")
Back to Journals » International Journal of Nanomedicine » Volume 20
Evaluation of Luteolin Nanosuspensions on Pharmacokinetics of Atorvastatin: Drug-Drug Interactions Using Rat Models
Authors Liang Y, Sun B, Yang M, Meng X, Wang M, Yang L, AI-Hamyari B, Zhang Y, Shen Y, Meng S
Received 2 October 2024
Accepted for publication 25 January 2025
Published 28 February 2025 Volume 2025:20 Pages 2557—2573
DOI https://doi.org/10.2147/IJN.S492141
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Farooq A. Shiekh
You Liang,* Bo Sun,* Min Yang, Xiaona Meng, Meng Wang, Lijuan Yang, Bandar AI-Hamyari, Yuqian Zhang, Yutong Shen, Shengnan Meng
Department of Pharmaceutics, School of Pharmacy, China Medical University, Shenyang, 110122, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shengnan Meng, Department of Pharmaceutics, School of Pharmacy, China Medical University, No. 77 Puhe Road, Shenyang North New Area, Shenyang, Liaoning Province, 110122, People’s Republic of China, Tel +86 024 31939448, Email [email protected]
Purpose: The co-administration of luteolin (LUT) and atorvastatin (ATV) may drive synergetic effects on against atherosclerotic cardiovascular disease (ASCVD). This study aims to explore the pharmacokinetic (PK) drug-drug interactions (DDIs) of LUT toward ATV and the influencing mechanisms involving CYP450s and OATPs, and using the physiologically based pharmacokinetic (PBPK) models extrapolated to humans to optimize the DDIs dosage regimens for subsequent research.
Methods: Luteolin nanosuspensions lyophilized powder (LUT-NS-LP) were prepared for improving LUT’s solubility and bioavailability, the effects of both LUT on the ATV CYP450s enzyme kinetics and LUT-NS-LP/LUT on the PK behavior of ATV in rats were further studied by UPLC. The DDI PBPK model of ATV and LUT-NS-LP was established with the hepatic CYP450s, OATPs, and enterohepatic circulation, and extrapolated to humans through a physiological allometric scaling process with parameter optimization and verified using clinical datasets obtained from various dosage regimens.
Results: LUT inhibited ATV as the non-competitive form in rat liver microsomes (RLMs). The Cmax and AUC (0-t) of ATV in the group receiving combined administration of LUT and LUT-NS-LP increased by 1.87-fold and 2.29-fold, 5.42-fold and 10.35-fold, respectively. The constructed PBPK models successfully predicted the PK DDIs between ATV and LUT in rats, demonstrating excellent performance. LUT might inhibit the hepatic CYP450s and OATPs activities to influence the PK behavior of ATV. The extrapolated human model could adequately describe and predict the systemic exposure of ATV in DDIs.
Conclusion: LUT nanosuspensions could significantly increase systemic exposure to ATV by inhibiting CYP450s and OATPs activities. The combined application strategy is suggested to run ATV in half of the highest dosage by guidelines. This study offers a valuable experimental foundation for the combined administration of statins with natural drugs and their nanoformulations, providing significant insights into the investigation of underlying mechanisms and potential clinical applications.
Plain Language Summary: In this study, we focused on how luteolin (LUT), a natural compound, interacts with atorvastatin (ATV), a commonly used drug for heart disease. We wanted to understand whether taking these two compounds together could alter ATV’s absorption and breakdown, which potentially influences its effectiveness and safety.
To improve LUT’s ability to work in the body, we prepared a special form that dissolves more easily, and tested how LUT impacted ATV’s behavior in rats and used advanced computer models to predict what might happen in humans.
We found that the cooperation of LUT and ATV, LUT significantly increased the levels of ATV in the bloodstream. This happens because LUT slows down the liver’s ability to dispose of ATV, leading to higher levels of the drug in the body. We confirmed these results in rats, and the computer models also supported the findings.
This study offers evidence that when LUT is combined with ATV, LUT increases the amount of ATV in the bloodstream, and a promising strategy by computer predictions for humans is that the dosage of ATV need to be adjusted to avoid potential side effects. Our research offers valuable insights into how natural compounds like LUT could be used alongside medications to improve treatments for heart disease in the future.
Keywords: luteolin, atorvastatin, drug-drug interactions, nanosuspension, PBPK, atherosclerotic cardiovascular disease
Graphical Abstract:

Introduction
Cardiovascular disease (CVD) is a major threat to human health and a leading cause of death globally, with most due to atherosclerotic cardiovascular disease (ASCVD).1 Despite continuous improvement in treatment methods, ASCVD still has a high morbidity and mortality rate, which is growing at an astonishing rate in developing countries.2 Therefore, the prevention and treatment of ASCVD remains a serious challenge and has become a public health problem worldwide that needs to be urgently addressed.
Statins are the first-line therapeutic agents for ASCVD that reduce cholesterol biosynthesis by inhibiting the activities of 3-hydroxy-3-methylglutaryl coenzyme A reductase.3 Prolonged exposure of arteries to elevated low-density lipoprotein (LDL-C) concentrations is known to be a major cause of ASCVD.4 But the use of statins alone is often insufficient for reducing LDL-C to target levels. In addition, some patients, particularly those with inherited disorders such as familial hypercholesterolemia, do not respond adequately to statins.5 Moreover, serious adverse effects such as myopathy, rhabdomyolysis, hepatotoxicity, etc., can prevent a significant number of patients from taking statins at the appropriate intensity.6 Due to the complex etiology of ASCVD, statins are often combined with non-statin lipid-lowering drugs and antithrombotic drugs for the treatment of ASCVD in some clinical trials.7 However, previous studies have shown that the combination of niacin and statin has limited additive effects on plaque regression in ASCVD.7 In addition, the side effects of fibrates and the high cost of PCSK9 antibodies have limited their widespread clinical use in combination with statins.7 Therefore, it is particularly important to develop new drug combinations against ASCVD.
Luteolin (LUT, Figure 1A) is a plant flavonoid extracted from natural herbs, fruits, and vegetables, and has been reported to exert potent anti-inflammatory, anti-oxidation, anti-cancer, and neuroprotective effects.8 Recently, the protective effects of LUT on CVD have been reported, and in a few of the studies involved in exploring the role of LUT in atherosclerosis and the associated vascular inflammation, the results showed that LUT could be a promising candidate molecule for atherosclerosis.9 Thus, the combination of LUT and statins might be synergizing the treatment of ASCVD. However, LUT belongs to BCS class II drugs with poor solubility and low bioavailability, which severely limits the cardiovascular protective effects. Nanosuspension technology could solve the problem of drugs which are poorly aqueous soluble and less bioavailability.10 Our novel research on preparing LUT into nanosuspension and combined application with stains shows enormous development potential and application prospects.
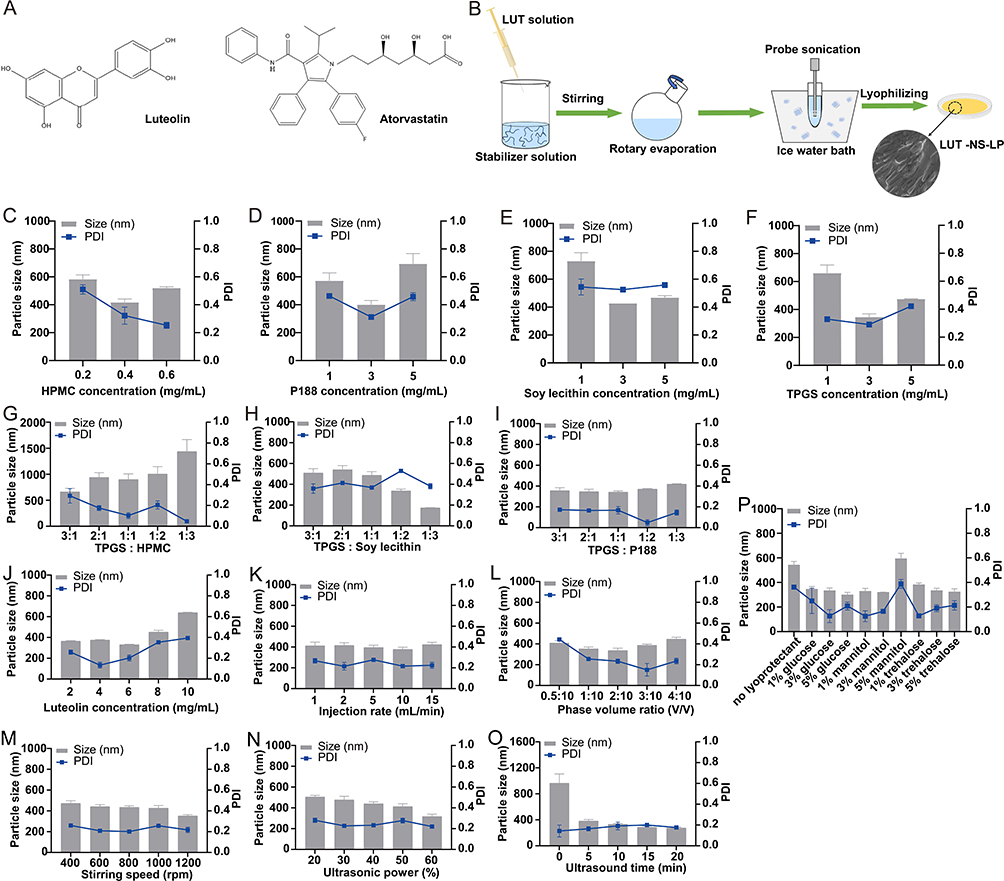 |
Figure 1 Formulation and process optimization of LUT-NS-LP. (A) Luteolin and atorvastatin. (B) Schematic illustration for the preparation process of LUT-NS-LP. Effects of the different stabilizers on the particle size and PDI of luteolin nanosuspension (C) HPMC, (D) P188, (E) Soy lecithin, (F) TPGS, (G) TPGS and HPMC, (H) TPGS and Soy lecithin, (I) TPGS and P188, Effects of different process factors on the particle size and PDI of luteolin nanosuspension (J) Luteolin concentration, (K) Injection rate, (L) Phase volume ratio, (M) Stirring speed, (N) Ultrasonic power, (O) Ultrasonic time. (P) Different lyoprotectant agent. |
Pharmacokinetic (PK) studies play a crucial role in evaluating the in vivo behavior of drugs. Numerous studies have demonstrated that the PK behavior of atorvastatin (ATV, Figure 1A), the most commonly prescribed statin, is subject to influence from various enzyme and transporter proteins, including the hepatic CYP450s and OATPs, also by the disposition of the enterohepatic circulation which are closely related to its curative effects and toxicity.11 However, there is a paucity of relevant literature concerning the investigation of the PK drug-drug interactions (DDIs) between ATV and LUT mediated by hepatic CYP450s and OATP transporters. Moreover, traditional studies typically require extensive and expensive experiments in vitro and in vivo. Therefore, it is a promising purpose to explore more faster and effective methods to reveal the DDIs.
Physiologically based on pharmacokinetic (PBPK) modeling is widely utilized in drug development. PBPK model can describe the absorption, distribution, metabolism, and excretion (ADME) of drugs within the body, elucidating their complex behaviors throughout these processes.12 This enables PBPK model to predict the systemic exposure and local tissue concentration of a compound in clinical settings through interspecies scaling and/or in vitro to in vivo extrapolation processes. A key advantage of PBPK model is its robust predictive capabilities, particularly in characterizing DDI mediated by metabolic enzymes or drug transporters. For example, Li et al developed a PBPK model for ATV to predict its DDI mediated by OATP transporters and CYP3A4 with fluconazole, palbociclib, diltiazem, and cyclosporine.13 Similarly, Nina et al established a whole-body PBPK model for rosuvastatin to study transporter-mediated DDIs.14 Therefore, the application of PBPK model to predict and elucidate the mechanisms of DDI is a highly efficient method. Furthermore, the PBPK model allows for forecasting the systemic exposure level of the drug in the human body through interspecies extrapolation, and delivers substantial guidance for clinical DDI practice.
This study aims to prepare LUT into nanosuspensions to hope to improve its solubility and bioavailability for driving well co-administration effects with ATV. The PK DDIs were focused on investigating through in vitro CYP450s-mediated enzyme kinetic and in vivo PK studies, then further constructed PBPK model united with ATV and luteolin nanosuspensions (LUT-NS), and extrapolate this model from rat to human through a physiological allometric scaling process with parameter optimization to recommend the DDI dosage regimens. Our research will provide a reference for the clinical co-administration of ATV and LUT nano-formulations.
Materials and Methods
Luteolin (LUT, purity > 98%) was purchased from Xi’an Green Biotechnology Co., Ltd. (Xi′an, China). Atorvastatin (ATV, purity > 98%) was purchased from Hubei Jiuzhou Kangda Biotechnology Co., Ltd. (Zaoyang, China). Testosterone was provided by Beijing Solabao Technology Co., Ltd. (Beijing, China). Tocopherol polyethylene glycol vitamin E succinate (TPGS) was obtained from Xi’an Healthful Biotechnology Co., Ltd. (Xi’an, China). Poloxamer 188 (P188), mannitol, and trehalose were procured from Luofu Pharmaceutical Technology Co., Ltd. (Shanghai, China). Hypromellose (HPMC), soy lecithin, and anhydrous glucose were purchased from Tianjin Guangfu Fine Chemical Research Institute. (Tianjin, China). All chemical reagents used were of analytical grade and used as received.
Preparation of Luteolin Nanosuspensions
LUT-NS was prepared by an anti-solvent precipitation-ultrasonication method (Figure 1B). To obtain LUT-NS-LP with desired pharmaceutics properties. The single factor method was used to screen the best formulation and process parameters with particle size, polydispersity index (PDI), and stability as key evaluation metrics, including the single stabilizer and combined ones (HPMC, P188, soy lecithin, TPGS, TPGS, and HPMC, TPGS and P188, TPGS and soy lecithin), the preparation process conditions (drug concentration, injection rate, phase volume ratio, stirring speed, ultrasound power, and ultrasound time) and type of lyoprotectant (glucose, mannitol, and trehalose). The LUT-NS-LP was prepared with optimized formulation and process. In brief, LUT was dissolved in absolute ethanol to obtain a 6 mg/mL solution. Then, 2 mL of the LUT solution was injected into 10 mL of this antisolvent containing stabilizers (0.1% w/v of P188 and 0.3% w/v of TPGS), and stirred at 1200 rpm for 10 minutes. Ethanol was removed by rotary evaporation. The suspension was collected and then sonicated in an ice-water bath. A freeze-drying method was used to solidify the nanosuspension for better stability, 3% mannitol was added to LUT-NS before pre-freezing at −80°C and freeze-drying for 48 hours.
Characterization of Luteolin Nanosuspensions
The particle size, PDI, and zeta potential of LUT-NS-LP were determined using dynamic light scattering (DLS) (PSS NICOMP, Santa Barbara, CA, US) at 25°C. And further characterized in terms of its physicochemical properties including morphology by Scanning electron microscope (SEM) (Hitachi X650, Tokyo, Japan), thermal properties by differential scanning calorimetry (DSC) and thermogravimetric (TG) (Mettler-Toledo International Inc., Switzerland), crystalline patterns by powder X-ray diffraction (PXRD) (Philips, Xper’t-Pro, Amsterdam, The Netherlands), and the presence of LUT and excipients were determined using Fourier Transform-Infrared Spectrometry (FT-IR) (Thermo Fisher Scientific, USA).
In vitro Drug Release Study
In the saturation solubility study, excess amounts of LUT crude powder and LUT-NS-LP were added into distilled water and then shaken for 72 h at 37°Ϲ. Dissolution test (TYPE DT 820, ERWEKA) was conducted using LUT-NS-LP, LUT crude powders, and physical mixture using the United States Pharmacopoeia (USP) apparatus II (paddles) at 100 rpm in sink situation. Equivalent to 15 mg LUT of nano-formulations were dispersed in 900 mL of release mediums at 37°C. The samples were collected at predetermined intervals and replaced with fresh medium. The experiments were carried out in triplicate. LUT concentrations were determined by UPLC as detailed in the Supplementary Text.
Pharmacokinetic Study
Six-week-old and specific pathogen-free male Sprague-Dawley (SD) rats (200–220g) were provided by Beijing HFK Bio-Technology Co., Ltd. (Beijing, China). The animal studies were approved by the Institutional Animal Care and Use Committee of China Medical University.
In the PK study, thirty rats were randomly divided into five groups (n = 6), including the LUT group (A), LUT-NS-LP group (B), ATV group (C), ATV + LUT group (D) and ATV + LUT-NS-LP group (E). Rats in the A, B, and C groups were orally given a single dose of LUT (200 mg/kg), LUT-NS-LP (equivalent to 200 mg/kg of LUT), and ATV (200 mg/kg), respectively. Group D and E were orally administered with 200 mg/kg ATV and 200 mg/kg LUT or LUT-NS-LP. Blood samples (0.3 mL) were collected via a jugular vein catheter using a disposable syringe before and at 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after administration. Plasma samples were separated after centrifugation at 2500 g for 15 min and stored at −80°C until analysis. The Supplementary Text provides the sample preparation and analytical method.
In vitro CytochromeP450-Metabolism of Atorvastatin and Luteolin by Rat Liver Microsomes
In order to study the interaction of ATV with LUT, two single concentrations of 5 μM and 10 μM LUT as specific interplayers, were investigated by enzyme kinetics methods using the CYP450-containing mixture of Rat Liver Microsomes (RLMs) at different ATV concentrations (1.25–160 μM). The incubation mixture (200 μL total volume) consisted of 0.1 M potassium phosphate buffer (pH 7.4), an NADPH-generating system (10 mM NADP+, 10 mM glucose-6-phosphate, 1 unit/mL of glucose-6-phosphate dehydrogenase, and 4 mM MgCl2), RLMs, ATV and LUT.15 Control incubations without LUT were also performed. After a 5-min pre-incubation at 37°C, the reactions were initiated by the addition of NADP+ and subsequently incubated for 20–30 min at 37°C in a shaking bath.15 After incubation, the reaction was terminated by the addition of 200 μL of acetonitrile, and the resulting mixture was kept on ice. The mixture was centrifuged at 20,000g for 20 min at 4°C. Aliquots of the supernatant were then taken for further UPLC analyses. All incubations throughout this study were conducted in triplicate.
The inhibition constant (Ki) values were determined by using various concentrations of ATV in the presence or absence of LUT with the program Prism (Version 9.3.0, GraphPad, San Diego, CA). Three kinetic models were used to calculate the Ki values by nonlinear regression using the equations for noncompetitive inhibition (Equation 1).16

where V is the velocity of the reaction, S and I are the substrate and inhibitor concentrations, respectively, Ki is the inhibition constant that describes the affinity of the inhibitor for the enzyme, and Km is the substrate concentration at half of the maximum velocity (Vmax) of the reaction. The goodness of the fits to the kinetic and inhibition models was assessed based on F statistics, R2 values, AIC (Akaike’s Information Criterion), parameter S.D. estimates, and 95% confidence intervals. The kinetic constants are reported as the mean ± S.D. of the parameter estimate.
Construction the Atorvastatin, Luteolin and Luteolin Nanosuspensions Lyophilized Powder of Physiologically Based Pharmacokinetic Models in Rats
The PBPK models for ATV, LUT, LUT-NS-LP, and their DDIs were constructed using the ADAPT version 5 (Biomedical Simulations Resource, University of Southern California, Los Angeles, CA) via incorporating their characteristics in vitro and in vivo (Figure S1 and Table 1). In brief, each model included the liver, kidney, lung, heart, spleen, intestine, muscle, and adipose, all organs except those mentioned were lumped into the remainder compartment to attain whole-body mass balance. Absorption was described using four transit compartments owing to the delayed absorption of drugs. For ATV, the liver was the primary metabolism organ and metabolized by CYP450s. The liver compartment was further subdivided into extrahepatic and hepatocellular compartments to incorporate the OATPs-mediated hepatic uptake process. Since the bile excretion of ATV was reported in rats,11 enterohepatic circulation was considered into the ATV model. Specially, for LUT-NS-LP modeling, its dissolution and release results in vitro were adopted into the modification of the Noyes-Whitney dissolution equation for evaluating its in vivo dissolution kinetics. All specific operations and differential equations are shown in the Supplementary Text.
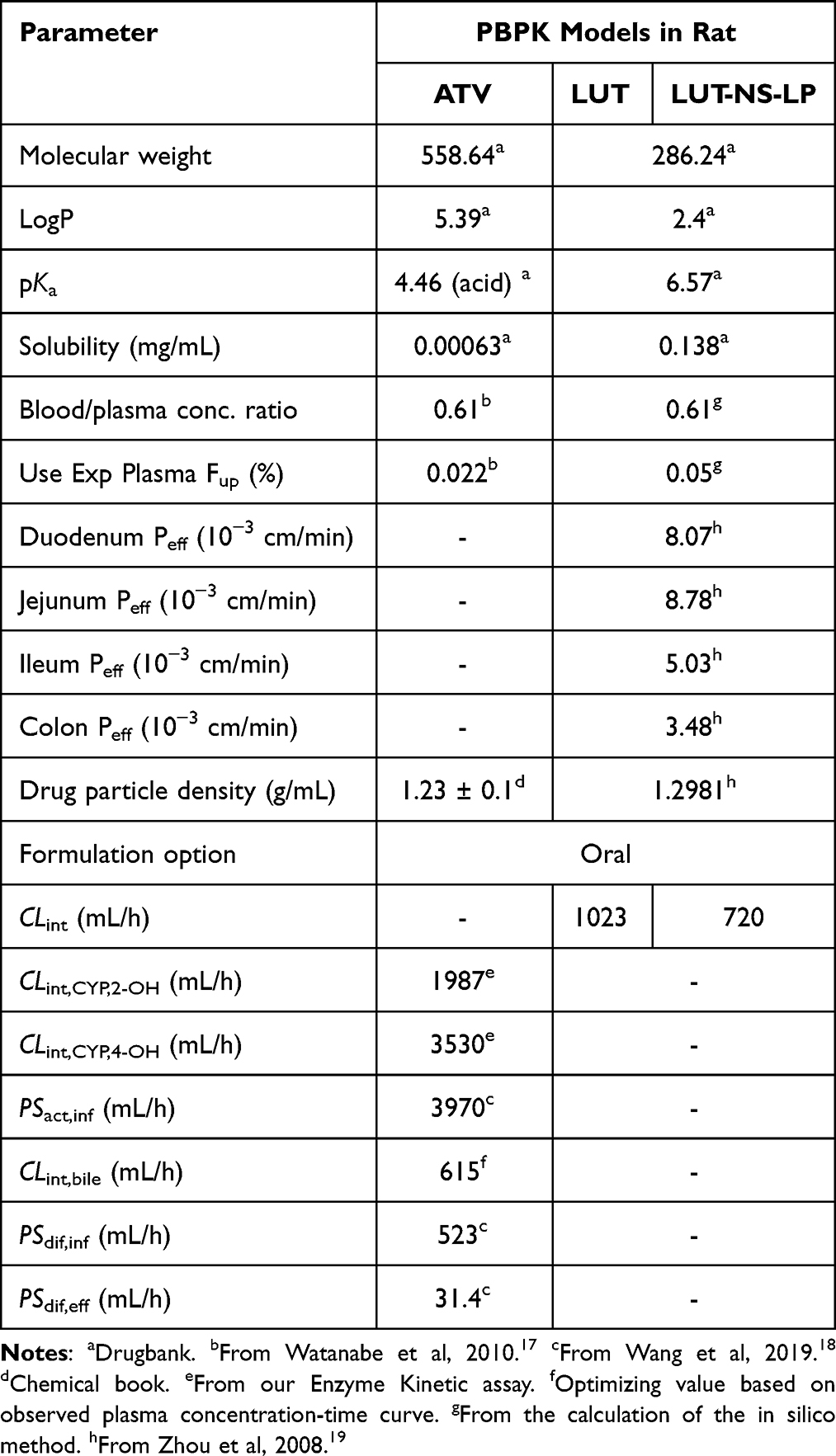 |
Table 1 Physical and Chemical Properties and Physiological Parameters of ATV, LUT, and LUT-NS-LP Used in the Rat PBPK Models |
The hepatic clearance is a critical factor in the construction of PBPK model of ATV. In our study, both CYP450s-mediated metabolism and OATPs-mediated transport were conducted as the key components affecting the hepatic clearance. To ensure the accuracy of the simulation, the methodology of “extended clearance concept (ECC)” as proposed by was distinctively followed,20 which quantitatively describes the influence of biomembrane permeability by taking into account the transmembrane permeation process in elimination organs.21 The detailed operations could be found in the Supplementary Text.
In the construction of the DDI models of ATV and LUT/LUT-NS-LP, combined with our own CYP450s-enzyme kinetics data and the published OATPs-mediated data for ATV.
For human models, rat physiological parameters (organ volume and blood flow) were replaced with corresponding values for humans (Table S1), which were obtained from the literature and scaled to the weights. Kp for the same type of tissues were assumed to be identical among the humans (Table S2). The parameters of metabolism and transport were estimated using the allometric equations with the allometric exponent at 0.75.22
Physiologically Based Pharmacokinetic Models Verification and Analysis
The model was evaluated based on AIC, visual inspection of the fitted profiles, and CV% of parameter estimates. To verify the model, simulated PK profiles of individual and co-administrated drugs were compared with both our own PK studies and the reported ones available in the literatures (listed in the Supplementary Text). When the predicted parameters were within a 2-fold error of the observed values (plasma concentrations, AUC(0-t), and Cmax), the model was considered to have acceptable accuracy of predictions.
A sensitivity analysis was conducted to assess the impact of model uncertainty parameters on the Cmax and AUC(0-t) of ATV in the DDI models. The examined model parameters included optimized values and also those expected to exert a great influence on Cmax and AUC(0-t). The normalized sensitivity coefficient (NSC) was calculated as the percentage change in the predicted AUC(0-t) and Cmax of ATV resulting from a 1% increase in the selected parameter. Parameters with absolute values of /NSC/ ≥ 0.2 were considered sensitive.23
Application of the Physiologically Based Pharmacokinetic - Drug-Drug Interactions Models in Humans
A new DDI model of ATV and LUT-NS-LP was further constructed based on the above models by ECC. According to the current clinical guidelines, the clinical dosages of ATV range from 10 to 80 mg, the maximum dosing regimen for ATV is 80 mg once daily, and 40 mg is the most commonly used one, so the 40 mg dose was fixed in our DDI simulation. However, there are no clinical reference doses available for LUT or LUT-NS-LP. The dose setting for LUT-NS-LP must be thought over to maintain the safety and the treatment effect of ATV, the strategy was adopted that the AUC(0-t) generated by the combination of LUT-NS-LP and ATV is equivalent to the one by the maximum dose of ATV (80 mg) alone.
Data and Statistical Analysis
The PK parameters were calculated by noncompartment analysis using the DAS 2.0 software (BioGuider Co., Shanghai, China). All results were expressed as mean ± standard deviation (SD). Statistical differences among groups were evaluated by the two-tailed Student’s t-test using GraphPad Prism software (San Diego, CA, USA). Values of P < 0.05 were considered statistically significant.
Results
Preparation and Characterization of Optimized Luteolin Nanosuspensions Lyophilized Powder
First, our study screened the types and amounts of stabilizers and investigated preparation process parameters to produce stable LUT-NS-LP. As shown in Figure 1C–P, the optimized LUT-NS-LP with the best particle size, PDI, and stability was prepared using 0.3%TPGS and 0.1%P188 as the co-stabilizers under the process factors with luteolin concentration of 6 mg/mL, injection rate of 10mL/min, phase volume ratio of 2:10, stirring speed of 1200 rpm, ultrasonic power of 60%, ultrasound time of 20 min, and 3% mannitol as lyoprotectant. The obtained nanosuspensions showed 276.5 nm with a narrow PDI value of 0.176 (Figure 2A), and the zeta potential was measured to be −27.99 mV. Meanwhile, we investigated the changes in particle size and PDI in 7 days, which presented the good stability of nanosuspensions (Figure 2B). In addition, LUT-NS-LP presented an irregular rod-like crystal shape in their morphology as observed by SEM (Figure 2C). According to PXRD results (Figure 2D), the characteristic peaks (2-theta of 14.6, and 21.7) of native LUT confirmed its crystalline solid state. The diffraction peak intensities of the LUT in LUT-NS-LP were reduced as compared to the physical mixture, which indicates that most of the drugs were transformed from crystalline to amorphous state during the manufacturing process. DSC thermograms of the LUT crude powder, blank excipients, physical mixture, and LUT-NS-LP are shown in Figure 2E. About the two endothermic peaks at 171.2°C and 340°C, the first peak could be the melting point of LUT crude powder, this peak still existed but with significantly lower intensity in the drug nanocrystal sample, indicating much loss of its crystallinity. The second peak was closer to the melting point of the excipients. The FT-IR spectra of LUT-NS-LP and its physical mixture were almost identical without any new characteristic absorption peaks (Figure 2F), confirming that the chemical structure of LUT did not change. The results of the TG analysis of the samples were shown in Figure 2G, the LUT crude powder was observed to lose weight from about 400°C, while the LUT-NS-LP began to lose weight from about 320°C. This might be because the particle size of the LUT-NS-LP was smaller compared with the native LUT led to its higher specific surface, which resulted in easier vaporization and quick thermal decomposition. These characteristics indicate that during the precipitation process of the nanosuspension, a significant amount of LUT transitions from a crystalline to an amorphous state.
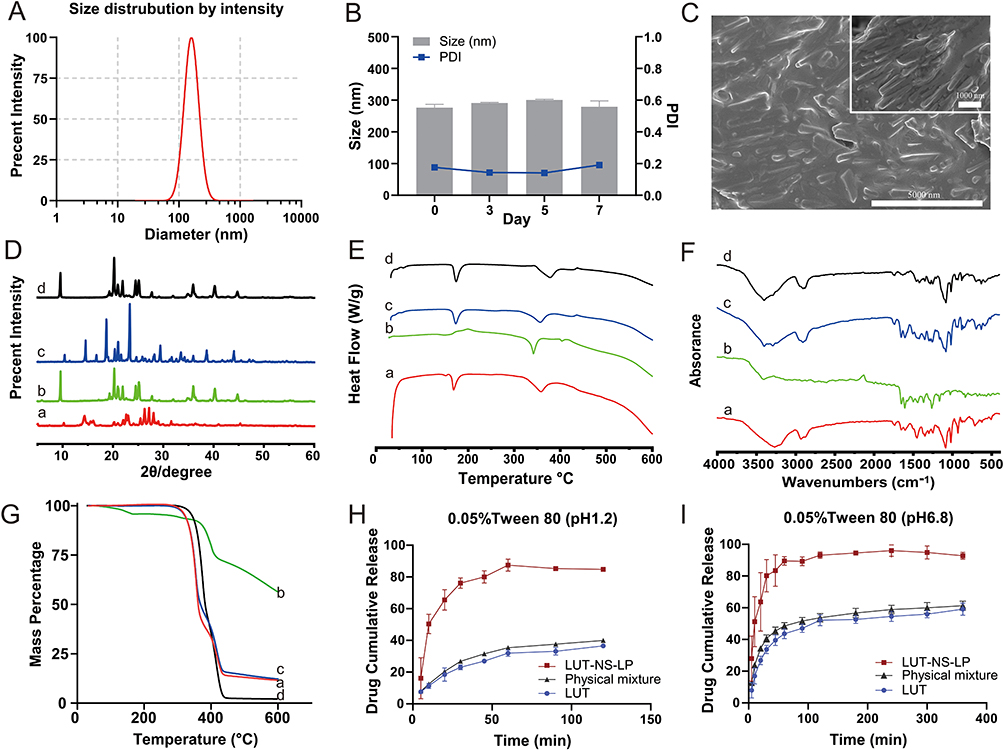 |
Figure 2 Characterization of LUT-NS-LP. (A) Particle size distribution, (B) short-term stability of LUT-NS, (C) SEM image, the curves of (D) PXRD, (E) DSC, (F) FT-IR, and (G) TG of different groups. (H) and (I) cumulative release of LUT-NS-LP, physical mixture, and luteolin crude powder. The buffer of pH 1.2 containing 0.05% Tween 80, and the buffer of pH 6.8 containing 0.05% Tween 80. Each value represents the mean ± S.D. (n=3). Notes: aLUT-NS-LP, bPhysical mixture, cBlank excipient, and dLUT crude powder. |
In vitro Dissolution
The saturated solubility of the LUT samples in water is listed in Table S3. In comparison to the crude powder, the solubility of LUT in water was significantly increased by 462 folds for the LUT-NS-LP. In vitro dissolution profiles of LUT, physical mixtures, and LUT-NS-LP in two phosphate buffers (pH 1.2 and pH 6.8) containing Tween 80 (0.05% w/v) were performed as shown in Figure 2H and I, respectively. The release data indicate that the release of the drug from the nanosuspensions was rather rapid. The optimized LUT-NS-LP showed 84.80 ± 1.06% drug release after 120 min compared to a crude powder with a release of 36.58 ± 0.99% in pH 1.2 phosphate buffer, whereas in phosphate buffers (pH 6.8), more than 90% of the drug was released from nanosuspension compared to the raw powder (59.10 ± 3.13%).
Pharmacokinetic Study
In Figure 3A, the plasma concentration-time curve of LUT and LUT-NS-LP is exhibited, with corresponding PK parameters summarized in Table 2. From the results, the main pharmacokinetic parameters (AUC(0-t) and Cmax) of LUT-NS-LP were increased by approximately 4.72-fold and 7.99-fold, respectively (P < 0.05).
 |
Figure 3 Plasma concentration-time profiles. (A) plasma concentration-time profiles of LUT (200 mg/kg) and LUT-NS-LP (equivalent to 200 mg/kg of LUT) in rats (mean ± SD, n = 6). (B) plasma concentration-time profiles of ATV after oral administration (200 mg/kg) combined with crude LUT (200 mg/kg) and LUT-NS-LP (equivalent to 200 mg/kg of LUT) in rats (mean ± SD, n = 6). |
The plasma concentration-time curve of ATV in the presence or absence of LUT or LUT-NS-LP is shown in Figure 3B, and the PK parameters are listed in Table 2. When co-administered with LUT or LUT-NS-LP, the AUC(0-t) of ATV increased by approximately 2.29-fold and 10.35-fold, respectively (P < 0.05), while the Cmax with LUT was observed with no significant difference, but that with LUT-NS-LP was increased from 2.63 ± 2.23 to 14.25 ± 3.65 μg/mL (P < 0.05). These results indicated a much higher increased system exposure level of ATV when co-administrated with LUT-NS-LP.
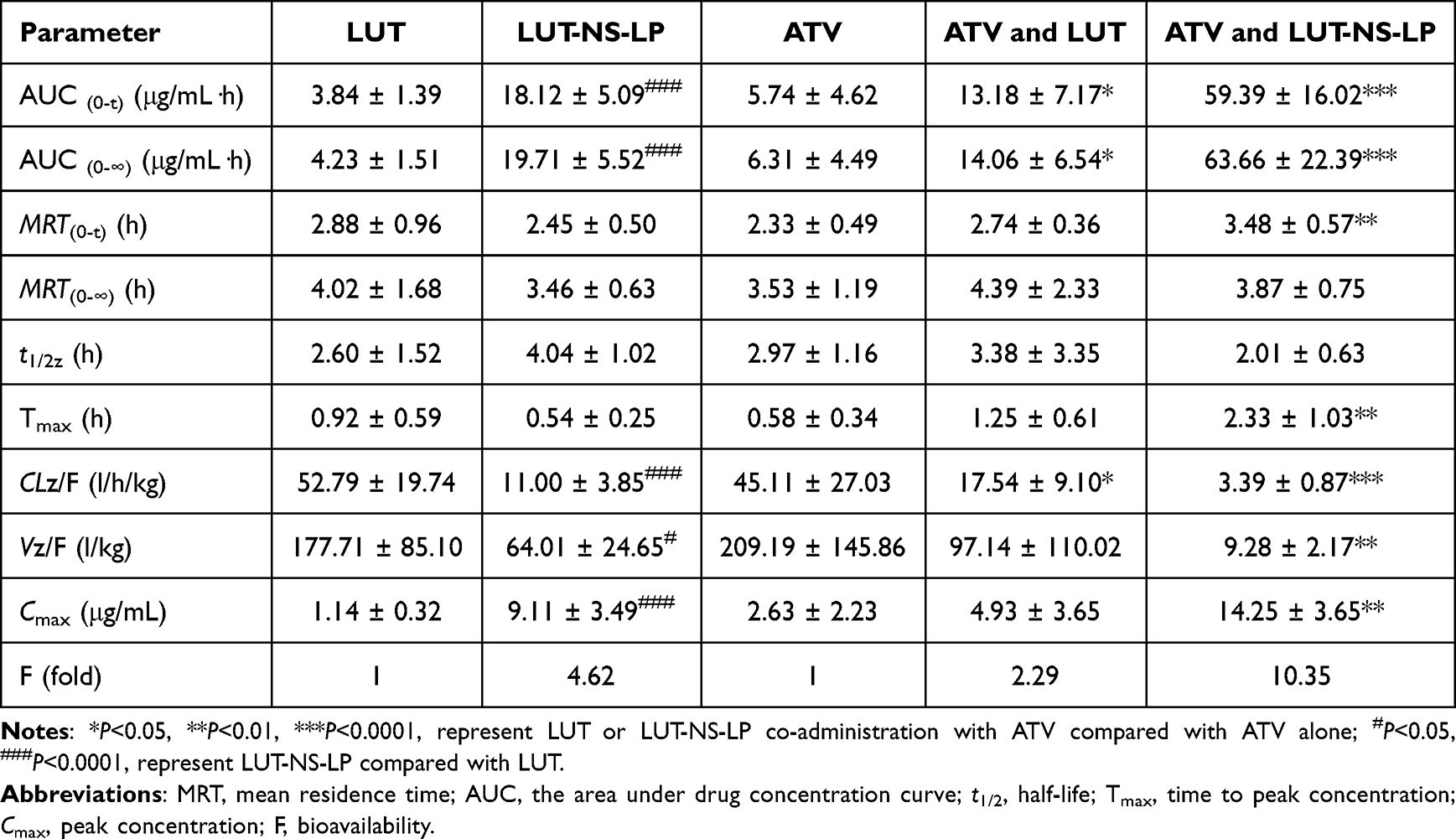 |
Table 2 Plasma Concentration-Time Profiles of LUT, LUT-NS-LP, and ATV After the Single Administration, and the Curves of ATV Co-Administrated With LUT or Its Nanosuspensions in Rats (Mean ± SD, n = 6) |
The Effects of Luteolin Towards Atorvastatin in CytochromeP450-Metabolism of Rat Liver Microsomes
LUT displayed strong inhibitory effects on the CYP450-mediated metabolites activities of 2-OH-ATV and 4-OH-ATV in RLMs. The inhibition kinetics curves of LUT towards ATV in the RLMs and those parameters were shown in Figure 4A and B and Table 3. And as shown in Figure 4C and D, LUT inhibited ATV as the noncompetitive form in the tested CYP450s mixture of the RLMs. These results would be further used as the internal data for the following studies of constructing the high-quality PBPK models of ATV and ATV united with LUT.
 |
Figure 4 The inhibitory effects of LUT (0–10μM) towards ATV in CYP450-metabolism of RLMs. Enzyme kinetics curves of the (A) 2-OH-ATV,(B) 4-OH-ATV. The Lineweaver-Burk plots of (C) 2-OH-ATV and (D) 4-OH-ATV. |
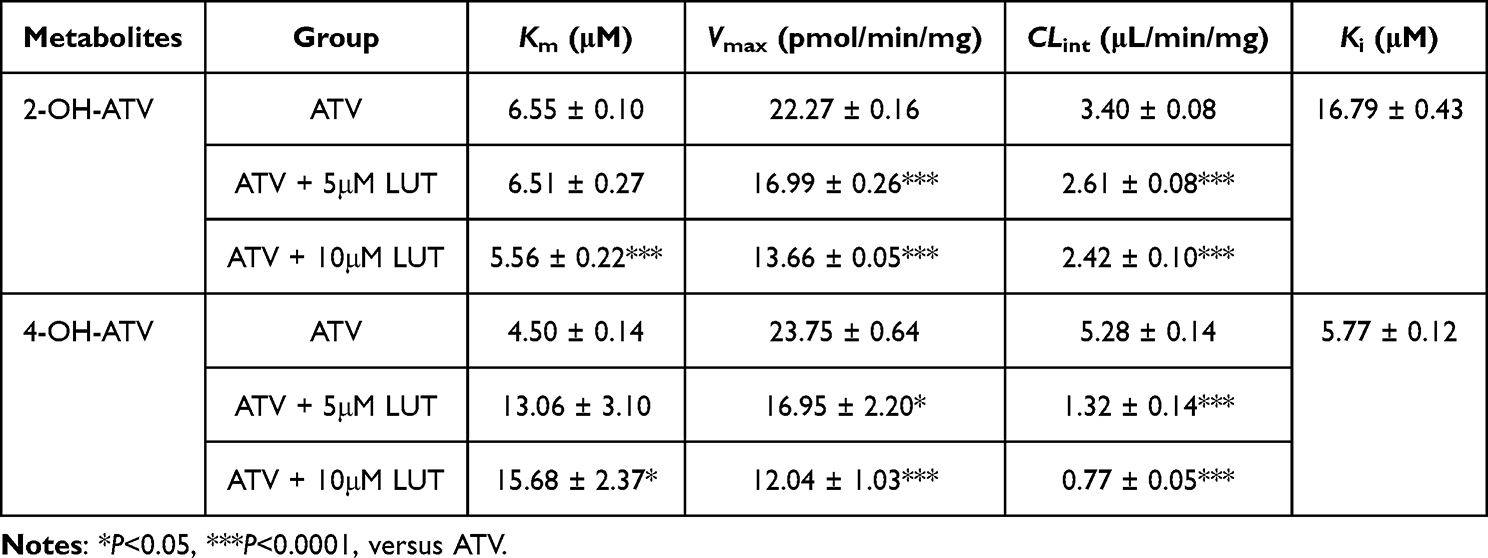 |
Table 3 Apparent Enzyme Kinetic Parameters of Metabolism of Co-Operating ATV and LUT in RLMs (Mean ± SD, n = 3) |
The Physiologically Based Pharmacokinetic Models in Rats
Our own PK parameters (ATV, LUT, and LUT-NS-LP) as observed values were adopted to compare with the predicted ones obtained from the PBPK models in rats (Table S4), and plotted in Figure 5A–C. It is clear the PK data were adequately modeled. The 10 ATV and 6 LUT articles related to the rat PK were collected for further assessing the above models, the predicted values and observed ones were close (Figure 5D–G). The models in ATV and LUT were validated by FE, AFE, and AAFE (Tables S5 and S6), and were all less than 2. It indicates all the models are accurate and reliable, and exhibited a favorable predictive capacity.
 |
Figure 5 ATV, LUT, and LUT-NS-LP model simulations and performance. Predicted and observed plasma concentration-time profiles of (A) ATV at 200 mg/kg, (B) LUT at 200 mg/kg, and (C) LUT-NS-LP at 200 mg/kg after oral administration in rats. Goodness-of-fit plots for the ATV and LUT models of predicted versus observed (D and F) plasma concentrations, (E–G) AUC(0-t), and Cmax values in rats. The analyzed studies were separated by training (A, B, and C from internal data) and test datasets (D, E, F, and G from external data). The solid line marks the line of identity. Dotted lines indicate 1.5-fold, and dashed lines indicate 2-fold deviation. Detailed information on all studies is listed in Table S4. Plasma concentration-time profiles in rats of (H) ATV (200 mg/kg) united with LUT (200 mg/kg) and (I) ATV (200 mg/kg) united with LUT-NS-LP (equivalent to 200 mg/kg of LUT) predicted by the DDI model driving in absence and presence of the influence from OATPs and/or CYP450s. |
Furthermore, the effects of LUT or LUT-NS-LP on the PK of ATV were predicted in three cases within the components for CYP450s and/or OATPs activities (Figure 5H and I). As shown in Table S4 indicated that when the double impacts of the hepatic CYP450s and OATPs were thought over, the predicted AUC(0-t) (13.05, 59.16) and Cmax (3.84, 12.34) of ATV aligned closely with the observed AUC(0-t) (13.18, 59.39) and Cmax (4.93, 14.25). By contrast, when we only considered the single influence of the hepatic CYP450s or OATPs, the prediction accuracy was reduced. This suggested that the co-operator of CYP450s and OATPs is the main cause of the DDIs of ATV and LUT. In addition, our parameters sensitive analysis results in rats showed that the OATPs are a sensitive factor influencing the ATV exposures by LUT (Figure S3A and S3B).
Extrapolation of Physiologically Based Pharmacokinetic Model to Humans and Application
The simulated plasma concentration-time profiles from the extrapolated ATV PBPK were well captured by the observed values from reported various dose clinical studies (Figure 6). Similarly, the extrapolated PBPK model of LUT showed good agreement with the only clinical studies involving a single oral dose of 20 mg/kg (Figure S2). The predicted AUC(0-t) and Cmax for all extrapolation models in humans within 2-fold range of the observed data (Table S7), which affirmed the robust predictive performance. To note that, similarly to the rat model, parameters associated with OATPs were found to be sensitive in the human model. But unlike in the rat model, the CYP450s-mediated metabolite of 2-OH-ATV is more sensitive than 4-OH-ATV for Cmax of ATV (Figure S3C and S3D).
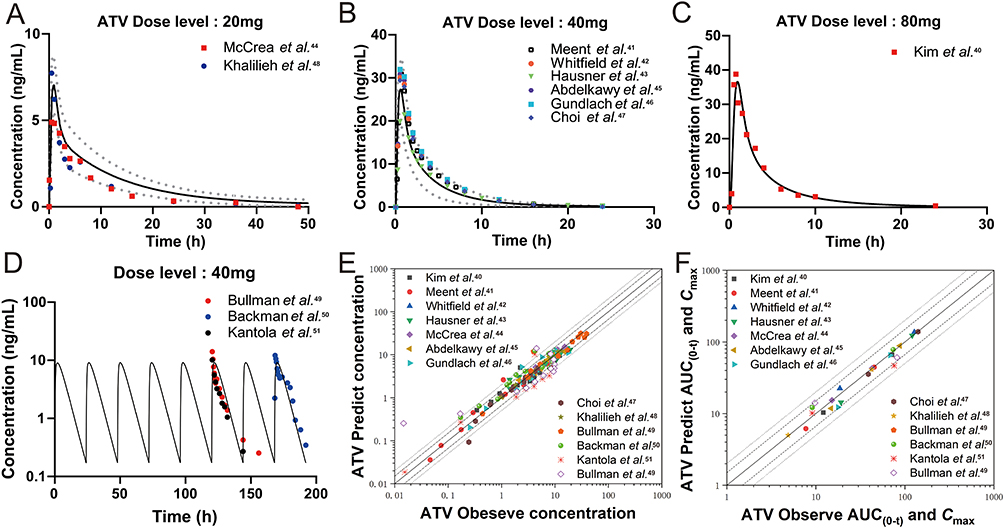 |
Figure 6 The extrapolated human PBPK model simulated the plasma concentration-time profiles of ATV at different dose levels. Simulated and observed mean plasma concentration-time profiles of a single oral dose of ATV at (A) 20 mg, (B) 40 mg, (C) 80 mg, (D) multiple oral doses of at 40 mg. Symbols represent the mean observed data obtained from reported clinical studies, black solid line represents the mean simulated results, and grey lines represent simulated 5th and 95th percentile of the predicted values. Goodness-of-fit plots for the ATV extrapolated PBPK model of predicted versus observed (E) plasma concentrations and (F) AUC(0-t) and Cmax values for the external data in clinical studies. The solid line marks the line of identity. Dotted lines indicate 1.5-fold, and dashed lines indicate 2-fold deviation. |
According to the strategy in Section of Application of the PBPK-DDI models in humans, it was found that when the LUT-NS-LP dose at 50 mg combined with 40 mg ATV, the co-operator AUC(0-t) (280.4 ng/mL·h) of ATV could achieve the similar AUC(0-t) value (282.9 ng/mL·h) of maximum dose ATV (80 mg), showed in Figure 7. This suggested that the ATV doses should be reduced by half when united with the LUT-NS-LP, for getting a safe and effective exposure level for ATV. This is a potential treatment protocol for clinical research in the future.
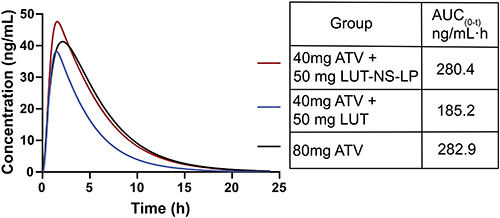 |
Figure 7 Predicted plasma concentration-time profiles of atorvastatin co-administrated with LUT-NS-LP in humans. The black line represents atorvastatin (80 mg) alone, the blue line represents atorvastatin (40 mg) united with LUT (50 mg), and the red line represents atorvastatin (40 mg) united with LUT-NS-LP (50 mg). The obtaining AUC(0-t) values with different co-administration plans are indicated on the right-hand side. |
Discussion
ATV is a classic first-line therapy for ASCVD, while recent reports suggest that LUT has notable lipid-lowering effects and may be a promising candidate molecule for it. Therefore, their combined utilization could offer synergistic treatment for ASCVD. Based on the ATV’s PK characteristics, its oral absolute bioavailability is approximately 14%, and it is commonly administered in its calcium salt form. LUT is a natural compound found in high amounts in plant-based diets, but it has low solubility and an oral absolute bioavailability under 5%, which can be enhanced within certain limits by nanotechnology without inducing adverse effects. Therefore, this study aims to improve the solubility and bioavailability of LUT to achieve good synergistic effects when combined with ATV.
This research employed an ultrasonic anti-solvent method to prepare LUT into nanosuspensions, which significantly improved its solubility and release profile. The substantial increase in the release rate can be attributed to the reduction in particle size afforded by nanotechnology, as described by the Noyes-Whitney equation, which increases the specific surface area of the drug and decreases the thickness of the diffusion layer.52 Based on the results of PXRD and DSC analyses the LUT-NS-LP are amorphous crystals, which typically have lower lattice energy than crystalline forms, thereby contributing to surface wettability and solubility. In addition, the prescribed stabilizers TPGS and P188 may enhance the solubility of LUT by further forming micelles. The LUT nanosuspensions possessed favorable characteristics, laying a solid foundation for subsequent DDI PK studies.
It is the first PK study, as far as we are aware, that ATV was combined with LUT-NS-LP to hope to achieve a synergistic effect on ASCVD. Our PK results confirmed that when LUT-NS-LP was co-administered with ATV, there were very significant changes for ATV in AUC(0-t) and Cmax. To explore the underlying mechanism, we first certified that ATV and LUT exist as obviously non-competitive inhibitory of enzyme kinetics in RLMs. Furthermore, numerous literatures also reported that ATV is transported into the hepatocytes via the OATP transporters on the basolateral membrane of the hepatocytes in the portal vein and subsequently metabolized in vivo mainly by the hepatic CYP450s.53 Moreover, some studies highlight a significant role of enterohepatic circulation in ATV disposition.54 Therefore, all of these factors are likely to be the available contributors to the DDI, which must be integrated to comprehensively assess the impact of LUT toward ATV.
The PBPK model provides an effective tool for drug development with various of functions, especially in evaluating DDIs. In the process of building DDI models, the construction of ATV models is vital. At present, seven PBPK models of ATV have been published, five models focusing on humans, and two models were established in rats. Our ATV model primarily focuses on one of the two rat PBPK modeling studies above, that of,18 who constructed a semi-PBPK model. This study has partially adopted his modeling concepts related to CYP3A and OATPs, while also incorporating the comprehensive tissue distribution and effects of enterohepatic circulation, for facilitating a more accurate description of the systemic exposure of ATV.
In addition, for the first time, the LUT PBPK model was established to accurately describe the behavior of LUT in rats. On this basis, a LUT-NS-LP PBPK model was specifically developed to detail the changes of LUT-NS-LP in vivo, but the model did not consider the potential lymphatic absorption for the LUT-NS-LP in the gastrointestinal tract.55 Because, LUT is a BCS class II drug that exhibits high permeability,19 the solubility of the drug is the key factor affecting its absorption. And it was confirmed that the solubility of LUT-NS-LP has been greatly improved in the release studies, even more, our PK studies have also shown that the AUC(0-t) of LUT-NS-LP has increased by 4.72-fold compared to native LUT. These indicated that LUT-NS-LP could be quickly released and easily permeated through the intestinal wall into the systemic circulation, be speculated that the absorption via permeation is significantly greater than lymphatic absorption.
Our DDI model about ATV and LUT-NS-LP was specially constructed following the ECC, which could more accurately predict whether transporters affect drug clearance.21 In the rat DDI simulated processing, it was discovered when the inhibition of ATV by LUT was only considered from single CYP450s or OATPs, the DDI model predictions fitted poorly and were lower than the observations, maybe underestimating the inhibitory effect of LUT on the metabolism or uptake of ATV. In contrast, when LUT simultaneously inhibits CYP450s and OATPs, the model’s capturing capability was enhanced obviously, the observed and predicted values of ATV were close, and exhibiting the model predictions were more accurate and reliable. Hence, we suggested that the combined inhibitory effect of LUT on both the CYP450s and the OATPs to ATV is the primary reason for the PK DDIs. Meanwhile, our parameter sensitivity analysis results advise that the role of OATPs-mediated transport is more significant than that of CYP450s-mediated metabolism. This finding is consistent with the observations reported.56,57
One of the most attractive features of the PBPK model is its scalability from one species to another species. In our studies, we first extrapolated the PBPK models of ATV and LUT from rats to humans and validated via clinical data, and then developed a DDI model for ATV and LUT-NS-LP. Since the information about two drugs in clinical research is poor, this DDI model is only used for the applied dosage recommendation for future research. By our predicted results, we recommend the combined doses for ATV and LUT-NS-LP were 40 mg and 50 mg, respectively. This co-administration dosage regimen which the applied dose for ATV was reduced by half, could ensure a similar therapeutic effect with the maximum dose ATV used alone by the guidelines, and also maintain ATV exposure level within the therapeutic window, thereby decreasing the toxic side effects caused by the high-dose ATV, might be drive a favorable synergistic effect with high efficiency and low toxicity on ASCVD. In the future, the DDI pharmacodynamics studies should be investigated and exposited.
Conclusion
In the current research, LUT-NS were prepared by ultrasound anti-solvent method, which significantly improved the solubility and in vitro release of LUT. Furthermore, assessed the enzyme kinetics interactions of ATV and LUT in RLMs and the PK interactions between LUT-NS-LP and ATV was investigated, revealing for the first time that LUT nanosuspension has a significant impact on the blood drug concentration of ATV. Furthermore, the PBPK model was used to predict the recommended doses for their clinical application. This study provides a theoretical and experimental foundation for the high efficiency and low toxicity co-application of statins with natural compounds and their nano-formulations.
Ethics Approval and Informed Consent
All animal experiments were approved by the Institutional Animal Care and Use Committee of China Medical University. The animal experiments were performed according to the guidelines of the committee in the experimental animal center of China Medical University.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Science Foundation of Liaoning Province (No.2020JH2/10300053).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Roeters van Lennep JE, Tokgözoğlu LS, Badimon L, et al. Women, lipids, and atherosclerotic cardiovascular disease: a call to action from the European Atherosclerosis Society. Eur Heart J. 2023;44(39):4157–4173. doi:10.1093/eurheartj/ehad472
2. Nedkoff L, Briffa T, Zemedikun D, et al. Global trends in atherosclerotic cardiovascular disease. Clin Ther. 2023;45(11):1087–1091. doi:10.1016/j.clinthera.2023.09.020
3. Cho L. A practical approach to the cholesterol guidelines and ASCVD prevention. Cleve Clin J Med. 2020;87(5 suppl 1):15–20. doi:10.3949/ccjm.87.s1.02
4. Ferhatbegović L, Mršić D, Kušljugić S, et al. LDL-C: the only causal risk factor for ASCVD. Why is it still overlooked and underestimated? Curr Atheroscler Rep. 2022;24(8):635–642. doi:10.1007/s11883-022-01037-3
5. Suryawanshi YN, Warbhe RA. Familial hypercholesterolemia: a literature review of the pathophysiology and current and novel treatments. Cureus. 2023;15(11):e49121. doi:10.7759/cureus.49121
6. Cai T, Abel L, Langford O, et al. Associations between statins and adverse events in primary prevention of cardiovascular disease: systematic review with pairwise, network, and dose-response meta-analyses. BMJ. 2021;374:374n1537. doi:10.1136/bmj.n1537
7. Beshir SA, Hussain N, Elnor AA, et al. Umbrella review on non-statin lipid-lowering therapy. J Cardiovasc Pharmacol Ther. 2021;26(5):437–452. doi:10.1177/10742484211002943
8. Taheri Y, Sharifi-Rad J, Antika G, et al. Paving luteolin therapeutic potentialities and agro-food-pharma applications: emphasis on in vivo pharmacological effects and bioavailability traits. Oxid Med Cell Longev. 2021;2021:1987588. doi:10.1155/2021/1987588
9. Ding X, Zheng L, Yang B, et al. Luteolin attenuates atherosclerosis via modulating signal transducer and activator of transcription 3-mediated inflammatory response. Drug Des Devel Ther. 2019;13:3899–3911. doi:10.2147/dddt.S207185
10. Priya TP, Manasa KS, Greeshmika C, et al. Nanosuspension technology: a review. 2021.
11. Balasubramanian R, Maideen NMP. HMG-CoA reductase inhibitors (statins) and their drug interactions involving CYP enzymes, P-glycoprotein and OATP transporters-an overview. Curr Drug Metab. 2021;22(5):328–341. doi:10.2174/1389200222666210114122729
12. Chow EC, Pang KS. Why we need proper PBPK models to examine intestine and liver oral drug absorption. Curr Drug Metab. 2013;14(1):57–79. doi:10.2174/138920013804545124
13. Li S, Yu Y, Jin Z, et al. Prediction of pharmacokinetic drug-drug interactions causing atorvastatin-induced rhabdomyolysis using physiologically based pharmacokinetic modelling. Biomed Pharmacother. 2019;119:109416. doi:10.1016/j.biopha.2019.109416
14. Hanke N, Gómez-Mantilla JD, Ishiguro N, et al. Physiologically based pharmacokinetic modeling of rosuvastatin to predict transporter-mediated drug-drug interactions. Pharm Res. 2021;38(10):1645–1661. doi:10.1007/s11095-021-03109-6
15. He W, Wu JJ, Ning J, et al. Inhibition of human cytochrome P450 enzymes by licochalcone A, a naturally occurring constituent of licorice. Toxicol In Vitro. 2015;29(7):1569–1576. doi:10.1016/j.tiv.2015.06.014
16. Liu Y, Ramírez J, House L, et al. Comparison of the drug-drug interactions potential of erlotinib and gefitinib via inhibition of UDP-glucuronosyltransferases. Drug Metab Dispos. 2010;38(1):32–39. doi:10.1124/dmd.109.029660
17. Watanabe T, Kusuhara H, Maeda K, et al. Investigation of the rate-determining process in the hepatic elimination of HMG-CoA reductase inhibitors in rats and humans. Drug Metab Dispos. 2010;38(2):215–222. doi:10.1124/dmd.109.030254
18. Wang Z, Yang H, Xu J, et al. Prediction of atorvastatin pharmacokinetics in high-fat diet and low-dose streptozotocin-induced diabetic rats using a semiphysiologically based pharmacokinetic model involving both enzymes and transporters. Drug Metab Dispos. 2019;47(10):1066–1079. doi:10.1124/dmd.118.085902
19. Zhou P, Li LP, Luo SQ, et al. Intestinal absorption of luteolin from peanut hull extract is more efficient than that from individual pure luteolin. J Agric Food Chem. 2008;56(1):296–300. doi:10.1021/jf072612
20. Yoshikado T, Yoshida K, Kotani N, et al. Quantitative analyses of hepatic OATP-mediated interactions between statins and inhibitors using PBPK modeling with a parameter optimization method. Clin Pharmacol Ther. 2016;100(5):513–523. doi:10.1002/cpt.391
21. Sugiyama Y, Aoki Y. A 20-year research overview: quantitative prediction of hepatic clearance using the in vitro-in vivo extrapolation approach based on physiologically based pharmacokinetic modeling and extended clearance concept. Drug Metab Dispos. 2023;51(9):1067–1076. doi:10.1124/dmd.123.001344
22. Hu ZY, Lu J, Zhao Y. A physiologically based pharmacokinetic model of alvespimycin in mice and extrapolation to rats and humans. Br J Pharmacol. 2014;171(11):2778–2789. doi:10.1111/bph.12609
23. Zhou K, Huo M, Ma W, et al. Application of a physiologically based pharmacokinetic model to develop a veterinary amorphous enrofloxacin solid dispersion. Pharmaceutics. 2021;13(5):602. doi:10.3390/pharmaceutics13050602
24. Yeom DW, Son HY, Kim JH, et al. Development of a solidified self-microemulsifying drug delivery system (S-SMEDDS) for atorvastatin calcium with improved dissolution and bioavailability. Int J Pharm. 2016;506(1–2):302–311. doi:10.1016/j.ijpharm.2016.04.059
25. Kwon J, Giri BR, Song ES, et al. Spray-dried amorphous solid dispersions of atorvastatin calcium for improved supersaturation and oral bioavailability. Pharmaceutics. 2019;11(9):461. doi:10.3390/pharmaceutics11090461
26. Alsmadi MM, Al-Daoud NM, Obaidat RM, et al. Enhancing atorvastatin in vivo oral bioavailability in the presence of inflammatory bowel disease and irritable bowel syndrome using supercritical fluid technology guided by wbPBPK modeling in rat and human. AAPS Pharm Sci Tech. 2022;23(5):148. doi:10.1208/s12249-022-02302-z
27. Sharma M, Mehta I. Surface stabilized atorvastatin nanocrystals with improved bioavailability, safety and antihyperlipidemic potential. Sci Rep. 2019;9(1):16105. doi:10.1038/s41598-019-52645-0
28. Kim JS, Kim MS, Park HJ, et al. Physicochemical properties and oral bioavailability of amorphous atorvastatin hemi-calcium using spray-drying and SAS process. Int J Pharm. 2008;359(1–2):211–219. doi:10.1016/j.ijpharm.2008.04.006
29. Li Z, Tao W, Zhang D, et al. The studies of PLGA nanoparticles loading atorvastatin calcium for oral administration in vitro and in vivo. Asian J Pharm Sci. 2017;12(3):285–291. doi:10.1016/j.ajps.2016.08.006
30. Ren Y, Li H, Liu X. Effects of Ginkgo leaf tablets on the pharmacokinetics of atovastatin in rats. Pharm Biol. 2019;57(1):403–406. doi:10.1080/13880209.2019.1622569
31. Sun S, Wang R, Fan J, et al. Effects of Danshen tablets on pharmacokinetics of atorvastatin calcium in rats and its potential mechanism. Pharm Biol. 2018;56(1):104–108. doi:10.1080/13880209.2018.1424209
32. Kong R, Zhu X, Meteleva ES, et al. Atorvastatin calcium inclusion complexation with polysaccharide arabinogalactan and saponin disodium glycyrrhizate for increasing of solubility and bioavailability. Drug Deliv Transl Res. 2018;8(5):1200–1213. doi:10.1007/s13346-018-0565-x
33. Kim MS, Jin SJ, Kim JS, et al. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2008;69(2):454–465. doi:10.1016/j.ejpb.2008.01.007
34. Lin LC, Pai YF, Tsai TH. Isolation of Luteolin and Luteolin-7-O-glucoside from Dendranthema morifolium Ramat Tzvel and their pharmacokinetics in rats. J Agric Food Chem. 2015;63(35):7700–7706. doi:10.1021/jf505848z
35. Chen X, Liu L, Sun Z, et al. Pharmacokinetics of luteolin and tetra-acetyl-luteolin assayed by HPLC in rats after oral administration. Biomed Chromatogr. 2010;24(8):826–832. doi:10.1002/bmc.1370
36. Shimoi K, Okada H, Furugori M, et al. Intestinal absorption of luteolin and luteolin 7-O-beta-glucoside in rats and humans. FEBS Lett. 1998;438(3):220–224. doi:10.1016/s0014-5793(98)01304-0
37. Tao Y, Su D, Li W, et al. Pharmacokinetic comparisons of six components from raw and vinegar-processed Daphne genkwa aqueous extracts following oral administration in rats by employing UHPLC-MS/MS approaches. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1079:34–40. doi:10.1016/j.jchromb.2018.02.005
38. Wang T, Xiao J, Hou H, et al. Development of an ultra-fast liquid chromatography-tandem mass spectrometry method for simultaneous determination of seven flavonoids in rat plasma: application to a comparative pharmacokinetic investigation of Ginkgo biloba extract and single pure ginkgo flavonoids after oral administration. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1060:173–181. doi:10.1016/j.jchromb.2017.05.021
39. Chen T, Li LP, Lu XY, et al. Absorption and excretion of luteolin and apigenin in rats after oral administration of chrysanthemum morifolium extract. J Agric Food Chem. 2007;55(2):273–277. doi:10.1021/jf062088r
40. Kim S, Seo JD, Yun YM, et al. Pharmacokinetics and genetic factors of atorvastatin in healthy Korean subjects. Front Genet. 2022;13:836970. doi:10.3389/fgene.2022.836970
41. Groenendaal-van de Meent D, den Adel M, Kerbusch V, et al. Effect of roxadustat on the pharmacokinetics of Simvastatin, rosuvastatin, and atorvastatin in healthy subjects: results from 3 Phase 1, open-label, 1-sequence, crossover studies. Clin Pharmacol Drug Dev. 2022;11(4):486–501. doi:10.1002/cpdd.1076
42. Whitfield LR, Porcari AR, Alvey C, et al. Effect of gemfibrozil and fenofibrate on the pharmacokinetics of atorvastatin. J Clin Pharmacol. 2011;51(3):378–388. doi:10.1177/0091270010366446
43. Hausner H, Derving Karsbøl J, Holst AG, et al. Effect of semaglutide on the pharmacokinetics of metformin, warfarin, atorvastatin and digoxin in healthy subjects. Clin Pharmacokinet. 2017;56(11):1391–1401. doi:10.1007/s40262-017-0532-6
44. McCrea JB, Menzel K, Adedoyin A, et al. Drug-drug interaction of letermovir and atorvastatin in healthy participants. Clin Pharmacol Drug Dev. 2022;11(4):420–428. doi:10.1002/cpdd.1071
45. Abdelkawy KS, Abdelaziz RM, Abdelmageed AM, et al. Effects of green tea extract on atorvastatin pharmacokinetics in healthy volunteers. Eur J Drug Metab Pharmacokinet. 2020;45(3):351–360. doi:10.1007/s13318-020-00608-6
46. Gundlach K, Wolf K, Salem I, et al. Safety of candesartan, amlodipine, and atorvastatin in combination: interaction study in healthy subjects. Clin Pharmacol Drug Dev. 2021;10(2):190–197. doi:10.1002/cpdd.787
47. Choi Y, Lee S, Jang IJ, et al. Pharmacokinetic interaction between fimasartan and atorvastatin in healthy male volunteers. Drug Des Devel Ther. 2018;12:2301–2309. doi:10.2147/dddt.S165171
48. Khalilieh S, Yee KL, Sanchez RI, et al. Results of a doravirine-atorvastatin drug-drug interaction study. Antimicrob Agents Chemother. 2017;61(2). doi:10.1128/aac.01364-16
49. Bullman J, Nicholls A, Van Landingham K, et al. Effects of lamotrigine and phenytoin on the pharmacokinetics of atorvastatin in healthy volunteers. Epilepsia. 2011;52(7):1351–1358. doi:10.1111/j.1528-1167.2011.03118.x
50. Backman JT, Luurila H, Neuvonen M, et al. Rifampin markedly decreases and gemfibrozil increases the plasma concentrations of atorvastatin and its metabolites. Clin Pharmacol Ther. 2005;78(2):154–167. doi:10.1016/j.clpt.2005.04.007
51. Kantola T, Kivistö KT, Neuvonen PJ. Effect of itraconazole on the pharmacokinetics of atorvastatin. Clin Pharmacol Ther. 1998;64(1):58–65. doi:10.1016/s0009-9236(98)90023-6
52. Kesisoglou F, Panmai S, Wu Y. Nanosizing--oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59(7):631–644. doi:10.1016/j.addr.2007.05.003
53. Hirota T, Fujita Y, Ieiri I. An updated review of pharmacokinetic drug interactions and pharmacogenetics of statins. Expert Opin Drug Metab Toxicol. 2020;16(9):809–822. doi:10.1080/17425255.2020.1801634
54. Black AE, Hayes RN, Roth BD, et al. Metabolism and excretion of atorvastatin in rats and dogs. Drug Metab Dispos. 1999;27(8):916–923. doi:10.1016/S0090-9556(24)15242-7
55. Rieux A, Fievez V, Garinot M, et al. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116(1):1–27. doi:10.1016/j.jconrel.2006.08.013
56. Duan P, Zhao P, Zhang L. Physiologically Based Pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug-drug Interactions (DDIs). Eur J Drug Metab Pharmacokinet. 2017;42(4):689–705. doi:10.1007/s13318-016-0383-9
57. Maeda K, Ikeda Y, Fujita T, et al. Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin Pharmacol Ther. 2011;90(4):575–581. doi:10.1038/clpt.2011.142
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.