")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 21
Genotypic and Phenotypic Characteristics of Pediatric X-Adrenoleukodystrophy in a Chinese Cohort
Authors Zhang Y, Shi X, Huang J, Wang C, Wu S, Tian G, Jia J, Chen Y
Received 20 November 2024
Accepted for publication 7 March 2025
Published 27 March 2025 Volume 2025:21 Pages 677—687
DOI https://doi.org/10.2147/NDT.S507632
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Yuanfeng Zhang,1 Xiaorong Shi,2 Jianjun Huang,1 Chunmei Wang,1 Shengnan Wu,3 Guoli Tian,4 Jia Jia,5 Yucai Chen1
1Department of Neurology, Shanghai Children’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200062, People’s Republic of China; 2Pediatric Department, The First Affiliated Hospital of Fujian Medical University, Fuzhou, 350004, People’s Republic of China; 3Molecular Diagnostic Laboratory, Shanghai Children’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200062, People’s Republic of China; 4Neonatal Screening Center, Shanghai Children’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200062, People’s Republic of China; 5Shanghai Engineering Research Center for Big Data in Pediatric Precision Medicine, Center for Biomedical Informatics, Shanghai Children’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200062, People’s Republic of China
Correspondence: Yucai Chen; Yuanfeng Zhang, Department of Neurology, Shanghai Children’s Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200062, People’s Republic of China, Tel +86 13512114542, Email [email protected]; [email protected]
Objective: This study aimed to investigate the clinical, phenotypic, and genotypic characteristics of pediatric patients with X-linked adrenoleukodystrophy (X-ALD).
Methods: Clinical and genetic data were retrospectively analyzed from 14 male pediatric patients with X-ALD (mean age: 6 years 11 months [SD: 1 year 9 months]; age range: 5– 10 years) from 14 unrelated families at a single center.
Results: All 14 patients were diagnosed with childhood cerebral adrenoleukodystrophy (CCALD). Initial clinical symptoms were atypical, primarily presenting as cognitive decline and neurological dysfunction, accompanied by elevated levels of very long-chain fatty acids (VLCFAs) in the blood. Brain magnetic resonance imaging (MRI) findings consistently showed characteristic white matter demyelination. Genetic analysis identified ABCD1 gene mutations in all pediatric patients, comprising 12 distinct known mutations. Among these, 9 cases involved mutations in exons 6 to 9, and 3 cases in exons 1 to 2. A total of 13 were missense mutations, while 1 was a coding mutation.
Conclusion: The findings indicate that early symptoms of X-ALD are often atypical. Blood VLCFA levels and ABCD1 gene mutation analysis play a crucial role in early diagnosis. Hematopoietic stem cell transplantation (HSCT) is an effective treatment for pediatric cases with early-stage CCALD.
Keywords: ABCD1 gene, hematopoietic stem cell transplantation, pediatric patients, very long chain fatty acids, X-adrenoleukodystrophy
Introduction
X-linked adrenoleukodystrophy (X-ALD, MIM #300100) is a peroxisomal disorder characterized by impaired β-oxidation, leading to the accumulation of very long-chain fatty acids (VLCFAs) in all tissues.1 X-ALD is an X-linked recessive condition that primarily affects males, while females typically remain asymptomatic carriers. Currently, there is no evidence to suggest known regional or ethnic differences in the incidence of X-ALD or its associated gene mutations. The disease arises from mutations in the ABCD1 gene, which consists of 10 exons on the Xq28 locus. The incidence of X-ALD ranges from 1 in 14,700 to 1 in 20,000 individuals.2–4 Clinical phenotypes of X-ALD vary, but the most common manifestations include progressive cognitive and motor decline, visual and auditory impairments, seizures, and spastic paralysis.5 Childhood cerebral ALD (CCALD) is the most prevalent subtype, with laboratory diagnostics centered on elevated very long-chain fatty acid (VLCFA) levels and mutations in the ABCD1 gene. CCALD is associated with a poor prognosis, with most pediatric patients succumbing to the disease by the age of 15.6,7
In the absence of effective drug therapies, allogeneic hematopoietic stem cell transplantation (HSCT) is the only effective intervention for X-ALD and is the preferred treatment for pediatric patients with early-stage CCALD.8 Current literature on X-ALD predominantly focuses on Western cohorts, with limited data on pediatric patients from China. This study addresses this gap by summarizing the clinical features, laboratory findings, and genetic profiles of 14 Chinese pediatric patients with X-ALD. By characterizing their unique phenotypic and genotypic profiles, we aim to enhance understanding of X-ALD in this underrepresented population, highlight potential regional differences, and inform tailored diagnostic and therapeutic strategies for early diagnosis and optimized management.
Materials and Methods
Patients
This retrospective analysis included 14 pediatric cases with X-ALD who were diagnosed or referred to the Department of Pediatrics at Shanghai Children’s Hospital and the First Affiliated Hospital of Fujian Medical University (Fuzhou) between June 2003 and December 2022. Diagnosis of X-ALD was confirmed based on the following criteria:9 (1) Clinical presentation, primarily in males, with symptoms including auditory and visual impairments, behavioral abnormalities, intellectual and motor dysfunction, personality changes, and neurological deficits, with or without adrenal insufficiency; (2) Brain magnetic resonance imaging (MRI) showing progressive demyelination of white matter; (3) Laboratory evidence of elevated serum VLCFA, along with abnormal levels of C26:0, C24:0/C22:0, C26:0/C22:0, adrenocorticotropic hormone (ACTH), and cortisol; (4) Conformation of an ABCD1 gene mutation through genetic testing.
Methods
Basic information, including name, sex, age of onset, clinical manifestations, electroencephalogram (EEG) findings, brain MRI results, laboratory tests, and ABCD1 gene mutation data, was gathered for comprehensive analysis.
VLCFA Test
Fasting whole blood was collected in EDTA tubes, and plasma was subsequently extracted for analysis. VLCFA levels were measured using gas chromatography-mass spectrometry (GC-MS).
ABCD1 Gene Mutation Detection and Analysis
DNA Extraction
Two milliliters of venous blood were collected from each pediatric patient and placed in EDTA anticoagulant tubes. Genomic DNA was extracted using the QIAamp DNA Blood Mini Kit, and its quality and concentration were assessed with a NanoDrop 2000 spectrophotometer (Thermo).
PCR Identification and Sanger Sequencing
Primers were designed using Oligo 7 software (primer sequences are shown in Table 1) and synthesized by Invitrogen Trading (Shanghai) Co., Ltd. PCR reagents were obtained from Takara Biotechnology (Dalian) Co., Ltd. Each 25 µL PCR reaction mixture contained 2.5 µL 10× PCR buffer, 2 µL dNTPs, 0.2 µL Taq DNA polymerase, 1 µL each of forward and reverse primers (20 µmol/L), 1 µL genomic DNA, and 17.3 µL ddH2O. PCR cycling conditions were as follows: initial denaturation at 95 °C for 5 minutes; denaturation at 95 °C for 30 seconds; a gradient reduction from 62 °C to 57 °C over 10 cycles (0.5 °C decrease per cycle); annealing at 57 °C for 30 seconds; extension at 72 °C for 25 cycles; followed by a final extension at 72 °C for 10 minutes. PCR products were purified using 1.5% agarose gel electrophoresis and sequenced. The sequencing results were aligned with the reference sequence to identify mutations in the ABCD1 gene.
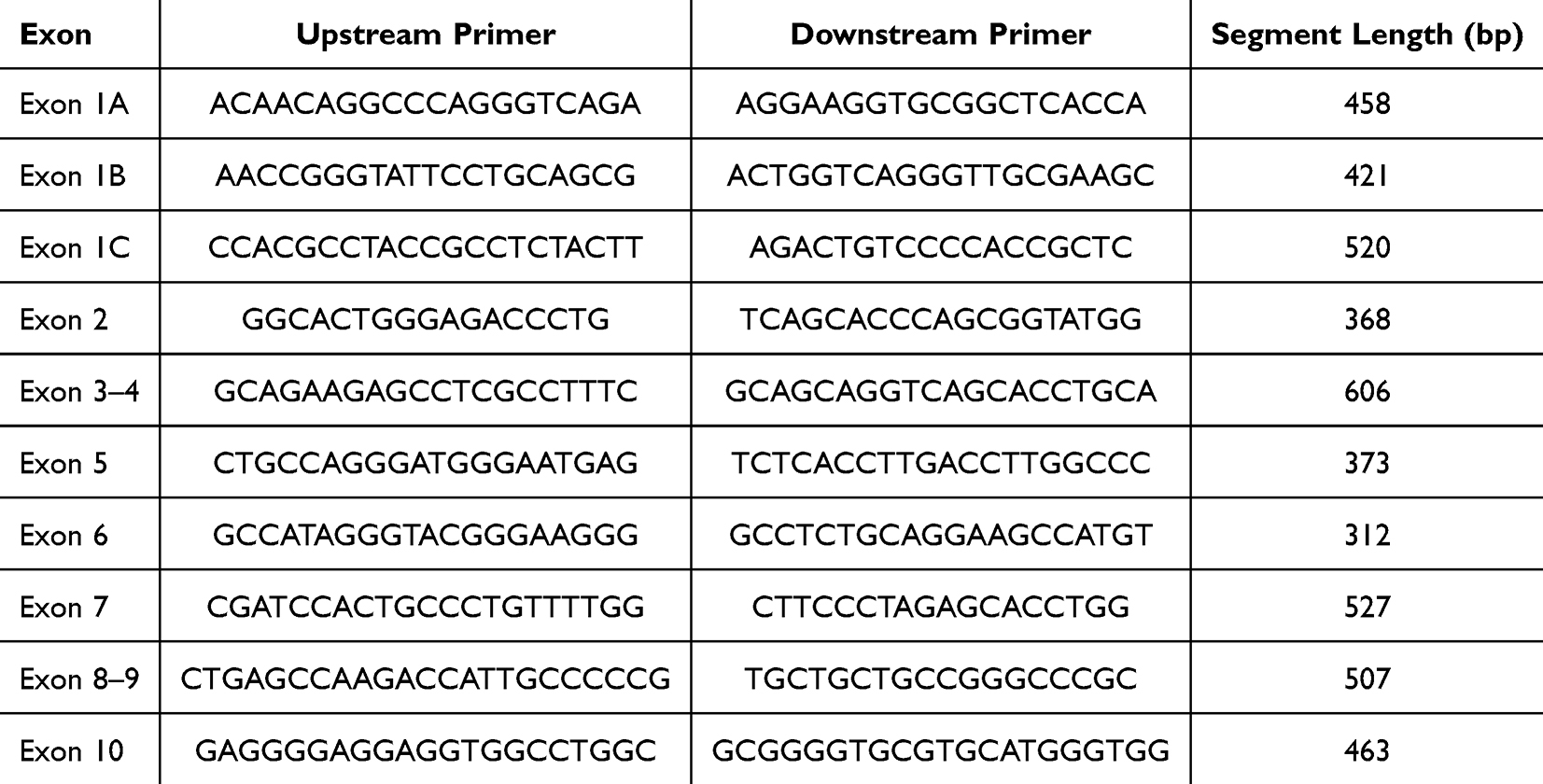 |
Table 1 Gene Primer Sequences |
Identified variants were validated through searches on established gene databases (http://www.hgmd.org/ and http://www.x-ald.nl/) and a review of relevant published literature.
Results
Basic Data
All 14 pediatric patients with X-ALD were male, with age of onset ranging from 5 to 10 years, and an average age at onset of 6 years and 11 months.
Clinical Characteristics
Detailed clinical characteristics of the 14 pediatric patients with X-ALD are shown in Table 2. All patients met the diagnostic criteria for CCALD. Initial symptoms in most cases included declining academic performance, inattention, and visual or auditory loss, while some presented with motor disorders, mental abnormalities, epilepsy, or unclear speech, with the disease progressing rapidly. Nine pediatric patients experienced progressive visual impairment, such as vision loss or visual field abnormalities, while three exhibited hearing loss. Intellectual and cognitive regression occurred in 10 cases, with one case showing mental abnormalities and another experiencing seizures. Early motor impairments included gait instability or limb weakness, which worsened as the disease advanced. Additionally, two patients displayed skin pigmentation. Chronic adrenocortical insufficiency was diagnosed in two pediatric patients, both requiring long-term treatment with hydrocortisone.
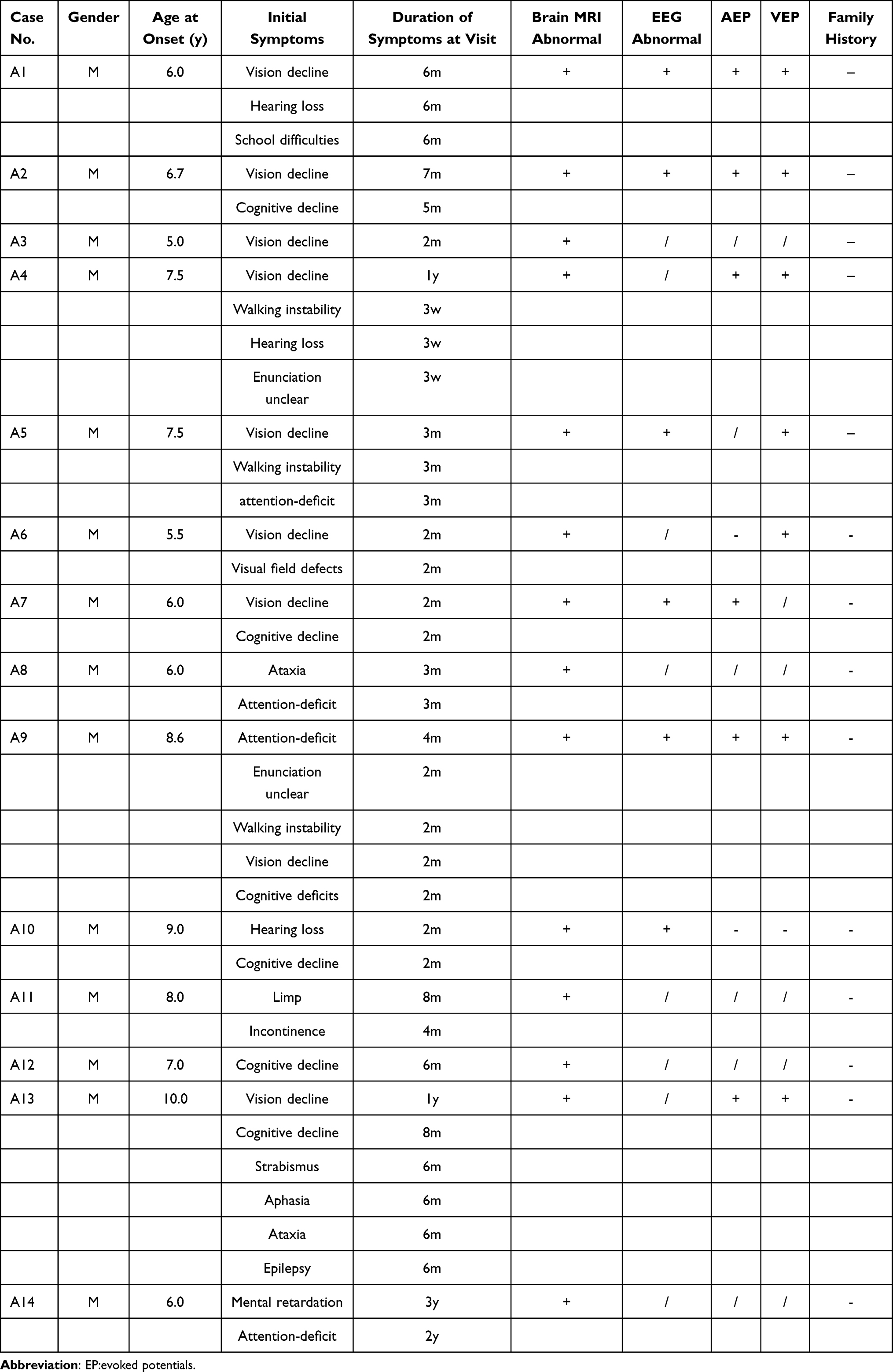 |
Table 2 Summary of Clinical Features of the 14 Children with X-ALD |
Features of Brain MRI
MRI of the brain in the 14 pediatric patients revealed characteristic “butterfly wing” patterns of white matter demyelination. Lesions were localized in the parietal and occipital lobes, gradually extending to the frontotemporal lobe, with frequent involvement of the brainstem and corpus callosum. MRI results indicated abnormal signal intensities, presenting as increased signals on T2-weighted and FLAIR sequences, decreased signals on T1-weighted sequences, and enhanced signals on T2 and FLAIR sequences, as illustrated in Figure 1.
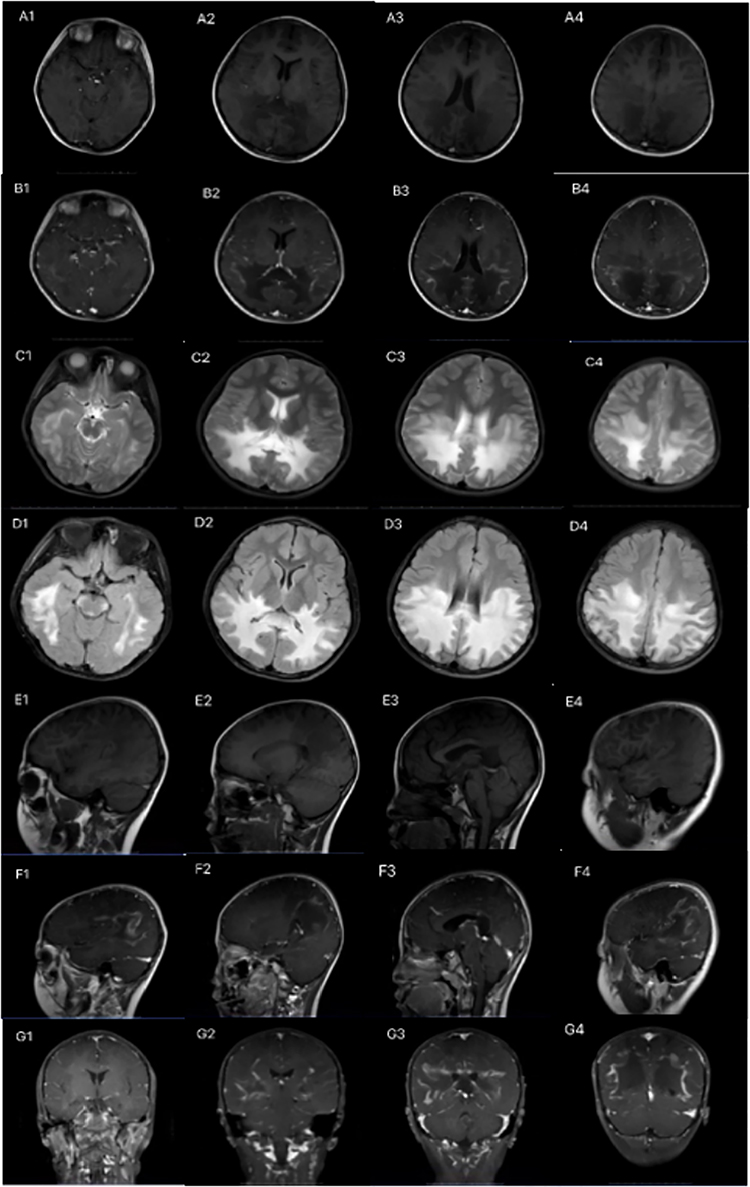 |
Figure 1 Brain MRI findings in pediatric patients with X-ALD. The images include: (A1–A4) Axial T1-weighted (T1W) images; (B1–B4) Axial enhanced T1W images; (C1–C4) Axial T2-weighted (T2W) images; (D1–D4) Axial T2 FLAIR images; (E1–E4) Sagittal T1W images; (F1–F4) Sagittal enhanced T1W images; and (G1–G4) Coronal enhanced T1W images. The MRI findings reveal symmetrical, patchy abnormal signals predominantly located in the white matter, particularly in the parietal-occipital region, brainstem, thalamus, and corpus callosum. These abnormalities appear as low intensity on T1W images and high intensity on T2W and T2-FLAIR images. Notably, the enhanced T1W images demonstrate marginal enhancement of the lesions. |
EEG Characteristics
Electroencephalogram (EEG) analysis in six pediatric patients revealed diffuse slow wave activity, particularly in the parietal and occipital regions.
Evoked Potential Abnormalities
Brainstem auditory evoked potential (BAEP) were abnormal in six cases, characterized by prolonged wave III latency (PL), as well as extended latencies for waves I–III (IPL) and I–V. Visual evoked potential (VEP) testing was conducted in eight cases, with seven cases exhibiting prolonged P-wave latency, along with poor differentiation and repeatability in bilateral flash visual evoked potentials (F-VEP).
Plasma VLCFA Level
All pediatric patients exhibited significantly elevated serum levels of VLCFA; C26:0, C24:0/C22:0, and C26:0/C22:0. Additionally, some patients exhibited abnormal levels of adrenocorticotropic hormone (ACTH) and cortisol (Table 3).
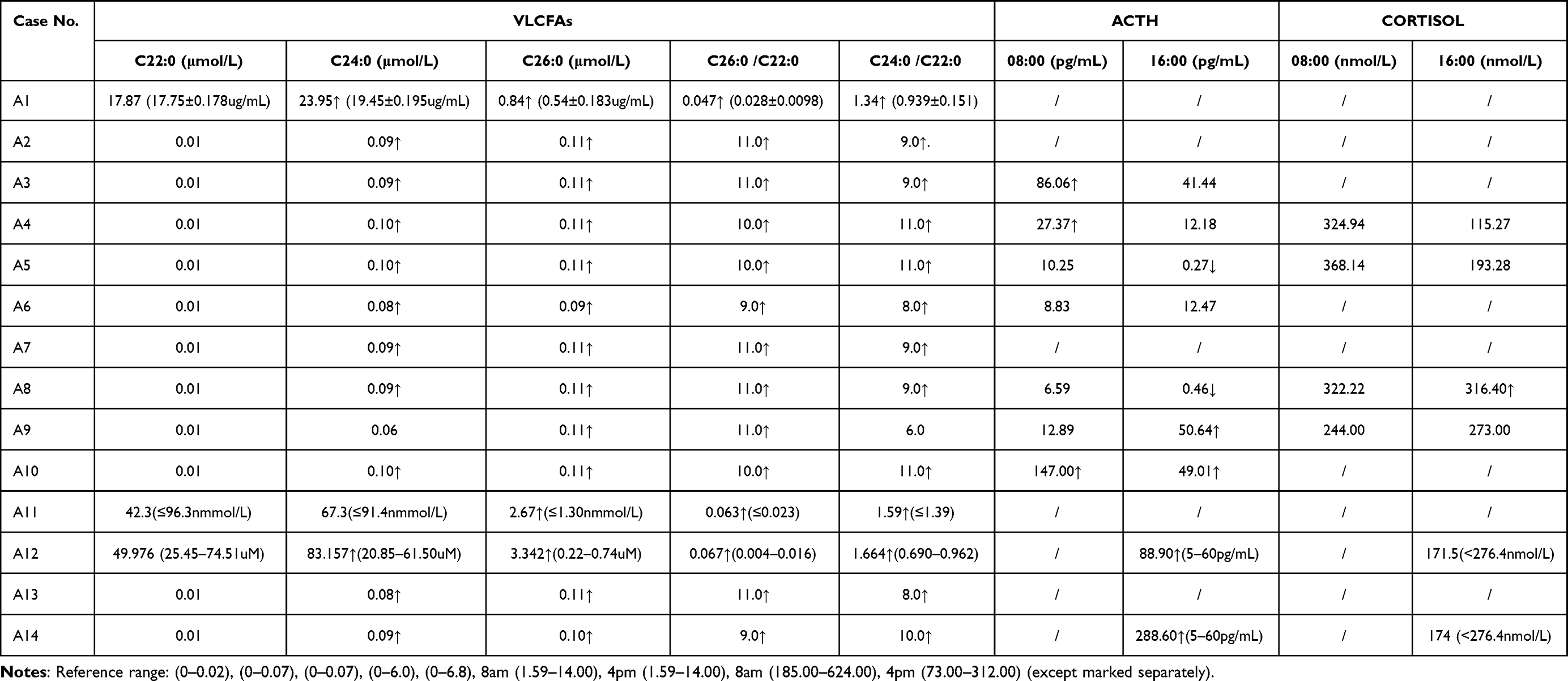 |
Table 3 Results of Laboratory Tests of 14 Children with X-ALD |
Mutation Data Analysis
Genetic testing performed on all 14 pediatric patients identified 12 distinct mutations in the ABCD1 gene (Table 4). The majority of these mutations were missense mutations, with one case presenting a frameshift mutation. Mutation sites were primarily concentrated in exon 6 (4 out of 14, 28.6%), followed by exons 1, 8, and 9. Figure 2 illustrates the gene locations and the corresponding coding amino acid domains for each mutation site. All identified mutations have been previously reported in the literature. The 14 pediatric patients were from 14 unrelated families.
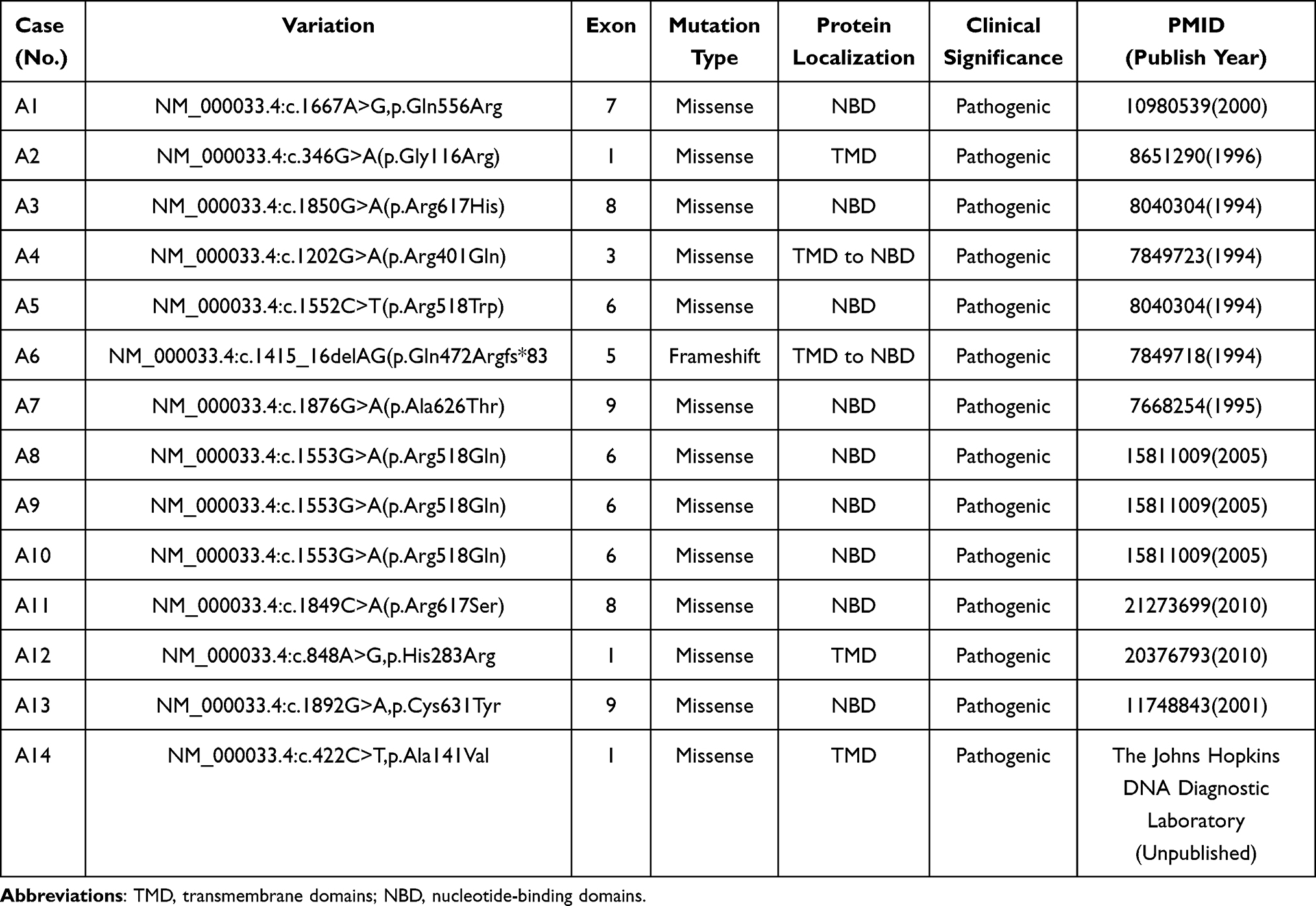 |
Table 4 ABCD1 Gene Mutations Identified in 14 Children with X-ALD |
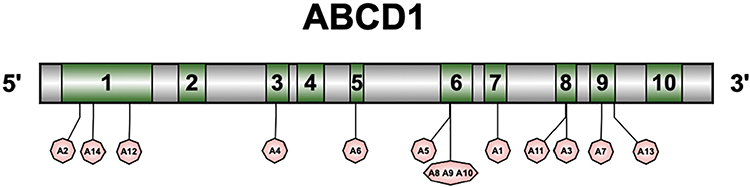 |
Figure 2 Diagram showing the location of each gene mutation locus in the 14 pediatric patients with X-ALD. |
Discussion
X-ALD is a peroxisomal metabolism disorder caused by mutations in the ABCD1 gene, located on Xq28. The gene spans 19.9 kb, consists of 10 exons, and encodes a peroxisomal transmembrane protein composed of 745 amino acids, functioning as an ATP-binding cassette (ABC) transporter known as adrenoleukodystrophy protein (ALDP).10 This protein shares structural similarities with the cystic fibrosis transmembrane conductance regulator (CFTR) protein.11,12 ALDP facilitates the entry of VLCFA into the peroxisome, likely in the form of coenzyme A (CoA) esters.13 Mutations in the ABCD1 gene may disrupt the normal transport of VLCFA, inhibiting their β-oxidation and subsequent degradation. The resulting accumulation of abnormal VLCFA in affected tissues, such as the central nervous system (CNS), testicular interstitial cells, and adrenal cortex, is believed to underlie the pathological changes associated with X-ALD.14 The distribution of ALDP corresponds to regions with high metabolic activity, including the heart, skeletal muscle, liver, and critical neural areas such as subcortical and cerebellar white matter, the hypothalamus, ACTH-producing cells in the pituitary gland, and the dorsal root ganglia (DRG).
As of December 2022, the ABCD1 variant database (https://adrenoleukodystrophy.info/) has documented 3,717 cases, which encompass 262 likely pathogenic variants, 41 benign variants, and 167 variants of uncertain significance (VUS). Among these, missense pathogenic variants represent the most prevalent, accounting for 63.7% of the total. Other types of variants include frameshift (15.6%), nonsense (9.3%), splice site (4.1%), amino acid insertions/deletions (3.4%), deletions of one or more exons (2.5%), and variants resulting in no translation initiation (1.5%).
Pathogenic variants in ABCD1 are predominantly localized within the transmembrane domain, comprising 47% of variants found in exons 1 and 2. This is followed by variants within the ATP-binding domain, representing 34% and primarily localized to exons 6–9. Additionally, exons 3 and 4 contribute 9% of the pathogenic variants, exon 5 contributes 7%, and exon 10 contributes 3%. The most frequently observed pathogenic variant is a 2-base pair deletion in exon 5, p.Gln472Argfs*83, identified in 177 cases accounting for 4.8% of all variants. Other prevalent pathogenic missense variants include p.Arg554His (n = 70, 1.9%), p.Arg660Trp (n = 64, 1.7%), p.Arg617His (n = 59, 1.6%), and p.Arg518Gln (n = 54, 1.5%).
In our study, we identified 12 distinct types of mutations in the ABCD1 gene, all previously reported in literature. Of these mutations, 9 were located in exons 6 to 9, and 3 were found in exons 1 and 2. Specific mutations included one case of the exon 5 mutation p.Gln472Argfs*83, one case of the exon 8 mutation p.Arg617His, one case of the exon 8 mutation p.Arg617Ser, and three cases of the exon 6 mutation p.Arg518Gln. These findings differ from existing datasets, which may be attributed to the limited sample size in our study.
X-ALD can manifest at any age, from childhood to adulthood, characterized by the progressive demyelination of white matter and adrenal insufficiency.15,16 The condition exhibits seven distinct phenotypes: CCALD, adolescent cerebral ALD, adult cerebral ALD (ACALD), adrenomyeloneuropathy (AMN), simple Addison’s disease type, asymptomatic type, and heterozygous type.
The earliest cases of X-ALD were documented in the late 19th century, when Heubner reported a case involving a young boy with rapid neurological deterioration, later classified as “diffuse sclerosis” upon autopsy.10 CCALD typically presents between the ages 4 and 8, with a peak incidence around age 7, and constitutes approximately 35% of all phenotypes within the ALD/AMN complex.17 The symptoms associated with cerebral ALD are contingent upon the location of the initial lesions. The primary manifestations include personality changes, intellectual and motor regression, and audio-visual dysfunction, with inattention frequently identified as the first symptom.
In the present study, all 14 pediatric involved CCALD, likely due to the focus on pediatric cases within our hospital and the limited sample size. The most common initial symptoms were decreased academic performance, inattention, as well as vision and hearing loss. Brain MRI imaging revealed typical “butterfly-wing” patterns of white matter demyelination. Laboratory tests showed elevated VLCFA levels, and evidence of adrenal cortical insufficiency, as indicated by increased ACTH levels. Timely hormone replacement therapy is essential for managing adrenal insufficiency. In this study, pediatric patients with adrenal insufficiency were administered oral hydroprednisone, and regular follow-up visits were scheduled with the endocrinology department. Mechanismly, the ABCD1 gene encodes a peroxisomal ABC transporter responsible for VLCFA degradation. Specific mutations in ABCD1 may variably impair this function, leading to differential VLCFA accumulation and contributing to phenotypic diversity in X-ALD. For example, missense mutations in critical domains could partially retain transporter function, resulting in milder phenotypes, while frameshift or nonsense mutations typically cause severe loss of function and more aggressive disease courses. Additionally, the phenotypic variability may also be influenced by genetic modifiers and environmental factors. Further studies are needed to elucidate the precise mechanisms underlying these genotype-phenotype correlations.
Currently, no specific pharmacological treatments are available for X-ALD. HSCT remains the most effective intervention for CCALD. HSCT can halt the progression of brain demyelination in pediatric patients diagnosed with early CCALD, but is ineffective for advanced CCALD and adult onset AMN.8,18 Consequently, early diagnosis is crucial for optimizing treatment outcomes. In 1990, Aubourg et al reported the first successful allogeneic HSCT in a pediatric patient with X-ALD.8 A retrospective study compared 30 patients with early cerebral ALD who did not undergo transplantation to 19 patients with early cerebral ALD who received transplantation, matching subjects based on neurological disability and MRI severity scores.19 The findings indicated that the five-year survival rate was significantly higher in the transplant group compared to the non-transplant group (95% vs 54%). As a potential alternative for pediatric patients with CCALD who lack an HLA-matched donor, gene therapy may represent a viable treatment option.20,21
The key to effectively managing X-ALD lies in early detection and treatment. However, the early clinical symptoms often lack specificity, complicating the diagnostic process. Therefore, neonatal screening is vital for these patients. In recent years, advancements in diagnostic techniques, including tandem mass spectrometry, gas chromatography-mass spectrometry, and next-generation sequencing, have facilitated rapid and accurate identification of X-ALD, enabling neonatal screening and prenatal diagnosis. At our hospital, early diagnosis is achieved through VLCFA detection in peripheral blood, providing essential information for subsequent diagnosis and treatment of X-ALD. In the United States, neonatal screening for X-ALD was added to the Recommended Uniform Screening Panel (RUSP) in 2016, supporting early detection of ABCD1 mutations.20,21
The characteristics of CCALD include inflammatory demyelination, typically starting in the occipitoparietal regions and progressing to the frontal or temporal lobes. The lesions sometimes extend to the brainstem, particularly affecting the pons.22 Brain MRI is the only method for identifying early CCALD. For children diagnosed with X-ALD, it is recommended to undergo head MRI scans every six months from ages 3 to 12, and annually before age 3 and after age 12.23 Furthermore, long-term clinical follow-up management is crucial for pediatric patients diagnosed with X-ALD. Engelen et al employed a consensus-based modified Delphi approach among 28 international ALD experts to formulate best-practice recommendations for the diagnosis, clinical surveillance, and treatment of pediatric patients ALD.24
Despite providing valuable insights into pediatric X-ALD, this study has notable limitations. The study’s small sample size limits statistical power and generalizability of findings, a common challenge in rare disease research. Additionally, the regional focus reduces population representativeness. Future larger, multicentric studies are needed to validate these results and enhance understanding of X-ALD’s diverse manifestations.
Conclusion
This study provides a comprehensive analysis of the clinical characteristics and genetic mutations in 14 pediatric cases of X-ALD, highlighting the atypical presentation of early symptoms, which often leads to diagnostic challenges. Our findings emphasize the importance of vigilant monitoring for male pediatric patients presenting with symptoms resembling ADHD, as these may be early indicators of X-ALD and could result in misdiagnosis if not properly evaluated. We confirm that timely detection through blood VLCFA measurement and brain MRI is critical for early diagnosis, with ABCD1 gene testing recommended as a definitive diagnostic tool, especially when VLCFA levels are inconclusive. Our results also underscore the necessity of genetic counseling based on ABCD1 mutation analysis to guide family planning and management. Given the complexities of X-ALD, we advocate for a multidisciplinary approach to diagnosis and treatment, with a focus on early identification and continuous follow-up to optimize the timing of interventions, such as HSCT, for eligible patients.
Abbreviations
X-ALD, X-adrenoleukodystrophy; CCALD, childhood cerebral adrenoleukodystrophy; MRI, magnetic resonance imaging; HSCT, Hematopoietic stem cell transplantation; VLCFA, Very long chain fatty acids; ACTH, adrenocorticotropic hormone; EEG, electroencephalogram; GC-MS, gas chromatography-mass spectrometry; VEP, Visual evoked potential.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
The study was conducted in accordance with the Declaration of Helsinki (as was revised in 2013). The study was approved by Ethics Committee of the Third Medical Center of the Shanghai Children’s Hospital, School of Medicine, Shanghai Jiao Tong University (No.2019R071-F03). All participants provided written informed consent for their involvement in the study. In cases where participants were under the age of 18, informed consent was additionally obtained from their parents or legal guardians.
Acknowledgments
We are grateful to the patients and their families for their participation in this study.
Funding
Shanghai key clinical specialty project (shslczdzk05705), Scientific Research Fund of China Association Against Epilepsy (CJ-A-2021-07), Hospital level project of Shanghai Children’s Hospital (2016YMS005), National Fund Cultivation Special Project of Shanghai Children’s Hospital (2021YGZQ05), Scientific Research Fund of China Association Against Epilepsy (CX-2022-013), Clinical Research Cultivation Special Project of Shanghai Children’s Hospital (2022YLYM07), Scientific Research Fund of China Association Against Epilepsy (CU-2023-036).
Disclosure
The authors declare that they have no competing interests.
References
1. Morita M, Imanaka T. Peroxisomal ABC transporters: structure, function and role in disease. Biochim Biophys Acta. 2012;1822(9):1387–1396. doi:10.1016/j.bbadis.2012.02.009
2. Bezman L, Moser AB, Raymond GV, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49(4):512–517. doi:10.1002/ana.101
3. Moser AB, Jones RO, Hubbard WC, et al. Newborn screening for X-Linked Adrenoleukodystrophy. Int J Neonatal Screen. 2016;2(4):15. doi:10.3390/ijns2040015
4. Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3(3):140–151. doi:10.1038/ncpneuro0421
5. Wiesinger C, Eichler FS, Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Appl Clin Genet. 2015;8:109–121. doi:10.2147/TACG.S49590
6. Igarashi M, Schaumburg HH, Powers J, Kishmoto Y, Kolodny E, Suzuki K. Fatty acid abnormality in adrenoleukodystrophy. J Neurochem. 1976;26(4):851–860. doi:10.1111/j.1471-4159.1976.tb04462.x
7. Mahmood A, Dubey P, Moser HW, Moser A. X-linked adrenoleukodystrophy: therapeutic approaches to distinct phenotypes. Pediatr Transplant. 2005;9(Suppl 7):55–62. doi:10.1111/j.1399-3046.2005.00447.x
8. Aubourg P, Blanche S, Jambaqué I, et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med. 1990;322(26):1860–1866. doi:10.1056/NEJM199006283222607
9. Moser HW, Raymond GV, Dubey P. Adrenoleukodystrophy: new approaches to a neurodegenerative disease. JAMA. 2005;294(24):3131–3134. doi:10.1001/jama.294.24.3131
10. Kemp S, Theodoulou FL, Wanders RJ. Mammalian peroxisomal ABC transporters: from endogenous substrates to pathology and clinical significance. Br J Pharmacol. 2011;164(7):1753–1766. doi:10.1111/j.1476-5381.2011.01435.x
11. Mosser J, Douar AM, Sarde CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:6414):726–730. doi:10.1038/361726a0
12. Holzinger A, Kammerer S, Berger J, Roscher AA. cDNA cloning and mRNA expression of the human adrenoleukodystrophy related protein (ALDRP), a peroxisomal ABC transporter. Biochem Biophys Res Commun. 1997;239(1):261–264. doi:10.1006/bbrc.1997.7391
13. van Roermund CW, Visser WF, Ijlst L, et al. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008;22(12):4201–4208. doi:10.1096/fj.08-110866
14. Kemp S, Wanders R. Biochemical aspects of X-linked adrenoleukodystrophy. Brain Pathol. 2010;20(4):831–837. doi:10.1111/j.1750-3639.2010.00391.x
15. Moser HW, Loes DJ, Melhem ER, et al. X-Linked adrenoleukodystrophy: overview and prognosis as a function of age and brain magnetic resonance imaging abnormality. A study involving 372 patients. Neuropediatrics. 2000;31(5):227–239. doi:10.1055/s-2000-9236
16. Kemp S, Huffnagel IC, Linthorst GE, Wanders RJ, Engelen M. Adrenoleukodystrophy - neuroendocrine pathogenesis and redefinition of natural history. Nat Rev Endocrinol. 2016;12(10):606–615. doi:10.1038/nrendo.2016.90
17. Raymond GV, Moser AB, Fatemi A. X-linked adrenoleukodystrophy. In GeneReviews (Last updated February 15, 2018). Available from: https://www.ncbi.nlm.nih.gov/books/NBK1315/.
18. Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118(7):1971–1978. doi:10.1182/blood-2011-01-329235
19. Mahmood A, Raymond GV, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol. 2007;6(8):687–692. doi:10.1016/S1474-4422(07)70177-1
20. A Summary of the Evidence and Advisory Committee Decision. Newborn Screening for X-linked Adrenoleukodystrophy. Available from: https://www.hrsa.gov/sites/default/files/hrsa/advisory- committees/heritable-disorders/rusp/x-ald-27-june-2018.pdf.
21. Kemper AR, Brosco J, Comeau AM, et al. Newborn screening for X-linked adrenoleukodystrophy: evidence summary and advisory committee recommendation. Genet Med. 2017;19(1):121–126. doi:10.1038/gim.2016.68
22. Liberato AP, Mallack EJ, Aziz-Bose R, et al. MRI brain lesions in asymptomatic boys with X-linked adrenoleukodystrophy. Neurology. 2019;92(15):e1698–e1708. doi:10.1212/WNL.0000000000007294
23. Mallack EJ, Turk BR, Yan H, et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: meta-analysis and consensus guidelines. J Inherit Metab Dis. 2021;44(3):728–739. doi:10.1002/jimd.12356
24. Engelen M, van Ballegoij WJC, Mallack EJ, et al. International recommendations for the diagnosis and management of patients with adrenoleukodystrophy: a consensus-based approach. Neurology. 2022;99(21):940–951. doi:10.1212/WNL.0000000000201374
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.