")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
Hub Genes PRPF19 and PPIB: Molecular Pathways and Potential Biomarkers in COPD
Authors Zhao J, Ge X, Li H, Jing G, Ma W, Fan Y, Chen J , Zhao Z, Hou J
Received 24 December 2024
Accepted for publication 3 June 2025
Published 11 June 2025 Volume 2025:20 Pages 1865—1880
DOI https://doi.org/10.2147/COPD.S511696
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Min Zhang
Jiale Zhao,1,2,* Xiahui Ge,3,* Hailong Li,4,* Genfei Jing,5,* Weirong Ma,2 Yuchun Fan,2 Juan Chen,2,6 Zhijun Zhao,7,8 Jia Hou2,6
1School of Clinical Medicine, Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 3Department of Respiratory and Critical Care Medicine, Shanghai Ninth People’s Hospital, Shanghai, People’s Republic of China; 4Department of Respiratory Medicine, Ningxia Hospital of Integrated Traditional Chinese and Western Medicine, Yinchuan, Ningxia, People’s Republic of China; 5Department of Respiratory and Critical Care Medicine, Yongning County People’s Hospital, Yinchuan, Ningxia, People’s Republic of China; 6Department of Key Laboratory of Ningxia Stem Cell and Regenerative Medicine, Institute of Medical Sciences, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 7Clinical Laboratory Center, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 8Ningxia Key Laboratory of Clinical and Pathogenic Microbiology, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jia Hou, Email [email protected] Zhijun Zhao, Email [email protected]
Background: Chronic Obstructive Pulmonary Disease (COPD), a complex respiratory disorder, results from genetic and environmental factors. Uncovering its genetic basis is vital for diagnostics and treatment. Robust genetic analysis is essential to establish a causal link.
Methods: Genome-wide DNA methylation analysis was performed using the Illumina Infinium HumanMethylation850 BeadChip in peripheral blood from 8 COPD patients and 8 healthy smoking controls. Differentially methylated genes (DMGs) were cross-analyzed with differentially expressed genes (DEGs) identified from the Gene Expression Omnibus (GEO) dataset GSE38974 (23 COPD, 9 controls). Weighted gene co-expression network analysis (WGCNA) and protein-protein interaction (PPI) networks were utilized to identify COPD-associated hub genes. Mendelian randomization (MR) analysis examined the causal relationship between hub genes and COPD. The expression of selected hub genes was validated through RT-qPCR (80 COPD, 62 controls), immunohistochemistry, and Western blot analyses (10 COPD and 10 controls).
Results: We found 10,593 DMGs and 646 DEGs associated with COPD. These genes were compared with WGCNA module genes, and the Protein-Protein Interaction (PPI) network interaction diagram was drawn, thereby identifying five Hub genes: PPIB, HSPA2, PRPF19, FKBP10 and DOHH. The expression levels of DOHH, FKBP10, PPIB and PRPF19 are higher in COPD, while the expression level of HSPA2 is lower. MR results indicate a potential causal relationship between PRPF19, PPIB and COPD. RT-qPCR, immunohistochemistry and Western blot experiments verified that the expression of PRPF-19 and PPIB was up-regulated in peripheral blood and lung tissue, which was consistent with the results of bioinformatics analysis.
Conclusion: Our findings suggest that PRPF19 and PPIB may serve as promising diagnostic biomarkers in COPD. Further studies are required to fully elucidate their roles in COPD pathogenesis.
Keywords: chronic obstructive pulmonary disease, epigenetic susceptibility, hub genes, protein-protein interaction, Mendelian randomization
Introduction
COPD is one of the leading causes of morbidity and mortality worldwide, affecting over 380 million people. It remains the third leading cause of death, responsible for approximately 3.23 million deaths annually, as reported by the World Health Organization (WHO).1 COPD is characterized by persistent airflow limitation and a progressive decline in lung function, significantly burdening healthcare systems globally with both direct medical costs and indirect expenses from loss of productivity. Although advancements in treatments have improved symptom management and reduced acute exacerbations, there remains a critical need for therapeutic breakthroughs to slow disease progression and reduce mortality.2
DNA methylation, a key epigenetic mechanism that regulates gene expression by adding a methyl group to the DNA molecule, plays a critical role in the interplay between genetic information, environmental factors, and the transcriptome.3 This link between DNA methylation and COPD has been extensively studied in recent years, with research demonstrating that changes in DNA methylation patterns of specific genes are associated with the development and progression of COPD.4,5 These modifications are caused by various factors such as oxidative stress, environmental and genetic factors like CS exposure, nutrition, genetic variation, and age.6 Studies have shown that DNA methylation modifications resulting from cigarette smoke exposure can impact cellular processes and contribute to the progression of COPD.7–9 Previously identified methylation-associated genes in COPD include AHRR, F2RL3, and CYP1A1, underscoring the potential utility of these epigenetic markers for disease diagnosis and prognosis.9–11
Recent studies have employed various DNA methylation profiling technologies such as the Illumina Infinium HumanMethylation850 BeadChip array, offering comprehensive coverage and high resolution, facilitating the identification of novel biomarkers. In addition, bioinformatics analyses like weighted gene co-expression network analysis (WGCNA) and protein-protein interaction (PPI) network analysis have been instrumental in uncovering critical genes and pathways involved in COPD. Moreover, Mendelian Randomization (MR), a method leveraging genetic variants as instrumental variables, provides robust causal inference between genetic variation and complex diseases, reducing confounding bias.12–14
This study utilized an integrative approach, combining genome-wide DNA methylation and gene expression data with WGCNA, PPI network, and MR analysis to identify and validate hub genes potentially central to COPD pathogenesis. Our comprehensive analysis highlights PRPF19 and PPIB as promising biomarkers associated with COPD, providing potential targets for future therapeutic strategies.
Materials and Methods
Patient Enrolment and Study Design
The study, approved by the Ethics Committee and Medical Faculty of Ningxia Medical University (approval no. 2020–678), adhered to the Declaration of Helsinki. Written consent was obtained from participants recruited at Ningxia Medical University Hospital, China. COPD patients in stable condition, meeting diagnostic criteria excluding severe alpha-1 antitrypsin deficiency, were selected, with exclusion criteria for other respiratory diseases, hypertension, diabetes, autoimmune diseases, and malignancies. Controls were age- and sex-matched smokers without COPD or chronic diseases (Table 1). Genome-wide DNA methylation analysis of >485,000 CpG sites used the Illumina Infinium Human Methylation 850 BeadChip array for 8 COPD and 8 controls. Their fresh peripheral blood was collected to extract mononuclear cells for sequencing. Subsequent RT-qPCR analysis validated identified differentially methylated genes in a larger cohort of 80 COPD patients and 62 smoking controls. The clinical characteristics of these subjects/patients are listed in Table 1.
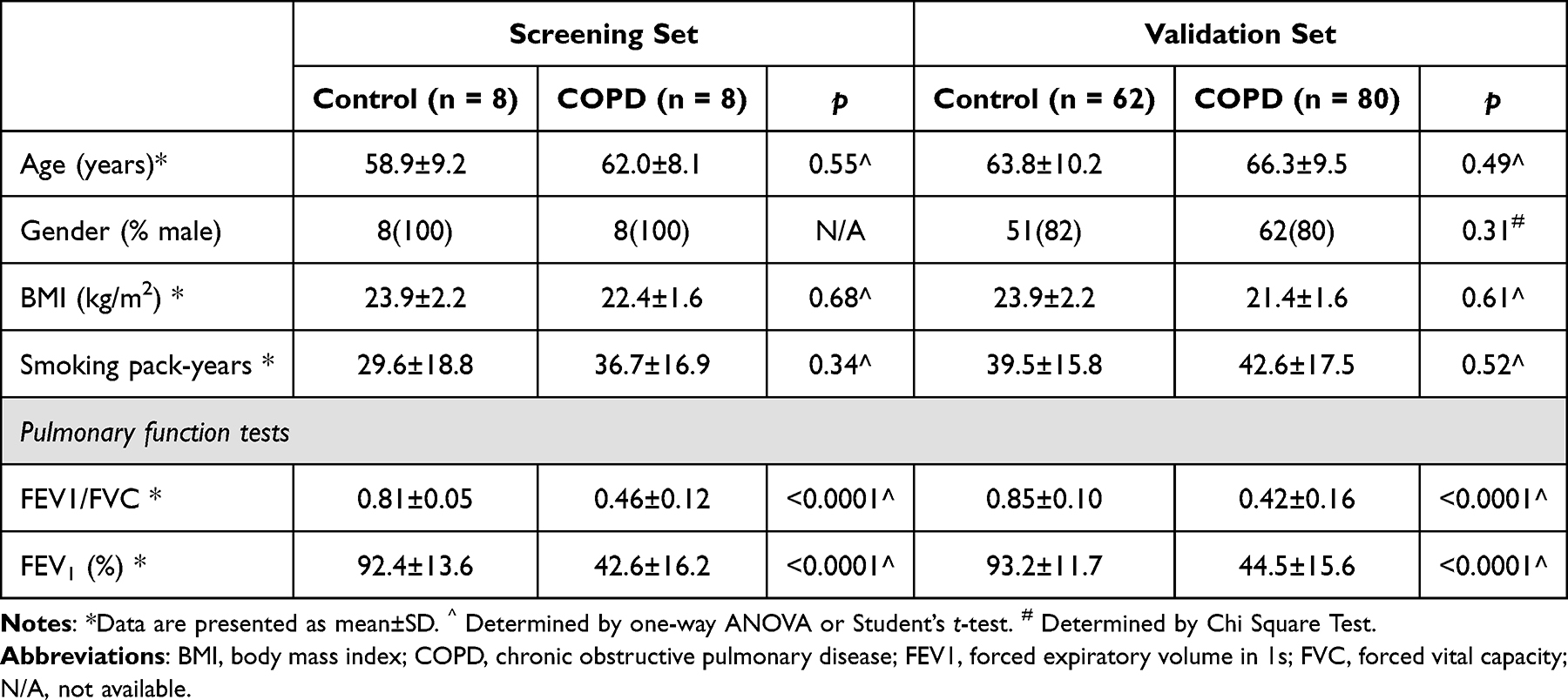 |
Table 1 General Clinical Characteristics of the Subjects in the Screening and Validation Sets |
At the same time, lung tissue samples were collected from the General Thoracic Surgery Department of our hospital. Non-cancerous lung tissues (paracancerous tissues), located at least 10 cm away from cancerous lesions, were surgically resected from patients undergoing pulmonary resection surgery. Among these patients, we selected 10 cases with normal lung function and 10 cases diagnosed with mild to moderate stable COPD before surgery. All tissue samples were immediately snap-frozen in liquid nitrogen upon collection and subsequently stored at −80 °C. These lung tissue samples were specifically utilized for validation experiments, including Western blot and immunohistochemistry analyses, to verify the protein expression of candidate hub genes. The detailed flowchart of this study is shown in Figure S1.
Data Source
Gene expression profile data were downloaded from GEO database. Eligible GEO datasets were selected according to the following inclusion criteria: 1) organism: Homo sapiens; 2) expression profiling by microarray; 3) samples: COPD and normal control samples. The expression profiles of lung tissue from 23 COPD patients and 9 healthy control subjects in the GSE38974 dataset were obtained in the study. Summary-level data for COPD (ukb-d-COPD_EARLYANDLATER) and hub genes were downloaded from the Integrative Epidemiology Unit (IEU). Genome-wide association study (GWAS) database (https://gwas.mrcieu.ac.uk/). The hub genes included HSPA2 (eqtl-a-ENSG00000126803), PRPF19 (eqtl-a-ENSG00000110107), FKBP10 (eqtl-a-ENSG00000141756), DOHH (eqtl-a-ENSG00000129932), and PPIB (eqtl-a-ENSG00000166794). The data for Mendelian randomization cohort validation were sourced from the FinnGen (https://www.finngen.fi/en/access_results) database. By searching for the outcome “COPD” in the FinnGen database. Search results: GWAS ID: finn - b - COPD_LATER; Ncase: 3087; Ncontrol: 212,197; Number of SNPs: 16,380,461; ethnicity: European.15
Combined Analysis of DNA Methylation and Transcriptome
Enrichment analysis of the sites and genes with differential methylation was conducted and enriched gene ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were obtained. We used the “IMA” R package to analyze the DMGs between COPD and control groups with p-value < 0.05. DEGs were identified between COPD and control groups with “limma” R package. Statistically significant DEGs were defined with adj. p-value < 0.05 and |log2FC| > 1 as the cut-off criterion. Next, in order to identify DEGs related to methylation, we used the “VennDiagram” R software package16 to overlap the differentially highly expressed genes and the differentially lowly expressed genes with the methylation-related genes respectively, so as to obtain the methylation-related highly expressed genes and lowly expressed genes.
GO and KEGG Enrichment Analysis of Methylation-Related Highly and Lowly Expressed Genes
GO term analysis and KEGG pathway analysis were performed using the R package “ClusterProfiler”17 to identify the functional roles of the methylated-related highly and lowly expressed genes. GO analysis included three categories: biological process (BP), cellular component (CC) and molecular function (MF). GO terms or KEGG pathways with adj. p-value < 0.05 were considered statistically significant.
Weighted Gene Co-Expression Network Construction and Analysis
The R package “WGCNA”18 was applied to find COPD-related modules and genes. Hierarchical clustering was performed on the sample to detect and eliminate outliers. Genes with similar expression patterns were assigned to a branch, and each branch represented a co-expression module. And we use the pickSoftThreshold function to find a soft threshold power β in accordance with standard scale-free networks. After calculating Pearson’s correlation coefficient, the key modules related to COPD were selected.
Construction of PPI Network and Identification of Hub Genes
The candidate hub genes were obtained by intersecting the methylated-related genes and module genes. The PPI network of candidate hub genes was constructed with the Search Tool for the Retrieval of Interacting Genes (STRING).19 The cut-off value of interaction score was set as 0.4 and isolated nodes in the network were removed. Subsequently, the hub genes in the core module were screened out by Molecular Complex Detection (MCODE) plug-in of Cytoscape based on the criteria of Degree Cutoff: 2, Node Score Cutoff: 0.2, K-Core: 2 and Max. Depth: 100. Correlation coefficients between genes were calculated by Spearman correlation analysis using the “corrplot” R package.20
Functional Similarity Analysis and GSVA of Hub Genes
The functional similarity among proteins was evaluated using the geometric mean of semantic similarities in GO through the “GOSemSim” package.21 To further explore the potential function of the selected hub genes in COPD, COPD samples were divided into two groups according to the median expression level of hub genes. We performed Gene set variation analysis (GSVA) for the high and low expression samples of hub genes. The “c2.cp.kegg.v7.5.1.symbols.gmt” in Molecular Signatures Database (MSigDB) was selected as the reference gene set.
Gene Set Enrichment Analysis (GSEA)
To further explore the potential function of the selected hub genes in COPD, GSEA was performed to analyze the enrichment of DEGs between high- and low-expression groups of hub genes. The “c2.cp.kegg.v7.5.1.symbols.gmt” in Molecular Signatures Database (MSigDB) was selected as the reference gene set. The GSEA on whole-genome expression was performed on the R package “clusterProfiler”.
Analysis of Immune Cell Characteristics
The single sample gene set enrichment analysis (ssGSEA) method was used to calculate the enrichment levels of immune cell infiltration in COPD cohort. ssGSEA was performed by “GSVA” R package.22 The difference in the immune cell infiltration between the COPD and normal groups was carried out using the Wilcoxon test (p-value < 0.05). Moreover, to further explore the relationship between hub genes and immune cells, we also analyzed the correlation between hub genes and differentially infiltrating immune cells.
Follow-Up MR Analyses
We accessed a large dataset on COPD (ukb-d-COPD_EARLYANDLATER) with a sample size of 361,194 individuals and 10,360,720 single nucleotide polymorphisms (SNPs). For the hub genes (HSPA2, PPIB, FKBP10, DOHH, and PRPF19), we obtained data on their expression levels and SNPs from relevant sources (eqtl-a-ENSG00000126803, eqtl-a-ENSG00000166794, etc). To select suitable instrumental variables, we employed the Two Sample MR package to read exposure factors and screen instrumental variables (IVs). The screening indicators included p-value thresholds (p < 5×10−6); linkage disequilibrium (LD) clumping, =TRUE, removing LD instrumental variables; r2=0.01; and region length (kb=10). These criteria ensured the selection of SNPs strongly associated with the hub genes, limiting potential bias. Besides, to further verify the causal relationship between the key genes and COPD, we conduct further verification and analysis based on the FinnGen database. Among them, the analysis methods were the same as those in the previous section.
Real Time Fluorescence Quantitative Reverse Transcription PCR
Extract total RNA from fresh peripheral blood samples using the Total RNA Extraction Kit (TIANGEN BIOTECH Co.DP-419, Ltd). Reverse transcribe RNA into cDNA using the universal cDNA synthesis kit (Takara Biomedical Technology Co. RR036A, Ltd). According to the manufacturer’s instructions (Takara Biomedical Technology Co. RR820A, Ltd), mix cDNA, TB Green Premium Ex Taq II, PCR Forward/Reverse Primer, and sterilized water in a 20µL mixed system. After sampling on a 96 well plate, detect CT values using a Roche light Cycle480 instrument. The RT-qPCR results were analyzed by using comparative (Ct (2−ΔΔCt)) method with three replicates. The mean ΔCt for non-COPD samples was calculated and ΔΔCts were calculated relative to the mean non-COPD ΔCt. Glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) was regarded as endogenous normalizer. The primer sequences utilized in this study were shown in Table 2.
 |
Table 2 Primer Sequence |
Immunohistochemistry (IHC) Assay
Formalin-fixed and paraffin-embedded lung tissue were cut into 5 μm thick sections. Then, routine deparaffinization and rehydration were performed. Repair the antigen in a pressure cooker for 15 minutes. After natural cooling, add endogenous peroxidase blocker and incubate at room temperature for 10 minutes. Then add 5% goat serum dropwise and block in a 37°C incubator for 30 minutes. Sections were incubated with specific antibody against PPIB(1:300; Art. No. 11607-1-AP; Proteintech, Inc.) or PRPF-19(1:200; Art. No. A12590; ABclonal, Inc.) overnight at 4°C refrigerator. The next day, add Ig enhancement solution to the slide and incubate it in a 37°C incubator for 30 minutes. Then add the secondary antibody and continue incubating it in a 37°C incubator for 30 minutes. Afterwards, the sections were treated with DAB for 2–3 min, repeat staining with hematoxylin, differentiate with dilute hydrochloric acid, dehydrate according to concentration gradient, and seal the film. Images of staining were captured under an inverted microscope (CKX41; Olympus Corporation) and were analyzed using Image J.
Western Blot Analysis
Total protein from lung tissue samples was extracted using RIPA lysis buffer (Thermo, USA) and quantified with the BCA Protein Assay Kit (KeyGene, China). Equal amounts of protein were separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). After blocking with 5% skim milk for 1.5 hours at room temperature, the membranes were incubated overnight at 4 °C with primary antibodies targeting PPIB/Cyclophilin B (Proteintech, 11607-1-AP), PRPF-19 (ABclonal, A12590), and GAPDH (Abcam, ab181602). This was followed by incubation with HRP-conjugated goat anti-rabbit secondary antibodies (ABclonal, AS014) for 1.5 hours at room temperature. Protein bands were visualized using an enhanced chemiluminescence system (Invigentech, USA). Chemiluminescent signals were detected with the Image system (GE Healthcare) and quantified using ImageJ software.
Results
Identification of Methylation Related DEGs
Genome-wide DNA methylation analysis of >485,000 CpG sites was performed using the Illumina Infinium HumanMethylation850 BeadChip array. In the methylation profiling dataset, we found 10,593 DMGs including 3540 hypermethylated genes and 7053 hypomethylated genes (Supplementary Table 1). The top 100 hypermethylated sites and top 100 hypomethylated sites were provided in Figure 1A. By analyzing the GSE38974 dataset, we found that 324 genes were up-regulated, and 322 genes were down-regulated in all 646 DEGs between COPD and control groups (Figure 1B). The expressions of the top 10 up-regulated genes and top 10 down-regulated genes (sorted by log2FC) were shown in Figure 1C. Then, using a Venn diagram, 81 methylation-related highly expressed genes (Figure 1D) and 39 methylation-related lowly expressed genes (Figure 1E) were revealed.
 |
Figure 1 Identification of methylation related DEGs. (A) Heatmap of differentially methylated loci. (B) Volcano plots of DEGs in the GSE38974 datasets and (C) Heatmap of top 10 up-regulated genes and top 10 down-regulated genes (sorted by log2FC). (D and E) The Venn diagram illustrates the overlap between differentially expressed genes and methylation-related genes. (D) Methylation-related genes that are differentially highly expressed. (E) Methylation-related genes that are differentially lowly expressed. |
Enrichment Analysis of Methylation-Related Highly and Lowly Expressed Genes
In order to deeply investigate the potential functions of these methylation-related genes, We performed GO enrichment analysis and KEGG pathway enrichment analysis for the methylation-related genes with high and low expression, which were obtained by overlapping methylation-related genes with DEGs through the Venn diagram. The results of GO enrichment indicated that methylated-related highly expressed genes were significantly enriched in “leukocyte migration (BP term)”, “secretory granule lumen (CC term)” and “calcium-dependent protein binding (MF term)” (Figure S2A), and methylated-related lowly expressed genes were significantly enriched in “transmembrane receptor protein serine/threonine kinase signaling pathway (BP term)” and “extracellular matrix structural constituent (MF term)” (Figure S2B). The result of KEGG pathway enrichment indicated that methylated-related highly expressed genes were mainly enriched in “viral protein interaction with cytokine and cytokine receptor”, “HIF-1 signaling pathway” and “cytokine-cytokine receptor interaction” (Figure S2C) and methylated-related lowly expressed genes were mainly enriched in “biosynthesis of unsaturated fatty acids” and “linoleic acid metabolism” (Figure S2D).
WGCNA and Identification of the Key Modules
To screen genes associated with COPD, we performed WGCNA. First, the Euclidean distance of the expression was used to perform hierarchical clustering for samples, and the result showed that no samples were outliers (Figure S3A). β = 10 was chosen to be the appropriate soft-thresholding value to ensure a scale-free analysis (Figure S3B). 25 modules were identified (Figure S3C), and during this process, the two modules with the strongest correlation with chronic obstructive pulmonary disease were selected. They were the blue module with the strongest positive correlation and the turquoise module with the strongest negative correlation (Figure S3D). Among them, the turquoise module contained 1882 genes, and the blue module contained 1587 genes. A total of 3469 genes were selected for subsequent analysis.
Identification of Hub Genes by PPI Network
56 overlapping genes from methylated-related genes and module genes were retained as candidate hub genes (Figure 2A). To study if there were protein interactions among these candidate hub genes, we constructed a PPI network based on the STRING database (Figure 2B). The core module was selected from the PPI network by using the MCODE plug-in of Cytoscape. And the 5 genes in the core module, namely PPIB, HSPA2, PRPF19, FKBP10 and DOHH, were identified as the hub genes of COPD (Figure 2C). DOHH, FKBP10, PPIB and PRPF19 were expressed higher, and HSPA2 was expressed lower in the COPD group compared to the control group (Figure 2D). Subsequently, the correlations among the hub genes were analyzed (Figure 2E). Clearly, DOHH, FKBP10, PPIB and PRPF19 were positively correlated with each other, and HSPA2 was negatively correlated with DOHH, FKBP10, PPIB and PRPF19. DOHH and PRPF19 had the highest positive correlation (cor = 0.81), and HSPA2 and PPIB had the highest negative correlation (cor = −0.73).
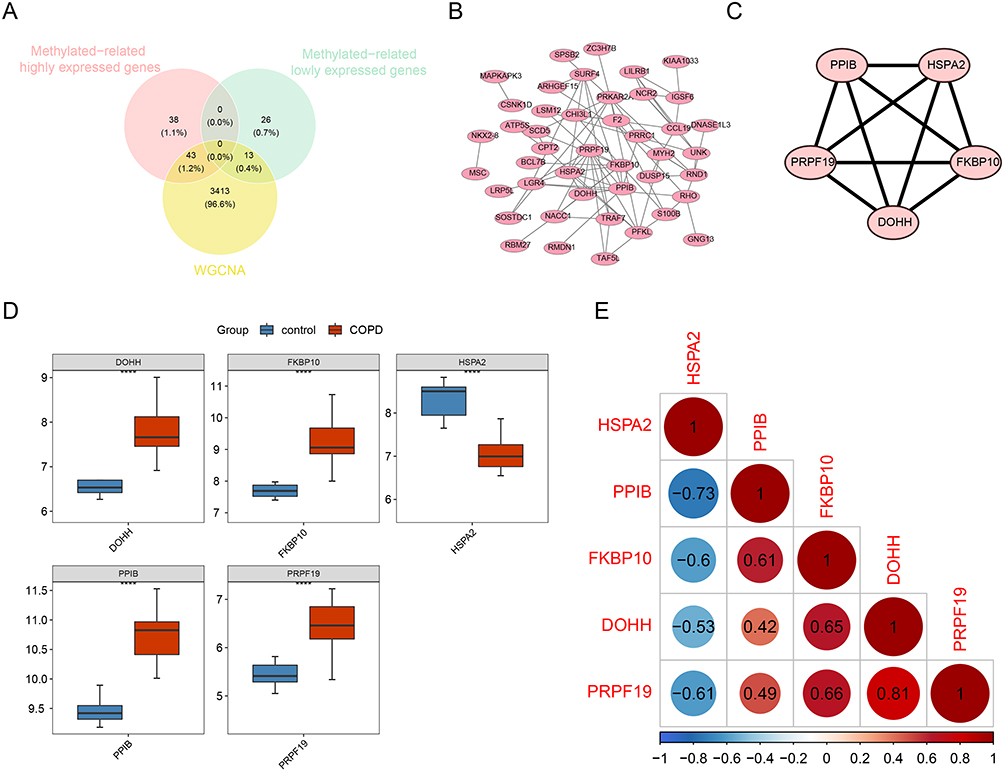 |
Figure 2 Identification of hub genes related to methylation in COPD. (A) Venn diagrams to indicate 56 shared genes from methylation-related DEGs and WGCNA network (43 methylation-related highly expressed genes and 13 methylation-related lowly expressed genes in COPD). (B and C) Visualization of the PPI network and top 5 hub genes. (D) Expression correlation analysis of hub genes. Red indicates a positive correlation and blue indicates a negative correlation. (E) Analysis of hub genes in COPD and control. P < 0.05 was considered to be a statistically significant difference, **** indicates statistical significance at p<0.0001. |
Functional Similarity Analysis and Gene Set Variation Analysis of Hub Genes
To identify important proteins among the hub genes, we ranked proteins based on the median of their functional similarities. PPIB, DOHH, and FKBP10 emerged as the top three proteins (Figure S4A). Moreover, through Gene Set Variation Analysis (GSVA) of samples with high and low expression of the hub genes, we observed differences in the “PANTOTHENATE_AND_COA_BIOSYNTHESIS” pathway between the high and low expression samples of DOHH, FKBP10, HSPA2, and PRPF19. Similarly, “GLYCOSYLPHOSPHATIDYLINOSITOL_GPI_ANCHOR_BIOSYNTHESIS” showed differences between the high and low expression samples of DOHH, FKBP10, PPIB, and PRPF19 (Figure S4B- F).
Infiltrating Immune Cell Analysis
Next, we analyzed the immune cells infiltration profile of COPD and control groups by ssGSEA. Figure 3A showed the distribution of 28 infiltrating immune cells in the COPD and normal samples. The result of the Wilcoxon test presented there were 9 types of immune cells with adjusted p-value < 0.05, which were natural killer cell, MDSC, activated dendritic cell, gamma delta T cell, memory B cell, type 17 T helper cell, effector memory CD4 T cell, natural killer T cell and macrophage. Moreover, the result of the correlations between hub genes and differentially infiltrating immune cells (Figure 3B) indicated that natural killer cell was negatively correlated with PPIB, DOHH, FKBP10 and PRPF19, and positively correlated with HSPA2. As contrast, the other cells were positively correlated with PPIB, DOHH, FKBP10 and PRPF19, and negatively correlated with HSPA2.
 |
Figure 3 The correlation between hub genes and immune cells. (A) Evaluation of 28 immune cells infiltration in ssGSEA analysis of COPD samples; (B) Correlation between hub genes and differential immune cell infiltration. Notes: * indicates p<0.05, ** indicates p<0.01. |
Causal Effects of Hub Genes on COPD
Our MR analysis identified two key genes, PRPF19 and PPIB, with significant causal associations with COPD. PRPF19 exhibited an inverse variance-weighted p-value of 0.012, while PPIB showed a p-value of <0.001. The scatter plots demonstrated a consistent negative correlation between the effects of instrumental variables (SNPs) on PRPF19 and PPIB and the occurrence of COPD, as observed using the IVW algorithm (Figure 4A and B). This suggests that variations in PRPF19 and PPIB expression are causally related to a reduced risk of COPD, indicating that these genes act as protective factors. The forest plots further confirmed the protective roles of PRPF19 and PPIB in COPD risk reduction (Figure 4C and D). IVW analysis revealed that both PRPF19 and PPIB significantly reduced the risk of COPD. The funnel plots displayed a symmetrical distribution of instrumental variables on both sides of the IVW line (Figure 4E and F), indicating that the MR analysis complied with Mendel’s second law of random assortment and did not exhibit systematic biases. Sensitivity analysis, including heterogeneity, pleiotropy, and leave-one-out tests, supported the reliability of our MR analysis (Figure 4G and H). We found no significant heterogeneity, pleiotropy, or substantial changes in the effects of instrumental variables, reinforcing the credibility of our findings. In addition, the cohort verification of the research results showed that PRPF19 was a safety factor (OR = 0.8349), which was consistent with the above - analyzed results. However, there was no causal effects between PPIB and COPD (OR=1.0591) (Supplementary Table 2). Besides, Scatter plots (Figure S5A) and forest plots (Figure S5B) also indicated that PRPF19 was a safety factor. Funnel plots (Figure S5C) showed that our results were in line with Mendel’s second law. Further sensitivity analysis revealed no heterogeneity (Supplementary Table 3) and pleiotropy (Supplementary Table 4). Meanwhile, leave - one - out plots (Figure S5D) demonstrated that the results were reliable and had good stability. In addition, MR steiger analysis (Supplementary Table 5) showed that the directionality of PRPF19 was correct and there was no reverse causality. All of these illustrate that our verification results are reliable.
 |
Figure 4 Univariate Mendelian analysis of PRPF19 and PPIB genes in relation to COPD. Scatterplots (A and B) show SNP effects on COPD. Colored lines represent different MR algorithms. Forest plots (C and D) display SNP effects; left of vertical line indicates reduced risk, right suggests increased risk. Funnel plots assess MR randomness (E and F), IVW line symmetry indicates random grouping. Sensitivity analysis (G and H) shows SNP removal’s minimal impact, ensuring MR reliability/stability. |
GSEA of Hub Genes
We conducted the GSEA on whole-genome data to further illustrate the biological functions of hub genes. The results of GSEA showed that the hub genes were closely associated with “Cytokine Cytokine-receptor interaction, Drug metabolism cytochrome-P450, JAK STAT signaling pathway, Metabolism of xenobiotics by cytochrome P450, Vascular smooth muscle contraction, Chemokine signaling pathway, Nod like receptor signaling pathway, Oxidative phosphorylation, Ribosome and so on, suggesting that these hub genes might participate in these processes (Figure S6).
Validation of Hub Genes as Biomarkers for COPD
The pulmonary expression of the identified hub genes was validated through immunohistochemistry on lung tissues obtained from both COPD patients and healthy subjects. Notably, PPIB and PRPF19 exhibited elevated protein expression levels in COPD cases, as illustrated in Figure 5. To evaluate the potential of these genes as biomarkers for COPD, we conducted RT-qPCR on peripheral blood samples from 80 COPD patients and 62 healthy controls. The results demonstrated significantly higher RNA expression of PPIB and PRPF19 in COPD patients (Figure 6A and C), and their areas under the ROC curves were above 0.6 and 0.7 respectively (Figure 6B and D), suggesting that genes PRPF-19 and PPIB have certain diagnostic value for COPD. Additionally, methylation analysis indicated that the methylation status of these two genes could effectively distinguish between COPD patients and healthy controls in peripheral blood. These findings suggest the diagnostic potential of PPIB and PRPF19 as biomarkers for COPD.
 |
Figure 5 Immunohistochemical validation of hub genes PPIB and PRPF19 expression in lung tissues. (A–D) Representative immunohistochemical staining images of PPIB and PRPF19 proteins in lung tissues from control subjects (A and C) and COPD patients (B and D). Magnification: 400×. (E and F) Quantitative analysis of immunohistochemistry results performed using ImageJ software. Data are presented as average optical density (AOD) values. Expression levels of PPIB (E) and PRPF19 (F) were significantly elevated in COPD patients compared with controls. Note: ****indicates statistical significance at p<0.0001. |
 |
Figure 6 Hub genes as Biomarkers for COPD Diagnosis. (A and B) The differential expression levels and ROC curve analysis of PPIB in peripheral blood samples of a COPD group (n = 80) and a control group (n = 62). (C and D) The differential expression levels and ROC curve analysis of PRPF19 in COPD and control group. Abbreviations: AUC, area under the curve; CI, confidence interval. Notes: ** indicates p<0.01, *** indicates p<0.001. |
Western Blot Analysis of PRPF19, PPIB, and Apoptosis-Related Proteins in COPD Lung Tissues
Western blot analysis was conducted to verify the up-regulation of PRPF19 and PPIB in lung tissues from COPD patients. As shown in the Western blot results, both PRPF19 and PPIB were significantly up-regulated in COPD lung tissues compared to controls (Figure 7A and B). Moreover, the analysis of apoptosis-related proteins demonstrated increased expression of Bcl-2, an anti-apoptotic protein, and decreased expression of Bax, a pro-apoptotic protein, in COPD lung tissues (Figure 7A and B). These findings suggest that both PRPF19 and PPIB are not only up-regulated in COPD but may also be associated with apoptosis regulation, as indicated by the shift toward anti-apoptotic signaling. GAPDH was used as the loading control for normalization, and quantification of the band intensities revealed statistically significant differences between COPD and control tissues (P < 0.05).
 |
Figure 7 PRPF19 and PPIB Expression and Their Role in Apoptosis Regulation in COPD Lung Tissues. (A) Representative Western blot images show increased expression of PRPF19 and PPIB, along with elevated Bcl-2 (anti-apoptotic) and decreased Bax (pro-apoptotic) protein levels in COPD lung tissues compared to controls, suggesting that PRPF19 and PPIB may contribute to apoptosis regulation by promoting cell survival. (B) Quantitative analysis of Western blot results demonstrating significant upregulation of PRPF19, PPIB, and Bcl-2, and downregulation of Bax in COPD samples (P < 0.05). GAPDH was used as the loading control. Note: *** indicates p<0.001, **** indicates p<0.0001. |
Discussion
COPD remains a significant global health burden, necessitating a deeper understanding of its molecular underpinnings for effective management and intervention strategies. In our study, we conducted a comprehensive analysis employing diverse bioinformatics approaches to unravel the intricate molecular landscape of COPD. By integrating genome-wide DNA methylation analysis, differential gene expression profiling, WGCNA, PPI network exploration, and MR analysis, we aimed to decipher the complex interplay of genetic and epigenetic factors contributing to COPD pathogenesis.
Our study on COPD patients’ genome-wide DNA methylation patterns revealed numerous DMGs, indicating epigenetic modifications associated with the disease. Intersection with DEGs identified a subset with altered expression and methylation, signifying the potential impact of epigenetic mechanisms in persistent inflammation and airway remodeling in COPD. Functional analysis highlighted their involvement in critical processes like leukocyte migration and cytokine signaling.23 Conversely, lowly expressed genes were enriched in transmembrane receptor protein signaling and extracellular matrix (ECM) structural constituents. ECM, comprising collagens, elastin, fibronectin, and laminins, plays a crucial role in COPD pathogenesis, where ECM breakdown contributes to airway remodeling,24 obstruction, and impaired lung function. Increased protease activity, like matrix metalloproteinases (MMPs),25 and decreased antiproteases, such as tissue inhibitor of metalloproteinases (TIMPs), are implicated in ECM breakdown and COPD pathogenesis.26
WGCNA identified modules of co-expressed genes associated with COPD, such as the positively correlated turquoise module and the negatively correlated blue module. Analysis within these modules revealed hub genes, including PPIB, HSPA2, PRPF19, FKBP10, and DOHH, which showed significant alterations in COPD patients compared to healthy controls in both gene expression and DNA methylation status. Recognizing these hub genes underscores their pivotal roles in COPD pathogenesis, highlighting their potential as diagnostic biomarkers. These genes have been reported to be involved in various biological processes, such as protein folding, RNA splicing, and post-translational modifications.27–31
There is limited information on the specific role of DOHH, FKBP10, PPIB, PRPF19, and HSPA2 in COPD, but some studies have reported potential associations between these genes and COPD. DOHH has been shown to be involved in the regulation of hypoxia-inducible factor 1 alpha (HIF1α) stability, which is a key player in the cellular response to hypoxia and is dysregulated in COPD.32 FKBP10 encodes a protein called FK506-binding protein 65 (FKBP65), which is involved in the maturation of collagen. Mutations in FKBP10 have been linked to osteogenesis imperfecta (OI), a genetic disorder characterized by fragile bones.33 Some studies have suggested that individuals with OI may be at increased risk for lung diseases,34 including COPD. PPIB encodes the peptidylprolyl isomerase B (PPIB) protein, which is involved in the folding and stabilization of proteins.35 Patients with mutations in the PPIB gene have been reported to have osteogenesis imperfecta along with recurrent pulmonary complications, indicating that PPIB may be involved in the development of lung disease.36 It has been reported that PRPF19 plays a role in regulating RNA splicing.37 Moreover, PRPF19 has been associated with several human disorders such as retinitis pigmentosa38 and malignant diseases.39,40 Although there is limited information on the specific role of PRPF19 in COPD, a GWAS identified a potential association between a variant in PRPF19 and lung function.41 HSPA2 encodes heat shock protein 70–2 (HSP70-2), which is involved in protein folding and stress responses. Some studies have suggested that HSPA2 may play a role in the development of lung cancer,42 but its role in COPD is not well understood.
Our study uncovered a unique correlation pattern between natural killer cells and hub genes, distinguishing them from other immune cells in COPD. Natural killer cells exhibited negative correlations with PPIB, DOHH, FKBP10, and PRPF19, and a positive correlation with HSPA2. This distinct association with hub genes in natural killer cells has not been reported in previous COPD studies. Our findings offer novel insights into the immune cell infiltration profile in COPD, emphasizing the importance of investigating interactions between immune cells and hub genes in the disease process. Further studies are required to validate these findings and elucidate the underlying mechanisms of immune cell dysfunction in COPD.
Our PPI network analysis supports the significance of identified hub genes, revealing a coordinated molecular mechanism in COPD development. The Mendelian randomization analysis highlights PRPF19 and PPIB as causal factors with protective effects. Despite higher expression being associated with increased protection against COPD, these genes are also elevated in COPD patients. This apparent contradiction may result from complex interactions, suggesting upregulation in response to COPD-related stressors or as a compensatory mechanism to counter disease progression. This finding emphasizes the intricate nature of COPD pathogenesis, where multiple factors interact, as per recent studies.
Our study further validated the up-regulation of PRPF19 and PPIB in COPD through Western blot analysis of lung tissues. The observed increase in the anti-apoptotic protein Bcl-2 and decrease in the pro-apoptotic protein Bax suggest a shift toward reduced apoptosis in these tissues. This may indicate a compensatory response to chronic cellular stress and inflammation in COPD, where certain cell populations attempt to survive despite ongoing tissue damage. The involvement of PRPF19 and PPIB in this apoptosis regulation highlights their potential role in modulating cell survival and disease progression. These findings align with the notion that COPD pathogenesis involves complex interactions between apoptosis and cell survival mechanisms, emphasizing the importance of targeting these pathways for therapeutic intervention.
However, there are also several limitations to this study. First, the sample size of the study is relatively small, especially the initial screening cohort consisting of only eight COPD patients and eight controls. This limited sample size may affect the generalizability of our findings and could introduce potential bias in identifying differentially methylated and expressed genes. Therefore, future studies with larger and more diverse populations are warranted to confirm our results, enhance statistical power, and provide stronger evidence for the roles of identified hub genes PRPF19 and PPIB as reliable biomarkers in COPD diagnosis and prognosis. Second, the study did not perform functional experiments to validate the potential biomarkers, and the clinical relevance of these biomarkers is yet to be determined.
In conclusion, our comprehensive analysis identifies PRPF19 and PPIB as important epigenetic and transcriptional regulators associated with COPD, highlighting their potential as biomarkers for disease diagnosis and prognosis. Future studies are warranted to validate their clinical relevance and elucidate the underlying molecular mechanisms, thereby laying a foundation for the development of targeted therapeutic strategies.
Abbreviations
COPD, Chronic obstructive pulmonary disease; WGCNA, Weighted gene co-expression network analysis; DMGs, Differential methylation genes; DEGs, Differentially expressed genes; PPI, Protein-protein interaction; MR, Mendelian Randomization; GO, Gene ontology; GEO, Gene Expression Omnibus; GWAS, Genome-Wide Association Study; GSEA, Gene Set Enrichment Analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; ssGSEA, Single-Sample Gene Set Enrichment Analysis; GSVA, Gene Set Variation Analysis; ECM, Extracellular Matrix; RT-qPCR, Real-Time Quantitative Polymerase Chain Reaction.
Data Sharing Statement
Data and materials are available upon request by contacting the correspondence author Jia Hou ([email protected]).
Ethics Approval and Consent to Participate
Research ethics approval was obtained from the Ethics Committee of General Hospital of Ningxia Medical University. All subjects provided written informed consent.
Declaration of Generative AI Use in Scientific Writing
In the preparation of this manuscript, chatGPT4 was employed exclusively for language editing to improve clarity and coherence. The scientific content, data analysis, interpretation, and conclusions were developed independently by the authors. All authors have reviewed and take full responsibility for the content of the publication.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work received support from the National Natural Science Foundation of China (Grant No. 81360008), Ningxia Key Research and Development Project (Grant No. 2021BEG03079), and the Ningxia Natural Science Foundation (Grants No. 2020AAC03404 and 2024AAC02075). Also used data from Vosa and co-authors and thank the eQTLGen consortium for data support.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Soriano JB, Kendrick PJ, Paulson KR. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Respir Med. 2020;8(6):585–596. doi:10.1016/S2213-2600(20)30105-3
2. Labaki WW, Rosenberg SR. Chronic obstructive pulmonary disease. Ann Intern Med. 2020;173(3):Itc17–itc32. doi:10.7326/AITC202008040
3. Do C, Shearer A, Suzuki M, et al. Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era. Genome Biol. 2017;18(1):120. doi:10.1186/s13059-017-1250-y
4. Comer BS, Ba M, Singer CA, Gerthoffer WT. Epigenetic targets for novel therapies of lung diseases. Pharmacol Ther. 2015;147:91–110. doi:10.1016/j.pharmthera.2014.11.006
5. Morrow JD, Make B, Regan E, et al. DNA methylation is predictive of mortality in current and former smokers. Am J Respir Crit Care Med. 2020;201(9):1099–1109. doi:10.1164/rccm.201902-0439OC
6. Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–447. doi:10.1161/CIRCGENETICS.116.001506
7. Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42(3):200–202. doi:10.1038/ng.535
8. Lee YJ, Choi S, Kwon SY, et al. A genome-wide association study in early COPD: identification of one major susceptibility loci. Int J Chron Obstruct Pulmon Dis. 2020;15:2967–2975. doi:10.2147/COPD.S269263
9. Cho MH, McDonald M-LN, Zhou X, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2(3):214–225. doi:10.1016/S2213-2600(14)70002-5
10. van der Plaat DA, de Jong K, Lahousse L, et al. Genome-wide association study on the FEV1/FVC ratio in never-smokers identifies HHIP and FAM13A. J Allergy Clin Immunol. 2017;139(2):533–540. doi:10.1016/j.jaci.2016.06.062
11. Wain LV, Shrine N, Artigas MS, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet. 2017;49(3):416–425. doi:10.1038/ng.3787
12. Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. doi:10.1093/ije/dyg070
13. Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res. 2017;26(5):2333–2355. doi:10.1177/0962280215597579
14. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98. doi:10.1093/hmg/ddu328
15. Võsa U, Claringbould A, Westra HJ, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53(9):1300–1310. doi:10.1038/s41588-021-00913-z
16. Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable venn and Euler diagrams in R. BMC Bioinf. 2011;12(1):35. doi:10.1186/1471-2105-12-35
17. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
18. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559. doi:10.1186/1471-2105-9-559
19. von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31(1):258–261. doi:10.1093/nar/gkg034
20. Zhang H, Liu R, Sun L, Guo W, Ji X, Hu X. Comprehensive analysis of gene expression changes and validation in hepatocellular carcinoma. Onco Targets Ther. 2021;14:1021–1031. doi:10.2147/OTT.S294500
21. Yu G, Li F, Qin Y, Bo X, Wu Y, Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26(7):976–978. doi:10.1093/bioinformatics/btq064
22. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14(1):7. doi:10.1186/1471-2105-14-7
23. Barnes PJ. Inflammatory endotypes in COPD. Allergy. 2019;74(7):1249–1256. doi:10.1111/all.13760
24. Liu G, Philp AM, Corte T, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther. 2021;225:107839. doi:10.1016/j.pharmthera.2021.107839
25. Brandsma CA, Van den Berge M, Hackett TL, Brusselle G, Timens W. Recent advances in chronic obstructive pulmonary disease pathogenesis: from disease mechanisms to precision medicine. J Pathol. 2020;250(5):624–635. doi:10.1002/path.5364
26. Demedts IK, Brusselle GG, Bracke KR, Vermaelen KY, Pauwels RA. Matrix metalloproteinases in asthma and COPD. Curr Opin Pharmacol. 2005;5(3):257–263. doi:10.1016/j.coph.2004.12.005
27. Epis MR, Giles KM, Kalinowski FC, Barker A, Cohen RJ, Leedman PJ. Regulation of expression of deoxyhypusine hydroxylase (DOHH), the enzyme that catalyzes the activation of eIF5A, by miR-331-3p and miR-642-5p in prostate cancer cells. J Biol Chem. 2012;287(42):35251–35259. doi:10.1074/jbc.M112.374686
28. Knüppel L, Heinzelmann K, Lindner M, et al. FK506-binding protein 10 (FKBP10) regulates lung fibroblast migration via collagen VI synthesis. Respir Res. 2018;19(1):67. doi:10.1186/s12931-018-0768-1
29. Valadares ER, Carneiro TB, Santos PM, Oliveira AC, Zabel B. What is new in genetics and osteogenesis imperfecta classification? Jornal de Pediatria. 2014;90(6):536–541. doi:10.1016/j.jped.2014.05.003
30. Yin J, Zhu JM, Shen XZ. New insights into pre-mRNA processing factor 19: a multi-faceted protein in humans. Biol Cell. 2012;104(12):695–705. doi:10.1111/boc.201200011
31. Scieglinska D, Krawczyk Z. Expression, function, and regulation of the testis-enriched heat shock HSPA2 gene in rodents and humans. Cell Stress Chaperones. 2015;20(2):221–235. doi:10.1007/s12192-014-0548-x
32. Dunham-Snary KJ, Wu D, Sykes EA, et al. Hypoxic pulmonary vasoconstriction: from molecular mechanisms to medicine. CHEST. 2017;151(1):181–192. doi:10.1016/j.chest.2016.09.001
33. Yüksel Ülker A, Uludağ Alkaya D, Elkanova L, et al. Long-term follow-up outcomes of 19 patients with osteogenesis imperfecta type XI and bruck syndrome type I caused by FKBP10 variants. Calcif Tissue Int. 2021;109(6):633–644. doi:10.1007/s00223-021-00879-4
34. Lafage-Proust MH, Courtois I. The management of osteogenesis imperfecta in adults: state of the art. Joint Bone Spine. 2019;86(5):589–593. doi:10.1016/j.jbspin.2019.02.001
35. Zhang Y, Liu L, Zhou M, et al. PPIB-regulated alternative splicing of cell cycle genes contributes to the regulation of cell proliferation. Am J Transl Res. 2022;14(9):6163–6174.
36. Rush ET, Caldwell KS, Kreikemeier RM, Lutz RE, Esposito PW. Osteogenesis imperfecta caused by PPIB mutation with severe phenotype and congenital hearing loss. J Pediatr Genet. 2014;3(1):29–34. doi:10.3233/PGE-14080
37. Yang M, Qiu Y, Yang Y, Wang W. An integrated analysis of the identified PRPF19 as an onco-immunological biomarker encompassing the tumor microenvironment, disease progression, and prognoses in hepatocellular carcinoma. Front Cell Dev Biol. 2022;10:840010. doi:10.3389/fcell.2022.840010
38. Mordes D, Yuan L, Xu L, Kawada M, Molday RS, Wu JY. Identification of photoreceptor genes affected by PRPF31 mutations associated with autosomal dominant retinitis pigmentosa. Neurobiol Dis. 2007;26(2):291–300. doi:10.1016/j.nbd.2006.08.026
39. Yu X, Chen P, Yi W, Ruan W, Xiong X. Identification of cell senescence molecular subtypes in prediction of the prognosis and immunotherapy of hepatitis B virus-related hepatocellular carcinoma. Front Immunol. 2022;13:1029872. doi:10.3389/fimmu.2022.1029872
40. Kessler T, Berberich A, Sadik A, et al. Methylome analyses of three glioblastoma cohorts reveal chemotherapy sensitivity markers within DDR genes. Cancer Med. 2020;9(22):8373–8385. doi:10.1002/cam4.3447
41. Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42(1):45–52. doi:10.1038/ng.500
42. Sojka DR, Gogler-Pigłowska A, Klarzyńska K, et al. HSPA2 chaperone contributes to the maintenance of epithelial phenotype of human bronchial epithelial cells but has non-essential role in supporting malignant features of non-small cell lung carcinoma, MCF7, and HeLa cancer cells. Cancers. 2020;12(10):2749. doi:10.3390/cancers12102749
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Causal Relationship Between Gastroesophageal Reflux Disease and Chronic Obstructive Pulmonary Disease: A Bidirectional Two-Sample Mendelian Randomization Study
Liu B, Chen M, You J, Zheng S, Huang M
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:87-95
Published Date: 10 January 2024
Exploring a Potential Causal Link Between Dietary Intake and Chronic Obstructive Pulmonary Disease: A Two-Sample Mendelian Randomization Study
Zhang C, Yu L, Xiong T, Zhang Y, Liu J, Zhang J, He P, Xi Y, Jiang Y
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:297-308
Published Date: 26 January 2024
Association of Chronic Obstructive Pulmonary Disease with Risk of Psychiatric Disorders: A Two-Sample Mendelian Randomization Study
Zhang Q, Zhang H, Xu Q
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:343-351
Published Date: 1 February 2024
A Bidirectional Mendelian Randomization Study Investigating the Causal Relationship Between Ankylosing Spondylitis and Chronic Obstructive Pulmonary Disease
Pan D, Dai X, Li P, Xue L
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:259-271
Published Date: 8 February 2025
Identifying Common Diagnostic Biomarkers and Therapeutic Targets between COPD and Sepsis: A Bioinformatics and Machine Learning Approach
Li X, Xiao Y, Yang M, Zhang X, Yuan Z, Zhang Z, Zhang H, Liu L, Zhao M
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:1761-1786
Published Date: 28 May 2025