")
Back to Journals » Cancer Management and Research » Volume 17
Gut Microbiome and Metabolite Characteristics Associated With Different Clinical Stages in Non-Small Cell Lung Cancer Patients
Authors Liu F, Lu X, Tang M, Chen Y, Zheng X
Received 20 October 2024
Accepted for publication 4 January 2025
Published 11 January 2025 Volume 2025:17 Pages 45—56
DOI https://doi.org/10.2147/CMAR.S499003
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Fan Liu,1 Xingbing Lu,2 Mengli Tang,2 Yuzuo Chen,2 Xi Zheng3,4
1Health Management Center, General Practice Medical Center, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 2Department of Laboratory Medicine, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 3Lung Cancer Center, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 4Department of Thoracic Surgery, West China Hospital, Sichuan University, Chengdu, People’s Republic of China
Correspondence: Xi Zheng, Lung Cancer Center, Department of Thoracic Surgery, West China Hospital, Sichuan University, Guoxue Lane 37, Chengdu, Sichuan Province, People’s Republic of China, Tel +86-28-86298113, Email [email protected]
Objective: Our research has pinpointed the gut microbiome’s role in the progression of various pathological types of non-small cell lung cancer (NSCLC). Nonetheless, the characteristics of the gut microbiome and its metabolites across different clinical stages of NSCLC are yet to be fully understood. The current study seeks to explore the distinctive gut flora and metabolite profiles of NSCLC patients across varying TNM stages.
Methods: The research team gathered stool samples from 52 patients diagnosed with non-small cell lung cancer (NSCLC) and 29 healthy individuals. Subsequently, they performed 16S rRNA gene amplification sequencing and untargeted gas/liquid chromatography-mass spectrometry metabolomics analysis.
Results: The study revealed that the alpha-diversity of the gut microbiome in NSCLC patients at different stages did not exhibit statistically significant differences. Notably, Lachnospira and Blautia were more abundant in healthy controls. The distribution of gut microbial species in patients with varying stages of NSCLC was uneven, with Bacteroides and Bacteroidaceae being most prevalent in stage T2, and Prevotella dominating in stage T4. Levels of Ruminococcus gnavus were notably elevated in stages N3 and M. The genus levels of Klebsiella, Parabacteroides, and Tannerellaceae were higher in stage II patients. Rodentibacter was the bacterium with increased levels in stage III NSCLC patients. Further metabolomics studies revealed significantly elevated levels of quinic acid and 3-hydroxybenzoic acid in the healthy control group. In contrast, Stage I+II non-small cell lung cancer (NSCLC) patients exhibited reduced levels of L-cystathionine. Notably, quinic acid, phthalic acid, and L-lactic acid were observed to be increased in Stage III+IV NSCLC patients.
Conclusion: Compared to the analysis of a single microbial dataset, this study provides deeper functional insights by incorporating comprehensive metabolomic profiling. This approach demonstrates that both the gut microbiome and associated metabolites are altered in NSCLC patients across different clinical stages. Our findings may offer novel perspectives on the pathogenesis of NSCLC at various TNM stages. Further research is warranted to validate and clinically apply these potential biomarkers.
Keywords: non-small cell lung cancer, gut microbiome, metabolites, clinical stages
Background
Lung cancer (LC) remains the most prevalent form of cancer worldwide, with approximately 2.5 million new cases diagnosed in 2022, representing 12.4% of all cancer diagnoses worldwide. Non-small cell lung cancer (NSCLC)1 constitutes the majority (85%) of these cases. In China, LC poses a significant public health challenge, accounting for over a million new cases and deaths annually.2 The insidious onset of LC symptoms often results in late-stage diagnosis, significantly contributing to its high mortality rate. The limited efficacy of current therapeutic modalities further exacerbates this issue. Contemporary clinical practice employs a range of therapeutic modalities for LC management, including surgery, chemotherapy, radiotherapy, targeted therapies, and emerging immunotherapeutic strategies. The selection of these therapeutic modalities is primarily guided by the TNM staging system for LC patients. Consequently, the accurate and early determination of the various clinical stages of LC is of significant importance in facilitating precise therapeutic interventions and optimizing patient outcomes.
In addition to established risk factors for cancer, including genetic predisposition, smoking, air pollution, and occupational exposure, recent research has identified potential contributions of microbiome dysbiosis to LC.3 Research has demonstrated that LC patients exhibiting Veillonella parvula (V. parvula) overabundance presented with increased tumor burden, decreased survival rates, and enhanced activation of interleukin-17 and markers for immune-checkpoint inhibitors (ICIs).4 These findings suggest a possible association between V. parvula overabundance and LC progression. In a study of 142 LC patients, Greathouse et al5 observed that the pulmonary microbiome associated with tumor tissues exhibited reduced microbial diversity and stability. This finding provides a basis for differential diagnosis of LC. Furthermore, significant correlation between characteristic gut microbiome profiles and LC. Zhuang et al6 reported that the intestinal microbiome of LC patients exhibited low bacterial community richness, homogeneity, and diversity. The researchers also identified Ruminococcus and Eubacterium as bacterial genera potentially implicated in LC development and as predictive indicators for chemotherapeutic efficacy in LC patients. Furthermore, Zhao et al7 demonstrated that Streptococcus mutans and Enterococcus casseliflavus are biomarkers in chemotherapy-responsive patients, suggesting their potential influence on treatment outcomes.
Our previous studies on the association between NSCLC and the gut microbiome and its metabolites revealed that Ruminococcus gnavus (R. gnavus) was significantly upregulated in patients with NSCLC, while Firmicutes and Lachnospira were decreased.8 Furthermore, distinct gut microbiome profiles were associated with different pathological types of LC, suggesting potential utility as novel biomarkers for early diagnosis and pathological staging of LC.8 Furthermore, our findings revealed a reduction in the levels of gut microbiome metabolites, including quinic acid (QA) and 3-hydroxybenzoic acid, which are closely associated with the characteristics of R. gnavus, in LC patients.8 However, existing literature on the correlation between the gut microbiome, its metabolites, and the TNM staging of NSCLC is limited, and the impact of these factors on NSCLC progression remains unclear. To address these knowledge gaps, we employed 16S rRNA gene sequencing and an untargeted metabolomic approach to analyze the structural composition and differentially enriched metabolites of the gut microbiome in NSCLC patients across various TNM stages. The objective of this study is to explore the characteristics of the gut microbiome and its metabolites in relation to different TNM stages of NSCLC patients.
Materials and Methods
Study Participants
A total of 81 stool samples were collected from 52 patients diagnosed with non-small cell lung cancer (NSCLC) and 29 age-matched healthy controls at the West China Hospital of Sichuan University. The median age of the NSCLC patient cohort was 56.9 years (Table 1). All NSCLC diagnoses were confirmed by histopathological examination, followed by postoperative TNM staging. Participants were excluded if they had any of the following conditions within one month prior to sample collection: congestive heart failure, respiratory failure, intestinal disease, renal failure, severe liver abnormalities, or recent use of probiotics or antibiotics. Additionally, only patients who received a first-time diagnosis of NSCLC, without prior medication directly related to this diagnosis, and without other oncological comorbidities were included in the study. No statistically significant differences were observed between the two groups regarding age, body mass index (BMI), or gender distribution (P > 0.05). Furthermore, there were no significant differences in medical history, smoking status, family health history, lifestyle, or dietary habits at baseline. This study was conducted in accordance with the ethical guidelines approved by the ethics committee of the West China Hospital of Sichuan University.
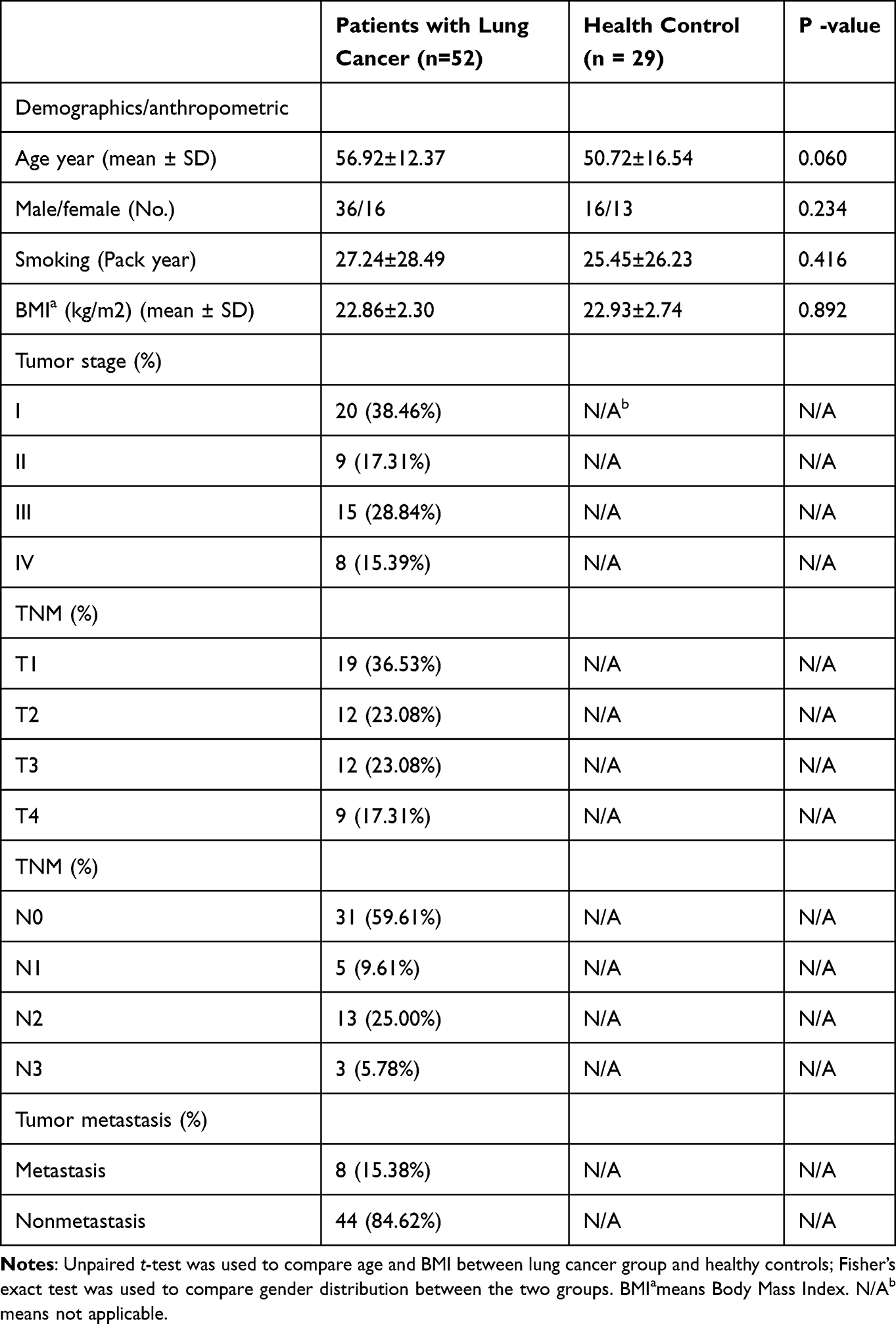 |
Table 1 Comparison of Clinical Information Between Lung Cancer Patients and Healthy Controls |
DNA Extraction and PCR Amplification
Stool samples were immediately snap-frozen post-collection and stored at −80°C. Bacterial DNA was extracted from cecal contents using the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. DNA concentration and integrity were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and agarose gel electrophoresis.8 A universal primer pair targeting the V3-V4 hypervariable region of the bacterial 16S rRNA gene was utilized for PCR amplification in 25 μL reactions. The forward primer sequence was 343F: 5′-TACGGRAGGCAGCAG-3′, and the reverse primer sequence was 798R: 5′-AGGTATCTAATCCT-3′.9 The reverse primers were appended with sample-specific barcodes, and both primers were linked to Illumina sequencing adapters.
Library Construction and Sequencing
The quality of the PCR amplicons was evaluated via agarose gel electrophoresis. Subsequently, PCR products were purified using AMPure XP beads (Agencourt), followed by an additional round of PCR amplification. Post-purification with AMPure XP beads, the final amplicons were quantified using the Qubit dsDNA Assay Kit (Thermo Fisher Scientific, USA). Concentrations were adjusted prior to sequencing. Sequencing was conducted on the Illumina NovaSeq 6000 platform, generating 250 bp paired-end reads. This work was performed at the sequencing facilities of Illumina Inc. (San Diego, CA, USA) and OE Biotech Company (Shanghai, China).8
Sample Preparation for Metabolome Profiling
Samples were thawed at room temperature after long-term storage at −80°C. For metabolomic analysis, 60 mg of fecal material was combined with 40 µL of L-2-chlorophenylalanine (0.06 mg/mL in methanol) as an internal standard in a 1.5 mL Eppendorf tube and vortexed. An additional 360 µL of ice-cold methanol/acetonitrile mixture (2:1v/v) was added. The extracts were subsequently subjected to centrifugation, freeze concentration, vacuum drying, and derivatization. The samples were analyzed via GC/LC-MS at ambient temperature.
Bioinformatic Analysis
Bioinformatic analysis of 16S rRNA sequencing results was conducted by OE Biotech Co., Ltd. (Shanghai, China). Raw sequencing data in FASTQ format underwent preprocessing using Cutadapt software for adapter identification and removal. Following trimming, paired-end reads were filtered for low-quality sequences, denoised, merged, and screened for chimeric reads using DADA2 with default QIIME2 parameters.10 Representative sequences and the ASV abundance table were generated by DADA2. Representative reads for each ASV were selected using the QIIME2 package and annotated through BLAST searches against the Silva database (version 138) using the QIIME2 feature classifier with default parameters.11 Alpha and beta diversity analyses were performed using QIIME2 software. Microbial diversity was estimated using R package-based PCoA analysis. Significant differences between groups were determined using ANOVA, Kruskal–Wallis, t-tests, and Wilcoxon tests. Taxonomic abundance spectra were compared using linear discriminant analysis effect size (LEfSe).8,11
Data Preprocessing and Statistical Analysis
For data preprocessing and statistical analysis, raw GC/LC-MS data in. D format were converted to .abf format using Analysis Base File Converter. The normalized data matrix obtained from MS-DIAL software was imported into R for Principal Component Analysis (PCA) to assess sample distribution and analytical stability. Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) was employed to identify metabolites that differed between groups. To avoid overfitting, 7-fold cross-validation and 200 response permutation tests (RPTs) were used to assess the quality of the models. The Variable Importance of Projection (VIP) values obtained from the OPLS-DA model were used to rank the overall contribution of the metabolites. Differential metabolites with VIP>1.0 and P<0.05 were screened.
Results
Features of Gut Microbiome Diversity
The study found that the differences in gut microbiome α-diversity between NSCLC patients at different TNM stages were not statistically significant. Furthermore, α-diversity did not show any correlation with the severity of TNM staging. It is noteworthy that the Chao1 and ACE levels of α-diversity in T2 patients were found to be lower than those observed in the T1 (p < 0.05) and HC groups (p < 0.01), respectively (Figure 1A and D). Furthermore, the Chao1 levels in N3 patients were observed to be lower than those observed in the HC groups (p < 0.01, Figure 1B), while the ACE levels were found to be smaller than those observed in the N0 and HC groups (p < 0.05, Figure 1E). Furthermore, the Chao1 levels in M patients were smaller than those observed in the HC groups (p < 0.05, Figure 1C), and the ACE levels were found to be lower than both the NM and HC group levels (p < 0.05, Figure 1F). Furthermore, a significant difference in β-diversity was observed between T- and M-stage NSCLC patients and healthy controls (p < 0.05, Figure 1G and H).
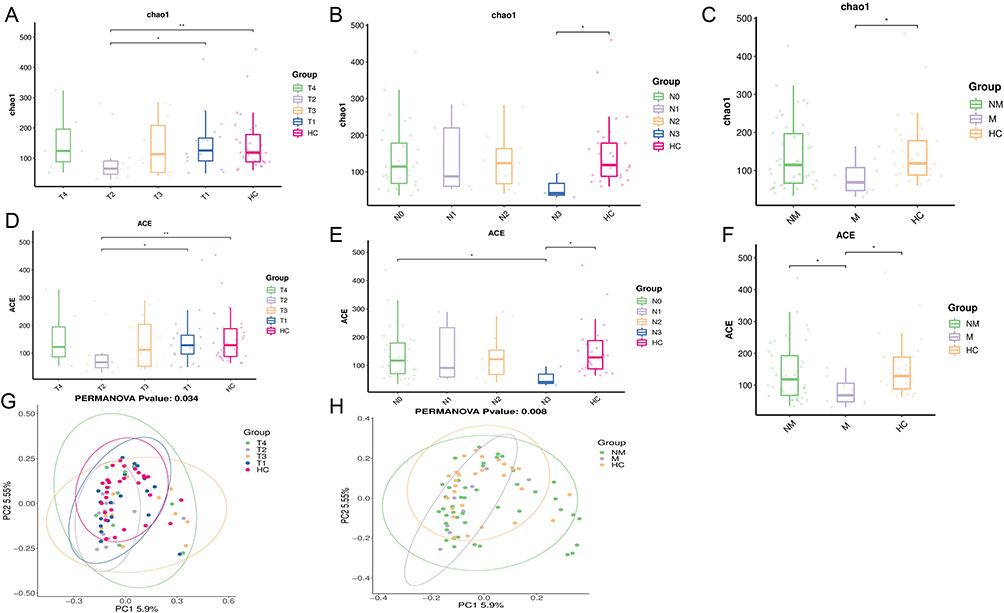 |
Figure 1 (A-C) Comparison of Chao1 richness, an indicator of alpha species diversity, between groups of lung cancer patients. (D-F) Comparison of ACE richness, an indicator of alpha species diversity, between groups of lung cancer patients. (G-H) β-diversity (expressed by principal coordinate analysis, PCoA) was significantly different between LC patients and healthy controls. HC means Healthy control group; The classification of T, N, and M staging was performed in accordance with the ninth edition of the TNM Classification of Lung Cancer. The T stage encompasses T0-T4, the N stage encompasses N0-N3, and the M stage encompasses both M and NM. *p < 0.05, **p < 0.01. |
Specific Species in Multi-Level Tests
Multilevel linear discriminant analysis (LDA) combined with biomarker effect size analysis (LEFSe) further identified specific dominant bacteria associated with different stages of NSCLC. The abundance of the gut metabolome was not uniformly distributed across the different stages of NSCLC patients, and the differences observed were not statistically significant. Notably, the highest abundance of Bacteroides and Bacteroidaceae was detected in stage T2, whereas the highest abundance of Prevotella was detected in stage T4 (Figure 2A-C). The highest colony abundance of Ruminococcus gnavus (R. gnavus) was detected in stages N3 and M (Figure 2E and G). Regarding clinical stage, the highest flora abundance of Klebsiella, Parabacteroides and Tannerellaceae was detected in lung cancer patients with stage II, whereas the highest abundance of Rodentibacter was detected in patients with stage III (Figure 2H and I). Lachnospira and Blautia were detected at the highest colony frequencies in healthy controls (Figure 2D and F).
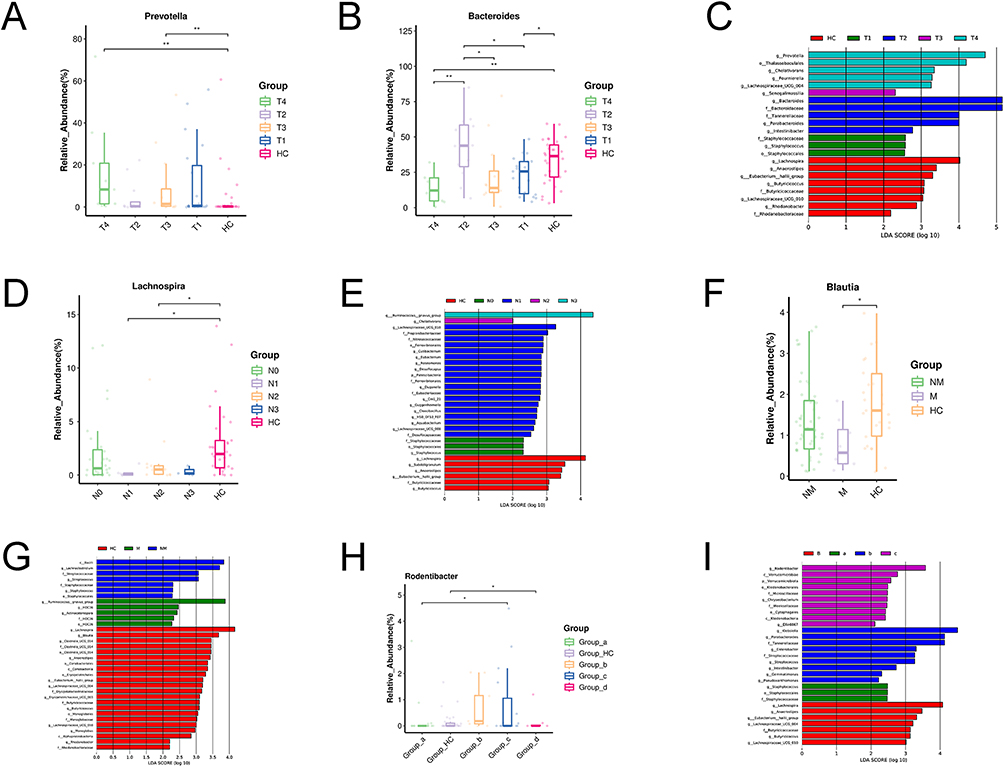 |
Figure 2 LEfSe discriminant analysis of species differences. (A-C) Discriminant analysis of LEfSe species difference between T stages and healthy group. (D and E) Discriminant analysis of LEfSe species difference between N stages and healthy group. (F and G) Discriminant analysis of LEfSe species difference between metastatic and non metastatic of LC. (H and I) A linear discriminant analysis (LDA) effect size (LEfSe) analysis was conducted to examine the abundance of gut microbiota in lung cancer by different TNM stages [stage I(a) vs stage II(b) vs stage III (c) vs stage IV (d)] and healthy group. HC means Healthy control group; *means 0.01≤P<0.05; **means 0.001≤P<0.01. The classification of T, N, and M staging was performed in accordance with the ninth edition of the TNM Classification of Lung Cancer. The T stage encompasses T0-T4, the N stage encompasses N-N3, and the M stage encompasses both M and NM. The TNM stages encompasses I, II, III, IV clinical stage. a means stage I; b means stage II; c means stage III; d means stage IV. |
Characterising the Gut Metabolome in NSCLC Patients
In this study, we integrated actual clinical diagnosis and treatment with the 9th edition of the lung cancer TNM staging guidelines recommended by the International Association for the Study of Lung Cancer (IASLC).12 Due to the discrepancy in the 5-year survival rate of NSCLC patients, the subjects were divided into two experimental groups: stage I+II and stage III+IV. The aim was to investigate two different levels of gut microbial metabolites by untargeted GC/LC-MS metabolic analysis. A permutation response test (RPT) was performed and the model fit (R2Y) and predictive ability (Q2Y) values indicated that the OPLS-DA model had good fit and predictive ability (Figure S1). The OPLS-DA plots generated by the supervised analysis function adequately reflected the differential metabolites between the LC and HC groups, and the model was valid and not overfitted (Figure 3A-D).
 |
Figure 3 The distribution of gut microbial metabolites in NSCLC patients at different TNM stages was analysed using orthogonal projections to latent structures–discriminant analysis (OPLS-DA) and volcano diagrams. (A and B) OPLS-DA results in stage I+II patients and healthy controls (GC-MS, LC-MS).(C and D) OPLS-DA results in stage I+II versus stage III+IV patients (GC-MS, LC-MS). (E and F) Volcano plots of stage I+II patients versus healthy controls (GC-MS, LC-MS). (G and H) Volcano plots of stage III+IV patients versus healthy controls (GC-MS, LC-MS). (I and J) Volcano plots of stage I+II patients versus stage III+IV (GC-MS, LC-MS). NSCLC means Non-small cell lung cancer. HC means Healthy control group; OPLS-DA means Orthogonal Partial Least-Squares-Discriminant Analysis; GC-MS means Gas chromatography-mass spectrometry technique; LC-MS means Liquid chromatography-mass spectrometry technique; Lung cancer staging according to the 9th edition of TNM, including clinical stages I, II, III, and IV. |
In this study, the default screening criteria for differential abundance metabolites were set as variable importance in the projection VIP >1.0, P <0.05, FC<0.5 or >2. These values were plotted as volcano plots and differential abundance metabolite heatmaps (Figure S2). The differential metabolites were predominantly downregulated in patients with I–IV stage of NSCLC compared to healthy controls (Figure 3E-H). Compared to patients with stage III+IV, GC/LC-MS-identified differential metabolites showed predominantly upregulated expression in the majority of patients with stage I+II disease (Figure 3I and J).
Potential Alteration in Functional Metabolic Pathways of NSCLC
Based on KEGG annotation results, we found that amino acid metabolism was significantly associated with metabolites of different abundances, including those involved in beta-alanine metabolism, phenylalanine, tyrosine, and tryptophan biosynthesis, vitamin digestion and absorption, and glycine, serine, and threonine metabolism, in both early and advanced NSCLC patients compared to healthy controls (p<0.05, Figure 4A). Furthermore, there were significant discrepancies in phenylalanine, tyrosine and tryptophan biosynthesis and metabolism between III+IV stage of NSCLC patients (p<0.05, Figure 4B). Compared with the III+IV group, the main pathways of glutathione metabolism, central carbon metabolism in cancer, ABC transporters and arginine biosynthesis showed significant differences in the I+II group (p<0.05, Figure 4C). In our study, we collated the faecal metabolic data with the aim of identifying key biomarkers and providing guidance on the pathogenesis and clinical research of NSCLC of different severity. As shown in the results (p <0.05, VIP>1.0), quinic acid and 3-hydroxybenzoic acid levels were significantly elevated in the healthy cohort (Figure 5A and B). The levels of L-cystathionine are reduced in patients with I+II stage of NSCLC. (Figure 5C). Comparison with healthy controls, quinolinic acid, phthalic acid and L-lactic acid were found to be upregulated in stage III+IV, in addition, phthalic acid levels were significantly higher in stage III+IV lung cancer patients than in stage I+II patients and healthy controls.(Figure 5D-F). It was observed that the majority of these NSCLC gut microbial metabolites had the effect of downregulating a number of metabolic pathways. In contrast, quinolinic acid was shown to upregulate the tryptophan pathway (Table S1).
 |
Figure 4 Enrichment analysis of GC/LC-MS metabolites in faecal samples from NSCLC with different TNM stages (A) I+II stages of patients and healthy controls (B) III+IV stages of patients and healthy controls. (C) I+II stages and III+IV stages of patients. GC-MS means Gas chromatography-mass spectrometry technique; LC-MS means Liquid chromatography-mass spectrometry technique; Lung cancer staging according to the 9th edition of TNM, including clinical stages I, II, III, and IV. NSCLC means Non-small cell lung cancer. |
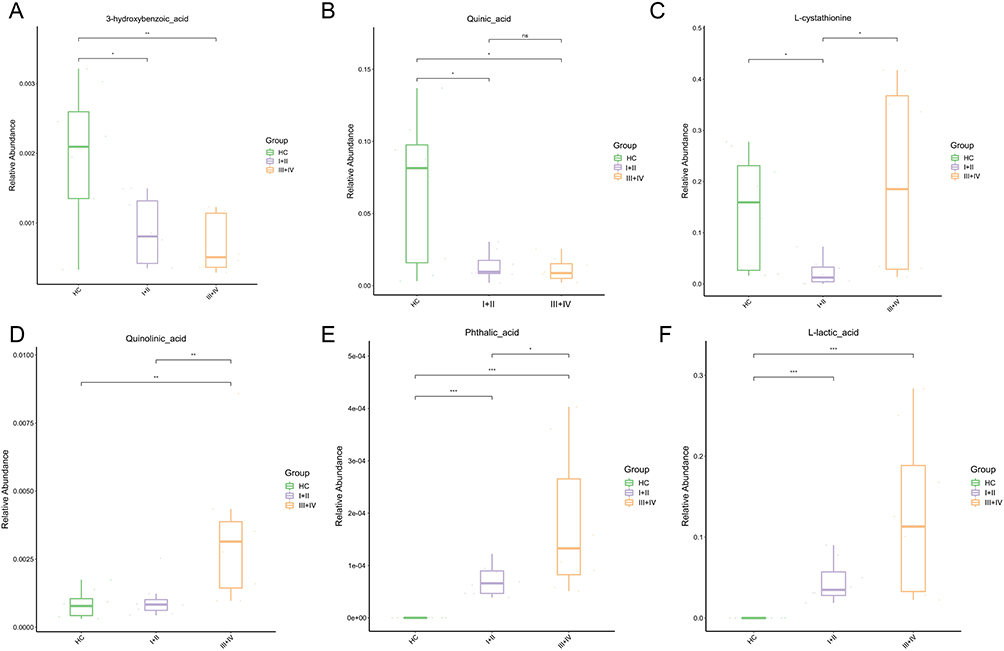 |
Figure 5 Analysis of differences in GC/LC-MS metabolites in fecal samples from NSCLC patients at different clinical stages. (A-D) Different stages of NSCLC compared to healthy people. (E and F) I+II stages and III+IV stages of NSCLC patients. *means 0.01≤P<0.05; **means 0.001≤P<0.01; ***means P<0.001; ns means P>0.05. Lung cancer staging according to the 9th edition of TNM, including clinical stages I, II, III, and IV. GC-MS means Gas chromatography-mass spectrometry technique; LC-MS means Liquid chromatography-mass spectrometry technique. NSCLC means Non-small cell lung cancer. |
Discussion
Our study reveals significant alterations in the community structure (measured by β-diversity) of the gut microbiome in NSCLC patients across the T and M stages. However, the microbial diversity (measured by α-diversity) did not show consistent changes corresponding to TNM progression, with reductions observed only in certain T2, N3, and M stages. These inconsistent biodiversity shifts may be attributed to limited sample size, modest patient numbers at each stage, and considerable inter-individual variability. Furthermore, a notable divergence in microbial profiles was observed between healthy controls and NSCLC patients. Lachnospira, a member of the phylum Firmicutes and a producer of short-chain fatty acids (SCFAs), was significantly reduced in NSCLC patients compared to healthy individuals. Lachnospira is generally considered a beneficial bacterium for the host, exhibiting anti-inflammatory properties, modulating host immune responses, and maintaining intestinal barrier integrity.13 Studies have shown that Lachnospira is significantly reduced in the gut microbiome of patients with colorectal cancer (CRC)14 and cervical cancer,15 suggesting its potential as a diagnostic biomarker. Our study revealed a significantly lower abundance of probiotic Lachnospira in NSCLC patients compared to healthy individuals. This reduction in Lachnospira, a source of SCFAs in the human gut, may contribute to NSCLC progression, aligning with previous findings in this field. However, some studies have reported increased Lachnospira abundance in patients with poor breast cancer prognosis.16 Bacteroides typically demonstrate symbiotic characteristics and may possess tumor-suppressive properties, potentially offering protection to humans. Reduced abundance of Bacteroides has been observed in patients with esophageal cancer,17 and higher levels are associated with improved responses to ICIs in NSCLC patients.18 Nevertheless, these organisms can also exhibit pathogenic potential. In contrast to HCs, Bacteroides are strongly associated with bile duct19 and grade II breast cancers,16 and are among the most enriched microbial species serving as gut microbiome markers for cancer diagnosis. This dual nature suggests that Bacteroides may play both beneficial and pathogenic roles in human health. Our study demonstrated a notable increase in the abundance of Bacteroides and Bacteroidaceae at the T2 stage in NSCLC patients. This observation may be attributed to dysregulated gut ecology stimulating Bacteroides to secrete toxins that promote malignant tumor development through cell proliferation and tumor inflammation, thus potentially contributing to tumor cell growth and pleural invasion.20 Although Bacteroides and Bacteroidaceae do not appear to be affected by the T-classification of lung cancer, it is hypothesized that their dysregulation may contribute to the pathogenesis of NSCLC.
Additionally, a significant increase in the incidence of R. gnavus was observed in the N3 and M stages of NSCLC patients. However, R. gnavus did not exhibit a consistent trend with increasing N grading, and further evidence is required to determine its potential as a characteristic intestinal microbial indicator for N3 and M stages. R. gnavus is a member of the phylum Firmicutes and was found to be significantly enriched in colorectal cancer patients who do not respond to cytotoxicity or targeted chemotherapy. This indicates that it may serve as a potential biomarker for monitoring treatment efficacy in colorectal cancer.21 R. gnavus has been identified as a distinctive taxon in patients with hepatocellular carcinoma, as opposed to intrahepatic cholangiocarcinoma patients.22 In vitro studies demonstrated that exposure of respiratory epithelial cells to Prevotella activates the ERK and PI3K signaling pathways, promoting tumor formation. It is hypothesized that Prevotella is generally associated with poor prognosis and tumor progression in lung cancer.23 Additionally, researchers observed that Prevotella levels were not significantly different between healthy subjects and patients diagnosed with hyperplastic/in situ carcinoma. Conversely, elevated levels were observed in patients diagnosed with minimally invasive adenocarcinoma or invasive adenocarcinoma.24 Moreover, it has been demonstrated that the genus Prevotella is the most prevalent in bronchoalveolar lavage fluid (BALF)25 and saliva26 of LC patients. In our study, patients with NSCLC exhibited the highest abundance of Prevotella at the T4 stage. However, the correlation between abundance and T-grade was not statistically significant.
Furthermore, our study revealed that Klebsiella, Parabacteroides, and Tannerellaceae exhibited significantly elevated abundance in stage II NSCLC patients, suggesting their potential utility as biomarkers for this stage. Conversely, Rodentibacter was identified as the most abundant in stage III. Klebsiella has been shown to be significantly increased in cancerous tissues of bladder cancer patients. The genotoxin produced by Escherichia coli (E. coli) can cause DNA double-strand breaks, induce genomic instability and cell cycle arrest, lead to chronic inflammation, and stimulate epithelial cell proliferation, thus contributing to tumor formation.27,8 These modifications may be directly or indirectly associated with the malignant progression of NSCLC. This study identifies intestinal metabolites affecting lung cancer progression as primarily involved in pathways associated with amino acid anabolism, vitamin digestion and absorption, and ABC transporter proteins.
Our analysis revealed that QA and 3-hydroxybenzoic acid were expressed in healthy individuals, while their levels were markedly reduced in NSCLC patients across stages I–IV. This observation suggests a potential inhibitory role of these metabolites in NSCLC progression. Previous studies have demonstrated the anticancer effects of QA and 3-hydroxybenzoic acid on LC.8 These metabolites are primarily involved in pathways associated with the biosynthesis of phenylalanine, tyrosine, and tryptophan throughout the LC progression. These pathways regulate cell survival through the conversion of tryptophan and nicotinamide.34 QA-16 (QA derivative) has demonstrated anticancer effects on glioblastoma, indicating its potential as a chemotherapeutic agent.34 The study revealed a positive correlation between the circulating levels of 3-hydroxybenzoic acid and the risk of developing colon cancer in men.35 L-cystathionine is an amino acid-containing non-protein sulfide that is produced primarily during the metabolic conversion of methionine to cysteine in humans.36 This study identified L-cystathionine as a crucial element in the metabolic pathways of glycine, serine, and threonine in patients with NSCLC. This finding suggests that the gut microbiome may exert a preventive and therapeutic effect on NSCLC by secreting L-cystathionine.
Inhibition of quinolinic acid secretion, a toxic metabolite, was demonstrated to be an effective treatment for invasive breast cancer, particularly in patients with triple-negative breast cancer.37 In vitro studies also demonstrated the antiproliferative and cytotoxic effects of quinolinic acid on melanoma cells, suggesting potential avenues for future therapeutic strategies when used in combination with other melanoma treatments.38 The results of our study indicated that quinolinic acid was significantly expressed and secreted in patients with stage III+IV lung cancer and was implicated in pathways associated with tryptophan metabolism in NSCLC. We hypothesized that tumor cells in NSCLC secrete quinolinic acid through high expression and utilize tryptophan and its metabolites to promote their growth and evade host defenses. However, this study did not identify a correlation between quinolinic acid secretion and progression in NSCLC patients. Further investigation with an expanded sample size and refined inclusion criteria is required to validate this finding.
As the severity of NSCLC increased, concentrations of phthalic acid and L-lactic acid metabolites increased significantly. These metabolites were observed to promote the progression of LC to stages III+IV, primarily through participation in ABC transporters and central carbon metabolism in cancer pathways. Phthalic acid is associated with an elevated risk of hormone receptor-negative breast cancer39 and potentially linked to prostate tumors in the US population,40 suggesting that phthalic acid-related metabolites can promote the development and progression of both breast and prostate cancers. L-lactic acid was found to be highly expressed in patients with high-grade squamous intraepithelial lesions (SILs) and cervical cancer.41 As a major circulating carbon metabolite, lactic acid regulates energy metabolism and signaling pathways in cancer cells, serving as both an energy source and a signaling molecule. High levels of L-lactic acid induce tumor metastasis and drug resistance.42 Compared to early-stage NSCLC patients, the gut microbiome accelerates NSCLC tissue metastasis by upregulating phthalic acid and L-lactic acid levels. Therefore, inhibition of the synthesis and secretion of phthalic acid and L-lactic acid metabolites may be a potential method for targeted cancer treatment, providing a new strategy for NSCLC treatment. Importantly, the differences in the metabolites did not correlate well with the differences in characteristic gut microbiome across NSCLC stages. This finding is consistent with our previous research, which showed that probiotics like Firmicutes and Lachnospira were positively associated with most metabolites, whereas opportunistic pathogens such as R. gnavus were negatively associated.8 In conclusion, the gut microbiome may play a critical role in NSCLC development by regulating metabolite secretion. However, more research is needed to determine whether there is a correlation between specific microbiome profiles and NSCLC staging. However, this study has several limitations. 1) The sample size of NSCLC patients was limited, which may affect the representativeness of the results. 2) While we identified correlations between the gut microbiome, its metabolites, and different TNM stages in NSCLC, no causality was established. 3) The study population had diverse genetic backgrounds, dietary habits, and environmental exposures. 4) Typical taxonomic levels in 16S rRNA gene sequencing analysis encompass phylum, class, order, family, and genus. Annotation of individual microorganisms is generally limited to the genus level. The 16S rRNA gene sequencing methodology possesses inherent limitations that may influence the accuracy of the results. 5) Additional histological and blood samples of NSCLC patients are needed for transcriptomic and metabolomic analyses to validate and investigate the interaction between the gut microbiome and NSCLC development.
Conclusion
This study represents the first investigation into the dysregulation of gut microbiota in NSCLC patients across various TNM stages, coupled with an analysis of alterations in gut metabolites. Our findings reveal a reduction in protective gut microbiota, such as Lachnospira, and an increase in microbiota that may facilitate disease progression, such as Prevotella. These results provide new insights into the pathogenesis of NSCLC at different TNM stages, suggesting that gut microorganisms and their metabolites may play a role in the development of stage-specific NSCLC. Furthermore, these findings indicate the potential of gut microbiota and associated metabolites as biomarkers for NSCLC patients at different TNM stages. This study provides an initial perspective on the potential correlation between lung cancer and gut microbes. Nevertheless, it is clear that additional research is necessary, which will be a protracted and arduous journey.
Data Sharing Statement
The NCBI Sequence Read Archive (SRA) PRJNA882575 contains the datasets presented in this study.
Ethics Statement
The study was free and comfortable for the volunteers, and the results will be used for scientific research. We declare that none of the authors have any conflicts of interest or financial relationships to disclose. This study was conducted in accordance with the recommendations of the Ethical Guidelines for Biomedical Research, West China Hospital, Sichuan University. The protocol was approved by the Ethics Review Committee of West China Hospital, Sichuan University (reference: 2021-1120, 04/11/2021). Written informed consent was obtained from all subjects in accordance with the Declaration of Helsinki. The rights of the subjects were adequately protected and there was no potential risk to the subjects.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The research was supported by grants from the Special Foundation for Natural Science Foundation of Sichuan (Grant No. 2023NSFSC1890).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Zhou Y, Zeng H, Liu K, et al. Microbiota profiles in the saliva, cancerous tissues and its companion paracancerous tissues among Chinese patients with lung cancer[J]. BMC Microbiol. 2023;23(1):237. doi:10.1186/s12866-023-02882-1
3. Mao Q, Jiang F, Yin R, et al. Interplay between the lung microbiome and lung cancer[J]. Cancer Lett. 2018;415:40–48. doi:10.1016/j.canlet.2017.11.036
4. Tsay JJ, Wu BG, Sulaiman I, et al. Lower airway dysbiosis affects lung cancer progression[J]. Cancer Discov. 2021;11(2):293–307. doi:10.1158/2159-8290.CD-20-0263
5. Greathouse KL, White JR, Vargas AJ, et al. Interaction between the microbiome and TP53 in human lung cancer[J]. Genome Biol. 2018;19(1):123. doi:10.1186/s13059-018-1501-6
6. Zhuang H, Cheng L, Wang Y, et al. Dysbiosis of the gut microbiome in lung cancer[J]. Front Cell Infect Microbiol. 2019;9:112. doi:10.3389/fcimb.2019.00112
7. Zhao Z, Fei K, Bai H, et al. Metagenome association study of the gut microbiome revealed biomarkers linked to chemotherapy outcomes in locally advanced and advanced lung cancer[J]. Thorac Cancer. 2021;12(1):66–78. doi:10.1111/1759-7714.13711
8. Lu X, Xiong L, Zheng X, et al. Structure of gut microbiota and characteristics of fecal metabolites in patients with lung cancer[J]. Front Cell Infect Microbiol. 2023;13:1170326. doi:10.3389/fcimb.2023.1170326
9. Nossa CW, Oberdorf WE, Yang L, et al. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome[J]. World J Gastroenterol. 2010;16(33):4135–4144. doi:10.3748/wjg.v16.i33.4135
10. Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2[J]. Nat Biotechnol. 2019;37(8):852–857. doi:10.1038/s41587-019-0209-9
11. Hill TC, Walsh KA, Harris JA, et al. Using ecological diversity measures with bacterial communities[J]. FEMS Microbiol Ecol. 2003;43(1):1–11. doi:10.1111/j.1574-6941.2003.tb01040.x
12. Rami-Porta R, Nishimura KK, Giroux DJ, et al. The international association for the study of lung cancer lung cancer staging project: proposals for revision of the TNM stage groups in the forthcoming (Ninth) Edition of the TNM classification for lung cancer[J]. J Thorac Oncol. 2024;19(7):1007–1027. doi:10.1016/j.jtho.2024.02.011
13. Silva YP, Bernardi A, Frozza RL. The role of short-chain fatty acids from gut microbiota in gut-brain communication[J]. Front Endocrinol. 2020;11:25. doi:10.3389/fendo.2020.00025
14. Fan JQ, Zhao WF, Lu QW, et al. Fecal microbial biomarkers combined with multi-target stool DNA test improve diagnostic accuracy for colorectal cancer[J]. World J Gastrointest Oncol. 2023;15(8):1424–1435. doi:10.4251/wjgo.v15.i8.1424
15. Sims TT, Colbert LE, Zheng J, et al. Gut microbial diversity and genus-level differences identified in cervical cancer patients versus healthy controls[J]. Gynecol Oncol. 2019;155(2):237–244. doi:10.1016/j.ygyno.2019.09.002
16. Jiang Y, Gong W, Xian Z, et al. 16S full-length gene sequencing analysis of intestinal flora in breast cancer patients in Hainan Province[J]. mol Cell Probes. 2023;71:101927. doi:10.1016/j.mcp.2023.101927
17. Deng Y, Tang D, Hou P, et al. Dysbiosis of gut microbiota in patients with esophageal cancer[J]. Microb Pathog. 2021;150:104709. doi:10.1016/j.micpath.2020.104709
18. Newsome RC, Gharaibeh RZ, Pierce CM, et al. Interaction of bacterial genera associated with therapeutic response to immune checkpoint PD-1 blockade in a United States cohort[J]. Genome Med. 2022;14(1):35. doi:10.1186/s13073-022-01037-7
19. Re OL, Lopez-Lopez V, Balaguer-Roman A, et al. New challenges in cholangiocarcinoma candidates for elective surgery: harnessing the microbiome dysbiosis[J]. Langenbecks Arch Surg. 2023;408(1):134. doi:10.1007/s00423-023-02867-8
20. Lopez LR, Bleich RM, Arthur JC. Microbiota effects on carcinogenesis: initiation, promotion, and progression[J]. Annu Rev Med. 2021;72(1):243–261. doi:10.1146/annurev-med-080719-091604
21. Kolisnik T, Sulit AK, Schmeier S, et al. Identifying important microbial and genomic biomarkers for differentiating right-versus left-sided colorectal cancer using random forest models[J]. BMC Cancer. 2023;23(1):647. doi:10.1186/s12885-023-10848-9
22. Pomyen Y, Chaisaingmongkol J, Rabibhadana S, et al. Gut dysbiosis in Thai intrahepatic cholangiocarcinoma and hepatocellular carcinoma[J]. Sci Rep. 2023;13(1):11406. doi:10.1038/s41598-023-38307-2
23. Gaeckle NT, Pragman AA, Pendleton KM, et al. The oral-lung axis: the impact of oral health on lung health[J]. Respir Care. 2020;65(8):1211–1220. doi:10.4187/respcare.07332
24. Qin X, Bi L, Yang W, et al. Dysbiosis of the gut microbiome is associated with histopathology of lung cancer[J]. Front Microbiol. 2022;13:918823. doi:10.3389/fmicb.2022.918823
25. Cheng C, Wang Z, Wang J, et al. Characterization of the lung microbiome and exploration of potential bacterial biomarkers for lung cancer[J]. Transl Lung Cancer Res. 2020;9(3):693–704. doi:10.21037/tlcr-19-590
26. Bingula R, Filaire E, Molnar I, et al. Characterisation of microbiota in saliva, bronchoalveolar lavage fluid, non-malignant, peritumoural and tumour tissue in non-small cell lung cancer patients: a cross-sectional clinical trial[J]. Respir Res. 2020;21(1):129. doi:10.1186/s12931-020-01392-2
27. Mansour B, Monyok A, Makra N, et al. Bladder cancer-related microbiota: examining differences in urine and tissue samples[J]. Sci Rep. 2020;10(1):11042. doi:10.1038/s41598-020-67443-2
28. Bossuet-Greif N, Vignard J, Taieb F, et al. The colibactin genotoxin generates DNA interstrand cross-links in infected cells[J]. mBio. 2018;9(2). doi:10.1128/mBio.02393-17
29. Qi YF, Sun JN, Ren LF, et al. Intestinal microbiota is altered in patients with gastric cancer from Shanxi Province, China[J]. Dig Dis Sci. 2019;64(5):1193–1203. doi:10.1007/s10620-018-5411-y
30. Bian Y, Chen X, Cao H, et al. A correlational study of Weifuchun and its clinical effect on intestinal flora in precancerous lesions of gastric cancer[J]. Chin Med. 2021;16(1):120. doi:10.1186/s13020-021-00529-9
31. Sheng QS, He KX, Li JJ, et al. Comparison of gut microbiome in human colorectal cancer in paired tumor and adjacent normal tissues[J]. Onco Targets Ther. 2020;13:635–646. doi:10.2147/OTT.S218004
32. Ren Z, Li A, Jiang J, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma[J]. Gut. 2019;68(6):1014–1023. doi:10.1136/gutjnl-2017-315084
33. Grenda A, Iwan E, Krawczyk P, et al. Attempting to identify bacterial allies in immunotherapy of NSCLC patients[J]. Cancers. 2022;14(24). doi:10.3390/cancers14246250
34. Murugesan A, Holmstedt S, Brown KC, et al. Design and synthesis of novel quinic acid derivatives: in vitro cytotoxicity and anticancer effect on glioblastoma[J]. Future Med Chem. 2020;12(21):1891–1910. doi:10.4155/fmc-2020-0194
35. Mori N, Murphy N, Sawada N, et al. Prediagnostic plasma polyphenol concentrations and colon cancer risk: the JPHC nested case-control study[J]. Clin Nutr. 2022;41(9):1950–1960. doi:10.1016/j.clnu.2022.06.041
36. Wang X, Wang Y, Zhang L, et al. L-cystathionine protects against homocysteine-induced mitochondria-dependent apoptosis of vascular endothelial cells[J]. Oxid Med Cell Longev. 2019;2019:1253289. doi:10.1155/2019/1253289
37. Sordillo LA, Sordillo PP. Suppression of kynurenine 3-monooxygenase as a treatment for triple-negative breast carcinoma[J]. Anticancer Res. 2023;43(12):5275–5282. doi:10.21873/anticanres.16731
38. Basson C, Serem JC, Hlophe YN, et al. An in vitro investigation of l-kynurenine, quinolinic acid, and kynurenic acid on B16 F10 melanoma cell cytotoxicity and morphology[J]. Cell Biochem Funct. 2023;41(7):912–922. doi:10.1002/cbf.3843
39. Wu AH, Franke AA, Wilkens LR, et al. Urinary phthalate exposures and risk of breast cancer: the Multiethnic Cohort study[J]. Breast Cancer Res. 2021;23(1):44. doi:10.1186/s13058-021-01419-6
40. Guo T, Meng X, Liu X, et al. Associations of phthalates with prostate cancer among the US population[J]. Reprod Toxicol. 2023;116:108337. doi:10.1016/j.reprotox.2023.108337
41. de Magalhaes C, Linhares IM, Masullo LF, et al. Comparative measurement of D- and L-lactic acid isomers in vaginal secretions: association with high-grade cervical squamous intraepithelial lesions[J]. Arch Gynecol Obstet. 2022;305(2):373–377. doi:10.1007/s00404-021-06258-6
42. Nguyen K, Chiou JY, Liu YC, et al. l-lactic acidosis confers insensitivity to PKC inhibitors by competing for uptake via monocarboxylate transporters[J]. J Cell Physiol. 2022;237(1):934–948. doi:10.1002/jcp.30570
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.