")
Back to Journals » Drug Design, Development and Therapy » Volume 18
Evaluation of Olaparib Tablet Safety and Pharmacokinetics in Healthy Chinese Male Subjects
Authors Dong R, Chen J, Guo N , Yang Y, Wu J, Wang X, Song Y, Zhang X
Received 5 June 2024
Accepted for publication 13 November 2024
Published 3 December 2024 Volume 2024:18 Pages 5529—5539
DOI https://doi.org/10.2147/DDDT.S481481
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ruihua Dong,1,* Jingcheng Chen,1,* Nini Guo,2 Yingying Yang,2 Jingxuan Wu,1 Xiaoru Wang,2 Yuqin Song,1 Xueyuan Zhang2
1Phase I Clinical Trial Laboratory, Beijing Friendship Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Clinical Department, CSPC Zhongqi Pharmaceutical Technology (SJZ) Co., LTD., Shijiazhuang, Hebei Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xueyuan Zhang, Clinical Department, CSPC Zhongqi Pharmaceutical Technology (SJZ) Co., LTD., Shijiazhuang, Hebei Province, 050035, People’s Republic of China, Tel +86-311-69085585, Email [email protected]
Purpose: To evaluate the safety and pharmacokinetics of olaparib tablet test formulation (T) and reference formulation (R) in healthy Chinese male subjects.
Subjects and Methods: This was a single-dose, randomized-sequence, two-way crossover study including three parts: part A: a safety exploration design in lower dose (n = 14, 100 mg), part B: a pivotal comparative pharmacokinetic (PK) trial under fast condition (n = 44, 150 mg) and part C: a pivotal comparative PK trial under food condition (n = 44, 150 mg). Blood samples were collected for 72 hours and the PK parameters of Cmax, AUC0-t, and AUC0-∞ were used to evaluate PK differences.
Results: PK analysis of the two olaparib formulations showed that the Geometric Least Squares Mean (GLSM) ratio 90% confidence intervals for pivotal fasting Cmax, AUC0-t, and AUC0-∞ were 94.82– 108.97%, 92.94– 104.28%, and 92.81– 103.85%, respectively, and pivotal fed Cmax, AUC0-t, and AUC0-∞ were 82.78– 100.97%, 91.59– 104.67%, and 92.17– 104.76%, respectively. The 90% confidence interval of the two preparations, Cmax, AUC0-t, and AUC0-∞, all fall within the equivalent range of 80– 125%. Both olaparib tablet formulations were well tolerated, with no serious adverse events (SAE) or adverse events (AE) causing withdrawal occurred.
Conclusion: Two types of olaparib tablets were bioequivalent under both fasting and fed condition, and were generally well tolerated in healthy Chinese male subjects.
Keywords: olaparib, safety, pharmacokinetics, bioequivalence, healthy Chinese male subjects
Introduction
Poly (ADP-ribose) polymerase (PARP) inhibitors is a type of small molecule inhibitor that specifically targets PARP1 and PARP2.1 PARP-1 and −2 are essential for protecting DNA from radioactive and monofunctional alkylating agent’s damage. One mechanism of action of PARP inhibitors is the inhibition of PARP1 and PARP2, which inhibits DNA single-strand breaks from being repaired, resulting in double-strand breaks (DSBs). Another mechanism by which PARP inhibitors cause DSBs is by trapping the PARP enzyme-DNA complex on single-strand breaks.1,2 Healthy cells can repair DSBs via the functional homologous recombination repair mechanism, so PARP inhibitors are ineffective in them. Cells with homologous recombination deficiencies (HRD), especially pathogenic breast cancer (BRCA)-1 or BRCA-2 mutations, are more susceptible to PARP inhibitors.2 PARP inhibitors can induce synthetic lethality in tumors harboring BRCA1/2 mutations or other homologous recombination repair (HRR) pathway deficiencies. They are promising for the treatment of several tumors.
In 2014, the European Medicines Agency (EMA) approved olaparib, the first PARP inhibitor, to treat ovarian cancer. In patients with platinum-sensitive, relapsed, high-grade epithelial ovarian cancer who had responded to platinum-based treatment, olaparib had a good anti-tumor effect.3 Patients with germline or somatic BRCA1/2 mutations showed the largest benefit, while those with wild-type BRCA showed less benefit.4 PARP inhibitors are currently approved for the treatment of a variety of tumors, including ovarian, breast, pancreatic, and prostate cancers. The indications for PARP inhibitors are rapidly expanding, starting with monotherapy in patients with BRCA mutations and progressing to patients with other HRD and no HRD, as well as combinations with immunotherapy, targeted therapies, radiotherapy, and chemotherapy.1,5
Olaparib’s PK profile in patients showed that it was rapidly absorbed after oral administration and that a high-fat diet delayed its absorption rate but did not significantly alter the extent of absorption of olaparib.6 Population pharmacokinetic analyses showed that patient age, gender, body weight, or race (including Caucasian, Chinese, and Japanese) were not significant covariates.6,7 The most common side effects of olaparib were gastrointestinal toxicity (first nausea mostly occurred within 1 month of treatment initiation and first vomiting occurred within 2 months of treatment initiation), hematologic toxicity (median time to first occurrence of anemia was approximately 4 weeks), and laboratory tests. In ovarian and prostate cancers, the safety and efficacy of the Chinese population were consistent with those of other populations, but the PK and safety profiles of the healthy Chinese population are unknown.6
Since it is generally believed that PK bioequivalence can be used as a replacement for therapeutic equivalence between formulations, our research focused on PK parameter endpoints rather than pharmacodynamics (PD) endpoints.8 With reference to the acceptable safety profile of the comparable drug fluzoparib in healthy subjects,9–14 the safety and pharmacokinetics of the test formulation and the reference formulation of olaparib tablets were evaluated in healthy male subjects in our study. Furthermore, we administered olaparib (T/R) with a high-fat diet or fasting to assess the influence of food on drug bioavailability in the fasted and fed states by comparing olaparib systemic exposures (Cmax, AUC0-t, and AUC0-∞). The availability of generic drugs as soon as possible after the patent protection period of the branded drug has expired will help reduce the burden on the healthcare system and on the patient’s family.
Materials and Methods
Materials
The test formulation (T) was produced by CSPC Ouyi Pharmaceutical Co., Ltd. (Specification: 100 mg, batch number: R27220203 and 150 mg, batch number: R28220201). The reference formulation (R) was produced by AbbVie Limited (Specification: 100 mg, batch number: RJ159 and 150 mg, batch number: RM646).
Ethics and Subjects
This study protocol and amendments were approved by the Bioethics Committee of Beijing Friendship Hospital, Capital Medical University and Center For Drug Evaluation (Approval No. 2022-P1-015-02). The trial was registered at www.chinadrugtrials.org.cn. (Registered number: CTR20221003) and ClinicalTrials.gov. (Registered number: NCT06360445). The International Conference on Harmonization Guidelines for Good Clinical Practice, the ethical standards of the Declaration of Helsinki, and the relevant domestic laws and regulations were followed in conducting this experiment. Before participating in this study, each individual provided written informed consent.
The subjects of this trial were healthy males aged 18–50 years, with a body mass index (BMI) in the range of 19–26 kg/m2 and weight ≥50 kg. A subject whose immediate family members had no breast, ovarian, pancreatic, prostate cancer, and other related diseases. A series of vital signs, physical and laboratory examinations including hematology, blood biochemistry, coagulation function, urinalysis, Chest Computed Tomography (CCT), and 12-lead electrocardiographic (ECG) examination were applied to determine the physical fitness of the subjects.
A significant medical history, a history of drug or food allergies, a history of drug abuse or a positive urine drug screening result, a history of alcoholism or a positive alcohol breath test, a history of blood donation, or a history of participation in another clinical trial within the previous 3 months were all considered exclusion criteria. Subjects who could not abstain from smoking and/or drinking during the study period, those who used contraindicated drugs (CYP3A4 inhibitors and inducers, p-gp inhibitors, or other drugs), and those who had surgery or plan to undergo surgery during the study period were excluded. Subjects with any clinically significant abnormalities, those who were unable to observe the dieting protocol of this study, or lactose intolerance were also excluded. Without a family plan or sperm donation plan, subjects are required to utilize effective contraception throughout the research and for 6 months following the last dose.
Study Design
A pilot study (Part A) and two pivotal studies (Part B and Part C) formed the clinical trial. The purpose of Part A was to determine the safety of olaparib (100 mg) in healthy subjects. In Part B and Part C, the maximum specification dose (150 mg) of olaparib was utilized to evaluate the safety and pharmacokinetics of the reference preparation and test preparation.
According to previous studies, the inter-individual variation in the AUC of Olaparib is 55–57%.15 Assuming intra-individual variation was 26% of AUC, at least 40 subjects were required for Part B and Part C to meet the equivalence interval of 80–125% (α was 0.05, 1-β was 90%). Considering a dropout rate of 10%, 44 healthy male volunteers were enrolled in Part B and Part C, respectively. Fourteen healthy male volunteers were included in Part A. No statistical assumptions were considered.
Subjects were randomly divided into sequence TR or sequence RT, and the trial was conducted in two periods with a 7-day washout period between doses. In each period, subjects received a single dose of olaparib (T or R) under fasting (Part A, Part B) or after consuming a high-fat breakfast (Part C). Figure 1 shows the main design and flow of this study. All subjects continued to fast for at least 4 h after the dose.
 |
Figure 1 Study flow chart. Flow chart of the study in part A (A). Flow chart of the study in part B (B). Flow chart of the study in part C (C). Abbreviation: N, the number of subjects. |
Blood samples for the determination of olaparib PK analysis were collected at 0 h, 0.25 h, 0.50 h, 1.0 h, 1.5 h, 2.0 h, 3.0 h, 4.0 h, 5.0 h, 6.0 h, 7.0 h, 8.0 h, 10 h,12 h, 24 h, 48 h and 72 h in each period. After centrifuging at 1700 g for 10 min at 2–8°C, the plasma was stored at −80°C and transported to Beijing Scinovo Laboratories Ltd., where concentrations of olaparib were determined by protein-precipitation extraction and analysis using high-performance liquid chromatography-tandem mass spectrometric (HPLC-MS/MS) detection (positive ion mode).
Bioanalytical Methods
The concentrations of olaparib in plasma in healthy subjects were measured by a newly developed and validated HPLC-MS/MS method. The calibration curve range was 10.0–5000 ng/mL. The lower limits of quantitation (LLOQ) of olaparib were 10.0 ng/mL. The intra-batch accuracy and precision were 0.37% to 7.83% and 5.55% (maximum precision) at the LLOQ and −7.83% to 3.94% and 4.21% (maximum precision) at other quality controls (QCs). The inter-batch accuracy and precision were 3.32% and 5.46% (maximum precision) at the LLOQ and −3.47% to 1.27% and 5.22% at other QCs.
PK Parameters and Statistical Analysis
The PK parameters of olaparib were calculated using a standard non-compartmental on Phoenix WinNonlin software (8.3.4, Pharsight Certara). The primary PK parameters are maximum plasma concentration (Cmax), AUC0-t, and AUC0-∞. The secondary PK parameters, including time of Cmax (Tmax), apparent terminal elimination rate (λz), t1/2, CL/F, and Vd/F were also analyzed.
To assess the Least Square Mean (LSM) difference values of the primary PK parameters and 90% confidence interval (90% CI) of the difference values between the tested preparation and the reference preparation, and then to obtain the Geometric Least squares mean (GLSM) ratio of the corresponding PK parameters and the 90% CI of the ratio by taking the opposition number.
If the 90% CI of the GLSM ratios of Cmax, AUC0-t, and AUC0–∞ were completely within the acceptance limit range of 80–125%, bioequivalence was accepted between the tested preparation and the reference preparation.
Safety Analysis
In this study, physical examinations, vital signs, laboratory tests, 12-lead ECGs, and CT scans were all monitored. Treatment-Emergent Adverse Event (TEAE), including all subjective symptoms reported by subjects and/or observed by investigators, and objective parameters monitored by investigators, were collected throughout the study. The Common Terminology Criteria for Adverse Events V5.0 (CTCAE 5.0) were used to grade adverse events.
Results
General Characteristics of the Subjects
A total of 14, 44, and 44 subjects were ultimately enrolled in Part A, Part B, and Part C, respectively. The above subjects were randomly divided into two sequences (TR or RT) at a ratio of 1:1 in the corresponding study. The baseline characteristics, including age, height, weight, or body mass index of the subjects were well-balanced between the two sequences in each study. The demographic and baseline characteristics of all subjects are presented in Table 1. One subject with a random number (C022, sequence TR) withdrew voluntarily from Part C after completing the blood sample collection of the first period, the other subjects completed correspondingly both treatment periods.
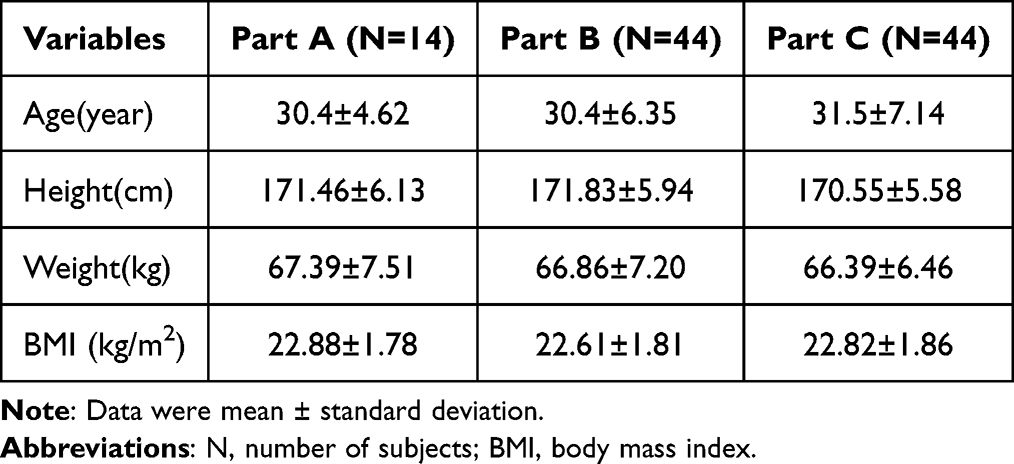 |
Table 1 Demographic Information and Baseline Information of Subjects in the Study |
PK Results
After a single dose orally, the test and reference drugs had similar mean plasma concentration–time curves profiles at fasting and fed status (Figure 2 and Table 2). The means ± SD of the primary PK parameters of the test and reference preparations in study B were as follows: Cmax were 2.98 ± 0.64 and 3.01 ± 0.96 μg/mL; AUC0-t were 12.39 ± 3.63 and 12.94 ± 4.88 h‧μg/mL, AUC0–∞ were 12.59 ± 3.69 and 13.17 ± 4.91 h‧μg/mL. The median (Min-Max) of Tmax were 1.00(0.5–3) h and 1.50(0.5–3) h.
 |
Table 2 The Main Pharmacokinetic Parameters of Olaparib After Taking the Test or Reference Drugs (Based on PKCS) |
 |
Figure 2 Mean plasma concentration–time curves of test formulation and reference formulation of olaparib at fasted (A) or fed status (B). Notes: Test: olaparib test formulation; Reference: olaparib reference formulation. |
The means ± SD of the primary PK parameters of the test and reference preparations in study C were as follows: Cmax were 2.40 ± 0.93 and 2.57 ± 0.83 μg/mL, AUC0-t were 12.26 ± 4.97 and 12.22 ± 4.06 h‧μg/mL, AUC0–∞ were 12.58 ± 5.04 and 12.50 ± 4.09 h‧μg/mL. The median (Min-Max) of Tmax were 2.50(1–5) h and 3.00(1–8) h.
In study B, the GLSM ratios (90%) of Cmax, AUC0-t, and AUC0–∞ between the test preparation and the reference preparation were 94.82%–108.97%, 92.94%–104.28%, and 92.81%–103.85%, respectively, within the equivalent interval 80.00%–125.00%. In study C, the GLSM ratios (90%) of Cmax, AUC0-t and AUC0–∞ between the test preparation and the reference preparation were 82.78%–100.97%, 91.59%–104.67%, and 92.17%–104.76%, respectively, also within the equivalent range of 80.00%–125.00%. All of the data indicated that the PK parameters of test preparation were similar to those of reference preparation, and the two preparations were bioequivalent (Table 3).
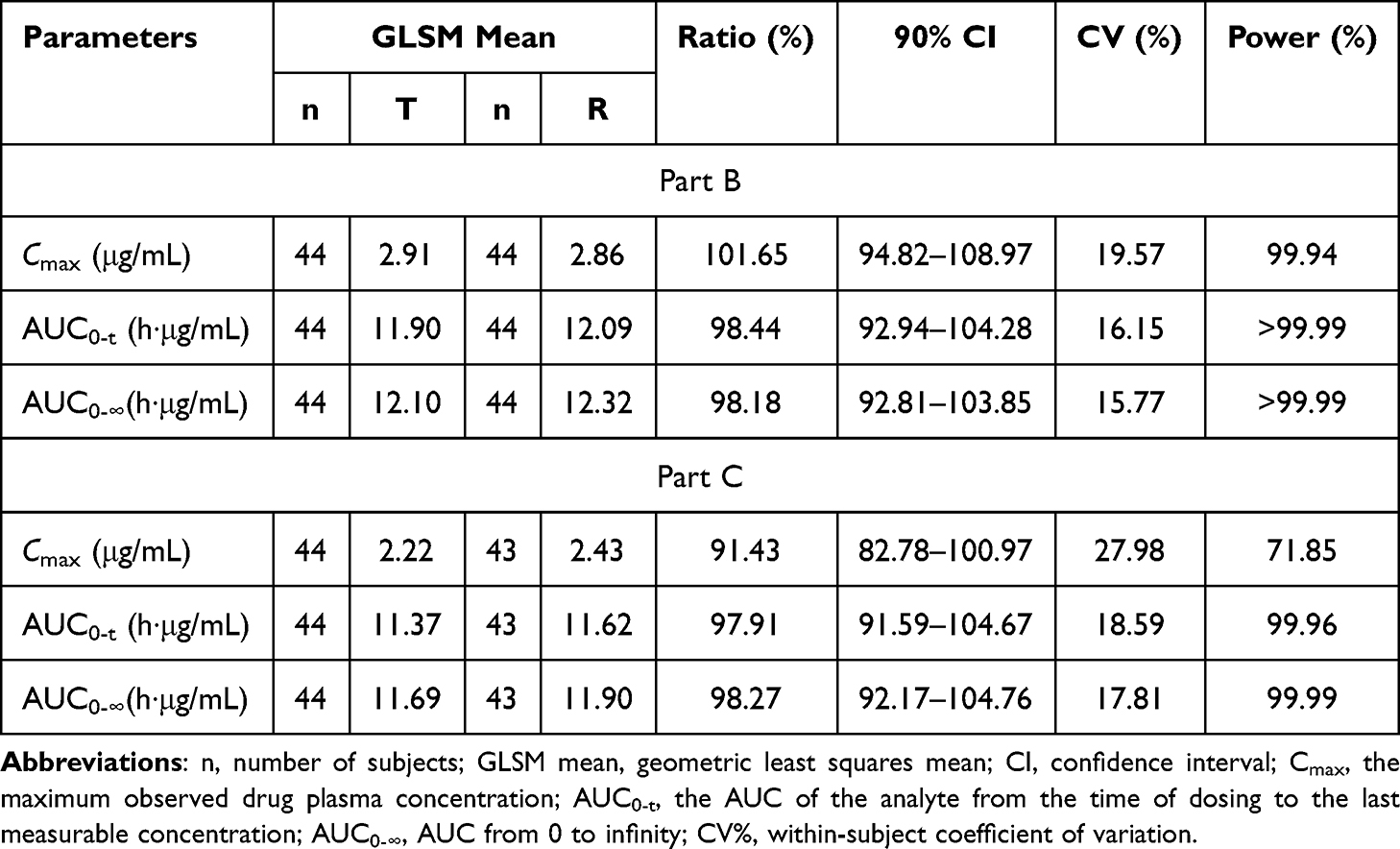 |
Table 3 Geometric Least Squares Mean Ratios (GLSMR) and 90% CIs for Cmax, AUC0–T, and AUC0–∞ Following Administration of Olaparib 150 mg (T/R) in Healthy Male Subjects (Based on BES) |
Effect of Food
For the test drug, the mean Tmax was about 1.28 h and 2.76 h under fasted and fed conditions; the mean Cmax was about 2.98 μg/mL and 2.40 μg/mL under fasted and fed conditions. Compared with fasting, Tmax was about 1.5 h later and Cmax was about 20% lower after a high-fat meal, the mean Tmax and Cmax were significantly different in the fed state compared with the fasting state (P < 0.05). But there was no significant difference in AUC0-t and AUC0-∞ (P > 0.05). For reference drug, mean Tmax was about 1.60 h and 2.72 h under fasted and fed conditions; mean Cmax was about 3.01 μg/mL and 2.57 μg/mL under fasted and fed conditions. Compared with fasting, Tmax was about 1.5 h later and Cmax was about 15% lower after a high-fat diet, the mean Tmax and Cmax were significantly different in the fed state compared with the fasted state (P < 0.05). But there was no significant difference in AUC0-t and AUC0-∞ (P > 0.05).
According to the summary of product characteristics of olaparib, when a high-fat, high-calorie meal combination with olaparib, the rate of absorption (Tmax delayed by 2.5 h) of olaparib slowed, the extent of olaparib absorption (mean AUC increased by approximately 8%) did not significantly alter. This study’s results were in compliance with the summary of product characteristics.
Safety
During Part A, Part B, and Part C, safety was monitored throughout each study. No SAE and TEAE that led to early withdrawal from the study were reported during all studies. All TEAEs were not treated with medication and spontaneously disappeared during the observation. All 102 subjects received at least one dose of the study drug and were included in the safety analyses.
In study A, 10 TEAEs were reported in 7 (50.0%) subjects. All TEAEs were adverse drug reactions (ADR), with the exception of one case (blood uric acid increased) that happened after leaving the hospital and was determined by the investigator to be unrelated to the drug. All TEAEs were grade 1. The results indicated that the safety of the reference preparation and the test preparation was similar and the two preparations were well tolerated in Chinese healthy male subjects (Table 4).
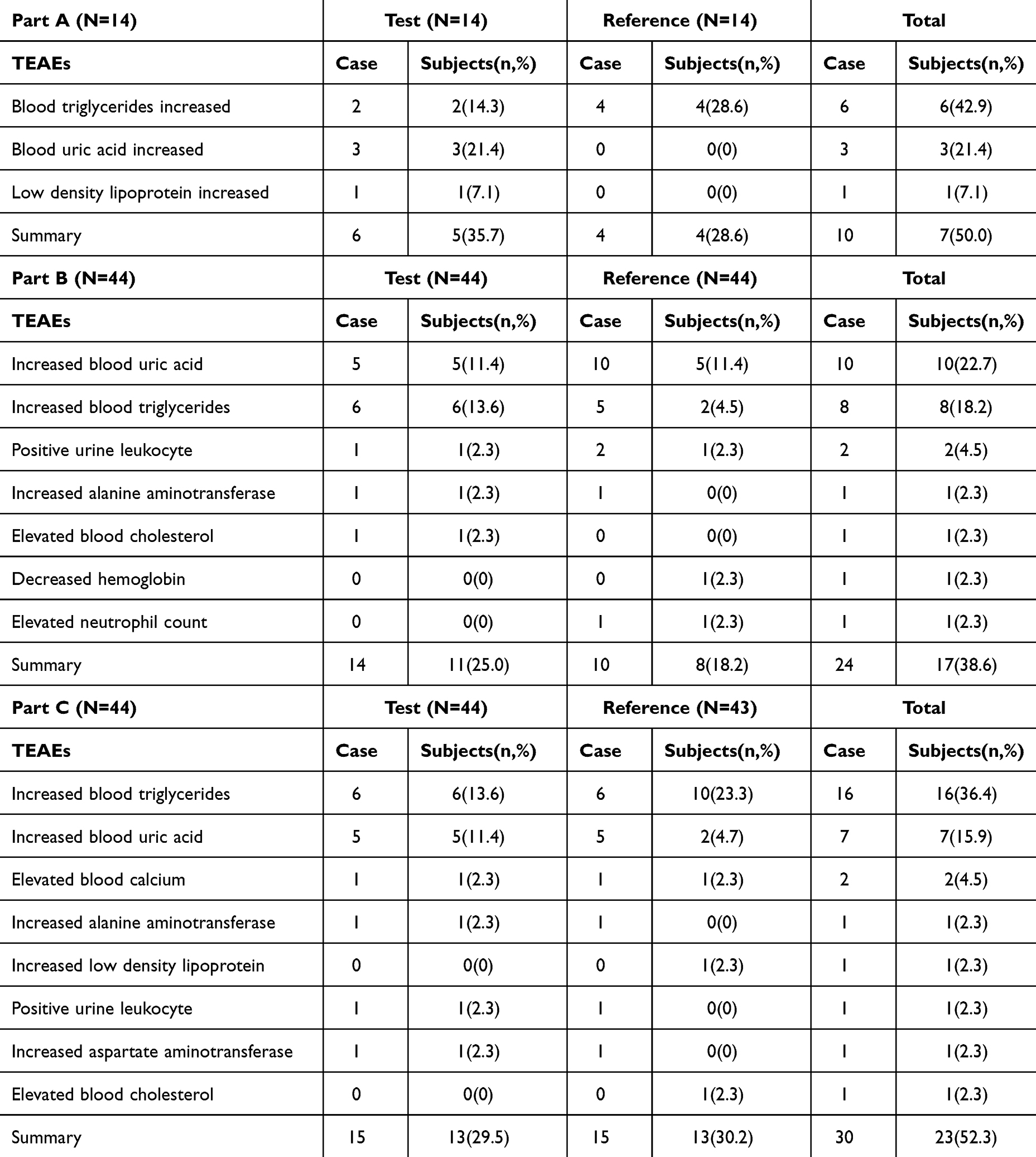 |
Table 4 TEAEs Receiving Olaparib 100 Mg and 150 Mg (T/R) in Healthy Male Subjects (SS) |
In study B, of the 44 subjects, a total of 17 (38.6%) subjects reported 24 TEAEs. Eleven (25.0%) of 44 subjects reported 14 TEAEs after taking the test preparation; 8(18.2%) of the subjects reported 10 TEAEs after taking the reference preparation. During the study, the most common TEAEs occurred in more than two subjects were increased blood uric acid, followed by increased blood triglycerides and positive urine leukocytes. All TEAEs were ADR. Among the 24 TEAEs, one (increased blood triglycerides) was grade 2 according to CTCAE guidelines, and the remainder were grade 1. The safety of the reference preparation and the test preparation was comparable (Table 4).
In study C, a total of 23 (52.3%) subjects reported 30 TEAEs, out of 44 subjects. Thirteen (29.5%) subjects reported 15 TEAEs after taking the test preparation, and 13 (30.2%) reported 15 TEAEs after taking the reference preparation. All TEAEs were ADR. Among the 30 TEAEs, one (increased blood triglycerides) was grade 3 according to CTCAE guidelines, and the remainder were grade 1 (Table 4).
Discussion
Olaparib is a selective inhibitor of PARP-1 and PARP-2 that is approved by many countries and effective in many cancer patients, particularly those harboring breast cancer gene (BRCA)1/2 mutations. There is currently a lack of research on the safety and tolerability of olaparib tablets in healthy subjects and along with evidence on the effects of food on drug bioavailability.
The safety and pharmacokinetics characteristics of olaparib preparation were studied in Chinese healthy male volunteers in this study, and the results showed that the test preparation and the reference preparation were well tolerated in healthy male volunteers, with no significant difference in safety profile, and the two preparations were bioequivalent in both fasting and fed states.
All TEAEs in this study were laboratory results, no gastrointestinal disorders occurred and all TEAEs were mild or moderate (grades 1 and 2) in severity, except for one subject who reported having blood triglycerides increased (grade 3), all TEAEs were treated without any therapeutic measures, and all recovered spontaneously at the end of the study. The safety profile of olaparib in healthy subjects in this study was consistent with that of fluzoparib in healthy subjects, with none of the SAEs occurring and being well tolerated.9–14
Although olaparib tablets were safe and well tolerated in the Chinese healthy population in this study, due to the limited sample size of this study, the safety and tolerability of olaparib tablets need to be supplemented and supported by more data from subsequent studies.
This study enrolled healthy individuals rather than cancer patients due to the following factors: the acceptable safety profile of fluzoparib, a PARP inhibitor approved by NMPA, in healthy subjects, the low mutation rate of BRCA1/2 in the Chinese healthy population, and the other considerations. First, six clinical pharmacology studies of fluzoparib in healthy individuals demonstrated that the drug was well tolerated by healthy individuals.9–14 Second, population screening research of 11386 Chinese Han individuals to discover BRCA variants revealed a prevalence of BRCA pathogenic variation of 0.38% in the Chinese Han general population.16 This was supported by the results that all BRCA gene mutations in 92 (90%) subjects who were able to participate in the follow-up of this study were negative. What is more, participants would not be allowed to participate if any member of their immediate family had prostate, breast, ovarian, pancreatic, or any other associated disease. These measures further decreased the likelihood of enrolling a carrier of a BRCA pathogenic mutation in this trial.17 Third, to better understand olaparib’s safety profile in the real world, we searched the FDA Adverse Event Reporting System, which is one of the world’s largest spontaneous adverse event reporting systems. 10,957 adverse events received that were related to olaparib from 2008 through the first quarter of 2023, according to a search of the FDA Adverse Event Reporting System. However, no adverse events were connected to olaparib’s genotoxicity.18 Fourth, this study is a single-dose PK study, and the clinical protocol requires strict contraception for 6 months after the last dose, which is longer than the required time of the FDA guideline for genotoxic antineoplastic drugs, which requires contraception for 3 months after the last dose,19 so our study can adequately protect the subjects’ safety. Fifth, the maximum dose administered in this study was 150 mg given as a single dose, which is lower than the clinically effective dose of olaparib, 300 mg given twice a day, and carries a decreased risk of side effects. Finally, when compared to patients, healthy individuals were the most homogeneous population, highlighting the PK difference between the test drug and the reference drug. Similarly, single-dose PK studies are more likely than multiple-dose PK studies to detect the difference between formulation technologies. Olaparib is a noncytotoxic medicine, and it is acceptable to use healthy volunteers in a single-dose PK study if subjects with a family history of cancer are excluded.
When considering the potential embryotoxicity of a drug, such as nintedanib esylate soft gelatin capsules and lenalidomide capsules, the FDA guidance on bioequivalence studies recommends that only male subjects be included in bioequivalence studies;20,21 the EMA bioequivalence guideline also states that bioequivalence studies may be conducted in either sex.22 Because population PK analysis revealed that gender was not a significant covariate and non-clinical research has suggested that olaparib may be embryotoxic, we decided to conduct the PK investigation in male individuals in this study, without influencing the assessment of the PK between formulations.
In Part B and Part C, after single-dose administration of olaparib (150 mg) to 87 evaluable healthy male volunteers in a fasting or fed state. The two olaparib (150 mg) formulations were both rapidly absorbed, the median Tmax of the test and reference preparations were 1 h and 1.5 h, respectively, at fasting, and 2.50 h and 3.00 h, respectively, at fed. The elimination profiles of the two preparations were also identical, whether fasting or fed state, as demonstrated by test preparation and reference preparation’s mean t1/2 values of 5.88 h and 5.94 h at fasting, respectively, and 6.39 h and 6.84 h at fed, respectively. Notably, the Tmax of olaparib was significantly prolonged in the fed state compared to the fasting state, but there was no significant change in Cmax or AUC, implying that food slows the rate of absorption (Tmax) and peak exposure to olaparib (Cmax), but has no effect on the extent of drug absorption (AUC), which is consistent with the specification results and the results of the food effect study in advanced solid tumors patients.6,23
The results from this study have demonstrated that the PK profile in Chinese healthy subjects is consistent with those previously reported for Chinese patient populations and Japanese patient populations after single dosing (median Tmax: Chinese healthy subjects, olaparib 150 mg 1.5 h; Chinese patients, olaparib 100 mg and 300 mg, 1.95 h, and 1.5 h, respectively; Japanese patients, olaparib 300 mg, 1.98 h; arithmetic means t1/2: Chinese healthy subjects, olaparib 150 mg 5.94 h; Chinese patients, olaparib 100 mg and 300 mg, 7.17 h and 6.52 h, respectively; Japanese patients, olaparib 300 mg, 9.43 h).23,24 Geometric means of Cmax and AUC after single administration of olaparib in healthy Chinese subjects, Chinese patients, and Japanese patients with comparable ranges of exposure after dose normalized to 100 mg (Figure 3).23,24 These observations are in line with the population PK analyses for pooled studies using both capsule and tablet formulations, which did not identify race as a significant covariate.7
 |
Figure 3 Comparison of geometric means(±SD) Cmax (A) and AUC (B) after a single administration of olaparib tablets with a dose normalized to 100 mg in healthy Chinese subjects, Chinese patients, and Japanese patients. Notes: 150 mg Chinese healthy subjects: a single dose of 150 mg olaparib tablet was administered to healthy Chinese subjects; 100 mg Chinese healthy subjects: a single dose of 100 mg olaparib tablet was administered to healthy Chinese subjects; 300 mg Japanese patients: a single dose of 300 mg olaparib tablet was administered to Japanese patients; 100 mg Chinese patients: a single dose of 100 mg olaparib tablet was administered to Chinese patients; 300 mg Chinese patients: a single dose of 300 mg olaparib tablet was administered to Chinese patients. |
The greatest intra-individual coefficient of variation (CV) for the primary parameters of olaparib was 19.57% and 27.98% in fasting and fed conditions in healthy subjects, respectively. Olaparib is not a highly variable drug. The mean (±SD) plasma drug concentration–time curves for each subject are shown in Figure 2A (fasted) and Figure 2B (fed). The two formulations are comparable at either a fasted or fed state because their 90% CIs for three primary PK parameters of Cmax, AUC0-t, and AUC0–∞ all fall within the pre-defined bioequivalence range of 80%–125%. The results showed that the test or reference drugs were bioequivalent.
Conclusion
Olaparib has been approved for the treatment of several tumor types and has demonstrated potential therapeutic outcomes, but there has been a lack of safety data for this product in healthy subjects. Olaparib was well tolerated in Chinese healthy subjects in this study, and there was no significant difference in safety between the test formulation and reference formulation. Besides, the two formulations of olaparib were bioequivalent to each other in both the fed and fasted status. Furthermore, high-fat food could slow the rate of absorption and peak exposure to olaparib but has no effect on the extent of drug absorption. The results of our study have significance not only for accelerating the marketing of generic drugs and reducing the burden of medication on patients, but also for providing a reference to regulatory authorities’ establishment of bioequivalence guidelines for this product.
Data Sharing Statement
The datasets generated during and/or analysed during the current study are available in the Centre for Drug Evaluation, NMPA repository, www.chinadrugtrials.org.cn., with the identification code CTR20221003 and, with the registered number: NCT06360445. The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethical Approval and Informed Consent
This study was performed in line with the principles of the Declaration of Helsinki. The study protocol, protocol amendments, and all applicable documents (including informed consent form) were reviewed and approved by the Bioethics Committee of Beijing Friendship Hospital, Capital Medical University. Informed consent was obtained from all individual participants included in the study.
Acknowledgments
The authors would like to thank all the subjects who participated in this trial, as well as those individuals who helped with the trial.
Funding
This work was supported by the sponsor of this study, CSPC Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., LTD.
Disclosure
The authors have no relevant financial or non-financial interests to disclose.
References
1. Ohmoto A, Yachida S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. OTT. 2017;10:5195–5208. doi:10.2147/OTT.S139336
2. Cook SA, Tinker AV. PARP inhibitors and the evolving landscape of ovarian cancer management: a review. BioDrugs. 2019;33(3):255–273. doi:10.1007/s40259-019-00347-4
3. Ledermann J, Harter P, Gourley C, et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N Engl J Med. 2012;366(15):1382–1392. doi:10.1056/NEJMoa1105535
4. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised Phase 2 trial. Lancet Oncol. 2014;15(8):852–861. doi:10.1016/S1470-2045(14)70228-1
5. Geenen JJJ, Linn SC, Beijnen JH, Schellens JHM. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin Pharmacokinet. 2018;57(4):427–437. doi:10.1007/s40262-017-0587-4
6. AstraZeneca Pharmaceuticals LP. LYNPARZA® (Olaparib) Tablets [Prescribing Information]. Wilmington: AstraZeneca Pharmaceuticals LP; 2023.
7. Zhou D, Li J, Bui K, et al. Bridging Olaparib Capsule and Tablet Formulations Using Population Pharmacokinetic Meta-analysis in Oncology Patients. Clin Pharmacokinet. 2019;58(5):615–625. doi:10.1007/s40262-018-0714-x
8. US Food and Drug Administration [homepage on the Internet]. Silver Spring: draft guidance for industry on bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA application. 2021. Available from: https://www.fda.gov/media/87219/download.
9. Li L, Xiang Y-X, Yang G-P, et al. Pharmacokinetic effects of proton pump inhibitors on the novel PARP inhibitor fluzoparib: a single-arm, fixed-sequence trial in male healthy volunteers. Invest New Drugs. 2021;39(3):796–802. doi:10.1007/s10637-020-01034-w
10. Jiang X, Tao Y, Liu Y, et al. A randomized, open‐label, two‐period crossover bridging study on fuzuloparib capsules of different specifications in healthy Chinese volunteers. Br J Clin Pharm. 2022;88(3):1087–1093. doi:10.1111/bcp.15035
11. Wu M, Li X, Sun J, Chen H, Ding Y. A phase I study of fluzoparib tablet formulation, an oral PARP inhibitor: effect of food on the pharmacokinetics and metabolism after oral dosing in healthy Chinese volunteers. Expert Opin Drug Metab Toxicol. 2021;17(4):503–508. doi:10.1080/17425255.2021.1881480
12. Chen X, Yang F, Zhao J, et al. Effect of fluconazole on the pharmacokinetics of fuzuloparib: an open-label, crossover study in Chinese healthy male volunteers. Cancer Chemother Pharmacol. 2022;89(1):141–148. doi:10.1007/s00280-021-04376-1
13. Hu C, Zhang Y, Pei T, Liu P, Zhang L. Itraconazole interferes in the pharmacokinetics of fuzuloparib in healthy volunteers. Cancer Chemother Pharmacol. 2023;91(6):523–529. doi:10.1007/s00280-023-04536-5
14. Hu L, Dou T, Sun Q, et al. Effect of a moderate CYP3A inducer efavirenz on the pharmacokinetics of fuzuloparib: an open-label, fixed sequence study in Chinese healthy male subjects. Invest New Drugs. 2023;41(2):276–283. doi:10.1007/s10637-023-01331-0
15. Plummer R, Swaisland H, Leunen K, et al. Olaparib tablet formulation: effect of food on the pharmacokinetics after oral dosing in patients with advanced solid tumours. Cancer Chemother Pharmacol. 2015;76(4):723–729. doi:10.1007/s00280-015-2836-2
16. Dong H, Chandratre K, Qin Y, et al. Prevalence of BRCA1 / BRCA2 pathogenic variation in Chinese Han population. J Med Genet. 2021;58(8):565–569. doi:10.1136/jmedgenet-2020-106970
17. Momozawa Y, Sasai R, Usui Y, et al. Expansion of cancer risk profile for BRCA1 and BRCA2 pathogenic variants. JAMA Oncol. 2022;8(6):871. doi:10.1001/jamaoncol.2022.0476
18. US Food and Drug Administration [homepage on the Internet]. Silver Spring: FAERS. 2023. Available from: https://www.fda.gov/drugs/questions-and-answers-fdas-adverse-event-reporting-system-faers/fda-adverse-event-reporting-system-faers-public-dashboard.
19. US Food and Drug Administration [homepage on the Internet]. Silver spring: oncology pharmaceuticals: reproductive toxicity testing and labeling recommendations. 2019. Available from: https://www.fda.gov/media/124829/download.
20. US Food and Drug Administration [homepage on the Internet]. Silver spring: draft guidance on nintedanib esylate. 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Nintedanib%20esylate_oral%20capsule_205832_RC09-15.pdf.
21. US Food and Drug Administration [homepage on the Internet]. Silver spring: draft guidance on lenalidomide. 2013. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Lenalidomide_draft_Oral%20cap_RLD%2021880_RC11-13.pdf.
22. European Medicines Agency [homepage on the Internet]. Amsterdam: guideline on the investigation of bioequivalence. 2010. Available from: https://www.ema.europa.eu/en/investigation-bioequivalence-scientific-guideline.
23. Yuan P, Shentu J, Xu J, et al. Pharmacokinetics and safety of olaparib tablets as monotherapy and in combination with paclitaxel: results of a Phase I study in Chinese patients with advanced solid tumours. Cancer Chemother Pharmacol. 2019;83(5):963–974. doi:10.1007/s00280-019-03799-1
24. Yonemori K, Tamura K, Kodaira M, et al. Safety and tolerability of the olaparib tablet formulation in Japanese patients with advanced solid tumours. Cancer Chemother Pharmacol. 2016;78(3):525–531. doi:10.1007/s00280-016-3106-7
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Evaluation of Pharmacokinetics and Safety with Bioequivalence of Ibuprofen Sustained-Release Capsules of Two Formulations, in Chinese Healthy Volunteers: Bioequivalence Study
Huang C, Yin Z, Yang Y, Mo N, Yang H, Wang Y
Drug Design, Development and Therapy 2023, 17:1881-1888
Published Date: 23 June 2023
Comparative Pharmacokinetics and Bioequivalence Evaluation of Two Formulations of Pramipexole Dihydrochloride Extended-Release Tablets in Healthy Chinese Subjects Under Fasted and Fed States: A Randomized, Open-Label, Single-Dose, Two-Period Crossover Clinical Trial
Yang L, Zhang L, Luo Z
Drug Design, Development and Therapy 2023, 17:2369-2381
Published Date: 15 August 2023
Effect of Food on the Pharmacokinetics of Tenofovir Amibufenamide: A Phase I, Randomized, Open-Label, Two-Period Crossover Trial in Healthy Adult Subjects

Liu J, Wu M, Kai J, Lin M, Zheng Y, Jiang Y, Huang Q, Zhai Y, Qiu Y
Drug Design, Development and Therapy 2023, 17:3061-3072
Published Date: 9 October 2023
The Pharmacokinetics, Safety and Tolerability of Aclidinium Bromide 400 μg Administered by Inhalation as Single and Multiple (Twice Daily) Doses in Healthy Chinese Participants
Li W, Daoud SZ, Trivedi R, Lukka PB, Jimenez E, Molins E, Stewart C, Bharali P, Garcia-Gil E
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:2725-2735
Published Date: 27 November 2023
Pharmacokinetics and Bioequivalence of Two Fixed-Dose Combination Tablets of Valsartan/Amlodipine (80/5 Mg) in Healthy Chinese Subjects
Tian M, Huang J, Chen Y, Jin Q, Jiang H, Shi C, Mei J, Xu M, Yu X, Yang S
Drug Design, Development and Therapy 2025, 19:11-22
Published Date: 3 January 2025