")
Back to Journals » International Journal of Nanomedicine » Volume 19
Fusing a Novel Anti-CTLA-4 Nanobody to the IgG1 Fc Region Strengthens Its Ability to Induce CD8+ T Cell-Mediated Immune Responses Against Solid Tumors
Authors Li TT, Yang JH, Jiang MJ, Cui HP, Yang XM, Lu XL, Liu AQ
Received 31 July 2024
Accepted for publication 5 November 2024
Published 21 November 2024 Volume 2024:19 Pages 12311—12321
DOI https://doi.org/10.2147/IJN.S480939
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Ting-ting Li,1 Jin-hua Yang,1 Meng-jie Jiang,1 Hao-peng Cui,1 Xiao-mei Yang,2 Xiao-ling Lu,2 Ai-qun Liu1
1Department of Gastroenterology and Respiratory Medicine & Endoscopy Center, Guangxi Medical University Cancer Hospital, Nanning, Guangxi, 530021, People’s Republic of China; 2Guangxi Key Laboratory of Nanobody Research, Guangxi Medical University, Nanning, Guangxi, 530021, People’s Republic of China
Correspondence: Ai-qun Liu; Xiao-ling Lu, Email [email protected]; [email protected]
Background: Our previously described a nanobody precisely targeting CTLA-4 and demonstrated that it can promote the antitumor response of adoptive T cells. Here we examined whether fusing it to the IgG1 Fc region would induce stronger, longer-lasting T-cell immune responses after exposure to the dendritic-tumor cell fusions.
Methods: The fusion of nanobody to Fc region was overexpressed in E. coli. Next, the proliferation, activation and cytotoxicity of the CD8+ T cells stimulated by the fusion protein and dendritic-tumor cell fusions was assessed in vitro, and the antitumor activity was evaluated in nude mice bearing xenografts of each type of solid tumor.
Results: Proliferation, activation and cytotoxicity of CD8+ T cells in vitro were significantly greater in the presence of the fusion protein than those in other groups. Consistently, different types of xenografts growth were significantly slower and animal survival significantly longer when the injected CD8+ T cells had been activated in vitro in the presence of the fusion protein.
Conclusion: Fusing our anti-CTLA-4 nanobody to the IgG1 Fc region potentiates its ability to induce strong, persistent CD8+ T cell responses against solid tumors in mice. This fusion strategy has the potential to realize clinical transformation and application to clinical treatment.
Keywords: nanobody, CTLA-4, therapeutic antibody derivative, cell fusion, adoptive immunotherapy
Introduction
Adoptive cell therapy, in which autologous or allogeneic CD8+ T cells are exposed to tumor antigens in vitro and then injected into cancer patients, has shown promise for eliciting strong therapeutic responses.1 Our previous work potentiated this type of therapy by taking advantage of the ability of dendritic cells to present tumor antigens effectively to T cells:2,3 we fused tumor cells with dendritic cells, then co-cultured the cell fusions with CD8+ T cells, which mounted strong anti-tumor responses in vitro as well as after injection into tumor-bearing mice.4 However, the anti-tumor responses in mice were transient, similar to results obtained with other types of adoptive cell therapy.1
In an effort to boost immune responses after injection into animals, we co-cultured the T cells and dendritic-tumor cell fusions with a nanobody that recognizes cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) on T cells, then we transferred the activated T cells into tumor-bearing mice.4,5 Our rationale was to prevent CTLA-4 from competing with CD28 for binding to antigen-presenting cells, because CTLA-4 inhibits T cell-mediated immune responses.6 The nanobody showed strong, specific binding, and it boosted the activation of T cells in co-cultures with dendritic-tumor cell fusions; but it failed to elicit persistently strong T cell responses in the animals.4 Nanobodies, although had been proven outstanding characteristics and shown weak immunogenicity, derived from camel heterologous proteins, humanized routinely is part of their development for future clinical translation.7,8
We wondered whether fusing the anti-CTLA-4 nanobody to an appropriate protein partner might prolong the activation of T cells in vivo during adoptive cell therapy. We took inspiration from the first monomeric nanobody formulation to be approved for clinical use, Envafolimab®, in which a nanobody against PD-L1 is fused to the Fc region of human immunoglobulin.9 We decided to fuse our anti-CTLA-4 nanobody to the Fc fragment of human immunoglobulin IgG1, because it is the most abundant and stable immunoglobulin in serum and binds strongly to Fc receptors.10 To elicit robust antibody- and complement-dependent immune responses.11,12
Materials and Methods
Cell Lines and Animals
Jurkat leukemia cells and MIHA immortalized human hepatocytes were purchased from Forte Cheung Biological Technology (Shanghai, China) and cultured in RPMI 1640 (Corning, USA) supplemented with 10% fetal bovine serum (Gibco, USA) and 1% penicillin‒streptomycin (Gibco). HepG2 hepatocellular carcinoma cells, MCF7 breast cancer cells and C8161 melanoma cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. Cultures were incubated at 37 °C in an atmosphere of 5% CO2.
Female BALB/c nude mice 4–6 weeks old and weighing 18–22 g were obtained from the Experimental Animal Center of Guangxi Medical University. Animals were housed in facilities free of specific pathogens at the Experimental Animal Center. All procedures on experimental animals including animal handling, welfare and medical treatments were approved by the Institutional Animal Care and Use Committee of Guangxi Medical University, and conducted by following the Guiding Opinions on the Ethical Treatment of Laboratory Animals issued by the Science and Technology Ministry of People’s Republic of China, and the National Standard Guidelines for Laboratory Animal Welfare and Ethical Review of the People’s Republic of China.
Production and Characterization of Anti-CTLA-4 Nanobody Fused to the Fc Region of Human IgG1
The preparation scheme for CTLA-4 Nb16-Fc is shown in Figure 1. The sequence of the anti-CTLA-4 nanobody that we previously selected from a library of the variable regions of heavy-chain IgG proteins from camel,7 which we thoroughly characterized for binding specificity and affinity and for ability to elicit T cell responses,4 was fused to the sequence of the Fc region of human IgG1 (GenBank accession AH007035.2). Between the two proteins we inserted a flexible pentapeptide linker with the sequence (Gly)4Ser, and we added a hexahistidine tag at the N-terminus to facilitate purification and detection. The plasmid was constructed through genetic engineering, subcloned into the expression vector pET-30a (Jinsirui, Nanjing, China), and transformed into E. coli. Bacterial cultures were inoculated into 1 L of terrific broth supplemented with 50 μg/mL kanamycin and grown at 37 °C for 8 h. Bacteria were harvested by centrifugation, resuspended in phosphate-buffered saline (PBS), ultrasonicated, and centrifuged again. The pellet (Inclusion Bodies) was dissolved in 8 M urea, purified on Ni2+-based affinity chromatography, refolded through dialysate of different concentrations, and finally concentrated on an ultrafiltration membrane with a molecular weight cut-off of 3 KDa. The same approach was used to prepare a fusion between a nanobody sequence that showed no appreciable binding to CTLA-4 and the Fc fragment of human IgG1. This fusion, hereafter referred to as CTRL Nb-Fc, served as an isotype control in certain experiments. Regarding protein overexpression in E. coli, we use non-heat source consumables throughout the process, and reused consumables are strictly sterilized to control endotoxin contamination.
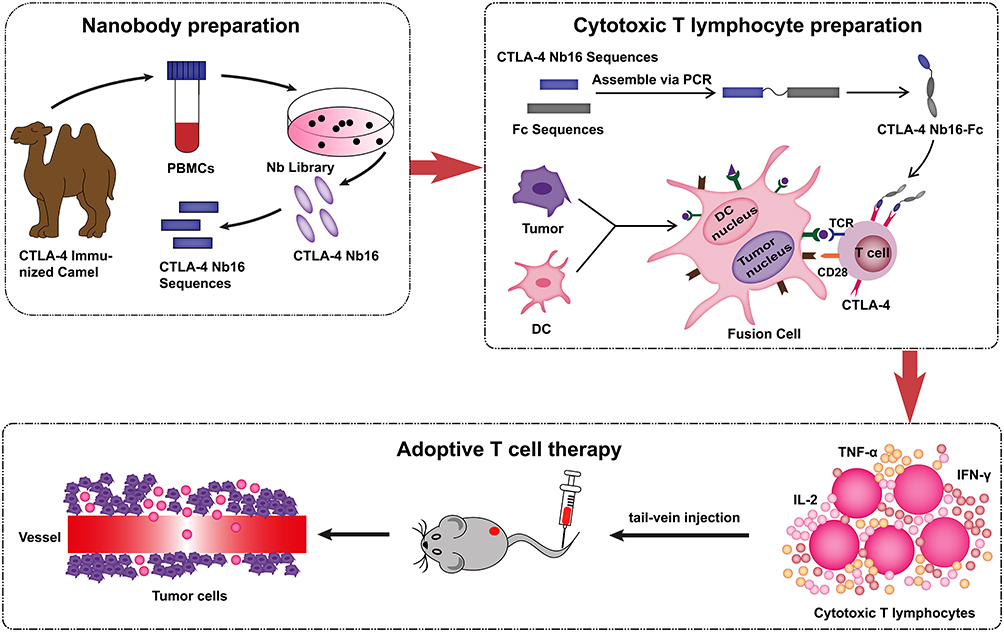 |
Figure 1 Schematic illustration of the preparation of CTLA-4 Nb16-Fc and DC/tumor fusion cells, and the CD8+ T lymphocytes induced by both of them enhanced antitumor activity of adoptive T cell treatment. |
The purity of the recombinant fusion was confirmed by analysis on 12% SDS-PAGE. Its ability to bind human CTLA-4 on its own was confirmed using an enzyme-linked immunosorbent assay, while its ability to bind CTLA-4 on the surface of activated CTLA-4+ cells was confirmed using flow cytometry (EPICS XL, Beckman Coulter, USA). In flow cytometry experiments, CTLA-4 Nb16-Fc was incubated with PHA‑stimulated T cells and Jurkat cells, respectively. The fusion was labeled for detection in flow cytometry by incubation at 4 °C for 30 min in the dark with phycoerythrin-conjugated antibody against the hexahistidine tag (BD Biosciences, NJ, USA). Cells were incubated with monoclonal antibody against CTLA-4 (BD Biosciences) as a positive control or with CTRL Nb-Fc as a negative control.
The cytotoxicity of the fusion protein was evaluated against cultures of primary human T cells or immortalized MIHA human hepatocytes. Cells in DMEM were seeded into 96-well plates (5 × 104 cells/well), incubated for 12 h, then exposed for 12–48 h to 1–10 μg of CTLA-4 Nb16-Fc. At different time points, cell viability was evaluated using the CCK-8 assay (Saint-bio, Shanghai, China), in which absorbance at 450 nm was measured using a microplate reader (ThermoFisher, USA).
Preparation of Dendritic-Tumor Cell Fusions
Peripheral blood was collected from healthy volunteers and subjected to density gradient centrifugation using a lymphocyte isolation medium (Solarbio, China). Peripheral blood mononuclear cells (PBMCs) were cultured at 37 °C for 4 h in RPMI 1640 (Gibco), after which cultures were washed to remove weakly adhering cells. The remaining adherent cells were incubated for 5 days in the presence of 1000 U/mL granulocyte-macrophage colony-stimulating factor (PeproTech, Rocky Hill, NJ, USA) and 500 U/mL recombinant human IL-4 (PeproTech) to induce their differentiation into dendritic cells. All experiments involving human samples were ethically approved and conducted in accordance with the Declaration of Helsinki and the guidelines established by the Institutional Review Board of Guangxi Medical University. All volunteers provided written informed consent to participate in the study.
These dendritic cells were fused with tumor cells as described.7 The dendritic cells were harvested, mixed 2:1 with HepG2, MCF7 or C8161 tumor cells for 5 min at 37 °C in prewarmed serum-free RPMI 1640 containing polyethylene glycol (Sigma, St Louis, MO, USA), diluted with prewarmed serum-free RPMI 1640, and centrifuged. The pellet was resuspended and incubated for 30 min at 37 °C in prewarmed serum-free RPMI 1640 containing 50 μg/mL type I collagen (Sigma). The suspended cells were washed three times in PBS through cycles of centrifugation and resuspension, then resuspended in RPMI 1640 containing 1000 U/mL granulocyte-macrophage colony-stimulating factor and 500 U/mL recombinant human IL-4, seeded into 12-well plates (1 × 106 cells/well) and incubated for 7 days.
The efficiency of fusion between dendritic and tumor cells was assessed by pre-labeling tumor cell membranes with the red fluorescent dye PKH26 (Solarbio, China) and dendritic cell membranes with the green fluorescent dye carboxyfluorescein succinimidyl ester (CFSE; Absin, China). Flow cytometry or fluorescence microscopy was used to assess the formation of orange cell fusions relative to the original red or green parental cells.
In vitro Activation of CD8+ T Cells
PBMCs were obtained as described above, then CD8+ T cells were isolated using a commercial kit (Invitrogen, Carlsbad, CA, USA). The cells were cultured for 24 h in RPMI 1640 containing 100 U/mL recombinant human IL-2 (PeproTech). Next the CD8+ T cells were co-cultured 10:1 with fusions of dendritic cells and each of the three types of tumor cells for 3 days in the presence of 5 μg/mL CTLA-4 Nb16-Fc and 10 U/mL of IL-2.
To assess the activation of T cells through this procedure, the CD8+ T cells were harvested, isolated from other cells using nylon cotton and incubated at 4 °C for 30 min in the dark with FITC-conjugated monoclonal antibody against human CD25 or CD69 (BD Biosciences), which serve as markers of T cell activation.13 The immunolabeled cells were analyzed by flow cytometry.
To assess the proliferation of T cells through the activation procedure, CD8+ T cells were labeled with 5 µM CFSE and co-cultured 10:1 for 5 days in the dark with fusions of dendritic cells with each of the three types of tumor cells in the presence of 5 µg/mL CTLA-4 Nb16-Fc. The attenuation of T cell fluorescence was analyzed using flow cytometry as an index of cell proliferation.
Anti-Tumor Responses of Activated CD8+ T Cells in vitro
CD8+ T cells that had been co-cultured for three days with dendritic-tumor cell fusions in the presence of CTLA-4 Nb16-Fc as described above were harvested and co-cultured 10:1 with each of the three types of tumor cells separately for 24 h. Levels of IL-2, IFN-γ, and TNF-α secreted by the CD8+ T cells into the culture medium were assayed using commercial enzyme-linked immunosorbent assays (Cusabio, China). Absorbance at 450 nm was determined using a Multiskan GO Scientific Microplate Reader (ThermoFisher).
In another experiment, activated CD8+ T cells were co-cultured at ratios of 1:1, 10:1 or 20:1 for 24 h in the dark with each of the three types of tumor cells that had been pre-labeled with CFSE. The tumor cells were harvested, stained for 10 min at room temperature with propidium iodide (Solarbio, China), and analyzed by flow cytometry to determine extent of apoptosis.
Anti-Tumor Responses of Activated CD8+ T Cells in vivo
Xenografts of HepG2, MCF7 or C8161 cells in female nude BALB/c mice were obtained by subcutaneously injecting tumor cells (2×106 cells, 100 µL) into the right armpit. When tumors had grown to approximately 100 mm3, animals were randomized to receive injections of activated CD8+ T lymphocytes (2 × 107 cells per injection) through the tail vein twice a week for two consecutive weeks (five mice per condition). The day of the first injection was defined as day 0. Tumor length and width were measured every three days. Then animals were sacrificed on day 30, and tumors were excised and weighed. In parallel, another set of five animals per condition were monitored for 100 days after the first injection in order to examine survival.
Toxicity of the CTLA-4 Nb16-Fc Fusion Protein in vivo
Mice were injected in the tail vein with CTLA-4 Nb16-Fc at a dose of 100 mg/kg in a volume of 100 μL of PBS, while same quantity mice were injected with the same volume of PBS. At one week after injection, animals were euthanized and tissue sections were prepared from the heart, liver, spleen, lung, and kidney. Sections were fixed, stained with hematoxylin-eosin, and analyzed for histopathology, including infiltration by immune cells.
Statistical Analysis
The data were presented as mean ± standard deviation. Significance was assessed via one-way analysis of variance (ANOVA) or repeated measures ANOVA with Tukey’s multiple-comparisons test. Differences in survival were assessed for significance using a two-sided Log-rank test. Statistical analyses were performed within GraphPad Prism 5.01 (GraphPad, USA). A p value of <0.05 was deemed statistically significant for all analysis (*p < 0.05; **p < 0.01; ***p< 0.001).
Results
Production and Characterization of CTLA-4 Nb16-Fc
The CTLA-4 Nb16-Fc fusion protein was constructed and expressed in E. coli (Figure 2a). The resulting purified protein showed the expected molecular weight around 39 kDa and purity exceeding 90% (Figure 2b). The purified fusion protein bound specifically to human CTLA-4 on its own (Figure 2c) or on the surface of activated primary T cells and Jurkat cells (Figure 2d-e). Exogenous CTLA-4 competed for the binding of the fusion protein to T cells or Jurkat cells, confirming the specificity of binding (Figure 2d-e). The fusion protein did not significantly reduce viability of MIHA cells (Figure 3a) or activated T cells (Figure 3b) in culture, nor did it induce obvious histopathology in the major organs of healthy mice (Figure 3c).
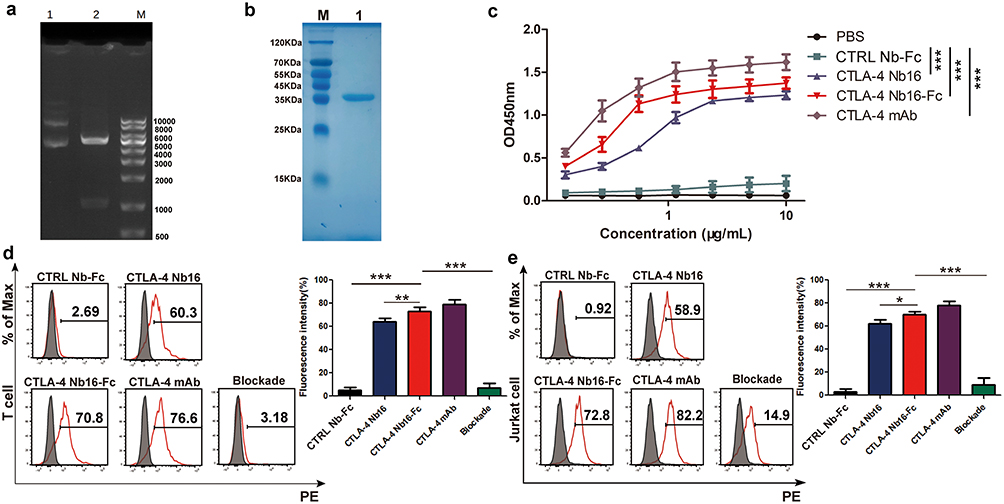 |
Figure 2 Production and characterization of CTLA-4 Nb16-Fc. (a) Agarose gel electrophoresis of the complete expression plasmid pET-30a (+)-CTLA-4 Nb16-Fc before (lane 1) and after (lane 2) digestion with NdeI and HindIII to excise the recombinant gene encoding the fusion protein. Lane (M) DNA markers. (b) Representative SDS-PAGE of purified CTLA-4 Nb16-Fc using affinity chromatography against the hexahistidine tag. Lane (M) Molecular weight markers. Lane 1: CTLA-4 Nb16-Fc fusion protein (~39 kDa). (c-e) Binding of purified CTLA-4 Nb16-Fc to (c) purified human CTLA-4 on its own in an enzyme-linked immunosorbent assay or to CTLA-4 on the surface of (d) activated T cells or (e) Jurkat cells by Flow cytometry. mAb (monoclonal antibody); CTRL Nb-Fc (a fusion between a nanobody that does not recognize CTLA-4 and the Fc region). Data come from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. |
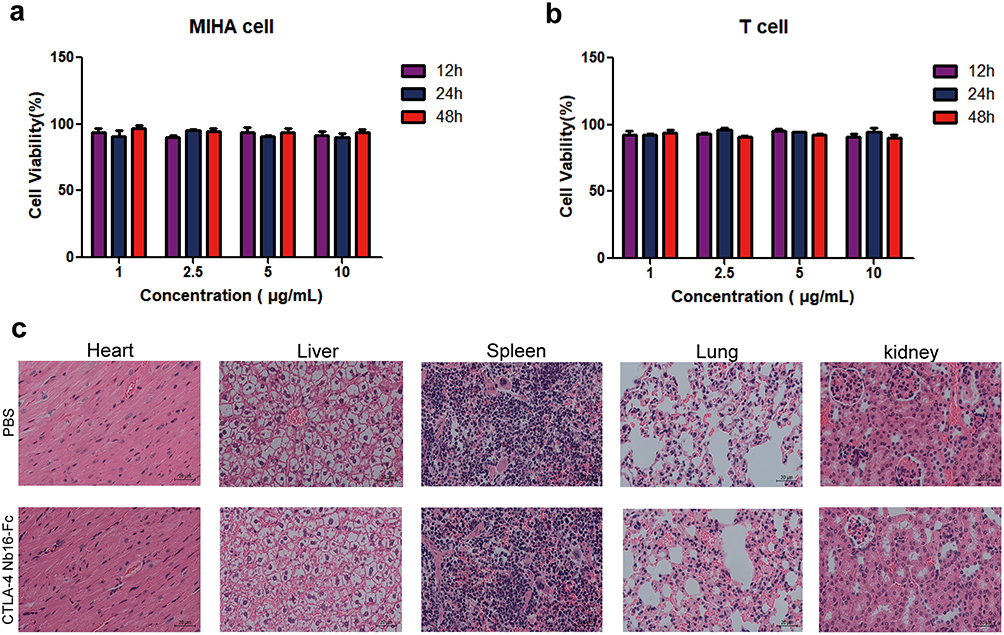 |
Figure 3 Toxicity of CTLA-4 Nb16-Fc in vitro and in vivo. (a-b) Cultures of (a) MIHA immortalized human hepatocytes or (b) primary human T cells were exposed to purified CTLA-4 Nb16-Fc for the indicated times, then cell viability was assayed using the CCK-8 kit. Data are mean ± SD of three independent experiments. (c) BALB/c mice were injected in the tail with CTLA-4 Nb16-Fc at a dose of 100 mg/kg or with vehicle only (phosphate-buffered saline, PBS). At one week after injection, animals were euthanized and tissue sections were prepared and stained with hematoxylin-eosin. Magnification, 200×. |
Characterization of Dendritic-Tumor Cell Fusions
Our optimized procedure of co-culturing dendritic cells with the three types of tumor cells led to fusion rates of 40–55% (Supplementary Figure 1a). Levels of CD80, CD86 and MHC-II were significantly higher on the surface of cell fusions than on the surface of unfused dendritic cells (Supplementary Figure 1b), implying that fusing the dendritic cells to tumor cells promotes the maturation of dendritic cells and enhances their ability to present antigens to T cells.
Ability of CTLA-4 Nb16-Fc to Enhance CD8+ T Cell Activation by Dendritic-Tumor Cell Fusions in vitro
The extent of activation of CD8+ T cells by dendritic-tumor cell fusions, which was measured in terms of expression of CD25 and CD69 on the T cell surface,13 was significantly higher in the presence of CTLA-4 Nb16-Fc than in the presence of the nanobody alone or in the absence of nanobody (Figure 4a-4b). Similar results were observed when extent of activation was measured in terms of CD8+ T cell proliferation (Figure 4c), production of the pro-inflammatory cytokines IL-2, IFN-γ, and TNF-α (Figure 5a), and ability to kill tumor cells after replating (Figure 5b).
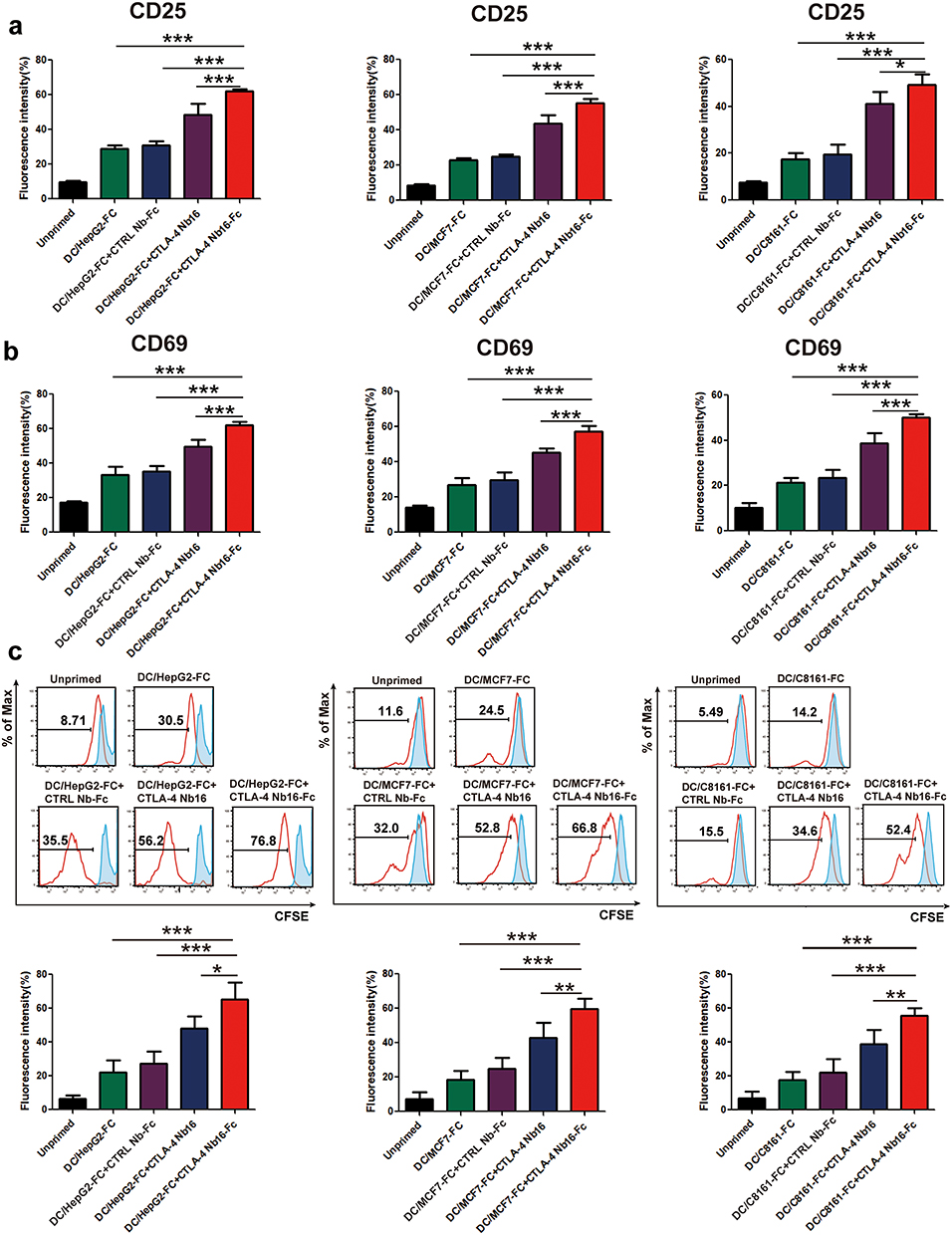 |
Figure 4 Influence of CTLA-4 Nb16-Fc on activation of CD8+ T cells in vitro by dendritic-tumor cell fusions, based on surface expression of activation markers and proliferation. CD8+ T cells were incubated with the dendritic-tumor cell fusions for three days in the presence of CTLA-4 Nb16-Fc, the camel nanobody lacking the Fc fusion partner (CTLA-4 Nb16), a fusion between a nanobody that does not recognize CTLA-4 and the Fc region (CTRL Nb-Fc), or no nanobody at all. “Unprimed” refers to monocultures of CD8+ T cells on their own. After incubation for three days, cultures were stained (a-b) with FITC-conjugated monoclonal antibody against CD25 or CD69. (c) The indicated CD8+ T cells were labeled with CFSE and co-cultured with the dendritic-tumor cell fusions at a ratio of 10:1 for 5 days, and cell proliferation was determined by flow cytometric assay of CFSE dilution. Data are from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. |
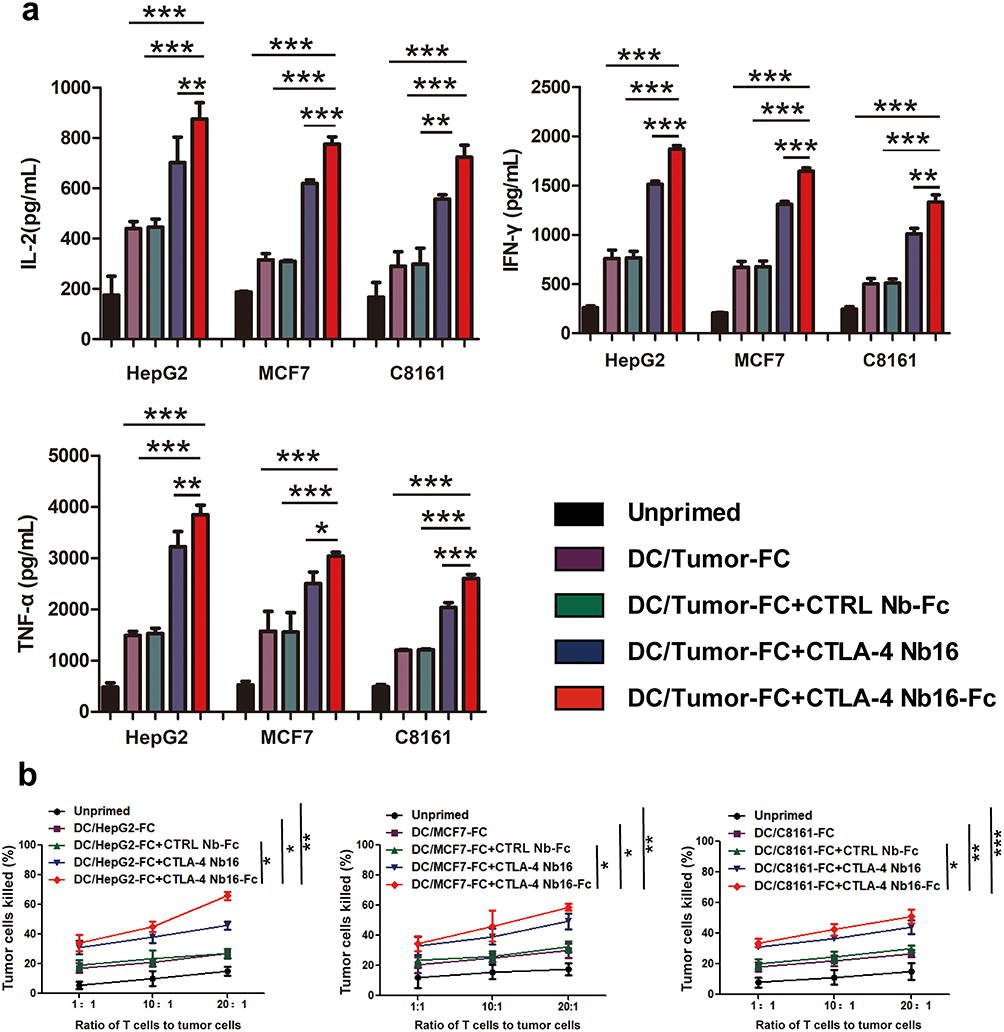 |
Figure 5 Influence of CTLA-4 Nb16-Fc on activation of CD8+ T cells in vitro by dendritic-tumor cell fusions, based on cytokine production and killing of tumor cells after replating. (a) CD8+ T cells were stimulated as described in Figure 4, then levels of IL-2, IFN-γ and TNF-α in the culture medium were assayed using commercial enzyme-linked immunosorbent assays. (b) CD8+ T cells were stimulated as described in Figure 4, harvested, and replated in co-cultures with each of the three types of solid tumors (HepG2, MCF7, C8161). The tumor cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) and propidium iodide, and the percentage of all tumor cells that were double-labeled (apoptotic) was determined. Data are from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. |
Ability of CTLA-4 Nb16-Fc to Enhance Anti-Tumor Responses by CD8+ T Cells in vivo
Through the experimental procedure schematized in Figure 6a, we found that, for all three types of solid tumor, in vitro activation of T cells in the presence of CTLA-4 Nb16-Fc led to significantly smaller tumors after injection of the T cells into tumor-bearing mice than in vitro activation in the presence of nanobody alone or in the absence of nanobody (Figure 6b–g). Similar results were observed when we tracked tumor volume during the 30-day treatment period (Figure 6h–j). Consistently, presence of CTLA-4 Nb16-Fc during in vitro activation led to significantly longer survival of tumor-bearing mice than activation in the presence of nanobody alone or without nanobody (Figure 6k–m).
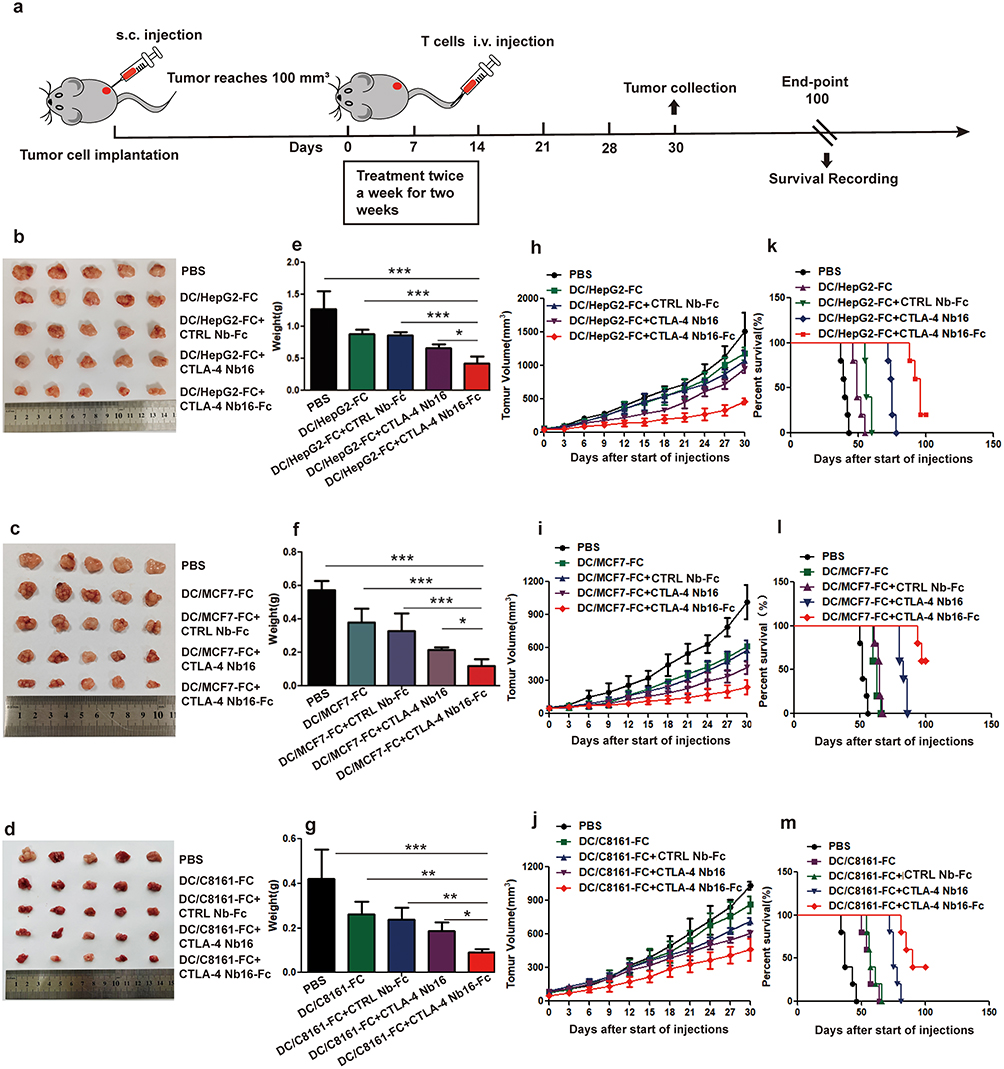 |
Figure 6 Influence of CTLA-4 Nb16-Fc during in vitro activation of CD8+ T cells by dendritic-tumor cell fusions on subsequent anti-tumor responses in mice. (a) Schematic of the experimental design. CD8+ T cells were stimulated as described in Figure 4, then injected intravenously (i.v.) into nude BALB/c mice during two weeks. On day 30 after the first injection, animals were sacrificed and tumors were excised and weighed. A parallel cohort of animals was allowed to live until 100 days, during which survival was monitored. “PBS” refers to animals that were injected with phosphate-buffered saline without T cells. (b–m) For each type of solid tumor, (b–d) representative photographs of excised tumors on day 30 after the start of injections were taken, (e–g) weight (mean ± SD) of excised tumors was determined (five animals per condition), (h–j) volume of tumors (mean ± SD) was measured throughout the treatment period and for two weeks afterward (five animals per condition), and (k–m) survival of animals out to 100 days after start of injections was evaluated using the Kaplan-Meier method. Survival was reported as the percentage of the original five animals that were still alive at the indicated time point. *p < 0.05, **p < 0.01, ***p < 0.001. |
Discussion
We previously showed that activating T cells in vitro by co-culturing them with fusions of dendritic and tumor cells can enhance anti-tumor responses after injection of the activated T cells into tumor-bearing animals.4 In that work, we were able to further enhance in vivo responses against three types of solid tumors by including an anti-CTLA-4 nanobody in the activation co-culture beforehand. However, even with the nanobody, the enhancement of anti-tumor responses in vivo was short-lived. Here we significantly strengthened the anti-tumor responses in vivo by fusing the Fc region of IgG1 to the nanobody that was included in the activation co-culture. Our work provides a strategy for increasing the efficacy of adoptive cell therapy against various types of solid tumors.
Consistent with several previous studies,14–16 our experiments show that fusing dendritic cells to tumor cells leads to stronger in vitro activation of T cells than unfused dendritic cells alone. Such fusions may present abundant antigen-MHC complexes to T cells more effectively through the formation of “immune synapses”, which may explain the enhanced T cell-mediated tumor cell killing observed here and in previous work.17,18
Why robust activation of CD8+ T cells in vitro can still lead to weak or transient anti-tumor responses in vivo remains unclear. The likely reason is complex immunosuppressive mechanisms in the tumor microenvironment,1,19 including signaling mediated by CTLA-4 or PD-1 on the surface of T cells.20,21 This may help to explain the anti-tumor efficacy of anti-CTLA-4 nanobodies for adoptive cell therapy, as we demonstrated here and in previous work.4 Future studies should continue to explore the immunosuppressive mechanisms in the microenvironment of solid tumors and develop ways to circumvent them.
One of the major obstacles to cancer immunotherapy is getting anti-tumor antibodies to penetrate deep within the tumor itself.22–24 Nanobodies, as the smallest protein moieties that can tightly and specifically bind antigens, may be more effective than monoclonal antibodies in this regard. Potentially even more effective, however, may be to treat tumors with appropriately activated T cells rather than with free antibodies. The in vitro activation of T cells must be optimized to induce sufficiently strong, long-lasting in vivo anti-tumor responses. Our work takes a step in this direction for the treatment of a potentially wide range of solid tumors.
Future preclinical studies should verify and extend our findings on CTLA-4 Nb16-Fc by exploring subcutaneous and intratumoral modes of administration as well as additional types of solid tumors, particularly in animals bearing patient-derived xenografts. Future work should elucidate how in vitro activation of T cells in the presence of CTLA-4 Nb16-Fc leads to persistent, strong anti-tumor response in vivo, and it should screen for different fusion partners for the nanobody that may lead to even greater improvements in anti-tumor efficacy. E. coli is extensively employed for the production of recombinant proteins, owing to its operational simplicity, cost-effectiveness, and remarkable expression levels. Currently, the exploration of immune checkpoint inhibitors, particularly those exemplified by PD-1 and PD-L1, has emerged as a prominent focal point in contemporary research. Furthermore, E. coli, recognized for its remarkable efficiency as a prokaryotic expression system, may hold significant promise for future applications in this domain.
Conclusion
Here we demonstrate that fusing the Fc region of human IgG1 to an anti-CTLA-4 nanobody can significantly improve anti-tumor responses by T cells in mouse models of three solid cancers following in vitro activation of the T cells in the presence of the nanobody. Our work suggests that appropriate fusion partners on nanobodies can boost their efficacy in adoptive cell therapy, which might relate to the extended half-life of the fusion protein and Fc-mediated anti-tumor immune response. Whether such fusions also allow nanobodies to kill tumor cells more effectively as a stand-alone immunotherapy should also be explored. What’s more, testing in more clinically relevant patient-derived xenograft models or investigating other solid tumor types could be very clinically significant.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
This project was supported by grants from the National Natural Scientific Foundation of China (82360559, 81560494), Guangxi Natural Science Foundation (2023GXNSFAA026298), Guangxi Key R & D Plan (AB18221084, AB20297021), Guangxi Medical and Health Key Cultivation Discipline Construction Project, and Fund for the Development and Promotion of Suitable Medical and Health Technologies in Guangxi (S2022107).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no competing interests related to this work.
References
1. Jafferji MS, Yang JC. Adoptive T-cell therapy for solid malignancies. Surg Oncol Clin N Am. 2019;28(3):465–479. doi:10.1016/j.soc.2019.02.012
2. Gerner MY, Casey KA, Kastenmuller W, et al. Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J Exp Med. 2017;214:3105–3122. doi:10.1084/jem.20170335
3. Sadeghzadeh M, Bornehdeli S, Mohahammadrezakhani H, et al. Dendritic cell therapy in cancer treatment; the state-of-the-art. Life Sci. 2020;254:117580. doi:10.1016/j.lfs.2020.117580
4. Tang Z, Mo F, Liu A, et al. A nanobody against cytotoxic T-lymphocyte associated antigen-4 increases the anti-tumor effects of specific CD8+ T cells. J Biomed Nanotechnol. 2019;15(11):2229–2239. doi:10.1166/jbn.2019.2859
5. Wan R, Liu A, Hou X, et al. Screening and antitumor effect of an anti CTLA 4 nanobody. Oncol Rep. 2018;39(2):511–518. doi:10.3892/or.2017.6131
6. Van Coillie S, Wiernicki B, Xu J. Molecular and cellular functions of CTLA-4. Adv Exp Med Biol. 2020;1248:7–32. doi:10.1007/978-981-15-3266-5_2
7. Liu M, Li L, Jin D, et al. Nanobody-A versatile tool for cancer diagnosis and therapeutics. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2021;13(4):e1697. doi:10.1002/wnan.1697
8. Rossotti MA, Bélanger K, Henry KA, Tanha J. Immunogenicity and humanization of single-domain antibodies. FEBS J. 2022;289(14):4304–4327. doi:10.1111/febs.15809
9. Li J, Deng Y, Zhang W, et al. Subcutaneous envafolimab monotherapy in patients with advanced defective mismatch repair/microsatellite instability high solid tumors. J Hematol Oncol. 2021;14(1):95. doi:10.1186/s13045-021-01095-1
10. Napodano C, Marino M, Stefanile A, et al. Immunological role of IgG subclasses. Immunol Invest. 2021;50(4):427–444. doi:10.1080/08820139.2020.1775643
11. Ingram JR, Blomberg OS, Rashidian M, et al. Anti-CTLA-4 therapy requires an Fc domain for efficacy. Proc Natl Acad Sci USA. 2018;115(15):3912–3917. doi:10.1073/pnas.1801524115
12. Sondermann P, Szymkowski DE. Harnessing Fc receptor biology in the design of therapeutic antibodies. Curr Opin Immunol. 2016;40:78–87. doi:10.1016/j.coi.2016.03.005
13. Gockel LM, Nekipelov K, Ferro V, Bendas G, Schlesinger M. Tumour cell-activated platelets modulate the immunological activity of CD4+, CD8+, and NK cells, which is efficiently antagonized by heparin. Cancer Immunol Immunother. 2022;71(10):2523–2533. doi:10.1007/s00262-022-03186-5
14. Orr S, Huang L, Moser J, et al. Personalized tumor vaccine for pancreatic cancer. Cancer Immunol Immunother. 2023;72(2):301–313. doi:10.1007/s00262-022-03237-x
15. Yan Y, Zeng S, Gong Z, Xu Z. Clinical implication of cellular vaccine in glioma: current advances and future prospects. J Exp Clin Cancer Res. 2020;39(1):257. doi:10.1186/s13046-020-01778-6
16. Pinho MP, Sundarasetty BS, Bergami-Santos PC, et al. Dendritic-tumor cell hybrids induce tumor-specific immune responses more effectively than the simple mixture of dendritic and tumor cells. Cytotherapy. 2016;18(4):570–580. doi:10.1016/j.jcyt.2016.01.005
17. Yu J, Sun H, Cao W, et al. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp Hematol Oncol. 2022;11(1):3. doi:10.1186/s40164-022-00257-2
18. Weng D, Calderwood SK, Gong J. A novel heat shock protein 70-based vaccine prepared from DC-tumor fusion cells. Methods Mol Biol. 2018;1709:359–369. doi:10.1007/978-1-4939-7477-1_26
19. Met Ö, Jensen KM, Chamberlain CA, et al. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. 2019;41(1):49–58. doi:10.1007/s00281-018-0703-z
20. Wang Y, Wang Y, Ren Y, et al. Metabolic modulation of immune checkpoints and novel therapeutic strategies in cancer. Semin Cancer Biol. 2022;86(Pt 3):542–565. doi:10.1016/j.semcancer.2022.02.010
21. Lucy Boyce K, Salama April KS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86–104. doi:10.3322/caac.21596
22. Uson Junior PLS, Liu AJ, Sonbol MB, Borad MJ, Bekaii-Saab TS. Immunotherapy and chimeric antigen receptor T-cell therapy in hepatocellular carcinoma. Chin Clin Oncol. 2021;10(1):11. doi:10.21037/cco-20-231
23. Ciliberto D, Caridà G, Staropoli N, et al. First-line systemic treatment for hepatocellular carcinoma: a systematic review and network meta-analysis. Heliyon. 2023;9(8):e18696. doi:10.1016/j.heliyon.2023.e18696
24. Keam SJ. Tremelimumab: first approval. Drugs. 2023;83(1):93–102. doi:10.1007/s40265-022-01827-8
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.