")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 17
Glutaminolysis is a Potential Therapeutic Target for Kidney Diseases
Authors Ou LP , Liu YJ, Qiu ST, Yang C , Tang JX, Li XY, Liu HF, Ye ZN
Received 6 May 2024
Accepted for publication 15 July 2024
Published 23 July 2024 Volume 2024:17 Pages 2789—2807
DOI https://doi.org/10.2147/DMSO.S471711
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Li-Ping Ou,* Yong-Jian Liu,* Shi-Tong Qiu,* Chen Yang, Ji-Xin Tang, Xiao-Yu Li, Hua-Feng Liu, Zhen-Nan Ye
Institute of Nephrology, and Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-Communicable Diseases, and Key Laboratory of Prevention and Management of Chronic Kidney Disease of Zhanjiang City, Affiliated Hospital of Guangdong Medical University, Zhanjiang, 524001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhen-Nan Ye; Hua-Feng Liu, Email [email protected]; [email protected]
Abstract: Metabolic reprogramming contributes to the progression and prognosis of various kidney diseases. Glutamine is the most abundant free amino acid in the body and participates in more metabolic processes than other amino acids. Altered glutamine metabolism is a prominent feature in different kidney diseases. Glutaminolysis converts glutamine into the TCA cycle metabolite, alpha-ketoglutarate, via a cascade of enzymatic reactions. This metabolic pathway plays pivotal roles in inflammation, maladaptive repair, cell survival and proliferation, redox homeostasis, and immune regulation. Given the crucial role of glutaminolysis in bioenergetics and anaplerotic fluxes in kidney pathogenesis, studies on this cascade could provide a better understanding of kidney diseases, thus inspiring the development of potential methods for targeted therapy. Emerging evidence has shown that targeting glutaminolysis is a promising therapeutic strategy for ameliorating kidney disease. In this narrative review, equation including keywords related to glutamine, glutaminolysis and kidney are subjected to an exhaustive search on Pubmed database, we identified all relevant articles published before 1 April, 2024. Afterwards, we summarize the regulation of glutaminolysis in major kidney diseases and its underlying molecular mechanisms. Furthermore, we highlight therapeutic strategies targeting glutaminolysis and their potential clinical applications.
Keywords: glutamine, glutaminolysis, kidney diseases, therapeutic target
Introduction: Definition and General Roles
Glutamine is considered as an unessential amino acid in physiological condition, and highly utilized in kidney, intestine, and immune compartment.1 Concentration of circulating glutamine fluctuates around 500 to 800 μM/L.2 As a ready source in a variety of biochemical processes, glutamine plays a fundamental role in cell metabolism.
Glutaminolysis is a metabolic pathway that involves glutamine uptake and catabolism to maintain bioenergetics and replenish macromolecular biosynthesis. As illustrated in Figure 1. glutaminolysis starts with the transportation of extracellular glutamine into the cytoplasm by membrane proteins (SLC1A5, SLC38A1, SLC38A2) and subsequently into the mitochondria by SLC1A5 var. In the mitochondria, glutamine (Gln) is broken down into glutamate (Glu) and ammonia by two isoenzymes of glutaminase, designated GLS1 and GLS2, which were initially found in the kidney and liver, respectively. GLS1 and GLS2 encode two transcripts. Isozymes of Gls1 are termed kidney glutaminase A (KGA) and glutaminase C (GAC), whereas those of GLS2 include liver glutaminase A (LGA) and glutaminase B (GAB). A portion of glutamate is then converted to α-ketoglutarate (α-KG) by two different pathways: the first through the activity of glutamate dehydrogenase (GDH) and the second through the activity of a group of aminotransferases, including glutamate-oxaloacetate transaminase (GOT), glutamate-pyruvate transaminase (GPT), and phosphoserine transaminase (PSAT).3
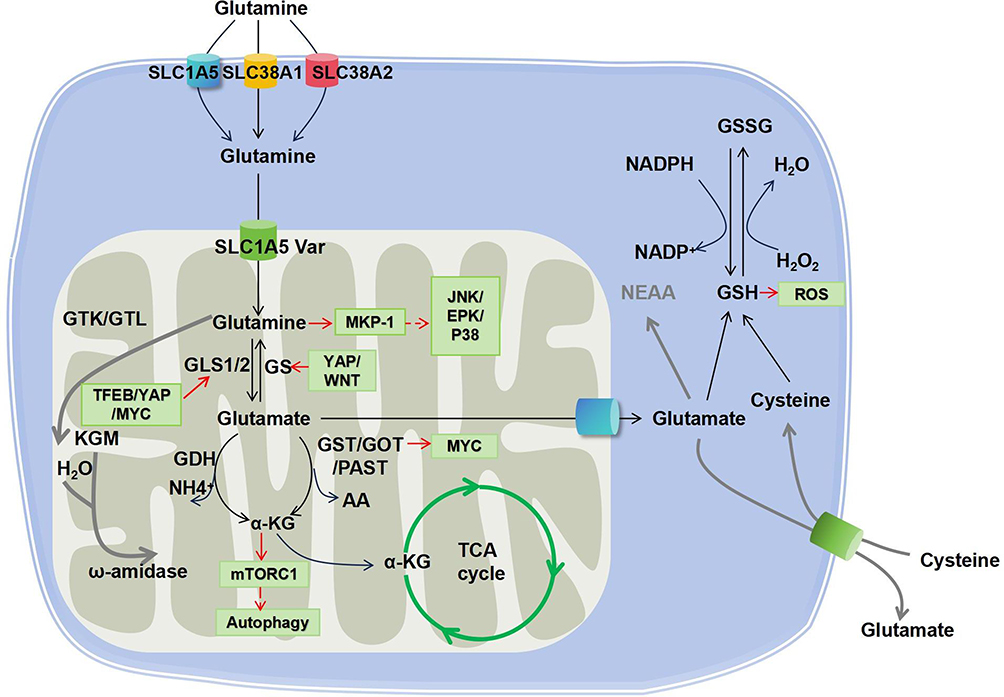 |
Figure 1 Glutaminolysis cascade and the interlinked signaling pathways. Glutamine is taken up into the cytosol by membrane transporters SLC1A5, SLC38A1 and SLC38A2. Afterwards, glutamine is transported to the mitochondrial matrix by SLC1A5 variants and further converted to glutamate by GLS, which is a rate-limiting reaction of glutaminolysis. Glutamate is then catalyzed by either GDH or transaminases (GOT, GST, or PAST) to generate α-KG. Besides, glutamine can also be metabolized by glutaminase II pathway mediated by enzymes including GTK (or GTL) and ω-amidase, as highlighted by the red line. Consequently, α-KG enters the TCA cycle. In addition, glutamate can be exported to the cytoplasm to participate in the glutathione metabolism (GSH/GSSG redox reaction). Abbreviations: GLS, glutaminase; GTK, glutamine transaminase K; GLT, glutamine transaminase L; KGM, α-ketoglutaramate; GS, glutamine synthetase; GOT, glutamate-oxaloacetate transaminase; GPT, glutamate–pyruvate transaminase; PAST, phosphoserine transaminase; α-KG, α-ketoglutarate; TCA, tricarboxylic acid; GSH, Glutathione; GSSG, Glutathione oxidized; SLC1A5, Solute Carrier Family 1 Member 5; SLC38A1, Solute Carrier Family 38 member 1; SLC38A2, Solute Carrier Family 38 member 2; SLC7A11, Solute Carrier Family 7 member 11. |
It is noteworthy that, while the above-described reaction is normally called the glutaminase I pathway, another cascade called the glutaminase II pathway (more recently referred to as the glutamine transaminase-ω-amidase (GTωA) pathway) also exists. In this reaction, glutamine is first transaminated to α-ketoglutaramate (KGM) by glutamine transaminase K (GTK) or glutamine transaminase L (GTL), and further hydrolyzed by ω-amidase to α-ketoglutarate.4,5 Glutaminolysis is known for its anaplerotic role in neoplastic cell proliferation. Moreover, emerging evidence has shown that it participates in many other pathological and physiological processes, including fibrogenesis, ischemic injury, oxidative insult, and aging, as well as in cells in demand for high energy, such as kidney and corneal endothelium.6,7
Glutaminolysis plays a crucial role in regulating redox homeostasis by supplying glutamate for the synthesis of glutathione (GSH), which serves as a protective mechanism against oxidative stress.8 However, it has also been reported that increased glutaminolysis can divert glutamine/glutamate from glutathione synthesis, resulting in oxidative insults.8 Moreover, ammonia is generated during the glutaminolysis of glutamate. Free ammonia is released from the breakdown of glutamine, which plays a crucial role in maintaining renal pH homeostasis, and subsequently contributes to cellular and systemic homeostasis.9 Glutaminolysis also plays a role in immune regulation. Inhibiting glutamine metabolism impacts the production of monocyte-derived cytokines (TNF, IL-6, and IL-1β) as well as T-cell-derived cytokines (IFN-γ and IL-22).10 Additionally, GLS1 can modulate immune cells, and its aberrant expression may contribute to various autoimmune diseases, including SLE.11,12 On the other hand, glutaminolysis is closely associated with epigenetic regulation and post-translational modification. Glutamine metabolism can enhance DNA methylation and histone acetylation,13,14 while interestingly, acetyl-CoA-mediated suppression of glutaminase activity has been observed.15
In conclusion, glutaminolysis plays a crucial role in maintaining the homeostasis of the body. Currently, research on glutaminolysis is mainly limited to various types of cancer, with relatively little in-depth study in kidney diseases. However, it has been reported that glutaminolysis is associated with renal fibrosis,16 diabetic nephropathy,17 acute kidney injury,18 systemic lupus erythematosus,19,20 renal cell carcinoma,21 anti-neutrophil cytoplasmic antibody-associated vasculitis (AAV), and nephrotic syndrome (NS)22 as well as polycystic kidney disease.23 Therefore, this paper will explore the current role of glutaminolysis in kidney diseases and related mechanistic studies to help develop new therapies for kidney diseases in the future.
Molecular Mechanisms: Key Factors and Signaling Pathways Related to Glutaminolysis
Upstream of Glutaminolysis
c-Myc
Myc, also known as c-Myc, serves as a transcription factor in various signaling pathways, including its involvement in glutaminolysis.24 Previous studies demonstrated a positive correlation between c-Myc and GLS1 expression. Wise et al discovered that upon introducing an inducible Myc transgene into mouse embryonic fibroblasts (MEF), GLS1 mRNA levels were noticeably increased owing to the induction of Myc expression.25 Similar findings have been observed in various cell lines, such as P493-6 and PC3,26 SW620 cells,27 U-1906 cells,28 and Myc-induced liver tumors in mice. Furthermore, c-MYC expression exerts influence the transcriptional level of GLS1 in both colorectal cancer and liver tumors.29
Gao et al discovered that Myc suppressed the activity of miR-23, which targets GLS, and concurrently upregulated the expression of mitochondrial glutamine synthetase to enhance glutaminolysis during their investigation into Myc-regulated mitochondrial proteins.26 However, they did not observe any indications of c-Myc transcriptionally regulating GLS1 despite the presence of a typical E-box sequence in its first intron.26 Secondly, they found that MYC controlled GLS1 expression post-transcriptionally through microRNAs miR-23a and miR-23b in PC3, P493-6, human lymphoid CB33 and breast cancer MCF-7 cells.26 In contrast, Haikala et al proposed that the impact of c-Myc on GLS1 expression was due to transcriptional regulation by binding to the promoter region near the 5’-UTR transcriptional initiation site.30 In summary, in addition to indirectly regulating GLS1 via miR-23a/b, MYC can directly influence its expression by interacting with the promoter region of GLS1.
In addition, the current research presents conflicting findings regarding the influence of MYC on GLS2 expression. Some studies have suggested a decrease in GLS2 protein levels in renal cell carcinoma induced by MYC,31 while an association between MYC and GLS2 has been observed in T cells.29
Additionally, the expression of glutamic acid dehydrogenase and glutamate-dependent aminotransferases is modulated by c-MYC. Shroff et al observed an elevation in GLUD1 protein levels in transgenic mice harboring human MYC-driven clear cell renal cell carcinoma as well as a reliance of GOT1 protein levels on MYC.32 The precise mechanism by which MYC regulates GLUD1 and GOT1 remains elusive; however, MYC has been suggested to exert its influence at the transcriptional level on GLUD1 and GOT1 expression.33
YAP
The Hippo signaling pathway plays multiple roles in renal physiology, including determining cell fate, tissue regeneration, and regulating kidney size to maintain homeostasis. YAP is a necessary protein involved in the transduction of the Hippo signal.34
YAP and GLS1
In liver fibrosis, upregulation of glutaminolysis is observed during transdifferentiation, which is driven by the induction of GLS1 mediated by YAP.35 The regulatory mechanism by which YAP controls GLS1 expression was investigated by Du et al who discovered that YAP collaborates with TAZ, a related transcription factor, to modulate the expression of genes containing TEAD binding sites.35 This finding aligns with that of Bertero et al, who confirmed the presence of TEAD/YAP-binding sites within the promoter region of GLS1 using ChIP-qPCR analysis.36 Additionally, QPCR experiments revealed that simultaneous knockout of YAP and TAZ resulted in a reduction in GLS1 expression in PAEC and PASMC. These results suggest a cooperative role of YAP and TAZ in the regulating of GLS1.36
YAP and Glutamine Synthetase
Transcriptomic and metabolomic analyses revealed that YAP1 influences glutamine metabolism by upregulating the expression and activity of glutamine synthetase (GS; encoded by GLUL).37 Moreover, GLUL activation facilitates the utilization of ammonia-bound nitrogen to produce glutamine as a precursor for nucleotide biosynthesis, thereby modulating nitrogen metabolism.37
YAP and GOT1
Yang et al examined the correlation between TAZ/YAP activity and genes involved in glutamine metabolism in breast cancer biopsies. They observed a robust positive association between TAZ/YAP activity and GOT1 and PSAT1 expression. Moreover, using ChIP analysis in MDA-MB-3 cells, they identified potential binding sites for YAP/TAZ and TEAD transcription factors within the enhancer region of GOT1 gene.38
In addition, YAP is closely related to myc signaling. For example, a recent study demonstrated that high blood glucose leads to hyperactivation of YAP, further stabilizing myc, and consequently resulting in abnormal alteration of mesangial cells in diabetic nephropathy.39
Wnt/β-Catenin Pathway
The Wnt/β-catenin pathway, known for its involvement in glutaminolysis40 34771718, plays a pivotal role in the regulation of this metabolic process.41 Previous studies have demonstrated that transgenic mice expressing activated β-catenin in the liver exhibit increased expression of three genes involved in glutaminolysis (GS, OAT, and GLT-1).42 These genes were specifically induced by the activation of the β-catenin pathway in the liver. Furthermore, Zhou et al discovered that during inflammatory injury, pulmonary endothelial cells release Rspondin3 to activate β-catenin signaling in pulmonary interstitial macrophages. Subsequently, this activation enhances glutaminolysis and improves the mitochondrial respiration.43 Collectively, these findings suggest that β-catenin target genes facilitating glutaminolysis.42 However, further research is required to elucidate the mechanisms by which Wnt signalling effectively regulates this process.
Interestingly, various studies have shown that glutamine breakdown can affect the β-catenin pathway. Wong et al discovered that glutaminolysis results in a reduction in DNA demethylation, leading to an increase in WNT signaling, stemness, and drug resistance.44 Furthermore, depriving cancer stem cells with glutamine caused an elevation in cellular reactive oxygen species (ROS) levels by inhibiting glutathione synthesis. This subsequently impedes the β-catenin pathway by inducing phosphorylation and degradation induction.45
TFEB
Transcription factor EB (TFEB), a member of the MiT/TFE family of basic helix-loop-helix leucine zipper transcription factors, is widely acknowledged as a pivotal regulator of lysosomal biogenesis.46 Research indicates that TFEB also participates in glutaminolysis. Ariano et al observed that silencing TFEB resulted in the suppression of glutaminolytic flux in melanoma tumors, as indicated by the reduced catalytic activity of GLS and the level of alpha-ketoglutarate.47 Another study performed in pancreatic cancer reported a downregulation in GLS mRNA transcription after knockdown of TFEB, and chromatin immunoprecipitation assays further revealed the existence of seven E-box motifs within the promoter region of GLS, which is a binding motif for TFEB.48
Downstream of Glutaminolysis
mTORC1
As is commonly acknowledged, the mTOR pathway is significant as a pivotal signaling intersection.49 It is currently hypothesized that mTORC1 activation is associated with glutaminolysis. Duran et al observed a decrease in mTORC1 activity when glutamine metabolism was inhibited by GLS inhibitors in U1OS and HeLa cells.50 They suggested that A-KG, a by-product of glutamine metabolism, activates mTORC1 and facilitates Rag-mediated GTP loading.50 In contrast, Bodineau et al proposed an alternative perspective, stating that mTORC1 is activated by glutamine. They observed a consistent increase in ATP levels after 4 h of treatment with glutamate and leucine in amino acid-deprived cells, leading to AMPK inhibition followed by mTORC1 activation.51 Furthermore, they investigated the impact of ASNS and GLS dual inhibition on mTORC1 using siRNA and BPTES, respectively, in U1OS cells and found that mTOR activity was inhibited only when both were present.51 This suggests that ASNS acts as an alternative pathway for glutamine, affecting both its metabolism and the activation of mTORC1.
In summary, mTORC1 can be activated via two distinct pathways involving glutamine, necessitating further investigation to determine their potential interactions. It is important to note that the relationship between glutamine and mTORC1 is not a straightforward one-way connection. Some studies have also indicated a reciprocal association between MTORC1 and glutamine breakdown, wherein MTORC1 enhances the activity of MYC-GLS and GLUD1, thereby facilitating glutamine decomposition.52,53
Mkp-1
Glutamine (Gln) is a protein with immunomodulatory properties that regulates oxidative stress, inflammation, and apoptosis. MKP-1 plays a crucial role in the inhibition of inflammation by controlling phosphorylation of proteins belonging to the MAPK family. It also contributes to the negative feedback regulation and maintains cellular homeostasis during signal transduction processes.54
Recent research indicates that Gln positively influences MKP-1 expression. For instance, Xuan et al suggested that glutamine might exert a protective effect against neuroinflammation by downregulating MAPK signaling through increased levels of MKP-1 in rats with brain injuries.54 Furthermore, Kim et al demonstrated that Gln modulates the MAPK pathway by upregulating MKP-1 in glutamine-deficient mice, thereby maintaining a balanced inflammatory response. This observation was also reported by Soo-Yeon Jeong55, Otgonzaya Ayush,56 and C-H Lee et al.57
There are diverse perspectives regarding the regulatory role of glutamine in MKP-1. Several studies have proposed that Gln inhibits JNK, ERK, and p38 phosphorylation by upregulating MKP-1, thereby mitigating inflammation.54,58 Conversely, Ayush et al postulated that Gln activates the Ras-ERK-MKP-1 pathway through initial Ca2+ activation as a precise mechanism for Gln-mediated MKP-1 regulation.56 Additionally, in their investigation of LPS-treated mice, Lee et al discovered an interaction between Gln and MKP-1 and demonstrated a physical association between MKP-1 and p-cPLA via immunoprecipitation and in situ orthogonal experiments. Further examination of various allergic asthma phenotypes revealed that Gln can directly deactivate cytosolic phospholipase a2 (cPLA2) through MKP-1-dependent cPLA2 dephosphorylation to alleviate inflammation.57 Interestingly, two viewpoints exist regarding the mechanism of Gln-induced cPLA inactivation. First, cPLA2 dephosphorylation is indirectly achieved through mkp −1 mediated p38 inactivation, which has been extensively studied;57,59 secondly, gln induces direct physical interaction between MKP-1 and p-cPLA2.57
Glutaminolysis and Autophagy
Because glutaminolysis provides metabolic and energy substrates, it is reasonable that autophagy is influenced by this reaction. However, glutaminolysis seems to regulate autophagy in both directions. Glutaminolysis activates mTORC1 and inhibits autophagy. In detail, AKG generated through glutaminolysis,51 Noteworthy, a recent study demonstrated that glutamine could also regulate mTORC1-autophagy independent of glutaminolysis, instead, glutamine is metabolized via asparagine synthesis and the GABA shunt to produce ATP and to inhibit AMPK.
Glutaminolysis in Epigenetic and Post-Translational Modifications
Epigenetic states and post-translational modifications are closely linked to glutaminolysis. A study in colorectal cancer showed that glutaminolysis increased DNA methylation due to the accumulation of succinate and a further reduction in 5-hydroxymethylcytosine-A marker of DNA demethylation, and consequently hypermethylation at CpG sites.44 In contrast, DNA hypermethylation repressed GLS2.13
In addition, since glutaminolysis yields intermediate metabolites such as acetyl-CoA, this acetyl carrier contributes to histone acetylation of RUNX2 promoters, as suggested by a study on calcific aortic valve disease suggested.14 Interestingly, the enzymatic activity of glutaminase is also regulated by acetylation, and enzyme oligomerization increases after acetylation, thereby inhibited activity.15 Afterwards, α-ketoglutarate (α-KG) is produced during glutaminolysis, and it has been proven to regulate gene expression by influencing the epigenetic structure in both normal and cancer cells.60,61 Initially, AKG acts as a cofactor for Jumonji histone demethylases (JHDMs), resulting in decreased JHDM activity when AKG levels are reduced, leading to increased histone methylation.61 Additionally, when AKG is transformed into 2-hydroxyglutaric acid by isocitrate dehydrogenase 1 and 2 following mutation induction, it inhibits DNA demethylation enzymes and histone methylation enzymes that result in lysine residues (K) 9, 27 DNA methylation and histone methylation.62,63
Considering that glutaminolysis is closely related to nutritional imbalance and oxidative stress, it is not surprising that glutaminolysis also regulates phosphorylation via mTOR and other signaling pathways.64 Interestingly, a recent study demonstrated that glutaminolysis mediates microtubule glutamylation in response to extracellular matrix stiffening in tumor microenvironment.65 Other epigenetic modifications are also involved in glutaminolysis, including succinylation,66 lactylation,67 ubiquitination,68 O-linked β-D-N-acetylglucosamine (O-GlcNAc).69
Other Signaling Pathways
Glutaminolysis is also involved in other signaling pathways. A study in senescent stem cells showed that JNK signaling is upstream of glutaminolysis and can upregulate GLS1 transcription.7 However, various studies have indicated that the activation of JNK results in an increase in the accumulation of ROS, a process that is countered by activated P53. Additionally, it has been observed that mutations in the P53 gene can either hinder or enhance the expression of Nrf2.70,71 To effectively eliminate ROS, both Nrf2 and P53 facilitate glutaminolysis, thereby providing glutamate and NADPH for GSH production.72
In addition, evidence has shown that glutaminolysis serves as an anti-apoptotic player. For example, in diabetes-induced podocyte injury, glutaminolysis can improve mitochondrial function and counteract apoptosis.72 In ischemia/reperfusion-induced acute kidney injury, apoptosis of tubular epithelial cells is prevented by glutamine catabolism.73 As indicated by the second Warburg-like effect, cancer cells rely heavily on glutaminolysis for their survival and migration. The underlying molecular pathways involve AMPK, mTOR, JNK etc.74,75
In summary, the upstream and downstream pathways involved in glutaminolysis are summarized in Figure 1.
Altered Glutaminolysis in Kidney Diseases
During intense periods of stress, glutamine is a conditionally essential amino acid. Alterations in glutaminolysis have frequently been reported in different kidney cell types, including podocyte,17 tubular epithelial cells,73,76 endothelial cells,77 fibroblasts, and kidney immune cells,78 etc. Generally, the effects of glutaminolysis seem context dependent.
Glutaminolysis and Fibrosis
Glutaminolysis has been frequently reported in organ fibrosis, including kidney.16 Most studies have reported that glutaminolysis supports fibrogenesis, and that the targeted cells are mainly fibroblasts and immune cells.6,79 Glutaminolysis regulates lysosomal function to maintain fibroblast homeostasis through the production of ammonium as a by-product.80 This reaction is necessary for the differentiation and persistence.81 Hence, glutaminolytic inhibitory conditions prevent the aggression of the fibrotic phenotype.
In macrophages, metabolic reprogramming to the Warburg Effect-like state, namely glutaminolysis, is critical for M1-like macrophage induction.82 Nevertheless, another study found that when macrophages were treated with helminth-derived peptides, proinflammatory responses were significantly inhibited, whereas glutaminolysis was upregulated.83 A similar situation was observed for the B cells. For example, several studies have suggested that glutaminolysis supports B cell proliferation and survival in a human B cell model of Burkitt lymphoma (P493) and in IL-10-expressing regulatory B cells, respectively.84 However, another study performed in B cells of lupus-prone mice showed that glutaminolysis requirement is not B cell-intrinsic.85
Mechanistically, glutaminolysis can be activated by TGF-β, which further facilitates the generation of extracellular matrix (ECM) generation, such as collagen.86
Glutaminolysis and Aging
Previous research has established a correlation between glutaminolysis and aging progression. To date, most studies have reported that glutaminolysis is positively associated with ageing. Using an RNAi screening strategy, Johmura et al found that the glutaminolysis pathway, particularly GLS1, is pivotal for the survival of senescent cells. Consistently, they demonstrated that the elimination of senescent cells or inhibition of glutaminolysis ameliorates age-related organ dysfunction.87 In addition, senescent cells are found to have impaired glycolysis, which leads to the use of glutamine as the primary energy source. This is accompanied by the production and accumulation of urea, which further aggravates the senescence phenotype. Debanik Choudhury et al found that blockade of GLS1 inhibited glutaminolysis and decreased urea, and significantly improved the phenotypes of senescent cells and premature aging mice. Moreover, Yangyang Zhang et al found that glutamine supplementation could attenuate the expression of aging-related proteins, inhibit aging in vivo and in vitro, and enhance autophagy.88
Regarding the process of aging caused by glutaminolysis, Choudhury et al put forward the hypothesized that an increase in glutamine synthetase (GLS1) activity results in a subsequent decrease in the expression of the urea transport protein SLC14A1. This leads to urea accumulation, which triggers mitochondrial dysfunction and DNA damage.7 Furthermore, the activation of JNK in senescent cells led to an increase in GLS1 transcription, resulting in enhanced glutaminolysis.7
Senescence is known to accelerate kidney disease and CKD,89 suggest possible glutaminolysis can be used as a therapeutic targets of kidney disease and chronic kidney disease (CKD). Current approaches to aging treatment involve the removal of senescent cells, but the associated soluble molecular pathways have yet to be identified. Therefore, we propose that targeting glutaminolysis and its regulatory signaling pathways, along with associated metabolites and by-products, could offer promising strategies for addressing cellular senescence.
Glutaminolysis and Diabetic Nephropathy
Recent epidemiological investigations, experimental research, and clinical trials have focused on the metabolic abnormalities associated to diabetic nephropathy. For example, a clinical epidemiological study showed that a decrease in urine glutamine is a risk factor for progression from moderate to severe albuminuria in DKD.90
Of particular significance is the pivotal involvement of podocyte metabolic reprogramming in driving its development.91,92 Previous studies have indicated that glutamine (Gln) plays a crucial role in maintaining the structure and function of podocytes. Similarly, Gln supplementation effectively decreased proteinuria and enhanced glomerular health in mice treated with lipopolysaccharide (LPS).93 Paradoxically, despite the advantages of incorporating glutamine supplements for managing oxidative stress and maintaining glucose balance, there have been reports of unfavorable reactions.94,95 According to Tatiana Carolina Alba-Loureiro et al, diabetic rats that received glutamine supplementation exhibited elevated levels of pro-inflammatory interleukin (IL)-1 β and IL-6 in the renal cortex, along with changes indicative of glomerulosclerosis.96
In addition, a study conducted by Jijia Hu et al revealed that the expression of GLS2 was reduced in db/db mice, leading to impaired glutaminolysis. However, LDH-1 activation resulted in upregulation of GLS2 expression, restoring glutamine metabolism and mitochondrial function. This subsequently leads to a reduction in podocyte apoptosis and ultimately mitigates the kidney injury caused by diabetic nephropathy.17 Unfortunately, the precise mechanism underlying the relationship between changes in glutaminolysis, and podocyte injury induced by diabetic nephropathy (DKD) remains unclear.17
Glutaminolysis and Acute Kidney Injury
The initial manifestation in acute kidney injury is the impairment of tubular epithelial cells, which triggers the release of inflammatory cytokines and chemotactic factors by leukocytes.97 This results in an excessive influx of neutrophils and other immune cells into the renal system, ultimately leading to renal dysfunction. The extent of this dysfunction is directly proportional to the severity of the tubular epithelial cell damage and neutrophil infiltration.98
Normally, only a small portion of glutamine is consumed by the kidney, but the demand for glutamine dramatically increases during the metabolic acidosis phase of AKI, and approximately one-third of the plasma glutamine is metabolized in the kidney.99 In line with this, a recent study found that GLS activity is upregulated in kidney T cells in ischemic AKI, and glutamine blockade by its antagonist JHU083 could ameliorate kidney injury.18
However, there are also reports suggesting that glutamine administration plays a protective role against AKI. A clinical trial claimed that markers of kidney damage, urinary [TIMP-2] *[IGFBP7], were significantly decreased after glutamine administration in cardiac surgery patients at a high risk for AKI.100 Another study has suggested that glutamine enhances the synthesis of heat shock proteins (HSPs), which may serve as a defense mechanism against cellular damage.101 Similarly, in a study conducted by Zhi-Yong Peng et al, it was observed that the administration of glutamine resulted in the upregulation of HSP70 and HSP16 expression. This leads to a reduction in inflammation and neutrophil infiltration, thereby protecting against nephrotoxic AKI.101 Moreover, a previous investigation demonstrated that the administration of a solitary dose of glutamine following the onset of sepsis led to a decrease in the expression of mediators linked to high mobility group box-1 (HMGB1) and a decline in oxidative stress within the kidneys.102 Kim et al discovered that glutamine mitigated tubular cell apoptosis during acute kidney injury by inhibiting c-Jun N-terminal kinase phosphorylation of 14-3-3 in an experimental rat model of myoglobinuria after intraperitoneal glutamine.103
Glutaminolysis and Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a long-term autoimmune disorder characterized by atypical T cell reactions to self-antigens, leading to the involvement of various organs.104 Lee et al discovered that individuals with SLE had reduced plasma glutamine levels compared to healthy individuals, and that there was an inverse relationship between disease activity and plasma glutamine levels.105 Shan et al discovered that Th17 cells exhibited higher levels of A-KG, suggesting increased glutaminolysis in Th17 cells.106 Moreover, SLE progression can be slowed by impeding glutaminolysis. In a study by Zhang et al, hindering glutaminolysis resulted in decreased kidney damage in MRL/lpr mice.107 Choi et al observed an increase in Tfh cells in individuals with SLE and lupus-prone mice. Additionally, they discovered that the utilization of DON to impede glutaminolysis resulted in a decrease in the generation of daDNA antibodies.108
Numerous studies have indicated the activation of the mTOR pathway in individuals diagnosed with systemic lupus erythematosus.109,110 The initial indication of mTOR’s role of mTOR in SLE was demonstrated by Warner et al, who effectively suppressed excessive T-cell activity and alleviated nephritis in rapamycin-treated lupus-prone MRL/lpr mice.111 A study conducted by Fernandez et al revealed a notable inhibitory effect of rapamycin on T cell activation in individuals diagnosed with systemic lupus erythematosus (SLE).112 Therefore, enhanced glutaminolysis in SLE might activate the mTOR pathway. Zhang et al discovered that when glutaminolysis was inhibited, downregulation of the mTOR/P70S6K/4EBP1 pathway was observed in MRL/lpr mice.107 However, previous studies have indicated that glutaminolysis can affect the differentiation of T cells.19 It has been observed that a decrease in glutamine levels or deficiency in the ASCT2 transporter can lead to a reduction in TH17 differentiation.113 Additionally, an investigation conducted by Michihito Kono et al discovered that inhibition of glutamine synthetase 1 could impede T cell differentiation among individuals with SLE.12
Glutaminolysis and Renal Cell Carcinoma
Glutaminolysis is also called the second Warburg-like effect, studies have shown that the level of glutaminolysis is significantly up-regulated in various cancers, including renal cell carcinoma,21 etc. In kidney cancer, one of the metabolic rewiring mechanisms is the significant reliance of tumor cells on glutamine.21 Unfortunately, the effects of alterations in tumor-specific glutamine metabolism on the nearby immune microenvironment remain unknown. Furthermore, the study conducted by Sarah J Ross et al revealed a correlation between MYC activation and heightened glutamine metabolism in renal cancer cells.114 Reinforcement of MYC triggers the upregulation of SLC1A5, leading to subsequent activation of the PI3K-Akt-mTOR signaling cascade.115
Other Kidney Diseases
Glutaminolysis is involved in the development of other kidney diseases. Interestingly, transcriptional profiling of kidney biopsy samples from patients showed that glutaminolysis is repressed in antineutrophil cytoplasmic antibody-associated vasculitis (AAV) and nephrotic syndrome (NS).22 Polycystic kidney disease (PKD), an inherited disorder characterized by multiple fluid-filled cysts in the kidneys, relies on glutamine metabolism to support cellular proliferation and growth.23
In summary, the glutamine metabolism and kidney disease mechanisms are summarized in Figure 2.
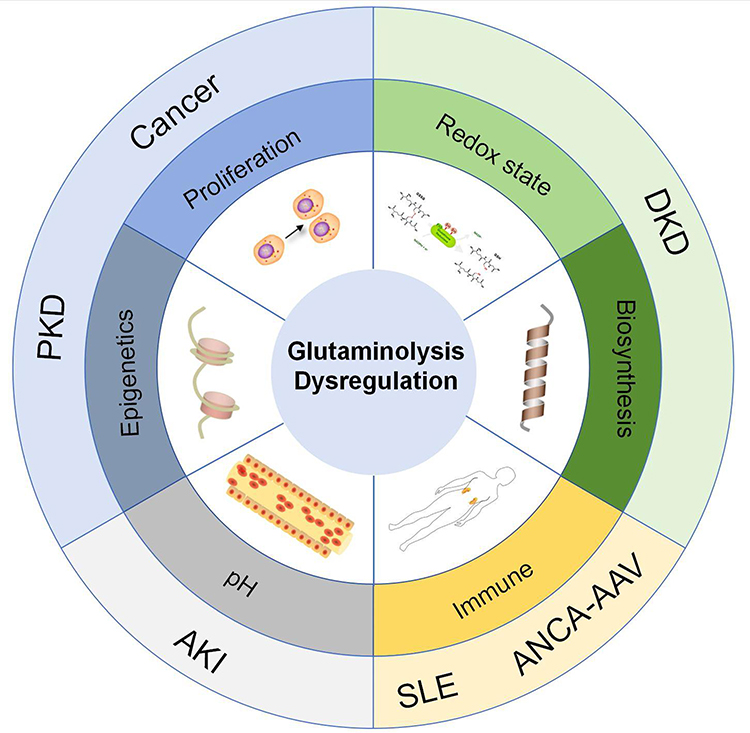 |
Figure 2 Glutamine metabolism and kidney disease mechanisms. Abbreviations: PKD, polycystic kidney disease; DKD, Diabetic Kidney Disease; AKI, Acute Kidney Injury; SLE, Systemic Lupus Erythematosus; ANCA-AAV, Anti-neutrophil cytoplasmic autoantibodies- Associated vasculitis. |
Therapeutic Strategies: Molecules Targeting Glutamine Metabolism and Glutaminolysis
Glutamine Analogs
Glutamine analogs have been developed to combat glutamine metabolism. These antagonists include 6-diazo-5-oxo-l-norleucine (DON),116 acivicin,117 and azaserine,117 1.5-N, N′-disubstituted-2-(substituted benzenesulphonyl) glutamamides.118 DON has long been known for its anticancer potency; however, its clinical translation is hindered by intolerable gastrointestinal toxicity. Persistent attempts have been made to optimize derivatives of DON, such as DRP-104, which is delivered specifically to tumors at much higher concentrations than to the plasma or gut119 and JHU083.120
Mechanistically, these glutamine analogs competitively inhibited glutamine-utilizing enzymes in a nonselective manner. The broad inhibition of glutamine metabolism surpassed the metabolic flexibility. According to two recent cancer studies, broad inhibition of glutamine metabolism with glutamine analogs seems to be a more effective strategy than the glutaminase inhibitors discussed below.121,122 However, some studies have demonstrated that glutamine supplementation protects against AKI and attenuates tubular apoptosis.73
Glutamine Depletion
Glutamine depletion is another strategy to suppress glutaminolysis. One reported drug is phenylbutyrate, which has been approved by the FDA for the treatment of urea cycle disorders. Mechanistically, phenylbutyrate is converted into phenylacetate via β-oxidation. Afterwards, phenylacetate is conjugated to glutamine to produce phenylacetylglutamine, which is catalyzed by the liver enzyme phenylacetyl-CoA. An in vivo study showed that this drug resulted in a significant reduction in plasma glutamine levels.123 Consistently, a recent study found that phenylbutyrate combined with renal injury in SLE mice.124
Inhibitors of Glutamine Transportes
Glutamine transporters are required in both plasma and mitochondrial membranes. Currently, the only known glutamine transporter in the mitochondrial inner membrane is SLC1A5 variant, while several exists in the plasma membrane, like SLC38A1, SLC38A2, and SLC1A5 are present in the plasma membrane. Therefore, targeting SLC1A5 variants may be an effective strategy to avoid compensation by other redundant transporters. SLC1A5 encodes the alanine-serine-cysteine transporter 2 (ASCT2). GPNA (L-γ-Glutamyl-p-nitroanilide) and V9302 (2-amino-4-bis (aryloxy benzyl) aminobutanoic acid) are potent and selective inhibitors of the glutamine (Gln) transporter ASCT2 (SLC1A5). V-9302 shows a 100-fold increase in SLC1A5 inhibitory potency over GPNA and exerts anticancer effects in various cancer cells.125 GPNA mainly inhibits the proliferation of different lung types,126–128 and endometrial cancers.129 GPNA also effectively inhibits T cell differentiation in oral planus moss130,131 and induces airway inflammation in asthma to a certain extent.132 In addition, new SLCA5 inhibitors have been gradually identified, but further research is needed. 20k and 25e, two novel Aminobutanoic Acid-Based compounds, identified as ASCT2 inhibitors that inhibit the growth of non-small cell carcinomas.133 2-substituted Nγ-glutamylanilides, a novel probe of ASCT2, exhibited a stronger inhibitory effect than GPNA but lacked testing in disease models.132 C118P, a novel ASCT2 Inhibitor, can inhibit glutamine metabolism and thus inhibit the proliferation of breast cancer cells.134
Interestingly, atractylenolide III, a sesquiterpenoid compound, can reduce liver fibrosis by inhibiting ASCT2-mediated glutaminolysis.135
Glutaminase Inhibitor
GLS-mediated deamination of glutamine to glutamate is the first rate-limiting step in glutaminolysis, making it an attractive target. The patented molecule bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES) is a potent and selective allosteric inhibitor of GLS1 and its truncated variant glutaminase C (GAC). However, owing to its poor solubility in water, great efforts on structural modifications have been made to improve its pharmacological utility without sacrificing its GLS inhibitory potency.136 CB-839, a derivative of BPTES, is thought to be one of the best-known GLS inhibitors and has been subjected to clinical trials for various cancers, including triple-negative breast cancer and renal cell carcinoma.137
Compound 968 is primarily considered to be an inhibitor of GLS and GAC.138 Further studies found that 968 also suppressed GLS2, with moderate (>3-fold) selectivity for GLS2.139 Other inhibitors are also emerging, such as caudatan A, a novel human kidney-type glutaminase inhibitor with tetracyclic flavan, isolated from Ohwia caudata.140
While targeting glutaminases, one concern is that the glutaminase II pathway may provide a mechanism that bypasses GLS inhibition. Inhibition of both GLS1/2 and the glutaminase II pathway deserves consideration as a novel therapeutic strategy, which is still lacking in kidney research.141 In addition, most studies have shown that high levels of GLS1 predict a poor prognosis of cancer, in contrast to GLS2, with more evidence suggesting a role as a tumor suppressor.142 Therefore, strategies that target these two glutaminases should be tested separately.
Glutamate Dehydrogenase (GDH) Inhibitors
To date, compounds targeting GDH are quite limited; the first reported compound was the green tea polyphenol EGCG.143 However, this effect is nonspecific, because EGCG inhibits a group of enzymes using NADPH as a cofactor. Additionally, it is poorly absorbed in the gut and quickly eliminated by the liver.144
Considering these disadvantages, Kang et al screened a library of FDA-approved small molecule compounds and identified purpurin as a GDH1-selective inhibitor. Moreover, its analog, R162, is a potent and cell-permeable inhibitor.145 Several other inhibitors, including ebselen and propylselen, have been identified. Ebselen inhibits GDH activity by binding to the catalytic site,146 whereas propylselen inhibits GDH activity by binding to the NADP+ binding site.147 Recently, Chang et al performed a molecular docking analysis of eight different plant-derived single compounds collected from the PubChem database to screen and select decursin (DN) and decursinol angelate (DA). DN and DA established a stable hydrogen bond interaction at the ADP activation site with the R400 and Y386 residues of GDH, thereby exerting an inhibitory effect on GDH activity.148 However, to date, none of these compounds has been subjected to clinical trials.149
Aminotransferase Inhibitors
In parallel with GDH-mediated glutamate consumption, aminotransferases, such as GPT, GOT, and PSAT, mediate the conversion of glutamate into alpha-ketoglutarate. Aminooxyacetate (AOA) is an inhibitor of glutamine‐utilizing transaminases.38 Studies have shown that the suppression of glutamate transaminase by AOA is sufficient to abolish the glutaminolysis-dependent phenotype. Therefore, targeting aminotransferase and GDH levels to inhibit glutamate conversion is a potential therapeutic strategy.
In conclusion, the molecules targeting glutamine metabolism and glutaminolysis are summarized in Table 1.
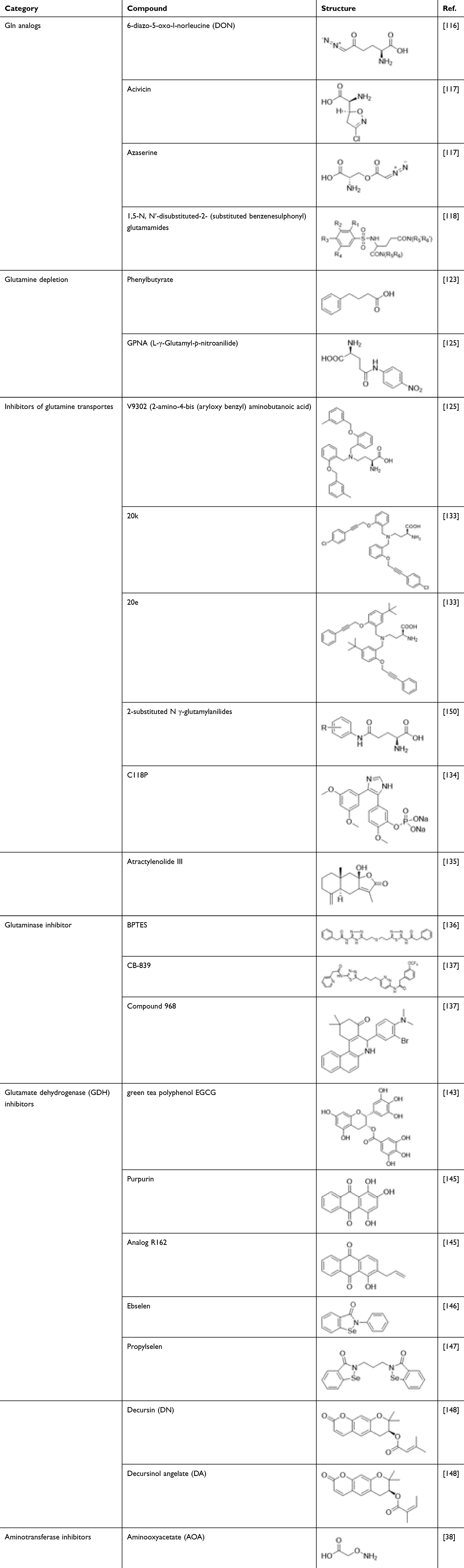 |
Table 1 Molecules Targeting Glutamine Metabolism and Glutaminolysis |
Conclusions and Perspectives
The development and progression of kidney disease are driven by multiple factors. Targeting metabolic pathways, particularly glutaminolysis and glutamine metabolism, has emerged as a promising therapeutic strategy. However, as reviewed above, glutaminolysis may function differently in a phase- and microenvironment-dependent manner. Therefore, further studies are urgently required to dissect its context-specific impact. In addition, while targeting glutaminolysis, metabolic flexibility should be considered as compensatory mechanisms may be induced to hamper therapeutic outcomes. Consequently, it is reasonable to combine multiple therapeutic targets to avoid metabolic adaptation, either in combination with other pathways or via the glutaminase II pathway. Besides, if glutamine supplementation is conducted in patients with reduced glutaminolysis, pharmacokinetic studies including drug delivery route, dosage need to be further evaluated. With the advance of metabolomics technology, understanding of the pleiotropic role of glutaminolysis in a specific context may be improved. At last, but not least, considering that glutaminolysis is a hallmark of cancer metabolism, so far, most of the above-mentioned molecules are investigated in cancer. Further investigations should be conducted in kidney diseases to better understand the underlying mechanisms and therapeutic efficacy.
Acknowledgment
The authors thank Dr. Bing-Chao Yan (Kunming Institute of Botany, CAS) for his assistance with the English editing and chemical structures in Table 1. We would also like to thank the anonymous reviewers for their valuable comments to improve the quality of this paper.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82100769), GuangDong Basic and Applied Basic Research Foundation (2024A1515011237), Science and Technology Planning Project of Zhanjiang (2021A05064, 2022A01158), Guangdong Provincial Key Laboratory of Autophagy and Major Chronic Non-Communicable Diseases (2022B1212030003), and National Clinical Key Specialty Construction Project (Institute of Nephrology, Affiliated Hospital of Guangdong Medical University).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–634. doi:10.1038/nrc.2016.71
2. Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. 2018;10(11). doi:10.3390/nu10111564
3. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. 2017;19:163–194. doi:10.1146/annurev-bioeng-071516-044546
4. Cooper AJ, Shurubor YI, Dorai T, et al. ω-Amidase: an underappreciated, but important enzyme in L-glutamine and L-asparagine metabolism; relevance to sulfur and nitrogen metabolism, tumor biology and hyperammonemic diseases. Amino Acids. 2016;48(1):1–20. doi:10.1007/s00726-015-2061-7
5. Guo L, Chen S, Ou L, Li S, Ye ZN, Liu HF. Disrupted alpha-ketoglutarate homeostasis: understanding kidney diseases from the view of metabolism and beyond. Diabetes Metab Syndr Obes. 2022;15:1961–1974. doi:10.2147/dmso.S369090
6. Xiang Z, Bai L, Zhou JQ, et al. Epigenetic regulation of IPF fibroblast phenotype by glutaminolysis. Mol Metab. 2023;67:101655. doi:10.1016/j.molmet.2022.101655
7. Choudhury D, Rong N, Ikhapoh I, et al. Inhibition of glutaminolysis restores mitochondrial function in senescent stem cells. Cell Rep. 2022;41(9):111744. doi:10.1016/j.celrep.2022.111744
8. Yiew NKH, Vazquez JH, Martino MR, et al. Hepatic pyruvate and alanine metabolism are critical and complementary for maintenance of antioxidant capacity and resistance to oxidative insult. Mol Metab. 2023;77:101808. doi:10.1016/j.molmet.2023.101808
9. Weiner ID, Hamm LL. Molecular mechanisms of renal ammonia transport. Annu Rev Physiol. 2007;69:317–340. doi:10.1146/annurev.physiol.69.040705.142215
10. Antunes D, Gonçalves SM, Matzaraki V, et al. Glutamine metabolism supports the functional activity of immune cells against aspergillus fumigatus. Microbiol Spectr. 2023;11(1):e0225622. doi:10.1128/spectrum.02256-22
11. Takahashi S, Saegusa J, Sendo S, et al. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther. 2017;19(1):76. doi:10.1186/s13075-017-1283-3
12. Kono M, Yoshida N, Maeda K, Suárez-Fueyo A, Kyttaris VC, Tsokos GC. Glutaminase 1 inhibition reduces glycolysis and ameliorates lupus-like disease in mrl/lpr mice and experimental autoimmune encephalomyelitis. Arthritis Rheumatol. 2019;71(11):1869–1878. doi:10.1002/art.41019
13. Szeliga M, Bogacińska-Karaś M, Kuźmicz K, Rola R, Albrecht J. Downregulation of GLS2 in glioblastoma cells is related to DNA hypermethylation but not to the p53 status. Mol, Carcinog. 2016;55(9):1309–1316. doi:10.1002/mc.22372
14. Zhong G, Su S, Li J, et al. Activation of Piezo1 promotes osteogenic differentiation of aortic valve interstitial cell through YAP-dependent glutaminolysis. Sci Adv. 2023;9(22):eadg0478. doi:10.1126/sciadv.adg0478
15. Zhang T, Cui Y, Wu Y, et al. Mitochondrial GCN5L1 regulates glutaminase acetylation and hepatocellular carcinoma. Clin Transl Med. 2022;12(5):e852. doi:10.1002/ctm2.852
16. Cai Y, Tian B, Deng Y, et al. Glutamine metabolism promotes renal fibrosis through regulation of mitochondrial energy generation and mitochondrial fission. Int J Biol Sci. 2024;20(3):987–1003. doi:10.7150/ijbs.89960
17. Hu J, Zhang Z, Hu H, et al. LRH-1 activation alleviates diabetes-induced podocyte injury by promoting GLS2-mediated glutaminolysis. Cell Prolif. 2023;56(11):e13479. doi:10.1111/cpr.13479
18. Lee K, Thompson EA, Gharaie S, et al. T cell metabolic reprogramming in acute kidney injury and protection by glutamine blockade. JCI Insight. 2023;8(12). doi:10.1172/jci.insight.160345
19. Kono M, Yoshida N, Maeda K, Tsokos GC. Transcriptional factor ICER promotes glutaminolysis and the generation of Th17 cells. Proc Natl Acad Sci U S A. 2018;115(10):2478–2483. doi:10.1073/pnas.1714717115
20. Sharma A, Lavie CJ, Sharma SK, et al. Duration of dual antiplatelet therapy after drug-eluting stent implantation in patients with and without acute coronary syndrome: a systematic review of randomized controlled trials. Mayo Clin Proc. 2016;91(8):1084–1093. doi:10.1016/j.mayocp.2016.06.004
21. Fu Q, Xu L, Wang Y, et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol. 2019;75(5):752–763. doi:10.1016/j.eururo.2018.09.030
22. Grayson PC, Eddy S, Taroni JN, et al. Metabolic pathways and immunometabolism in rare kidney diseases. Ann Rheum Dis. 2018;77(8):1226–1233. doi:10.1136/annrheumdis-2017-212935
23. Flowers EM, Sudderth J, Zacharias L, et al. Lkb1 deficiency confers glutamine dependency in polycystic kidney disease. Nat Commun. 2018;9(1):814. doi:10.1038/s41467-018-03036-y
24. Kühtreiber WM, Takahashi H, Keefe RC, et al. BCG vaccinations upregulate myc, a central switch for improved glucose metabolism in diabetes. iScience. 2020;23(5):101085. doi:10.1016/j.isci.2020.101085
25. Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105(48):18782–18787. doi:10.1073/pnas.0810199105
26. Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5. doi:10.1038/nature07823
27. Kim JH, Lee KJ, Seo Y, et al. Effects of metformin on colorectal cancer stem cells depend on alterations in glutamine metabolism. Sci Rep. 2018;8(1):409. doi:10.1038/s41598-017-18762-4
28. Munksgaard Thorén M, Vaapil M, Staaf J, et al. Myc-induced glutaminolysis bypasses HIF-driven glycolysis in hypoxic small cell lung carcinoma cells. Oncotarget. 2017;8(30):48983–48995. doi:10.18632/oncotarget.16904
29. Wu H, Li Z, Yang P, Zhang L, Fan Y, Li Z. PKM2 depletion induces the compensation of glutaminolysis through β-catenin/c-Myc pathway in tumor cells. Cell Signal. 2014;26(11):2397–2405. doi:10.1016/j.cellsig.2014.07.024
30. Haikala HM, Marques E, Turunen M, Klefström J. Myc requires RhoA/SRF to reprogram glutamine metabolism. Small GTPases. 2018;9(3):274–282. doi:10.1080/21541248.2016.1224287
31. Shroff EH, Eberlin LS, Dang VM, et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A. 2015;112(21):6539–6544. doi:10.1073/pnas.1507228112
32. Freire WB. Hemoglobin as a predictor of response to iron therapy and its use in screening and prevalence estimates. Am J Clin Nutr. 1989;50(6):1442–1449. doi:10.1093/ajcn/50.6.1442
33. Białopiotrowicz E, Noyszewska-Kania M, Kachamakova-Trojanowska N, et al. Serine biosynthesis pathway supports MYC-miR-494-EZH2 feed-forward circuit necessary to maintain metabolic and epigenetic reprogramming of Burkitt lymphoma cells. Cancers (Basel). 2020;12(3):580. doi:10.3390/cancers12030580
34. Habshi T, Shelke V, Kale A, Lech M, Gaikwad AB. Hippo signaling in acute kidney injury to chronic kidney disease transition: current understandings and future targets. Drug Discov Today. 2023;28(8):103649. doi:10.1016/j.drudis.2023.103649
35. Du K, Hyun J, Premont RT, et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology. 2018;154(5):1465–1479.e13. doi:10.1053/j.gastro.2017.12.022
36. Bertero T, Oldham WM, Cottrill KA, et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. 2016;126(9):3313–3335. doi:10.1172/jci86387
37. Cox AG, Hwang KL, Brown KK, et al. Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat Cell Biol. 2016;18(8):886–896. doi:10.1038/ncb3389
38. Yang CS, Stampouloglou E, Kingston NM, Zhang L, Monti S, Varelas X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 2018;19(6). doi:10.15252/embr.201643577
39. Choi S, Hong SP, Bae JH, et al. Hyperactivation of YAP/TAZ drives alterations in mesangial cells through stabilization of N-myc in diabetic nephropathy. J Am Soc Nephrol. 2023;34(5):809–828. doi:10.1681/asn.0000000000000075
40. Vallée A, Lecarpentier Y, Vallée JN. The Key Role of the WNT/β-catenin pathway in metabolic reprogramming in cancers under normoxic conditions. Cancers (Basel). 2021;13(21). doi:10.3390/cancers13215557
41. El-Sahli S, Xie Y, Wang L, Liu S. Wnt signaling in cancer metabolism and immunity. Cancers (Basel). 2019;11(7):904. doi:10.3390/cancers11070904
42. Cadoret A, Ovejero C, Terris B, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002;21(54):8293–8301. doi:10.1038/sj.onc.1206118
43. Zhou B, Magana L, Hong Z, et al. The angiocrine Rspondin3 instructs interstitial macrophage transition via metabolic-epigenetic reprogramming and resolves inflammatory injury. Nat Immunol. 2020;21(11):1430–1443. doi:10.1038/s41590-020-0764-8
44. Wong CC, Xu J, Bian X, et al. In colorectal cancer cells with mutant KRAS, SLC25A22-mediated glutaminolysis reduces DNA Demethylation to Increase WNT signaling, stemness, and drug resistance. Gastroenterology. 2020;159(6):2163–2180.e6. doi:10.1053/j.gastro.2020.08.016
45. Liao J, Liu PP, Hou G, et al. Regulation of stem-like cancer cells by glutamine through β-catenin pathway mediated by redox signaling. Mol Cancer. 2017;16(1):51. doi:10.1186/s12943-017-0623-x
46. Brunialti E, Rizzi N, Pinto-Costa R, et al. Design and validation of a reporter mouse to study the dynamic regulation of TFEB and TFE3 activity through in vivo imaging techniques. Autophagy. 2024:1–16. doi:10.1080/15548627.2024.2334111
47. Ariano C, Costanza F, Akman M, et al. TFEB inhibition induces melanoma shut-down by blocking the cell cycle and rewiring metabolism. Cell Death Dis. 2023;14(5):314. doi:10.1038/s41419-023-05828-7
48. Kim JH, Lee J, Cho YR, et al. TFEB supports pancreatic cancer growth through the transcriptional regulation of glutaminase. Cancers (Basel). 2021;13(3). doi:10.3390/cancers13030483
49. Nowis D, Malenda A, Furs K, et al. Statins impair glucose uptake in human cells. BMJ Open Diabetes Res Care. 2014;2(1):e000017. doi:10.1136/bmjdrc-2014-000017
50. Durán RV, Oppliger W, Robitaille AM, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47(3):349–358. doi:10.1016/j.molcel.2012.05.043
51. Bodineau C, Tomé M, Courtois S, et al. Two parallel pathways connect glutamine metabolism and mTORC1 activity to regulate glutamoptosis. Nat Commun. 2021;12(1):4814. doi:10.1038/s41467-021-25079-4
52. Csibi A, Fendt SM, Li C, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153(4):840–854. doi:10.1016/j.cell.2013.04.023
53. Csibi A, Lee G, Yoon SO, et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014;24(19):2274–2280. doi:10.1016/j.cub.2014.08.007
54. Xuan C, Cui H, Jin Z, et al. Glutamine ameliorates hyperoxia-induced hippocampal damage by attenuating inflammation and apoptosis via the MKP-1/MAPK signaling pathway in neonatal rats. Front Pharmacol. 2023;14:1096309. doi:10.3389/fphar.2023.1096309
55. Jeong SY, Im YN, Youm JY, Lee HK, Im SY. l-glutamine attenuates DSS-induced colitis via induction of MAPK phosphatase-1. Nutrients. 2018;10(3):288. doi:10.3390/nu10030288
56. Ayush O, Jin ZW, Kim HK, Shin YR, Im SY, Lee HK. Glutamine up-regulates MAPK phosphatase-1 induction via activation of Ca(2+)→ ERK cascade pathway. Biochem Biophys Rep. 2016;7:10–19. doi:10.1016/j.bbrep.2016.05.011
57. Lee CH, Kim HK, Jeong JS, et al. Mechanism of glutamine inhibition of cytosolic phospholipase a2 (cPLA2): evidence of physical interaction between glutamine-Induced mitogen-activated protein kinase phosphatase-1 and cPLA2. Clin Exp Immunol. 2015;180(3):571–580. doi:10.1111/cei.12585
58. Ko HM, Oh SH, Bang HS, et al. Glutamine protects mice from lethal endotoxic shock via a rapid induction of MAPK phosphatase-1. J Immunol. 2009;182(12):7957–7962. doi:10.4049/jimmunol.0900043
59. Ayush O, Lee CH, Kim HK, Im SY, Cho BH, Lee HK. Glutamine suppresses DNFB-induced contact dermatitis by deactivating p38 mitogen-activated protein kinase via induction of MAPK phosphatase-1. J Invest Dermatol. 2013;133(3):723–731. doi:10.1038/jid.2012.373
60. Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518(7539):413–6. doi:10.1038/nature13981
61. Tran TQ, Hanse EA, Habowski AN, et al. α-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat Cancer. 2020;1(3):345–358. doi:10.1038/s43018-020-0035-5
62. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:7274):739–44. doi:10.1038/nature08617
63. Seltzer MJ, Bennett BD, Joshi AD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981–8987. doi:10.1158/0008-5472.Can-10-1666
64. Chen PR, Lucas CG, Spate LD, Prather RS. Glutaminolysis is involved in the activation of mTORC1 in in vitro-produced porcine embryos. Mol Reprod Dev. 2021;88(7):490–499. doi:10.1002/mrd.23516
65. Torrino S, Grasset EM, Audebert S, et al. Mechano-induced cell metabolism promotes microtubule glutamylation to force metastasis. Cell Metab. 2021;33(7):1342–1357.e10. doi:10.1016/j.cmet.2021.05.009
66. Zhao S, Wang JM, Yan J, et al. BAG3 promotes autophagy and glutaminolysis via stabilizing glutaminase. Cell Death Dis. 2019;10(4):284. doi:10.1038/s41419-019-1504-6
67. Li X, Yang Y, Zhang B, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. 2022;7(1):305. doi:10.1038/s41392-022-01151-3
68. Han T, Wang P, Wang Y, et al. FAIM regulates autophagy through glutaminolysis in lung adenocarcinoma. Autophagy. 2022;18(6):1416–1432. doi:10.1080/15548627.2021.1987672
69. Swamy M, Pathak S, Grzes KM, et al. Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol. 2016;17(6):712–720. doi:10.1038/ni.3439
70. Tung MC, Lin PL, Wang YC, et al. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget. 2015;6(39):41692–41705. doi:10.18632/oncotarget.6150
71. Walerych D, Lisek K, Sommaggio R, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol. 2016;18(8):897–909. doi:10.1038/ncb3380
72. Chen D, Tavana O, Chu B, et al. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol Cell. 2017;68(1):224–232.e4. doi:10.1016/j.molcel.2017.09.009
73. Thomas K, Zondler L, Ludwig N, et al. Glutamine prevents acute kidney injury by modulating oxidative stress and apoptosis in tubular epithelial cells. JCI Insight. 2022;7(21). doi:10.1172/jci.insight.163161
74. Yuan L, Sheng X, Clark LH, et al. Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am J Transl Res. 2016;8(10):4265–4277.
75. Yuan L, Sheng X, Willson AK, et al. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr Relat Cancer. 2015;22(4):577–591. doi:10.1530/erc-15-0192
76. Rinschen MM, Harder JL, Carter-Timofte ME, et al. VPS34-dependent control of apical membrane function of proximal tubule cells and nutrient recovery by the kidney. Sci Signal. 2022;15(762):eabo7940. doi:10.1126/scisignal.abo7940
77. Oberkersch RE, Pontarin G, Astone M, et al. Aspartate metabolism in endothelial cells activates the mTORC1 pathway to initiate translation during angiogenesis. Dev Cell. 2022;57(10):1241–1256.e8. doi:10.1016/j.devcel.2022.04.018
78. Basso PJ, Andrade-Oliveira V, Câmara NOS. Targeting immune cell metabolism in kidney diseases. Nat Rev Nephrol. 2021;17(7):465–480. doi:10.1038/s41581-021-00413-7
79. Xue X, Zeng X, Wu X, Mu K, Dai Y, Wei Z. SIRT4 protects against intestinal fibrosis by facilitating GLS1 degradation. Matrix Biol. 2023;122:33–45. doi:10.1016/j.matbio.2023.08.001
80. Xiong J, Luu TTT, Venkatachalam K, Du G, Zhu MX. Glutamine produces ammonium to tune lysosomal pH and regulate lysosomal function. Cells. 2022;12(1). doi:10.3390/cells12010080
81. Gibb AA, Huynh AT, Gaspar RB, et al. Glutamine uptake and catabolism is required for myofibroblast formation and persistence. J Mol Cell Cardiol. 2022;172:78–89. doi:10.1016/j.yjmcc.2022.08.002
82. Jiang Q, Qiu Y, Kurland IJ, et al. Glutamine is required for M1-like polarization of macrophages in response to mycobacterium tuberculosis infection. mBio. 2022;13(4):e0127422. doi:10.1128/mbio.01274-22
83. Quinteros SL, von Krusenstiern E, Snyder NW, Tanaka A, O’Brien B, Donnelly S. The helminth derived peptide FhHDM-1 redirects macrophage metabolism towards glutaminolysis to regulate the pro-inflammatory response. Front Immunol. 2023;14:1018076. doi:10.3389/fimmu.2023.1018076
84. Liu JQ, Geng XR, Hu TY, et al. Glutaminolysis is required in maintaining immune regulatory functions in B cells. Mucosal Immunol. 2022;15(2):268–278. doi:10.1038/s41385-021-00481-9
85. Choi SC, Li W, Zhang X, et al. Pharmacologically inferred glycolysis and glutaminolysis requirement of b cells in lupus-prone mice. J Immunol. 2022;208(9):2098–2108. doi:10.4049/jimmunol.2100356
86. Lombardi AA, Gibb AA, Arif E, et al. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat Commun. 2019;10(1):4509. doi:10.1038/s41467-019-12103-x
87. Johmura Y, Yamanaka T, Omori S, et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science. 2021;371:6526):265–270. doi:10.1126/science.abb5916
88. Zhang Y, Huang Z, Han W, et al. Glutamine suppresses senescence and promotes autophagy through glycolysis inhibition-mediated AMPKα lactylation in intervertebral disc degeneration. Commun Biol. 2024;7(1):325. doi:10.1038/s42003-024-06000-3
89. Huang Y, Wang B, Hassounah F, et al. The impact of senescence on muscle wasting in chronic kidney disease. J Cachexia, Sarcopenia Muscle. 2023;14(1):126–141. doi:10.1002/jcsm.13112
90. Pena MJ, Lambers Heerspink HJ, Hellemons ME, et al. Urine and plasma metabolites predict the development of diabetic nephropathy in individuals with Type 2 diabetes mellitus. Diabet Med. 2014;31(9):1138–1147. doi:10.1111/dme.12447
91. Liu S, Yuan Y, Xue Y, Xing C, Zhang B. Podocyte injury in diabetic kidney disease: a focus on mitochondrial dysfunction. Front Cell Dev Biol. 2022;10:832887. doi:10.3389/fcell.2022.832887
92. Yoshimura Y, Nishinakamura R. Podocyte development, disease, and stem cell research. Kidney Int. 2019;96(5):1077–1082. doi:10.1016/j.kint.2019.04.044
93. Altintas MM, Moriwaki K, Wei C, et al. Reduction of proteinuria through podocyte alkalinization. J Biol Chem. 2014;289(25):17454–17467. doi:10.1074/jbc.M114.568998
94. Chen Q, Kirk K, Shurubor YI, et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018;27(5):1007–1025.e5. doi:10.1016/j.cmet.2018.03.002
95. van Kuilenburg ABP, Tarailo-Graovac M, Richmond PA, et al. Glutaminase deficiency caused by short tandem repeat expansion in GLS. N Engl J Med. 2019;380(15):1433–1441. doi:10.1056/NEJMoa1806627
96. Alba-Loureiro TC, Ribeiro RF, Zorn TM, Lagranha CJ. Effects of glutamine supplementation on kidney of diabetic rat. Amino Acids. 2010;38(4):1021–1030. doi:10.1007/s00726-009-0310-3
97. Cao Y, Chen X, Zhu Z, et al. STING contributes to lipopolysaccharide-induced tubular cell inflammation and pyroptosis by activating endoplasmic reticulum stress in acute kidney injury. Cell Death Dis. 2024;15(3):217. doi:10.1038/s41419-024-06600-1
98. Huang W, Wang BO, Hou YF, et al. JAML promotes acute kidney injury mainly through a macrophage-dependent mechanism. JCI Insight. 2022;7(14). doi:10.1172/jci.insight.158571
99. Zhao L, Hao Y, Tang S, Han X, Li R, Zhou X. Energy metabolic reprogramming regulates programmed cell death of renal tubular epithelial cells and might serve as a new therapeutic target for acute kidney injury. Front Cell Dev Biol. 2023;11:1276217. doi:10.3389/fcell.2023.1276217
100. Weiss R, Meersch M, Gerke M, et al. Effect of glutamine administration after cardiac surgery on kidney damage in patients at high risk for acute kidney injury: a randomized controlled trial. Anesth Analg. 2023;137(5):1029–1038. doi:10.1213/ane.0000000000006288
101. Peng ZY, Zhou F, Wang HZ, et al. The anti-oxidant effects are not the main mechanism for glutamine’s protective effects on acute kidney injury in mice. Eur J Pharmacol. 2013;705(1–3):11–19. doi:10.1016/j.ejphar.2013.02.028
102. Hu YM, Pai MH, Yeh CL, Hou YC, Yeh SL. Glutamine administration ameliorates sepsis-induced kidney injury by downregulating the high-mobility group box protein-1-mediated pathway in mice. Am J Physiol Renal Physiol. 2012;302(1):F150–8. doi:10.1152/ajprenal.00246.2011
103. Kim YS, Jung MH, Choi MY, et al. Glutamine attenuates tubular cell apoptosis in acute kidney injury via inhibition of the c-Jun N-terminal kinase phosphorylation of 14-3-3. Crit Care Med. 2009;37(6):2033–2044. doi:10.1097/CCM.0b013e3181a005ba
104. Tsokos GC, Lo MS, Costa Reis P, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12(12):716–730. doi:10.1038/nrrheum.2016.186
105. Lee HT, Lin CS, Pan SC, et al. Alterations of oxygen consumption and extracellular acidification rates by glutamine in PBMCs of SLE patients. Mitochondrion. 2019;44:65–74. doi:10.1016/j.mito.2018.01.002
106. Shan J, Jin H, Xu Y. T cell metabolism: a new perspective on Th17/treg cell imbalance in systemic lupus erythematosus. Front Immunol. 2020;11:1027. doi:10.3389/fimmu.2020.01027
107. Zhang X, Wang G, Bi Y, Jiang Z, Wang X. Inhibition of glutaminolysis ameliorates lupus by regulating T and B cell subsets and downregulating the mTOR/P70S6K/4EBP1 and NLRP3/caspase-1/IL-1β pathways in MRL/lpr mice. Int Immunopharmacol. 2022;112:109133. doi:10.1016/j.intimp.2022.109133
108. Choi SC, Titov AA, Abboud G, et al. Inhibition of glucose metabolism selectively targets autoreactive follicular helper T cells. Nat Commun. 2018;9(1):4369. doi:10.1038/s41467-018-06686-0
109. Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. 2016;12(3):169–182. doi:10.1038/nrrheum.2015.172
110. Tsokos GC. Systemic lupus erythematosus in 2015: cellular and metabolic requirements of effector T cells. Nat Rev Rheumatol. 2016;12(2):74–76. doi:10.1038/nrrheum.2015.178
111. Warner LM, Adams LM, Sehgal SN. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheum. 1994;37(2):289–297. doi:10.1002/art.1780370219
112. Lai ZW, Borsuk R, Shadakshari A, et al. Mechanistic target of rapamycin activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus erythematosus. J Immunol. 2013;191(5):2236–2246. doi:10.4049/jimmunol.1301005
113. Nakaya M, Xiao Y, Zhou X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40(5):692–705. doi:10.1016/j.immuni.2014.04.007
114. Ross SJ, Critchlow SE. Emerging approaches to target tumor metabolism. Curr Opin Pharmacol. 2014;17:22–29. doi:10.1016/j.coph.2014.07.001
115. Rathmell JC. T cell Myc-tabolism. Immunity. 2011;35(6):845–846. doi:10.1016/j.immuni.2011.12.001
116. Lemberg KM, Vornov JJ, Rais R, Slusher BS. We’re Not ”DON” Yet: optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther. 2018;17(9):1824–1832. doi:10.1158/1535-7163.Mct-17-1148
117. Chittur SV, Klem TJ, Shafer CM, Davisson VJ. Mechanism for acivicin inactivation of triad glutamine amidotransferases. Biochemistry. 2001;40(4):876–887. doi:10.1021/bi0014047
118. Srikanth K, Debnath B, Jha T. Syntheses, biological evaluation and QSAR study on antitumor activity of 1,5-N,N’-disubstituted-2-(substituted benzenesulphonyl) glutamamides. Bioorg Med Chem. 2002;10(6):1841–1854. doi:10.1016/s0968-0896(02)00007-x
119. Rais R, Lemberg KM, Tenora L, et al. Discovery of DRP-104, a tumor-targeted metabolic inhibitor prodrug. Sci Adv. 2022;8(46):eabq5925. doi:10.1126/sciadv.abq5925
120. Rais R, Jančařík A, Tenora L, et al. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J Med Chem. 2016;59(18):8621–8633. doi:10.1021/acs.jmedchem.6b01069
121. Encarnación-Rosado J, Sohn ASW, Biancur DE, et al. Targeting pancreatic cancer metabolic dependencies through glutamine antagonism. Nat Cancer. 2024;5(1):85–99. doi:10.1038/s43018-023-00647-3
122. Recouvreux MV, Grenier SF, Zhang Y, et al. Glutamine mimicry suppresses tumor progression through asparagine metabolism in pancreatic ductal adenocarcinoma. Nat Cancer. 2024;5(1):100–113. doi:10.1038/s43018-023-00649-1
123. Thibault A, Samid D, Cooper MR, et al. Phase I study of phenylacetate administered twice daily to patients with cancer. Cancer. 1995;75(12):2932–2938. doi:10.1002/1097-0142(19950615)75:12<2932::AID-CNCR2820751221>3.0.CO;2-P
124. Bonnemaison ML, Marks-Nelson ES, Boesen EI. Sodium 4-phenylbutyrate treatment protects against renal injury in NZBWF1 mice. Clin Sci (Lond). 2019;133(2):167–180. doi:10.1042/cs20180562
125. Schulte ML, Fu A, Zhao P, et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. 2018;24(2):194–202. doi:10.1038/nm.4464
126. Hassanein M, Hoeksema MD, Shiota M, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013;19(3):560–570. doi:10.1158/1078-0432.Ccr-12-2334
127. Silva C, Andrade N, Rodrigues I, Ferreira AC, Soares ML, Martel F. The pro-proliferative effect of interferon-γ in breast cancer cell lines is dependent on stimulation of ASCT2-mediated glutamine cellular uptake. Life Sci. 2021;286:120054. doi:10.1016/j.lfs.2021.120054
128. Wang W, Pan H, Ren F, Chen H, Ren P. Targeting ASCT2-mediated glutamine metabolism inhibits proliferation and promotes apoptosis of pancreatic cancer cells. Biosci Rep. 2022;42(3). doi:10.1042/bsr20212171
129. Marshall AD, van Geldermalsen M, Otte NJ, et al. ASCT2 regulates glutamine uptake and cell growth in endometrial carcinoma. Oncogenesis. 2017;6(7):e367. doi:10.1038/oncsis.2017.70
130. Li W, Ling Z, Wang J, et al. ASCT2-mediated glutamine uptake promotes Th1 differentiation via ROS-EGR1-PAC1 pathway in oral lichen planus. Biochem Pharmacol. 2023;216:115767. doi:10.1016/j.bcp.2023.115767
131. Lu J, Su Z, Li W, et al. ASCT2-mediated glutamine uptake of epithelial cells facilitates CCL5-induced T cell infiltration via ROS-STAT3 pathway in oral lichen planus. Int Immunopharmacol. 2023;119:110216. doi:10.1016/j.intimp.2023.110216
132. Kim JM, Im YN, Chung YJ, et al. Glutamine deficiency shifts the asthmatic state toward neutrophilic airway inflammation. Allergy. 2022;77(4):1180–1191. doi:10.1111/all.15121
133. Qin L, Cheng X, Wang S, et al. Discovery of novel aminobutanoic acid-based ASCT2 inhibitors for the treatment of non-small-cell lung cancer. J Med Chem. 2024;67(2):988–1007. doi:10.1021/acs.jmedchem.3c01093
134. Lyu XD, Liu Y, Wang J, et al. A novel ASCT2 Inhibitor, C118P, blocks glutamine transport and exhibits antitumour efficacy in breast cancer. Cancers (Basel). 2023;15(20). doi:10.3390/cancers15205082
135. Wang F, Li Z, Chen L, et al. Inhibition of ASCT2 induces hepatic stellate cell senescence with modified proinflammatory secretome through an IL-1α/NF-κB feedback pathway to inhibit liver fibrosis. Acta Pharm Sin B. 2022;12(9):3618–3638. doi:10.1016/j.apsb.2022.03.014
136. Shukla K, Ferraris DV, Thomas AG, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012;55(23):10551–10563. doi:10.1021/jm301191p
137. Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. doi:10.1158/1535-7163.Mct-13-0870
138. Wang JB, Erickson JW, Fuji R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18(3):207–219. doi:10.1016/j.ccr.2010.08.009
139. Lukey MJ, Cluntun AA, Katt WP, et al. Liver-type glutaminase GLS2 is a druggable metabolic node in luminal-subtype breast cancer. Cell Rep. 2019;29(1):76–88.e7. doi:10.1016/j.celrep.2019.08.076
140. Sun Y, Feng X, Liu X, et al. Caudatan A, an undescribed human kidney-type glutaminase inhibitor with tetracyclic flavan from Ohwia caudata. Phytochemistry. 2018;152:22–28. doi:10.1016/j.phytochem.2018.04.013
141. Udupa S, Nguyen S, Hoang G, et al. Upregulation of the glutaminase II pathway contributes to glutamate production upon glutaminase 1 inhibition in pancreatic cancer. Proteomics. 2019;19(21–22):e1800451. doi:10.1002/pmic.201800451
142. Suzuki S, Venkatesh D, Kanda H, et al. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res. 2022;82(18):3209–3222. doi:10.1158/0008-5472.Can-21-3914
143. Li C, Li M, Chen P, et al. Green tea polyphenols control dysregulated glutamate dehydrogenase in transgenic mice by hijacking the ADP activation site. J Biol Chem. 2011;286(39):34164–34174. doi:10.1074/jbc.M111.268599
144. Smith TJ. Green Tea Polyphenols in drug discovery - A success or failure? Expert Opin Drug Discov. 2011;6(6):589–595. doi:10.1517/17460441.2011.570750
145. Jin L, Li D, Alesi GN, et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 2015;27(2):257–270. doi:10.1016/j.ccell.2014.12.006
146. Jin Y, Li D, Lu S, et al. Ebselen reversibly inhibits human glutamate dehydrogenase at the catalytic site. Assay Drug Dev Technol. 2018;16(2):115–122. doi:10.1089/adt.2017.822
147. Hou W, Lu S, Zhao H, et al. Propylselen inhibits cancer cell growth by targeting glutamate dehydrogenase at the NADP(+) binding site. Biochem Biophys Res Commun. 2019;509(1):262–267. doi:10.1016/j.bbrc.2018.12.117
148. Chang SN, Keretsu S, Kang SC. Evaluation of decursin and its isomer decursinol angelate as potential inhibitors of human glutamate dehydrogenase activity through in silico and enzymatic assay screening. Comput Biol Med. 2022;151(Pt B):106287. doi:10.1016/j.compbiomed.2022.106287
149. Bian Y, Hou W, Chen X, Fang J, Xu N, Ruan BH. Glutamate dehydrogenase as a promising target for hyperinsulinism hyperammonemia syndrome therapy. Curr Med Chem. 2022;29(15):2652–2672. doi:10.2174/0929867328666210825105342
150. Schulte ML, Dawson ES, Saleh SA, Cuthbertson ML, Manning HC. 2-Substituted Nγ-glutamylanilides as novel probes of ASCT2 with improved potency. Bioorg Med Chem Lett. 2015;25(1):113–116. doi:10.1016/j.bmcl.2014.10.098
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.