Improved anticancer effects of albumin-bound paclitaxel nanoparticle via augmentation of EPR effect and albumin-protein interactions using S-nitrosated human serum albumin dimer. Biomaterials. 2017;140:162–169. doi:10.1016/j.biomaterials.2017.06.021
33. Yang M, Gu Y, Yang D, et al. Development of triptolide-nanoemulsion gels for percutaneous administration: physicochemical, transport, pharmacokinetic and pharmacodynamic characteristics. J Nanobiotechnol. 2017;15(1):88. doi:10.1186/s12951-017-0323-0
34. Kyrylkova K, Kyryachenko S, Leid M, et al. Detection of apoptosis by TUNEL assay. Methods Mol Biol. 2012;887:41–47.
35. Chung HJ, Kim HJ, Hong ST. Tumor-specific delivery of a paclitaxel-loading HSA-haemin nanoparticle for cancer treatment. Nanomedicine. 2020;23:102089. doi:10.1016/j.nano.2019.102089
36. Li F, Yuan H, Zhang H, et al. Neonatal Fc receptor (FcRn) enhances tissue distribution and prevents excretion of nab-paclitaxel. Mol Pharm. 2019;16(6):2385–2393. doi:10.1021/acs.molpharmaceut.8b01314
37. Pandya AD, Jäger E, Bagheri FS, et al. Paclitaxel-loaded biodegradable ROS-sensitive nanoparticles for cancer therapy. Int J Nanomed. 2019;14:6269–6285. doi:10.2147/IJN.S208938
38. Verco J, Johnston W, Baltezor M, et al. Pharmacokinetic profile of inhaled submicron particle paclitaxel (NanoPac(®)) in a rodent model. J Aerosol Med Pulm Drug Deliv. 2019;32(2):99–109. doi:10.1089/jamp.2018.1467
39. Gardner ER, Dahut W, Figg WD. Quantitative determination of total and unbound paclitaxel in human plasma following Abraxane treatment. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;862(1–2):213–218. doi:10.1016/j.jchromb.2007.12.013
40. Kaddoumi A, Gill KK, Elfakhri K, et al. Development and qualification of an LC-MS/MS method for investigating the biological implications of micelle entrapped paclitaxel in cell culture and rats. Biomed Chromatogr. 2017;31(9):10. doi:10.1002/bmc.3960
41. Yin T, Wang Y, Chu X, et al. Free adriamycin-loaded pH/reduction dual-responsive hyaluronic acid-adriamycin prodrug micelles for efficient cancer therapy. ACS Appl Mater Interfaces. 2018;10(42):35693–35704. doi:10.1021/acsami.8b09342
42. Okamoto Y, Taguchi K, Sakuragi M, et al. Preparation, characterization, and in vitro/in vivo evaluation of paclitaxel-bound albumin-encapsulated liposomes for the treatment of pancreatic cancer. ACS Omega. 2019;4(5):8693–8700. doi:10.1021/acsomega.9b00537
43. Liao J, Zheng H, Fei Z, et al. Tumor-targeting and pH-responsive nanoparticles from hyaluronic acid for the enhanced delivery of doxorubicin. Int J Biol Macromol. 2018;113:737–747. doi:10.1016/j.ijbiomac.2018.03.004
44. Han M, Vakili MR, Soleymani AH, et al. Mitochondrial delivery of doxorubicin via triphenylphosphine modification for overcoming drug resistance in MDA-MB-435/DOX cells. Mol Pharm. 2014;11(8):2640–2649. doi:10.1021/mp500038g
45. Gu Y, Ma J, Fu Z, et al. Development of novel liposome-encapsulated combretastatin A4 acylated derivatives: prodrug approach for improving antitumor efficacy. Int J Nanomed. 2019;14:8805–8818. doi:10.2147/IJN.S210938
46. Pozzi D, Colapicchioni V, Caracciolo G, et al. Effect of polyethylene glycol (PEG) chain length on the bio-nano-interactions between PEGylated lipid nanoparticles and biological fluids: from nanostructure to uptake in cancer cells. Nanoscale. 2014;6(5):2782–2792. doi:10.1039/c3nr05559k
47. Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15(7–8):457–464. doi:10.1080/10611860701539584
48. Singh P, Gupta U, Asthana A, et al. Folate and folate-PEG-PAMAM dendrimers: synthesis, characterization, and targeted anticancer drug delivery potential in tumor bearing mice. Bioconjug Chem. 2008;19(11):2239–2252. doi:10.1021/bc800125u
49. Wu MY, Lu JH. Autophagy and macrophage functions: inflammatory response and phagocytosis. Cells. 2019;9(1):70. doi:10.3390/cells9010070
50. Lai X, Wang S, Hu M, et al. Dual targeting single arrow: neutrophil-targeted sialic acid-modified nanoplatform for treating comorbid tumors and rheumatoid arthritis. Int J Pharm. 2021;607:121022. doi:10.1016/j.ijpharm.2021.121022
51. Qiu Q, Li C, Song Y, et al. Targeted delivery of ibrutinib to tumor-associated macrophages by sialic acid-stearic acid conjugate modified nanocomplexes for cancer immunotherapy. Acta Biomater. 2019;92:184–195. doi:10.1016/j.actbio.2019.05.030
52. Shi L, Wang Y, Wang Q, et al. Transforming a toxic drug into an efficacious nanomedicine using a lipoprodrug strategy for the treatment of patient-derived melanoma xenografts. J Control Release. 2020;324:289–302. doi:10.1016/j.jconrel.2020.05.025
53. Hatakeyama H, Akita H, Kogure K, et al. [Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid]. Yakugaku Zasshi. 2007;127(10):1549–1556. Danish. doi:10.1248/yakushi.127.1549
54. Mantovani A, Sica A, Sozzani S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. doi:10.1016/j.it.2004.09.015
55. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi:10.1016/j.immuni.2010.05.007
56. Xu J, Wang J, Qiu J, et al. Nanoparticles retard immune cells recruitment in vivo by inhibiting chemokine expression. Biomaterials. 2021;265:120392. doi:10.1016/j.biomaterials.2020.120392
57. Shi J, Kantoff PW, Wooster R, et al. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108
58. Van der Meel R, Sulheim E, Shi Y, et al. Smart cancer nanomedicine. Nat Nanotechnol. 2019;14(11):1007–1017. doi:10.1038/s41565-019-0567-y
59. Liu W, Liu J, Salt LJ, et al. Structural stability of liposome-stabilized oil-in-water Pickering emulsions and their fate during in vitro digestion. Food Funct. 2019;10(11):7262–7274. doi:10.1039/C9FO00967A
60. Meng H, Leong W, Leong KW, et al. Walking the line: the fate of nanomaterials at biological barriers. Biomaterials. 2018;174:41–53. doi:10.1016/j.biomaterials.2018.04.056
61. Nowak M, Klink M. The role of tumor-associated macrophages in the progression and chemoresistance of ovarian cancer. Cells. 2020;9(5):1299. doi:10.3390/cells9051299
62. She Z, Zhang T, Wang X, et al. The anticancer efficacy of pixantrone-loaded liposomes decorated with sialic acid-octadecylamine conjugate. Biomaterials. 2014;35(19):5216–5225. doi:10.1016/j.biomaterials.2014.03.022
63. Tubin S, Khan MK, Gupta S, et al. Biology of NSCLC: interplay between cancer cells, radiation and tumor immune microenvironment. Cancers. 2021;13(4):775. doi:10.3390/cancers13040775
64. Sun J, Song Y, Lu M, et al. Evaluation of the antitumor effect of dexamethasone palmitate and doxorubicin co-loaded liposomes modified with a sialic acid-octadecylamine conjugate. Eur J Pharm Sci. 2016;93:177–183. doi:10.1016/j.ejps.2016.08.029
65. Zhang TY, Huang B, Wu HB, et al. Synergistic effects of co-administration of suicide gene expressing mesenchymal stem cells and prodrug-encapsulated liposome on aggressive lung melanoma metastases in mice. J Control Release. 2015;209:260–271. doi:10.1016/j.jconrel.2015.05.007
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.