")
Back to Journals » Journal of Inflammation Research » Volume 17
Identification of Macrophage-Related Biomarkers for Abdominal Aortic Aneurysm Through Combined Single-Cell Sequencing and Machine Learning
Authors Yao G , Hu X, Song D, Yao J, Chen D , Luan T, Zhao Y
Received 16 October 2024
Accepted for publication 10 December 2024
Published 13 December 2024 Volume 2024:17 Pages 11009—11027
DOI https://doi.org/10.2147/JIR.S499593
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Guoqing Yao,1,* Xuemei Hu,2,* Daqiang Song,3 Jin Yao,4 Deqing Chen,2 Tiankuo Luan,5 Yu Zhao1
1Department of Vascular Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 2Department of Endocrinology, The People’s Hospital of Rongchang District, Chongqing, 402460, People’s Republic of China; 3Chongqing Key Laboratory of Molecular Oncology and Epigenetics, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 4School of Acu-Mox and Tuina, Chengdu University of Traditional Chinese Medicine, Chengdu, 610075, People’s Republic of China; 5Department of Anatomy, Chongqing Medical University, Chongqing, 400016, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tiankuo Luan, Department of Anatomy, Chongqing Medical University, Chongqing, 400016, People’s Republic of China, Email [email protected] Yu Zhao, Department of Vascular Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China, Email [email protected]
Purpose: The relationship between macrophages and the progression of abdominal aortic aneurysms (AAA) remains unclear, and effective biomarkers are lacking. In this study, we elucidated the mechanism whereby macrophages promote AAA development and identified associated biomarkers, with the goal of developing new targeted therapies and improving patient outcomes.
Patients and Methods: Differential expression analysis, weighted gene co-expression network analysis, and single-cell analysis were used to identify macrophage-related genes in an AAA dataset. Machine learning algorithms identified THBS1, HCLS1, DMXL2, and ZEB2 as key macrophage-related genes upregulated in AAA; these four hub genes were then used to construct a nomogram as an auxiliary tool for clinical diagnosis. Subsequent downstream single-cell and CellChat analyses were conducted to observe the interactions between macrophages and fibroblasts and analyze potential pathways.
Results: Single-cell validation confirmed enhanced THBS1 expression in macrophages in AAA. CellChat analysis revealed enhanced interactions between macrophages and fibroblasts in AAA through THBS1–CD47 signaling. Finally, an analysis of clinical samples from patients with AAA confirmed the high expression of THBS1 and CD47 in AAA and that THBS1 promotes the progression of AAA through the TNF-NFκB signaling pathway. Our findings reveal the THBS1–CD47 signaling pathway as a critical mechanism in macrophage-driven AAA progression, highlighting THBS1’s potential as a therapeutic target.
Conclusion: Our findings highlight THBS1 as a potential driver of macrophage-mediated AAA formation and an important biomarker for AAA diagnosis. The study results would help in improving treatment outcomes in patients with AAA. These findings provide a foundation for the development of diagnostic tools and targeted therapies for AAA, potentially improving early detection and patient outcomes.
Keywords: abdominal aortic aneurysm, macrophage, single-cell sequencing, machine learning algorithm, cell chat
Graphical Abstract:

Introduction
Abdominal aortic aneurysm (AAA) is a cardiovascular condition characterized by a pathological dilation of the aorta exceeding 50%, which can pose a major risk of aortic rupture and hemorrhage.1 Notably, AAA is often clinically silent until rupture.2 It is closely associated with high morbidity and mortality in older adults owing to aortic enlargement and rupture.3 As a chronic inflammatory disease, AAA is characterized by the infiltration of inflammatory cells throughout the arterial wall.1 The pathological process of AAA is also linked to changes in and loss of vascular smooth muscle cells (VSMCs) as well as remodeling of the extracellular matrix.4 Current treatments are limited to open or endovascular surgical repair in eligible patients. Continuous monitoring of the biological activity of AAA is crucial for mitigating the high mortality and morbidity associated with rupture. However, few effective biomarkers exist to predict AAA progression, and no therapies are available to prevent it.5,6 Previous studies investigating elastin peptides and type III procollagen amino-terminal pro-peptide as potential biomarkers in AAA have not been further developed because of their low sensitivity and specificity.7 Therefore, understanding the potential mechanisms underlying AAA progression is essential for accurately identifying appropriate therapeutic targets.
Macrophages within the arterial wall play a pivotal and multifaceted role in the pathogenesis of AAA.8 Moreover, macrophages are key participants in the immune system that exhibit phenotypic plasticity and functions under various pathophysiological conditions.9 Classically activated (M1) macrophages produce high levels of pro-inflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and inducible nitric oxide synthase (iNOS), which exacerbate local inflammation, recruit more immune cells, and promote the growth of AAA.10 In contrast, M2 macrophages are associated with wound healing and reduced inflammation. In the arterial wall, AAA progression is initiated when M1 macrophages respond to environmental stimuli and sustain persistent inflammation by producing proteolytic enzymes and pro-inflammatory mediators, leading to an M1/M2 imbalance.11 Elevated levels of pro-inflammatory cytokines have been detected in tissues from AAA animal models, which promote changes in the VSMC phenotype and elastin degradation.12–14 Therefore, inhibition of the expression of inflammatory cytokines, such as iNOS and IL-1β, can significantly reduce AAA formation.15,16 The small molecule activator SIRT1 enhances cellular p65 protein deacetylation, thereby inhibiting TNF-α-induced transcriptional activation of NF-κB, a key transcription factor for M1 macrophage activation.17 Macrophage-specific Sirt1 knockout AAA mice showed that Sirt1 deficiency promoted M1 polarization and reduced M2 polarization, accelerating AAA progression.18 However, despite our understanding of the crucial role of macrophages in AAA, identifying macrophage-related biomarkers and enhancing their clinical application remain challenging.
Following continuous advances in high-throughput sequencing technologies, the use of transcriptome analysis and single-cell RNA sequencing have become increasingly common for studying the relationship between immune cells and diseases.19 Weighted gene co-expression network analysis (WGCNA) is a systems biology approach designed to investigate gene interaction patterns across multiple samples. This method enables the identification of highly co-expressed gene clusters and facilitates the discovery of potential biomarker genes or therapeutic targets by aiding in analyzing the relationships between these gene sets and their associations with clinical traits.20 Furthermore, machine learning is increasingly being applied in the biomedical field for diagnosing and predicting disease outcomes.21,22 Together, these technologies can be used to analyze datasets to uncover disease mechanisms and guide clinical decision-making.
In this study, we investigated variations in the expression of genes associated with AAA and identified potential diagnostic and therapeutic targets using data from publicly available AAA databases, single-cell sequencing, WGCNA, and differential expression analysis. Furthermore, we applied machine learning algorithms, including support vector machine-recursive feature elimination (SVM-RFE), least absolute shrinkage and selection operator (LASSO), and random forest (RF), to pinpoint hub genes involved in AAA. Finally, through downstream single-cell analysis, verification of biomarkers, and identification of potential pathways, we elucidated the roles of hub genes in AAA. The biomarkers identified in this study reflect the pathological characteristics of different patients and can guide personalized treatment strategies. Additionally, tissue-level studies are more specific than circulating markers, as they reflect the biological characteristics of local lesions, thus addressing the limitations of existing screening technologies.
Materials and Methods
We retrieved two AAA RNA array datasets (GSE47472 and GSE57691) and one single-cell RNA sequencing dataset (GSE166676) from the Gene Expression Omnibus (GEO) database. The two RNA array datasets were used as the training set for model development. Additionally, we included another RNA array dataset (GSE98278) as an independent validation set to further assess the robustness and generalizability of the identified biomarkers. The GEO database is accessible at http://www.ncbi.nlm.nih.gov/geo. The transcriptome datasets were annotated and normalized using their respective GPL platforms. The information of 56 patients from the AAA group and 17 patients from the normal group was included in the transcriptome data for subsequent differential expression analysis and WGCNA. Detailed characteristics of the four datasets are provided in Table S1. All scripts used for data preprocessing, WGCNA analysis, and machine learning in this study are publicly available on the GitHub repository at https://github.com/guoqyao/cqmugqy.git.
Single-Cell Quality Control and Dimensionality Reduction
We identified cells that expressed 200–10,000 genes. We excluded the cells in which mitochondrial or ribosomal gene expression exceeded 20%. After identifying 2000 highly variable genes for subsequent analysis, we selected principal component (PC) 14 based on the PC clustering heatmap to generate a clustering tree. A resolution of one was chosen for dimensionality reduction, and the “UMAP” plot was used for visualization and annotation. Twenty clusters were identified.
Differential Gene Analysis and Single-Cell Annotation
Next, we analyzed the differentially expressed genes (DEGs) within each cluster based on the clustering results and performed manual single-cell annotation according to the expression of known marker genes in different cell types. We then used the “FindAllMarkers” function from the Seurat R package to filter the top five DEGs for each cell type. Based on manual annotations, we further identified DEGs in myeloid immune cell populations.
DEG Identification and WGCNA
The merged transcriptome datasets were used for differential expression analysis. We employed the “limma” R package to perform this analysis and visualized the results using heat maps. By applying the filtering criteria (|logFC| > 0.585, adjusted P < 0.05), we identified a set of DEGs, which were subsequently displayed in a volcano plot. We performed WGCNA on the merged transcriptome dataset to calculate gene significance. We divided the genes into different modules and identified the modules most closely associated with AAA.
Functional Enrichment Analysis of AAA-Related Genes
We intersected DEGs from single-cell myeloid immune cells, DEGs from transcriptome analysis, and gene clusters related to AAA from WGCNA. This intersection yielded overlapping disease-related genes for subsequent analyses. Next, to explore the functions and pathways of AAA-related genes, we used the “ClusterProfiler” R package for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment.
Screening of Biomarkers by Machine Learning
SVM-RFE, LASSO, and RF were used to screen for AAA biomarkers. LASSO logistic regression was performed using the “glmnet” R package, whereas RF analysis was conducted with the “RandomForest” R package. The “caret” package was employed for RF analysis with 10× cross-validation to select significant genes. We constructed the SVM classifier using the “e1071” R package. We then identified overlapping genes across these three classification models for further evaluation of their effectiveness as diagnostic markers. A diagnostic nomogram was constructed for disease prediction models. Additionally, receiver operating characteristic (ROC) analysis was used for validation; the predictive power of the algorithms was measured using area under the curve values.
Immune Infiltration Analysis
We employed the CIBERSORT analytical technique to evaluate immune cell infiltration levels in the transcriptome datasets by comparing AAA samples with normal controls. The analysis was conducted with the “PERM” parameter set to 1000 and a significance threshold of P < 0.05.
CellChat and Single-Cell Sequencing Analysis
We utilized the “CellChat” R package to analyze and visualize intercellular communication among various cell subpopulations in AAA. Ligand-receptor interaction analysis was conducted using a normalized expression matrix obtained from Seurat. Known ligand-receptor pairs were sourced from CellChatDB, a literature-supported database detailing ligand-receptor interactions in both mice and humans. Initially, we identified overexpressed ligands and receptors across different cell types, followed by the inference of communication probabilities by calculating interactions associated with each signaling pathway.
Based on the single-cell annotation results, we extracted the macrophage and fibroblast subpopulations separately to validate the expression of key genes and then visualized their differential expression using heatmaps and violin plots. Additionally, differential gene analysis was performed on macrophage and fibroblast subpopulations based on THBS1 and CD47 expression, respectively. GO and KEGG enrichment analyses were performed to reveal changes in the cellular functions and pathways of DEGs.
Cell Culture
THP-1 cells were obtained from the National Cell Bank of China, Chinese Academy of Sciences, Shanghai, China. THP-1 cells were cultured in RPMI Medium 1640, containing 10% fetal bovine serum, penicillin, and streptomycin, at 37°C in a humidified atmosphere of 5% CO2. 293T cells were cultured in DMEM high-glucose medium supplemented with 10% fetal bovine serum. THP-1 cells were incubated with 12-myristate 13-acetate (PMA, 100 ng/mL) for 24 h, after which the medium was replaced, and the cells were treated with lipopolysaccharide (LPS, 100 ng/mL) and interferon (IFN)-γ (20 ng/mL) for 24 h before proceeding with further experiments. All the reagents mentioned above were obtained from Solarbio, China.
Primer Design and Lentiviral Packaging
Primers were designed, annealed, and ligated into the AgeI and EcoRI restriction sites of the pLKO.1-puro vector to insert the shRNA sequences, resulting in the construction of shTHBS1 plasmid. Lipo6000 was used as the transfection reagent, and sh-THBS1, psPAX.2, and pMD2.G were added to actively growing 293T cells at a ratio of 4:3:1 for lentiviral packaging. After 72 h, viral supernatant was collected using a 0.45 μm filter. The viral supernatant and polybrene (10 μg/mL) were added to THP-1 cells in a six-well plate for infection, followed by puromycin (2 μg/mL) selection. All reagents mentioned above were purchased from Beyotime, China. The knockdown sequences are as follows: Sh-1: 5′-CCGG-TGACATCAGTGAGACCGATTT-CTCGAG-AAATCGGTCTCACTGATGTCA-TTTTT-3′; Sh-2: 5′-CCGG-GTAGGTTATGATGAGTTTAAT-CTCGAG-ATTAAACTCATCATAACCTAC-TTTTT-3′; Sh-3: 5′-CCGG-AGACATCTTCCAAGCATATAA-CTCGAG-TTATATGCTTGGAAGATGTCT-TTTTT-3′.
Quantitative PCR (qPCR) Analysis
Six tissue samples were obtained from the First Affiliated Hospital of Chongqing Medical University (three AAA and three normal tissue samples). This study was approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (K2024-221-01). Patients provided informed consent following the principles of the Declaration of Helsinki. Total RNA was extracted from AAA tissues and THP-1 cells using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. The qPCR procedure was carried out as described previously.23 The primer sequences used were as follows: GAPDH (Forward: 5′-GGACCTGACCTGCCGTCTAG-3′, Reverse: 5′-GTAGCCCAGATGCCCTTGA-3′), THBS1 (Forward: 5′-GCCATCCGCACTAACTACATT-3′, Reverse: 5′-TCCGTTGTGATAGCATAGGGG-3′), CD47 (Forward: 5′-TCCGGTGGTATGGATGAGAAA-3′, Reverse: 5′-ACCAAGGCCAGTAGCATTCTT-3′), synthesized by BGI (Shenzhen, China). GAPDH was used as an internal control. Relative expression levels were calculated using the 2^(-ΔCt) method. Statistical significance was set to P < 0.05.
Western Blotting
THP-1 cells were lysed using a protein lysis buffer containing protease and phosphatase inhibitors (Beyotime, China). After centrifugation, the supernatant was collected, and the protein concentration was measured using the BCA assay. Equal amounts of total protein from different samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% skimmed milk and then incubated with appropriate primary antibodies. After treatment with the appropriate HRP-conjugated secondary antibodies and washing the membrane, chemiluminescence (Bio-Rad, USA) was used to detect the signal as described previously.24 We assessed the expression levels of p65, phospho-p65, IκBα, phospho-IκBα, and β-actin. All antibodies were obtained from Proteintech, China.
Statistical Analysis
Statistical analyses were conducted using the R and SPSS software. A two-tailed Student’s t-test was used to validate significant differences. Statistical significance was set at P < 0.05.
Results
The workflow of this study is shown in Figure 1.
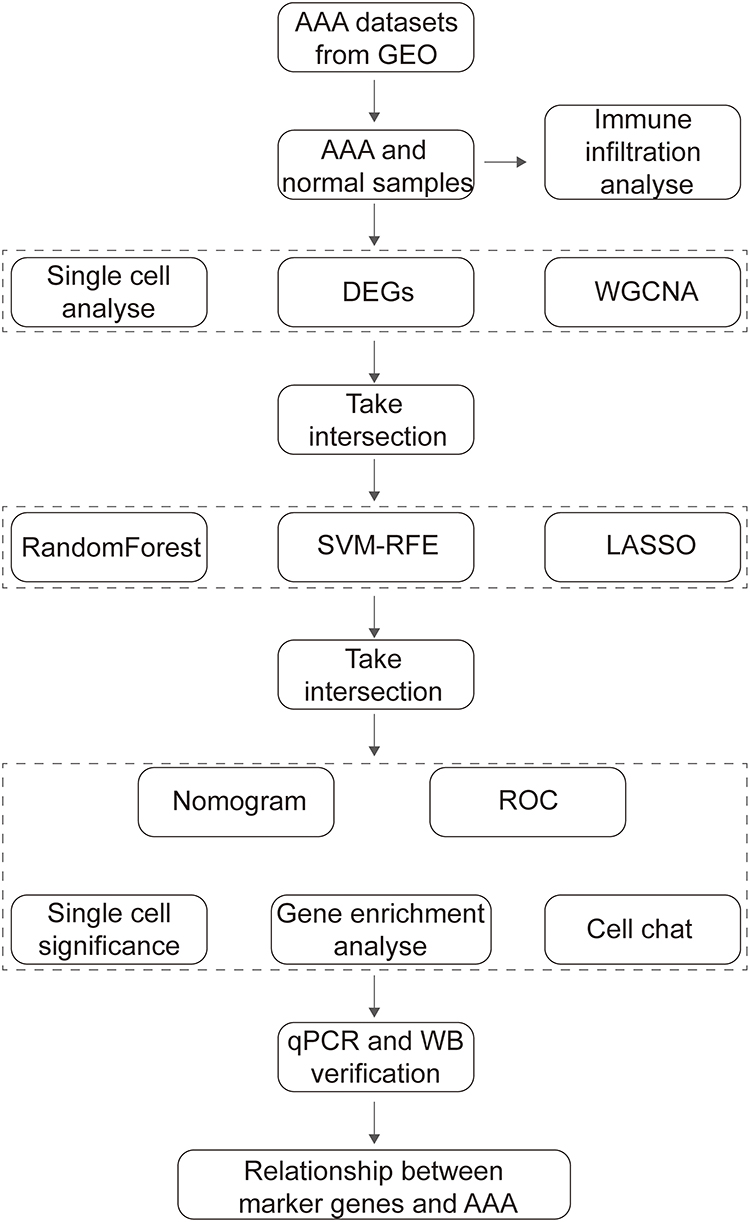 |
Figure 1 Flowchart of the study process. |
Single-Cell Quality Control and Clustering
The R package “Seurat” was used to analyze a single-cell dataset. The violin plots in Figure S1A display the filtered single-cell RNA characteristics, RNA quantity, mitochondrial RNA proportion, and ribosomal RNA proportion. According to the correlation analyses between the RNA quantity and RNA characteristics, mitochondrial gene proportion, and ribosomal gene proportion (Figure S1B), RNA quantity was highly correlated with RNA characteristics but not with the mitochondrial RNA proportion of mtRNA or RNA. Subsequently, we identified 2000 highly variable genes (highlighted in red in Figure S1C). Four AAA datasets and two normal arterial tissue datasets were combined; after data normalization and dimensionality reduction using PC analysis, we visualized the results using a “UMAP” plot (Figure S1D). Based on the PC analysis heatmap and JackStraw analysis (Figure S1E and F), PC = 14 was identified as the key PC for cell clustering (Figure 2A). Cell clustering analysis was executed employing the “FindNeighbors” and “FindClusters” functions from the “Seurat” package, with a resolution of one, and the results were displayed in a “UMAP” plot. The cells were categorized into 20 clusters (Figure 2B). Subsequently, we used the “FindAllMarkers” function from the “Seurat” package to identify DEGs in each cluster. The 20 cell clusters were annotated into 13 cell types based on cell-specific genes (Figure 2C), including neutrophils (S100A9, S100A8, and LYZ), macrophages (FCGR3A, CD68, and CD86), epithelial cells (EPCAM), monocytes (C1QA, CD14, and C1QB), fibroblasts (COL1A1), B cells (CD38 and MK167), NK cells (NKG7), T cells (CD3D and CD3E), endothelial cells (VWF), exocrine gland cells (PNLIP), VSMCs (LTBP1 and CNN1), mast cells (MS4A2 and FCER1A), and keratinocytes (CALML3) (Figure 2D). We calculated the proportion of all cells from different patients with AAA, noting the variability in the proportion of different cell types across patients. The DEGs for each cell type are shown in Figure 2E. The top five genes with the smallest P-values for each cell subpopulation are shown in Figure 2F. Finally, we identified 791 differential genes from myeloid immune cells using single-cell sequencing data (excluding mast cells).
 |
Figure 2 Single-cell clustering and annotation of abdominal aortic aneurysms (AAA). (A) Cluster tree visualization of clustering with PC = 14. (B) “UMAP” plot showing the distribution of clusters after dimensionality reduction. (C) Single-cell manual annotation of marker genes and their expression. (D) “UMAP” plot displaying single-cell annotation results. (E) Bar plot showing the relative proportions of single-cell subgroups in different AAA tissue samples. (F) Heatmap displaying differential genes across single-cell subgroups in AAA. Gray dashed lines represent myeloid immune cell subgroups. |
Differential Gene Analysis and WGCNA
Two AAA-related datasets (GSE47472 and GSE57691) collected from the GEO database were merged and normalized to reduce errors in the subsequent analyses. Differential analysis between the AAA and normal groups was performed using the criteria of adjusted P < 0.05 and |logFC| > 0.585, resulting in the selection of 1800 DEGs for further analysis (Figure 3A). A heatmap showing the top 30 most significantly upregulated genes and the top 15 most downregulated genes is presented in Figure 3B.
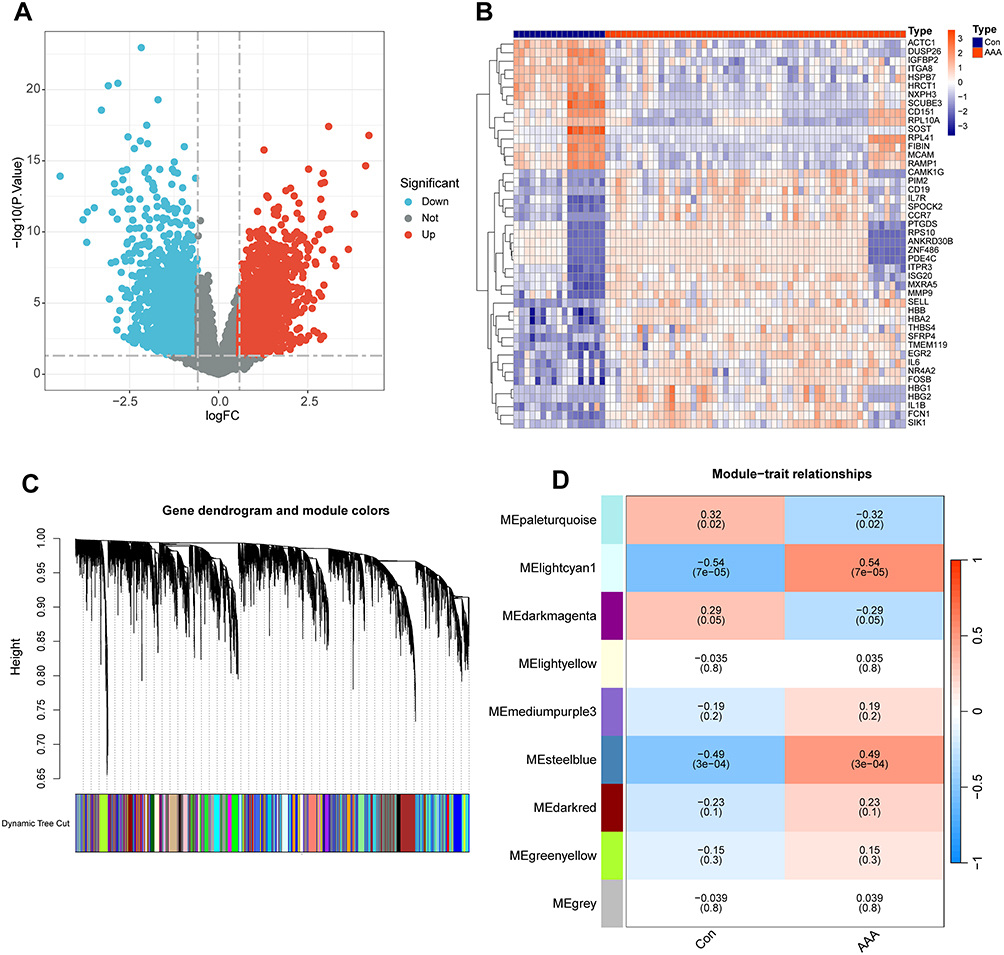 |
Figure 3 Differential gene visualization and weighted gene co-expression network analysis (WGCNA). (A) Volcano plot of differentially expressed genes in abdominal aortic aneurysms (AAA). (B) Heatmap showing the most significantly specific differential genes between AAA and normal abdominal aorta. (C) Genes with strong correlations are aggregated into the same module, with different modules shown in different colors. (D) Correlation analysis between WGCNA modules and AAA. |
We simultaneously performed WGCNA on the merged dataset. First, we used the “flashClust” package to perform clustering analysis with a threshold of 110; this clustering contained 49 samples. Next, using the “WGCNA” package’s “pickSoftThreshold” function, we filtered the power parameter range from 1 to 30 and chose a power of b = 9 (scale-free R2 = 0.9) as the soft threshold for constructing the scale-free network. We set the minimum number of genes per module to 50 and clustered the highly correlated genes into the same module (Figure 3C). To merge similar modules, a threshold of 0.6 was applied, leading to the identification of nine modules, each comprising genes with similar co-expression patterns (Figure 3D). Module-trait association analysis indicated that several modules were associated with AAA, with the lightcyan1 and steelblue modules showing strong associations and containing 1838 genes.
Identification and Validation of Hub Genes
Venn diagrams were used to aggregate the differential genes identified through single-cell myeloid immune cell analysis, DEG analysis, and WGCNA, resulting in a total of 41 genes (Figure 4A). GO and KEGG analyses were performed to explore the potential biological significance of these genes. The GO results were categorized into three domains (Figure S2A). The biological pathways involved included antigen processing and the presentation of peptides or polysaccharide antigens via major histocompatibility complex (MHC) class II molecules. Cellular component analysis indicated that MHC may be involved in this process. Molecular functions included binding of the MHC class II protein complex. The KEGG results were primarily focused on phagosome and allograft rejection; the genes most closely related to the top five KEGG pathways are displayed in Figure S2B and C.
 |
Figure 4 Machine learning selection of hub genes for abdominal aortic aneurysms (AAA). (A) Venn diagram identifying macrophage-related genes associated with AAA. (B) Identification of feature hub genes using the SVM-RFE algorithm. (C) Random Forest tree, where the x-axis represents the number of trees, and the y-axis represents the error rate. Red, green, and black dots represent AAA samples, normal samples, and all samples, respectively. (D) and (E) Eleven candidate genes obtained through LASSO regression and 10-fold cross-validation. (F) X-axis represents gene importance, and Y-axis represents hub genes. (G) Venn diagram identifying hub genes selected using machine learning algorithms. (H) Correlations between hub genes. |
Next, we integrated the results from the three machine learning algorithms. SVM-RFE selected twelve genes that resulted in the highest model accuracy (Figure 4B). RF ranked the genes according to disease relevance and selected the top genes that stabilized the decision tree with the smallest error (Figure 4C). Using linear regression, LASSO identified eleven important genes (Figure 4D and E). Finally, the four most important hub genes were identified (Figure 4G). The RF results also indicated that these four genes have relatively high importance (Figure 4F). Among them, THBS1 emerged as the most important. The correlation analysis of these four hub genes revealed that THBS1 was significantly negatively correlated with HCLS1 and DMXL2, whereas ZEB2 was minimally correlated with the other three genes (Figure 4H).
Accordingly, we established an AAA-related nomogram model based on four hub genes (THBS1, HCLS1, DMXL2, and ZEB2) as a simplified and reliable diagnostic tool for clinicians (Figure 5A). The accuracy of the nomogram model based on hub genes was close to the actual positive rate (Figure S3A). Both decision curve and clinical impact curve analyses significantly supported the ability of the nomogram model to identify AAA (Figure S3B and C). Additionally, ROC curves showed excellent classification performance for the hub genes, with high area under the curve values (Figure 5B–E). Violin plots illustrated the expression of the four hub genes in the normalized dataset, showing significant differences between AAA and normal tissues (Figure 5F–I).
 |
Figure 5 Construction of the hub-gene diagnostic clinical model. (A) Nomogram showing the prognostic value of THBS1, HCLS1, DMXL2, and ZEB2 in patients with abdominal aortic aneurysms (AAA). (B–E): ROC analysis results for THBS1, HCLS1, DMXL2, and ZEB2. (F–I): Violin plots showing the expression differences of THBS1, HCLS1, DMXL2, and ZEB2 in AAA and normal arterial tissues. **** P < 0.0001. |
Finally, we validated the identified hub genes in an independent dataset (GSE98278). The results showed that THBS1 was significantly upregulated in AAA tissues and exhibited strong diagnostic performance (Figure S4A and E). However, the other three genes (HCLS1, DMXL2, and ZEB2) did not show statistically significant differences in expression or diagnostic utility in the validation dataset (Figure S4B-D and F–H).
Differences in Immune Characteristics of Patients with AAA
Here, we used the CiberSort algorithm to investigate immune cell infiltration in AAA and normal samples. Figure 6A shows the relative proportions of immune cell infiltration in AAA and normal tissues. Compared with the normal group, patients with AAA showed a significant increase in B cells naive, T cells CD4 memory activated, T cells CD4 naive, NK cells resting, monocytes, macrophages M0, and neutrophils (Figure 6B). In contrast, the normal group exhibited increased infiltration rates of NK cells activated, macrophages M1, macrophages M2, and dendritic cells resting.
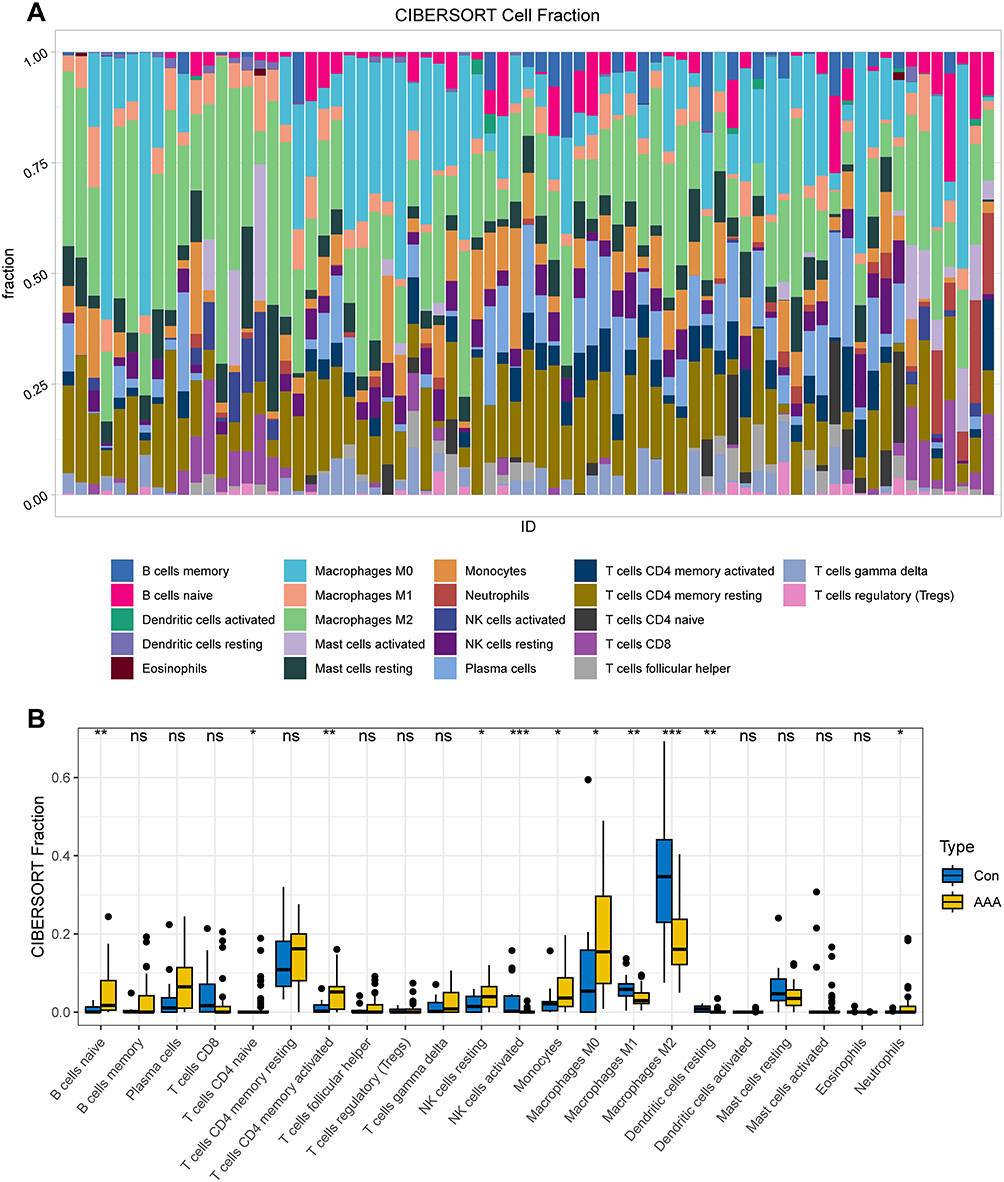 |
Figure 6 Immune infiltration analysis of abdominal aortic aneurysms (AAA). (A) Heatmap showing the relative proportions of immune cell infiltration in AAA and normal arterial samples. (B) Boxplot displaying the differential analysis results of immune cells in AAA and normal arterial samples. * P < 0.05; ** P < 0.01; *** P < 0.001. |
Single-Cell Downstream and CellChat Analyses
We identified AAA-related hub genes (THBS1, HCLS1, DMXL2, and ZEB2) via machine learning for validation using single-cell datasets. A violin plot showing the expression levels of these hub genes in different cell subgroups is presented in Figure 7A. We extracted macrophage subgroups from single-cell data using the “subset” function in the “Seurat” package and visualized the expression of hub genes in AAA and normal tissues using “UMAP” plots (Figure 7B). Differential analysis indicated that THBS1 and DMXL2 expression was increased in the macrophage subgroups of AAA, whereas ZEB2 expression was increased in the macrophage subgroups of normal arteries (Figure 7C). However, HCLS1 showed no significant differences between the AAA and normal tissue macrophage subgroups.
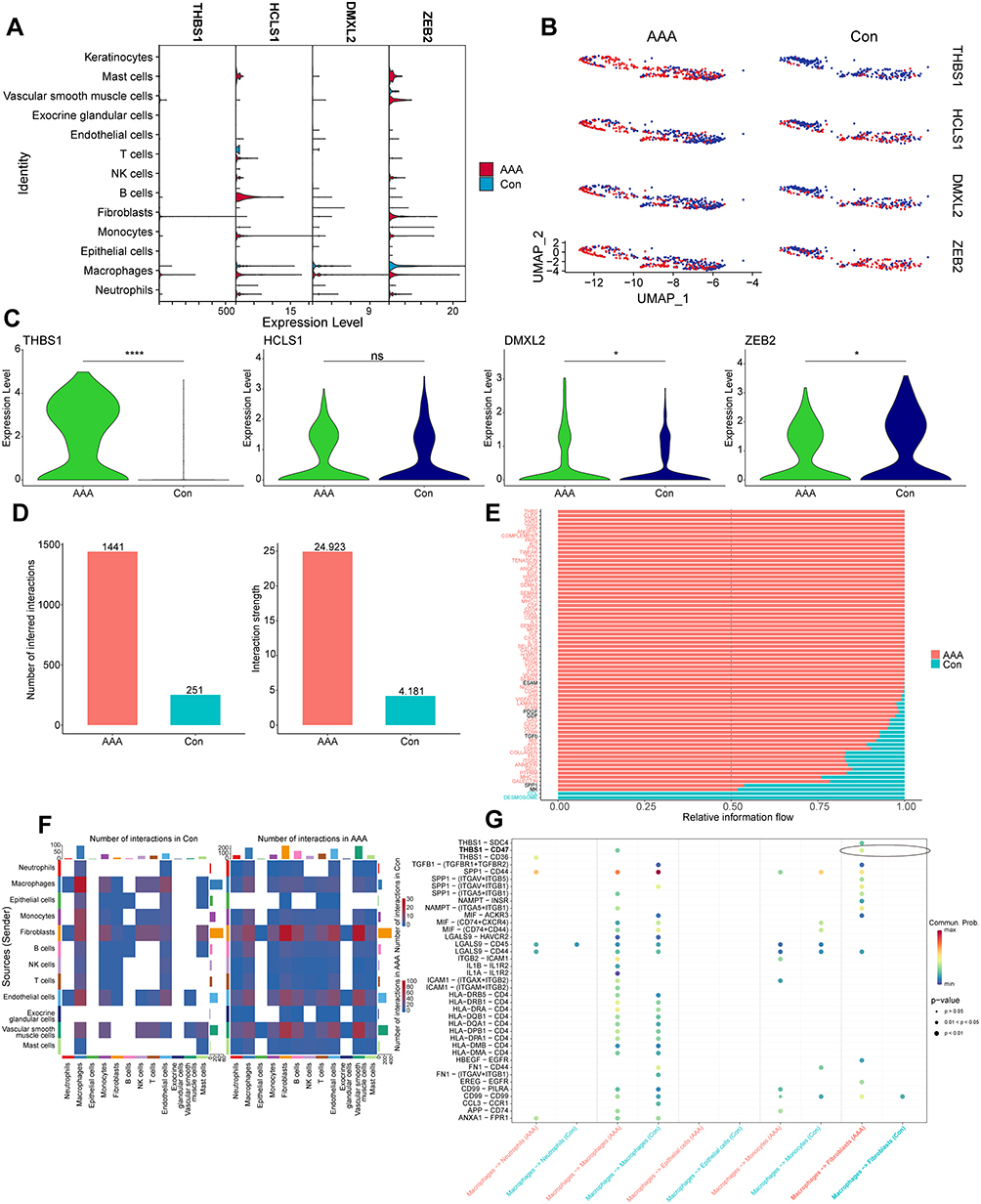 |
Figure 7 Single-cell subgroup and CellChat analysis. (A) Violin plot showing the expression of hub genes in single-cell subgroups of abdominal aortic aneurysms (AAA) and normal samples. (B) “UMAP” plot displaying the expression and distribution of hub genes in macrophage subgroups of AAA and normal samples. Red dots represent single cells with positive gene expression. (C) Violin plot showing the differences in hub-gene expression levels in macrophage subgroups of AAA and normal samples. (D) Bar plot showing differences in the number and weight of cell interactions between AAA and normal samples. (E) Relative differences in gene signaling pathways between AAA and normal samples. (F) Heatmap showing cell interactions between single-cell subgroups in AAA and normal arterial tissues. Deeper red indicates stronger interaction. (G) Differences in gene signaling interactions between macrophage subgroups and other cell subgroups in AAA and normal samples. * P < 0.05; **** P < 0.0001. |
To elucidate the potential cell communication and state transitions in AAA, we analyzed intercellular communication networks from single-cell transcriptome data using the “CellChat” software package. The number and weight of cells were higher in AAA tissues than in normal tissues (Figure 7D). We then summarized the communication probabilities in the information network to compare the overall information flow between AAA and normal arterial tissues. Results revealed that the THBS, CLEC, and CD45 signaling pathways were more abundant in AAA tissues (red), whereas the DESMOSOME and CCL signaling pathways were more prevalent in normal arterial tissues (green) (Figure 7E). The heat map in Figure 7F shows the differences in interactions between different cells in AAA and normal vascular tissues. AAA tissues exhibited increased incoming signals from fibroblasts, endothelial cells, and VSMCs but reduced outgoing signals from macrophages. Further comparison of ligand–receptor communication between macrophages and other cells in AAA and normal arterial tissues revealed that THBS1–CD47 communication was highly active between macrophages and fibroblasts (Figure 7G).
To understand CD47 expression in AAA fibroblasts, we extracted the fibroblast subgroups and displayed them using “UMAP” plots (Figure 8A). Differential analysis showed that CD47 was highly expressed in AAA fibroblast subgroups compared with that in normal arterial tissues (Figure 8B). As THBS1–CD47 plays a key role in AAA macrophage subgroups affecting fibroblast subgroups, we categorized macrophages and fibroblasts into high- and low-expression groups according to the median expression of THBS1 and CD47 to analyze biological differences between the two subgroups (Figure 8D and F). Results indicated that the proportion of high THBS1-expressing macrophages and high CD47-expressing fibroblasts was higher in AAA tissues than in normal arterial tissues (Figure 8C and E).
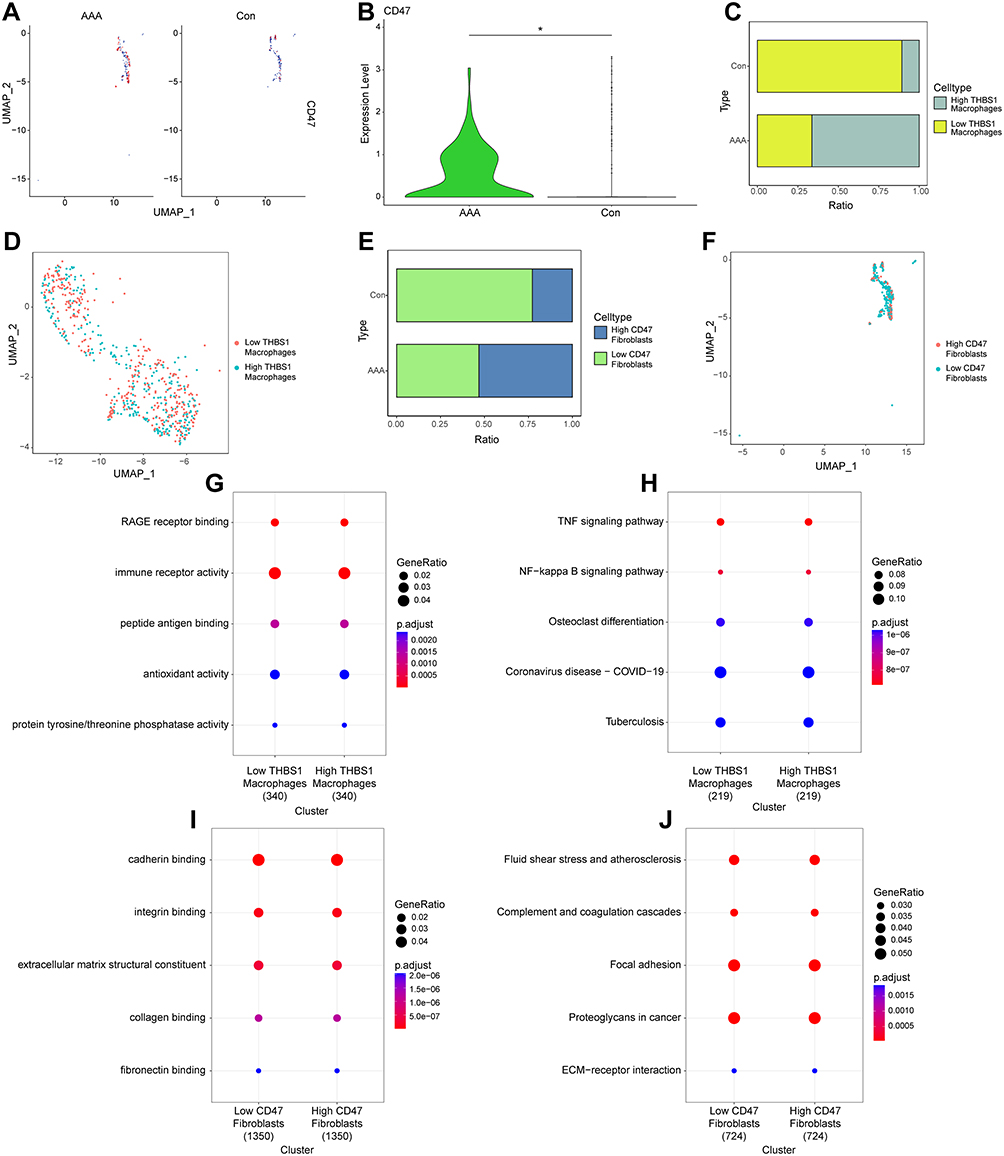 |
Figure 8 Pathway analysis of macrophage and fibroblast subgroups at the single-cell level. (A and B): “UMAP” plot and differential analysis of CD47 expression in fibroblast subgroups. (C and D): Macrophages categorized into two groups based on THBS1, visualized using bar plots and “UMAP” plots. (E and F): Fibroblasts categorized into two groups based on CD47, visualized using bar plots and “UMAP” plots. (G and H): GO and KEGG pathway enrichment analysis for high-THBS1 and low-THBS1 macrophage groups. (I and J): GO and KEGG pathway enrichment analysis for high-CD47 and low-CD47 fibroblast groups. |
Differential analysis was performed on the two groups of cells within each subgroup, followed by GO and KEGG analyses of the DEGs. GO analysis showed that the high THBS1-expressing macrophage subgroup was associated with RAGE receptor binding and immune receptor activity, whereas the high CD47-expressing fibroblast subgroup was associated with cadherin and integrin binding (Figure 8G and I). KEGG analysis revealed that the high THBS1-expressing macrophage subgroup was involved in activation of the TNF and NF-kappa B signaling pathways, whereas the high CD47-expressing fibroblast subgroup was involved in the activation of fluid shear stress, atherosclerosis, and complement and coagulation cascades (Figure 8H and J).
Verification of Biomarkers and Investigation of the NF-κB Pathway
Figure 9A, B shows the expression levels of THBS1 and CD47 detected via qPCR in six tissue samples (three AAA and three normal samples). Both THBS1 and CD47 were significantly upregulated in AAA tissues (P < 0.01). Additionally, the results shown in Figure 8H indicate that THBS1 may affect the TNF-NFκB signaling pathway. We stimulated THBS1-knockdown THP-1 cells with LPS and INF-γ to observe whether THBS1 influences the progression of AAA through the TNF-NFκB signaling pathway. Results showed that upon the inhibition of THBS1 expression, the expression of p-IκBα and p-P65 significantly decreased (Figure 9C–F).
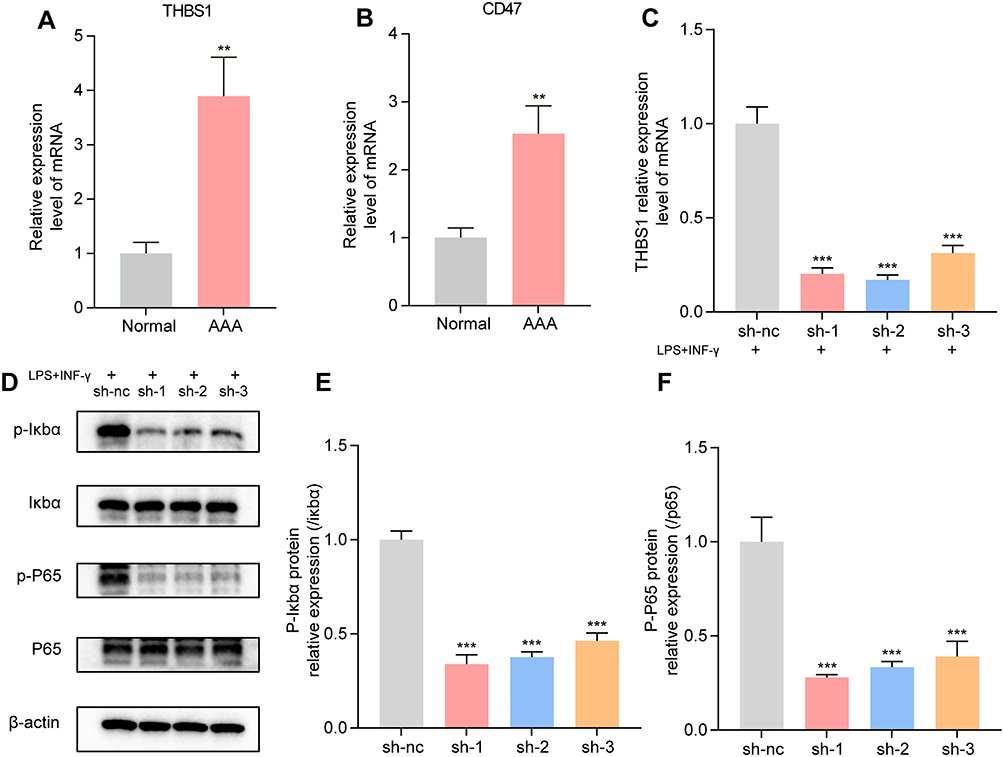 |
Figure 9 Verification of Biomarkers and Pathway. (A) THBS1 is significantly upregulated in abdominal aortic aneurysms (AAA) tissues. (B) CD47 is significantly upregulated in AAA tissues. (C) qPCR results showing a decrease in THBS1 expression after knockdown. (D-F): Western blot analysis showing changes in the expression of p-IκBα, IκBα, p-P65, and P65 proteins in THP-1 cells. **: P < 0.01; ***: P < 0.001. |
Discussion
AAA is a life-threatening condition wherein arterial rupture leads to a very high mortality rate.3 Owing to a limited understanding of AAA progression and its molecular mechanisms, current treatment options are restricted to surgical interventions, with no effective nonsurgical methods available to delay or even reverse AAA progression. Therefore, identifying effective biomarkers for early diagnosis is crucial for timely intervention and improving AAA patient outcomes. Macrophages are key participants of the immune system that exhibit phenotypic plasticity and functional diversity under various pathophysiological conditions.25,26 Macrophages also play a critical role in the pathogenesis of AAA as key mediators of inflammation.27 In this study, we explored the pivotal role of macrophages in the progression of AAA by identifying and analyzing key genes linked to macrophage activity. Our objective was to elucidate the underlying biological mechanisms, thereby providing a reference for the development of innovative diagnostic and therapeutic strategies.
Machine learning algorithms offer various advantages for accurately predicting the core molecules involved in disease processes and identifying potential clinical features in biomedical research.28 Thus, we employed machine learning algorithms to identify THBS1, HCLS1, DMXL2, and ZEB2 as potential auxiliary diagnostic biomarkers for AAA. HCLS1 is a regulatory factor that encodes immune system cells and is known to be associated with immune response, lymphocyte activation, and migration.29 Its role in the migration of immune cells, particularly T cells, may be linked to the inflammatory microenvironment in AAA. DMXL2 is a protein that regulates various biological processes, particularly cell adhesion and signal transduction.30 The occurrence of AAA involves the dedifferentiation and migration of VSMCs, processes that may be influenced by the regulation of DMXL2 in intracellular inflammatory signaling pathways. ZEB2 is a transcription factor that helps maintain the tissue-specific identity of macrophages.31 In different immune microenvironments, it preserves macrophage function by regulating the expression of key transcription factors, thereby promoting disease progression.
Single-cell analysis and validation in an independent dataset further revealed that THBS1 is closely associated with macrophages in the pathogenesis of AAA, highlighting its potential as a universal biomarker. Thrombospondin-1 (THBS1), also known as TSP1, is a homotrimeric glycoprotein released by activated platelets, playing a crucial role in fibrin clotting responses following tissue injury.32 THBS1 is broadly expressed across a variety of cell types.33,34 Previous studies have reported that THBS1 binds to CD47 to promote the development of acute kidney injury and that blocking THBS1 signaling can improve renal interstitial fibrosis.35 Moreover, THBS1 is upregulated in mouse models of sepsis-induced acute kidney injury, and the excessive release of inflammatory mediators is a major trigger.36 Macrophages are present in both human aortic aneurysm lesions and animal AAA models.37,38 High levels of pro-inflammatory cytokines are persistently detected in AAA tissues, which promote the degradation of VSMCs, and the extracellular matrix.38 We found that THBS1 was highly expressed in the macrophage subgroups within AAA tissues, confirming that the occurrence of AAA is closely related to the extensive infiltration of high THBS1-expressing macrophages. At the single-cell level, we classified the macrophages into subgroups based on THBS1 expression and divided them into high and low THBS1-expressing macrophages according to the median expression. The proportion of high THBS1-expressing macrophages was higher in AAA tissues than in normal tissues, which further confirmed the importance of THBS1 in promoting the release of macrophage inflammatory mediators. In the presence of elevated THBS1 levels, inflammatory pathways, including the TNF-NFκB signaling pathway, were upregulated in macrophages. However, when THBS1 is knocked down, the TNF-NFκB signaling pathway is significantly inhibited. Considering the established role of macrophages in orchestrating inflammatory responses, which are pivotal in several immune-mediated pathophysiologies, we speculate that elevated THBS1 expression in macrophages may enhance their pro-inflammatory characteristics.
Upon comparing the differences in intercellular communication networks between normal tissues and AAA samples, we found that macrophage communication with fibroblasts was significantly increased in AAA. Additionally, the upregulation of THBS1 in macrophages promoted the expression of CD47 in fibroblasts. Fibroblasts, as the primary components of large- and medium-sized arteries, contribute to vascular adventitia, along with other cells and numerous extracellular matrix components.39 During oxidative stress and damage repair, fibroblasts can migrate to the subendothelial layer, promoting the formation of new endothelium and transformation into myofibroblasts in the adventitia.40 Under AAA pathological conditions, macrophages accumulate in the affected areas and release various cytokines that stimulate fibroblasts to release proteins involved in extracellular remodeling, thereby exacerbating inflammation.38 In aneurysm tissues, fibroblasts are involved not only in tissue repair and extracellular matrix metabolic homeostasis but also in immune system regulation.41 Increased collagen fiber synthesis is a major characteristic of active fibroblast. The expression of type I/III collagen cross-links is prominent in human AAA tissues; however, the reduced expression of total collagen markers suggests a possible defect in the biosynthesis of new collagen.41 CD47 is a widely expressed cell receptor known for its immunoregulatory functions.42 By interacting with its ligands, including THBS1, signal regulatory protein alpha (SIRPα), integrins, and SH2 domain-containing protein tyrosine phosphatase substrate 1 (SIRPα), CD47 regulates macrophage phagocytosis, neutrophil migration, and the activation of dendritic cells and T cells. Senescent and dysfunctional fibroblasts exhibit high CD47 expression, which results in the inhibition of macrophage-mediated phagocytosis and clearance.43 Therefore, we believe that CD47 upregulation may lead to the reduced clearance of abnormal fibroblasts by macrophages, potentially exacerbating AAA progression.
In this study, we utilized a comprehensive set of bioinformatic tools to reveal the complex relationship between macrophage function and AAA pathogenesis, providing new insights into this complex disease. We identified THBS1 as a highly precise biomarker for the accurate diagnosis of AAA, which represents a potential therapeutic target for delaying AAA progression. Furthermore, specific pathways regulating macrophage activity, such as TNF-NFκB signaling, are known to play crucial roles in inflammation and tissue remodeling within the aortic wall. Interventions targeting these pathways could modulate macrophage function, reducing inflammation and slowing AAA progression. These potential therapeutic strategies not only highlight the relevance of our findings but also open new avenues for AAA management. Despite these advances, our study has certain limitations, including the reliance on existing sequencing data, insufficient external validation, and an incomplete understanding of the underlying biological mechanism. In future studies, we will continue to explore the crosstalk between macrophages and fibroblasts, as well as the specific pathways regulating macrophage behavior, to better understand their role in AAA development.
Conclusion
In summary, we conducted a comprehensive study on the relationship between macrophage-related biomarkers and AAA and identified the key genes involved in the process. These key genes were then subjected to integrated analysis using machine learning algorithms and single-cell sequencing. Finally, we identified THBS1 as a potential biomarker of AAA formation facilitated by macrophages. This study has important implications for achieving timely intervention and improved outcomes in patients with AAA.
Data Sharing Statement
The data presented in the study are included in the article or in the Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Approval
This study was approved by the Ethics Committee of the First Hospital of Chongqing Medical University (K2024-221-01). Patients provided informed consent following the principles of the Declaration of Helsinki.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Disclosure
The authors report no conflicts of interest in this work. This paper has been uploaded to ResearchSquare as a preprint: https://www.researchsquare.com/article/rs-4977816/v1.
References
1. Gao J, Cao H, Hu G, et al. The mechanism and therapy of aortic aneurysms. Signal Transduct Target Ther. 2023;8(1):55. doi:10.1038/s41392-023-01325-7
2. Kuivaniemi H, Ryer EJ, Elmore JR, et al. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther. 2015;13(9):975–987. doi:10.1586/14779072.2015.1074861
3. Nordon IM, Hinchliffe RJ, Loftus IM, et al. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2010;8(2):92–102. doi:10.1038/nrcardio.2010.180
4. Ma D, Zheng B, Suzuki T, et al. Inhibition of KLF5–Myo9b–RhoA pathway–mediated podosome formation in macrophages ameliorates abdominal aortic aneurysm. Circ Res. 2017;120(5):799–815. doi:10.1161/circresaha.116.310367
5. Kokje VBC, Hamming JF, Lindeman JHN. Editor’s choice – pharmaceutical management of small abdominal aortic aneurysms: a systematic review of the clinical evidence. Eur J Vasc Endovascular Surg. 2015;50(6):702–713. doi:10.1016/j.ejvs.2015.08.010
6. Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. 2018;16(4):225–242. doi:10.1038/s41569-018-0114-9
7. Torres-Fonseca M, Galan M, Martinez-Lopez D, et al. Fisiopatología del aneurisma de aorta abdominal: biomarcadores y nuevas dianas terapéuticas [Pathophisiology of abdominal aortic aneurysm: biomarkers and novel therapeutic targets]. Clínica e Investigación en Arteriosclerosis. 2019;31(4):166–177. Spanish. doi:10.1016/j.arteri.2018.10.002
8. Yuan Z, Lu Y, Wei J, et al. Abdominal aortic aneurysm: roles of inflammatory cells. Front Immunol. 2021;11(609161). doi:10.3389/fimmu.2020.609161
9. Murray Peter J, Allen Judith E, Biswas Subhra K, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
10. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi:10.1038/nri2448
11. Hou N, Zhou H, Li J, et al. Macrophage polarization and metabolic reprogramming in abdominal aortic aneurysm. Immun Inflamm Dis. 2024;12(11). doi:10.1002/iid3.1268
12. Bai L, Ge L, Zhang B, et al. CtBP proteins transactivate matrix metalloproteinases and proinflammatory cytokines to mediate the pathogenesis of abdominal aortic aneurysm. Exp Cell Res. 2022;421(2):113386. doi:10.1016/j.yexcr.2022.113386
13. Fu H, Q-r S, Zhao Y, et al. Activating α7nAChR ameliorates abdominal aortic aneurysm through inhibiting pyroptosis mediated by NLRP3 inflammasome. Acta Pharmacol Sin. 2022;43(10):2585–2595. doi:10.1038/s41401-022-00876-9
14. Stepien KL, Bajdak-Rusinek K, Fus-Kujawa A, et al. Role of extracellular matrix and inflammation in abdominal aortic aneurysm. Int J Mol Sci. 2022;23(19):11078. doi:10.3390/ijms231911078
15. Oller J, Méndez-Barbero N, Ruiz EJ, et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nature Med. 2017;23(2):200–212. doi:10.1038/nm.4266
16. Da Ros F, Carnevale R, Cifelli G, et al. Targeting interleukin-1β protects from aortic aneurysms induced by disrupted transforming growth factor β signaling. Immunity. 2017;47(5):959–973. doi:10.1016/j.immuni.2017.10.016
17. Ryffel B, Yang H, Zhang W, et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS One. 2012;7(9). doi:10.1371/journal.pone.0046364
18. Zhang Z, Xu J, Liu Y, et al. Mouse macrophage specific knockout of SIRT1 influences macrophage polarization and promotes angiotensin II-induced abdominal aortic aneurysm formation. J Genet Genome. 2018;45(1):25–32. doi:10.1016/j.jgg.2018.01.002
19. Stubbington MJT, Rozenblatt-Rosen O, Regev A, et al. Single-cell transcriptomics to explore the immune system in health and disease. Science. 2017;358:6359):58–63. doi:10.1126/science.aan6828
20. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559. doi:10.1186/1471-2105-9-559
21. Gutiérrez-Gómez L, Vohryzek J, Chiêm B, et al. Stable biomarker identification for predicting schizophrenia in the human connectome. NeuroImage Clin. 2020;27. doi:10.1016/j.nicl.2020.102316
22. Wang H, Yang F, Luo Z. An experimental study of the intrinsic stability of random forest variable importance measures. BMC Bioinf. 2016;17(1):60. doi:10.1186/s12859-016-0900-5
23. Chang J-W, Liu S-C, Lin -Y-Y, et al. Nesfatin-1 stimulates CCL2-dependent monocyte migration and M1 macrophage polarization: implications for rheumatoid arthritis therapy. Int J Bio Sci. 2023;19(1):281–293. doi:10.7150/ijbs.77987
24. Yao G, Li H, Zuo X, et al. Oscillatory shear stress promotes vein graft intimal hyperplasia via NADPH oxidase-related pathways. Front Surg. 2023;10(1073557). doi:10.3389/fsurg.2023.1073557
25. Xue S, Su Z, Liu D. Immunometabolism and immune response regulate macrophage function in atherosclerosis. Ageing Res Rev. 2023;90(101993):101993. doi:10.1016/j.arr.2023.101993
26. Sheu KM, Hoffmann A. Functional hallmarks of healthy macrophage responses: their regulatory basis and disease relevance. Ann Rev Immunol. 2022;40(1):295–321. doi:10.1146/annurev-immunol-101320-031555
27. Raffort J, Lareyre F, Clément M, et al. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14(8):457–471. doi:10.1038/nrcardio.2017.52
28. Handelman GS, Kok HK, Chandra RV, et al. eDoctor: machine learning and the future of medicine. J Internal Med. 2018;284(6):603–619. doi:10.1111/joim.12822
29. Aslam Y, Williamson J, Romashova V, et al. Human cytomegalovirus upregulates expression of HCLS1 resulting in increased cell motility and transendothelial migration during latency. iScience. 2019;20:60–72. doi:10.1016/j.isci.2019.09.016
30. Esposito A, Falace A, Wagner M, et al. Biallelic DMXL2 mutations impair autophagy and cause ohtahara syndrome with progressive course. Brain. 2019;142(12):3876–3891. doi:10.1093/brain/awz326
31. Scott CL, T’Jonck W, Martens L, et al. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity. 2018;49(2):312–325.e5. doi:10.1016/j.immuni.2018.07.004
32. Gutierrez LS, Gutierrez J. Thrombospondin 1 in metabolic diseases. Front Endocrinol. 2021;12(638536). doi:10.3389/fendo.2021.638536
33. Tabib T, Huang M, Morse N, et al. Myofibroblast transcriptome indicates SFRP2hi fibroblast progenitors in systemic sclerosis skin. Nat Commun. 2021;12(1):4384. doi:10.1038/s41467-021-24607-6
34. Dylag AM, Haak J, Warren R, et al. Low-dose hyperoxia primes airways for fibrosis in mice after influenza A infection. Am J Physiol Lung Cell Mol Physiol. 2021;321(4):L750–L763. doi:10.1152/ajplung.00289.2020
35. Julovi SM, Sanganeria B, Minhas N, et al. Blocking thrombospondin-1 signaling via CD47 mitigates renal interstitial fibrosis. Lab Invest. 2020;100(9):1184–1196. doi:10.1038/s41374-020-0434-3
36. Sun J, Ge X, Wang Y, et al. USF2 knockdown downregulates THBS1 to inhibit the TGF-β signaling pathway and reduce pyroptosis in sepsis-induced acute kidney injury. Pharmacol Res. 2022;176(105962):105962. doi:10.1016/j.phrs.2021.105962
37. Zhao G, Lu H, Chang Z, et al. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiol Res. 2021;117(5):1402–1416. doi:10.1093/cvr/cvaa214
38. Wagenhäuser MU, Mulorz J, Krott KJ, et al. Crosstalk of platelets with macrophages and fibroblasts aggravates inflammation, aortic wall stiffening, and osteopontin release in abdominal aortic aneurysm. Cardiol Res. 2024;120(4):417–432. doi:10.1093/cvr/cvad168
39. Gao J-P, Guo W. Mechanisms of abdominal aortic aneurysm progression: a review. Vascu Med. 2021;27(1):88–96. doi:10.1177/1358863x211021170
40. Fu L, Zhao H, Xiang Y, et al. Reactive oxygen species-evoked endoplasmic reticulum stress mediates 1-nitropyrene-induced epithelial-mesenchymal transition and pulmonary fibrosis. Environ Pollut. 2021;283(117134):117134. doi:10.1016/j.envpol.2021.117134
41. Weng Y, Lou J, Bao Y, et al. Single-cell RNA sequencing technology revealed the pivotal role of fibroblast heterogeneity in angiotensin ii-induced abdominal aortic aneurysms. DNA Cell Biol. 2021;41(5):495–520. doi:10.21203/rs.3.rs-970793
42. Hayat SMG, Bianconi V, Pirro M, et al. CD47: role in the immune system and application to cancer therapy. Cell Oncol. 2019;43(1):19–30. doi:10.1007/s13402-019-00469-5
43. Schloesser D, Lindenthal L, Sauer J, et al. Senescent cells suppress macrophage-mediated corpse removal via upregulation of the CD47-QPCT/L axis. J Cell Biol. 2023;222(2):e202207097. doi:10.1083/jcb.202207097
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Netrin-1 Monoclonal Antibody-Functionalized Nanoparticle Loaded with Metformin Prevents the Progression of Abdominal Aortic Aneurysms

Zhang Z, Zhuang J, Sun D, Ding Q, Zheng H, Li H, Zhang X, Du Y, Ma T, Meng Q
International Journal of Nanomedicine 2023, 18:627-639
Published Date: 6 February 2023
IL7-IL7R Interaction Mediates Fibroblast-Driven Macrophage-to-Osteoclast Differentiation in Periodontitis

Huang P, Gao L, Guan J, Li Y, Jia Y, Zeng Z, Chen Y, Wang L, Li W, Wang Y, Yang B
Journal of Inflammation Research 2025, 18:6105-6122
Published Date: 11 May 2025