")
Back to Journals » Drug Design, Development and Therapy » Volume 19
From Oral to Sublingual: A Redefined Avanafil Tablet with a Breakthrough in Bioavailability and First-Pass Metabolism Avoidance
Authors Asar TO , Al-hejaili OD, El-Sawy HS , Abd-Allah FI, Omar AM , Ahmed TA, El-Say KM
Received 2 November 2024
Accepted for publication 27 March 2025
Published 3 April 2025 Volume 2025:19 Pages 2551—2576
DOI https://doi.org/10.2147/DDDT.S504291
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Georgios Panos
Turky Omar Asar,1 Omar D Al-hejaili,2 Hossam S El-Sawy,3,4 Fathy I Abd-Allah,5 Abdelsattar M Omar,6 Tarek A Ahmed,2 Khalid M El-Say2
1Department of Biology, College of Science and Arts at Alkamil, University of Jeddah, Jeddah, Saudi Arabia; 2Department of Pharmaceutics, Faculty of Pharm Saudi Arabia Acy, King Abdulaziz University, Jeddah, 21589, Saudi Arabia; 3Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Egyptian Russian University, Cairo, 11829, Egypt; 4College of Pharmacy, Kut University, Wasit, 52001, Iraq; 5Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Al-Azhar University, Cairo, 11651, Egypt; 6Department of Pharmaceutical Chemistry, Faculty of Pharmacy, King Abdulaziz University, Jeddah, 21589, Saudi Arabia
Correspondence: Khalid M El-Say, Department of Pharmaceutics, Faculty of Pharmacy, King Abdulaziz University, Jeddah, Saudi Arabia, Tel +966582934511 ; +96626400000 Ext. 20073, Fax + 96626951696, Email [email protected]
Introduction: Avanafil (AVA) is a very efficient phosphodiesterase type 5 inhibitor for the treatment of erectile dysfunction. However, it has limited bioavailability when taken orally and considerable first-pass metabolism. Enhancing its solubility and choosing an alternative delivery route may enhance its effectiveness and duration of action.
Methods: Eight complex formulations were elaborated and analyzed at various ratios using different polyethylene glycols and hydroxypropyl-beta-cyclodextrin (HP-β-CD). Sublingual tablets containing AVA were designed and optimized using the Quality-by-design approach. The tablets’ pre-compression and post-compression properties were evaluated. The in-vivo pharmacokinetic behavior of the optimized tablet was assessed and compared with that of the commercial oral tablets in human volunteers.
Results: The HP-β-CD–AVA inclusion complex (1:1 molar ratio) showed an optimum solubilization capacity with an amount suitable for incorporation into sublingual tablets. The total amounts of superdisintegrants and Plasdone XL and the percentage of starch significantly influenced the length of time it took for 80% of the AVA to be released from the sublingual tablets, the tablet hardness, and the length of time for tablet disintegration. The optimized AVA sublingual tablet exhibited a 5.98-fold increase in the AVA mean residence time over the commercial tablet, with greater plasma exposure over 72 hours and 1356.42% relative bioavailability.
Conclusion: The sublingual tablets of the solubility-enhanced HP-β-CD–AVA inclusion complex represent a promising strategy to improve AVA bioavailability and bypass the first-pass effect. Furthermore, their extended activity offers potential clinical benefits, particularly for ED patients, such as ease of administration and reduced side effects.
Keywords: erectile dysfunction, avanafil, inclusion complex, sublingual tablets, pharmacokinetic study
Graphical Abstract:

Introduction
Sexual intimacy, mental health, and physical life satisfaction are all impacted by sexual health, which is a significant component of overall health. According to a study, sexual dysfunction can affect up to 50% of both men and women, particularly as they age.1 In a global study of sexually active respondents, 49% of women and 43% of men reported having at least one sexual problem. Early ejaculation was the most prevalent problem among men (24%), followed by trouble getting or keeping an erection (17%).2 Erectile dysfunction (ED) is a male sexual impotence disorder that affects approximately one-third of men over 40 years of age in the USA. It is one of the most common disorders influencing male sexual function.3,4 Recent research in Middle Eastern and Arab nations has revealed that the incidence of ED exceeded 40% and was associated with many risk factors, including aging, smoking, decreased daily exercise, obesity, and complications resulting from diabetes mellitus.5,6 In addition, more than 70% of the subjects in a multi-clinical cross-sectional investigation carried out in Saudi Arabia showed intermediate acute symptoms of ED.7 Although ED is not frequently thought of as a condition with a mortality rate, it is closely linked to many major health problems. It has a high probability of being detrimental to the psychological welfare of men and, therefore, has a significant impact on the quality of life of men.8
Regaining the ability to get and keep an efficient penile erection, besides reducing and monitoring risk factors for an organic cardiovascular disorder, are the foremost objectives of ED therapy. The condition’s underlying cause must be identified, addressed, and treated to effectively treat ED rather than only treating symptoms.9 According to the recommendations of the American Urological Association, phosphodiesterase type 5 inhibitors (PDE5-Is) are endorsed for ED management as a first-line treatment.10,11 Several international pharmaceutical regulatory bodies, including the European Medicines Agency (EMA) in 2013 and the United States Food and Drug Administration (US FDA) in 2012, approved the use of avanafil (AVA) as a second-generation PDE5-I for ED.12 AVA has minimal pharmacodynamic effects on other PDE subtypes because it has a strong selectivity for PDE5, resulting in fewer adverse effects than other PDE-Is.13 AVA has an action duration of up to 12 hours and a short half-life of 3 to 5 hours,14 which allows the patient to take AVA orally before engaging in sexual activity.11,15
Because of the convenience, simplicity, and low cost of the popular conventional oral drugs, oral delivery is the main administration strategy for many drugs, especially for chronic conditions such as ED. Any oral medication’s pharmacological activity depends on its solubility and permeability in vivo after administration.16 Unfortunately, the Biopharmaceutical Classification System categorizes AVA as a class II active agent, indicating that AVA has poor solubility in water and high permeability. Hence, the rate-limiting step in AVA absorption is its disintegration. It also undergoes an extensive first-pass metabolism, which lowers its oral bioavailability.17 AVA absorption is also influenced by the concomitant intake of food, which delays the time it takes to reach its maximum blood level.12 Therefore, altering the solubility of AVA and choosing a different delivery strategy may have a favorable effect on its bioavailability.
Oro-mucosal delivery systems have been designed and formulated to overcome the drawbacks of the low bioavailability of standard oral dosage forms.18 Sublingual drug delivery is one promising example of an oro-mucosal delivery system. The sublingual mucosa is a desirable channel for medication delivery since it is a richly vascularized area.19 It offers a needle-free method with an area of 26.5 cm2 for medication absorption and passage into the bloodstream.20 Using the sublingual approach as a route of administration protected the administered drug significantly from the degrading effect of first-pass metabolism, resulting in a profile of high bioavailability.21 Additionally, it is a simple way to provide medication for toddlers, psychiatric and elderly patients, and those with trouble swallowing or no access to water.22 The higher overall permeability of the buccal and sublingual mucosa, compared with the other mucosa in the mouth, is another beneficial characteristic for systemic drug transfer via those routes. The oral mucosa has 10 times the permeability of water over the skin, which means its permeability may reach 4000 times that of the skin.23
In formulations of sublingual tablets, rapid disintegration and a short dissolution time may limit the rate at which drugs reach systemic circulation.21 Therefore, sublingual drugs must be stable and soluble to penetrate the mucosal barrier and enter the systemic circulation. Co-crystallization, nanoparticle formulations, surfactants, lipid-based nanocarriers, and cyclodextrin (CD) inclusion complexation are only a few of the pharmaceutical strategies utilized to increase the solubility of poorly water-soluble drugs.24,25 By creating host‒guest inclusion complexes linked by weak intermolecular interactions, CD-based inclusion complexation facilitates the permeability and absorption of the guest lipophilic medication.16 CDs, which were discovered by Antoine Villiers in 1891, are cyclic oligosaccharides produced by CD-glycosyltransferase from starch or starch derivatives.26 CDs have a hydrophilic surface and a lipophilic core, and their molecular structures resemble a truncated cone.27 When CD drug complexation occurs, the molecules of many medications can be completely or partially fitted into the CD’s cavity.28 Because of the ability of CD‒drug inclusion complexes to significantly affect the physicochemical properties of the guests (drugs), CDs have a wide range of commercial and pharmaceutical benefits, including enhancing the solubility, dissolution, and stability of a drug, aiding in the processing of formulations; reducing the volatility of a substance; and masking the side effects of loaded drugs.29 CD‒drug inclusion complexes can also have sophisticated controlled-release profiles.24,26 Kfoury et al found that the controlled release of essential oil and its therapeutic impact was prolonged when the essential oil under investigation was enclosed in a CD.30 A controlled-release approach with drugs that require frequent administration (as well as drugs with a short half-life, such as AVA) can potentially improve the drugs’ therapeutic effects and patient compliance compared with conventional formulations. In pharmaceutical applications, there are three categories of CDs devised according to their ring structures, namely, alpha-CD (α-CD), beta-CD (β-CD), and gamma-CD (γ-CD), which have 6, 7, or 8 glucose units, respectively.31 β-CD and its hydrophilic derivatives, such as hydroxypropyl-β-CD (HP-β-CD), are the first-choice candidates in pharmaceutical formulations due to their appropriate cavity sizes and inexpensive cost.32 HP-β-CD has higher aqueous solubility than β-CD at 25 °C, reaching approximately 600 mg/mL.27
In conclusion, it is crucial to create novel delivery systems that can deliver therapeutics with increased bioavailability and efficiency, decreased dosages and unwanted adverse effects, enhanced patient compliance and convenience, and increased commercialization potential. Consequently, a novel technique has been implemented to produce an AVA delivery system that can achieve high therapeutic efficiency and circumvent the first-pass effect by increasing the solubility of AVA, which will aid in achieving these objectives. Hence, we intended in this research to design an optimized sublingual tablet of an AVA complex with an appropriate solubility enhancer (HP-β-CD) to increase the drug’s solubility, circumvent the hepatic first-pass effect, and improve the drug’s bioavailability and effectiveness.
Materials and Methods
Materials
AVA was purchased from Jinlan Pharm-Drugs Technology Co., Ltd. (Hangzhou, China), while the Spectrum Chemical Manufacturing Corporation (Gardena, CA, USA) was the source of polyethylene glycol 4000 (PEG 4000). HP-β-CD, mannitol, PEG 2000, PEG 6000, and starch were purchased from Sigma-Aldrich (St. Louis, MO, USA). Explotab (sodium starch glycolate) was obtained from JRS Pharma (Rosenberg, Germany). Plasdone XL (homopolymer of N-vinylpyrrolidone) was provided by ISP (Baar, Switzerland). Whittaker Clark and Daniels (South Plainfield, NJ, USA) and Winlab Laboratory Chemical Reagents (Leicestershire, UK) were the sources of talc powder and magnesium stearate. Acetonitrile, ethanol, and methanol were obtained from Merck (Darmstadt, Germany). Honeywell Riedel-de Haën AG (Seelze, Germany) supplied the other solvents used in this work.
Solubility of Various AVA Solid Dispersions
Various solubility enhancers were used in this investigation to increase AVA’s solubility. PEG 2000, PEG 4000, PEG 6000, and HP-β-CD were used to create various solid dispersions in a 1:1 or 2:1 molar ratio with AVA. AVA (25 mg) was dissolved in 5 mL of dimethyl sulfoxide (DMSO). Another 5 mL of DMSO was used to dissolve each solubility enhancer separately in the required quantity. The two portions were vortexed after mixing for 30 minutes, and the resultant dispersion was placed in a Büchi-M/HB-140 rotary evaporator (Flawil, St. Gallen, Switzerland), where it was vacuum-dried at 47.5 ± 2.5 °C at 22 rpm. Despite the fact that using DMSO as a solvent was quite challenging, we implemented an extended drying process after the initial round of evaporation in the rotary vacuum. Initially, the rotary evaporator was set to 50 °C with a deep vacuum to remove the bulk of the DMSO for 6 hours. Following this, the samples were further dried under high vacuum conditions at 40 °C for an additional 24 hours. This two-step process ensured that any residual DMSO was eliminated. A loss-on-drying confirmatory test was performed for each AVA dispersion preparation, and each test revealed that residual DMSO did not exceed 0.5% in all of the AVA dispersions. This confirmed the validity of this drying process and its compliance with the ICH guidelines for residual solvents of class 3.33 The dry mass for each solid dispersion of an AVA complex was removed from the round flask of the rotary evaporator, and the resulting solid dispersion of each AVA complex was kept in a desiccator overnight for characterization.34
As previously mentioned, a solubility analysis for the generated solid dispersions was carried out separately in distilled water (the full details are provided in earlier research studies).35,36 In screw-capped vials, distilled water (10 mL) and an excess of AVA (either the pure form or a newly created AVA complex) were added. For 48 hours, the screw-capped vials were shaken constantly in a shaking water bath with a thermostat (Model 1031; GFL Corporation, Burgwedel, Germany). At a maximum wavelength of 285 nm, samples from the vials were collected and evaluated spectrophotometrically in triplicate. The AVA complex with the greatest solubilization capacity in water was identified and chosen for additional research.
Powder X-Ray Diffraction
Powder X-ray diffraction (PXRD) was employed to analyze the crystalline structure of the pure AVA form and the AVA complex with the highest solubility in water. This technique helps identify the material’s physical state (crystalline or amorphous), which is crucial for understanding solubility and bioavailability. The diffractograms were obtained using the Ultima IV diffractometer (Rigaku Inc., Tokyo, Japan).37 For the measurements, the samples were placed in a holder and analyzed under standard operating conditions of 40 kV voltage and 35 A current, which ensure consistent and reliable data. A copper x-ray source (Cu Kα radiation with a wavelength of 1.54050 Å) was used to generate the diffraction patterns. The samples were scanned over a small angle increment of 0.1 degrees with a 1-second dwell time at each angle (2θ), providing high-resolution data to characterize the crystalline phases accurately.
Box–Behnken Design for AVA Sublingual Tablets
The AVA sublingual tablet formulations were created using the chosen AVA complex. The Box-Behnken design (BBD) assessed the impact of the total percentage of superdisintegrant as X1, the percentage of Plasdone XL in the disintegrant mixture as X2, and the percentage of starch in the diluent mixture as X3 on the quality attributes of the AVA sublingual tablets.38–40 Three center points were included in the 15 trial runs. The total percentage of superdisintegrant (X1) was 8% to 12%; the X2 was 25% to 50%, and the X3 was 20% to 40% (Table 1). Three designated quality attributes (responses) were considered when employing the three-level, three-factor BBD model. The tablet hardness (N) was Y1, the disintegration time (in seconds) was Y2, and the time for 80% of the AVA to be released from the sublingual tablets (in minutes) was Y3. As stated in Table 1, the objectives of the BBD were to minimize all other examined responses and to achieve 29.5 N for Y1 (ie, the minimum acceptable hardness for sublingual tablets). Table 2 displays the precise levels of the formulation’s components and the identified results of the responses.
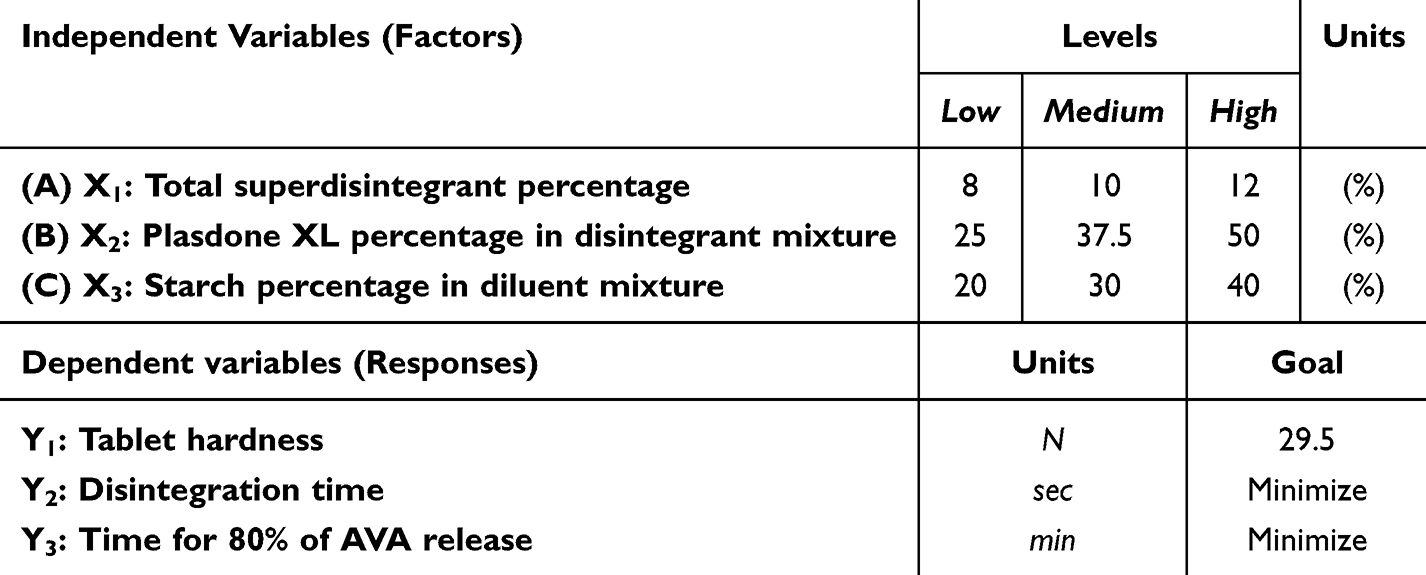 |
Table 1 Box-Behnken Design Independent Levels/Variables With the Designated Dependent Variables and Chosen Goals of AVA Sublingual Tablet Formulations |
 |
Table 2 Levels of Factors for AVA Sublingual Tablets and Observed Values of the Studied Responses Using Box-Behnken Design |
The statistical analysis was conducted using the Statgraphics Centurion 18 program from Statgraphics Technologies, Inc. (The Plains, VA, USA) to ascertain the effects of these independent variables/factors on the dependent variables/responses. A lack-of-fit test was also performed to confirm the model’s suitability for reflecting the responses’ data.35,41–43
Formulation of AVA Sublingual Tablets
Fifteen formulations of sublingual tablets were generated with various types and amounts of ingredients, as shown in Table 3. The chosen well-dried AVA inclusion complex was in a predetermined quantity (equivalent to 25 mg AVA) in each AVA sublingual tablet. Taking into consideration the amounts of factors X1, X2, and X3, as specified in Table 2 by the BBD, predetermined quantities of Explotab, Plasdone XL, and starch were added to each AVA sublingual tablet, along with 30 mg mannitol. After being completely mixed and constantly triturated for 10 minutes using a mortar and pestle, the mixture was dried in an Isotherm forced convection laboratory oven (Esco Lifesciences, Singapore) for 2 hours at 60 °C and put through a 20-mesh sieve. Each powder excipient was de-lumped using a 40-mesh sieve. The de-lumped powders were added and vigorously blended for 15 minutes using a capacity Philips electric mixer (Amsterdam, Netherlands). Talc powder and magnesium stearate (0.5% w/w for each) were added to the mixture and mixed for 3 minutes after being de-lumped in a 40-mesh sieve. AVA sublingual tablets were created using an Erweka double-punch tablet press (Heusenstamm, Germany) equipped with 9-mm flat-face round punches with a compression force of 10 KN.37
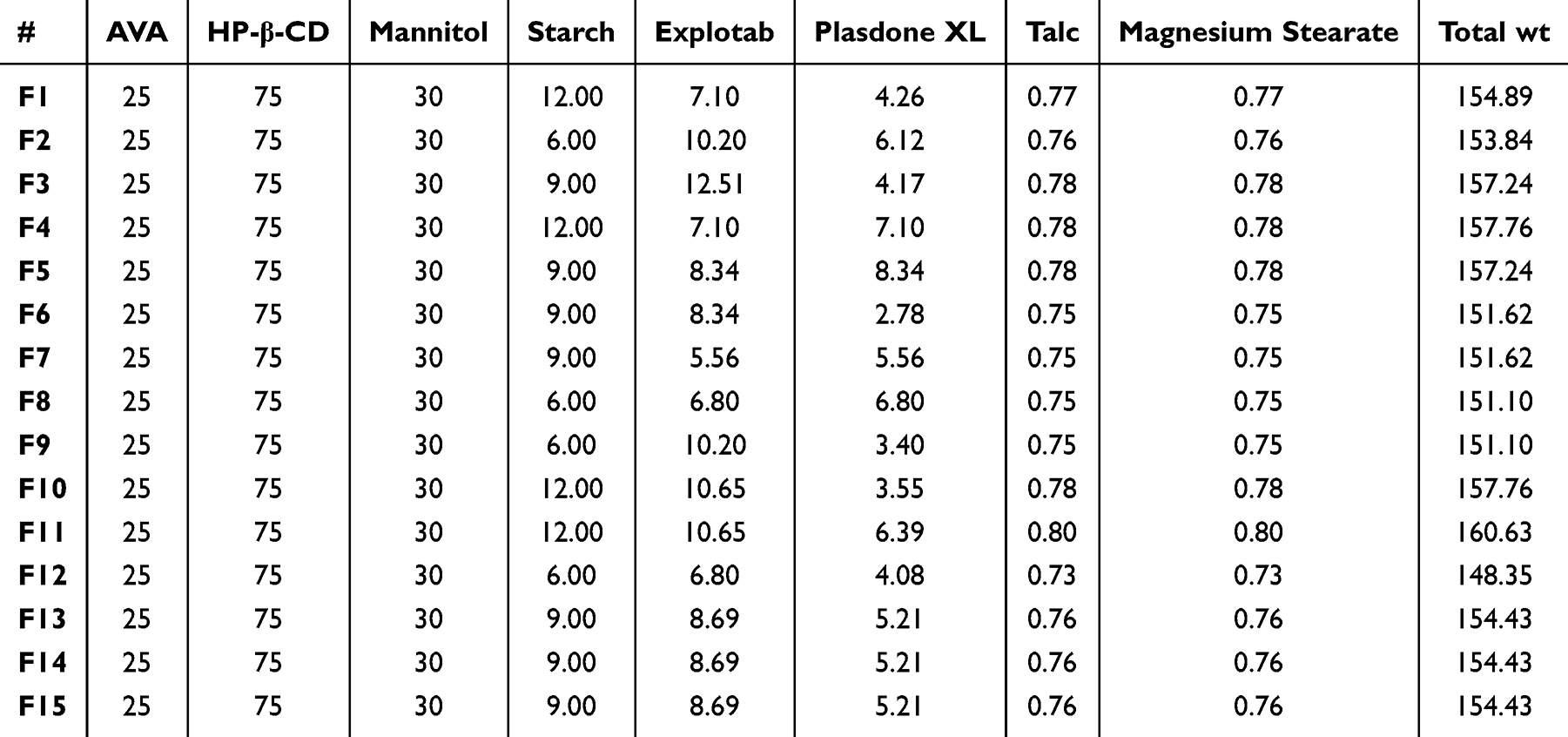 |
Table 3 Composition and Amounts of Ingredients (Mg) of AVA Sublingual Tablet Formulations |
Characterization of BBD-Elaborated AVA Sublingual Tablets
The prepared AVA sublingual tablets were inspected for the quality of their attributes, including thickness, weight uniformity, AVA content, and friability, along with the hardness of the tablet, its disintegration time, and the in-vitro dissolution/release pattern of AVA, following USP specifications, after being visually inspected for any flaws that had surfaced during the compression process.44
The drug content of the manufactured BBD AVA sublingual tablets was assessed three separate times using a spectrophotometric assay at λmax 285 nm. The average weight of 20 tablets (ie, the starting/pre-test weight) of each sublingual formulation was measured using an electronic balance. The tablets were then taken to the friabilator, placed in the drum, and rotated. The tablets were removed after 100 revolutions, cleaned of dust, and weighed again (ie, the post-test weight), and the percentage of friability (%) was thus computed (Eq. 1).

Hardness of Tablets
Each tablet’s breaking point and any changes noted in its structural integrity when packed, transported, stored, and handled before use were tested using a hardness tester. Twenty sublingual tablets from each formulation were placed one at a time between the hardness tester’s moving and stationary probes. The reading recorded by the tester was the force required for tablet breakdown, and that value was used to determine the tablet’s hardness.37
Disintegration Time
Employing a disintegration test apparatus (PharmaTest PTZ3, Hainburg, Germany), the disintegration time for each produced AVA sublingual tablet was measured. Each formulation’s six tablets (n = 6) were added one at a time to the apparatus, which contained a phosphate buffer of 6.8 pH containing 1% sodium lauryl sulfate at 37 ± 0.5 °C. The sublingual tablet’s total fragmentation point was the time at which disintegration was recorded.45
In-Vitro Dissolution Study
The USP Dissolution Test Apparatus Type II (Paddle Type), DT 700 LH equipment (Erweka GmbH, Heusenstamm, Germany) was used to examine the release of AVA from the manufactured sublingual tablets. Six replicates were made for each sublingual tablet formulation. To ensure the sink conditions, each tablet was placed in 900 mL of a dissolution medium of phosphate buffer of 6.8 pH containing 1% sodium lauryl sulfate.46 The test was run at a paddle speed of 50 rpm at a temperature of 37 ± 0.5 °C. Using a Jenway UV-Vis spectrophotometer (Staffordshire, UK), the withdrawn samples were diluted with a buffer and thoroughly filtered via a 0.45-µm Millipore filter; then, they were assessed using the same spectrophotometric assay used for the drug content test. At 5, 10, 15, 20, 30, 45, and 60 minutes, samples of 5 mL were removed from the medium and replaced with the same volume of fresh medium. The point at which 80% of the AVA (Y3) in the sublingual tablets had been released was determined.
Prediction and Characterization of the Optimized AVA Sublingual Tablets
The variance and multiple response optimization were analyzed to forecast the optimized AVA sublingual tablet using Statgraphics Centurion XVIII Software, with a license purchased from StatPoint, Inc. (Warrenton, VA, USA). The AVA release profile from the optimized sublingual tablet’s in-vitro dissolution study, the tablet hardness, and the tablet disintegration time were examined. This optimized formulation was also assessed using differential scanning calorimetry (DSC) and Fourier transform infrared spectroscopy (FT-IR). It was further evaluated in vivo with human volunteers to determine its pharmacokinetic behavior compared with a commercial AVA oral tablet.
FT-IR and DSC of The Optimized AVA Sublingual Tablets
FT-IR spectroscopy was utilized to investigate potential molecular interactions between AVA and the solubility enhancer used in the inclusion complex, as well as the individual additives and ingredients incorporated into the optimized sublingual tablet. This technique helps identify specific functional groups and bonding changes that may occur during formulation, providing insights into compatibility and interaction between components. The spectra were recorded using a Nicolet iS10 FT-IR spectrometer (Thermo Scientific Inc., Waltham, MA, USA), enabling a detailed assessment of the molecular structure and chemical stability of the formulation.47
DSC was employed to evaluate the AVA thermal profile, as well as the selected inclusion complex of AVA and the optimized AVA sublingual tablet and the individual additives/ingredients used for elaborating the optimized AVA sublingual tablet, via DSC 8000 (PerkinElmer, Inc., Waltham, MA, USA). In aluminum pans, the sample (5 mg) was heated at a rate of 10 °C/min while under the influence of nitrogen flowing at 100 mL/min. The temperature range was set up from 25 °C to 400 °C.48,49
In-Vivo Pharmacokinetic Evaluation with Healthy Volunteers
This study compared the optimized AVA sublingual tablet (test) with the commercial AVA oral tablet (reference) via an in-vivo pharmacokinetic assessment in 12 healthy human volunteers.
Study Design and Ethical Considerations
The study used a randomized parallel design (open-label, one-period, single-dose, one-treatment) while fasting. The 72-hour study periods were preceded by 14 days of screening. Two 25-mg optimized AVA sublingual tablets were administered sublingually to the individuals (test). Along with water, the same dosage of a single commercial oral tablet (50 mg) was administered orally and used as the reference drug. The Code of Ethics of the World Medical Association (Declaration of Helsinki) and the International Conference on Harmonization (ICH) of Good Clinical Practices (GCP) were both followed when conducting the study. Regulations set forth by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) were followed throughout the study. The experiment was conducted in Cairo, Egypt, at a certified research and development facility (International Centre for Bioavailability, Pharmaceutical, and Clinical Research; ICBR). The Institutional Review Board (IRB) of the Independent Ethics Committee (IEC) of the ICBR officially assessed the intended study’s goal, design, conduct, and analysis and accorded the Ethical Approval Code (RESH-007) for the proposed study protocol.
Twelve healthy Egyptian male volunteers were assigned to two groups, with six in each group. Signed informed consent forms were obtained from the volunteers before they took part in this investigation. The inclusion of representative human populations was made according to the Recommendations for the Conduct, Reporting, Editing, and Publication of Scholarly Work in Medical Journals. The selected subjects were deemed healthy based on their vital signs, physical examinations, complete medical histories, and extensive laboratory assessments (eg, urinalysis and hematological and biochemical tests). The sample size was determined using a power analysis to ensure adequate statistical power for detecting significant differences in Cmax, AUC, and MRT between the optimized sublingual formulation and the reference oral tablet. The calculation was based on previously published pharmacokinetic variability data for Avanafil, targeting an 80% power (1–β) and a significance level of 0.05. A total of 12 healthy volunteers (6 per group) were enrolled. This parallel-group design adhered to regulatory recommendations for bioequivalence studies, which advise a minimum of 12 subjects per treatment group for a parallel design.50,51 Although parallel designs generally require more subjects than crossovers due to higher inter-subject variability, the chosen sample size was considered adequate based on the large expected formulation effect and practical considerations, achieving approximately 80% power at α = 0.05. They were thoroughly followed by doctors before, during, and after the study period, and they also had tests for viral infections. Any participant in the trial had to have fasted for at least 12 hours before receiving the study’s tablets. Volunteers were kept in-house for 72 hours before and after drug administration so that routine blood samples could be taken at a scheduled time (as detailed under the Blood Sampling section).
Selection Criteria of Subjects
The study enrolled healthy adult volunteers aged 21–30 years with a body mass index (BMI) within the normal range (20–30 kg/m²), and their average height was 172 ± 5.3 cm to minimize variability in pharmacokinetic outcomes due to age, body composition, or underlying health conditions. Participants were nonsmokers with no history of chronic illness, drug allergies, or recent medication use that could interfere with the study results. Additional exclusion criteria included pregnancy, lactation, and participation in another clinical trial within the past 30 days.
The participants were divided into two groups: the first group received the commercial AVA oral tablet Spedra manufactured by Menarini International Operations Luxembourg S.A. (Luxembourg), and the second group received the optimized AVA sublingual tablet.
Sampling Procedure
Before and at 0.08, 0.17, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 48, and 72 hours after administration of the oral tablet or the sublingual tablets, 5-mL blood samples were drawn into heparinized tubes. After being centrifuged at 3000 rpm for 5 minutes, plasma samples were separated and stored at –20 °C pending analysis.
High-Performance Liquid Chromatography
In the Egyptian Research and Development Company labs, a high-performance liquid chromatography (HPLC) method was developed to measure amounts of AVA in the plasma samples. The technique was validated following the FDA Bio-analytical Method Validation Guidelines 2003.52 The method’s linearity was examined for a concentration range of 10 to 600 ng/mL of AVA, with a regression coefficient (R2) of 0.998. The internal standard that was used was azithromycin. According to the established guidelines, the technique validation findings were acceptable The proposed method was demonstrated to have acceptable reproducibility, sensitivity, and precision. The HPLC-MS/MS system utilized the Agilent Series 1200 from Agilent Technologies, Inc. (Santa Clara, CA, USA). The system consisted of an autosampler (G1329A), a quaternary pump (G1311A), a vacuum degasser (G1322), and an ESI electrospray ionization ion source with Mass Hunter software. The mobile phase comprised 1:1 acetonitrile:ammonium formate (10 mol) and had a flow rate of 1 mL/min. An Intersil ODS-3 reverse phase column from GL Sciences (Tokyo, Japan) was used at 25 °C.
Assessment of Pharmacokinetic Data
An add-in tool (PKSolver) was used to evaluate the pharmacokinetic data and analyze the pharmacokinetic characteristics of AVA using a noncompartmental model. The area under the plasma concentration–time curve (AUC0–t) and the plasma AVA maximum concentration (Cmax) and time (Tmax) were computed. Separate computations were carried out to determine the mean residence time (MRT), the terminal elimination rate constant (Kel), and the elimination half-life (t½). The drug’s apparent volume of distribution during the terminal phase (Vd) and apparent whole-body clearance (Cl) were also calculated. Finally, the relative bioavailability of the improved AVA sublingual tablet (AUCsublingual/AUCoral × 100) was determined.53 Plasma samples were collected from 12 volunteers at 18 time points post-administration (5 minutes to 72 hours), allowing for a robust assessment of drug absorption, distribution, and elimination.
Statistical Assessment
GraphPad Prism (version 8.4; San Diego, CA, USA) was used for all statistical analyses. The solubility study data were assessed via one-way ANOVA with Tukey–Kramer post-hoc test (significance level p < 0.05). Regression analysis was performed to model the impact of independent formulation factors on response variables. For the in vivo study, a Holm–Sidak multiple-comparisons two-way ANOVA was employed to compare AVA plasma concentrations at each time point between the two groups. An unpaired two-tailed t-test was used to evaluate differences in pharmacokinetic parameters between the sublingual and oral groups (significance set at p < 0.05). Before statistical comparisons, all pharmacokinetic parameters (Cmax, AUC, etc) were log-transformed, in line with EMA guidelines requiring parametric analysis on log-transformed data.50 Normality of the log-transformed values was assessed using the Shapiro–Wilk test and visual Q–Q plot inspection, given the small sample size (n = 6 per group). Homogeneity of variances was evaluated with Levene’s test. If unequal variances were detected, a Welch’s adjusted t-test was planned in place of the standard Student’s t-test to ensure robust comparison.
Results and Discussion
We have employed two different approaches in this research work. The first involved the enhancement of AVA’s solubility by complexing it with a solubility enhancer such as HP-β-CD. The second step was choosing a delivery method, such as the sublingual route, that would circumvent AVA degradation by high levels of gastric acid or by the hepatic first-pass metabolism.
Explotab/Plasdone XL superdisintegrants were combined in the sublingual tablet formulation to help the AVA inclusion complex and cause the ultimate rapid release of AVA beneath the tongue, enhancing absorption. All the previously mentioned variables contributed to improving AVA’s bioavailability.
Solubilization Capacity and Suitable Amount of The Elaborated AVA Complexes
The solubility enhancers HP-β-CD and PEGs were chosen in accordance with previously verified studies on the quality of solubilization they produced, which would enhance AVA’s solubility.54 Besides their influences on solubility, their applicability in the utilized amount of each solubility enhancer was considered. The total weight of a sublingual tablet should not exceed 150 to 160 mg, so the weight of the solubility enhancer:AVA complex had to be 90 to 110 mg (including the 25-mg target dose of AVA in the sublingual tablets); this weight would allow for the incorporation of the other ingredients essential for the manufacture of the sublingual tablets containing this complex.
The solubility of pure AVA in distilled water was found to be 0.003 mg/mL. This value indicated that AVA was nearly practically insoluble based on the USP definition of practically insoluble substances (ie, more than 10,000 mL was needed to dissolve 1 g). Figure 1 shows the solubility of AVA in distilled water after complexation with PEG 2000, PEG 4000, PEG 6000, or HP-β-CD in a 1:1 or 2:1 molar ratio. An HP-β-CD:AVA (2:1) inclusion complex had the greatest capacity for the solubilization of AVA, at 0.058 mg/mL. However, utilizing such an amount of HP-β-CD (approximately 150 mg) in an HP-β-CD:AVA (2:1) inclusion complex in a sublingual tablet with the 25-mg target dose of AVA would be impossible. The HP-β-CD:AVA (1:1) inclusion complex exhibited the second highest capacity of solubilization of AVA, at 0.043 mg/mL, with no significant difference from the HP-β-CD:AVA (2:1) inclusion complex (p = 0.24). In addition, 75 mg of the HP-β-CD was considered more suitable than its corresponding amount in the HP-β-CD:AVA (2:1) inclusion complex for incorporation into sublingual tablets.
 |
Figure 1 Top panel; Solubility evaluation of AVA complexes with different types and ratios of solubility enhancers in distilled water, ns indicates non-significance, while *, **, and *** indicate significant differences at P < 0.05, P < 0.01, and P < 0.001, respectively. Bottom panel; PXRD patterns of pure AVA, its inclusion complex with HP-β-CD (1:1 ratio), and pure HP-β-CD. |
It was also noticed that the PEG 4000:AVA (2:1) solid dispersion had significantly enhanced solubility of the AVA (0.03 mg/mL), with no significant difference from the HP-β-CD:AVA (1:1) inclusion complex (p = 0.3337). However, the amount of PEG 4000 needed (360 to 410 mg) made its incorporation into a PEG 4000:AVA (2:1) solid dispersion for the sublingual tablets unfeasible.
The utilization of HP-β-CD for enhancing AVA aqueous solubility was proven superior to other types of CDs and solubility enhancers. Many earlier research works focused on various AVA preparations for different pharmaceutical and preclinical/clinical applications that displayed significant loading capacities and entrapment efficiencies for AVA.7,9,17,52,54–59 Only two of them had been investigated for the solubility of AVA.
Fahmy et al studied the solubility of AVA in various oils, surfactants, and cosurfactants to design a nano-emulsifying drug delivery system with a rapid onset of action and enhanced bioavailability, but the aqueous solubility of AVA was not studied in that research.17 In another research work, Soliman et al investigated the effect of AVA inclusion with β-CD (at an equimolar ratio) on its aqueous solubility, along with other polymers. Their studies revealed that the AVA-β-cyclodextrin polyvinyl pyrrolidone K-30 (7% w/w) inclusion complex exhibited the highest level of AVA aqueous solubility (36.29 µg/mL ± 0.88, n = 2) compared with other ternary AVA-β-cyclodextrin-polymer inclusion complexes.54 Interestingly, after we had performed an unpaired t-test with Welch’s correction (SDs not equal), our HP-β-CD:AVA (1:1) inclusion complex was found to exhibit a significantly higher solubilization capacity of AVA (0.043 mg/mL ± 0.007, n = 3) than the β-CD:AVA (1:1) inclusion complex reported by Soliman et al (p = 0.0109).
Therefore, because the solubilization capacity of the HP-β-CD:AVA (1:1) inclusion complex significantly exceeded the capacities of other solubility enhancers (Figure 1) and also because the amount needed for inclusion was suitable, this inclusion complex was selected for further investigation.
PXRD with Confirmed Solubilization/Amorphization of The HP-Β-CD AVA Inclusion Complex
Using PXRD, which identified the retention of the solubility enhancer’s peaks as well as the disappearance of specific AVA peaks in the inclusion complex, the crystalline states of both the HP-β-CD:AVA (1:1) inclusion complex and pure AVA were identified. The XRD pattern seen in Figure 1 representing the pure AVA shows significant, extremely intense diffraction peaks at 7.5, 13.5, 18, 19, and 21 degrees, demonstrating that the pure AVA had a crystalline structure.
The decrease in the number and strength of the recognizable peaks in the inclusion complex’s PXRD pattern in Figure 1 illustrates the transformation of AVA to the amorphous form. The decreased crystallinity in this formulation may have been due to AVA solubilization and/or inclusion within the HP-β-CD architecture.35,54,60 However, the presence of residual crystalline AVA (compared with that in the pattern displayed by an HP-β-CD diffractogram) indicates that the formation of the inclusion complex predominantly led to partial amorphization and enhanced solubilization rather than complete amorphization. As confirmed in the previous section, AVA’s observed amorphization/solubilization in the inclusion complex increased its apparent solubility. Therefore, the HP-β-CD:AVA (1:1) inclusion complex was chosen for inclusion in the design of the sublingual tablet formulations.
Elaboration and Assessment of The BBD AVA Sublingual Tablets
Fifteen AVA sublingual tablets were made using the HP-β-CD:AVA inclusion complex and the suggested formulations from the BBD (Table 2). Figure 2 presents the in-vitro research on the dissolution of the prepared BBD formulations.
 |
Figure 2 In vitro study of AVA’s release/dissolution pattern from BBD sublingual tablets. |
The AVA content of all formulations tested for quality assurance in the production of AVA sublingual tablets ranged from 87.9% in F5 to 99.2% in F7. These results, produced using the standard USP criteria, demonstrated weight uniformity across all formulations.44 Furthermore, no discernible variation in formulation thickness was discovered. With friability and hardness varying from 0.021% to 0.804% and 19 to 55.67 N, respectively, all sublingual formulations met the BP friability test restrictions (< 1%). The results of the friability and hardness tests confirmed that the mechanical properties were satisfactory and the breaking strengths were good in the manufactured sublingual tablets.
In addition, the disintegration times ranged from 26.3 seconds for F2 to 95.3 seconds for F12. The initial release percentages (at 5 minutes) from the in-vitro dissolution research (Figure 2) ranged from 22.013% for F1 to 43.42% for F15, while the cumulative release percentages (at 60 minutes) ranged from 80.05% for F7 to 98.473% for F3. The time for 80% of the AVA to be released from the sublingual tablets was measured graphically from the in-vitro dissolution profiles of the AVA sublingual tablets (Figure 2); they ranged from 16.5 minutes for F3 to 60 minutes for F7.
The following sections describe how we predicted the optimized formulation following a comprehensive examination and justification of the selected quality factors.
Optimization of BBD AVA Sublingual Tablets
Efforts to adapt existing products and invent updated formulation architectures have substantially used response surface methodology. It interprets the dependent variables over a formulation’s independent factors from the produced polynomial equations to forecast an optimized formulation to meet the specified goals.41 This study used response surface methods combined with BBD to ascertain how the selected parameters (X1, X2, and X3) affected the variables of the analyzed responses (Y1, Y2, and Y3). Table 2 lists the independent variables (factors) for all suggested formulations, accompanied by the observed values of the dependent responses.
Explotab, Plasdone XL, and starch were chosen to contribute to scheming the selected factors for optimizing the BBD responses (regarding hardness, disintegration point, and drug release patterns of the AVA sublingual tablet formulations). Because mannitol dissolves easily in water and has a pleasant, cool feeling in the mouth, it was employed as a diluent in our sublingual tablets. Unfortunately, mannitol is a sugar alcohol and often has issues of having low compactability and being difficult to form into a tablet, with capping and sticking.61 Besides, variations in the level of mannitol can influence the disintegration time.62
Likewise, certain talc and magnesium stearate amounts can affect tablet disintegration and drug release profiles.63,64 Therefore, and because we did not want to lessen our focus by studying too many factors simultaneously, we fixed the levels of mannitol, talc, and magnesium stearate and the HP-β-CD:AVA (1:1) inclusion complex ratio. This helped us focus only on the effects of the chosen factors on the selected responses for this research. Future investigations of the levels/ratios of these ingredients are crucial.
The next sections use a three-level/three-factor model of BBD to elucidate the tablet hardness, disintegration time, and time for 80% of AVA release from the sublingual tablets (ie, these are the chosen dependent variables). This model was applied to investigate, enhance, and evaluate the interactions and quadratic impacts of the selected factors on the investigated dependent variables. The generated polynomial equations and a summary of the lack-of-fit test data are discussed in the following sections.
Lack-of-Fit Test
This test demonstrates how accurate the facts of the selected model are. The model appeared to have a 95% confidence level when the p-value for the lack-of-fit test was greater than or equal to 0.05.65,66 The summary of statistics for the lack-of-fit test (Table 4) showed that the Y1 and Y2 variables lacked a minimal fit (p ˃ 0.05). Despite a p-value of 0.0338 of the lack-of-fit test for Y3, the employed model maximized the desirability function for an overall optimum desirability value of 0.9493. These findings supported the assertion that the quadratic model could be considered valid for displaying the observed data of all the examined responses in this study.
 |
Table 4 Lack of Fit Test Summary Statistics for BBD Responses |
Quantification of The Estimated Effects of Factors
The ANOVA was utilized to clarify the polynomial equations that signified the detected dependent variables and the relevance-focused mathematical modeling of the designated factors. Table 5 displays the estimated effects of variables alongside the corresponding p-values from the ANOVA for each tested response of the AVA sublingual tablets. A factor was considered significant if its influence was greater than zero and the corresponding p-value was equal to or less than 0.05. A positive sign indicated a factor’s synergistic effect, while a negative sign indicated its antagonistic effect. Figure 3 shows the Pareto charts of responses and the contour response surface plots. Table 5 shows the estimated effects of the variables and the corresponding p-values.
 |
Table 5 Estimated Effects of Factors Alongside Related P-values for the Responses of AVA Sublingual Tablets, Along With Equations (Eqs. 2–4) Describing the Analytical Results of the Multiple Linear Regression |
 |
Figure 3 Top panel; Pareto charts representing the significant effects of all factors on tablet hardness, disintegration time, and time for 80% of AVA release from sublingual tablets. Bottom panel; 3-D contour-response surface graphs for the influence of the studied factors on tablet hardness, disintegration time, and time for 80% of AVA release from sublingual tablets. |
The findings showed that the total superdisintegrant percentage utilized to formulate the sublingual tablets (X1) had a significant antagonistic influence on the tablet hardness (Y1), the disintegration time (Y2), and the time needed for 80% of the AVA to be released (Y3); the corresponding p-values were 0.0286, 0.0003, and 0.0007, respectively. The amount of Plasdone XL in the disintegrant mixture (X2) was found to have a significant synergistic effect on both Y2 and Y3 at p-values of 0.0108 and 0.0073, respectively. The same significant positive effect was found with the quadratic term of X2 (X22) for Y2 and Y3, at p-values of 0.0068 and 0.0232, respectively. Furthermore, the percentage of starch in the diluent mixture (X3) significantly influenced Y1 via a positive effect, with a p-value of 0.018. The quadratic term of X3 (X32) had a significant positive impact on Y1 and Y2 (at p-values of 0.0264 and 0.0047, respectively), while it had a significant negative influence on Y3 (at a p-value of 0.0481). Moreover, Y3 was synergistically affected (at a p-value of 0.0371) by the interaction term between X1 and X2 (X1X2).
On the other hand, it was noticed that the quadratic term of X1 (X12), the interaction term between X1 and X3 (X1X3), and the interaction term between X2 and X3 (X2X3) did not significantly influence any of the detected responses (Table 5).
After investigating and evaluating the effects of the independent variables studied on the selected dependent variables, mathematical modeling was performed for each response. Equations 2 to 4 were developed to describe the analytical results of the multiple linear regression (Table 5).
Quantified Effects on The Tablet Hardness (Y1)
The hardness test is crucial for determining the mechanical strength of tablet formulations. The hardness data for the HP-β-CD–AVA inclusion complex sublingual tablets are shown in Table 2. For F2 and F1, the findings varied from 19 to 55.67 N.
Data displayed in the Pareto chart and 3-D contour response surface graph for Y1 (see Figure 3), alongside the ANOVA results from Table 5, reveal that the tablet hardness (Y1) was significantly influenced by the synergistic effect of the percentage of starch in the diluent mixture (X3) and the quadratic term of X3 (X32), with p-values of 0.018 and 0.0264, respectively. As the level of X3 increased from 20% to 40% at 12% for X1 and 37.5% for X2, Y1 also increased from 19 N for F2 to 39 N for F11. This trend was also confirmed by a decrease in tablet hardness from 51.67 N for F4 to 43.67 N for F8, as X3 declined from 40% to 20% at 10% for X1 and 50% for X2.
To some extent, the increased hardness of tablets with an increasing percentage of starch was expected and in accordance with earlier studies.67,68 Since starch can be used as a binder and diluent, increasing its percentage in the diluent mixture would potentially increase the diluent’s binding capacity and, thus, the tablet’s hardness. However, the explanation of this effect is not as simple as one might think because many other intercorrelated factors are also present in a diluent mixture. Some of these include the source of the starch itself, the relative amount/percentage of the disintegrants, and the physicochemical properties of all the additives other than starch, such as mannitol, Explotab, and Plasdone XL.67–70 To conclude, the major observed trend was that an increasing concentration of starch increased the hardness of the elaborated tablets.
In contrast, Y1 was significantly affected by the antagonistic effect of the total percentage of superdisintegrant (X1), with a p-value of 0.0286 (Table 5 and Figure 3). The data displayed for F1 and F11 in Table 2 show that the increase in this percentage from 8% to 12% was accompanied by a decline in the tablet hardness from 55.67 N to 39 N, respectively, at fixed levels of X2 and X3. In addition, a reduction in the X1 level from 12% to 8% was accompanied by an increase in the tablet hardness from 27.5 N to 39 N in F3 and F6, respectively, at constant levels of X2 and X3.
The decreased tablet hardness with an increase in the total percentage of the superdisintegrant (which mainly occurred for the Explotab) agreed with similar findings in earlier studies.71,72
Quantified Effects on The Disintegration Time (Y2) and Time for 80% of AVA to Be Released (Y3)
The times of disintegration and dissolution are critical factors for drugs delivered sublingually. A short disintegration and fast dissolution time are essential to ensure that a drug reaches the systemic circulation from the sublingual vasculature rather than the unintended swallowed parts of insufficient disintegrated tablets.73
Table 2 shows that the disintegration time (Y2) ranged from 26.3 seconds for F2 to 95.3 seconds for F12, while the time for 80% of the AVA to be released (Y3) ranged from 16.5 minutes for F3 to 60 minutes for F7. Figure 3 and Table 5 show that Y2 and Y3 were greatly affected by the significant antagonistic influence of the total percentage of the superdisintegrant (X1), with p-values of 0.0003 and 0.0007, respectively. From the data shown for F1 and F11 in Table 2, it is apparent that the increase in X1 from 8% to 12% was associated with a decrease in Y2 from 91.7 to 33 seconds, respectively, as well as a concomitant decrease in Y3 from 45.5 to 26 minutes, respectively, at constant levels of X2 and X3. Furthermore, the decrease in the X1 level from 12% for F3 to 8% for F6 was accompanied by an elevation in Y2 from 30.3 to 79 seconds, respectively, along with an increase of Y3 from 16.5 to 55.5 minutes, respectively, at constant X2 and X3 levels.
These findings indicated that increasing the total percentage of superdisintegrant (Explotab/Plasdone XL mixture) yielded sublingual tablets with a short disintegration/dissolution time and a rapid release of AVA from the tablets; this was in accordance with a previous study confirming their superiority as superdisintegrants.74
Unexpectedly, the percentage of the Plasdone XL disintegrant mixture (X2) had a significant synergistic effect on Y2 and Y3 (see Table 5 and Figure 3), with p-values of 0.0108 and 0.0073, respectively. This effect was confirmed by the significant synergistic effect of the quadratic term of X2 (X22) on Y2 and Y3, at p-values of 0.0068 and 0.0232, respectively. Table 2 shows that the increase in X2 from 25% for F10 to 50% for F4 was accompanied by an increase in Y2 from 65.33 to 76.7 seconds, respectively, and an increase in Y3 from 36 to 42 minutes, respectively, at a 10% level of X1 and a 40% level of X3. Furthermore, the decrease in the X2 level from 50% for F7 to 25% for F6 was accompanied by a decrease in Y2 from 94.3 to 79 seconds, respectively, along with a decrease in Y3 from 60 to 55.5 minutes, respectively, at fixed levels of X1 and X3 levels (8% and 30%, respectively).
These outcomes could be due to an interruptive incident that resulted from utilizing both the Explotab and Plasdone XL superdisintegrants. It was most likely due to the difference in the physicochemical natures of the polymeric Plasdone XL and the starch-based Explotab and the disintegration mechanism for each. Plasdone XL is characterized by high compactability and low density, which increase the probability of a more even distribution in the matrix of the formulated tablet and higher capillary action besides its swelling ability.75 Explotab mainly exerts its disintegrative activity via its swelling ability.76 In addition, increasing levels of either Explotab or Plasdone XL in certain combinations may reverse their activity, leading to a longer disintegration time and higher hardness values,77,78 leading to a longer time for drug release from the tablets.79
All these outcomes may emphasize the importance of implementing quality-by-design approaches to set the design’s selected goals and obtain an optimized formulation with the targeted outcomes. The predicted result would be that the optimized AVA sublingual tablet would have the highest percentage of total superdisintegrant, with the lowest level of Plasdone XL and the optimal level of starch in the diluent mixture.
Formulation and Characterization of The Predicted Optimized AVA Sublingual Tablet
The study’s desired outcome was met when an AVA sublingual tablet had undergone multiple response optimization. The parameters that would result in the best outcomes were first predicted and then confirmed. The hope was that a tablet could be formulated in which each factor would improve the desirability function relative to the design objectives.80 The optimized levels of factors were determined. The optimized sublingual tablet had a level of X1 of 12%, X2 of 25.1%, and X3 of 34.34%. The optimized AVA sublingual tablet had a desirability value of 0.9493, and it was developed and assessed to determine the best corresponding dependent variables, as described in the preceding sections. The values for Y1, Y2, and Y3 were found to be 31.33 N, 33.5 seconds, and 20.7 minutes, respectively, with minimal residuals from their corresponding forecasted values from the design, which were 29.5 N, 30.23 seconds, and 20.54 minutes, respectively.
FTIR and DSC of The Optimized AVA Sublingual Tablets, Confirming AVA Solubilization and the Absence of Interchemical Reactions
The FT-IR spectra of the materials used to formulate the optimized AVA sublingual tablet, as well as the HP-β-CD:AVA (1:1) inclusion complex and pure AVA powder, are shown in Figure 4. Identification of an AVA analogue was made when the absorption-specific peaks of the pure AVA were detected in the range of 1660 to 440 cm‒1. The carbonyl group of the amide bond (HN-C=O) became evident when the AVA absorption band was 1656.53 cm‒1. Another drug peak was found at 1426.76 cm‒1, associated with the C-N stretching vibration; another band at 743.35 cm‒1 indicated the benzene ring.81 The absorption bands of both the HP-β-CD:AVA (1:1) inclusion complex and the optimized AVA sublingual tablet did not interfere with many of the distinctive peaks of the pure AVA or the absorption bands of each ingredient used in the sublingual tablet (Figure 4), indicating that there were no chemical interactions (ie, no new covalent bonds) between the drug and the ingredients used to make the optimized AVA sublingual tablets,35 particularly the peaks representing the carbonyl group of the amide bond and the C-N stretching vibration. However, notable changes were seen in the bands corresponding to the O-H stretching vibrations (3200 to 3600 cm‒1), as well as the C-H stretching vibrations of AVA (2930 and 2862 cm‒1) in the absorption bands of the HP-β-CD:AVA (1:1) inclusion complex and the optimized AVA sublingual tablets, respectively. Because the formation of an inclusion complex typically involves non-covalent interactions such as hydrogen bonding, van der Waals forces, and hydrophobic interactions, the shifts in IR band positions could be attributed to these intermolecular interactions.82 For example, hydrogen bonding between the hydroxyl groups of HP-β-CD and the functional groups of AVA could cause changes in the bonds’ vibrational frequencies. Although the presence of multiple components could have diluted the signal of the AVA, the inclusion complex formation and corresponding shifts were still discernible.
 |
Figure 4 Left panel; FTIR Spectra of the ingredients utilized in formulating the optimized sublingual tablet. Right panel; FTIR Spectra of pure AVA, AVA inclusion complex with HP-β-CD (1:1 ratio), and the optimized sublingual tablet. |
The DSC thermograms of additives utilized in the optimized sublingual tablet and the raw AVA powder, the HP-β-CD:AVA (1:1) inclusion complex, and the optimized AVA sublingual tablet are shown in Figure 5. The thermogram of the pure AVA had a clear endothermic peak at 162 °C, which corresponded to the substance’s melting temperature and showed that the AVA was crystalline. The characteristic peak of the pure AVA vanished from the DSC thermograms of the HP-β-CD:AVA (1:1) inclusion complex before reappearing at 161 °C in the thermograms of the optimized AVA sublingual tablet; this was referred to as the characteristic peak of the mannitol at the same temperature (Figure 5).
 |
Figure 5 Left panel; DSC Thermograms of the ingredients utilized in formulating the optimized sublingual tablet. Right panel; DSC Thermograms of pure AVA, AVA inclusion complex with HP-β-CD (1:1 ratio), and the optimized sublingual tablet. |
The partial amorphization/solubilization of AVA in the inclusion complex (described earlier by PXRD) caused us to expect a peak for AVA in the DSC thermograms. Its absence can be interpreted in several ways. The amount of crystalline AVA detected by PXRD might have been so small that it fell below the level of detection of the DSC. In addition, the amount of crystalline AVA could have been so well dispersed in the inclusion complex that it reduced the sensitivity of the DSC in detecting its melting point. Moreover, the interaction with HP-β-CD and other excipients of the sublingual tablet (especially mannitol) might have led to the suppression or broadening of the melting point signal, making it undetectable by the DSC analysis parameters used; this is most likely what happened.35,37
Upsurge of Bioavailability and Pharmacokinetic Parameters with Prolonged Activity of The Optimized AVA Sublingual Tablet in Healthy Human Volunteers
The pharmacokinetic parameters of the clinical study are shown in Table 6. Figure 6 depicts the plasma concentration–time profiles for the optimized AVA sublingual tablet versus the commercial AVA oral tablet.
 |
Table 6 In-Vivo PK Parameters of the Optimized AVA Sublingual Tablet and Commercial AVA Oral Tablet (n = 6). The Dose of AVA in Both Groups Was 50 mg |
 |
Figure 6 The mean-plasma concentration-time profiles of AVA after a single administration of two 25 mg optimized AVA sublingual tablets and one 50 mg commercial AVA oral tablet, *, **, ***, and **** indicate significant differences at P < 0.05, P < 0.01, P < 0.001, and P < 0.0001, respectively. |
The results showed that the optimized AVA sublingual tablet’s maximum plasma concentration (Cmax) was 337.69 ± 94.29 ng/mL. The peak plasma concentration was attained at 4.67 ± 3.06 hours (Tmax). In contrast, the commercial oral tablet had a Cmax of 295 ± 65.38 ng/mL after 0.67 ± 0.29 hours. Although the optimized AVA sublingual tablet’s Cmax did not differ significantly from that of the commercial oral tablet (p > 0.05), it did take 3 hours longer for the optimized sublingual tablet to achieve peak plasma concentration. The optimized AVA sublingual tablet generated noticeably greater AVA plasma concentrations than the commercial oral tablet from 3 hours up to 48 hours after administration, verifying its extended therapeutic effect duration. Surprisingly, the optimized AVA sublingual tablet had a greater AUC than the commercial AVA oral tablet; its relative AVA exposure (AUC) was 1356.42% of that of the commercial oral tablet. These Cmax and AUC results indicate substantial differences between the formulations. For Cmax, Cohen’s d was 0.53 (95% confidence interval: –0.76 to 1.81), suggesting a moderate effect size with considerable uncertainty due to the small sample. For AUC, the difference corresponds to an extremely large effect (Cohen’s d = 4.85, 95% CI: 3.57–6.14; Hedges’ g = 4.48), far exceeding conventional bioequivalence limits. The clearance of the optimized AVA sublingual tablet was 12.94 times lower than that of the commercial AVA tablet, according to the unpaired t-test (p = 0.0002). This showed that the MRT0–∞ of the optimized AVA sublingual tablet was 5.98-fold greater than the MRT0–∞ of the commercial AVA oral tablet (p = 0.025). In regulatory terms, such a large increase in systemic exposure lies outside the typical 80–125% bioequivalence acceptance range,50 confirming that the sublingual and oral products are not bioequivalent. This outcome was expected given that the sublingual formulation was designed to enhance AVA bioavailability rather than match the reference product markedly.
Two theories could explain the optimized AVA sublingual tablet’s enhanced absorption, bioavailability, and duration. The first involved using an HP-β-CD inclusion complex to increase the drug’s solubilization, which is the amount of AVA that could be absorbed. A drug’s solubility is known to be the primary rate-limiting step for the absorption of Biopharmaceutical Classification System (BCS) class II medicines.83,84 Therefore, enhancing the drug’s solubility would increase its absorption. The second theory relies on the oro-mucosal route for administration, which has a comparatively small surface area for absorption compared with the oral route.85 This benefit would be intentionally employed to control the amount of drug absorbed and lessen the likelihood that high plasma concentrations of AVA would occur rapidly, besides avoiding the extensive hepatic first-pass effect.
Nevertheless, a short disintegration and rapid dissolution time ensure that drugs entering the sublingual vasculature reach the systemic circulation rather than the portions of the tablet that are insufficiently disintegrated and end up being swallowed. In light of this, using sublingual tablets with a modified release pattern might be considered a second rate-limiting step for absorption.86 Thus, a 20-minute release time for 80% of AVA from the optimized sublingual tablet (achieved with the optimized Explotab/Plasdone XL content) would allow the AVA–HP-β-CD inclusion complex to provide a 72-hour duration of AVA action. This outcome would be in accordance with earlier studies on the effect of HP-β-CD drug complexes in prolonging the duration of action.87,88 These outcomes could be anticipated to maximize the benefits of administering AVA via the sublingual route, particularly by enhancing AVA’s solubility and avoiding its hepatic metabolism.
In general, the use of HP-β-CD complexation drastically enhanced drug bioavailability.89,90 In the oral route of administration, the high solubility and rapid dissolution of the drug–HP-β-CD complex likely enhanced its absorption in the gastrointestinal tract, potentially leading to higher bioavailability for BCS class II drugs such as AVA.91 However, this does not fully explain the extended half-life. In our case, the sublingual administration of the AVA–HP-β-CD inclusion complex effectively caused AVA to be absorbed directly into the systemic circulation and then to have a slower rate of clearance/elimination, thereby circumventing the first-pass effect of the oral route and improving bioavailability further. Nikolić et al confirmed this by reporting that sublingual/buccal administration of tablets based on β-CD complexes led to effective absorption of the active moieties into the systemic circulation, followed by gradual elimination (thus avoiding a rapid first-pass loss).92
Once the AVA–HP-β-CD complex is absorbed, its distribution can be influenced by its interaction with plasma proteins and tissue binding. The inclusion complex with HP-β-CD may enhance the protein binding of AVA. Increased protein binding can reduce the concentration of free (unbound) drug, leading to a slower elimination rate and prolonged half-life. This concept aligns with the observed extended half-life in our study.93 It has also been reported that CDs can alter the distribution profile by forming inclusion complexes, which may result in a more extensive distribution phase and prolonged presence in tissues.94 For example, intravenous dexamethasone and baicalein were found to have relative bioavailabilities of 1.22 and 1.25, respectively, and dexamethasone showed a lower clearance when administered as HP-β-CD complex formulations.95,96
A modified/prolonged release time for the drug–HP-β-CD inclusion complex can also affect a drug’s metabolism. CDs protect the drug from premature degradation, potentially leading to a slower metabolic rate, prolonged drug delivery, and higher efficacy.93,97,98 An earlier study reported that reducing camptothecin’s lactone ring hydrolysis from 40% to 6.7%, due to stabilizing the compound in HP-β-CD cavities, led to a notable upsurge in antitumor activity in many tumor cell lines.99 The drug’s excretion rate may also be influenced by complexation with CDs, as the complex can alter renal or biliary excretion patterns, resulting in a prolonged half-life.91
The optimized AVA sublingual tablet’s ~4-hour lag in Tmax (time to peak) compared to the commercial oral tablet’s quick onset suggests that the sublingual formulation can provide substantially better long-term pharmacological control of AVA. This feature is especially advantageous for conditions that require a long duration of drug action. ED is a chronic/persistent disorder that may involve 3 to 12 months of treatment.100,101 Because of the novel approach’s upsurge in bioavailability and prolonged action, patient adherence, convenience, and compliance in ED therapy are expected to improve. The current study demonstrates the effectiveness of the sublingual formulation at a laboratory scale. However, challenges such as batch uniformity, process reproducibility, and regulatory compliance will need to be addressed for large-scale manufacturing. Additionally, long-term stability under various storage conditions has not been fully evaluated; future research should include stability testing to ensure the formulation maintains its efficacy and shelf life over time.
In light of these findings, several important considerations and future research directions are recommended. First, the extended mean residence time observed in this study may lead to drug accumulation with repeated dosing, particularly in populations with compromised drug clearance. Therefore, pharmacokinetic monitoring and appropriate dose adjustments will be crucial in clinical practice to avoid potential safety risks. The study’s limitations should also be acknowledged, including the small sample size (which impacts statistical power and generalizability) and the need for larger-scale studies to validate these findings. Given the single-dose nature of the study, future research should explore multiple-dose regimens to reflect steady-state pharmacokinetics better. Moreover, the controlled experimental conditions (eg, a 12-hour fast before drug administration) differ from real-life scenarios where drug absorption and metabolism may vary. Future studies should account for these variables by including diverse patient populations and evaluating drug performance under more representative conditions. These steps will help refine the formulation and improve its clinical applicability.
It is important to note that our optimized AVA sublingual tablet is the first AVA dosage form to utilize the sublingual route. To date, six patents102–107 and nine research studies7,9,17,52,55–59 have focused on various AVA preparations for different pharmaceutical and preclinical/clinical applications. Still, none has provided a dosage form for the sublingual delivery route. However, a buccal formulation of AVA patented in 2023 achieved an enhancement in bioavailability over its reference oral tablet, with a Cmax of 314.14 ng/mL and an AUC0–72 of 11,044.78 ng·h/mL.102 By comparison, our optimized AVA sublingual tablet showed an even greater enhancement in AVA bioavailability, with an AUC0–72 of 12,727.5 ng·h/mL and a Cmax of 337.69 ng/mL, as well as 123.73% relative bioavailability, compared with the patented buccal tablet. Furthermore, our optimized AVA sublingual tablet reached its Cmax after 4.67 hours versus 8.67 hours for the buccal tablet, indicating a longer duration of action via the more convenient sublingual route. Additionally, no earlier research has demonstrated the remarkable upsurge in bioavailability and 72-hour prolonged activity that our optimized AVA sublingual tablet achieved.
Conclusions
Based on the results of this investigation, we conclude that the inclusion complex with HP-β-CD successfully increased the solubility of AVA, and that combining this inclusion complex with a sublingual formulation yielded tablets with desirable qualities such as acceptable hardness, rapid disintegration and dissolution, and improved pharmacokinetic properties, as confirmed by the Box–Behnken design. Although AVA has low oral bioavailability and a short half-life, the optimized sublingual tablet demonstrated a prolonged half-life (27.79 ± 11.72 hours) and mean residence time (41.895 ± 16.51 hours). The Tmax and AUC0–72 values for this sublingual tablet were also significantly extended, at 4.667 ± 3.06 hours and 12,727.5 ± 3641.87 ng·h/mL, respectively. There was no discernible increase in Cmax compared to the commercial oral tablets. These pharmacokinetic results indicate the enhanced bioavailability, extended duration of action, and improved safety profile of the sublingual tablets. As a result, this sublingual delivery method may be considered a therapeutically appropriate platform for treating erectile dysfunction in the future. However, rigorous experimental validation is essential to confirm that the 1:1 hP-β-CD:AVA inclusion complex is indeed optimal for complex formation and solubility enhancement. Therefore, we suggest additional experiments be performed to optimize the HP-β-CD:AVA ratio for complex formation (using spectroscopic analyses such as UV-Vis, FT-IR, NMR, as well as phase solubility and thermodynamic studies). We strongly believe that further investigations will support the optimal HP-β-CD:AVA ratio for developing effective inclusion complexes and that further pharmacokinetic/toxicokinetic studies will lead to more effective and safer sublingual tablets or even alternative dosage forms.
Data Sharing Statement
The data presented in this study are available in this article. Any other data will be made available upon request.
Informed Consent Statement
Informed consent was obtained from all participants included in the study.
Institutional Review Board Statement
The experiment was conducted in Cairo, Egypt, at a certified research and development facility (International Centre for Bioavailability, Pharmaceutical, and Clinical Research; ICBR). The Institutional Review Board (IRB) in the Independent Ethics Committee (IEC) of ICBR officially assessed the intended study’s goal, design, conduct, and analysis. The proposed study protocol was accorded the Ethical Approval Code (RESH-007). The International Conference on Harmonization (ICH) of Good Clinical Practices (GCP) and the Declaration of Helsinki were adhered to when conducting the study. Regulations set forth by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) were adhered to throughout the study’s execution.
Funding
This work was funded by the University of Jeddah, Jeddah, Saudi Arabia, under grant No. (UJ-23-SRP-6). The authors, therefore, thank the University of Jeddah for its technical and financial support.
Disclosure
The authors declare no conflict of interest.
References
1. Yu Z, Niu J, Wang C. The prevalence and risk factors of sexual dysfunction in the elderly in Southern China. Int J Gene Med. 2024;17(May):2355–2360. doi:10.2147/ijgm.s462124
2. Wang H, Chang H, Wang A, Guo J, Wang F, Chung E. A narrative review of acupuncture for sexual dysfunction: perspective of traditional Chinese medicine. Transl Androlo Urol. 2024;13(11):2587–2600. doi:10.21037/tau-24-409
3. Selvin E, Burnett AL, Platz EA. Prevalence and risk factors for erectile dysfunction in the US. Am J Med. 2007;120(2):151–157. doi:10.1016/j.amjmed.2006.06.010
4. Mulhall JP, Luo X, Zou KH, Stecher V, Galaznik A. Relationship between age and erectile dysfunction diagnosis or treatment using real-world observational data in the USA. Int J Clin Pract. 2016;70(12):1012–1018. doi:10.1111/ijcp.12908
5. Fahmy UA. Nanoethosomal transdermal delivery of vardenafil for treatment of erectile dysfunction: optimization, characterization, and in vivo evaluation. Drug Des Devel Ther. 2015;9(January):6129–6137. doi:10.2147/DDDT.S94615
6. El-Sakka AI. Erectile dysfunction in Arab countries. part I: prevalence and correlates. Arab Journal of Urology. 2012;10(2):97–103. doi:10.1016/j.aju.2012.01.004
7. Alamoudi AA, Ahmed OAA, El-Say KM. Investigating the potential of transdermal delivery of avanafil using vitamin E-TPGS based mixed micelles loaded films. Pharmaceutics. 2021;13(5):739. doi:10.3390/pharmaceutics13050739
8. Hackett G, Kirby M, Wylie K, et al. British society for sexual medicine guidelines on the management of erectile dysfunction in men—2017. J Sex Med. 2018;15(4):430–457. doi:10.1016/j.jsxm.2018.01.023
9. El-Say KM, Al-hejaili OD, Alamoudi AA, Ahmed OAA, El-Say KM. Transdermal film loaded with avanafil ultra-deformable nanovesicles to enhance its percutaneous absorption and bioavailability. AAPS Pharm Sci Tech. 2022;23(1):1–14. doi:10.1208/S12249-021-02195-4
10. Evans J, Burke R. Avanafil for treatment of erectile dysfunction: review of its potential. Vascular Health and Risk Management. 2012;8:517–523. doi:10.2147/VHRM.S26712
11. Burke RM, Evans JD. Avanafil for treatment of erectile dysfunction: review of its potential. Vascular Health and Risk Management. 2012;8(1):517–523. doi:10.2147/VHRM.S26712
12. Hosny KM, Ahmed OAA, Fahmy UA, Alkhalidi HM. Nanovesicular systems loaded with a recently approved second generation type-5 phospodiesterase inhibitor (avanafil): i. Plackett-Burman screening and characterization. J Drug Delivery Sci Technol. 2018;43:154–159. doi:10.1016/j.jddst.2017.10.009
13. Corona G, Rastrelli G, Burri A, Jannini EA, Maggi M. The safety and efficacy of Avanafil, a new 2 generation PDE5i: comprehensive review and meta-analysis. Expert Opin Drug Saf. 2016;15(2):237–247. doi:10.1517/14740338.2016.1130126
14. Alwaal A, Al-Mannie R, Carrier S. Future prospects in the treatment of erectile dysfunction: focus on avanafil. Drug Des Devel Ther. 2011;5:435–443. doi:10.2147/DDDT.S15852
15. Kyle JA, Brown DA, Hill JK. Avanafil for Erectile Dysfunction. Ann. Pharmacother. 2013;47(10):1312–1320. doi:10.1177/1060028013501989
16. Mendes C, Buttchevitz A, Kruger JH, et al. Inclusion complexes of hydrochlorothiazide and β-cyclodextrin: physicochemical characteristics, in vitro and in vivo studies. Eur. J. Pharm. Sci. 2016;83:71–78. doi:10.1016/j.ejps.2015.12.015
17. Fahmy UA, Ahmed OAA, Hosny KM. Development and evaluation of avanafil self-nanoemulsifying drug delivery system with rapid onset of action and enhanced bioavailability. AAPS Pharm Sci Tech. 2015;16(1):53–58. doi:10.1208/s12249-014-0199-3
18. Singh SK, Sameer AA. Development and characterization of sublingual tablet of Lisinopril. Asian Pac J Trop Biomed. 2012;2(3 SUPPL):S1711–S1719. doi:10.1016/S2221-1691(12)60483-3
19. Dos Chaves P, Ourique S, Frank LA AF, Pohlmann AR, Guterres SS, Beck RCR. Carvedilol-loaded nanocapsules: mucoadhesive properties and permeability across the sublingual mucosa. Eur. J. Pharm. Biopharm. 2017;114:88–95. doi:10.1016/j.ejpb.2017.01.007
20. Wang Z, Chow MSS. Overview and appraisal of the current concept and technologies for improvement of sublingual drug delivery. Therapeutic Delivery. 2014;5(7):807–816. doi:10.4155/tde.14.50
21. Bredenberg S, Duberg M, Lennernäs B, et al. In vitro and in vivo evaluation of a new sublingual tablet system for rapid oromucosal absorption using fentanyl citrate as the active substance. Eur. J. Pharm. Sci. 2003;20(3):327–334. doi:10.1016/j.ejps.2003.07.002
22. Shelke V, Mutha S. Formulation and evaluation of lansoprazole sublingual tablet. Journal of Research in Pharmacy. 2020;24(2):264–276. doi:10.35333/jrp.2020.143
23. Lesch CA, Squier CA, Cruchley A, Williams DM, Speight P. The permeability of human oral mucosa and skin to water. Journal of Dental Research. 1989;68(9):1345–1349. doi:10.1177/00220345890680091101
24. Salústio PJ, Pontes P, Conduto C, et al. Advanced technologies for oral controlled release: cyclodextrins for oral controlled release. AAPS Pharm Sci Tech. 2011;12(4):1276–1292. doi:10.1208/s12249-011-9690-2
25. Conceicao J, Adeoye O, Cabral-Marques HM, Lobo JMS. Cyclodextrins as drug carriers in pharmaceutical technology: the state of the art. Curr. Pharm. Des. 2018;24(13):1405–1433. doi:10.2174/1381612824666171218125431
26. Conceição J, Adeoye O, Cabral-marques HM, Manuel J, Lobo S. Cyclodextrins as excipients in tablet formulations. Drug Discovery Today. 2018;23(6):1274–1284. doi:10.1016/j.drudis.2018.04.009
27. Loh GOK, Tan YTF, Peh KK. Enhancement of norfloxacin solubility via inclusion complexation with β-cyclodextrin and its derivative hydroxypropyl-β-cyclodextrin. Asian J. Pharm. Sci. 2016;11(4):536–546. doi:10.1016/j.ajps.2016.02.009
28. Jug M, Bećirević-Laćan M. Influence of hydroxypropyl-β-cyclodextrin complexation on piroxicam release from buccoadhesive tablets. Eur. J. Pharm. Sci. 2004;21(2–3):251–260. doi:10.1016/j.ejps.2003.10.029
29. Li X, Jin Z, Wang J. Complexation of allyl isothiocyanate by α- and β-cyclodextrin and its controlled release characteristics. Food Chem. 2007;103(2):461–466. doi:10.1016/j.foodchem.2006.08.017
30. Kfoury M, Auezova L, Greige-Gerges H, Fourmentin S. Promising applications of cyclodextrins in food: improvement of essential oils retention, controlled release and antiradical activity. Carbohydr. Polym. 2015;131:264–272. doi:10.1016/j.carbpol.2015.06.014
31. Badr-eldin SM, Ahmed TA, Ismail HR. Aripiprazole-cyclodextrin binary systems for dissolution enhancement: effect of preparation technique, cyclodextrin type and molar ratio. Iranian Journal of Basic Medical Sciences. 2013;16(6):1223–1231.
32. Liu L, Zhu S. Preparation and characterization of inclusion complexes of prazosin hydrochloride with β-cyclodextrin and hydroxypropyl-β-cyclodextrin. Journal of Pharmaceutical & Biomedical Analysis. 2006;40(1):122–127. doi:10.1016/j.jpba.2005.06.022
33. ICH-Q3C-(R8). International Council for Harmonisation of Technical Requirements for Impurities: Guideline for Residual Solvents. 2021.
34. Sinha S, Ali M, Baboota S, Ahuja A, Kumar A, Ali J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS Pharm Sci Tech. 2010;11(2):518–527. doi:10.1208/s12249-010-9404-1
35. El-Say KM, Al-hejaili OD, El-Sawy HS, et al. Incorporating sodium deoxycholate endorsed the buccal administration of avanafil to heighten the bioavailability and duration of action. Drug Delivery Transl Res. 2023. doi:10.1007/s13346-023-01314-x
36. Al-Subaie MM, Hosny KM, El-Say KM, Ahmed TA, Aljaeid BM. Utilization of nanotechnology to enhance percutaneous absorption of Acyclovir in the treatment of herpes simplex viral infections. Int j Nanomed. 2015;10:3973–3985. doi:10.2147/IJN.S83962
37. El-Say KM, Alotaibi FO, Alhakamy NA, Omar AM, El-Say KM. Clinical pharmacokinetic evaluation of optimized liquisolid tablets as a potential therapy for male sexual dysfunction. Pharmaceutics. 2020;12(12):1–23. doi:10.3390/pharmaceutics12121187
38. Desoqi MH, El-Sawy HS, Kafagy E, Ghorab M, Gad S. Fluticasone propionate–loaded solid lipid nanoparticles with augmented anti-inflammatory activity: optimisation, characterisation and pharmacodynamic evaluation on rats. Journal of Microencapsulation. 2021;38(3):177–191. doi:10.1080/02652048.2021.1887383
39. Kassem MA, El-Sawy HS, Abd-Allah FI, Abdelghany TM, El-Say KM. Maximizing the therapeutic efficacy of imatinib mesylate–loaded niosomes on human colon adenocarcinoma using box-behnken design. J Pharmaceut Sci. 2017;106(1):111–122. doi:10.1016/j.xphs.2016.07.007
40. Soliman MS, Abd-Allah FI, Hussain T, Saeed NM, El-Sawy HS. Date seed oil loaded niosomes: development, optimization and anti-inflammatory effect evaluation on rats. Drug Dev. Ind. Pharm. 2018;44(7):1185–1197. doi:10.1080/03639045.2018.1438465
41. El-Say KM, Ahmed TA, Aljefri AH, El-Sawy HS, Fassihi R, Abou-Gharbia M. Oleic acid–reinforced pegylated polymethacrylate transdermal film with enhanced antidyslipidemic activity and bioavailability of atorvastatin: a mechanistic ex-vivo/in-vivo analysis. Int J Pharm. 2021;608(August):121057. doi:10.1016/j.ijpharm.2021.121057
42. El-Say KM, Ahmed TA, El-Sawy HS. Transdermal film formulations and methods of use. 2021.
43. Gardouh AR, Ewedah TM, Abd-allah FI, Ghorab MM, Omran MM, El-sawy HS. Enhanced efficacy, cellular uptake, and antiangiogenic activity of the optimized imatinib mesylate-loaded proniosomal-derived nanovesicles. J Drug Delivery Sci Technol. 2021;61:102267. doi:10.1016/j.jddst.2020.102267
44. Yan X, Gurtler J, Fratamico P, et al. Comprehensive approaches to molecular biomarker discovery for detection and identification of cronobacter spp. (Enterobacter sakazakii) and salmonella spp. Appl. Environ. Microbiol. 2011;77(5):1833–1843. doi:10.1128/AEM.02374-10
45. El-Say KM, Ahmed TA, Ahmed OAA, Elimam H. Enhancing the hypolipidemic effect of simvastatin in poloxamer-induced hyperlipidemic rats via liquisolid approach: pharmacokinetic and pharmacodynamic evaluation. AAPS Pharm Sci Tech. 2020;21(6):223. doi:10.1208/s12249-020-01754-5
46. Ahmed TA, Alotaibi HA, Alharbi WS, Safo MK, El-Say KM. Development of 3D-printed, liquisolid and directly compressed glimepiride tablets, loaded with black seed oil self-nanoemulsifying drug delivery system: in vitro and in vivo characterization. Pharmaceuticals. 2022;15(1):1–14. doi:10.3390/ph15010068
47. Silva CO, Rijo P, Molpeceres J, et al. Polymeric nanoparticles modified with fatty acids encapsulating betamethasone for anti-inflammatory treatment. Int J Pharm. 2015;493(1–2):271–284. doi:10.1016/j.ijpharm.2015.07.044
48. Mulik RS, Mönkkönen J, Juvonen RO, Mahadik KR, Paradkar AR. ApoE3 mediated polymeric nanoparticles containing curcumin: apoptosis induced in vitro anticancer activity against neuroblastoma cells. Int J Pharm. 2012;437(1–2):29–41. doi:10.1016/j.ijpharm.2012.07.062
49. Al-Zuhairy SAS, Teaima MH, Shoman NA, Elasaly M, El-Nabarawi MA, El-Sawy HS. PEGylated Tween 80-functionalized chitosan-lipidic nano-vesicular hybrids for heightening nose-to-brain delivery and bioavailability of metoclopramide. Drug Delivery. 2023;30(1):2189112. doi:10.1080/10717544.2023.2189112
50. International Council for Harmonisation of Technical requirements for Pharmaceuticals for Human Use. Bioequivalence for immediate-release solid oral dosage forms M13A 2024. Available from: https://database.ich.org/sites/default/files/ICH_M13A_Step2_draft_Guideline_2022_1125.pdf.
51. Ewp. Committee for medicinal products for human use (chmp) guideline on the investigation of bioequivalence discussion in the joint efficacy and quality working group adoption rev. 1 by chmp for release for consultation end of consultation rev. 1 (deadline for comments). 1997. Available from: http://www.ema.europa.eu.
52. Aldawsari H, Fahmy U, Abd-Allah F, Ahmed O. Formulation and optimization of avanafil biodegradable polymeric nanoparticles: a single-dose clinical pharmacokinetic evaluation. Pharmaceutics. 2020;12(6):596. doi:10.3390/pharmaceutics12060596
53. Abo-EL-Sooud K. Absolute and Relative Bioavailability. Drug Discovery and Evaluation: Methods. 2018;1–7. doi:10.1007/978-3-319-56637-5_16-1
54. Soliman KA, Ibrahim HK, Ghorab MM. Effect of different polymers on avanafil–β-cyclodextrin inclusion complex: in vitro and in vivo evaluation. Int J Pharm. 2016;512(1):168–177. doi:10.1016/j.ijpharm.2016.08.044
55. Ahmed OAA, Badr-Eldin SM. Development of an optimized avanafil-loaded invasomal transdermal film: ex vivo skin permeation and in vivo evaluation. Int J Pharm. 2019;570:118657. doi:10.1016/j.ijpharm.2019.118657
56. Kurakula M, Naveen NR, Patel B, Manne R, Patel DB. Preparation, optimization and evaluation of chitosan-based avanafil nanocomplex utilizing antioxidants for enhanced neuroprotective effect on PC12 cells. Gels. 2021;7(3):96. doi:10.3390/gels7030096
57. Soliman KAB, Ibrahim HK, Ghorab MM. Formulation of avanafil in a solid self-nanoemulsifying drug delivery system for enhanced oral delivery. Eur. J. Pharm. Sci. 2016;93:447–455. doi:10.1016/j.ejps.2016.08.050
58. Gardouh AR, Elhusseiny S, Gad S. Mixed avanafil and dapoxetin hydrochloride cyclodextrin nano-sponges: preparation, in-vitro characterization, and bioavailability determination. J Drug Delivery Sci Technol. 2022;68(December 2021):103100. doi:10.1016/j.jddst.2022.103100
59. Kurakula M, Ahmed OAA, Fahmy UA, Ahmed TA. Solid lipid nanoparticles for transdermal delivery of avanafil: optimization, formulation, in-vitro and ex-vivo studies. J Liposome Res. 2016;26(4):288–296. doi:10.3109/08982104.2015.1117490
60. Asbahr ACC, Franco L, Barison A, Silva CWP, Ferraz HG, Rodrigues LNC. Binary and ternary inclusion complexes of finasteride in HPβCD and polymers: preparation and characterization. Bioorg. Med. Chem. 2009;17(7):2718–2723. doi:10.1016/j.bmc.2009.02.044
61. Takeuchi H, Nagira S, Yamamoto H, Kawashima Y. Die wall pressure measurement for evaluation of compaction property of pharmaceutical materials. Int J Pharm. 2004;274(1–2):131–138. doi:10.1016/j.ijpharm.2004.01.008
62. Katsuno E, Tahara K, Takeuchi Y, Takeuchi H. Orally disintegrating tablets prepared by a co-processed mixture of micronized crospovidone and mannitol using a ball mill to improve compactibility and tablet stability. Powder Technol. 2013;241:60–66. doi:10.1016/j.powtec.2013.03.008
63. El-Malah Y, Nazzal S. Effect of Eudragit® RS 30D and talc powder on verapamil hydrochloride release from beads coated with drug layered matrices. AAPS Pharm Sci Tech. 2008;9(1):75–83. doi:10.1208/s12249-007-9008-6
64. Billany MR, Richards JH. Batch variation of magnesium stearate and its effect on the dissolution rate of salicylic acid from solid dosage forms. Drug Dev. Ind. Pharm. 1982;8(4):497–511. doi:10.3109/03639048209022117
65. Harbi I, Aljaeid B, El-Say KM, Zidan AS. Glycosylated sertraline-loaded liposomes for brain targeting: qbd study of formulation variabilities and brain transport. AAPS Pharm Sci Tech. 2016;17(6):1404–1420. doi:10.1208/s12249-016-0481-7
66. Liu J, Wang J, Leung C, Gao F. A multi-parameter optimization model for the evaluation of shale gas recovery enhancement. Energies. 2018;11(3):654. doi:10.3390/en11030654
67. Bayor MT, Tuffour E, Lambon PS. Evaluation of starch from new sweet potato genotypes for use as a pharmaceutical diluent, binder or disintegrant. J Appl Pharm Sci. 2013;5(7):807–816. doi:10.7324/JAPS.2013.38.S4
68. Manek RV, Builders PF, Kolling WM, Emeje M, Kunle OO. Physicochemical and binder properties of starch obtained from Cyperus esculentus. AAPS Pharm Sci Tech. 2012;13(2):379–388. doi:10.1208/s12249-012-9761-z
69. Hill PM. Effect of compression force and corn starch on tablet disintegration time. J Pharmaceut Sci. 1976;65(11):1694–1697. doi:10.1002/jps.2600651134
70. Moore F, Okelo G, Colón I, Kushner J. Improving the hardness of dry granulated tablets containing sodium lauryl sulfate. Int J Pharm. 2010;400(1–2):37–41. doi:10.1016/j.ijpharm.2010.08.024
71. Mehta S, De Beer T, Remon JP, Vervaet C. Effect of disintegrants on the properties of multiparticulate tablets comprising starch pellets and excipient granules. Int J Pharm. 2012;422(1–2):310–317. doi:10.1016/j.ijpharm.2011.11.017
72. El-Say KM, Alamri SH, Alsulimani HH, et al. Incorporating valsartan in sesame oil enriched self-nanoemulsifying system-loaded liquisolid tablets to improve its bioavailability. Int J Pharm. 2023;639(January):122966. doi:10.1016/j.ijpharm.2023.122966
73. Rathbone MJ, Hadgraft J. Absorption of drugs from the human oral cavity. Int J Pharm. 1991;74(1):9–24. doi:10.1016/0378-5173(91)90403-B
74. Köster C, Kleinebudde P. Evaluation of binders in twin-screw wet granulation – optimal combination of binder and disintegrant. Eur. J. Pharm. Biopharm. 2023;186(7):55–64. doi:10.1016/j.ejpb.2023.03.003
75. Kurakula M, GSNK R. Pharmaceutical assessment of polyvinylpyrrolidone (PVP): as excipient from conventional to controlled delivery systems with a spotlight on COVID-19 inhibition. J Drug Delivery Sci Technol. 2020;60(June):102046. doi:10.1016/j.jddst.2020.102046
76. Dashevsky A, Mohamad A. Development of pulsatile multiparticulate drug delivery system coated with aqueous dispersion Aquacoat® ECD. Int J Pharm. 2006;318(1–2):124–131. doi:10.1016/j.ijpharm.2006.03.022
77. Chebli C, Cartilier L. Cross-linked cellulose as a tablet excipient: a binding/disintegrating agent. Int J Pharm. 1998;171(1):101–110. doi:10.1016/S0378-5173(98)00161-6
78. Szakonyi G, Zelkó R. Prediction of oral disintegration time of fast disintegrating tablets using texture analyzer and computational optimization. Int J Pharm. 2013;448(2):346–353. doi:10.1016/j.ijpharm.2013.03.047
79. Pathan LS, Modi CD, Gohel MC, Udhwani NH, Thakkar VT, Rana HB. Introducing novel hybridization technique for solubility enhancement of Bosentan formulation. Food Hydrocoll Health. 2022;2(7):100055. doi:10.1016/j.fhfh.2022.100055
80. Kapoor B, Gupta R, Gulati M, Singh SK, Khursheed R, Gupta M. The why, where, who, how, and what of the vesicular delivery systems. Adv. Colloid Interface Sci. 2019;271:101985. doi:10.1016/j.cis.2019.07.006
81. Attia KAM, Mohamad AA, Emara MS, et al. Second derivative synchronous fluorescence determination of avanafil in the presence of its acid-induced degradation product aided by powerful Lean Six Sigma tools augmented with D-optimal design. RSC Adv. 2021;11(7):3834–3842. doi:10.1039/D0RA08216C
82. Steiner T, Saenger W. Weak hydrogen bonding in cyclodextrin complex stabilisation. In: Tsoucaris G, editor. Current Challenges on Large Supramolecular Assemblies. Dordrecht: Springer; 1999:375–383. doi:10.1007/978-94-011-5284-6_26
83. Arzani G, Haeri A, Daeihamed M, Bakhtiari-Kaboutaraki H, Dadashzadeh S. Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge. Int j Nanomed. 2015;10:4797–4813. doi:10.2147/IJN.S84703
84. Davanço MG, Campos DR, de O Carvalho P. In vitro – in vivo correlation in the development of oral drug formulation: a screenshot of the last two decades. Int J Pharm. 2020;580:119210. doi:10.1016/j.ijpharm.2020.119210
85. El-Say KM, Ahmed TA. Buccal route of drug delivery. In: Talevi A editor. The ADME Encyclopedia. Springer International Publishing; 2022:222–231. doi:10.1007/978-3-030-84860-6_12
86. Cardot JM, Garrait G, Beyssac E. Use of IVIVC to optimize generic development. Dissolution Technologies. 2015;22(2):44–48. doi:10.14227/DT220215P44
87. Jang J, Yaksh TL, Hill HF. Use of 2-hydroxypropyl-β-cyclodextrin as an intrathecal drug vehicle with opioids. J Pharmacol Exp Ther. 1992;261(2):592–600. doi:10.1016/S0022-3565(25)11080-X
88. Gidwani B, Vyas A. A comprehensive review on cyclodextrin-based carriers for delivery of chemotherapeutic cytotoxic anticancer drugs. Biomed Res. Int. 2015;2015:198268. doi:10.1155/2015/198268
89. Topuz F. Rapid sublingual delivery of piroxicam from electrospun cyclodextrin inclusion complex nanofibers. ACS Omega. 2022;7(39):35083–35091. doi:10.1021/acsomega.2c03987
90. Sarabia-Vallejo Á, Del M Caja M, Olives AI, Martín MA, Menéndez JC. Cyclodextrin inclusion complexes for improved drug bioavailability and activity: synthetic and analytical aspects. Pharmaceutics. 2023;15(9):2345. doi:10.3390/pharmaceutics15092345
91. Jones DS, Dressman JB, Loftsson T, Moya-Ortega MD, Alvarez-Lorenzo C, Concheiro A. Pharmacokinetics of cyclodextrins and drugs after oral and parenteral administration of drug/cyclodextrin complexes. J Pharm Pharmacol. 2016;68(5):544–555. doi:10.1111/jphp.12427
92. Nikolić V, Ilić-Stojanović S, Petrović S, Tačić A, Nikolić L. Administration routes for nano drugs and characterization of nano drug loading. In: Characterization and Biology of Nanomaterials for Drug Delivery. Elsevier; 2019:587–625. doi:10.1016/B978-0-12-814031-4.00021-0
93. Soares AF, de A Carvalho R, Veiga F. Oral administration of peptides and proteins: nanoparticles and cyclodextrins as biocompatible delivery systems. Nanomedicine. 2007;2(2):183–202. doi:10.2217/17435889.2.2.183
94. Yankovsky I, Bastien E, Yakavets I, et al. Inclusion complexation with β-cyclodextrin derivatives alters photodynamic activity and biodistribution of meta-tetra(hydroxyphenyl)chlorin. Eur. J. Pharm. Sci. 2016;91:172–182. doi:10.1016/j.ejps.2016.06.012
95. Dietzel K, Estes KS, Brewster ME, Bodor NS, Derendorf H. The use of 2-hydroxypropyl-β-cyclodextrin as a vehicle for intravenous administration of dexamethasone in dogs. Int J Pharm. 1990;59(3):225–230. doi:10.1016/0378-5173(90)90113-I
96. Liu J, Qiu L, Gao J, Jin Y. Preparation, characterization and in vivo evaluation of formulation of baicalein with hydroxypropyl-β-cyclodextrin. Int J Pharm. 2006;312(1–2):137–143. doi:10.1016/j.ijpharm.2006.01.011
97. Wankar J, Kotla NG, Gera S, Rasala S, Pandit A, Rochev YA. Recent advances in host–guest self‐assembled cyclodextrin carriers: implications for responsive drug delivery and biomedical engineering. Adv. Funct. Mater. 2020;30(44):1909049. doi:10.1002/adfm.201909049
98. Hundshammer C, Schön C, Kimura M, et al. Enhanced metabolic bioavailability of tetrahydrocurcumin after oral supplementation of a γ-cyclodextrin curcumin complex. Journal of Functional Foods. 2021;79:104410. doi:10.1016/j.jff.2021.104410
99. González-Ruiz V, Cores Á, Martín-Cámara O, et al. Enhanced stability and bioactivity of natural anticancer topoisomerase i inhibitors through cyclodextrin complexation. Pharmaceutics. 2021;13(10):1609. doi:10.3390/pharmaceutics13101609
100. Kedia GT, Ückert S, Assadi-Pour F, Kuczyk MA, Albrecht K. Avanafil for the treatment of erectile dysfunction: initial data and clinical key properties. Ther Adv Urol. 2013;5(1):35–41. doi:10.1177/1756287212466282
101. Katz EG, Tan RBW, Rittenberg D, Hellstrom WJ. Avanafil for erectile dysfunction in elderly and younger adults: differential pharmacology and clinical utility. Therapeutics Clin Risk Manag. 2014;10(1):701–711. doi:10.2147/TCRM.S57610
102. Al-hejaili OD, El-Say KM, Ahmed TA, El-Sawy HS, Abd-Allah FI. Buccal formulation of avanafil with enhanced bioavailability and prolonged duration. 2023;1.
103. El-Say KM, Al-hejaili OD, Alamoudi AA, Ahmed OAA. Transfersome-containing transdermal film formulations and methods of use. United States Patent and Trademark Office. 2021.
104. Mosli HAM, Atteiah SAG, Omar KMH, El-Bassossy HMAEM, Helal MHAM. In situ gel loaded with phosphodiesterase type V inhibitors nanoemulsion. United States Patent Application Publication. 2015.
105. Broman CT, Sheu E. Orally disintegrating dosage form for administration of avanafil, and associated methods of manufacture and use. 2019.
106. Fossel ET, Gutman L. Transdermal delivery of sildenafil and other phosphodiesterase type 5 inhibitors. United States Patent Application Publication. 2016.
107. Ahmed OAA, Badr-Eldin SM. Development of an optimized avanafil-loaded invasomal transdermal film. United States Patent and Trademark Office. 2020.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.