")
Back to Journals » International Journal of Nanomedicine » Volume 20
Functional Liposomes Improve the Oral Absorption of Lurasidone Hydrochloride by Overcoming Multiple Absorption Barriers and Eliminating Food Effect
Authors Song T, Wang W, Wu Y, Liu C, Yuan L, Sun Z, Zhang J , Sun Y
Received 15 January 2025
Accepted for publication 7 April 2025
Published 16 April 2025 Volume 2025:20 Pages 4883—4901
DOI https://doi.org/10.2147/IJN.S512876
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Tingting Song,1 Wenyu Wang,1 Yan Wu,2 Chaolong Liu,1 Lu Yuan,1 Zhihong Sun,1 Jingjing Zhang,1 Yong Sun1
1Department of Pharmaceutics, School of Pharmacy, Qingdao University, Qingdao, Shandong, 266071, People’s Republic of China; 2Department of Obstetrics and Gynecology, The Affiliated Qingdao Hiser Hospital of Qingdao University, Qingdao Hospital of Traditional Chinese Medicine, Qingdao, Shandong, 266033, People’s Republic of China
Correspondence: Yong Sun, Department of Pharmaceutics, School of Pharmacy, Qingdao University, Qingdao, Shandong, 266071, People’s Republic of China, Email [email protected]
Purpose: Mental illness is the leading cause of the global burden of non-fatal disease. Lurasidone hydrochloride (LSD) is an important antipsychotic drug, but has poor water solubility and low oral bioavailability (9– 19%). Additionally, LSD exhibits twice the positive food effect, meaning that patients need to consume 350 kcal when taking the medication, which leads to reduced adherence. In this study, we developed oral LSD liposome enteric-coated capsules to eliminate the food effect and improve the oral bioavailability of LSD.
Methodsc: Firstly, liposomes were prepared by cethanocl injection cmethod, and their morphology, particle size, polydispersity index, encapsulation efficiency, drug loading capacity, stability and in vitro release were characterized. Subsequently, the mucous permeability and transepithelial transport capacity of p-R8-DOCA-Lipos in intestinal epithelial cells were investigated, and the in vivo pharmacokinetics and biosafety of LSD liposome enteric-coated capsules were further studied.
Results: p-R8-DOCA-Lipos had uniform morphology (particle size~112 nm), high encapsulation efficiency and drug loading capacity, and good stability in SIF. Cellular studies have shown that pHPMA gradually dissolved as it penetrated the mucus layer, and exposed R8-DOCA-Lipos facilitated cellular uptake. The cellular uptake and cumulative transepithelial transport of p-R8-DOCA-Lipos were 4.96 and 3.80 times higher than those in the solution group, respectively. The endocytosis of p-R8-DOCA-Lipos were mainly mediated by clathrin, caveolin and ASBT. Intracellular tracing showed that p-R8-DOCA-Lipos could achieve lysosomal escape, and ER and GA pathways were involved in their intracellular transport. In vivo pharmacokinetic studies have shown that AUC0-t of p-R8-DOCA-Lipos under fasted and fed conditions were similar to that of LSD suspension under fed conditions, which reduced the food effect of LSD and improved patient compliance. Finally, they had good biosafety after continuous oral administration.
Conclusion: Therefore, p-R8-DOCA-Lipos may be a promising strategy for overcoming multiple gastrointestinal barriers to improve oral absorption of LSD.
Keywords: oral delivery, liposomes, lurasidone hydrochloride, mucus permeation, transcytosis, bioavailability
Graphical Abstract:

Introduction
Lurasidone hydrochloride (LSD) is an atypical oral antipsychotic drug used to treat schizophrenia and bipolar disorders. Preclinical studies have shown that LSD acts as a combined antagonist of serotonin 5-hydroxytryptamine 2A (5-HT2A) and dopamine D2, improving the negative symptoms of psychosis and reducing the side effects of the extrapyramidal system.1,2 However, LSD is a BCS Class II drug with high intestinal permeability and poor solubility. The dissolution rate of conventional tablets after oral administration limits their absorption, resulting in low and variable bioavailability (9–19%).3
Because psychosis is a chronic disease, oral administration is the preferred approach. However, the presence of food may affect the oral bioavailability of certain drugs.4 Postprandial changes in local gastrointestinal pH significantly affect the dissolution and absorption of pH-dependent drugs, such as LSD, which is most soluble at pH 3.8.5,6 Previous studies have shown that the Cmax and AUC of LSD are 3 and 2 times higher, respectively, than those in the fasted state. Thus, it is recommended to take LSD with food at least 350 kcal, which means that there is a positive food effect.7 However, relying on food consumption to increase drug absorption is unstable and highly dependent on the patient compliance.8 Therefore, a more effective formulation strategy is necessary to improve oral bioavailability of LSD, independent of food.
The gastrointestinal tract (GIT) has multiple absorption barriers, the first of which is the harsh gastric acidic environment and various hydrolases, which cause drug instability and degradation.9 Subsequently, the mucus layer composed of a network of mucins rapidly renews, preventing further penetration of the drug delivery system. Intestinal epithelial cells are the last barrier for drug absorption into systemic circulation, including apical endocytosis, intracellular transport, and basolateral exocytosis. Recently, the mucus layer hindering the absorption of oral nanoparticles (NPs) has attracted increasing attention. Viral surfaces are densely wrapped in cationic and anionic groups, which can freely pass through the mucus layer.10 Inspired by their unique surface properties and the negatively charged and hydrophobic surface properties of the mucus layer, NPs with hydrophilicity and neutral charge are conducive to escaping the mucous capture. NPs modified with “mucus-inert” coatings such as N-(2-hydroxypropyl) methacrylamide copolymer (pHPMA) derivatives or polyethylene glycol (PEG) have been reported to promote mucus penetration.11,12 However, NPs with hydrophobicity and positive charge are more easily taken up by epithelial cells, meaning that they have exactly opposite surface properties and can effectively overcome mucus layer and epithelium barriers, such as some core-shell or zwitterion NPs.13
To date, various nanotechnology strategies have been used to improve the oral bioavailability of drugs with physicochemical properties similar to LSD, such as lipid-based formulations, including solid lipid nanoparticles,14 self-nano emulsion drug delivery systems,15 and liposomes,16 etc. Nano formulations have many advantages over conventional formulations, such as good biocompatibility, protection of encapsulated drugs from enzymatic degradation in the GIT, and improved drug solubility. The physicochemical properties of NPs are optimized by surface modification with cell-penetrating peptides (CPPs) or specific receptors/transporters to enhance the transepithelial transport of drugs. Most of these strategies focused on the apical endocytosis. Previous studies have shown that although the cellular uptake of NPs is significantly enhanced, their transepithelial transport is significantly lower than cellular uptake,17 and how NPs are transported within cells and their basolateral exocytosis remain unclear. Under physiological conditions, nutrients and endogenous substances are absorbed into the bloodstream through various transport pathways, such as the highly efficient and specific enterohepatic circulation of bile acids mediated by apical sodium-dependent bile acid transporter (ASBT) and cytoplasmic ileal bile acid-binding protein (IBABP), to overcome multiple absorption barriers, especially to avoid lysosomal degradation. Studies have shown that deoxycholic acid-coupled NPs enhance the oral bioavailability of insulin through ASBT-mediated endocytosis.18 In addition to the mucus-inert surface properties, viruses can invade host cells by fusing their envelopes with the host cells or forming membrane pores through special proteins.19 Similarly, the CPP octa-arginine R8 can promote cellular uptake of various substances by opening membrane pores.20 Lipid-based formulations can reduce the effect of food on drugs absorption and improve their oral bioavailability.21–23
In this study, liposomes composed of internal hydrophilic space and external hydrophobic lipid bilayer were selected as drug carriers, modified with deoxycholic acid (DOCA) and internalized into the epithelium via ASBT-mediated endocytosis pathway, promoting lysosomal escape and basolateral exocytosis. Simultaneously, the liposomes were modified with R8 peptide to obtain cationic liposomes (R8-DOCA-lipos) and promote drug absorption. The mucus-inert hydrophilic material pHPMA with opposite charges adsorbed on the surface of R8-DOCA-Lipos to form hydrophilic and neutrally charged p-R8-DOCA-Lipos, which were encapsulated into enteric-coated capsules to improve the oral bioavailability of LSD by overcoming the mucus layer and epithelial barriers. We assumed that pHPMA gradually dissociated during mucus permeation of p-R8-DOCA-Lipos to expose positively charged R8-DOCA-Lipos, solving the dilemma of mucus penetration and transcellular transport, and thus improving the oral bioavailability of LSD. Compared with the LSD suspension, p-R8-DOCA-Lipos can eliminate the food effect of LSD. Mucous permeability of liposomes was studied using a mucus-secreting cell model, and the cellular uptake, intracellular tracking, and efficiency of transepithelial transport of liposomes were investigated. Finally, the in vivo intestinal absorption and pharmacokinetics of liposomes were studied, laying a foundation for future applications.
Material and Methods
Materials
LSD was supplied by Lunan Pharmaceutical Group (Linyi, Shandong, China). Egg yolk lecithin was provided by AVT Pharmaceutical Technologies Co. Ltd. (Shanghai, China). Deoxycholic acid (DOCA), Glycyl-glycine (GG), methacryloyl chloride, pepsin, pancreatin and N-(2-hydroxypropyl) methacrylamide (HPMA) was purchased from Aladdin Biochemical Technology Co. Ltd. (Shanghai, China). DSPE-PEG2000-FITC, DSPE-PEG2000-NH2, DSPE-PEG2000-Mal, and Cys-R8 were acquired from Xi’an Ruixi Biotechnology Co., Ltd. (Xian, Shanxi, China). Cy3-conjugated Goat Anti-Rabbit IgG (H+L), ASBT Polyclonal antibody, and ZO-1 Polyclonal antibody were obtained from Wuhan Sanying Biotechnology Co., Ltd. (Wuhan, Hubei, China). ER-Tracker Red, Golgi-Tracker Red and Lyso-Tracker Red, 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) were acquired from Beyotime Biotechnology (Shanghai, China). Fetal bovine serum (FBS), Penicillin/streptomycin, and Dulbecco’s Modified Eagle’s medium (DMEM) were acquired from Wuhan Pricella Biotechnology Co., Ltd. (Wuhan, Hubei, China). All other reagents and solvents used were of analytical grade.
Synthesis of DSPE-PEG2000-DOCA Conjugates and DSPE-PEG2000-R8 Conjugates
DSPE-PEG2000-DOCA was synthesized through an amidation reaction.24 Deoxycholic acid (19.6 mg), N-hydroxysuccinimide (NHS) (58.0 mg), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) (191.7 mg) were placed in a 50 mL round-bottom flask and filled with nitrogen. The reactants were ultrasonically dissolved in dimethylformamide (DMF) (10 mL) and activated for 1 h at room temperature. DSPE-PEG2000-NH2 (137.1 mg in 10 mL PBS) solution was then added dropwise to the above solution and stirred at room temperature for 24 h. Finally, the product was obtained by dialysis in ethanol-water (4/1) for 2 d, freeze-dried, and its structure was confirmed by 1H nuclear magnetic resonance (1H NMR).
DSPE-PEG2000-R8 was synthesized by Michael addition between the maleimide (Mal) groups of DSPE-PEG2000-Mal and the thiol (SH) groups of Cys-R8.25 DSPE-PEG2000-Mal (101.3 mg) and Cys-R8 peptide (70.0 mg) were dissolved in dimethyl sulfoxide (DMSO) and reacted at room temperature for 24 h. The product was obtained by dialysis with deionized water, freeze-dried, and characterized by 1H NMR spectroscopy.
Preparation of Liposomes
DOCA-R8-Lipos were prepared by ethanol injection method.26 Briefly, EPC, cholesterol, DSPE-PEG2000-DOCA, DSPE-PEG2000-R8 and LSD were dissolved in 1 mL absolute ethanol to obtain the lipid phase, which was injected into 5 mL deionized water, and stirred at 60°C water bath temperature for 30 min. Then ethanol was evaporated by a rotary evaporator to prepare cationic liposomes. Finally, the particle size of the liposomes was reduced by liposome extruder.
To prepare p-DOCA-R8-Lipos, an equal volume of DOCA-R8-Lipos was added to an aqueous solution of pHPMA polymer and stirred for 30 min at room temperature. The pHPMA polymer was synthesized using a previously described method.27
Additionally, the fluorescent dye coumarin 6 (Cou-6) was introduced into the lipid phase using the same method used to track liposomes, and the double fluorescence-labeled liposomes were labeled with FITC and RITC. Dissolving RITC in water and introducing DSPE-PEG2000-FITC into the lipid phase to obtain double fluorescence-labeled liposomes, and unencapsulated fluorescent dyes were removed by dialysis.
In order to avoid the degradation of gastric acid, the above liposomes were freeze-dried and sucrose was used as a cryoprotectant. Briefly, the liposome suspension was mixed with 9% sucrose solution (v/v 1/1) for 30 min and frozen overnight at −20°C. Subsequently, the frozen liposomes were freeze-dried in a freeze dryer (Epsilon 2–4 LSCplus, Christ, Germany).
Characterization of Liposomes
The liposome suspension was diluted 10 times for measurement. The average particle size, polydispersity index (PDI), and zeta potential were measured using a Zetasizer NanoZS90 instrument (Malvern Instruments Ltd., Malvern, UK) at 25°C, scattering angle of 90°, refraction coefficient of 1.33 and equilibrium for 2 min. Then, they were negatively stained with phosphotungstic acid and dried at room temperature to observe their morphology before and after lyophilization under a transmission electron microscope (TEM, JEM-2100Plus, JEOL, Japan). After removing the unencapsulated LSD by centrifugation (3000 rpm, 10 min), the drug content encapsulated in the liposomes was quantitatively detected by high-performance liquid chromatography (HPLC, Agilent 1260, USA).23 A 5 μL sample was injected into the Agilent 20RBAX SB-C18 (4.6 mm × 150 mm, 5 μm) column. The detection wavelength was 230 nm, and the average retention time was 6.1 min. The mobile phase consists of acetonitrile and 0.1% trifluoroacetic acid aqueous solution (45:55 v/v) at a flow rate of 1 mL/min and maintained at 30°C. The encapsulation efficiency (EE) and loading capacity (LC) were calculated using the following formulae:13
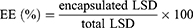

To further study the interaction between pHPMA and liposomes, fluorescence resonance energy transfer (FRET) technology was used. Using RITC and FITC as FRET pairs, pHPMA was labeled with RITC (RITC-pHPMA), and liposomes were labeled with FITC (FITC-Lipos). RITC-pHPMA was synthesized using a previously reported method.18 FITC-Lipos were prepared using the above method. The emission spectra in the wavelength range of 500–800 nm were recorded using a fluorescence spectrophotometer (F-7100, Hitachi, Japan) at an excitation wavelength of 450 nm. FRET efficiency (E) was calculated as follows:
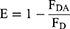
where FD is the fluorescence intensity of FITC-Lipos without RITC-pHPMA, and FDA is the fluorescence intensity of FITC-Lipos in double fluorescence-labeled liposomes.
Stability Study
To investigate the stability of liposomes in the GIT, freshly prepared liposomes were evenly mixed with simulated gastric fluid (SGF, pH 1.2, containing 80 mm HCl, 35 mm NaCl, 0.3% (w/v) pepsin) and simulated intestinal fluid (SIF, pH 6.8, containing 15 mm NaOH,50 mm KH2PO4, 1.0% (w/v) pancreatin) and incubated in a constant temperature orbital shaker (37°C, 100 rpm). Changes in the drug content and particle size of liposomes were determined at predetermined time points according to the above method.
In vitro Release Study of Enteric-Coated Capsules
The in vitro release of enteric-coated capsules containing 20 mg liposome freeze-dried powder in 5 mL SGF (pH 1.2) and SIF (pH 6.8) was studied by incubation in a constant-temperature shaker (37°C, 100 rpm). Samples were collected at set time points, disrupted with a mobile phase to release LSD, and analyzed by HPLC.
Cell Culture
Mucus-secreting HT29-MTX-E12 (E12) and human colon adenocarcinoma (Caco-2) cells were used in this study. Caco-2 cells were obtained from Wuhan Pricella Biotechnology Co. Ltd. (Wuhan, Hubei, China). The HT29-MTX-E12 cell line was acquired from Shanghai Fuheng Biotechnology Co. Ltd. (Shanghai, China). Caco-2/E12 (7/3) cells were co-cultured in a complete medium in an incubator with 5% CO2 at 37°C. Caco-2/E12 co-cultured cells were cultured on the polycarbonate membrane of 12 wells/plate transwell filter inserts for 3 weeks to construct a complete cell monolayer. Caco-2 cell monolayers were also established using the same procedure.
Mucus Penetration of Liposomes
After Caco-2/E12 cell monolayers were starved for 30 min, fluorescence-labeled liposomes were added to the apical side and incubated in a cell incubator for 2 h. Subsequently, the apical solution was discarded, incubated with WGA Alexa 555 (10 μg/mL) for 20 min, washed three times with HBSS, and the nuclei were stained with Hoechst 33342 for 15–30 min. The transwell membranes were cut off and inverted in special confocal culture dishes, sealed with 90% glycerol, and photographed using the Z-stack mode of CLSM to observe the mucus penetration of the liposomes.
Caco-2/E12 cell monolayers were incubated with p-R8-DOCA-Lipos, composed of FITC-Lipos and RITC-pHPMA, for 2 h. Subsequently, the apical solution was discarded, the cell monolayers were washed with HBSS, and nuclei were stained with Hoechst 33342 (1 μg/mL). The structural changes of p-R8-DOCA-Lipos during mucus penetration were observed layer-by-layer (0–40 μm) using a confocal laser scanning microscope (CLSM, STELLARIS 5, Leica, Germany).
To measure the binding rate of mucin to liposomes, fluorescently labeled liposomes were dispersed in mucin solutions of different concentrations, vortexed, and incubated in a shaker at 37°C for 30 min. The mucin-liposome aggregates were then separated by centrifugation at 1500 rpm for 5 min, leaving unbound liposomes in the supernatant. The supernatant was collected, and DMSO was added to disrupt the liposomes. Initial liposomes (not incubated with mucin) were treated in the same way, and fluorescence intensity was determined at 490 nm using a microplate reader (Infinite F200 PRO, Tecan Austria GmbH).
To investigate the transcellular transport efficiency of LSD, 200 μL of medium was taken out from the basolateral side at 1 h in “Transepithelial transport of liposomes” and the drug concentration was detected by HPLC to calculate the apparent permeability coefficient (Papp). Simultaneously, NAC (10 mM in PBS) was used to remove mucus to study how mucus affects the transcellular transport of liposomes. Papp was calculated as follows:28

where Area represents the membrane area, VA represents the volume of the basolateral chamber, and time represents the transmembrane transport time of the formulations.
In vitro Cytotoxicity of Liposomes
The cytotoxicity of the liposomes in vitro was evaluated by MTT assay. Caco-2 cells/E12 cells were cultured in 96-well plates until adherence. The culture medium was discarded and different concentrations of p-R8-DOCA-Lipos were added and incubated at 37°C for 24 h. Then, 100 μL of MTT solution was added to each well and incubated at 37°C for 4 h. DMSO was added and incubated in a shaker for 10 min to dissolve formazan. The absorbance was determined at 490 nm using a microplate reader, and cell viability was evaluated using untreated cells, with 100% cell viability as the negative control.
Lactate dehydrogenase (LDH) release assay can also be used to evaluate the cytotoxicity of liposomes in vitro. Caco-2/E12 co-cultured cells were seeded in 96-well plates for 2 d and incubated with different concentrations of p-R8-DOCA-Lipos at 37°C for 4 h. Subsequently, the supernatant was added to a new 96-well plate and the samples were analyzed using an LDH cytotoxicity assay kit. Cytotoxicity was assessed by comparing the differences in LDH release between untreated and treated cells.
Cellular Uptake of Liposomes
Cellular uptake of the various liposomes was qualitatively analyzed using CLSM. Caco-2/E12 co-cultured cells were seeded in confocal special culture dishes. After cell adherence, various liposomes containing 4 μg/mL Cou-6 were added and incubated in an incubator for 2 h. Then the cells were fixed with 4% paraformaldehyde at room temperature for 10 min. DAPI was added to stain the nuclei, and cellular uptake of various liposomes was observed using CLSM.
Cellular uptake of various liposomes was further quantitatively determined using flow cytometry (CytoFLEXS, Beckman Coulter, USA). Caco-2/E12 co-cultured cells were cultured in 6-well plates and incubated with various liposomes containing the same concentration of Cou-6 for 2 h. Then the cells were digested, centrifuged, and collected to detect intracellular fluorescence intensity.
To clarify the cellular uptake pathway of liposomes, Caco-2/E12 co-cultured cells were pretreated with amiloride (a caveolin-mediated endocytic inhibitor), sodium taurocholate (TCA, ASBT competitive inhibitor), chlorpromazine (a clathrin-mediated endocytic inhibitor), nystatin (a macropinocytosis-mediated endocytic inhibitor), and low temperature (4°C) for 1 h. The cells were then incubated with various liposomes containing the same concentration of Cou-6 for 2 h. The intracellular fluorescence intensity was measured using flow cytometry.
Expression of ASBT Protein
Caco-2 cells were cultured in culture dishes and transwell filter inserts in an incubator for 3 days and 3 weeks, respectively. The cells were then washed with PBS, lysed with cell lysis buffer, and centrifuged at 12,000 rpm at 4°C for 15 min to collect the supernatant, which was boiled for 15 min to denature proteins. The samples were subjected to gel electrophoresis and membrane transfer assay. Bands were cut according to the molecular weight of ASBT and blocked at room temperature for 2 h. They were then incubated with ASBT primary antibodies at 4°C overnight, and then incubated with Cy3-labeled secondary antibodies at room temperature for 1h. Finally, these bands were developed using Pico and ECL substrate solutions and imaged using a gel imager (ChemiDocTM XRS+, Bio-Rad, USA). ASBT-mediated endocytosis of liposomes was provided in the Supplementary Materials.
Intracellular Tracking of Liposomes
To study the intracellular fate of liposomes, we observed their co-localization with the Golgi apparatus (GA), lysosomes, and endoplasmic reticulum (ER) using CLSM. After incubation with fluorescently labeled liposomes for 1 and 2 h, Caco-2/E12 co-cultured cells were stained with Golgi-Tracker Red, Lyso-Tracker Red, and ER-Tracker Red for 30 min. Finally, Hoest33342 solution was added to stain the nucleus and co-localization was observed using CLSM.
Measurements of the Transepithelial Electrical Resistance
The transepithelial electrical resistance (TEER) value represents the degree of tight connections between cells and was measured using a resistance test instrument to determine the integrity of the cell monolayers (Millicell ERS-2, Millipore). When the TEER value is in the range of 500–800 Ω·cm2, cell monolayers can be used to study the transcellular transport. The TEER values after treatment with various formulations were measured at 30, 60, 90, 120 and 150 min to record their changes over time. The effect of transcellular transport of liposomes on the tight junction structure of Caco-2/E12 cell monolayers was studied by immunofluorescence staining of the tight junction marker protein ZO-1, which was provided in the Supplementary Materials.
Transepithelial Transport of Liposomes
Caco-2/E12 cell monolayers were washed three times with HBSS and starved for 30 min. Subsequently, various liposomes containing 0.1 mg/mL LSD were added to the apical side, and 200 μL of medium was taken out from the basolateral side at 0.25, 0.5, 1, 2, and 4 h and immediately supplemented with 200 μL HBSS. The drug content in the basolateral chamber was analyzed to calculate the cumulative transepithelial transport curve.
To study liposome integrity after transcellular transport, Caco-2/E12 cell monolayers were starved for 30 min and incubated with FITC- and RITC- labeled p-R8-DOCA-Lipos. After incubation for 8h, the basolateral medium was collected and the fluorescence emission spectra were recorded.
In situ Intestinal Absorption of Liposomes
The jejunal intestinal segments were ligated into small loops of 2 cm length, and fluorescence-labeled R8-DOCA-Lipos and p-R8-DOCA-Lipos were injected into the intestinal cavity. Subsequently, intestinal segments were placed back into the abdominal cavity for 30 min. Each intestinal segment was cut, washed with PBS, fixed with 4% paraformaldehyde for more than 4 h, dehydrated with a precooled 30% sucrose solution overnight, and sliced using a freezing microtome. After staining the nucleus with DAPI and sealing the slides with 90% glycerol, the absorption of liposomes in the intestinal villi was observed using CLSM.
Fast/Fed in vivo Pharmacokinetic Studies
All experimental procedures followed the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Ethics Committee of Qingdao University (QDU-AEC-20240947). To investigate the absorption of LSD suspension and freeze-dried LSD liposomes enteric-coated capsules in the fed and fasted states, healthy Sprague-Dawley rats (200 ± 20 g) obtained from Beijing Huafukang Biotechnology Co., Ltd. (Beijing, China) were randomly divided into 4 groups (n = 5). In the fasted study, rats were fasted overnight, during which they were allowed to drink freely and eat 2 h after administration.
LSD was suspended in water containing 0.5% w/v carboxymethylcellulose, vortexed, and ultrasonicated to obtain an LSD suspension. The rats were orally administered the LSD suspension and freeze-dried LSD liposome enteric-coated capsules at a dose of 4.2 mg/kg LSD. Subsequently, approximately 0.4 mL of blood was collected from the retro-orbital plexus of rats before and 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 h after oral administration and placed in a pre-heparinized centrifuge tube. The upper plasma was separated by centrifugation at 4000 rpm at 4°C for 10 min and frozen at −80°C to analyze the drug content.
Analysis of Plasma Samples
A standard solution of LSD was prepared in the concentration range of 0.1–300 ng/mL. Then, a series of standard solutions (4 μL) were added to the blank plasma of rats (76 μL) and vortexed for 30s. Then, 50 μL of the above plasma was taken and added to 100 μL of the internal standard solution (IS, Ziprasidone hydrochloride in acetonitrile, 6 ng/mL) and vortexed for 30s. The samples were centrifuged at 12,000 rpm for 10 min to precipitate plasma proteins and filtered through a 0.22 μm PTFE membrane. Plasma drug concentrations were detected by liquid chromatography-mass spectrometry (LC-MS, Shimadzu 8050 mass spectrometry, Japan). A 2 µL sample was injected into the Agilent EcliosePlusC18 (1.8µm, 3 mm×50 mm) column and maintained at 30°C. Gradient elution was performed with the mobile phase consisting of (A) 0.1% formic acid aqueous solution and (B) 0.1% formic acid in acetonitrile at a flow rate of 0.3 mL/min. Multiple Reaction Monitoring (MRM) mode was used for quantitative determination: m/z 413.2→m/z 194.1 for ZIP (IS) and m/z 493.4→m/z 166.2 for LSD.
50 μL of the thawed unknown plasma sample was added to 100 μL of internal standard solution (in acetonitrile), mixed for 30s, and treated as described above. Plasma drug concentration-time curves and pharmacokinetic parameters (including Cmax, T1/2, Tmax, and AUC) of LSD were analyzed using DAS 2.0 software and the non-compartment model.
Safety Studies of Liposomes
To ensure the safety of the formulations, freeze-dried LSD liposome enteric-coated capsules were orally administered to healthy Sprague Dawley rats for 14 consecutive days. Subsequently, the stomachs and intestines of the sacrificed rats were fixed, gradually dehydrated, and embedded. The tissues were then further dewaxed, hydrated, and stained with hematoxylin and eosin solution. Histological and morphological differences between the treated and untreated groups were observed under an inverted fluorescence microscope (Ti2-U, Nikon, Japan). Simultaneously, blood samples of rats after long-term administration were collected for hematological and biochemical tests.
Statistical Analysis
The experimental data were expressed as mean ± standard deviation (SD). Student’s t-test was used to analyze the differences between the two groups, and one-way analysis of variance (ANOVA) was used to analyze the differences between the multiple groups. P < 0.05 was considered statistically significant.
Results and Discussion
Synthesis of DSPE-PEG2000-DOCA Conjugates and DSPE-PEG2000-R8 Conjugates
DSPE-PEG2000-DOCA was synthesized by coupling the carboxyl group of DOCA with the amino group of DSPE-PEG2000-NH2 via EDC and NHS activation. The chemical structures of the DSPE-PEG2000-DOCA conjugates were confirmed using 1H NMR spectroscopy (Figure 1A). The three methyl characteristic peaks of DOCA appeared at δ 0.6–1 ppm,29 confirming the successful synthesis of DSPE-PEG2000-DOCA.
 |
Figure 1 1H NMR spectra of (A) DSPE-PEG2000-DOCA conjugates and (B) DSPE-PEG2000-R8 conjugates in DMSO-d6. |
Octa-arginine R8 is a typical CPP, and its cationic properties are conducive to promoting cellular uptake.30 The R8 peptide was coupled with DSPE-PEG2000-Mal to synthesize DSPE-PEG2000-R8, which was inserted into the phospholipid bilayer of liposomes to prepare cationic liposomes. The characteristic peaks of the peptide at δ 6–9 ppm were confirmed using 1H NMR spectroscopy, indicating that the R8 peptide was successfully coupled to DSPE-PEG2000-Mal (Figure 1B).
Preparation and Characterization of Liposomes
The p-R8-DOCA-Lipos were prepared using a two-step method. First, DSPE-PEG2000-R8 was added to the lipid phase to prepare cationic liposomes by ethanol injection. The surface of the liposomes was coated with negatively charged pHPMA through electrostatic interactions to prepare p-R8-DOCA-Lipos (Figure 2A).31 The 1H NMR spectra of the pHPMA polymer were shown in Figure S1–S3 (Supplementary Materials). p-R8-DOCA-Lipos with hydrophilicity and neutral charges were prepared by adjusting the amount of pHPMA. The EE and LC of the liposomes were shown in Table 1. Dynamic light scattering results showed that the particle size of R8-DOCA-Lipos was ~102.9 nm and the surface zeta potential was +15.4 mV, whereas the particle size of p-R8-DOCA-Lipos was ~112.2 nm and the surface zeta potential was −6.8 mV (Figure 2B). Compared with R8-DOCA-Lipos, the particle size of p-R8-DOCA-Lipos increased, and the surface charge changed from positive to negative, indicating that anionic pHPMA was successfully coated on the surface of the liposomes. As observed by TEM, R8-DOCA-Lipos and p-R8-DOCA-Lipos were spheroidal with a typical phospholipid bilayer structure, and pHPMA was coated on the surface of the liposomes (Figure S4 in the Supplementary Materials and Figure 2C).
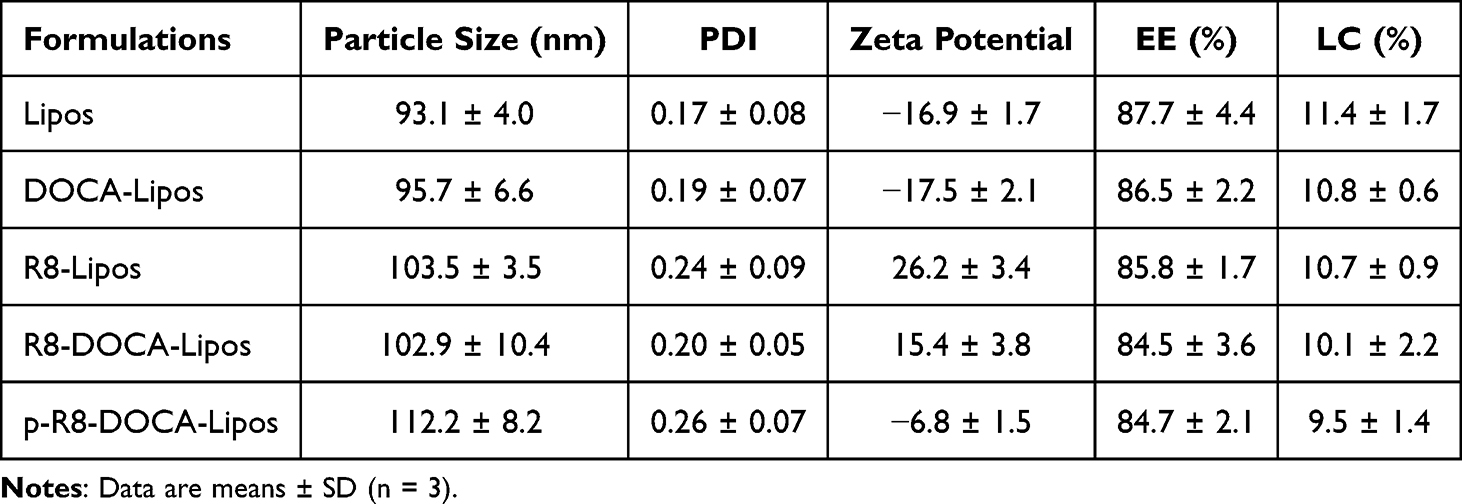 |
Table 1 Characterization of Liposomes |
 |
Figure 2 Preparation and characterization of LSD@p-R8-DOCA-Lipos. (A) Schematic diagram of LSD@p-R8-DOCA-Lipos. (B) Particle size distribution of LSD@p-R8-DOCA-Lipos. (C) TEM images of LSD@p-R8-DOCA-Lipos. |
The structure of pHPMA-coated liposomes was further confirmed by FRET technology.32 The 1H NMR spectra of RITC-pHPMA were shown in Figure S5 (Supplementary Materials). Double fluorescence-labeled liposomes were prepared with a RITC-pHPMA coating and FITC-Lipos cores, which can form FRET pairs. The results showed that the emission intensity of FITC decreased at 517 nm, whereas the emission intensity of RITC increased at the corresponding wavelength (Figure 3A), indicating that FRET phenomenon occurred and the FRET efficiency was 47.2%. LSD@p-R8-DOCA-Lipos composed of a PHPMA coating and an R8-DOCA-Lipos core were successfully prepared.
 |
Figure 3 (A) Fluorescence emission spectra of p-R8-DOCA-Lipos, FITC-Lipos and RITC-pHPMA at 450 nm excitation. (B and C) The stability of p-R8-DOCA-Lipos in SGF (pH 1.2) and SIF (pH 6.8). Data are means ± SD (n = 3). (D) In vitro release curve of freeze-dried liposomes enteric-coated capsules in simulated gastrointestinal fluid. Data are means ± SD (n = 3). |
Stability Study and in vitro Release Study
The stability of p-R8-DOCA-Lipos in SGF and SIF was shown in Figure 3B and C. The drug content of p-R8-DOCA-Lipos decreased and the particle size increased in SGF, which may be due to the long-term incubation in SGF destroyed the integrity of phospholipid bilayer, leading to its instability in SGF, while the drug content and particle size of p-R8-DOCA-Lipos in SIF did not change significantly, indicating that they were stable in SIF. To avoid degradation of the gastric acid environment, the liposome freeze-dried powder was encapsulated in enteric-coated capsules.33 The particle size of p-R8-DOCA-Lipos did not change much at 4°C and 25°C, but the EE of p-R8-DOCA-Lipos decreased slightly at 25°C within 7 d, indicating that they had good storage stability at 4°C. (Supplementary Materials, Figure S6). The release curves in the simulated gastrointestinal fluid showed that the enteric-coated capsules remained intact in the stomach, and released liposomes in the intestinal tract (Figure 3D).
Mucus Penetration Ability of Liposomes
To observe the process of liposomal mucous penetration more directly,34 Caco-2/E12 cell monolayers were incubated with various liposomes to reconstruct 3D fluorescence images (Figure 4A). Positively charged R8-DOCA-Lipos showed less co-localization within the cell monolayers due to electrostatic interactions with mucin, while p-R8-DOCA-Lipos could effectively penetrate the mucus layer and enhance cellular uptake, and showed obvious co-localization within the cell monolayers, indicating that pHPMA promoted faster mucus penetration of liposomes.
 |
Figure 4 Mucus penetration ability of liposomes. (A) 3D fluorescence images of R8-DOCA-Lipos and p-R8-DOCA-Lipos permeating the mucus layer on the Caco-2/E12 cell monolayers. Blue: Nucleus stained with Hoechst 33342. Green: Cou-6-loaded formulation. Red: Mucus stained with WGA Alexa 555. (B) CLSM images of the structural changes of p-R8-DOCA-Lipos across the Caco-2/E12 cell monolayers. |
Double fluorescence-labeled p-R8-DOCA-Lipos were scanned layer by layer to observe their structural changes during mucus penetration (Figure 4B). The results showed that pHPMA and liposomes co-localized in the upper layer of the mucus, indicating that p-R8-DOCA-Lipos existed in their intact form. Further scanning showed that the red fluorescence of pHPMA gradually decreased, while the green fluorescence of liposomes was observed in the mucus layer and cell monolayers, indicating that pHPMA gradually degraded as it penetrated the mucus, and the exposed positively charged liposomes were effectively absorbed by epithelial cells.
Hydrophobic and positively charged NPs can be adhered to and be removed by the mucus.35 Previous studies have shown that NPs with a hydrophilic surface and a neutral or slightly negative charge can effectively reduce interactions with the mucus layer.36 Based on this study, p-R8-DOCA-Lipos with hydrophilicity and neutral charges can reduce the electrostatic and hydrophobic interactions with the mucus layer. The binding rates of liposomes with different concentrations of mucin were shown in Figure 5A. The binding rate of p-R8-DOCA-Lipos incubated with 0.5% (w/v) mucin for 30 min was only 12.65%, which was not significantly different from that of Lipos group, but significantly lower than that of R8-DOCA-Lipos group.
 |
Figure 5 (A) The binding rates of Lipos, R8-DOCA-Lipos and p-R8-DOCA-Lipos with different concentrations of mucus. Data are means ± SD (n = 3), **P < 0.01, ***P < 0.001, ns, nonsignificant, compared with p-R8-DOCA-Lipos group. (B) Papp of LSD in various formulations incubated with the Caco-2/E12 cell monolayers for 1 h. Data are means ± SD (n = 3), *P < 0.05, ns, nonsignificant. (C and D) MTT assay and LDH assay of Caco-2/E12 cells incubated with different concentrations of p-R8-DOCA-Lipos. Data are means ± SD (n = 6). |
Transcellular transport of LSD on cell monolayers was evaluated. To investigate the effect of mucus on the Papp of LSD, a group of cells was pretreated with NAC to remove mucus,37 and the Papp measurement results of LSD were shown in Figure 5B. The Papp of both formulations was higher than that of Solution group (P < 0.001). In addition, the Papp of p-R8-DOCA-Lipos in the pretreated and untreated groups was approximately 3.07 times and 3.18 times of that in Solution group, respectively. The presence of mucus reduced the transport of R8-DOCA-Lipos but did not significantly affect the transport of p-R8-DOCA-Lipos.
In vitro Cytotoxicity and Cellular Uptake Studies
First, safety tests of p-R8-DOCA-Lipos showed that they were not cytotoxic within a certain concentration range, which further indicated that they were not taken up by cells by destroying the cell membrane structure (Figure 5C and D). The cellular uptake of various Cou-6-loaded formulations was shown in Figure 6A–C. The green fluorescence intensity of p-R8-DOCA-Lipos was the strongest, and the mean fluorescence intensity (MFI) was 4.96 times and 1.79 times that of Cou-6 Solution and R8-DOCA-Lipos, respectively, while there was no significant difference between the MFI of R8-Lipos and R8-DOCA-Lipos. These results suggested that as the pHPMA coating degraded during mucus penetration, the exposed core R8-DOCA-Lipos could significantly promote the cellular uptake of p-R8-DOCA-Lipos, consistent with the results in Figure 4B.
 |
Figure 6 Studies on the cellular uptake of liposomes. (A) CLSM images of Caco-2/E12 cells incubated with Cou-6 Solution (1), Cou-6@Lipos (2), Cou-6@DOCA-Lipos (3), Cou-6@R8-Lipos (4), Cou-6@R8-DOCA-Lipos (5) and Cou-6@p-R8-DOCA-Lipos (6). The nucleus was stained with DAPI (blue). (B and C) The quantitative determination of cellular uptake of various formulations using flow cytometry. Data are means ± SD (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001, compared with the Cou-6 Solution group. |
Different uptake mechanisms lead to different intracellular fates of NPs. Caveolin-mediated endocytic pathway involves ER and GA, which can avoid lysosomal degradation.38 Uptake inhibition experiments showed that the cellular uptake of R8-DOCA-Lipos were mainly through clathrin- and caveolin-mediated endocytic pathways, and ASBT-mediated energy-dependent active endocytosis also participated in this process (Figure 7A).
 |
Figure 7 (A) The inhibitory effects of chlorpromazine, nystatin, amiloride, sodium taurocholate and low temperature (4°C) pretreated Caco-2/E12 co-cultured cells for 1 h on p-R8-DOCA-Lipos endocytosis by flow cytometry. Data are means ± SD (n = 3), *P < 0.05, **P < 0.01, ***P < 0.001, compared with the control group. (B–D) CLSM images of liposomes (green) co-localized with lysosomes (red), ER (red) and GA (red). |
Intracellular Tracking of Liposomes
After endocytosis, NPs are first wrapped in endosomes, which are responsible for transporting the NPs along microtubules within the cell and eventually maturing into lysosomes.39 Typically, NPs can be delivered to lysosomes via the endolysosomal pathway, where the acidic environment and enzymes can degrade NPs.40 To determine the intracellular fate of liposomes, the co-localization of liposomes with various organelles were observed using CLSM.41,42 As the incubation time increased from 1h to 2h, the co-localization of the green fluorescence of Cou-6 with the red fluorescence of lysosomes decreased, while the co-localization with the red fluorescence of ER and GA increased, suggesting that liposomes could achieve lysosomal escape and transport through the ER and GA pathways (Figure 7B–D).
Expression of ASBT Protein
ASBT protein is an important protein that mediates bile salt reabsorption on the surface of intestinal epithelial cells.43 It has been reported that Caco-2 cells can express ASBT protein,44 and Western blot analysis was used to detect the expression of ASBT protein in Caco-2 cells cultured for 3 days and 3 weeks. As shown in Figure 8A and B, ASBT protein was expressed in Caco-2 cells, and its expression in Caco-2 cells cultured for 3 weeks was approximately 1.9 times that in Caco-2 cells cultured for 3 days.
 |
Figure 8 (A and B) The expression of ASBT protein on Caco-2 cells cultured for 3 days and 3 weeks by western blot analysis. Tubulin served as the control group. Data are means ± SD (n = 3), **P < 0.01. |
To further confirm the role of ASBT in the cellular uptake of liposomes, the distribution of ASBT in Caco-2 cell monolayers and the co-localization of ASBT with liposomes was observed by CLSM.45 The results of the control group showed that ASBT was distributed on the cell membrane, which was consistent with ASBT being a membrane protein. (Supplementary Materials, Figure S7). Compared with the Lipos group, DOCA-Lipos were significantly co-localized with ASBT. These results suggested that DOCA-modified liposomes were taken up by the cells via the ASBT-mediated endocytic pathway.
In vitro Transcellular Transport Studies of Liposomes
To investigate the effect of transcellular transport of liposomes on the tight junction structure of Caco-2/E12 cell monolayers, the tight junction marker protein ZO-1 was stained by immunofluorescence.46 After incubation with p-R8-DOCA-Lipos for 1 h, the red fluorescence of ZO-1 was not significantly different from that of the control group, suggesting that the tight junction structure did not change significantly (Supplementary Materials, Figure S8). Additionally, the integrity of the tight junction structure was further confirmed by measuring the TEER values.47 It was found that the TEER values of the cell monolayers incubated with various formulations showed no significant changes (Figure 9A). These results suggested that the translocation of liposomes on the cell monolayers was not achieved by opening tight junctions between cells but by the transcellular transport pathway.
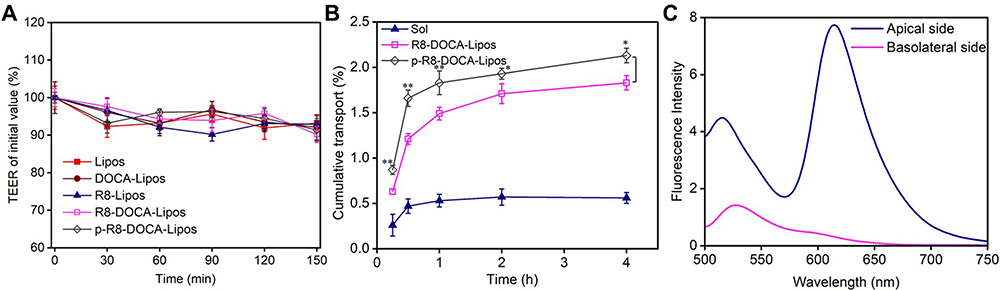 |
Figure 9 In vitro transcellular transport of liposomes. (A) The changes of TEER values at different time points after treatments with various formulations. Data are means ± SD, n = 3. (B) The transcellular transport kinetics curves of LSD in various formulations incubated with the Caco-2/E12 cell monolayers for 0.25, 0.5, 1, 2, 4 h. Data are means ± SD, n = 3, *P < 0.05, **P < 0.01, compared with R8-DOCA-Lipos. (C) Fluorescence emission spectra of p-R8-DOCA-Lipos in the apical and basolateral medium of cell monolayers. |
The cumulative transepithelial transport of p-R8-DOCA-Lipos was also higher than that of R8-DOCA-Lipos (Figure 9B), indicating that the pHPMA coating facilitated mucus penetration of p-R8-DOCA-Lipos and promoted their transepithelial transport. The integrity of p-R8-DOCA-Lipos during transcellular transport was further investigated by FRET technique. As shown in Figure 9C, there was an obvious FRET phenomenon in the apical medium of the cell monolayers (blue curve), whereas the FRET phenomenon disappeared in the medium collected from the basolateral side (red curve), indicating that exocytosis of p-R8-DOCA-Lipos was not in their complete form, and that LSD was released from the basolateral side into the bloodstream in molecular form.
In situ Intestinal Absorption and Safety Studies
Based on the good mucus penetration and transcellular transport abilities of p-R8-DOCA-Lipos in vitro, we investigated whether they could overcome mucus layer and epithelial cell barriers through in situ absorption in rat intestines.48 As shown in Figure 10A, compared with R8-DOCA-Lipos, p-R8-DOCA-Lipos had wider distribution and deeper penetration in mucus layer. This could be attributed to the presence of the pHPMA coating, which promoted mucus penetration of p-R8-DOCA-Lipos, and as the pHPMA coating gradually separated, the exposed core R8-DOCA-Lipos facilitated transcellular transport. These results were consistent with those of the in vitro experiments described above. Subsequently, long-term safety of the formulations was evaluated. The histological morphology of the stomachs and intestines in the freeze-dried liposome enteric-coated capsule group showed no significant changes (Figure 10B). The physiological and biochemical parameters of the rats orally administered freeze-dried liposome enteric-coated capsules were not significantly different from those of the control group, indicating that freeze-dried liposome enteric-coated capsules had good long-term safety. (Supplementary Materials, Tables S1 and S2).
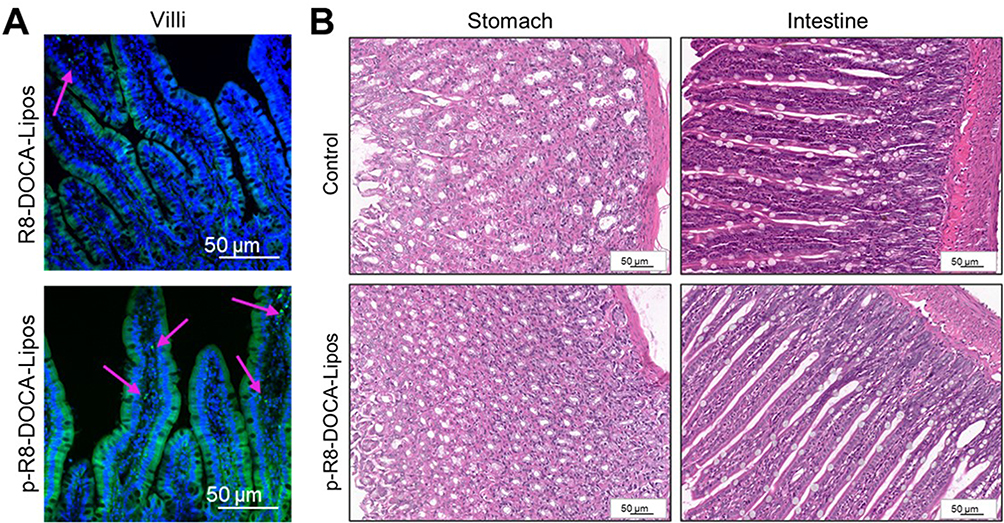 |
Figure 10 (A) The distribution of R8-DOCA-Lipos and p-R8-DOCA-Lipos in rat intestinal villi. Green: Cou-6-loaded formulation. Blue: Intestinal villi nuclei stained with DAPI. The red arrow represents the absorption point. (B) The histological images of stomachs and intestines observed using an inverted fluorescence microscope after 14 consecutive days of oral administration. n = 5. |
In vivo Pharmacokinetics
After oral administration of LSD suspension and freeze-dried LSD liposomes enteric-coated capsules, the plasma drug concentration-time curves of Sprague-Dawley rats in the fasted and fed states were shown in Figure 11. The corresponding pharmacokinetic parameters were shown in Table 2. The AUC0-t and Cmax of the LSD suspension administered orally to Sprague-Dawley rats in the fed state (677.81 ± 97.49 ng. h/mL and 148.65 ± 7.14 ng/mL) were significantly increased compared with those in the fasted state (274.89 ± 30.11 ng. h/mL and 55.23 ± 11.24 ng/mL, P < 0.001). These results suggested that food can influence the oral absorption of LSD suspensions. The AUC0-t and Cmax of the freeze-dried LSD liposome enteric-coated capsules in the fasted state (690.74 ± 86.19 ng. h/mL and 76.81 ± 7.38 ng/mL) had no significant difference with those in the fed state (662.84 ± 73.94 ng. h/mL and 82.75 ± 8.85 ng/mL) or those of LSD suspension in the fed state (P >0.05). The results showed that freeze-dried LSD liposome enteric-coated capsules significantly enhanced the absorption of LSD in the fasted state and eliminated the food effect, which could be attributed to the fact that p-R8-DOCA-Lipos penetrated the mucus layer and promoted transcellular transport of drugs. The maximum plasma drug concentration time (Tmax) and elimination half-life (T1/2) of the LSD suspension and freeze-dried LSD liposome enteric-coated capsules were not significantly different between the fasted and fed groups (P > 0.05), and the Tmax and T1/2 of freeze-dried LSD liposome enteric-coated capsules were longer than LSD suspension.
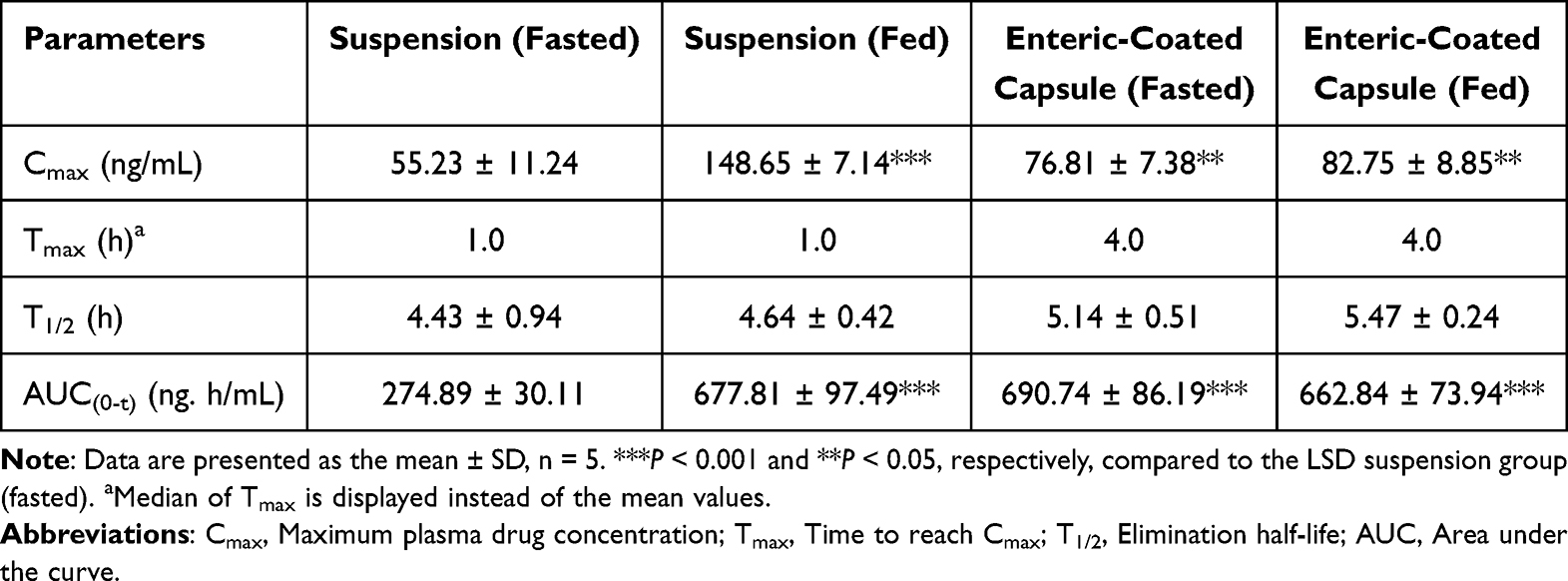 |
Table 2 Pharmacokinetic Parameters of Sprague-Dawley Rats After Oral Administration of Various LSD Formulations at a Dose of 4.2 mg/kg |
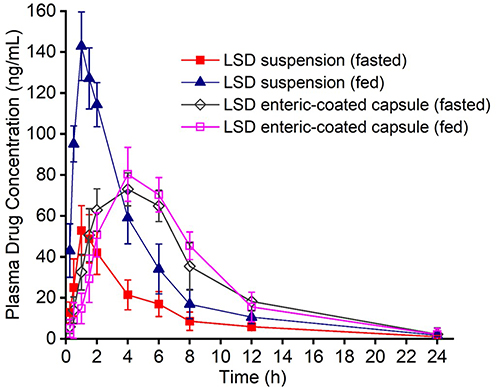 |
Figure 11 The plasma drug concentration-time curves of fasted and fed Sprague-Dawley rats after oral administration of LSD suspension and freeze-dried LSD liposomes enteric-coated capsules at a dose of 4.2 mg/kg. Data are means ± SD, n = 5. |
Conclusion
In this study, we prepared LSD liposomes enteric-coated capsules with good mucus penetration and intestinal epithelial absorption to improve the oral delivery of LSD. p-R8-DOCA-Lipos had a small particle size and narrow distribution, and their hydrophilicity and neutral charges were conducive to rapid mucus penetration. The “mucus-inert” material pHPMA gradually degraded during mucus penetration, and the exposed positively charged R8-DOCA-Lipos could promote transepithelial transport through ASBT-mediated endocytosis and R8 peptide-mediated high cellular uptake. In addition, p-R8-DOCA-Lipos could escape from lysosomal compartments to avoid lysosomal degradation and were transported via the ER/GA pathway, promoting basolateral exocytosis. Fasted/fed in vivo pharmacokinetic studies indicated that LSD liposome enteric-coated capsules could enhance the oral absorption of LSD and eliminate the food effect, thereby improving patient compliance. These results suggested that liposome enteric-coated capsules may be a promising strategy for enhancing the oral absorption of drugs with physicochemical properties similar to those of LSD.
Acknowledgments
The authors are very grateful for the support of Qingdao Key Technology Research and Industrialization Demonstration Project, and the project number is 22-3-3-hygg-25-hy.
Funding
This research was funded by the Key Technology Research and Industrialization Demonstration Project of Qingdao (22-3-3-hygg-25-hy).
Disclosure
The authors declare no conflict of interest.
References
1. Percelay S, Since M, Lagadu S, Freret T, Bouet V, Boulouard M. Antipsychotic lurasidone: behavioural and pharmacokinetic data in C57BL/6 mice. Pharmacol Biochem Behav. 2020;194:172933. doi:10.1016/j.pbb.2020.172933
2. Greenberg WM, Citrome L. Pharmacokinetics and Pharmacodynamics of Lurasidone Hydrochloride, a Second-Generation Antipsychotic: a Systematic Review of the Published Literature. Clin Pharmacokinet. 2017;56(5):493–503. doi:10.1007/s40262-016-0465-5
3. Wang J, Huang BB, Dai J, Chen GG, Ren LL. Inclusion complex of lurasidone hydrochloride with Sulfobutylether-β-cyclodextrin has enhanced oral bioavailability and no food effect. Am J Transl Res. 2022;14(3):1495–1506.
4. Patel MH, Mundada VP, Sawant KK. Fabrication of solid lipid nanoparticles of lurasidone HCl for oral delivery: optimization, in vitro characterization, cell line studies and in vivo efficacy in schizophrenia. Drug Dev Ind Pharm. 2019;45(8):1242–1257. doi:10.1080/03639045.2019.1593434
5. Sharma S, Bhatt S, Saini V. Formulation Development and Evaluation of Novel Vesicular Carrier for Enhancement of Bioavailability of Poorly Soluble Drug. Pharm Nanotechnol. 2021;9(1):70–82. doi:10.2174/2211738508999201123213232
6. AboulFotouh K, Allam AA, El-Badry M, El-Sayed AM. Role of self-emulsifying drug delivery systems in optimizing the oral delivery of hydrophilic macromolecules and reducing interindividual variability. Colloids Surf B Biointerfaces. 2018;167:82–92. doi:10.1016/j.colsurfb.2018.03.034
7. Miao Y, Sun J, Chen G, Lili R, Ouyang P. Enhanced oral bioavailability of lurasidone by self-nanoemulsifying drug delivery system in fasted state. Drug Dev Ind Pharm. 2015;42(8):1234–1240. doi:10.3109/03639045.2015.1118496
8. Meola TR, Joyce P, Wignall A, Bremmell KE, Prestidge CA. Harnessing the potential of nanostructured formulations to mimic the food effect of lurasidone. Int J Pharm. 2021;608:121098. doi:10.1016/j.ijpharm.2021.121098
9. Wang A, Yang T, Fan W, et al. Protein Corona Liposomes Achieve Efficient Oral Insulin Delivery by Overcoming Mucus and Epithelial Barriers. Adv Healthc Mater. 2018;8(12). doi:10.1002/adhm.201801123
10. Zhang Y, Xiong MT, Ni XM, et al. Virus-Mimicking Mesoporous Silica Nanoparticles with an Electrically Neutral and Hydrophilic Surface to Improve the Oral Absorption of Insulin by Breaking Through Dual Barriers of the Mucus Layer and the Intestinal Epithelium. ACS Appl Bio Mater. 2021;13(15):18077–18088. doi:10.1021/acsami.1c00580
11. Shan W, Zhu X, Liu M, et al. Overcoming the Diffusion Barrier of Mucus and Absorption Barrier of Epithelium by Self-Assembled Nanoparticles for Oral Delivery of Insulin. ACS Nano. 2015;9(3):2345–2356. doi:10.1021/acsnano.5b00028
12. Liu M, Zhang J, Zhu X, et al. Efficient mucus permeation and tight junction opening by dissociable “mucus-inert” agent coated trimethyl chitosan nanoparticles for oral insulin delivery. J Control Release. 2016;222:67–77. doi:10.1016/j.jconrel.2015.12.008
13. Li C, Yuan L, Zhang X, et al. Core-shell nanosystems designed for effective oral delivery of polypeptide drugs. J Control Release. 2022;352:540–555. doi:10.1016/j.jconrel.2022.10.031
14. Dening TJ, Rao S, Thomas N, Prestidge CA. Oral nanomedicine approaches for the treatment of psychiatric illnesses. J Control Release. 2016;223:137–156. doi:10.1016/j.jconrel.2015.12.047
15. Basalious EB, Abdallah Ahmed M. Phospholipid based self-nanoemulsifying self-nanosuspension (p-SNESNS) as a dual solubilization approach for development of formulation with diminished food effect: fast/fed in vivo pharmacokinetics study in human. Eur J Pharm Sci. 2017;109:244–252. doi:10.1016/j.ejps.2017.08.017
16. Kim KS, Na K, Bae YH. Nanoparticle oral absorption and its clinical translational potential. J Control Release. 2023;360:149–162. doi:10.1016/j.jconrel.2023.06.024
17. Wu L, Bai Y, Wang L, et al. Promoting apical-to-basolateral unidirectional transport of nanoformulations by manipulating the nutrient-absorption pathway. J Control Release. 2020;323:151–160. doi:10.1016/j.jconrel.2020.04.013
18. Fan W, Xia D, Zhu Q, et al. Functional nanoparticles exploit the bile acid pathway to overcome multiple barriers of the intestinal epithelium for oral insulin delivery. Biomaterials. 2018;151:13–23. doi:10.1016/j.biomaterials.2017.10.022
19. Yang T, Wang A, Nie D, et al. Ligand-switchable nanoparticles resembling viral surface for sequential drug delivery and improved oral insulin therapy. Nat Commun. 2022;13(1):1. doi:10.1038/s41467-022-34357-8
20. Wu J, Zheng Y, Liu M, Shan W, Zhang Z, Huang Y. Biomimetic Viruslike and Charge Reversible Nanoparticles to Sequentially Overcome Mucus and Epithelial Barriers for Oral Insulin Delivery. ACS Appl Mater Interfaces. 2018;10(12):9916–9928. doi:10.1021/acsami.7b16524
21. Nora GI, Venkatasubramanian R, Strindberg S, et al. Combining lipid based drug delivery and amorphous solid dispersions for improved oral drug absorption of a poorly water-soluble drug. J Control Release. 2022;349:206–212. doi:10.1016/j.jconrel.2022.06.057
22. Patel MH, Sawant KK. Self microemulsifying drug delivery system of lurasidone hydrochloride for enhanced oral bioavailability by lymphatic targeting: in vitro, Caco-2 cell line and in vivo evaluation. Eur J Pharm Sci. 2019;138:105027. doi:10.1016/j.ejps.2019.105027
23. Meola TR, Paxton K, Joyce P, Schultz HB, Prestidge CA. The effect of drug ionization on lipid-based formulations for the oral delivery of anti-psychotics. ADMET DMPK. 2020;8(4):437–451. doi:10.5599/admet.830
24. Li J, Huo M, Wang J, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33(7):2310–2320. doi:10.1016/j.biomaterials.2011.11.022
25. Chakma P, Digby ZA, Via J, Shulman MP, Sparks JL, Konkolewicz D. Tuning thermoresponsive network materials through macromolecular architecture and dynamic thiol-Michael chemistry. Polym Chem. 2018;9(38):4744–4756. doi:10.1039/c8py00947c
26. Japiassu KB, Fay F, Marengo A, et al. Interplay between mucus mobility and alveolar macrophage targeting of surface-modified liposomes. J Control Release. 2022;352:15–24. doi:10.1016/j.jconrel.2022.10.006
27. Ferri M, Dash M, Cometa S, De Giglio E, Sabbatini L, Chiellini F. Preparation and characterization of hybrid nanoparticles based on chitosan and poly(methacryloylglycylglycine). J Nanopart Res. 2014;16(5). doi:10.1007/s11051-014-2422-2
28. Yu Y, Xing L, Li L, Wu J, He J, Huang Y. Coordination of rigidity modulation and targeting ligand modification on orally-delivered nanoparticles for the treatment of liver fibrosis. J Control Release. 2022;341:215–226. doi:10.1016/j.jconrel.2021.11.026
29. Dong Y, He Y, Fan D, Wu Z. Preparation of pH-sensitive chitosan-deoxycholic acid-sodium alginate nanoparticles loaded with ginsenoside Rb1 and its controlled release mechanism. Int J Biol Macromol. 2023;228:234. doi:10.1016/j.ijbiomac.2023.123736
30. Xing L, Zheng Y, Yu Y, et al. Complying with the physiological functions of Golgi apparatus for secretory exocytosis facilitated oral absorption of protein drugs. J Mater Chem B. 2021;9(6):1707–1718. doi:10.1039/d0tb02848g
31. Zhang Y, Xiong GM, Ali Y, Boehm BO, Huang YY, Venkatraman S. Layer-by-layer coated nanoliposomes for oral delivery of insulin. Nanoscale. 2021;13(2):776–789. doi:10.1039/d0nr06104b
32. Zhang Q, Li Y, Wang S, et al. Chitosan-based oral nanoparticles as an efficient platform for kidney-targeted drug delivery in the treatment of renal fibrosis. Int J Biol Macromol. 2024;256:128315. doi:10.1016/j.ijbiomac.2023.128315
33. Chen KH, Miao YB, Shang CY, et al. A bubble bursting-mediated oral drug delivery system that enables concurrent delivery of lipophilic and hydrophilic chemotherapeutics for treating pancreatic tumors in rats. Biomaterials. 2020;255:120157. doi:10.1016/j.biomaterials.2020.120157
34. Li J, Qiang H, Yang W, et al. Oral insulin delivery by epithelium microenvironment-adaptive nanoparticles. J Control Release. 2022;341:31–43. doi:10.1016/j.jconrel.2021.11.020
35. Bao C, Liu B, Li B, et al. Enhanced Transport of Shape and Rigidity-Tuned α-Lactalbumin Nanotubes across Intestinal Mucus and Cellular Barriers. Nano Lett. 2020;20(2):1352–1361. doi:10.1021/acs.nanolett.9b04841
36. Zhou W, Li B, Min R, et al. Mucus-penetrating dendritic mesoporous silica nanoparticle loading drug nanocrystal clusters to enhance permeation and intestinal absorption. Biomater Sci. 2023;11(3):1013–1030. doi:10.1039/d2bm01404a
37. Liu L, Tian C, Dong B, et al. Models to evaluate the barrier properties of mucus during drug diffusion. Int J Pharm. 2021;599:120415. doi:10.1016/j.ijpharm.2021.120415
38. Miao R, Jin F, Wang Z, et al. Oral delivery of decanoic acid conjugated plant protein shell incorporating hybrid nanosystem leverage intestinal absorption of polyphenols. Biomaterials. 2022;281:121373. doi:10.1016/j.biomaterials.2022.121373
39. Ejazi SA, Louisthelmy R, Maisel K. Mechanisms of Nanoparticle Transport across Intestinal Tissue: an Oral Delivery Perspective. ACS Nano. 2023;17(14):13044–13061. doi:10.1021/acsnano.3c02403
40. Zheng Y, Xing L, Chen L, et al. Tailored elasticity combined with biomimetic surface promotes nanoparticle transcytosis to overcome mucosal epithelial barrier. Biomaterials. 2020;262:120323. doi:10.1016/j.biomaterials.2020.120323
41. Li X, Jafari SM, Zhou F, et al. The intracellular fate and transport mechanism of shape, size and rigidity varied nanocarriers for understanding their oral delivery efficiency. Biomaterials. 2023;294:121995. doi:10.1016/j.biomaterials.2023.121995
42. Xi Z, Ahmad E, Zhang W, et al. Dual-modified nanoparticles overcome sequential absorption barriers for oral insulin delivery. J Control Release. 2022;342:1–13. doi:10.1016/j.jconrel.2021.11.045
43. Nallamothu B, Kuche K, Ghadi R, Chaudhari D, Jain S. Enhancing oral bioavailability of insulin through bilosomes: implication of charge and chain length on apical sodium-dependent bile acid transporter (ASBT) uptake. Int J Biol Macromol. 2023;252. doi:10.1016/j.ijbiomac.2023.126565
44. Han Y, Liu W, Chen L, et al. Effective oral delivery of Exenatide-Zn(2+) complex through distal ileum-targeted double layers nanocarriers modified with deoxycholic acid and glycocholic acid in diabetes therapy. Biomaterials. 2021;275:120944. doi:10.1016/j.biomaterials.2021.120944
45. Al-Hilal TA, Chung SW, Alam F, et al. Functional transformations of bile acid transporters induced by high-affinity macromolecules. Sci Rep. 2014;4(1):4163. doi:10.1038/srep04163
46. Chai G, Xu Y, Chen S, et al. Transport Mechanisms of Solid Lipid Nanoparticles across Caco-2 Cell Monolayers and their Related Cytotoxicology. ACS Appl Mater Interfaces. 2016;8(9):5929–5940. doi:10.1021/acsami.6b00821
47. Han S, Xin P, Guo Q, Cao Z, Huang H, Wu J. Oral Delivery of Protein Drugs via Lysine Polymer‐Based Nanoparticle Platforms. Adv Healthc Mater. 2023;12(23). doi:10.1002/adhm.202300311
48. Wang X, Zheng Y, Qiu L, et al. Evaluation and antitumor mechanism of functionalized chitosan-based polymeric micelles for oral delivery of paclitaxel. Int J Pharm. 2022;625:122138. doi:10.1016/j.ijpharm.2022.122138
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Nanometerizing Taxifolin Into Selenized Liposomes to Ameliorate Its Hypoglycemic Effect by Optimizing Drug Release and Bioavailability
Qi C, Xing H, Ding N, Feng W, Wu Y, Zhang X, Yu Y
International Journal of Nanomedicine 2025, 20:2225-2240
Published Date: 21 February 2025