")
Back to Journals » Journal of Pain Research » Volume 18
Exploring the Interaction Mechanisms of Antibody-Mediated Immune Responses with Gout and Rheumatoid Arthritis Through a Bidirectional Two-Sample Mendelian Randomization Study
Authors Zhou H , Liu Q , Lv D, Zheng Y , Xing Z
Received 16 December 2024
Accepted for publication 9 May 2025
Published 27 May 2025 Volume 2025:18 Pages 2707—2738
DOI https://doi.org/10.2147/JPR.S508813
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor King Hei Stanley Lam
Hanyu Zhou,1 Qiaolin Liu,1 Duanbo Lv,1 Yinghao Zheng,1 Zhenlong Xing2
1Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China; 2Department of Joint Orthopedics, Guangdong Provincial Hospital of Integrated Traditional and Western Medicine, Foshan, Guangdong, People’s Republic of China
Correspondence: Zhenlong Xing, Department of Joint Orthopedics, Guangdong Provincial Hospital of Integrated Traditional and Western Medicine, No. 16, South Fifth Road, Guicheng Street, Foshan, Guangdong, People’s Republic of China, Tel +86 15015578028, Email [email protected]
Purpose: Gout and Rheumatoid arthritis (RA) are two prevalent non-infectious inflammatory joint diseases that can occur independently or concurrently. The effects and mechanisms related to antibody-mediated immune responses and both Gout and RA remain unclear. The research seeks to investigate the potential causal association and offer a novel perspective for their prevention and treatment strategies.
Methods: The study employed the bidirectional two-sample Mendelian randomization (MR) analysis for investigation. Datasets comprising 46 antibody-mediated immune responses, as well as those for Gout and RA, were curated from published genome-wide association studies (GWAS). For the causality analysis, methods such as Inverse Variance Weighted (IVW), Weighted Median, Simple Mode, MR-Egger, and Weighted Mode were utilized. We chose MR pleiotropy residual sum and outlier (MR-PRESSO), IVW, MR-Egger, and Leave-one-out for sensitivity analysis to enhance the reliability of the results.
Results: We meticulously excluded the results that exhibited pleiotropy and instability. Finally, four antibody-mediated immune responses have been found as causal factors in the development of Gout: Anti-chlamydia trachomatis IgG seropositivity, Anti-human herpes virus 6 IE1B IgG seropositivity, Helicobacter pylori GroEL antibody levels, and Polyomavirus 2 JC VP1 antibody levels; Two antibody-mediated immune responses influence RA causally: BK polyomavirus VP1 antibody levels, and Helicobacter pylori Catalase antibody levels. In the reverse analysis, three antibody-mediated immune responses could be influenced by Gout: BK polyomavirus VP1 antibody levels, Chlamydia trachomatis tarp-D F2 antibody levels, and Varicella zoster virus glycoproteins E and I antibody levels; Two antibody-mediated immune responses could be causally affected by RA: Anti-human herpes virus 7 IgG seropositivity, and Merkel cell polyomavirus VP1 antibody levels.
Conclusion: The research indicated that antibody-mediated immune responses establish a causal link with this two non-infectious inflammatory joint diseases: Gout and RA, offering new avenues and perspectives for the future prophylaxis and treatment of diseases from an immunological standpoint.
Keywords: antibody-mediated immune responses, rheumatoid arthritis, Gout, Mendelian randomization
Introduction
RA, a chronic inflammatory autoimmune disorder, impacts approximately 1% of the population globally, predominantly striking the individuals aged between 30 and 50, with a higher incidence among the elderly. The incidence among women is roughly two-three times that of men.1,2 RA primarily affects joint, leading to persistent inflammation and injury. The pathological features revolve around the joint synovitis, characterized by synovial cell proliferation, fibrosis, the infiltration of inflammatory cells, and formation of pannus, which results in the degradation of bone and cartilage tissues.3 Symptoms typically manifest as symmetrical joint pain, swelling, stiffness. Additionally, RA can affect various extra-articular tissues and organs, including the heart, blood vessels, lungs, kidneys, and gastrointestinal tract.4,5 Cardiovascular disease represents a major contributor to mortality rates among individuals suffering from RA. A meta-analysis conducted in 2012, encompassing 14 studies and 41,490 patients, revealed that individuals suffer from RA are confronted with a 48% elevated risk of cardiovascular disease.6 Innate and adaptive immunity are pivotal in the advancement of RA. Dysregulated humoral immunity overactivates T and B lymphocytes. Dendritic cells serve as the primary antigen-presenting cells, playing a crucial role in presenting antigens to T cells and providing co-stimulatory signals to initiate their activation.7 This contributes to various inflammatory mediators’ production, like interleukin(IL)-17A, IL-17F, and IL-228. B cells become hyperactive and produce autoantibodies: anti-citrullinated protein antibodies and rheumatoid factor, which led to the formation of immune complexes and trigger the inflammation.9
The key factors driving the development of Gout are the chronic accumulation of monosodium urate (MSU) crystals and hyperuricemia.10 It is an inflammatory arthritis associated with metabolic and immune factors, with a prevalence ranging from 1% to 6.8%. The number of patients affected is on the rise annually.11 This is a recurrent, self-limiting condition characterized by symptoms including redness, swelling, pain, and impaired function in one or more joints. It may also involve the formation of tophi, which can result in the deterioration of joint bones and cartilage. In certain instances, it also contribute to the cardiovascular, renal, and various other systemic diseases.12,13 MSU crystals activate adaptive and innate immunity in the context of hyperuricemia, stimulating macrophages, innate lymphocytes, dendritic cells, T cells, and so forth, which triggers to the activation of the NLRP3 inflammasome and mediates a cascade of inflammatory responses.14 Gout and RA can manifest either separately or concurrently, exerting a more significant psychosocial impact on patients.
Gout and RA have been confirmed as non-infectious inflammatory joint disorders linked to autoimmunity. These diseases exert significant impacts not only on local tissues but also on the body as a whole, extending beyond the joints. However, certain researches indicated that the incidence of Gout and RA is connected with infections caused by infectious agents such as bacteria or viruses. Xie D15 discovered that individuals with Gout are at a higher likelihood of contracting SARS-CoV-2 and face a heightened risk of severe complications compared to those without Gout. Porphyromonas gingivalis significantly affects the onset and progression of RA by expressing the peptidyl arginine deiminase enzyme.16 RA patients exhibited higher titers of EA, EBNA, and VCA antibodies.17 A meta-analysis that included 5 Chinese studies and 1 American study indicated a positive correlation between autoimmune diseases and an increased risk of COVID-19.18 Butler-Laporte et al conducted a study utilizing serum samples from 9724 individuals, selecting 13 pathogens for a GWAS.19 Previous studies on infectious diseases associated with RA and Gout are limited and primarily observational, with the causal relationship between them remaining unclarified. This study innovatively investigates whether other infectious diseases have effects on the pathogenesis of RA and Gout, based on the identified research gap, and discusses how these infectious, antibody-mediated immune responses participate in and influence these two non-infectious diseases.
MR employs genetic variation associated with exposure as an instrumental variable (IV) to infer causal relationships with outcomes, mitigating the impact of confounders and reverse causal bias.20 MR is more convenient and cost-effective than randomized controlled trials (RCT). It leverages the principle that alleles are randomly allocated during the meiosis, emulating the randomization process of the RCT.21 To exclude the influence of confounders, we utilized MR to establish a direct association between exposure and outcome. The study is grounded in GWAS datasets, using a bidirectional two-sample MR to explore the causal relationship between antibody-mediated immune responses and the progression of both Gout and RA. The findings are anticipated to offer a more comprehensive understanding of the etiological mechanisms behind Gout and RA, and to provide a novel perspective and direction for the prevention of these diseases.
Methods
Study Design
The study conducted by using the “TwoSampleMR” package in R Studio (2024.09.0+375), R (version 4.4.1), with a significance level of P<0.05. We designed a bidirectional two-sample MR analysis process model (refer to Figure 1) to assess the association between the antibody-mediated immune responses and both Gout and RA. First, we designated antibody-mediated immune responses as “exposure”, with Gout and RA serving as “outcome”. We employed two-sample MR to analyze the impact of antibody-mediated immune response on Gout and RA. Then, using reverse two-sample MR, we designated Gout and RA to “exposure” and antibody-mediated immune responses to “outcome”, to explore the influence of Gout and RA on antibody-mediated immune responses.
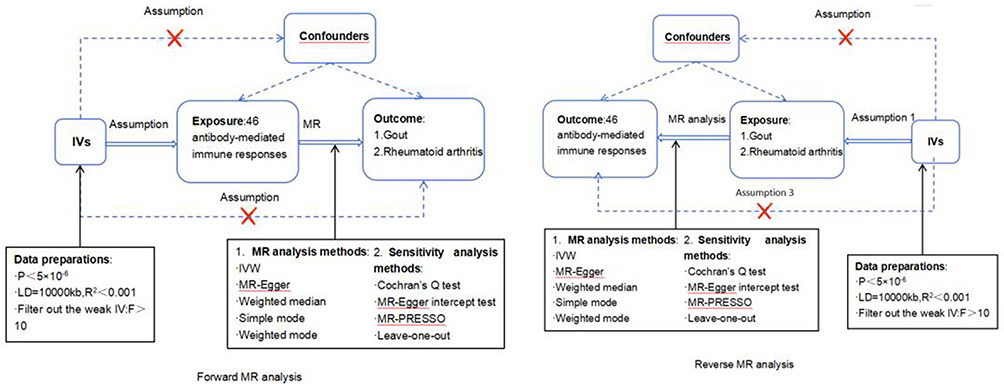 |
Figure 1 Overall design of the MR analysis. Abbreviations: IVs, instrumental variable; IVW, inverse-variance weighted; MR, Mendelian randomization; MR-PRESSO, Mendelian randomization-pleiotropy residual sum and outlier; SNPs, single-nucleotide polymorphisms. |
Data Sources
The study selected exposure and outcome from different studies based on two different individuals in Finland and the UK to minimize sample overlap and ensure the robustness of the findings. Summary data related to antibody-mediated immune responses were sourced from the GWAS Catalog (https://www.ebi.ac.uk/gwas/) (accessed on November 8, 2024). Butler-Laporte et al utilized the UK Biobank to perform serologic measurements on 9724 adults of European descent, selecting 13 pathogens for 46 GWAS (GWAS ID: GCST90006884 to GCST90006909) [18]. The datasets for RA (finngen_R11_M13_RHEUMA) and Gout (finngen _ R11 _ M 13 _ Gout) are from the FinnGen biobank analysis round 11 (https://www.finngen.fi/en) (accessed on November 9, 2024). Gout is characterized by “ A condition characterized by painful swelling of the joints, which is caused by deposition of urate crystals”. RA is described as “A chronic, systemic autoimmune disorder characterized by inflammation in the synovial membranes and articular surfaces. It manifests primarily as a symmetric, erosive polyarthritis that spares the axial skeleton and is typically associated with the presence in the serum of rheumatoid factor”. Other autoimmune joint diseases, drug-induced joint pain, infectious joint pain, degenerative bone and joint diseases, and other datasets that do not meet the definition have all been excluded. These datasets are derived from European descent samples. The RA dataset encompassed 302614 samples (14818 cases and 287796 controls). Furthermore, the Gout dataset included 298684 samples (10888 cases and 287796 controls). The participants’ genetic backgrounds in this study were confined to European ancestry, including individuals of both sexes. The original studies of the selected GWAS datasets were approved by the ethics committee, and all the participants in the original research obtained informed consent. The datasets used in this study were publicly accessible, therefore ethical approval was deemed unnecessary. (The detailed GWAS can be found in Table 1)
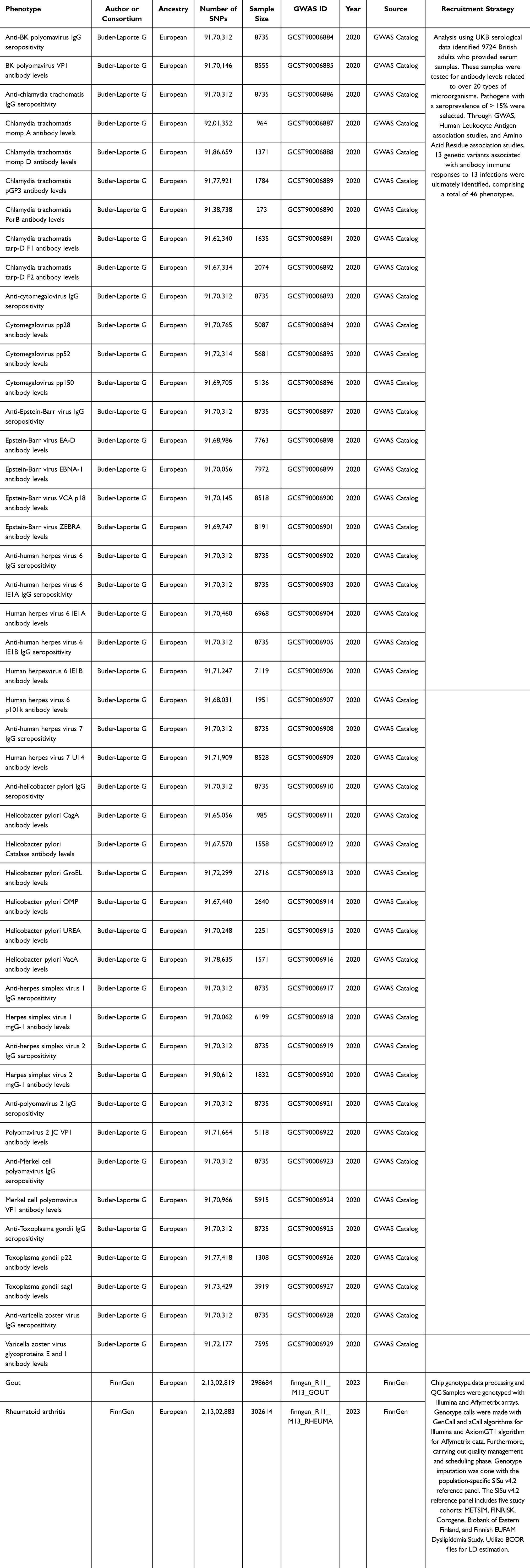 |
Table 1 GWAS data sources for instrumental variables selection. |
Selection of Instrumental Variables
To ensure effective IVs, this study strictly adheres to three core assumptions: (1) the correlation hypothesis: selected IVs has a robust correlation with exposure factors; (2) the independence assumption: there’s no correlation exists between IVs and any confounders; (3) the exclusion of the restrictive assumption: IVs are exclusively allowed to influence the outcome via exposure factors, without directly affecting on the outcome itself. We adjusted the threshold for P<5×10−6 to obtain adequate exposure-related SNPs. We remove the linkage disequilibrium (r2<0.001,10,000 kb) to reduce the superposition effect of the associated SNPs. We utilized Geneatlas (http://geneatlas.roslin.ed.ac.uk/) to search for SNP names and manually screened them to ensure that the selected IVs were not correlated with confounding factors. In addition, to minimize the bias from genotyping techniques, the palindromic SNP was excluded. To evaluate the robustness of IVs, the F-statistics was computed using the formula (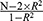 ), where N represents the sample size, R2 represents the proportion of variance in the exposure explained by the SNP.22 The formula of R2 incorporates effect allele frequency (EAF), standard error (SE), and effect size (β):
), where N represents the sample size, R2 represents the proportion of variance in the exposure explained by the SNP.22 The formula of R2 incorporates effect allele frequency (EAF), standard error (SE), and effect size (β): 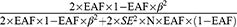 To prevent bias, weak IVs with an F-statistic less than 10 were filtered out.
To prevent bias, weak IVs with an F-statistic less than 10 were filtered out.
MR Analysis
Forward MR Analysis
In the forward analysis, antibody-mediated immune responses served as exposure, with Gout and RA as outcome. IVW provides the most reliable assessment of causality.23 Consequently, we chose IVW as main analysis method. Additionally, the weighted median, MR-Egger, weighted mode, and simple mode were utilized as secondary supplements and tests. We calculate effect size of β, odds ratio(OR) and 95% confidence intervals(CI) to analyze the association between the antibody-mediated immune responses and both Gout and RA. If the IVW analysis are significant, it indicates the presence of a causal relationship. When two or more analytical methods yield significant results, it indicates that the findings are comparatively stable and reliable.24 Furthermore, we visualize the effects of the five analytical methods through Scatter plots and Forest plots, and the fitting results intuitively reflect the trend of the impact.
Reverse MR Analysis
In the reverse analysis, same analytical methods were applied, including IVW, weighted median, MR-Egger, weighted mode, and simple mode. Gout and RA were served as exposure while antibody-mediated immune responses considered as outcome. This reverse analysis can also function as validation to exclude potential reverse causality.
Sensitivity Analysis
After analyzing the results, we ascertain the reliability of the findings by conducting sensitivity analysis. Initially, we quantified the heterogeneity of genetic variations using the Cochran Q test, where P>0.05 states that heterogeneity was not significantly detected. Subsequently, we employed MR-egger intercept analysis to estimate horizontal pleiotropy. The P> 0.05 indicates that pleiotropy was not detected, and the results were reliable. Additionally, we used MR-PRESSO to further investigate pleiotropy. The MR-Egger can also be applied to analyze the heterogeneity among genetic variants. Finally, we used the Leave-one-out method to assess whether any single SNP significantly impacts the results, thereby verifying the reliability of the results.
Results
Select Instrumental Variables
SNP linked to antibody-mediated immune responses, Gout, and RA were identified as IVs. First, we pinpointed 654 SNPs associated with 46 antibody-mediated immune responses (refer to Supplementary Table 1). Then we identified 68 SNPs correlated with Gout and 102 SNPs related with RA (refer to Supplementary Table 2). The F statistics for all instrumental variables surpassed 10 (ranging from 20.740 to 216.331). (The process of selecting and filtering SNPs can be found in Figure 2).
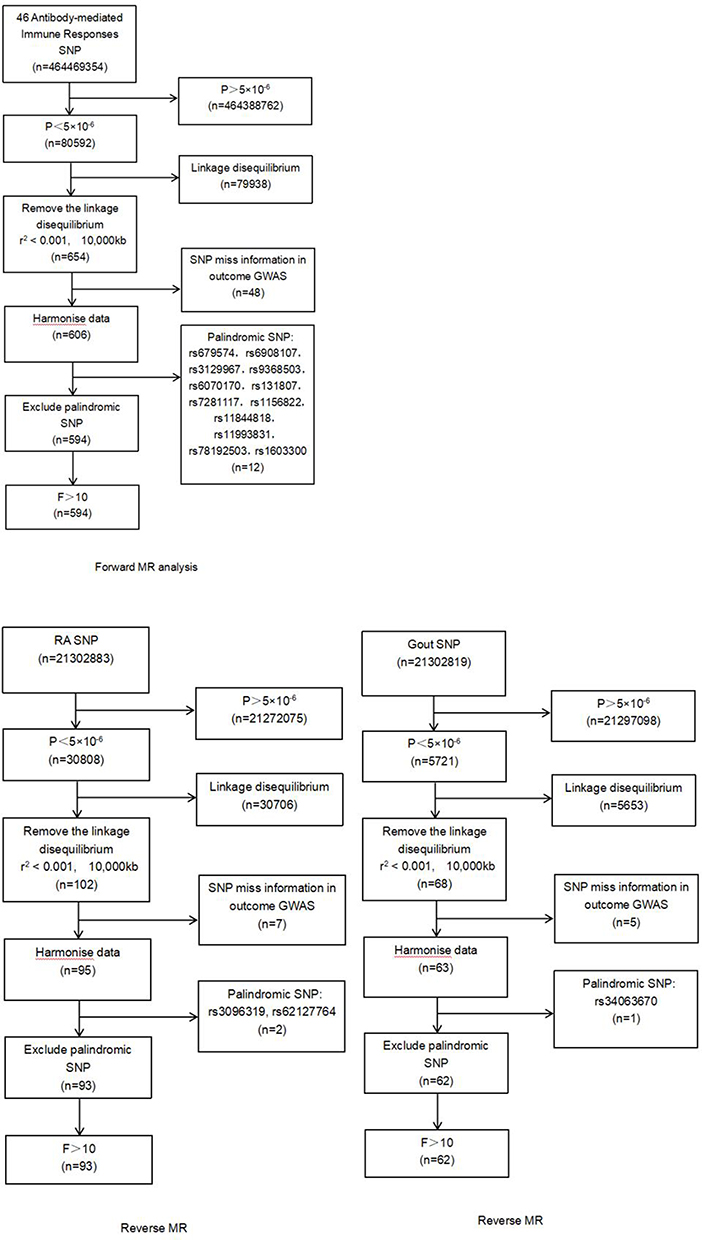 |
Figure 2 The process of selecting and filtering SNPs. Abbreviations: RA, Rheumatoid Arthritis; SNPs, single-nucleotide polymorphisms. |
The Causal Relationship Between Antibody-Mediated Immune Responses and Gout, RA
OR<1 indicates that exposure may reduce the risk of the outcome occurring. In forward MR, it represents a negative correlation between antibody-mediated immune responses and the occurrence of Gout or RA, meaning it would decrease the incidence of Gout or RA. In reverse MR, it indicates a negative correlation between Gout or RA and antibody-mediated immune responses, which would lower the levels of corresponding antibodies. OR>1 suggests that exposure may increase the risk of the outcome occurring. In forward MR, it signifies a positive correlation between antibody-mediated immune responses and the occurrence of Gout or RA, meaning it would increase the likelihood of Gout or RA. In reverse MR, it represents a positive correlation between Gout or RA and antibody-mediated immune responses, which would raise the levels of corresponding antibodies.
As depicted in Table 2 and Figure 3, the IVW analysis reveal Anti-chlamydia trachomatis IgG seropositivity (OR=0.951, 95% CI =0.915–0.989, P=0.012), Anti-human herpes virus 6 IE1B IgG seropositivity (OR=0.940, 95% CI =0.895–0.988, P=0.015), Helicobacter pylori GroEL antibody levels (OR=0.916, 95% CI =0.841–0.998, P=0.045) were inversely associated with Gout, representing that these antibody-mediated immune responses are protective factors for Gout in certain genetic circumstances, potentially decreasing the likelihood of Gout attacks. While Polyomavirus 2 JC VP1 antibody levels (OR = 1.122, 95% CI =1.036–1.215, P=0.005) exhibited a positive correlation with Gout, indicating that the immune response mediated by this antibody is a risk factor for Gout attacks (refer to Figures 4 and 5). No significant heterogeneity or horizontal pleiotropy (both P> 0.05) were found in subsequent sensitivity tests (refer to Table 3), and the Leave-one-out revealed that the results were relatively stable, implying their reliability (refer to Figure 6). After the analysis of IVW and Weighted median, Anti-human herpes virus 6 IE1B IgG seropositivity showed a more stable negative causality with Gout, whereas Polyomavirus 2 JC VP1 antibody levels represent a more stable positive causality.
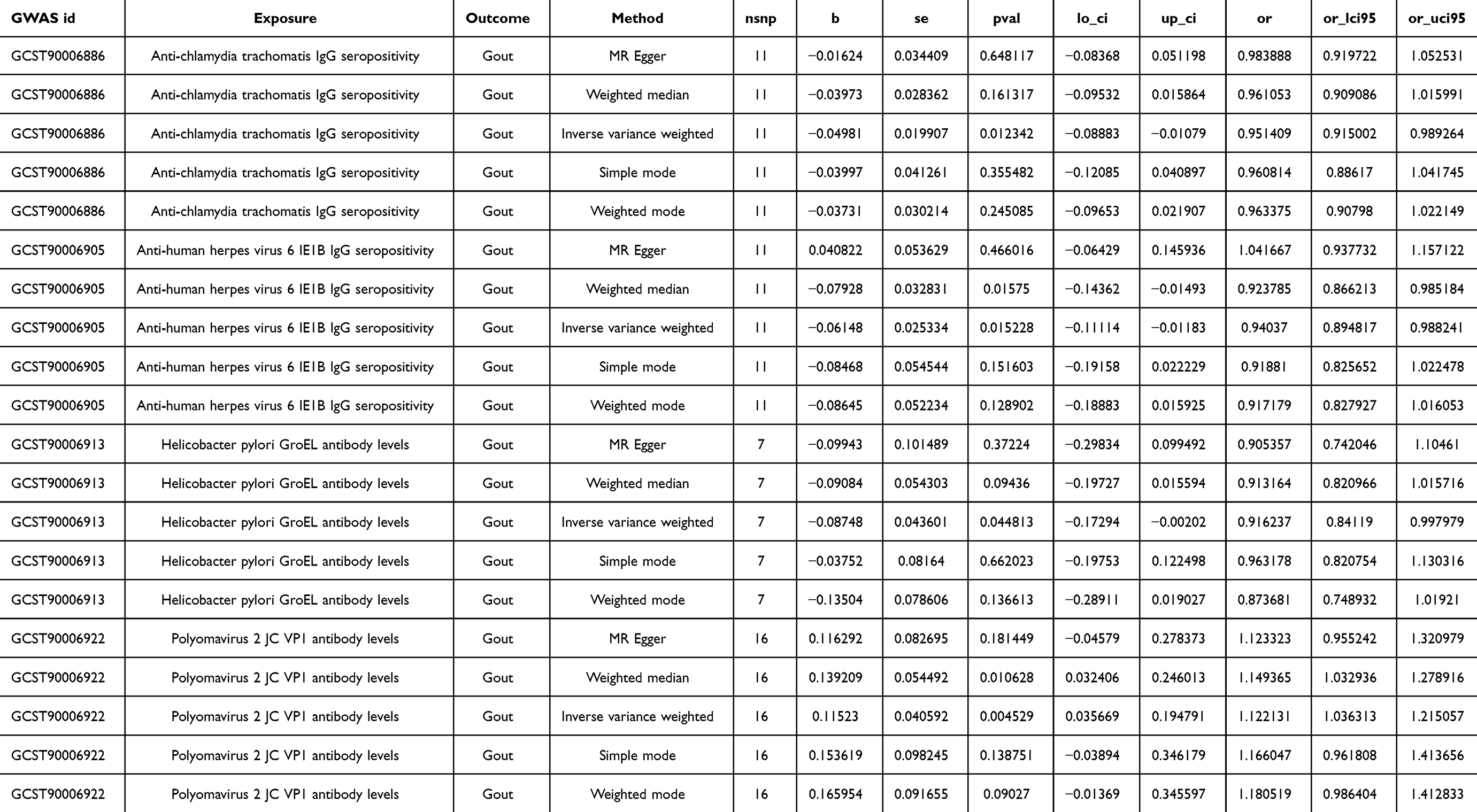 |
Table 2 MR Results of Causal Effects Between Antibody-Mediated Immune Responses and Gout |
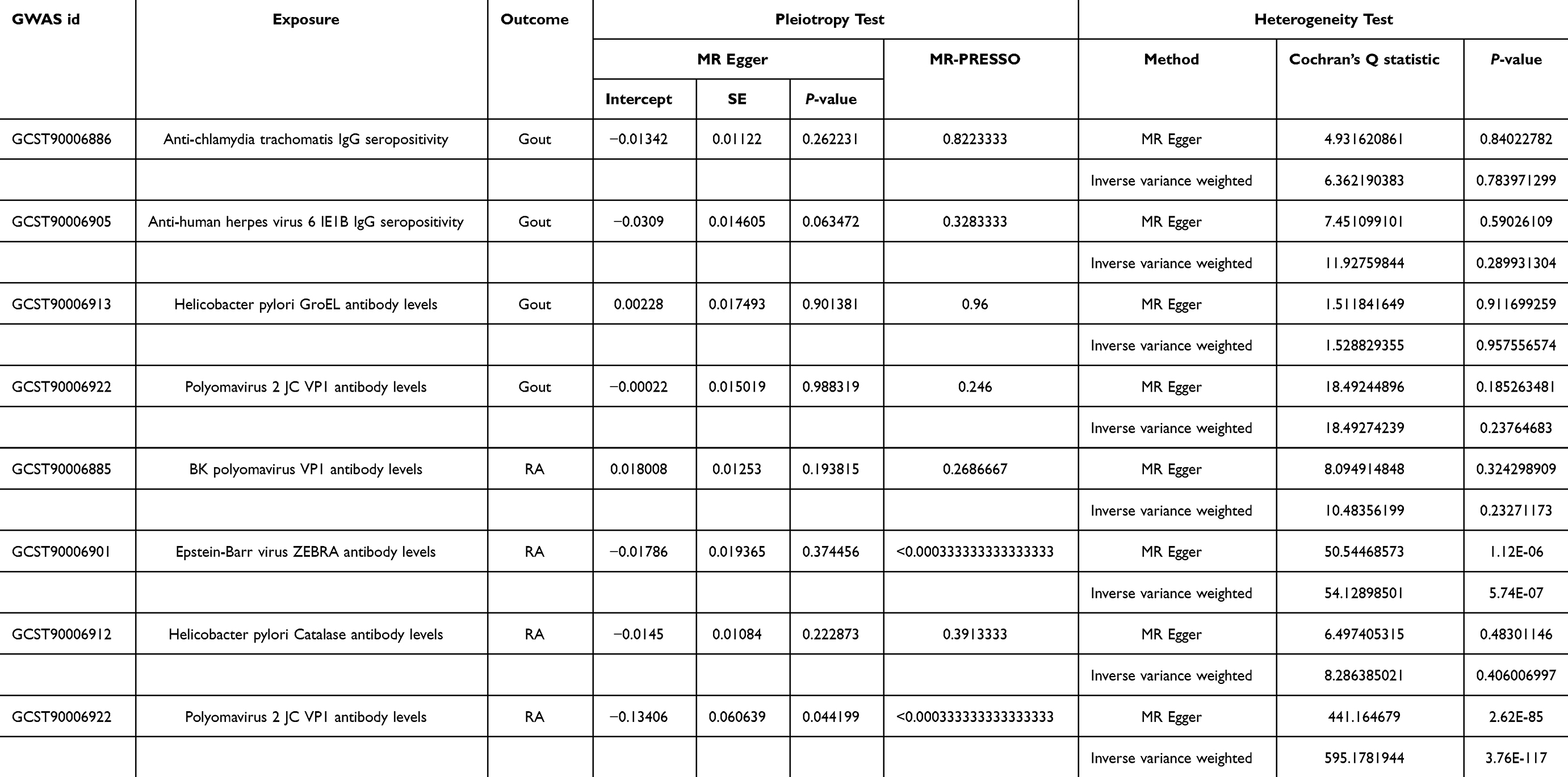 |
Table 3 Mendelian Randomization Sensitivity Analysis of Antibody-Mediated Immune Responses |
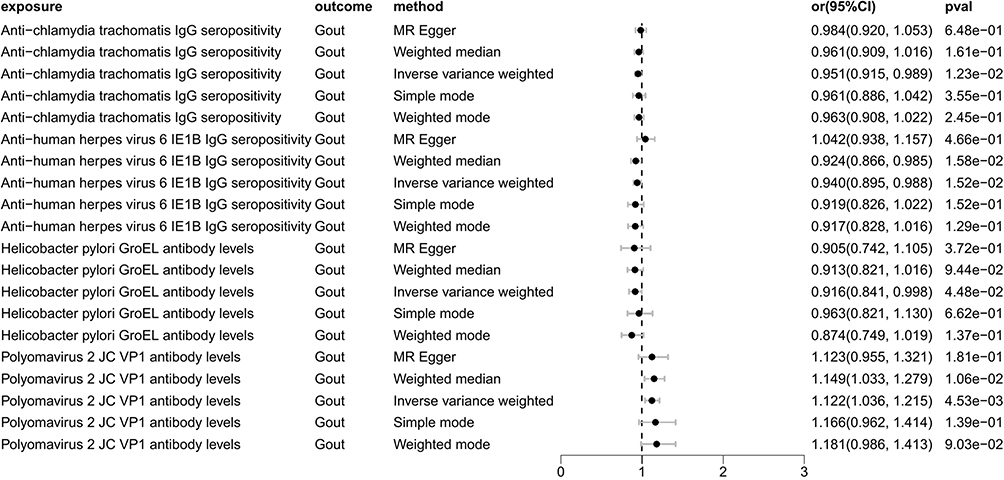 |
Figure 3 Forest plot visualization of the causal effect of antibody-mediated immune responses on Gout. Abbreviations: or, odds ratio; CI, confidence interval. |
 |
Figure 4 Scatter plots for the effect of antibody-mediated immune responses on Gout. (a) Analysis for “Anti-chlamydia trachomatis IgG seropositivity” on “Gout”. (b) Analysis for “Anti-human herpes virus 6 IE1B IgG seropositivity” on “Gout”. (c) Analysis for “Helicobacter pylori GroEL antibody levels” on “Gout”. (d) Analysis for “Polyomavirus 2 JC VP1 antibody levels” on “Gout”. Abbreviations: SNP, single-nucleotide polymorphism. |
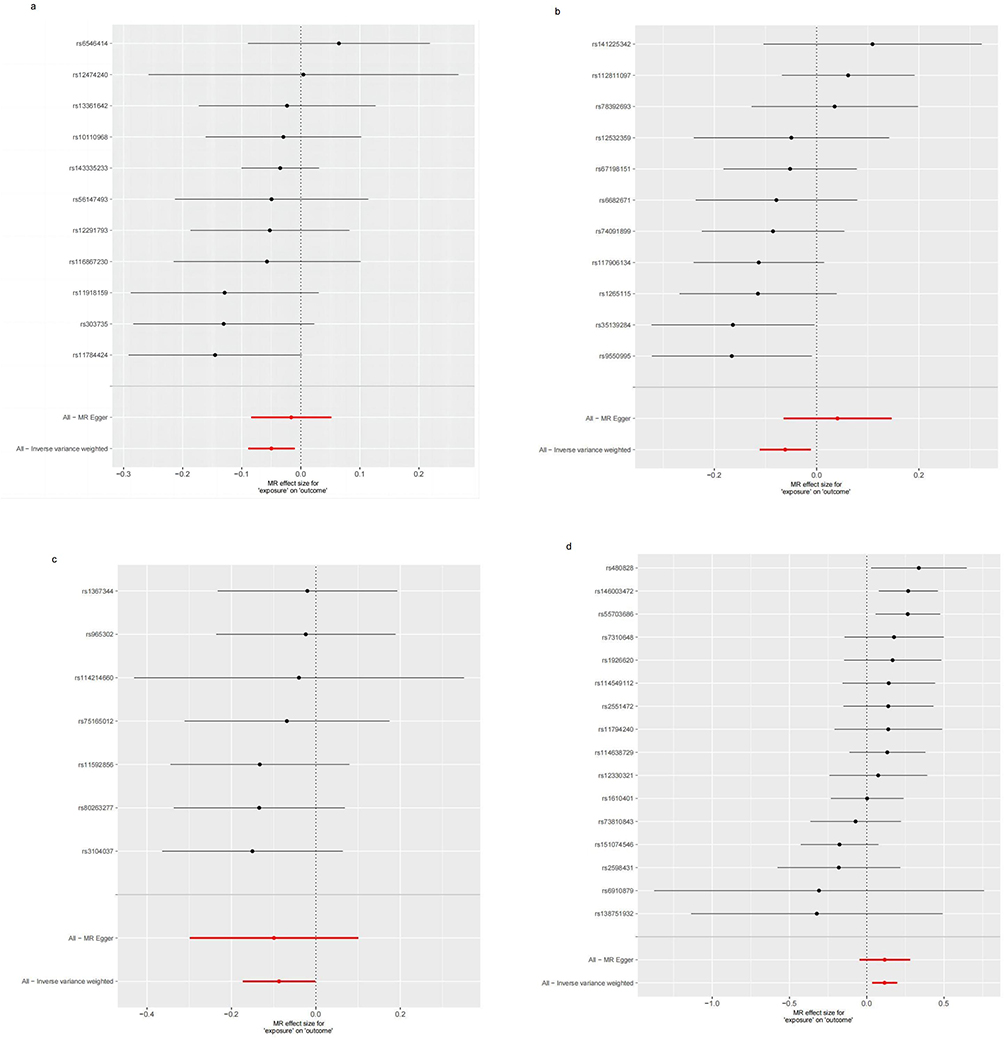 |
Figure 5 Forest plots for the effect of antibody-mediated immune responses on Gout. (a) Analysis for “Anti-chlamydia trachomatis IgG seropositivity” on “Gout”. (b) Analysis for “Anti-human herpes virus 6 IE1B IgG seropositivity” on “Gout”. (c) Analysis for “Helicobacter pylori GroEL antibody levels” on “Gout”. (d) Analysis for “Polyomavirus 2 JC VP1 antibody levels” on “Gout”. Abbreviations: SNP, single-nucleotide polymorphism. |
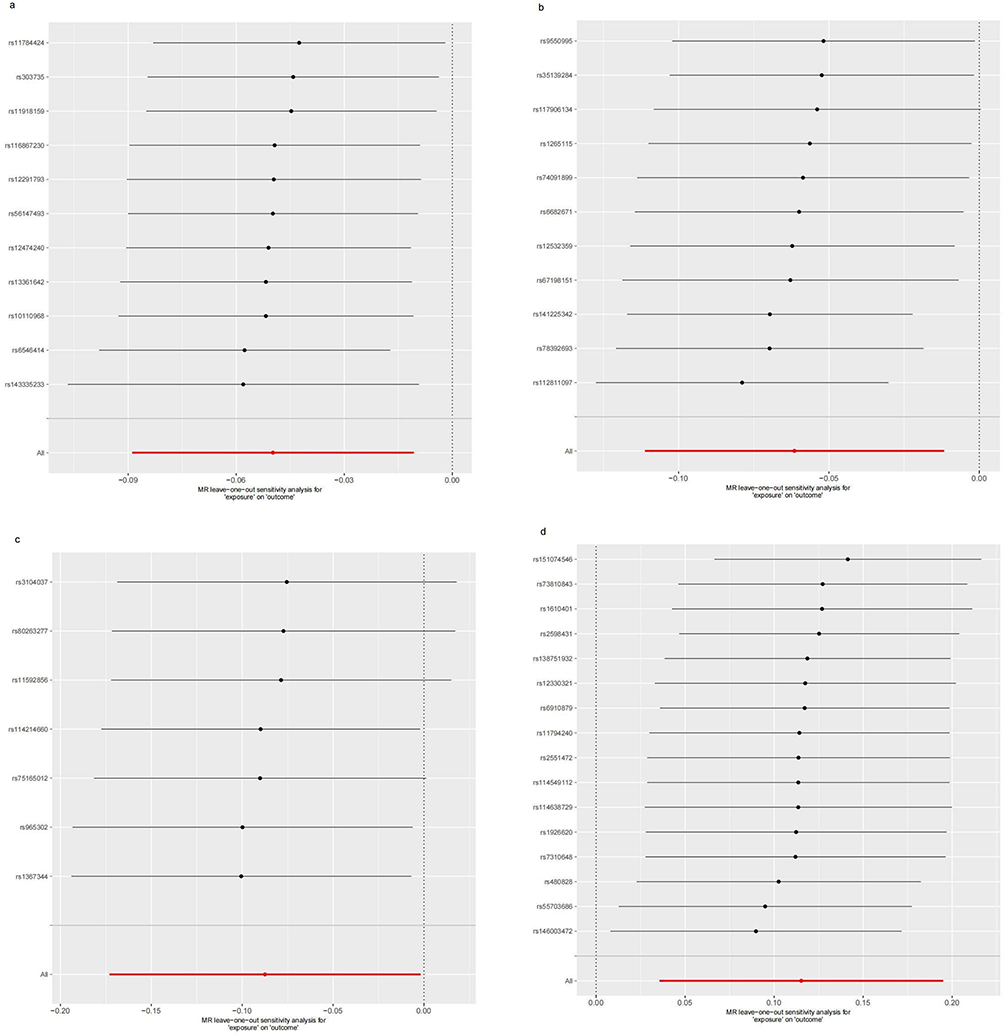 |
Figure 6 Leave-one-out sensitivity analysis for antibody-mediated immune responses on Gout. (a) Analysis for “Anti-chlamydia trachomatis IgG seropositivity” on “Gout”. (b) Analysis for “Anti-human herpes virus 6 IE1B IgG seropositivity” on “Gout”. (c) Analysis for “Helicobacter pylori GroEL antibody levels” on “Gout”. (d) Analysis for “Polyomavirus 2 JC VP1 antibody levels” on “Gout”. Abbreviations: MR, Mendelian randomization. |
As pictured in Figure 7, the IVW analysis revealed that BK polyomavirus VP1 antibody levels (OR = 0.825, 95% CI =0.727–0.937, P=0.003), Epstein-Barr virus ZEBRA antibody levels (OR=0.859, 95% CI =0.756–0.976, P=0.020), Helicobacter pylori Catalase antibody levels (OR=0.957, 95% CI =0.919–0.996, P=0.029) were inversely correlated with RA. This suggests that these antibody-mediated immune responses are important protective factors in RA. Conversely, Polyomavirus 2 JC VP1 antibody levels (OR=1.484, 95% CI =1.025–2.148, P=0.037) was positively associated with RA, implying the hazards for RA (refer to Table 4, Figures 8 and 9). In the sensitivity test, heterogeneity was observed in the results of Epstein-Barr virus ZEBRA antibody levels and Polyomavirus 2 JC VP1 antibody levels (P <0.05). The global test of MR-PRESSO revealed pleiotropy.(P <0.05). The remaining results did not exhibit significant heterogeneity or horizontal pleiotropy (both P> 0.05) (refer to Table 3). After employing the Leaf-one-out method, no single SNP substantially alter the results, indicating that the results were relatively stable (refer to Figure 10). The analysis of IVW, weighted median, weighted mode and MR-Egger revealed that the positive causality of BK polyomavirus VP1 antibody levels on RA is more stable.
 |
Table 4 MR Results of Causal Effects Between Antibody-Mediated Immune Responses and RA |
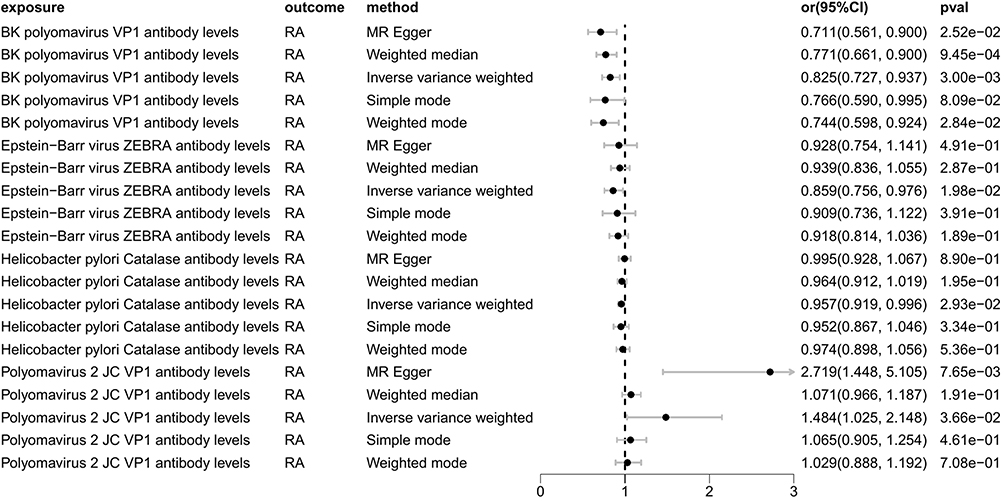 |
Figure 7 Forest plot visualization of the causal effect of antibody-mediated immune responses on RA. Abbreviations: or, odds ratio; CI, confidence interval; RA, Rheumatoid Arthritis. |
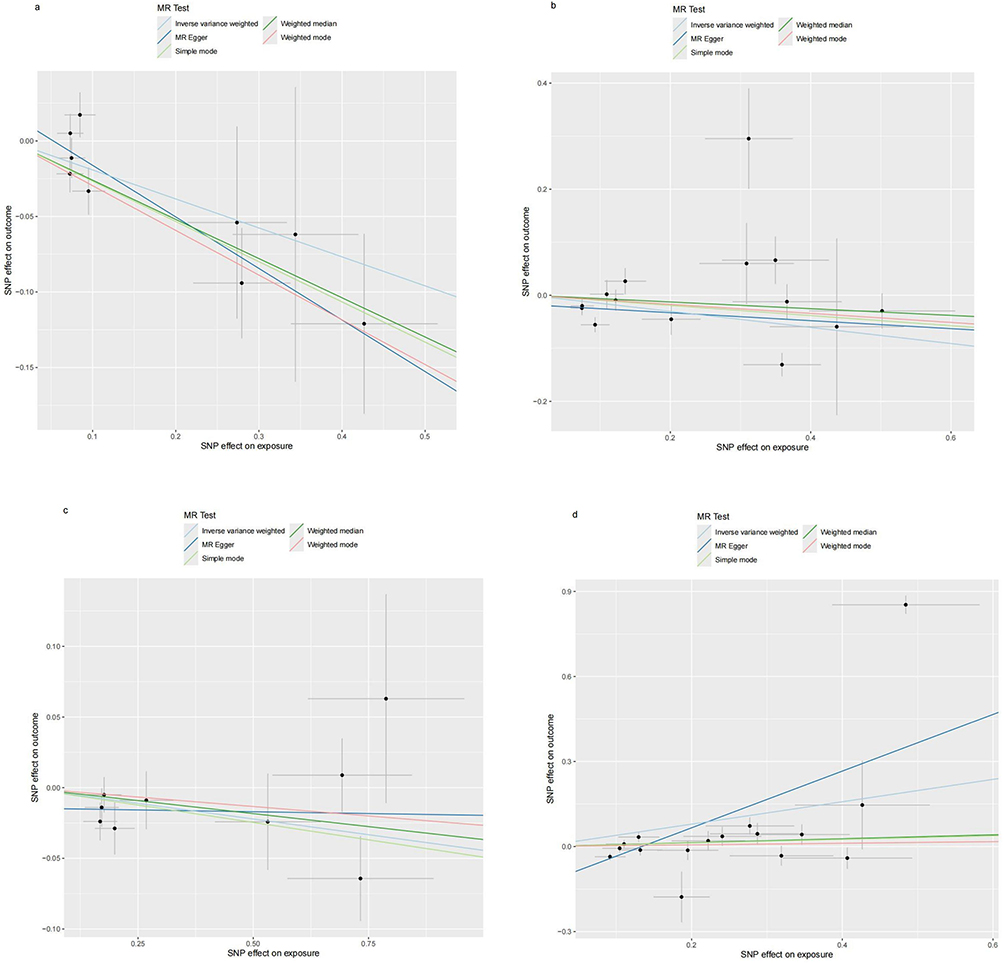 |
Figure 8 Scatter plots for the effect of antibody-mediated immune responses on RA. (a) Analysis for “BK polyomavirus VP1 antibody levels” on “RA”. (b) Analysis for “Epstein-Barr virus ZEBRA antibody levels” on “RA”. (c) Analysis for “Helicobacter pylori Catalase antibody levels” on “RA”. (d) Analysis for “Polyomavirus 2 JC VP1 antibody levels” on “RA”. Abbreviations: SNP, single-nucleotide polymorphism; RA, Rheumatoid Arthritis. |
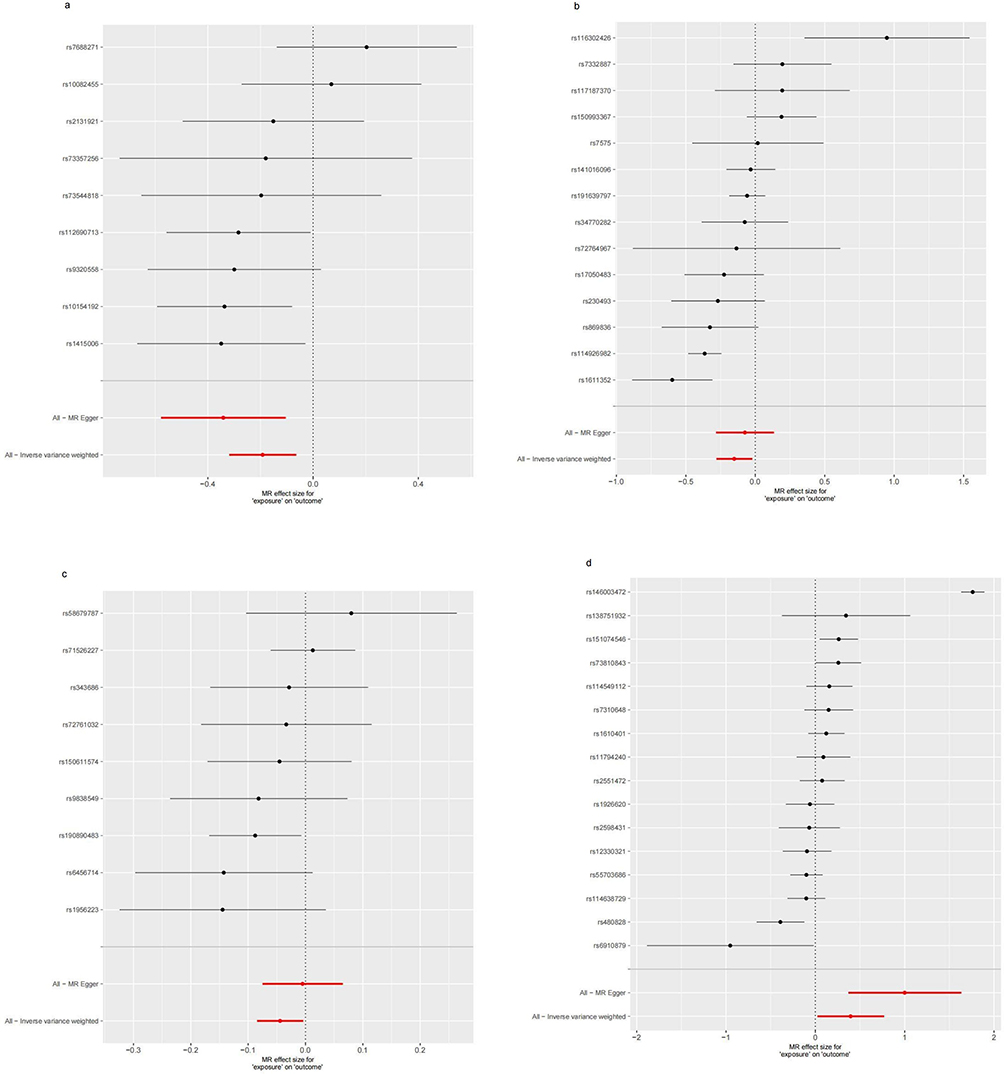 |
Figure 9 Forest plots for the effect of antibody-mediated immune responses on RA. (a) Analysis for “BK polyomavirus VP1 antibody levels” on “RA”. (b) Analysis for “Epstein-Barr virus ZEBRA antibody levels” on “RA”. (c) Analysis for “Helicobacter pylori Catalase antibody levels” on “RA”. (d) Analysis for “Polyomavirus 2 JC VP1 antibody levels” on “RA”. Abbreviations: SNP, single-nucleotide polymorphism; RA, Rheumatoid Arthritis. |
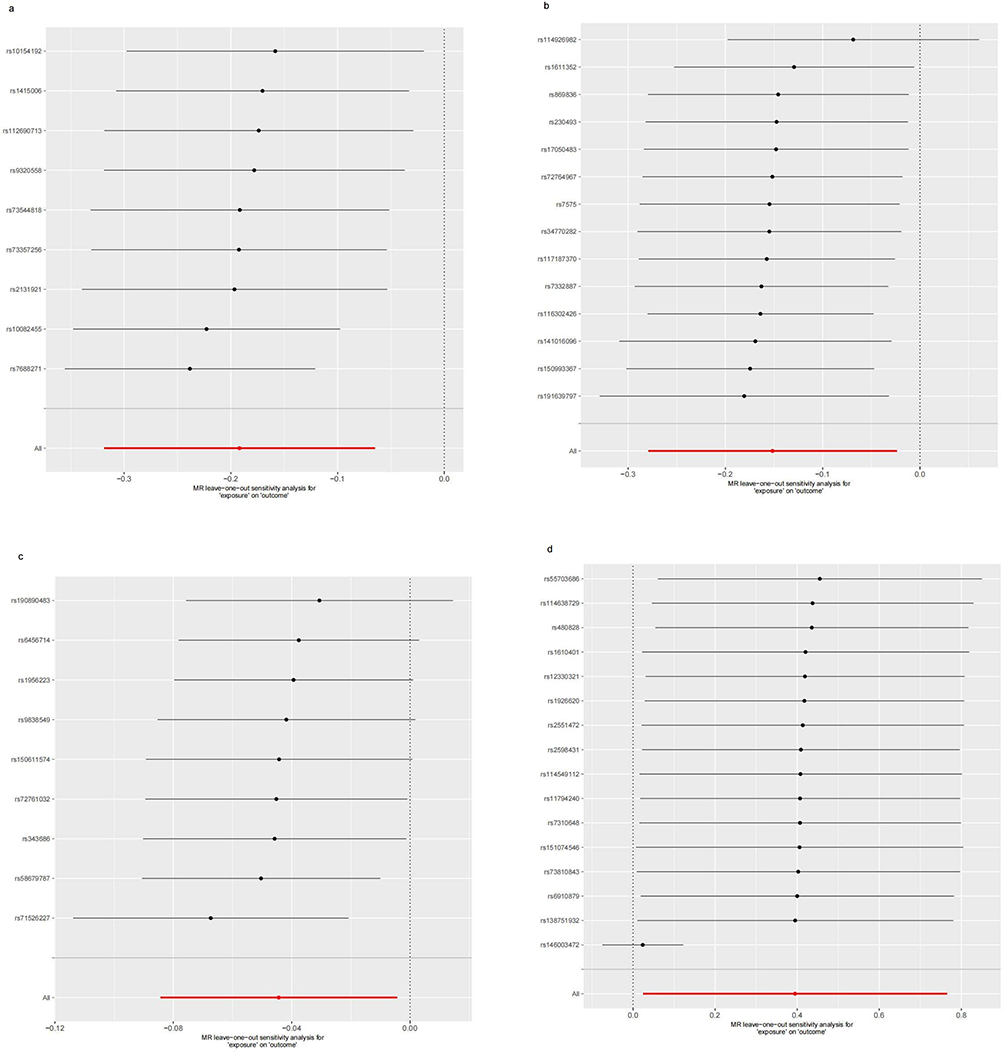 |
Figure 10 Leave-one-out sensitivity analysis for antibody-mediated immune responses on RA. (a) Analysis for “BK polyomavirus VP1 antibody levels” on “RA”. (b) Analysis for “Epstein-Barr virus ZEBRA antibody levels” on “RA”. (c) Analysis for “Helicobacter pylori Catalase antibody levels” on “RA”. (d) Analysis for “Polyomavirus 2 JC VP1 antibody levels” on “RA”. Abbreviation: MR, Mendelian randomization. |
The Causal Relationship Between Gout, RA and Antibody-Mediated Immune Responses
The IVW analysis indicates that Gout is causally associated with four antibody-mediated immune responses, while RA is causally linked with five antibody-mediated immune responses. As illustrated in Figure 11, Gout positively correlates with BK polyomavirus VP1 antibody levels (OR = 1.057, 95% CI =1.007–1.109, P=0.025), Chlamydia trachomatis tarp-D F2 antibody levels (OR=1.113, 95% CI =1.006–1.231, P=0.038), Varicella zoster virus glycoproteins E and I antibody levels (OR=1.061, 95% CI =1.009–1.117, P=0.022). This suggests that the onset of Gout, influenced by genetic variants, will bolster the relevant antibody-mediated immune responses. A negative correlation with Human herpes virus 6 p101k antibody levels (OR = 0.897, 95% CI =0.811–0.993, P=0.037) was also observed, suggesting that Gout attenuates the antibody-mediated immune response (refer to Table 5, Figures 12 and 13). No significant heterogeneity or pleiotropy (P> 0.05) was found (refer to Table 6). Upon assessing the sensitivity of Gout to Human herpes virus 6 p101k antibody levels’ causal relationship by using Leave-one-out method, a SNP (rs117581227) was identified, and its removal resulted a substantial alteration of the result (PIVW=0.161). This indicates that the result may be unstable yet the remaining causality was considered reliable through Leave-one-out analysis (refer to Figure 14). After the analysis of IVW, weighted median and MR Egger, Gout exhibited greater stability in its association with BK polyomavirus VP1 antibody levels and Chlamydia trachomatis tarp-D F2 antibody levels.
 |
Table 5 MR Results of Causal Effects Between Gout and Antibody-Mediated Immune Responses |
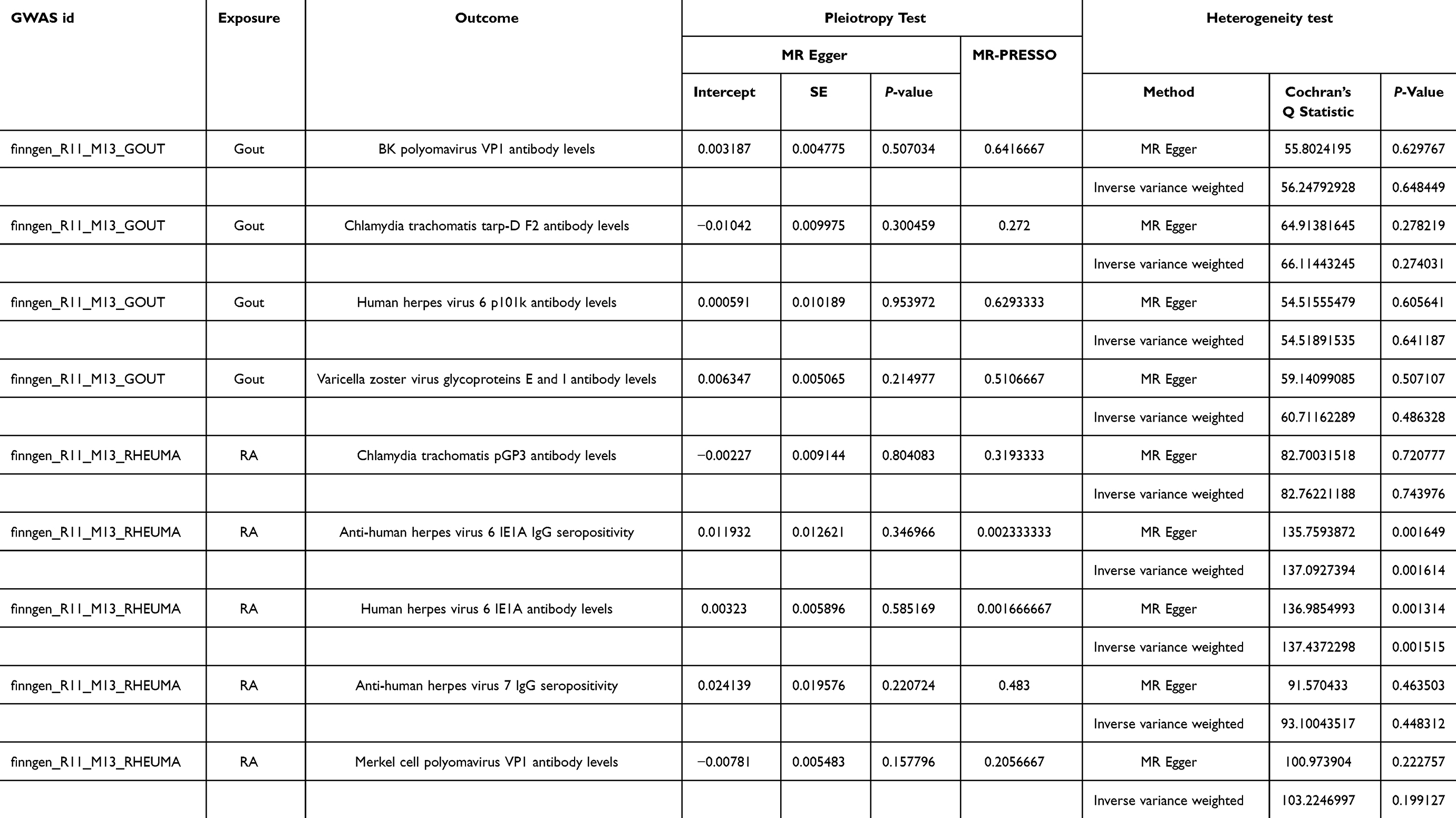 |
Table 6 Mendelian Randomization Sensitivity Analysis of Gout and RA |
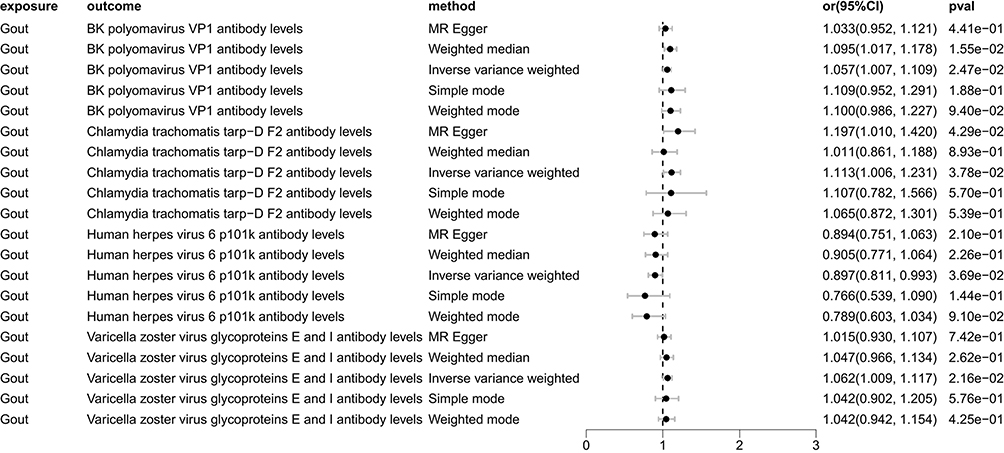 |
Figure 11 Forest plot visualization of the causal effect of Gout on antibody-mediated immune responses. Abbreviations: or, odds ratio; CI, confidence interval. |
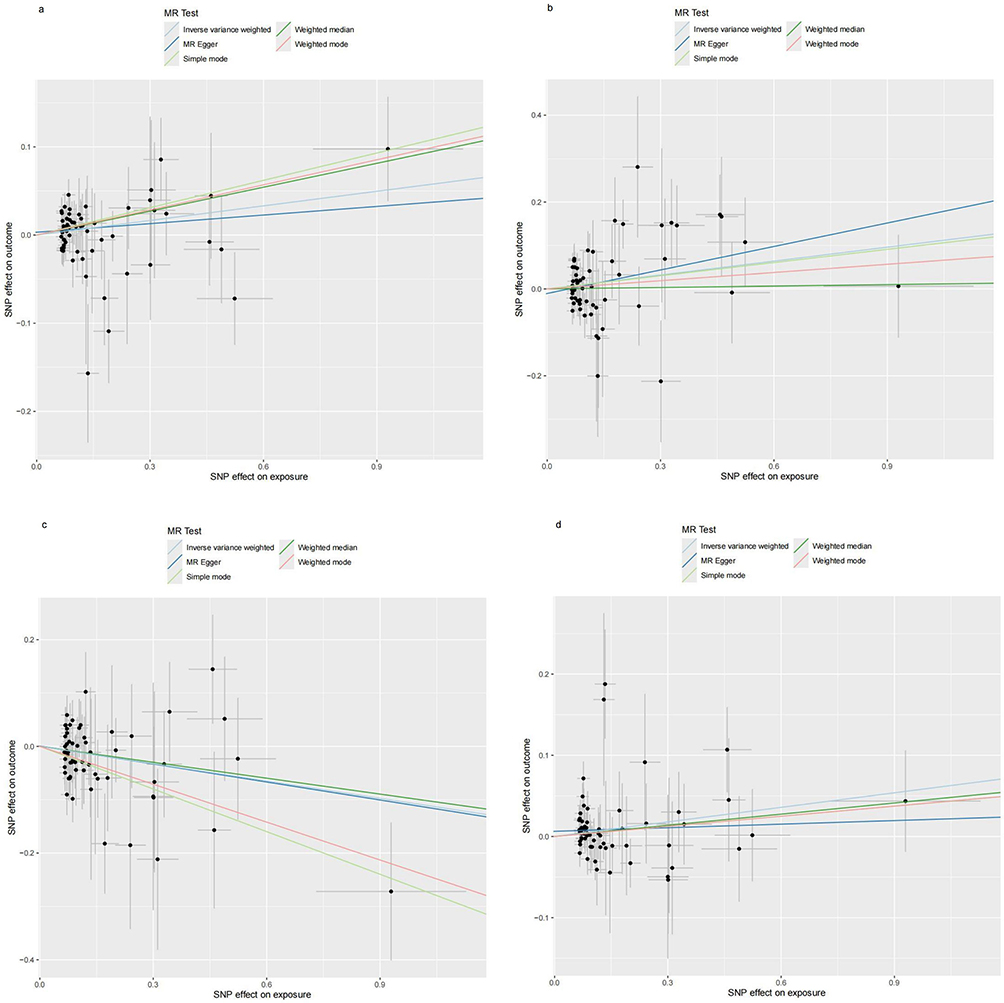 |
Figure 12 Scatter plots for the effect of Gout on antibody-mediated immune responses. (a) Analysis for “Gout” on “BK polyomavirus VP1 antibody levels”. (b) Analysis for “Gout” on “Chlamydia trachomatis tarp-D F2 antibody levels”. (c) Analysis for “Gout” on “Human herpes virus 6 p101k antibody levels”. (d) Analysis for “Gout” on “Varicella zoster virus glycoproteins E and I antibody levels”. Abbreviation: SNP, single-nucleotide polymorphism. |
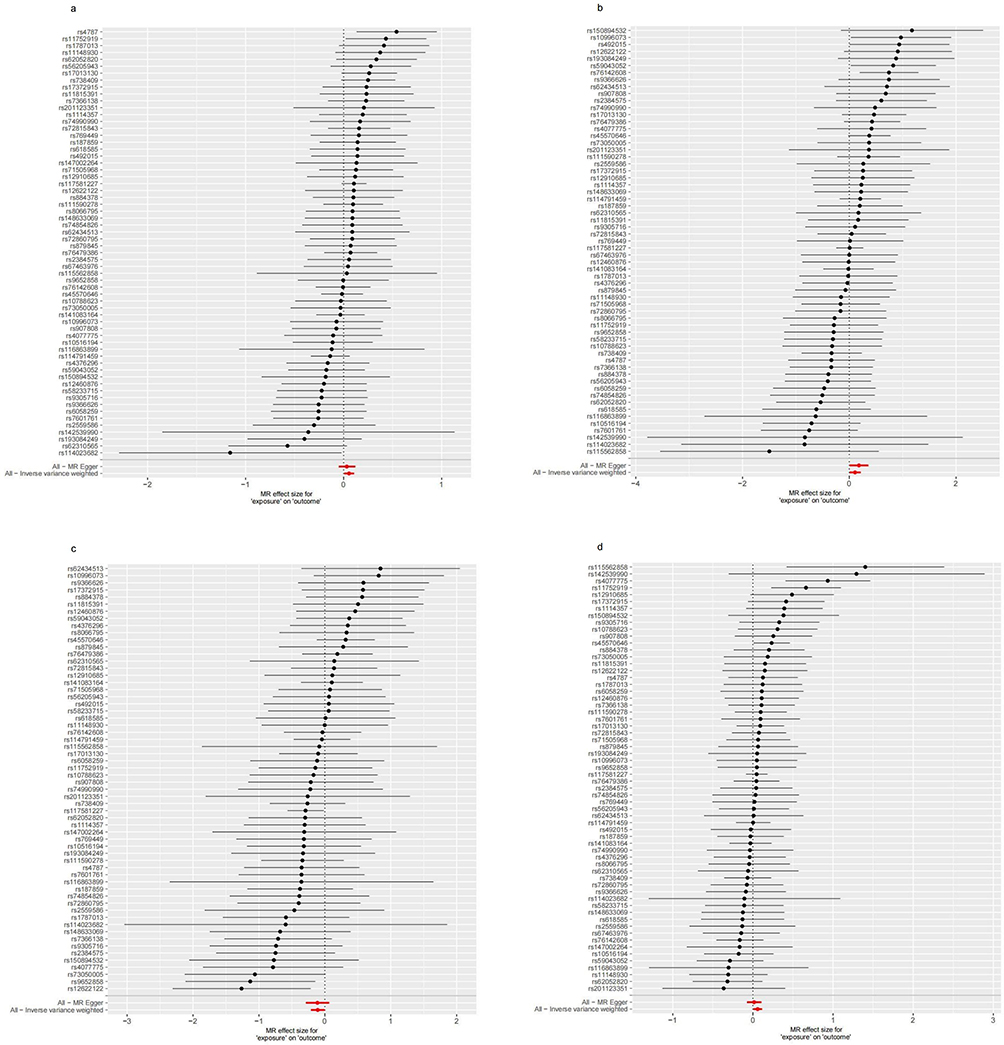 |
Figure 13 Forest plots for the effect of Gout on antibody-mediated immune responses. (a) Analysis for “Gout” on “BK polyomavirus VP1 antibody levels”. (b) Analysis for “Gout” on “Chlamydia trachomatis tarp-D F2 antibody levels”. (c) Analysis for “Gout” on “Human herpes virus 6 p101k antibody levels”. (d) Analysis for “Gout” on “Varicella zoster virus glycoproteins E and I antibody levels”. Abbreviation: SNP, single-nucleotide polymorphism. |
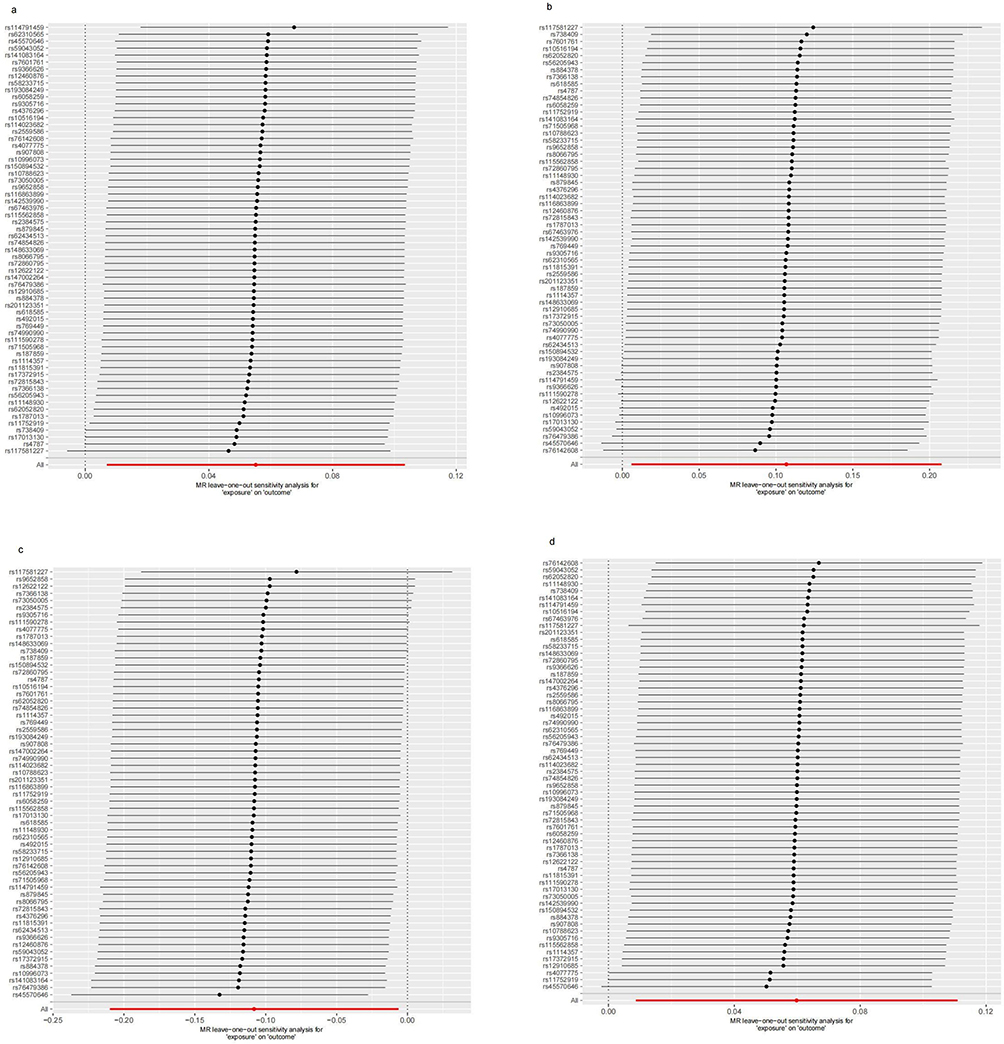 |
Figure 14 Leave-one-out sensitivity analysis for Gout on antibody-mediated immune responses. (a) Analysis for “Gout” on “BK polyomavirus VP1 antibody levels”. (b) Analysis for “Gout” on “Chlamydia trachomatis tarp-D F2 antibody levels”. (c) Analysis for “Gout” on “Human herpes virus 6 p101k antibody levels”. (d) Analysis for “Gout” on “Varicella zoster virus glycoproteins E and I antibody levels”. Abbreviation: MR, Mendelian randomization. |
As shown in Figure 15, RA will increase the Chlamydia trachomatis pGP3 antibody levels (OR = 1.109, 95% CI =1.008–1.221, P=0.034), Anti-human herpes virus 6 IE1A IgG seropositivity (OR=1.165, 95% CI =1.021–1.329, P=0.023), Human herpes virus 6 IE1A antibody levels (OR=1.063, 95% CI =1.000–1.131, P=0.048), Anti-human herpes virus 7 IgG seropositivity (OR=1.271, 95% CI =1.035–1.561, P=0.022), Merkel cell polyomavirus VP1 antibody levels (OR=1.093, 95% CI =1.032–1.158, P=0.002). Importantly, no negative causal relationship was found between RA and other immune responses (refer to Table 7, Figures 16 and 17). Further sensitivity analysis reveals that Anti-human herpes virus 6 IE1A IgG seropositivity and Human herpes virus 6 IE1A antibody levels exhibit heterogeneous and pleiotropy (P <0.05) (refer to Table 6). The Leave-one-out analysis indicates a SNP (rs189189451) that modifies the causal relationship between RA and Chlamydia trachomatis pGP3 antibody levels upon its elimination (PIVW=0.224), indicating the instability of this analysis. The remaining results appeared relatively stable (refer to Figure 18). The analysis of IVW, MR-Egger, weighted median confirms that the positive causal relationship between RA and Merkel cell polyomavirus VP1 antibody levels is more robust.
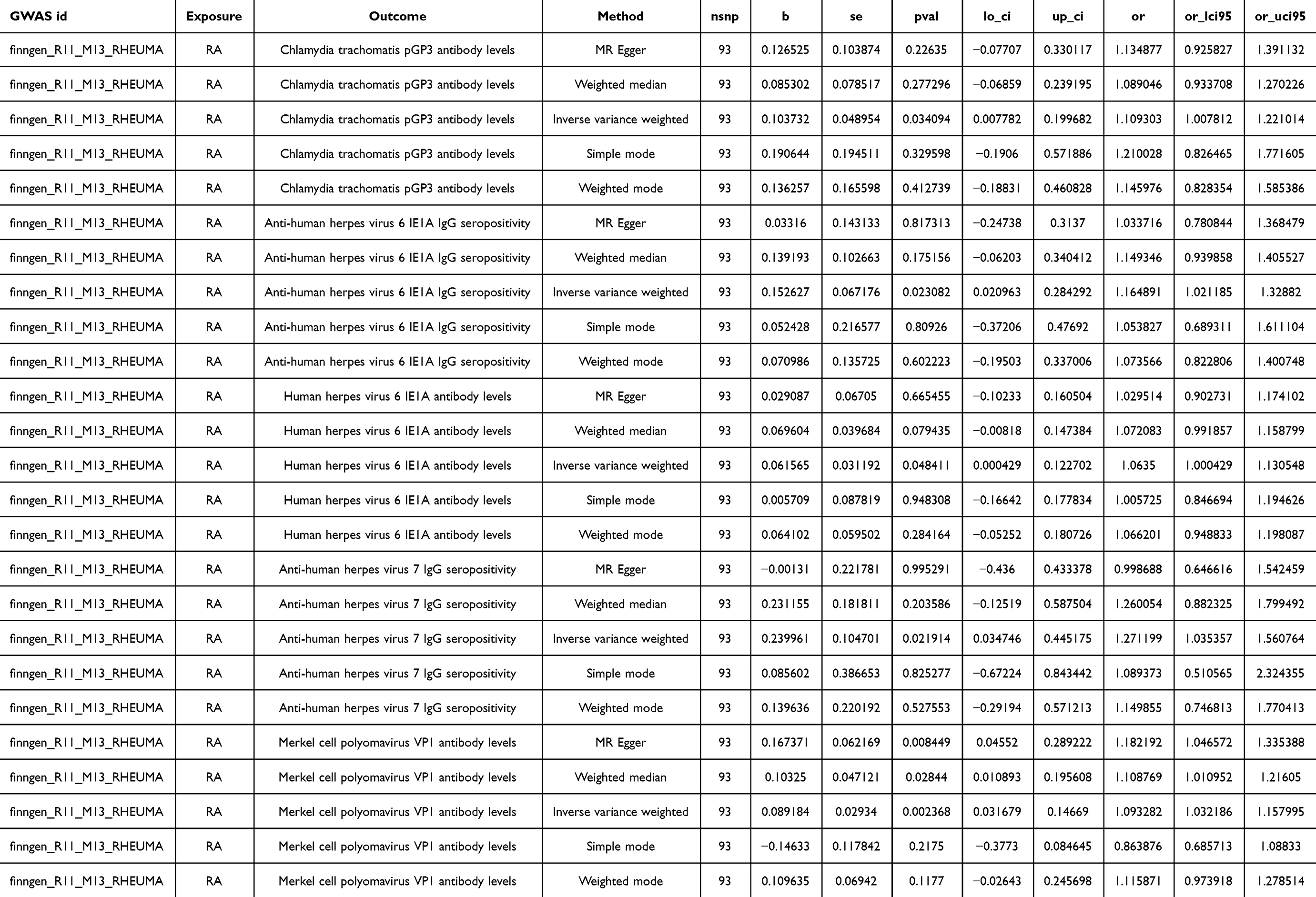 |
Table 7 MR Results of Causal Effects Between RA and Antibody-Mediated Immune Responses |
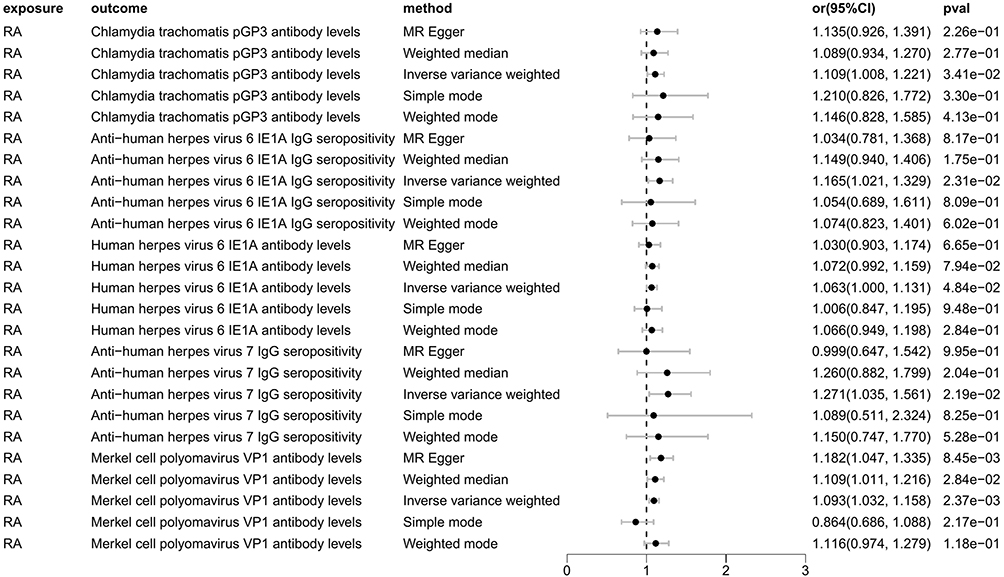 |
Figure 15 Forest plot visualization of the causal effect of RA on antibody-mediated immune responses. Abbreviations: or, odds ratio; CI, confidence interval; RA, Rheumatoid Arthritis. |
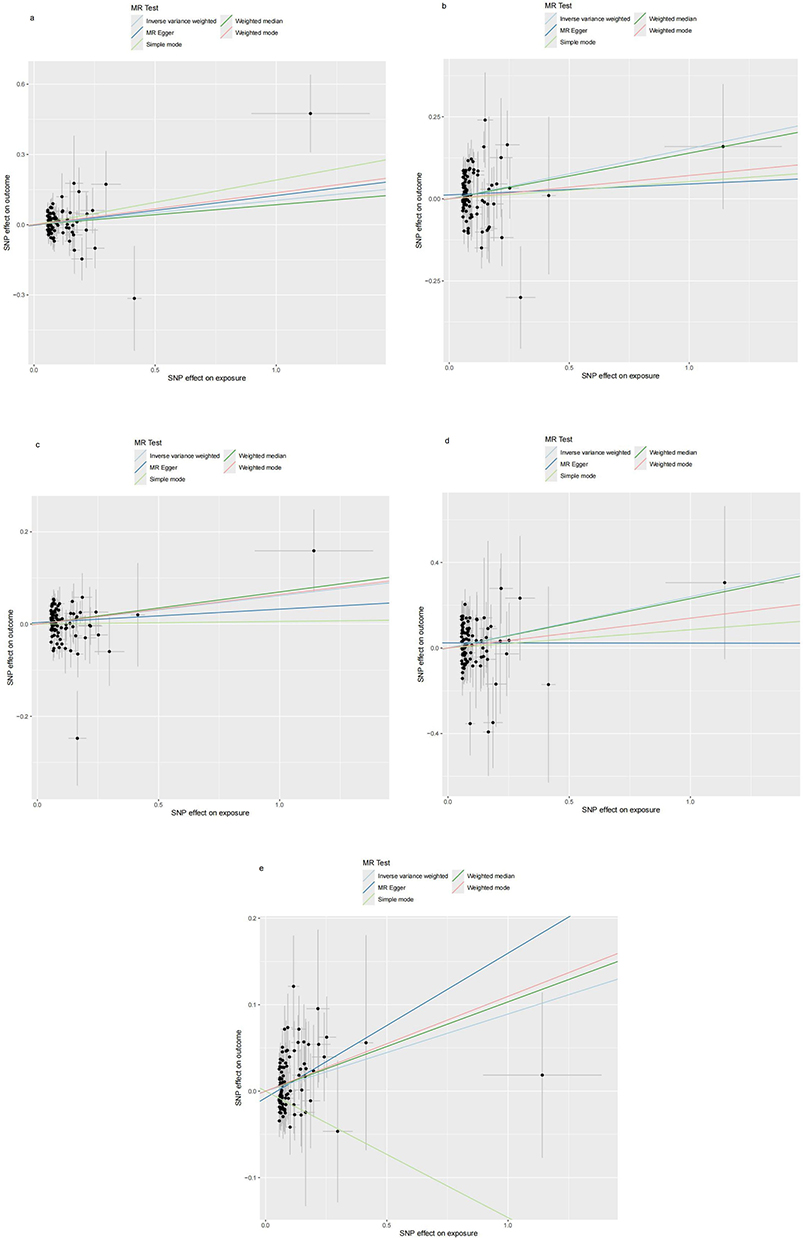 |
Figure 16 Scatter plots for the effect of RA on antibody-mediated immune responses. (a) Analysis for “RA” on “Chlamydia trachomatis pGP3 antibody levels”. (b) Analysis for “RA” on “Anti-human herpes virus 6 IE1A IgG seropositivity”. (c) Analysis for “RA” on “Human herpes virus 6 IE1A antibody levels”. (d) Analysis for “RA” on “Anti-human herpes virus 7 IgG seropositivity” (e) Analysis for “RA” on “Merkel cell polyomavirus VP1 antibody levels”. Abbreviations: SNP, single-nucleotide polymorphism; RA, Rheumatoid Arthritis. |
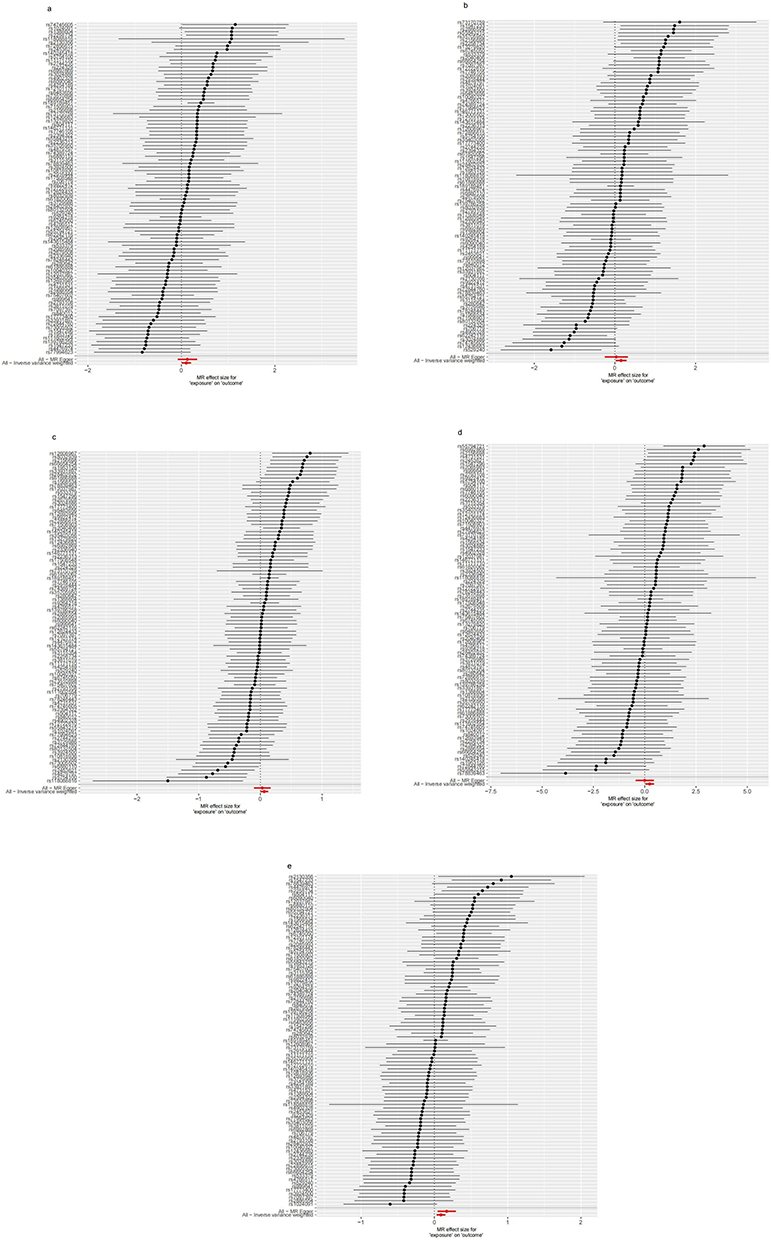 |
Figure 17 Forest plots for the effect of RA on antibody-mediated immune responses. (a) Analysis for “RA” on “Chlamydia trachomatis pGP3 antibody levels”. (b) Analysis for “RA” on “Anti-human herpes virus 6 IE1A IgG seropositivity”. (c) Analysis for “RA” on “Human herpes virus 6 IE1A antibody levels”. (d), Analysis for “RA” on “Anti-human herpes virus 7 IgG seropositivity”. (e) Analysis for “RA” on “Merkel cell polyomavirus VP1 antibody levels”. Abbreviations: SNP, single-nucleotide polymorphism; RA, Rheumatoid Arthritis. |
 |
Figure 18 Leave-one-out sensitivity analysis for RA on antibody-mediated immune responses. (a) Analysis for “RA” on “Chlamydia trachomatis pGP3 antibody levels”. (b) Analysis for “RA” on “Anti-human herpes virus 6 IE1A IgG seropositivity”. (c) Analysis for “RA” on “Human herpes virus 6 IE1A antibody levels”. (d) Analysis for “RA” on “Anti-human herpes virus 7 IgG seropositivity”. (e) Analysis for “RA” on “Merkel cell polyomavirus VP1 antibody levels”. Abbreviation: MR, Mendelian randomization. |
Discussion
In this research, we used the most extensive infectious disease-related GWAS data for a bidirectional two-sample MR to evaluate the interaction relationship with non-infectious inflammatory joint diseases. In particular, we identified four antibody-mediated immune responses that could potentially play a causative role in Gout, and two antibody-mediated immune responses that may potentially cause RA. We focus on some stable and significant analytical results, among which the more significant analysis suggest that Anti-human herpes virus 6 IE1B IgG seropositivity appears to serve as a protective factor against Gout, while Polyomavirus 2 JC VP1 antibody levels pose a risk for Gout, higher BK polyomavirus VP1 antibody levels have been identified as a protective factor for RA. In the reverse MR analysis, we also discovered that Gout has a significant positive causality with BK polyomavirus VP1 antibody levels, as well as Chlamydia trachomatis tarp-D F2 antibody levels. Furthermore, we observed that RA has a positive correlation with Merkel cell polyomavirus VP1 antibody levels. Although the correlation between antibody levels and disease outcomes remains to be exactly elaborated, existing studies provide a theoretical basis for the correlation.
Human herpes virus 6 (HHV-6) is a virus associated with roseola rash in infants. More than 90% individuals infected with HHV-6B are under the age of three and enter the latent phase following primary infection, resulting in lifelong latent infection.25,26 The study found that B cells can induce the transformation of effector T cells, leading to an increase in the activation of Treg cell populations and a decrease in Th1 and Th17 cells.27–29 Existing studies have shown that HHV-6 can trigger specific Treg cells to inhibit immune responses, which may serve as a mechanism for immune evasion. Viral-specific Treg cells can directly inhibit effector T cell and impair the function of dendritic cells (DCs), thereby affecting both innate and adaptive immunity.30 Treg cells can secrete high levels of IL-10 and TGF-β to participate in the suppression of inflammation and immunity, driving the transformation of T cells into Treg cells.31,32 MUS induced changes in the Th 17/Treg ratio expressed in Gout model mice are similar to the process of Gout’s onset.33 Wang et al also discovered that Treg cells inhibited the progression of Gout while Th17 and Th1 cells exert a promoting effect.34 Our study pointed that the HHV-6B-related antibody-mediated immune response acts as a protective factor against Gout. Consequently, we hypothesize that the protective mechanism might be inducted by an immune escape process mediated by HHV-6B Treg cells, resulting in a cascade of immunosuppressive effects. Nevertheless, the precise mechanisms necessitate further investigation in both in vitro and population-based studies.
Our research analysis found that immunity from JC polyomavirus (JCPyV) virus infection is a risk factor for Gout. VP1, the major capsid protein of JCPyV, is associated with immunodeficiency patients being more susceptible to the reactivation of the virus. Its infection is closely related to CD4+ and CD8+ T cells.35,36 CD8+ T cells differentiate into effector T cells with the assistance of CD4 + T cells, and CD8 + cytotoxic T lymphocytes can control viruses by eliminating the target cells.37 The differentiation of both CD4T and CD8T is regulated by B cells.38 Research has found the increase in CD8+ T cell expression within the synovial tissue of individuals suffering from Gout.39 GuH et al discovered that hyperactivated CD8T could be a primary mechanism for the deposition of MSU crystals, contributing to the development of Gout.40 This aligns with our findings. These results underscore the connection between JCPyV and Gout, indicating that preventing JCPyV may help to reduce the onset of Gout. However, the specific molecular relationship between infection and disease is still not clearly elucidated and needs further exploration, which also provides a new direction to prevent Gout.
BK polyomavirus (BK PyV) is a member of polyomavirus family. The capsid protein VP1 is crucial in facilitating the viral entry into cells.41 Our study found that the elevated levels of BK PyV antibody offer some protective effect against RA. Antibodies are mainly produced by B cells, and certain studies have indicated that B cells facilitate the emergence of Treg cells.42–44 Treg cells can maintain immune self-tolerance, effectively inhibit the proliferation of effector cells, and secrete IL-10 and TGF- β. Moreover, Treg cells can exert bystander inhibition by non-specific suppressing immune response against non-cognate antigens.45 They can also specifically infiltrate inflammatory sites such as synovium.46 Some studies have used the adoptive transfer of polyclonal Treg cells to treat autoinflammatory diseases and reduce the incidence of the disease.47–49 This also suggests that we could focus more on this immunotherapy mechanism.
On the other hand, Gout will elevate the increase in BK PyV, Chlamydia trachomatis tarp-D F2 antibody levels. RA leads to elevated increased Merkel cell polyomavirus VP1 antibody levels. This finding suggests that the two non-infectious inflammatory joint diseases play a certain modulatory role on the systemic immune response, potentially impacting the progression of certain infectious diseases. While there is no conclusive evidence to establish a clear association, and further research is needed to clarify the relationship.
We have innovatively discovered that antibodies to infectious diseases may be related to Gout and RA, which will draw the attention of clinicians in the diagnosis and treatment of related diseases. For instance, patients infected with Polyomavirus 2 JC VP1 are more prone to Gout, and these patients may need to strictly control uric acid and diet. Anti-human herpes virus 6 IE1B IgG seropositivity and BK polyomavirus VP1 antibody levels can be somewhat helpful in preventing Gout and RA, but there are currently no vaccines available related to HHV-6B and BKPyV. This also calls for more scholars to actively engage in vaccine research and development. Gout and RA can also cause an increase in antibodies for certain infectious diseases, indicating that the likelihood of patients with Gout or RA contracting these diseases is reduced. This discovery updates our understanding and can assist clinicians in conducting preliminary exclusionary diagnoses for related patients.
In addition to the aforementioned relatively stable and significant findings, the IVW analysis results also indicated several secondary significant outcomes. In the forward MR analysis, anti-Chlamydia trachomatis IgG seropositivity and Helicobacter pylori GroEL antibody levels were identified as protective factors for Gout, reducing its likelihood of occurrence. While Helicobacter pylori Catalase antibody levels served as a protective factor for RA, decreasing the probability of its development. In the reverse MR analysis, Gout episodes may elevate Varicella zoster virus glycoproteins E and I antibody levels, and the onset of RA might increase Anti-human herpes virus 7 IgG seropositivity. Since these results only demonstrated significance under IVW analysis, we still need to maintain a cautious attitude when interpreting these results, and further animal or clinical studies are necessary to validate these conclusions.
Limitations does have in this study. First, the GWAS datasets were selected from individuals of European ancestry, which means there is a lack of analysis pertaining to Asia, Africa and other populations. Consequently, the findings are not universally applicable and necessitate additional research and analysis to confirm their validation. Second, when screening for IVs, we adopted the less strict threshold of P <5×10−6 due to the insufficient of SNPs. This may impacted the estimated causal effects, so we anticipate incorporating more complete statistics and verification in future experiments to make the result analysis more reliable. In addition, the GWAS of Gout and RA did not clearly discriminate their onset and remission period, which hinders complete classification and may introduce bias into the results. Thus, the results should be interpreted with caution, and future experimental analyses may allow for stratified MR from the ictal and remission periods.
Conclusion
In conclusion, our study innovatively established a connection between infectious and non-infectious diseases, providing evidence for early screening of individuals at risk versus. We have finally identified 4 antibody-mediated immune responses that affect Gout, 2 antibody-mediated immune responses that influence RA, 3 antibody mediated immune responses influenced by Gout, and 2 antibody-mediated immune responses affected by RA. This proved that the antibody mediated immune responses are associated with these two non-infectious inflammatory joint diseases. Interestingly, we have found that different infectious antibodies may have varying causal effects on Gout. We propose an innovative conclusion that this may involve different immune mechanisms, including immune escape of HHV-6B and overactivation of JC polyomavirus CD8T cells. Our research findings provide new directions for future experiments, which can focus on the comorbidity and immunological perspectives, paying attention to immune escape mechanisms, CD8T cells, and Treg cells. Existing research reports are limited, and we hope to have further clinical observational studies or immunological research to support our findings and perspectives. In the results of the inverse relationship, there is no clear theoretical or experimental evidence to support our views, thus the impact of Gout and RA on infectious antibodies cannot be clearly defined at present. Future comparative experiments are needed to demonstrate whether patients with gout and RA are more likely to have high antibodies related to infectious diseases and the underlying immune mechanisms, which current research cannot explain.
Abbreviations
RA, Rheumatoid Arthritis; MR, Mendelian Randomization; GWAS, genome-wide association studies; IVW, Inverse Variance Weighted; MR-PRESSO, MR pleiotropy residual sum and outlier; IV, instrumental variable; RCT, randomized controlled trials; OR, odds ratio; CI, confidence intervals; HHV-6, Human herpes virus 6; DCs, dendritic cells; JCPyV, JC polyomavirus; BK PyV, BK polyomavirus.
Data Sharing Statement
Public databases used in this study are available at https://www.ebi.ac.uk/gwas/ and https://www.finngen.fi/en. The analysis data and results of this study are detailed in Table 1-7, Supplementary Tables 1 and 2, and Figures 1-18. Reasonable requests are supported, and data that supports the conclusion of this article can be obtained from the author ([email protected]).
Ethics Approval and Consent to Participate
Our GWAS analysis study has been approved for exemption from ethical review by the IRB (Ethics Committee of Guangdong Provincial Hospital of Integrated Traditional and Western Medicine), and we have included a statement of approval in the attachment (details of ethics committee approval). According to the National Health Commission of the People’s Republic of China, the Ministry of Education, the Ministry of Science and Technology, the State Administration of Traditional Chinese Medicine issued the “Notice on the issuance of Human Life Sciences and medical research ethical Review Measures” Article 32: Using legally available public data, or by observing and does not interfere with the study on the data of public behavior of exempt from ethical review (https://www.gov.cn/zhengce/zhengceku/2023-02/28/content_5743658.htm).
Acknowledgments
We are grateful to Butler-Laporte et al authors, FinnGen consortium, GWAS Catalog for publicly available data.
Disclosure
The authors declare that there is no potential conflict of interest in this study.
References
1. Cross M, Smith E, Hoy D, et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1316–1322. Epub 2014 Feb 18. PMID: 24550173. doi:10.1136/annrheumdis-2013-204627
2. Kumar LD, Karthik R, Gayathri N, Sivasudha T. Advancement in contemporary diagnostic and therapeutic approaches for rheumatoid arthritis. Biomed Pharmacother. 2016;79:52–61. doi:10.1016/j.biopha.2016.02.001
3. Edilova MI, Akram A, Abdul-Sater AA. Innate immunity drives pathogenesis of rheumatoid arthritis. Biomed J. 2021;44(2):172–182. Epub 2020 Jul 8. PMID: 32798211; PMCID: PMC8178572. doi:10.1016/j.bj.2020.06.010
4. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis [published correction appears in Lancet. 2016 Oct 22;388(10055):1984. doi: 10.1016/S0140-6736(16)30794-2]. Lancet. 2016;388(10055):2023–2038. doi:10.1016/S0140-6736(16)30173-8
5. van Delft MAM, Huizinga TWJ. An overview of autoantibodies in rheumatoid arthritis. J Autoimmun. 2020;110:102392. doi:10.1016/j.jaut.2019.102392
6. Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2012;71(9):1524–1529. Epub 2012 Mar 16. PMID: 22425941. doi:10.1136/annrheumdis-2011-200726
7. Lebre MC, Jongbloed SL, Tas SW, Smeets TJ, McInnes IB, Tak PP. Rheumatoid arthritis synovium contains two subsets of CD83-DC-LAMP- dendritic cells with distinct cytokine profiles. Am J Pathol. 2008;172(4):940–950. doi:10.2353/ajpath.2008.070703
8. Corvaisier M, Delneste Y, Jeanvoine H, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol. 2012;10(9):e1001395. doi:10.1371/journal.pbio.1001395
9. Jiang Q, Yang G, Liu Q, Wang S, Cui D. Function and Role of regulatory T cells in rheumatoid arthritis. Front Immunol. 2021;12:626193.PMID: 33868244; PMCID: PMC8047316. doi:10.3389/fimmu.2021.626193
10. Dehlin M, Jacobsson L, Roddy E. Global epidemiology of Gout: prevalence, incidence, treatment patterns and risk factors. Nat Rev Rheumatol. 2020;16(7):380–390. Epub 2020 Jun 15. PMID: 32541923. doi:10.1038/s41584-020-0441-1
11. Dalbeth N, Choi HK, Joosten LAB, et al. Gout. Nat Rev Dis Primers. 2019;5(1):69. PMID: 31558729. doi:10.1038/s41572-019-0115-y
12. Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. Lancet. 2021;397(10287):1843–1855. Epub 2021 Mar 30. Erratum in: Lancet. 2021 May 15;397(10287):1808. doi: 10.1016/S0140-6736(21)01010-2. PMID: 33798500. doi:10.1016/S0140-6736(21)00569-9
13. Liu W, Peng J, Wu Y, et al. Immune and inflammatory mechanisms and therapeutic targets of Gout: an update. Int Immunopharmacol. 2023;121:110466.Epub 2023 Jun 11. PMID: 37311355. doi:10.1016/j.intimp.2023.110466
14. Yu H, Xue W, Yu H, et al. Single-cell transcriptomics reveals variations in monocytes and Tregs between Gout flare and remission. JCI Insight. 2023;8(23):e171417. PMID: 38063198; PMCID: PMC10795830. doi:10.1172/jci.insight.171417
15. Xie D, Choi HK, Dalbeth N, et al. Gout and excess risk of severe SARS-CoV-2 infection among vaccinated individuals: a general population study. Arthritis Rheumatol. 2023;75(1):122–132. Epub 2022 Nov 30. PMID: 36082457; PMCID: PMC9537980. doi:10.1002/art.42339
16. Leech MT, Bartold PM. The association between rheumatoid arthritis and periodontitis. Best Pract Res Clin Rheumatol. 2015;29(2):189–201. Epub 2015 May 1. PMID: 26362738. doi:10.1016/j.berh.2015.03.001
17. Toussirot E, Roudier J. Pathophysiological links between rheumatoid arthritis and the Epstein-Barr virus: an update. Joint Bone Spine. 2007;74(5):418–426. Epub 2007 Jun 8. PMID: 17625943. doi:10.1016/j.jbspin.2007.05.001
18. Liu M, Gao Y, Zhang Y, Shi S, Chen Y, Tian J. The association between severe or dead COVID-19 and autoimmune diseases: a systematic review and meta-analysis. J Infect. 2020;81(3):e93–e95. Epub 2020 Jun 2. PMID: 32502509; PMCID: PMC7264926. doi:10.1016/j.jinf.2020.05.065
19. Butler-Laporte G, Kreuzer D, Nakanishi T, Harroud A, Forgetta V, Richards JB. Genetic determinants of antibody-mediated immune responses to infectious diseases agents: a genome-wide and HLA association study. Open Forum Infect Dis. 2020;7(11):ofaa450. PMID: 33204752; PMCID: PMC7641500. doi:10.1093/ofid/ofaa450
20. Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: a review. Res Synth Methods. 2019;10(4):486–496. Epub 2019 Apr 23. PMID: 30861319; PMCID: PMC6973275. doi:10.1002/jrsm.1346
21. Larsson SC, Butterworth AS, Burgess S. Mendelian randomization for cardiovascular diseases: principles and applications. Eur Heart J. 2023;44(47):4913–4924. PMID: 37935836; PMCID: PMC10719501. doi:10.1093/eurheartj/ehad736
22. Burgess S, Thompson SG; CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–764. Epub 2011 Mar 16. PMID: 21414999. doi:10.1093/ije/dyr036
23. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–1163. PMID: 17886233. doi:10.1002/sim.3034
24. Fu J, Qin Y, Xiao L, Dai X. Causal relationship between gut microflora and dementia: a Mendelian randomization study. Front Microbiol. 2024;14:1306048.PMID: 38287957; PMCID: PMC10822966. doi:10.3389/fmicb.2023.1306048
25. Rasa S, Nora-Krukle Z, Henning N, et al; European Network on ME/CFS (EUROMENE). Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J Transl Med. 2018;16(1):268. PMID: 30285773; PMCID: PMC6167797. doi:10.1186/s12967-018-1644-y
26. Lusso P. HHV-6 and the immune system: mechanisms of immunomodulation and viral escape. J Clin Virol. 2006;37 Suppl 1:S4–10.PMID: 17276368. doi:10.1016/S1386-6532(06)70004-X
27. Lu J, Cen Z, Tang Q, Dong J, Qin L, Wu W. The absence of B cells disrupts splenic and myocardial Treg homeostasis in coxsackievirus B3-induced myocarditis. Clin Exp Immunol. 2022;208(1):1–11. PMID: 35262174; PMCID: PMC9113299. doi:10.1093/cei/uxac015
28. Lu Y, Liu F, Li C, Chen Y, Weng D, Chen J. IL-10-producing B cells suppress effector T cells activation and promote regulatory t cells in crystalline silica-induced inflammatory response in vitro. Mediators Inflamm. 2017;2017:8415094.Epub 2017 Aug 2. PMID: 28831210; PMCID: PMC5558645. doi:10.1155/2017/8415094
29. Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD-L1hi B cells are critical regulators of humoral immunity. Nat Commun. 2015;6:5997.PMID: 25609381. doi:10.1038/ncomms6997
30. Wang F, Chi J, Peng G, et al. Development of virus-specific CD4+ and CD8+ regulatory T cells induced by human herpesvirus 6 infection. J Virol. 2014;88(2):1011–1024. Epub 2013 Nov 6. PMID: 24198406; PMCID: PMC3911638. doi:10.1128/JVI.02586-13
31. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174(4):1783–1786. PMID: 15699103. doi:10.4049/jimmunol.174.4.1783
32. Fastenackels S, Bayard C, Larsen M, et al. Phenotypic and functional differences between human herpesvirus 6- and human cytomegalovirus-specific T cells. J Virol. 2019;93(13):e02321–18. PMID: 30996090; PMCID: PMC6580948. doi:10.1128/JVI.02321-18
33. Dai XJ, Tao JH, Fang X, et al. Changes of Treg/Th17 ratio in spleen of acute gouty arthritis rat induced by MSU crystals. Inflammation. 2018;41(5):1955–1964. PMID: 30039428. doi:10.1007/s10753-018-0839-y
34. Wang B, Chen S, Qian H, et al. Role of T cells in the pathogenesis and treatment of Gout. Int Immunopharmacol. 2020;88:106877.Epub 2020 Aug 14. PMID: 32805695. doi:10.1016/j.intimp.2020.106877
35. Neu U, Maginnis MS, Palma AS, et al. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe. 2010;8(4):309–319. PMID: 20951965; PMCID: PMC2957469. doi:10.1016/j.chom.2010.09.004
36. Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5(3):e1000363. Epub 2009 Mar 27. PMID: 19325891; PMCID: PMC2655709. doi:10.1371/journal.ppat.1000363
37. Beltrami S, Gordon J. Immune surveillance and response to JC virus infection and PML. J Neurovirol. 2014;20(2):137–149. Epub 2013 Dec 3. PMID: 24297501; PMCID: PMC3972311. doi:10.1007/s13365-013-0222-6
38. Durali D, de Goër de Herve MG, Gasnault J, Taoufik Y. B cells and progressive multifocal leukoencephalopathy: search for the missing link. Front Immunol. 2015;6:241.PMID: 26042124; PMCID: PMC4437032. doi:10.3389/fimmu.2015.00241
39. Chang JG, Tu SJ, Huang CM, et al. Single-cell RNA sequencing of immune cells in patients with acute Gout. Sci Rep. 2022;12(1):22130. PMID: 36550178; PMCID: PMC9772586. doi:10.1038/s41598-022-25871-2
40. Gu H, Yu H, Qin L, et al. MSU crystal deposition contributes to inflammation and immune responses in Gout remission. Cell Rep. 2023;42(10):113139. Epub 2023 Sep 26. PMID: 37756161. doi:10.1016/j.celrep.2023.113139
41. Ambalathingal GR, Francis RS, Smyth MJ, Smith C, Khanna R. BK polyomavirus: clinical aspects, immune regulation, and emerging therapies. Clin Microbiol Rev. 2017;30(2):503–528. PMID: 28298471; PMCID: PMC5355639. doi:10.1128/CMR.00074-16
42. Ray A, Basu S, Williams CB, Salzman NH, Dittel BN. A novel IL-10-independent regulatory role for B cells in suppressing autoimmunity by maintenance of regulatory T cells via GITR ligand. J Immunol. 2012;188(7):3188–3198. Epub 2012 Feb 24. PMID: 22368274; PMCID: PMC3311743. doi:10.4049/jimmunol.1103354
43. Walters SN, Webster KE, Daley S, Grey ST. A role for intrathymic B cells in the generation of natural regulatory T cells. J Immunol. 2014;193(1):170–176. Epub 2014 May 28. PMID: 24872190. doi:10.4049/jimmunol.1302519
44. Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Malignant B cells skew the balance of regulatory T cells and TH17 cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2009;69(13):5522–5530. Epub 2009 Jun 9. PMID: 19509224; PMCID: PMC2764404. doi:10.1158/0008-5472.CAN-09-0266
45. Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164(1):183–190. PMID: 10605010. doi:10.4049/jimmunol.164.1.183
46. van Herwijnen MJ, Wieten L, van der Zee R, et al. Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis. Proc Natl Acad Sci U S A. 2012;109(35):14134–14139. Epub 2012 Aug 13. PMID: 22891339; PMCID: PMC3435203. doi:10.1073/pnas.1206803109
47. Ohata J, Miura T, Johnson TA, Hori S, Ziegler SF, Kohsaka H. Enhanced efficacy of regulatory T cell transfer against increasing resistance, by elevated Foxp3 expression induced in arthritic murine hosts. Arthritis Rheum. 2007;56(9):2947–2956. PMID: 17763426. doi:10.1002/art.22846
48. Desreumaux P, Foussat A, Allez M, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology. 2012;143(5):1207–1217.e2. Epub 2012 Aug 8. PMID: 22885333. doi:10.1053/j.gastro.2012.07.116
49. McLarnon A. IBD: regulatory T-cell therapy is a safe and well-tolerated potential approach for treating refractory Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2012;9(10):559. Epub 2012 Aug 28. PMID: 22926154. doi:10.1038/nrgastro.2012.167
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Association Between Autoimmune Diseases and Sarcopenia: A Two-Sample Mendelian Randomization Study
Su Q, Jin C, Yang Y, Wang J, Wang J, Zeng H, Chen Y, Zhou J, Wang Y
Clinical Epidemiology 2023, 15:901-910
Published Date: 25 August 2023
Hospital-Treated Infections and 15-year Incidence of Musculoskeletal Disorders: A Large Population-Based Cohort Study
Gao Y, McGagh D, Ding L, Hong S, Ouyang Z, Wei J, Zeng C, Lei G, Xie J
Clinical Epidemiology 2025, 17:251-264
Published Date: 11 March 2025
Exploring the Causal Relationship Between Osteoporosis and Rheumatoid Arthritis: A Bidirectional Mendelian Randomization Study
Li J, Bao L, Dai C, He M
Orthopedic Research and Reviews 2025, 17:147-157
Published Date: 13 April 2025
The Impact of Triglycerides on Rheumatoid Arthritis: Risk Factor and Mendelian Randomization Study
Liu S, Liang Q
Open Access Rheumatology: Research and Reviews 2025, 17:101-115
Published Date: 23 May 2025