")
Back to Journals » Journal of Pain Research » Volume 18
Hyperalgesic Priming in the Transition From Acute to Chronic Pain: Focus on Different Models and the Molecular Mechanisms Involved
Authors Zhang M , Li N, Zhao S, Feng X
Received 29 December 2024
Accepted for publication 15 March 2025
Published 21 March 2025 Volume 2025:18 Pages 1491—1501
DOI https://doi.org/10.2147/JPR.S514851
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wendy Imlach
Mi Zhang,1 Ningbo Li,2 Shuai Zhao,1 Xiaobo Feng1,3
1Department of Anesthesiology, Zhongnan Hospital, Wuhan University, Wuhan, 430071, People’s Republic of China; 2Department of Anesthesiology and Pain Medicine, Hubei Key Laboratory of Geriatric Anesthesia and Perioperative Brain Health, and Wuhan Clinical Research Center for Geriatric Anesthesia, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430030, People’s Republic of China; 3Department of Pain Medicine, Zhongnan Hospital, Wuhan University, Wuhan, 430071, People’s Republic of China
Correspondence: Xiaobo Feng, Email [email protected] Shuai Zhao, Email [email protected]
Abstract: Poorly treated acute pain can develop into chronic pain, resulting in significant impairment of patients’ quality of life. The hyperalgesic priming model is commonly used to study how acute pain transforms into chronic pain. Inflammatory factors, small molecules, opioid receptor agonists, chemotherapy drugs, and stress serve as initiating factors in the hyperalgesic priming model. Various signaling pathways such as PKCϵ, MOR and ephrin-B2 pathways, and sexual differences also contribute to the transformation process of chronic pain. In this review, we examine various hyperalgesic priming models and their underlying molecular mechanisms. By thoroughly investigating these molecular mechanisms, researchers can more precisely identify the critical nodes involved in pain transformation, thereby developing more targeted treatment strategies.
Keywords: transition from acute to chronic pain, hyperalgesic priming, experimental models, molecular pathways, sex differences
Introduction
Pain is closely related to the disease’s occurrence, development, and outcome. Acute pain is a recent, short-lived pain with a clear cause. It is associated with emergency or elective surgery, trauma, burns, childbirth, natural disasters, and war.1 Poor management of acute pain can lead to adverse effects, including increased morbidity, impaired physical function, and severe pain that may develop into chronic pain, significantly reducing patients’ quality of life.2 For example, a study reported that 11 years after injury, nearly 52% of patients were suffering from chronic pain caused by burns.3 Furthermore, the economic burden attributable to work disability caused by chronic pain is estimated to reach billions of dollars, significantly exceeding the financial impact of cancer, cardiovascular diseases, and diabetes.4 Despite the widespread occurrence of persistent pain, little is known about the underlying factors that contribute to the development of chronic pain. Currently, management for chronic pain includes exercise and psychotherapy, non-opioid pharmacological treatment, opioids, surgical treatment, and integrative treatments.5 While current treatments can slow the progress of chronic pain, exploring the mechanisms behind the transition from acute to chronic pain is essential for developing more effective therapies.
Hyperalgesic priming is a pain condition wherein repeated or persistent nociceptive stimuli induce structural and functional alterations in the central nervous system (CNS), leading to an increased sensitivity to subsequent painful stimuli. These modifications may encompass changes in synaptic strength, neurotransmitter release dynamics, and receptor sensitivity.6–8 Hyperalgesic priming is considered a pivotal mechanism underlying the progression of chronic pain, comprising three distinct phases: 1) The acute phase is a brief hypersensitivity reaction caused by various nociceptive compounds, which can resolve within a week; 2) The primed phase can last for at least three weeks, during which the animal is not allergic; 3) Chronic phase can be induced by plantar injection of prostaglandin E2 (PGE2) or other substances, lasting for at least two months.7,9–12 In short, hyperalgesic priming represents a state in which the initial injury has “primed” the nervous system, making it more sensitive to subsequent painful stimuli. As hyperalgesia priming occurs before and may lead to chronic pain, using priming animal models has become the primary method for studying the transition from acute to chronic pain states.13–15 Given the lack of a comprehensive summary of various commonly used priming animal models, in this review, we primarily discuss relevant animal models of hyperalgesia priming. We will summarize the priming states induced by various stimuli and explain the mechanisms involved.
The Establishment of Hyperalgesia Priming Model
Various pain models have been developed to investigate the mechanisms of chronic pain, including those that utilize chronic inflammation induced by complete Freund’s adjuvant or carrageenan.16,17 The mechanisms of acute pain and chronic pain can be challenging to differentiate, making it unclear how acute pain transitions into chronic pain. The concept of hyperalgesic priming emerged from an original discussion aimed at understanding the mechanisms of acute and chronic pain separately.9 Researchers induced acute pain using carrageenan to investigate whether acute inflammation increases sensitivity to inflammatory mediators. Five days after the initial injection, when PGE2, 5-hydroxytryptamine (5-HT), and the A2 adenosine receptor agonist CGS-21680 were injected at the same site, it was observed that the ability of carrageenan to prolong hyperalgesia induced by inflammatory mediators persisted for up to three weeks. This was in comparison to the control group that did not receive carrageenan prior to the injection of inflammatory mediators, such as PGE2.9 This animal model was defined as “hyperalgesic priming”.18,19 Based on this, Levine et al first demonstrated that acute inflammatory pain increases pain sensitivity, and chronic pain can persist after re-stimulation.
Typical Stimuli of PEG2 in the Chronic Phase of Hyperalgesic Priming
To investigate the transition mechanism from acute to chronic pain, researchers used PGE2 injection at the same site as the previous injury, consistent with methods typically employed in hyperalgesic priming studies (Table 1). A single infusion of PGE2 sensitizes nociceptors, reducing the mechanical threshold within 30 minutes and returning to baseline after 60 minutes. However, in the primed model, the sensitization from PGE2 lasts longer than four hours.6 This priming effect was also confirmed by in vivo single-fibre electrophysiology.20
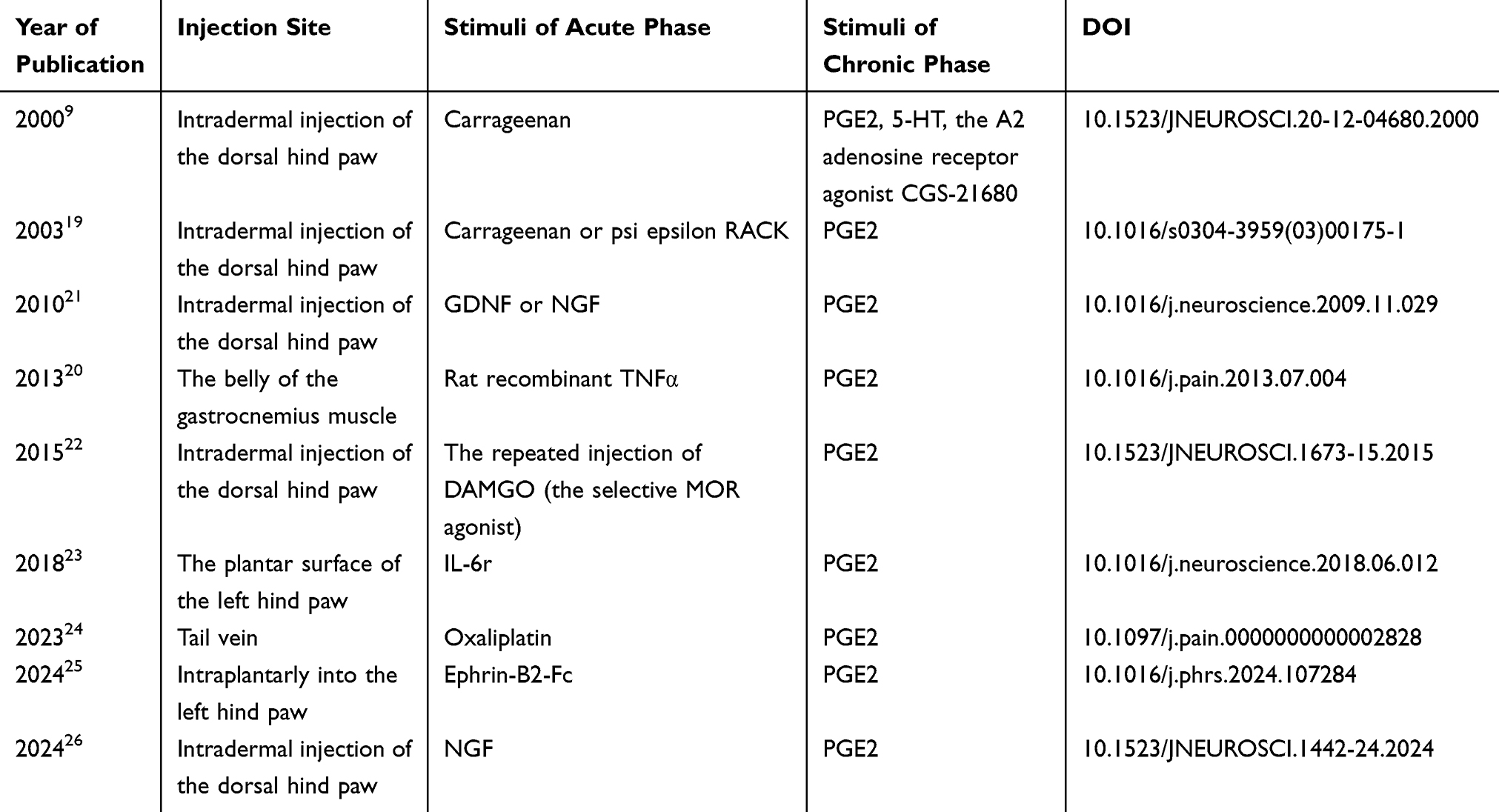 |
Table 1 Key Articles Discussing PGE2 as a Prolonged Trigger for Hyperalgesic Priming |
Carrageenan-Induced Inflammation Primes
Chronic inflammatory pain is crucial to hyperalgesic priming, with carrageenan serving as a classic substance that induces this type of pain. The method for establishing hyperalgesic priming involves injecting 5 μL of a 1% carrageenan solution into the dorsal paw intradermally. Five days later, 100 ng of PGE2 is administered at the same site. A mechanical pain test is then performed to assess changes in pain sensitization.9,27 Early studies indicated that the activation of protein kinase C epsilon (PKCε) in the peripheral terminal of the nociceptor plays a crucial role in this model of hyperalgesic priming.6,9,18 Although PKCε is expressed in almost all dorsal root ganglion(DRG) neurons,28–31 the regulation of hyperalgesic priming is limited to the isolectin B4-positive (IB4 (+)) nociceptors.21,32 In the hyperalgesic priming model, the cytoplasmic polyadenylation element binding protein (CPEB) is a key component downstream of PKCε in the cellular signaling pathway that triggers the induction of priming, playing a role in the process of chronic pain development.33 Nearly all neurons that express CPEB are positive for IB4, and the intrathecal administration of antisense oligodeoxynucleotide (ODN) targeting CPEB mRNA significantly reduced PGE2-induced hyperalgesia in rats treated with carrageenan. Additionally, the selective PKCε agonist can be inhibited by the intrathecal administration of antisense ODN targeting CPEB mRNA for 7 consecutive days, further supporting the idea that priming results from PKCε-induced activation of CPEB.33
TNF-α regulates acute hyperalgesia by indirectly activating the release of downstream prostaglandins and sympathetic amines.34–36 In hyperalgesic priming, TNF-α is involved by directly activating the TNFR1 receptor, which in turn activates PKCε. This indicates that blocking prostaglandin synthesis may not prevent chronic hyperalgesic priming. Additionally, it suggests that the cellular mechanisms responsible for initiating inflammation-induced chronic hyperalgesia are different from those involved in acute hyperalgesia.27
Injected Repeatedly the Selective Mu-Opioid Receptor (MOR) Agonist DAMGO to Induce Primes
The hyperalgesic priming model, induced by inflammatory factors and PGE2, provides insights into the mechanism of acute-to-chronic pain transformation. However, the occurrence and development of chronic pain are not limited to inflammatory induction but also include other mechanisms, such as opioid-induced hyperalgesia (OIH). Previous studies have shown that a single injection of DAMGO does not affect nociceptive pain thresholds.37,38 In contrast, repeated injections of DAMGO can trigger hyperalgesia and induce a potentially hyperreactive state to subsequent injections of nociceptive mediators, known here as type II hyperalgesic priming.39,40 The operation method was as follows: DAMGO (1 μg) was injected subcutaneously into the dorsum of the rat’s hind paw every hour for three consecutive hours. The change in mechanical pain sensitization was assessed after the fourth injection of DAMGO. Subsequently, PGE2 (100 ng) was injected into the same site, and the degree of hyperalgesia was evaluated again 30 minutes and 4 hours later.22
This model is similar to the classical hyperalgesic priming model (type I) in that it can induce prolonged mechanical hyperalgesia by co-injection of carrageenan and PGE2.9 The differences between these two types involve a change in signaling by PKA that contributes to type II priming instead of PKCε. Type I priming is mediated by IB4+ nociceptors, while type II priming is mediated by IB4− nociceptors.22,32 Furthermore, bisexual dimorphism is present in type I, whereas type II priming can be induced in both males and females.19,22 Recent studies have reported sex differences in the biological mechanisms of hyperalgesic priming. At the central nervous system (CNS) level, PGE2-induced hyperalgesic priming activates microglia in the spinal cord of both females and males, whereas P2X3/4 antagonists and p38 inhibitors only alleviate hyperalgesic priming in male mice.23 It was discovered that the expression of prolactin receptors in Nav1.8+ neurons plays a role in triggering pain specifically in female mice.41 While these studies offer a theoretical foundation for understanding how acute pain can transform into chronic pain, much of the existing research on chronic pain still primarily focuses on male animals.42 The ongoing discussion of gender differences is essential for advancing personalized pain treatment.
Small Molecules Induce Primes
Recent studies have introduced hyperalgesic priming induced by small molecules, in conjunction with PEG2, alongside the classical inflammatory substances and the opioid-induced hyperalgesic priming model previously mentioned. Previous studies have shown that Ephrin-B ligands are expressed in DRG neurons and other peripheral cell types43–45 and regulate some painful diseases, like pancreatic cancer46 and rheumatoid and osteoarthritis.47 However, the mechanism of the ephrin-B2-EphB signaling pathway in pain remains unclear. David et al established the hyperalgesic priming model by injecting ephrin-B2-Fc in the back paw first and PGE2 at the same site 3 days later. The researchers found that ephrin-B2 directly affects mouse and human sensory neurons, promoting nociceptor plasticity through MNK-eIF4E signaling during hyperalgesic priming, with noted gender differences in response to ephrin-B2. Female mice could only exhibit hyperalgesic priming when 100 ng of ephrin-B2-Fc was combined with PGE, while male mice could respond to 10 ng of ephrin-B2-Fc. However, the specific mechanism remains to be clarified. Furthermore, this study introduces a novel approach whereby PGE2 can serve as a stimulus to assess priming in the supradural migraine model, a concept not previously addressed in earlier research.25
Recent studies have examined chronic pain through the lens of metabolomics and cellular metabolism.48–50 Melemedjian et al developed a model of hyperalgesia priming through the co-stimulation of nerve growth factor (NGF) and PGE2. They discovered that compensatory alterations in glutamine metabolism, mediated by NGF via the ASCT2 transporter, are fundamental to the transition from acute to chronic pain states from a metabolic viewpoint. This finding highlights a new therapeutic target for pain management.26
Chemotherapeutic Drugs Induce Primes
With the increasing incidence of cancer, chemotherapy-induced peripheral neuropathy(CIPN) can occur in about 25% to 30% of patients treated with chemotherapy,51,52 resulting in a painful state lasting months to years after the end of treatment, implying a transition from acute to chronic pain in these patients.53 Studies have shown that subcutaneous injection of PGE2 at 21, 42, and 60 days after oxaliplatin administration can prolong the duration of hyperalgesia. In addition, oxaliplatin combined with PGE2 caused the hyperalgesic priming to remain unattenuated at day 60, compared with oxaliplatin administration alone.24 This model can be used as a reference for initiating hyperalgesic priming induced by chemotherapeutic drugs and also provides a novel direction for the mechanism study of CIPN.
Opioid analgesics are the primary treatment for patients with CIPN, but the effectiveness of opioids remains unsatisfactory due to complications like OIH.54,55 To investigate the role of the MOR in oxaliplatin-induced hyperalgesia priming, researchers measured hyperalgesic priming by assessing the prolongation of PGE2-induced hyperalgesia in CIPN. They found that hyperalgesic priming was significantly reduced in rats treated with MOR antisense oligonucleotide (AS-ODN) compared to the control group. This suggests that hyperalgesic priming induced by oxaliplatin is dependent on the activation of MOR.56 By studying this model and its administration, the involvement of other drug receptors in hyperalgesic priming caused by chemotherapy drugs can be further examined to generate strategies for preventing and addressing the chronic nature of CIPN.
Bee Venom Induces Primes
Bee venom is a biotoxin produced by glands in bees’ abdomens. It is a colorless, acidic substance with a distinctive odor and bitter taste.57 In recent years, bee venom has been used to treat various diseases, including stimulating acupuncture points in patients and addressing chronic and autoimmune conditions.58–60 Bee venom offers a range of benefits, including cellular protection, antioxidant properties, and antibacterial, antiviral, anti-inflammatory, neuroprotective, anti-arthritis, anti-metastasis, and anticancer effects. However, its use is not widely promoted due to its double-edged sword nature, which can also cause pain and inflammation.61,62
Bee venom is a widely used toxic substance that is employed to induce acute inflammatory pain.62 A single injection of bee venom into the plantar surface of the hind paw of the rat can trigger short-term mechanical hyperalgesia.63 To explore how bee venom influences the shift from acute pain to chronic pain, Chen et al developed a hyperalgesic priming model.64 They achieved this by injecting bee venom and PEG2 into the plantar surface of the rat’s hind paw, utilizing a modeling approach that combines inflammatory stimulants with PEG2. The specific practice involves injecting bee venom into the skin of the rat’s sole. Seven days later, an intraplantar injection of PEG2 is administered at the same site. This procedure results in lasting mechanical hyperalgesia compared to the control group. By extending the time frame, it was found that the pain stimulation effect caused by bee venom could last for 21 days or more.64
Since the SDF1-CXCR4 signaling pathway is involved in regulating various chronic pain conditions,65 including pain caused by spared nerve injury,66 sciatic nerve injury,67 and bone cancer.68 By establishing the above model, Chen et al further confirmed that the SDF1-CXCR4 signaling pathway regulates the transition from acute pain to chronic pain.64 The creation of this model enhances the understanding of chronic pain and offers a direction for researching the mechanisms involving biotoxins.
Acid-Induced Chronic Model of Hyperalgesia Priming Originating from the Musculoskeletal System
Musculoskeletal diseases are often accompanied by varying degrees of pain. According to statistics, about 1.75 billion people worldwide suffer from chronic musculoskeletal pain, like fibromyalgia, which seriously affects people’s daily activities.69 Earlier models of musculoskeletal pain primarily considered short-term hyperalgesia until 2001,70,71 when researchers introduced an animal model that simulates chronic musculoskeletal pain.72 The acidity of tissues is positively correlated with the level of pain.73 Research on humans and animals has shown that injecting acidic solutions into muscles or tissues can induce pain.73–75 Maximum activation of nociceptors occurs at a pH level of 5.2, while sustained activation is evident at a pH of 6.0.76 The researchers first injected 100 μL of preservative-free, low-pH sterile saline into the gastrocnemius muscle of the rats. This injection was repeated with the same volume and pH of sterile saline on days 2, 5, and 10 at the same site. Their findings showed that repeated injections of low-pH saline into the gastrocnemius induced persistent and widespread mechanical hyperalgesia without causing movement disorders or peripheral tissue damage. Significant reductions in bilateral mechanical withdrawal thresholds were observed only on days 2 and 5 following injections with pH 4.0 saline, and not on day 10. These results indicate that the timing of repeated injections can influence the duration of the body’s pain response.72 Since then, a comprehensive, long-lasting model of musculoskeletal pain has been developed, providing a foundation for further research on how acute pain evolves into chronic pain in this context (Table 2). This model is commonly used to replicate chronic widespread pain (CWP) or fibromyalgia syndrome (FMS) in humans.
 |
Table 2 Critical Articles Discussing Acid-Induced Hyperalgesic Priming |
In the above model, administering a series of analgesics to the entire body revealed that morphine has a dose-dependent effect on mechanical hypersensitivity, particularly resulting in long-term reductions. NS1209 (a selective, competitive and potent AMPA receptor antagonist), ketamine, methetine, regabine and flupirtine were second only to morphine, but lamotrigine, carprofen and diazepam showed no significant effect, indicating that peripheral inflammatory mediators did not significantly regulate the pathophysiological processes in this model.77 This finding aligns with the observation of fewer histological changes at the injection site after repeated injections of acid saline, suggesting that central regulation is involved in the development of acid-induced muscle pain.72 Moreover, pregabalin, a selective calcium channel blocker, also reduces acid-induced muscle pain,81 further enriching the study of drug effects in this model.
Chronic musculoskeletal pain is more common in women,82–85 and acid-induced muscle hyperalgesia is also a sex-dependent manner. In male mice, hyperalgesia returned to baseline levels by day 14, while in female mice, it lasted up to 42 days.86 Further research found that removing the ovaries alleviated mechanical allodynia induced by repeated acid solution injection, which could be reversed by intrathecal supplementation with the ovarian hormone 17 beta-oestradiol.79 At the same time, repeated acid solution injection induced the activation of ERK in the spinal dorsal horn, and inhibition of ERK activation could reverse the abnormal mechanical pain.79 These findings suggest the role of ovarian hormone and spinal cord ERK activation in developing chronic muscle pain induced by repeated acid solution injection and provide insight into the mechanisms of sex hormone regulation at the CNS level.
The earlier study confirmed the activation of ERK in the dorsal horn of the spinal cord associated with acid-induced chronic pain, identifying that the active ERK cells were predominantly spinal neurons or astrocytes.79 Recent studies further determined that the spinal cord p-ERK positive cells in this hyperalgesic priming model are mainly vesicular glutamate transporter-2 positive neurons. Additionally, the p-ERK signaling pathway can further activate spinal cord astrocytes through the involvement of glutamate transporters. Activated astrocytes, in turn, affect spinal cord neurons by releasing D-serine, an intercellular interaction involved in the regulation of acid-induced muscle pain.80 Further research is needed to clarify how astrocytes contribute to this type of pain through D-serine secretion, which should also be explored in relation to other pain types.
In addition to the role of the sex hormone-nervous system circulatory interaction in acid-induced muscle pain, researchers have found that local immune cells are also involved in regulating this pain model. Research indicates that eliminating resident macrophages in muscle tissue or suppressing their function can alleviate acid-related muscle pain, suggesting that resident macrophages in muscle tissue are involved in developing acid-induced chronic muscle pain.78 Further studies found that the acid-induced resident macrophages secrete cytokines, including IL-4, IL-6, GM-CSF, TNF-α, IL-2, IL-5, IL-10, and IFN-γ, suggesting that these cytokines regulate the development of chronic muscle pain.78 Despite the ongoing research, the mechanisms by which resident macrophages regulate the up-pathway remain unclear. Future studies may provide a better understanding of the regulatory interactions between immune cells and sensory nerves in this model.
Stress-Induced Hyperalgesic Priming for Migraine-Like Pain
In 2021, about 3.4 billion people worldwide were affected by neurological disorders, with migraines being the third most common, significantly impacting health and well-being.87,88 Recent studies have found that migraine patients have a lower pain threshold compared to individuals without migraines, making them more susceptible to pain attacks.89 Previous attempts to induce migraines in healthy volunteers using stimulants have overlooked the specific vulnerabilities of migraine sufferers.90,91 In contrast, research involving stimulation studies on individuals prone to migraines is becoming more relevant and realistic.92,93
Stress is a common factor that induces migraines, and repeated stress can lead to an increase in migraine attacks, which raises the risk of transforming episodic migraines into chronic migraines.94,95 Additionally, inhaling umbellulone can trigger cluster headaches.96,97 Since prior studies have not investigated umbellulone in the absence of headaches, Frank et al combined stress with umbellulone to develop a new chronic transformation model of migraine-like pain.98 The researchers exposed mice to restraint stress (RS) for two hours each day over three days to initiate a potentially sensitive pain condition. Pain sensitivity was assessed on days 3, 5, 7, 9, 11, 14, and 16 following the initial RS. Baseline measurements of tactile stimuli were collected on days 16, 18, and 20 or days 16, 17, and 18 after the first RS and UMB inhalation under isoflurane anesthesia or isoflurane alone. After UMB exposure, the response frequency to tactile stimuli was tested every 30 minutes and every hour until reaching 5–6 hours, and again 24 hours after exposure. The findings revealed that repeated RS heightened pain sensitivity in both female and male rats following exposure to UMB. Previous studies indicate that painful behavior does not occur without inflammatory stimulation before inhaling substances containing UMB, suggesting that the trigeminal nervous system requires sensitization.99 This model can be used to investigate stress-induced hyperalgesic priming for migraine-like pain.98
Frequent migraine episodes indicate a priming phenomenon that increases susceptibility to later stimuli due to central and possibly peripheral sensitization.89 This “two-hit” hyperalgesia priming model may contribute to transitioning from episodic migraine to chronic migraine through central sensitization.6 In this modality, exposure to subliminal UMB stimulates the TRPA1 receptor in the trigeminal afferent nerve.98 Stress can activate the kappa opioid receptor (KOR), and KOR antagonists effectively block pain induced by stress.100,101 Administering KOR antagonists before initiating RS can prevent pain from subsequent exposure to UMB events.98 This suggests that KOR antagonists may have the potential to prevent and treat chronic migraines. Further investigation revealed that kappa opioid agonists exhibit sex-based differences in a “two-hit” priming strategy, with PRL/PRLR signaling involvement identified exclusively in females.102 Additionally, stress-induced activation of KOR in the hypothalamus raised circulating prolactin levels, which reduced trigeminal pain sensitivity.103 These mechanisms are linked to higher prolactin levels in circulation and may contribute to the increased prevalence of migraine and higher stress responses in women.102–105 Current clinical trials are investigating the effectiveness of KOR antagonists in preventing migraines in both sexes,106,107 while prolactin and prolactin receptor antibodies may help improve treatment for migraines and other stress-related neurological disorders specifically in women. Future research could explore the mechanisms behind hormone-induced sex differences in migraines, providing a foundation for personalized treatment.
Limitations of Translating the Above Models to Clinical Settings
Despite significant advancements in studying the transition from acute to chronic pain using animal models, the clinical translation of hyperalgesic priming remains limited owing to its intricate molecular mechanisms. For example, the nervous systems of rodents are still quite different from those of humans, especially in higher pain-processing areas such as the cerebral cortex. Animal models often ignore the psychological, social, and environmental factors that contribute to pain, which play an important role in the human experience of pain. Analgesics that are effective in animal models may not work well or even produce adverse reactions in human clinical trials, which may be related to the pharmacokinetic and pharmacodynamic differences between the drugs in humans and animals. Future research needs to develop more complex and integrated animal models that combine multidisciplinary approaches (such as neuroimaging, genetics, and behavioral science) to better simulate the complexity and diversity of chronic pain in humans.
Conclusion
This review of hyperalgesic priming synthesizes existing literature to elucidate the intricate mechanisms of pain sensitization and its manifestations under various pathological conditions, thereby enhancing our understanding of pain perception and regulation. It underscores how prior pain experiences can potentiate subsequent pain responses, offering a novel perspective on the development and maintenance of chronic pain. Furthermore, the review identifies limitations in current research and suggests future directions, including the exploration of more effective intervention strategies to prevent or reverse pain sensitization. This is crucial for developing innovative methods to treat chronic pain and improving patient quality of life.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (grant number 82401415).
Disclosure
The authors declare that they have no competing financial interests or personal relationships that could have influenced the work reported in this paper.
References
1. Brennan F, Carr DB, Cousins M. Pain management: a fundamental human right. Anesth Analg. 2007;105(1):205–221. doi:10.1213/01.ane.0000268145.52345.55
2. Gan TJ. Poorly controlled postoperative pain: prevalence, consequences, and prevention. J Pain Res. 2017;10:2287–2298. doi:10.2147/JPR.S144066
3. Dauber A, Osgood PF, Breslau AJ, et al. Chronic persistent pain after severe burns: a survey of 358 burn survivors. Pain Med. 2002;3(1):6–17. doi:10.1046/j.1526-4637.2002.02004.x
4. Dey S, Sanders AE, Martinez S, et al. Alternatives to opioids for managing pain. In: StatPearls. Treasure Island (FL): StatPearls Publishing LLC; 2025.
5. Cohen SP, Vase L, Hooten WM. Chronic pain: an update on burden, best practices, and new advances. Lancet. 2021;397(10289):2082–2097. doi:10.1016/S0140-6736(21)00393-7
6. Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32(12):611–618. doi:10.1016/j.tins.2009.07.007
7. Melemedjian OK, Asiedu MN, Tillu DV, et al. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J Neurosci. 2010;30(45):15113–15123. doi:10.1523/JNEUROSCI.3947-10.2010
8. Chivers SB, Andrade MA, Hammack RJ, et al. Peripheral macrophages contribute to nociceptor priming in mice with chronic intermittent hypoxia. Sci Signal. 2024;17(847):eadn8936. doi:10.1126/scisignal.adn8936
9. Aley KO, Messing RO, Mochly-Rosen D, et al. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20(12):4680–4685. doi:10.1523/JNEUROSCI.20-12-04680.2000
10. Navratilova E, Porreca F. Reward and motivation in pain and pain relief. Nat Neurosci. 2014;17(10):1304–1312. doi:10.1038/nn.3811
11. Ferrari LF, Araldi D, Levine JD. Distinct terminal and cell body mechanisms in the nociceptor mediate hyperalgesic priming. J Neurosci. 2015;35(15):6107–6116. doi:10.1523/JNEUROSCI.5085-14.2015
12. Ferrari LF, Bogen O, Reichling DB, et al. Accounting for the delay in the transition from acute to chronic pain: axonal and nuclear mechanisms. J Neurosci. 2015;35(2):495–507. doi:10.1523/JNEUROSCI.5147-13.2015
13. Dina OA, Green PG, Levine JD. Role of interleukin-6 in chronic muscle hyperalgesic priming. Neuroscience. 2008;152(2):521–525. doi:10.1016/j.neuroscience.2008.01.006
14. Khasar SG, Burkham J, Dina OA, et al. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28(22):5721–5730. doi:10.1523/JNEUROSCI.0256-08.2008
15. Moy JK, Kuhn JL, Szabo-Pardi TA, et al. eIF4E phosphorylation regulates ongoing pain, independently of inflammation, and hyperalgesic priming in the mouse CFA model. Neurobiol Pain. 2018;4:45–50. doi:10.1016/j.ynpai.2018.03.001
16. Yonehara N, Kudo T, Iwatsubo K, et al. A possible involvement of the central endorphin system in the autoanalgesia induced by chronic administration of Freund’s adjuvant solution in rats. Pain. 1983;17(1):91–98. doi:10.1016/0304-3959(83)90131-8
17. Tanaka M, Cummins TR, Ishikawa K, et al. SNS Na+ channel expression increases in dorsal root ganglion neurons in the carrageenan inflammatory pain model. Neuroreport. 1998;9(6):967–972. doi:10.1097/00001756-199804200-00003
18. Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113(1–2):185–190. doi:10.1016/j.pain.2004.10.021
19. Joseph EK, Parada CA, Levine JD. Hyperalgesic priming in the rat demonstrates marked sexual dimorphism. Pain. 2003;105(1–2):143–150. doi:10.1016/S0304-3959(03)00175-1
20. Hendrich J, Alvarez P, Joseph EK, et al. Electrophysiological correlates of hyperalgesic priming in vitro and in vivo. Pain. 2013;154(10):2207–2215. doi:10.1016/j.pain.2013.07.004
21. Ferrari LF, Bogen O, Levine JD. Nociceptor subpopulations involved in hyperalgesic priming. Neuroscience. 2010;165(3):896–901. doi:10.1016/j.neuroscience.2009.11.029
22. Araldi D, Ferrari LF, Levine JD. Repeated mu-opioid exposure induces a novel form of the hyperalgesic priming model for transition to chronic pain. J Neurosci. 2015;35(36):12502–12517. doi:10.1523/JNEUROSCI.1673-15.2015
23. Paige C, Maruthy GB, Mejia G, et al. Spinal inhibition of P2XR or p38 signaling disrupts hyperalgesic priming in male, but not female, mice. Neuroscience. 2018;385:133–142. doi:10.1016/j.neuroscience.2018.06.012
24. Staurengo-Ferrari L, Araldi D, Green PG, et al. Neuroendocrine mechanisms in oxaliplatin-induced hyperalgesic priming. Pain. 2023;164(6):1375–1387. doi:10.1097/j.pain.0000000000002828
25. David ET, Yousuf MS, Mei H-R, et al. ephrin-B2 promotes nociceptive plasticity and hyperalgesic priming through EphB2-MNK-eIF4E signaling in both mice and humans. Pharmacol Res. 2024;206:107284. doi:10.1016/j.phrs.2024.107284
26. Haque MM, Kuppusamy P, Melemedjian OK. Glutamine oxidation in mouse dorsal root ganglia regulates pain resolution and chronification. J Neurosci. 2024;44(47):e1442242024. doi:10.1523/JNEUROSCI.1442-24.2024
27. Parada CA, Yeh JJ, Joseph EK, et al. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. 2003;17(9):1847–1852. doi:10.1046/j.1460-9568.2003.02626.x
28. Cesare P, Dekker LV, Sardini A, et al. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23(3):617–624. doi:10.1016/S0896-6273(00)80813-2
29. Hundle B, McMahon T, Dadgar J, et al. Overexpression of epsilon-protein kinase C enhances nerve growth factor-induced phosphorylation of mitogen-activated protein kinases and neurite outgrowth. J Biol Chem. 1995;270(50):30134–30140. doi:10.1074/jbc.270.50.30134
30. Khasar SG, Lin Y-H, Martin A, et al. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24(1):253–260. doi:10.1016/S0896-6273(00)80837-5
31. Summer GJ, Puntillo KA, Miaskowski C, et al. TrkA and PKC-epsilon in thermal burn-induced mechanical hyperalgesia in the rat. J Pain. 2006;7(12):884–891. doi:10.1016/j.jpain.2006.04.009
32. Joseph EK, Levine JD. Hyperalgesic priming is restricted to isolectin B4-positive nociceptors. Neuroscience. 2010;169(1):431–435. doi:10.1016/j.neuroscience.2010.04.082
33. Bogen O, Alessandri-Haber N, Chu C, et al. Generation of a pain memory in the primary afferent nociceptor triggered by PKCε activation of CPEB. J Neurosci. 2012;32(6):2018–2026. doi:10.1523/JNEUROSCI.5138-11.2012
34. Cunha FQ, Poole S, Lorenzetti BB, et al. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol. 1992;107(3):660–664. doi:10.1111/j.1476-5381.1992.tb14503.x
35. Woolf CJ, Allchorne A, Safieh‐Garabedian B, et al. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br J Pharmacol. 1997;121(3):417–424. doi:10.1038/sj.bjp.0701148
36. Poole S, Lorenzetti BB, Cunha JM, et al. Bradykinin B1 and B2 receptors, tumour necrosis factor alpha and inflammatory hyperalgesia. Br J Pharmacol. 1999;126(3):649–656. doi:10.1038/sj.bjp.0702347
37. Levine JD, Taiwo YO. Involvement of the mu-opiate receptor in peripheral analgesia. Neuroscience. 1989;32(3):571–575. doi:10.1016/0306-4522(89)90279-0
38. Taiwo YO, Levine JD. Direct cutaneous hyperalgesia induced by adenosine. Neuroscience. 1990;38(3):757–762. doi:10.1016/0306-4522(90)90068-F
39. Aley KO, Green PG, Levine JD. Opioid and adenosine peripheral antinociception are subject to tolerance and withdrawal. J Neurosci. 1995;15(12):8031–8038. doi:10.1523/JNEUROSCI.15-12-08031.1995
40. Joseph EK, Reichling DB, Levine JD. Shared mechanisms for opioid tolerance and a transition to chronic pain. J Neurosci. 2010;30(13):4660–4666. doi:10.1523/JNEUROSCI.5530-09.2010
41. Paige C, Barba-Escobedo PA, Mecklenburg J, et al. Neuroendocrine mechanisms governing sex differences in hyperalgesic priming involve prolactin receptor sensory neuron signaling. J Neurosci. 2020;40(37):7080–7090. doi:10.1523/JNEUROSCI.1499-20.2020
42. Yang J, Xie Y-F, Smith R, et al. Discordance between preclinical and clinical testing of NaV1.7-selective inhibitors for pain. Pain. 2024;166(3):481–501. doi:10.1097/j.pain.0000000000003425
43. Kobayashi H, Kitamura T, Sekiguchi M, et al. Involvement of EphB1 receptor/EphrinB2 ligand in neuropathic pain. Spine. 2007;32(15):1592–1598. doi:10.1097/BRS.0b013e318074d46a
44. Song XJ, Zheng J-H, Cao J-L, et al. EphrinB-EphB receptor signaling contributes to neuropathic pain by regulating neural excitability and spinal synaptic plasticity in rats. Pain. 2008;139(1):168–180. doi:10.1016/j.pain.2008.03.019
45. Zhao J, Yuan G, Cendan CM, et al. Nociceptor-expressed ephrin-B2 regulates inflammatory and neuropathic pain. Mol Pain. 2010;6:77. doi:10.1186/1744-8069-6-77
46. Wangzhou A, Paige C, Neerukonda SV, et al. A ligand-receptor interactome platform for discovery of pain mechanisms and therapeutic targets. Sci Signal. 2021;14(674). doi:10.1126/scisignal.abe1648
47. Kuo D, Ding J, Cohn IS, et al. HBEGF(+) macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci Transl Med. 2019;11(491). doi:10.1126/scitranslmed.aau8587
48. Aroke EN, Powell-Roach KL. The metabolomics of chronic pain conditions: a systematic review. Biol Res Nurs. 2020;22(4):458–471. doi:10.1177/1099800420941105
49. Haque MM, Kuppusamy P, Melemedjian OK. Disruption of mitochondrial pyruvate oxidation in dorsal root ganglia drives persistent nociceptive sensitization and causes pervasive transcriptomic alterations. Pain. 2024;165(7):1531–1549. doi:10.1097/j.pain.0000000000003158
50. Kuppusamy P, Haque MM, Traub RJ, et al. Targeting metabolic pathways alleviates bortezomib-induced neuropathic pain without compromising anticancer efficacy in a sex-specific manner. Front Pain Res. 2024;5:1424348. doi:10.3389/fpain.2024.1424348
51. Colvin LA. Chemotherapy-induced peripheral neuropathy: where are we now? Pain. 2019;160(Suppl 1):S1–s10. doi:10.1097/j.pain.0000000000001540
52. Park SB, Lin CS, Krishnan AV, et al. Long-term neuropathy after oxaliplatin treatment: challenging the dictum of reversibility. Oncologist. 2011;16(5):708–716. doi:10.1634/theoncologist.2010-0248
53. Price TJ, Basbaum AI, Bresnahan J, et al. Transition to chronic pain: opportunities for novel therapeutics. Nat Rev Neurosci. 2018;19(7):383–384. doi:10.1038/s41583-018-0012-5
54. Chou R, Turner JA, Devine EB, et al. The effectiveness and risks of long-term opioid therapy for chronic pain: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med. 2015;162(4):276–286. doi:10.7326/M14-2559
55. Shah A, Hoffman EM, Mauermann ML, et al. Incidence and disease burden of chemotherapy-induced peripheral neuropathy in a population-based cohort. J Neurol Neurosurg Psychiatry. 2018;89(6):636–641. doi:10.1136/jnnp-2017-317215
56. Araldi D, Staurengo-Ferrari L, Bogen O, et al. Mu-Opioid Receptor (MOR) dependence of pain in chemotherapy-induced peripheral neuropathy. J Neurosci. 2024;44(42).
57. Vahidinia Z, Barati S, Azami Tameh A, et al. Bee venom as a promising therapeutic strategy in central nervous system diseases. Neuropeptides. 2024;107:102451. doi:10.1016/j.npep.2024.102451
58. Sung SH, Lee G. Bee venom acupuncture effects on pain and its mechanisms: an updated review. Toxins. 2021;13(9):608. doi:10.3390/toxins13090608
59. Małek A, Strzemski M, Kurzepa J, et al. Can bee venom be used as anticancer agent in modern medicine? Cancers. 2023;15(14). doi:10.3390/cancers15143714
60. El-Seedi HR, Eid N, Abd El-Wahed AA, et al. Honey bee products: preclinical and clinical studies of their anti-inflammatory and immunomodulatory properties. Front Nutr. 2021;8:761267. doi:10.3389/fnut.2021.761267
61. Stela M, Cichon N, Spławska A, et al. Therapeutic potential and mechanisms of bee venom therapy: a comprehensive review of apitoxin applications and safety enhancement strategies. Pharmaceuticals. 2024;17(9):1211. doi:10.3390/ph17091211
62. Chen J, Lariviere WR. The nociceptive and anti-nociceptive effects of bee venom injection and therapy: a double-edged sword. Prog Neurobiol. 2010;92(2):151–183. doi:10.1016/j.pneurobio.2010.06.006
63. Chen J, Luo C, Li H-L, et al. Primary hyperalgesia to mechanical and heat stimuli following subcutaneous bee venom injection into the plantar surface of hindpaw in the conscious rat: a comparative study with the formalin test. Pain. 1999;83(1):67–76. doi:10.1016/S0304-3959(99)00075-5
64. Yang F, Sun W, Luo WJ, et al. SDF1-CXCR4 signaling contributes to the transition from acute to chronic pain state. Mol Neurobiol. 2017;54(4):2763–2775. doi:10.1007/s12035-016-9875-5
65. Luo X, Wang X, Xia Z, et al. CXCL12/CXCR4 axis: an emerging neuromodulator in pathological pain. Rev Neurosci. 2016;27(1):83–92. doi:10.1515/revneuro-2015-0016
66. Knerlich-Lukoschus F, von der Ropp-Brenner B, Lucius R, et al. Spatiotemporal CCR1, CCL3(MIP-1α), CXCR4, CXCL12(SDF-1α) expression patterns in a rat spinal cord injury model of posttraumatic neuropathic pain. J Neurosurg Spine. 2011;14(5):583–597. doi:10.3171/2010.12.SPINE10480
67. Dubový P, Klusáková I, Svíženská I, et al. Spatio-temporal changes of SDF1 and its CXCR4 receptor in the dorsal root ganglia following unilateral sciatic nerve injury as a model of neuropathic pain. Histochem Cell Biol. 2010;133(3):323–337. doi:10.1007/s00418-010-0675-0
68. Hu XM, Liu Y-N, Zhang H-L, et al. CXCL12/CXCR4 chemokine signaling in spinal glia induces pain hypersensitivity through MAPKs-mediated neuroinflammation in bone cancer rats. J Neurochem. 2015;132(4):452–463. doi:10.1111/jnc.12985
69. Cieza A, Causey K, Kamenov K, et al. Global estimates of the need for rehabilitation based on the Global Burden of Disease study 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2021;396(10267):2006–2017. doi:10.1016/S0140-6736(20)32340-0
70. Mense S. Nociception from skeletal muscle in relation to clinical muscle pain. Pain. 1993;54(3):241–289. doi:10.1016/0304-3959(93)90027-M
71. Schaible HG, Grubb BD. Afferent and spinal mechanisms of joint pain. Pain. 1993;55(1):5–54. doi:10.1016/0304-3959(93)90183-P
72. Sluka KA, Kalra A, Moore SA. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle Nerve. 2001;24(1):37–46. doi:10.1002/1097-4598(200101)24:1<37::AID-MUS4>3.0.CO;2-8
73. Issberner U, Reeh PW, Steen KH. Pain due to tissue acidosis: a mechanism for inflammatory and ischemic myalgia? Neurosci Lett. 1996;208(3):191–194. doi:10.1016/0304-3940(96)12576-3
74. Hamamoto DT, Ortiz-González XR, Honda JM, et al. Intraplantar injection of hyaluronic acid at low pH into the rat hindpaw produces tissue acidosis and enhances withdrawal responses to mechanical stimuli. Pain. 1998;74(2–3):225–234. doi:10.1016/S0304-3959(97)00185-1
75. Steen KH, Reeh PW. Sustained graded pain and hyperalgesia from harmless experimental tissue acidosis in human skin. Neurosci Lett. 1993;154(1–2):113–116. doi:10.1016/0304-3940(93)90184-M
76. Steen KH, Reeh PW, Anton F, et al. Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J Neurosci. 1992;12(1):86–95. doi:10.1523/JNEUROSCI.12-01-00086.1992
77. Nielsen AN, Mathiesen C, Blackburn-Munro G. Pharmacological characterisation of acid-induced muscle allodynia in rats. Eur J Pharmacol. 2004;487(1–3):93–103. doi:10.1016/j.ejphar.2004.01.017
78. Gong WY, Abdelhamid RE, Carvalho CS, et al. Resident macrophages in muscle contribute to development of hyperalgesia in a mouse model of noninflammatory muscle pain. J Pain. 2016;17(10):1081–1094. doi:10.1016/j.jpain.2016.06.010
79. Chang JH, Tsai S-Y, Zeng Y-J, et al. Ovarian hormone-dependent and spinal ERK activation-regulated nociceptive hypersensitivity in female rats with acid injection-induced chronic widespread muscle pain. Sci Rep. 2019;9(1):3077. doi:10.1038/s41598-019-39472-z
80. Abdelaziz MA, Chen W-H, Chang Y-W, et al. Exploring the role of spinal astrocytes in the onset of hyperalgesic priming signals in acid-induced chronic muscle pain. PNAS Nexus. 2024;3(9):pgae362. doi:10.1093/pnasnexus/pgae362
81. Yokoyama T, Maeda Y, Audette KM, et al. Pregabalin reduces muscle and cutaneous hyperalgesia in two models of chronic muscle pain in rats. J Pain. 2007;8(5):422–429. doi:10.1016/j.jpain.2006.11.007
82. Bennett R. Myofascial pain syndromes and their evaluation. Best Pract Res Clin Rheumatol. 2007;21(3):427–445. doi:10.1016/j.berh.2007.02.014
83. Hsu ES. Acute and chronic pain management in fibromyalgia: updates on pharmacotherapy. Am J Ther. 2011;18(6):487–509. doi:10.1097/MJT.0b013e3181d6b6d4
84. Karibe H, Goddard G, Gear RW. Sex differences in masticatory muscle pain after chewing. J Dent Res. 2003;82(2):112–116. doi:10.1177/154405910308200207
85. Troeltzsch M, Troeltzsch M, Cronin RJ, et al. Prevalence and association of headaches, temporomandibular joint disorders, and occlusal interferences. J Prosthet Dent. 2011;105(6):410–417. doi:10.1016/S0022-3913(11)60084-X
86. Gregory NS, Gibson-Corley K, Frey-Law L, et al. Fatigue-enhanced hyperalgesia in response to muscle insult: induction and development occur in a sex-dependent manner. Pain. 2013;154(12):2668–2676. doi:10.1016/j.pain.2013.07.047
87. Leone M. Globalisation of the pharmacological treatment of migraine. Lancet Neurol. 2024;23(12):1179–1180. doi:10.1016/S1474-4422(24)00427-7
88. Brennan KC, Pietrobon D. A systems neuroscience approach to migraine. Neuron. 2018;97(5):1004–1021. doi:10.1016/j.neuron.2018.01.029
89. Peng KP, May A. Migraine understood as a sensory threshold disease. Pain. 2019;160(7):1494–1501. doi:10.1097/j.pain.0000000000001531
90. Tvedskov JF, Iversen HK, Olesen J, et al. Nitroglycerin provocation in normal subjects is not a useful human migraine model? Cephalalgia. 2010;30(8):928–932. doi:10.1111/j.1468-2982.2009.02014.x
91. Amin FM, Asghar MS, Guo S, et al. Headache and prolonged dilatation of the middle meningeal artery by PACAP38 in healthy volunteers. Cephalalgia. 2012;32(2):140–149. doi:10.1177/0333102411431333
92. Thomsen LL, Kruuse C, Iversen HK, et al. A nitric oxide donor (nitroglycerin) triggers genuine migraine attacks. Eur J Neurol. 1994;1(1):73–80. doi:10.1111/j.1468-1331.1994.tb00053.x
93. Kunkler PE, Ballard CJ, Oxford GS, et al. TRPA1 receptors mediate environmental irritant-induced meningeal vasodilatation. Pain. 2011;152(1):38–44. doi:10.1016/j.pain.2010.08.021
94. Borsook D, Maleki N, Becerra L, et al. Understanding migraine through the lens of maladaptive stress responses: a model disease of allostatic load. Neuron. 2012;73(2):219–234. doi:10.1016/j.neuron.2012.01.001
95. Haque B, Rahman KM, Hoque A, et al. Precipitating and relieving factors of migraine versus tension type headache. BMC Neurol. 2012;12:82. doi:10.1186/1471-2377-12-82
96. Benemei S, Appendino G, Geppetti P. Pleasant natural scent with unpleasant effects: cluster headache-like attacks triggered by Umbellularia californica. Cephalalgia. 2010;30(6):744–746. doi:10.1111/j.1468-2982.2009.01988.x
97. Nassini R, Materazzi S, Vriens J, et al. The ‘headache tree’ via umbellulone and TRPA1 activates the trigeminovascular system. Brain. 2012;135(Pt 2):376–390. doi:10.1093/brain/awr272
98. Kopruszinski CM, Navratilova E, Swiokla J, et al. A novel, injury-free rodent model of vulnerability for assessment of acute and preventive therapies reveals temporal contributions of CGRP-receptor activation in migraine-like pain. Cephalalgia. 2021;41(3):305–317. doi:10.1177/0333102420959794
99. Hawkins JL, Cornelison LE, Blankenship BA, et al. Vagus nerve stimulation inhibits trigeminal nociception in a rodent model of episodic migraine. Pain Rep. 2017;2(6):e628. doi:10.1097/PR9.0000000000000628
100. Xie JY, De Felice M, Kopruszinski CM, et al. Kappa opioid receptor antagonists: a possible new class of therapeutics for migraine prevention. Cephalalgia. 2017;37(8):780–794. doi:10.1177/0333102417702120
101. Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi:10.1016/j.brainres.2009.08.062
102. Kopruszinski CM, Watanabe M, Martinez AL, et al. Kappa opioid receptor agonists produce sexually dimorphic and prolactin-dependent hyperalgesic priming. Pain. 2023;164(6):e263–e273. doi:10.1097/j.pain.0000000000002835
103. Watanabe M, Kopruszinski CM, Moutal A, et al. Dysregulation of serum prolactin links the hypothalamus with female nociceptors to promote migraine. Brain. 2022;145(8):2894–2909. doi:10.1093/brain/awac104
104. Goldfarb EV, Seo D, Sinha R. Sex differences in neural stress responses and correlation with subjective stress and stress regulation. Neurobiol Stress. 2019;11:100177. doi:10.1016/j.ynstr.2019.100177
105. Verma R, Balhara YP, Gupta CS. Gender differences in stress response: role of developmental and biological determinants. Ind Psychiatry J. 2011;20(1):4–10. doi:10.4103/0972-6748.98407
106. Guerrero M, Urbano M, Kim E-K, et al. Design and synthesis of a novel and selective Kappa Opioid Receptor (KOR) Antagonist (BTRX-335140). J Med Chem. 2019;62(4):1761–1780. doi:10.1021/acs.jmedchem.8b01679
107. Rorick-Kehn LM, Witkin JM, Statnick MA, et al. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology. 2014;77:131–144. doi:10.1016/j.neuropharm.2013.09.021
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.