")
Back to Journals » International Journal of Nanomedicine » Volume 19
Improved Antitumor Efficiency of N4-Tetradecyloxycarbonyl Gemcitabine-Loaded Liposomes for Pancreatic Cancer Chemotherapy
Authors Wang D, Wang X , Li Y, Wang X, Wang X, Su J, Wang A, Lv K, Liu M, Xia G
Received 7 July 2024
Accepted for publication 23 November 2024
Published 11 December 2024 Volume 2024:19 Pages 13391—13410
DOI https://doi.org/10.2147/IJN.S485861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Jie Huang
Dan Wang,* Xiaobo Wang,* Yan Li, Xiaowei Wang, Xuelei Wang, Jiayi Su, Apeng Wang, Kai Lv, Mingliang Liu, Guimin Xia
Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, 100050, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guimin Xia; Apeng Wang, Email [email protected]; [email protected]
Background: Gemcitabine (Gem) is one of the first-line chemotherapy drugs for pancreatic cancer treatment. However, its short half-life in plasma and adverse effects limited its broader application.
Methods: A novel Gem derivative (N4-tetradecyloxycarbonyl gemcitabine, tcGem) was synthesized and encapsulated into liposomes (LipotcGem) to overcome the above shortcomings.
Results: LipotcGem has been successfully formulated, with the average size of 115 nm, zeta potential values of − 36 mV, encapsulation efficiency of up to 98%, and drug loading capacity of 8.1%. Compared to Gem, LipotcGem improved in vitro antitumor activity significantly, as evidenced by the lower IC50, the higher percentage of apoptotic cells, the stronger ability to inhibit cell migration and invasion due to the higher cellular accumulation (100 times). Additionally, the endocytosis of LipotcGem was mainly mediated by caveolae, and was then processed in the lysosome, where tcGem was released and hydrolyzed into Gem. LipotcGem inhibited tumor growth by 70% in subcutaneous xenograft model and 90% in orthotopic xenograft model, respectively. LipotcGem suppressed tumor metastasis and prolonged survival without perceptible systemic toxicity, which may be caused by the longer t1/2 in vivo (3.5 times, 5.23 vs 1.46 h) and more enrichment in tumor tissue (750 times).
Conclusion: LipotcGem significantly increased the anti-tumor efficiency and decreased the toxicity for chemotherapy of pancreatic cancer.
Keywords: pancreatic cancer, gemcitabine, N4-tetradecyloxycarbonyl gemcitabine, liposomes
Graphical Abstract:

Introduction
Pancreatic cancer is one of the most aggressive malignant tumors, with a 5-year survival rate of approximately 10%.1,2, Surgery excision is the common approach to eradicate pancreatic cancer, but it is necessary to combine this with postoperative chemotherapy to eliminate residual lesions and reduce the recurrence rate.3,4 Given that the pancreas is located in a concealed position physiologically and patients typically do not exhibit symptoms at the early stages, the majority of them are diagnosed at advanced stages or with metastasis initially, thus missing the opportunity for surgical intervention.5–7 Hence, nonsurgical treatments, including chemotherapy, are of great significance.
Gemcitabine (Gem) is a first-line chemotherapy for pancreatic cancer.8–10 However, its very short plasma half-life, resulting from rapid deamination into inactive 2’, 2’-difluorodeoxyuridine by cytidine deaminase, limits its broader application.11–14 High doses and prolonged infusions have to be adopted in clinical practice to achieve therapeutic drug concentrations of Gem, but this regimen is often associated with severe side effects. Two major strategies have been reported to address these issues, lipophilic derivatives and drug-loaded nano-formulations.
Numerous nano-formulations for Gem have been published recently,15–17 including albumin nanoparticles,18,19 mixed micelles formulated via TPGS/DSPE-PEG20 or PEG-b-PLA,21 chitosan nanoparticles22 and more. Among these, liposomes stand out as one of the most promising candidate carriers for clinical applications, offering several advantages: biocompatibility, biodegradability, extension of circulation time, prevention of drug from being metabolized, and most importantly, good stability along with a straightforward process for large-scale production.23
Some researchers encapsulated Gem in the inner core of liposomes to prevent from catabolic effects.24–27 Unfortunately, due to Gem’s high water solubility, it is not suitable for direct encapsulation into liposomes with high encapsulation efficacy, which is essential for antitumor efficiency and industrial development. Inspired by the structure of phospholipids, we first designed and synthesized a novel N4-tetradecyloxycarbonyl Gem (tcGem). A myristyl group was directly linked to the 4-amino position of Gem to facilitate liposomal formulation, prolong the half-life, target tumors and improve antitumor efficacy.
Therefore, this study concentrated on the development of tcGem loaded liposomes (LipotcGem) after the optimization of formulation and preparation processes. The antitumor activity was assessed in vitro by evaluating its capacity to inhibit cell growth, induce cellular apoptosis, cause cell cycle arrest, and prevent tumor cell migration and invasion. The mechanism of action of the liposomes was also explored. In vivo assessments were conducted on BALB/c or tumor bearing nude mice, including studies on pharmacodynamics, pharmacokinetics (PK) and tissue distribution. The aim is to provide an improved Gem formulation with high efficiency and low toxicity for chemotherapy of pancreatic cancer.
Materials and Methods
Materials
Gemcitabine Hydrochloride for Injection (Gem, Batch No.19061111) was purchased from Yangtze River Pharmaceutical Co., Ltd. (Zhejiang, China). 2’,2’-difluoro-2’-deoxycytidine-3’,5’-dibenzoate (>96%, Cat. No. 01023855) and Myristoyl Chloroformate (>95%, Cat. No. 01018240) were bought from Beijing Ouhe Technology Co., Ltd. (Beijing, China). 1.2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC, >99%, Cat. No. S01003), 1.2-dipalmitoyl-rac-glycero-3-phosphocholine (DPPC, >99%, Cat. No. S01004), 1.2-distearoyl-sn-glycero-3-phosphocholine (DSPC, >99%, Cat. No. S01005), 1.2-Dimyristoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DMPG, >99%, Cat. No. S02002), 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE- PEG2K, >99%, Cat. No. F01008) and Cholesterol (Chol, >99%, Cat. No. O01001) were purchased from A.V.T. Co., Ltd. (Shanghai, China). L-α-phosphatidylcholine, hydrogenated (Soy) (HSPC, >98%, Cat. No. 840058P) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Cell cycle and apoptosis analysis kit (Cat. No. C1052), Annexin V-FITC apoptosis detection kit (Cat. No. C1062M), 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide and 1.1’-dioctadecyl 3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI, Cat. No. C1036), crystal violet (Cat. No. C0121) and Bicinchoninic Acid Assay Kit (Cat. No. P0011) were bought from Beyotime Biotechnology (Shanghai, China). InStabTM phosphatase inhibitor cocktail (Cat. No. 20109ES) and LysoTracker Green DND-26 (Cat. No. 40738ES) were bought from Yeasen Biotechnology Co., Ltd. (Shanghai, China). Amiloride (Cat. No. A0080) and phenylmethanesulfonyl fluoride (Cat. No. IP0280) were bought from Solarbio Life Science (Beijing, China). Genistein (Cat. No. G106673) and Chlorpromazine (Cat. No. C131611) were bought from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Anti-caspase 3 (Cat. No. 9662), anti-cleaved caspase 3 (Asp175) (Cat. No. 9661) antibodies were bought from Cell Signaling Technology (Beverly, MA, USA). Anti-GAPDH antibody (Cat. No. TA-08) was bought from ZSGB-BIO (Beijing, China). Tetrahydrouridine (THU, Cat. No. HY-15345A) was purchased from MedChemExpress (Monmouth Junction, New Jersey, USA). Purinomycin (Cat. No. ant-pr-1) and Diphenylterazine (Cat. No. CTCC-luc-001) were purchased from Meisen Cell Technology Co., Ltd (Zhejiang, China). All other chemicals and reagents were commercially available and used as received.
Cells and Animals
Human pancreatic carcinoma cell lines AsPC-1, PANC −1, BxPC-3, Mia PaCa-2, and SU.86.86 were purchased from National Infrastructure of Cell Line Resource, Chinese Academy of Medical Sciences (Beijing, China). AsPC-1-luc2 (AsPC-1-luc) cell line was purchased from Meisen Cell Technology Co., Ltd (Zhejiang, China). AsPC-1, PANC-1 and SU.86.86 cells were cultured in RPMI-1640 (HyClone, USA), while BxPC-3 and Mia PaCa-2 were cultured in DMEM (HyClone, USA), both supplemented with 10% (v/v) fetal bovine serum (Gibco, Grand Island, NY, USA), 1% sodium pyruvate (Invitrogen, Carlsbad, CA, USA) and 1% nonessential amino acids (Invitrogen, Carlsbad, CA, USA) at 37°C in a humidified atmosphere with 5% CO2. AsPC-1-luc cells were cultured in RPMI-1640 (HyClone, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco, Grand Island, NY, USA) and 0.5 μg/mL purinomycin.
BALB/c nude mice (female, 18–20 g) were purchased from HFK Bioscience Co., Ltd. (Beijing, China). The animals were housed in the specific pathogen-free animal facility with free access to food and water. All experimental procedures were strictly performed according to the Guidelines for Ethical Review of Laboratory Animal Welfare in China (GB/T35892-2018) and were approved by Institutional Animal Care and Use Committee of Institute of Medicinal Biotechnology [License Number: SYXK (Jing) 2022–0023].
Synthesis of tcGem
Detailed synthetic pathways to tcGem were outlined in Figure 1A. To a solution of benzoyl (Bz) protected Gem (1, 9.42 g, 0.02 mol) in dichloromethane (DCM, 75 mL) were added triethylamine (Et3N, 3.03g, 0.03 mol) and tetradecyl chloroformate (6.62 g, 0.24 mol) successively. After stirring for 2 h at room temperature, the mixture was washed with saturated brine and then saturated sodium bicarbonate solution, dried over anhydrous sodium sulfate, and then filtered. The filtrate was concentrated under reduced pressure to give crude product 2. A mixture of the above crude 2, NaOH (0.1 g) and MeOH (50 mL) was stirred for 0.5 h at room temperature, and then filtered. The filtrate was concentrated under reduced pressure, and the residue was purified by silica gel column (DCM: MeOH = 20: 1) to yield target compound tcGem (5.16 g, 51.3% for two steps) as a white solid. The structure was confirmed by 1H-NMR, 13C-NMR and MS-ESI.
Figure 1 Continued. Figure 1 Synthesis of tcGem, preparation and characterization of tcGem-loaded liposomes. (A) Synthesis route of tcGem. (B) Optimization of phospholipids. Size, PDI, and zeta potential values of tcGem-loaded liposomes with different phospholipids. (C) Optimization of mass ratios of DMPC to tcGem. Size, PDI, zeta potential values, and tcGem concentrations of tcGem-loaded liposomes during 40-day storage. (D) Optimization of the prescribed amount of Chol. Size, PDI, zeta potential values, and tcGem concentrations of tcGem-loaded liposomes prepared with 0.9, 1.8, 2.7 mg Chol. (E) Optimization of the prescribed amount of DSPE-PEG2K. Size, PDI, zeta potential values, and tcGem concentrations of tcGem-loaded liposomes prepared with 1.5, 3.0, 4.5 mg DSPE-PEG2K. (F) TEM image (Scale bar, 100 nm). (G) size and distribution, and the upper left images represent the physical picture (left) and the picture of the Tyndall effect of LipotcGem suspension (right). (H) zeta potential and (I) in vitro release of LipotcGem. Data were presented as mean ± SD (n = 3).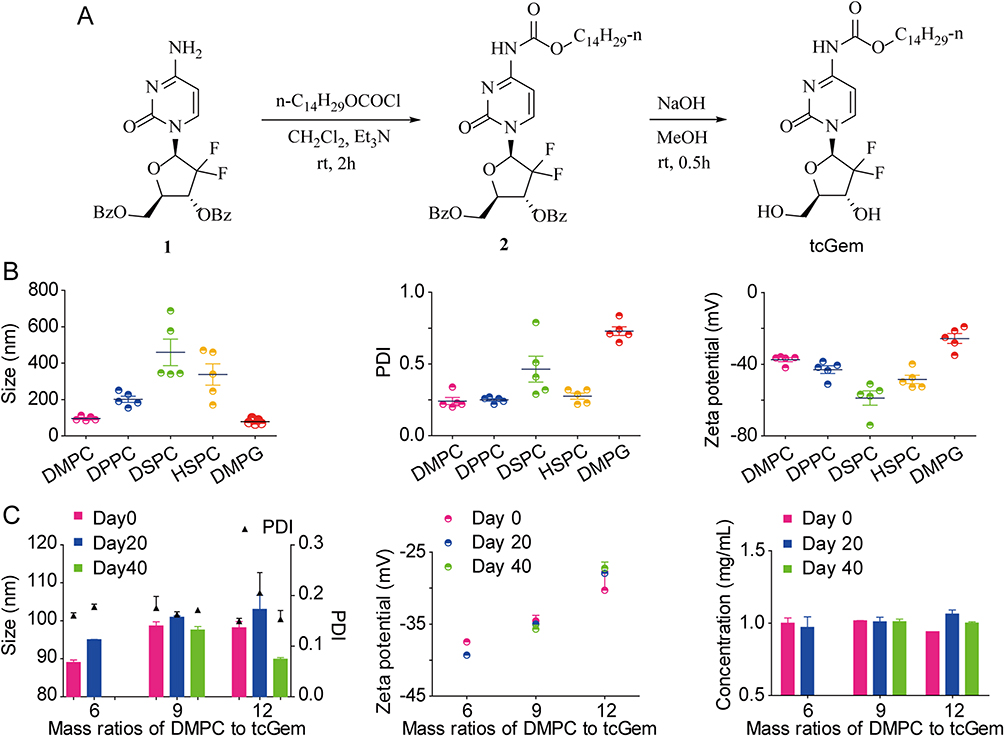

Preparation of tcGem-Loaded Liposomes
The tcGem-loaded liposomes were prepared by the film dispersion method. Briefly, 18.0 mg DMPC (or DPPC, or DSPC, or HSPC, or DMPG), 3.0 mg DSPE-PEG2K, 1.8 mg Chol, 2.0 mg tcGem were dissolved in chloroform, and the solution was concentrated at 45°C by rotary evaporation. The resulted thin and uniform film was subjected to hydration at 45°C for 60 min in 2 mL 5% glucose and then sonicated for 30 min at 100 W. Finally, LipotcGem (1.0 mg/mL tcGem) suspensions were obtained.
The DiI labeled Liposomes without tcGem (DiI-Liposome) were prepared by adding 0.3 mg DiI into chloroform, with the remaining steps following the previously mentioned protocol. The un-encapsulated free DiI was removed by centrifugation (65,000 g for 2 h at 4°C). The obtained DiI-Liposomes may also dissolve some free DiI, so an additional centrifugation (8000 g for 20 min at 4°C) with an ultrafiltration membrane (molecular weight cutoff 10 kDa) was performed to obtain supernatant control.
Characterization of LipotcGem
The average size and PDI of LipotcGem were measured by dynamic light scattering and the zeta potentials were evaluated by electrophoretic light scattering using a Malvern Zetasizer (Nano-ZS, Malvern, UK) at 25°C. The morphological images of LipotcGem were observed by transmission electron microscopy (TEM, JEM-1200EX, JEOL) following negative staining with 2% uranyl acetate solution, and by scanning electron microscopy (SEM, JSM-7900F, JEOL) following coating with platinum by a vacuum evaporator after preparing the sample on metallic studs with double-sided conductive tape.28
The determination of tcGem in LipotcGem was performed by high performance liquid chromatography (HPLC) with a diode-array detector (LC-20A, SHIMADZU, Japan). The LipotcGem suspension was mixed with 20 times the volume of methanol, and then centrifuged at 12,000 × g for 10 min at 4°C, finally the supernatant was injected into HPLC system with an InerstilTM ODS-SP (4.6 × 150 mm, 5 μm, GL Sciences, Japan). The mobile phase consisted of 80% methanol - 20% PBS (pH 2.4–2.6) at a flow rate of 1.0 mL/min. The ultraviolet detection wavelength was set at 243 nm. The limits of detection and quantification were 5.2 ng and 16.8 ng, respectively. Finally, the following equations were used to calculate encapsulation efficiency (EE, %) and drug-loading efficiency (DL, %), respectively.
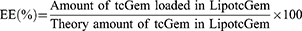
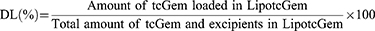
In vitro Release Study
The in vitro release profile of LipotcGem was investigated using the dialysis method. Briefly, 0.5 mL LipotcGem or free tcGem DMSO solution was sealed in a dialysis bag with the molecular weight cutoff of 300 kDa (Cat. No. HF131450), then immersed in 50 mL of PBS at pH 7.4 supplemented with 50% serum (or in PBS at pH 7.4 and pH 5 containing 1% Tween-80) at 37°C at 100 rpm (THZ-D desktop constant temperature oscillator, China) for 72 h. At predetermined time intervals of 8, 12, 24, 48, and 72 h, 1 mL of the receiving media was withdrawn and replenished promptly with the equal volume of medium. Then 200 μL receiving media was mixed with 800 μL methanol, vortexed, centrifuged at 12,000 × g for 10 min at 4°C, finally the supernatant was determined by the HPLC method described above. The Cumulative release (%) was calculated as followed.
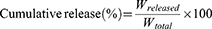
In vitro Cytotoxicity
The in vitro cytotoxicity of Gem, tcGem and LipotcGem was assessed by 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay on AsPC-1, PANC-1, BxPC-3, Mia PaCa-2, and SU.86.86 cells. The cells were seeded on 96-well plates at a density of 2×104 per well for AsPC-1, and 1×104 for PANC-1, BxPC-3, Mia PaCa-2, and SU.86.86, and allowed to adhere for 24 h in a 5% CO2 atmosphere at 37°C. Then, the medium was replaced with fresh medium containing Gem, tcGem, or LipotcGem at a series of concentrations (0.01, 0.1, 1, 10 and 100 μM). After 48 h incubation, 20 μL of the MTT solution (5 mg/mL in PBS) was added. After another 4 h incubation at 37 °C, the MTT solution was replaced with 100 μL DMSO. Finally, the optical density (OD) was determined using an EPOCH12 microplate reader (BioTek, USA) at 490 nm (ODtest). Cells in drug-free medium were taken as the negative control (ODcontrol), while wells containing only drug-free medium without cells were served as the blank (ODblank). Cell viability (%) was calculated according to the following equation. The half-maximal inhibitory concentration (IC50) values were calculated with nonlinear regression analysis by GraphPad Prism 5.0 software.29
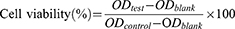
Cellular Uptake and Endocytic Mechanism
The internalization of Liposome was investigated with the help of DiI probe, with excitation/emission wavelengths of 550 nm / 575 nm, due to the similar lipophilic properties of tcGem and DiI. AsPC-1 cells were seeded on 12-well plates (5 × 105 cells per well) and cultured overnight. The cells were then exposed to DiI-Liposome for 4 h (or 8h, or 12 h) at 37°C. Afterwards, the cells were washed and stained with LysoTracker Green DND-26, with excitation/emission wavelengths of 488 nm / 510 nm, and observed under a confocal laser scanning microscope (CLSM, LSM 710, ZEISS, Germany). The cells treated with the supernatant served as the negative control. Meantime, the treated cells were collected and measured by a flow cytometer (BD Biosciences, San Jose, 207 CA, USA).
AsPC-1 cells pretreated with endocytic inhibitors for 40 min at 37°C, 100 μΜ genistein, or 80 μΜ amiloride (EIPA), or 60 μΜ chlorpromazine (CPZ), were used to investigate the endocytic pathway of DiI-Liposome. After pretreated, the cells were exposed to DiI-Liposome for another 4 h at 37°C, at the presence of inhibitors. Finally, the cellular mean fluorescent intensity (MFI) was measured using a flow cytometer.
Cellular accumulation of LipotcGem was detected by liquid chromatography-tandem mass spectrometry (LC-MS/MS). AsPC-1 cells were seeded on 10 cm dishes at a density of 5×106 and cultured overnight, then exposed to Gem or LipotcGem at 10 μM for 12, 24, or 48 h. The cells were subsequently washed, collected, counted, and centrifuged to remove the medium, followed by ultrasonic disruption at 4°C in 500 μL methanol-water (1: 1, v: v) solution. A 30 μL homogenate and 120 μL acetonitrile (containing amiodarone as internal standard at 20 ng/mL) were mixed, vortexed, centrifuged at 12,000 × g for 10 min at 4°C, and the supernatant was injected into the Shimadzu 20A HPLC system coupled with an AB SCIEX QTRAP 5500 system (Toronto, Canada), containing an electrospray ionization source. The compound was separated using an ACQUITY UPLC BEH C8 column (2.1 mm × 100 mm, 1.7 μm) with a gradient elution at a flow rate of 0.2 mL/min. Mobile phase consisted of water (0.1% formic acid) (A)-acetonitrile (0.1% formic acid) (B), with the following gradient: 0.0–2.0 min (3% B), 2.0–3.0 min (3–95% B) and 3.0–8.0 min (95% B). For positive ion mode, the multiple reaction monitoring (MRM) transitions were set at m/z 264.2→112.2 for Gem, m/z 504.2→156.0 for tcGem, and 646.0→100.0 for amiodarone. Declustering potential, entrance potential, collision energy and collision cell exit potential were set as 80.0, 10.0, 16.0, and 13.0, respectively. Data acquisition and quantification were performed with Analyst software (Version, 1.6.2, AB SCIEX).
Lysosomal Function
To investigate lysosomal function, the AsPC-1 cells were pretreated with 30 mM NH4Cl for 20 min at 37°C to weaken the acidic environment of lysosomes, then exposed to 10 μΜ LipotcGem for 12 h at 37°C. The cells were then divided into two parts, one part was replaced with fresh medium, while the other was replaced with the medium containing the same concentration of NH4Cl for an additional 24 h. The cellular concentrations of Gem and tcGem were analyzed by LC-MS/MS as previously described.
Cell Apoptosis Assay
The apoptotic effect on AsPC-1 cells was assessed using the Annexin V-FITC and PI double staining method as previously described.30 The procedure was as follows: AsPC-1 cells were seeded on 6-well plates (5 × 105 cells per well) and cultured for 24 h at 37°C. After treatment with 10 μM Gem, tcGem or LipotcGem for 48 h, the cells were subsequently processed according to the instructions of the Annexin V-FITC apoptosis detection kit and measured by a flow cytometer. The percentages of cells in early and late apoptosis were analyzed with FlowJo.v10.8.1.
Cell Migration Assay
The in vitro wound scratch assay was conducted to evaluate the cell migration ability after administration.31,32 AsPC-1 cells were seeded on 12-well plates (2 × 106 cells per well) and allowed to form the confluent monolayer. Then several straight lines were scratched using a sterile 200 μL micropipette tip to create a cell-free zone into which cells at the edges of the wound can migrate. Then cells were subsequently washed several times and treated with 3% serum medium containing 10 μM Gem, tcGem, or LipotcGem for 24 or 48 h. The scratches were photographed at 0, 24 and 48 h post wounding using an inverted microscope (Carl Zeiss AG, Oberkochen, Germany). The wound width was measured by Image J software and wound healing (%) was calculated by the following formula.
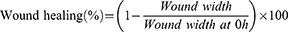
Cell Invasion Assay
The transwell chambers with 8-μm pores (Costar, corning, Fisher Scientific, Cat. No. 140629) were used to perform cell invasion assay. Specifically, AsPC-1 cells were seeded on the upper chamber (1 × 104 / chamber) in serum-free medium, and 600 μL complete medium was added to the lower chamber. The cells were then treated with 1 μM Gem, tcGem, or LipotcGem. After 48 h exposure, the upper surface of membrane was cleaned with cotton swabs. The invading cells adhered to the lower surface of the chamber were stained with 0.5% crystal violet, and subsequently observed under a bright-field microscope.33,34 The number of invading cells per field was counted using Image J software.
Cell Cycle
AsPC-1 cells were seeded on 6-well plates (5 × 105 cells per well), and then exposed to Gem, tcGem, or LipotcGem at the concentration of 10 μM for 48 h. Afterwards, the cells were collected, fixed with cold 70% ethyl alcohol overnight, centrifuged, washed, and stained with propidium iodide containing RNase A in the dark at room temperature. Cells were measured by flow cytometry and analyzed using FlowJo.v10.8.1.
Western Blot
AsPC-1 cells were seeded on 12-well plates (1 × 105 cells per well), cultured overnight, then treated with 10 μM Gem, tcGem, or LipotcGem for 24 h. The cell lysates were made using RIPA lysis buffer for 30 min-incubation in an ice bath, containing phenylmethanesulfonyl fluoride and InstabTM phosphatase inhibitor cocktail, followed by centrifugation (4°C, 12,000 × g, 15 min). The total protein concentration was detected by Bicinchoninic Acid Assay Kit. The lysates were then mixed with 4 × loading buffer and boiled for 10 min. Equal amounts of proteins in lysates were electrophoresed by 10% SDS-PAGE and transferred to Polyvinylidene Fluoride membranes which were blocked with 5% skimmed milk at room temperature for 2 h and immunoblotted with primary antibody overnight at 4°C, followed by another 1 h incubation with secondary antibody. The protein bands were revealed by a chemiluminescent HRP substrate (Millipore, Billerica, MA, USA, Cat. No. WBKLS0500), visualized with a ChemiDocTM imaging system (Bio-RAD Laboratories, Hercules, CA, USA) and semi-quantitatively analyzed by Image J 1.41o software.
Pharmacodynamics in Nude Mice
BALB/c nude mice were subcutaneously inoculated with 5×106 AsPC-1 cells into the right flank. Until the tumor volume was 100 mm3 or so, the mice were numbered, sorted according to tumor volume, and randomly assigned into three groups to ensure a similar tumor volume (~100 mm3) at the initial. The mice received intravenous administration of 5% glucose as a Control, 10 mg/kg Gem as a positive control, or 5 mg/kg LipotcGem as a treatment group every two other days (Q3D × 6). Tumor volume and body weight were measured throughout the treatment. After the treatment, the heart, liver, spleen, lung, kidney and tumor were harvested and sectioned for Hematoxylin-eosin (H&E) staining.
The orthotropic model of pancreatic cancer was also established as previously described.35,36 Briefly, each mouse was anesthetized and a 1-cm longitudinal skin incision was made on the left upper region of the abdomen, the peritoneum was opened and the pancreas was well exposed. 25 μL suspension containing 3×106 AsPC-1-luc cells was injected into the pancreas, holding for 5 min before closing the openings using Matrigel. The pancreas was gently returned to the abdominal cavity, and the surgical opening was closed.
7 days after engraftment, the luciferase substrate was injected intraperitoneally, and the mice were scanned by IVIS Spectrum in vivo imaging system (PerkinElmer, Waltham, MA, USA) within 5 min. According to the intensity of bioluminescence, the mice were divided into three groups and administrated intravenously with either 5% glucose, 10 mg/kg Gem, or 5 mg/kg LipotcGem (Q3D × 6), respectively. Body weight and health status of the mice were recorded throughout the treatment, and in vivo imaging was scanned on Day 17 and Day 27. After the final scanning, the mice were dissected under anesthesia. The heart, liver, spleen (the pancreatic tumor was adhered to the spleen), lung, kidney and intestine were collected and scanned ex vivo. Meanwhile, blood was collected for the hematological and biochemical analyzation by a hematological counter (ABX Pentra 60, Horiba Medical, Montpellier, France) and an automatic biochemical analyzer (7100, Hitachi, Tokyo, Japan), respectively.
H&E Staining
The tissues were fixed in 10% neutral formalin at room temperature overnight, dehydrated in ascending grades of ethanol, and embedded in paraffin. Tissue blocks were cut at 4 μm, deparaffinized, stained with hematoxylin and eosin and observed under a microscope.
Pk
The healthy male BALB/c mice were randomly assigned to receive 10 mg/kg Gem or 19 mg/kg LipotcGem via the tail vein. Blood samples were then collected at 0.03, 0.25, 0.5, 1, 2, 4, 8, 12, 24 and 48 h, respectively, at the presence of tetrahydrouridine (THU, 50 μg/mL, prevent Gem from deamination by cytidine deaminase ex vivo37) and heparin. The samples were centrifuged (4000 × g, 10 min, 4°C). A total of 30 μL supernatant was mixed with 120 μL acetonitrile via vortex, followed another centrifugation (12,000 × g, 10 min, 4°C), and the supernatant was used for quantitative analysis. The amounts of Gem in Gem group, Gem and tcGem in LipotcGem group were detected by LC-MS/MS as mentioned above. The pharmacokinetic parameters were analyzed by Phoenix winnonlin 6.4 software.
Tissue Distribution
BALB/c nude mice were subcutaneously inoculated with 5×106 AsPC-1 cells into the right flank. Once the tumor volume reached approximately 300–500 mm3, the mice were randomly assigned into two groups, and administered one bolus injection of 10 mg/kg Gem or 19 mg/kg LipotcGem. Then the heart, liver, spleen, lung, kidney, pancreas and tumor were collected at the predetermined time points (0.5, 4, 8, 12 and 24 h) after cardiac perfusion. After grinding, the concentrations of Gem and tcGem in samples were detected by LC-MS/MS as mentioned above.
Statistical Analysis
Results were expressed as mean ± standard deviation (SD). Statistical significance was performed using GraphPad Prism software 5.0 version (GraphPad Software Company, San Diego, California, America). Two-group comparison was performed using tow-tailed Student’s t-test. p < 0.05 was regarded as statistical significance, indicated as *p < 0.05, **p < 0.01, ***p < 0.001 and no significant (ns).
Results and Discussions
Synthesis and Characterization of tcGem
Target compound tcGem was successfully synthesized and characterized (Figure 1A and Figure S1-S4). Mp:120–122°C,  41.5° (c 6.6 mg / mL, CH2Cl2). 1H NMR (500M, CD3OD): δ 8.30 (d, J = 7.2 Hz, 1H), 7.34 (d, J = 6.8 Hz, 1H), 6.25 (brs, 1H), 4.32–4.26 (m, 1H), 4.20–4.17 (m, 2H), 3.98–3.96 (m, 2H), 3.81 (d, J = 12.5 Hz, 1H), 1.69 (q, J = 7.2 Hz, 2H), 1.41–1.28 (m, 22H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.41, 154.01, 153.21, 144.30, 122.95 (t, J = 258.8 Hz), 94.86, 84.06 (t, J = 31.8 Hz), 81.03, 68.38 (t, J = 22.4 Hz), 65.35, 58.79, 39.52, 31.31, 29.07, 29.03, 28.95, 28.73, 28.61, 28.19, 25.16, 22.11, 13.95. MS-ESI (m/z): 504.2 (M+H) +.
41.5° (c 6.6 mg / mL, CH2Cl2). 1H NMR (500M, CD3OD): δ 8.30 (d, J = 7.2 Hz, 1H), 7.34 (d, J = 6.8 Hz, 1H), 6.25 (brs, 1H), 4.32–4.26 (m, 1H), 4.20–4.17 (m, 2H), 3.98–3.96 (m, 2H), 3.81 (d, J = 12.5 Hz, 1H), 1.69 (q, J = 7.2 Hz, 2H), 1.41–1.28 (m, 22H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.41, 154.01, 153.21, 144.30, 122.95 (t, J = 258.8 Hz), 94.86, 84.06 (t, J = 31.8 Hz), 81.03, 68.38 (t, J = 22.4 Hz), 65.35, 58.79, 39.52, 31.31, 29.07, 29.03, 28.95, 28.73, 28.61, 28.19, 25.16, 22.11, 13.95. MS-ESI (m/z): 504.2 (M+H) +.
Preparation and Characterization of tcGem-Loaded Liposomes
To obtain excellent tcGem-loaded liposomes with small particle size, low polydispersity index (PDI), and suitable zeta potential, five phospholipids (DMPC, DPPC, DSPC, HSPC, and DMPG, 18.0 mg) were screened first under the conditions of 2.0 mg of tcGem, 1.8 mg of cholesterol (Chol), and 3.0 mg of DSPE-PEG2K. The results showed that the liposomes prepared using DMPC were optimal in terms of size, PDI, and zeta potential (Figure 1B), which was likely attributed to the identical length of alkane chains in the molecular structure of DMPC and tcGem. Subsequently, the mass ratios of DMPC to tcGem, the prescribed amounts of Chol and DSPE-PEG2K were also optimized based on the 40-day stability test data of the prepared liposomes with DMPC (Figure 1C-E).
The data revealed that the composition of tcGem-loaded liposomes was optimal with a mass ratio of 9: 1 for DMPC to tcGem, 1.8 mg of Chol, and 3.0 mg of DSPG-PEG2K. In summary, the excellent tcGem-loaded liposomes (referred as LipotcGem) were composed of tcGem: DMPC: Chol: DSPE-PEG2K with a mass ratio of 2: 18: 1.8: 3, featuring an average size of 115 nm (Figure 1G), a zeta potential value of –36 mV (Figure 1H), EE exceeding 98%, DL of 8.1%, a single spherical shape with core-shell structure (Figure 1F and Figure S5), and slow drug release in PBS at pH 7.4 containing 50% serum (Figure 1I), or PBS at pH 7.4 and at pH 5 containing 1% Tween-80 (Figure S6), in comparison to free tcGem.
Improved Antitumor Activity in vitro of LipotcGem
AsPC-1, PANC-1, BxPC-3, Mia PaCa-2, and SU.86.86, as typical and common pancreatic cancer cell lines, are widely used to evaluate antitumor activity in vitro and to understand the underlying mechanism. AsPC-1 is a widely utilized cell line known for its high metastatic potential.38–41 To comprehensively assess the in vitro activity and elucidate mechanism of LipotcGem, experiments of cytotoxicity, cell apoptosis, cell cycle arrest, cell migration and invasion, cellular uptake, endocytosis, subcellular localization, intracellular accumulation and drug release were conducted.
Improved Cytotoxicity
The IC50 values of tcGem and LipotcGem were 1.28 μM and 2.58 μM, respectively, indicating the antitumor activity of tcGem and LipotcGem was significantly greater than that of Gem (Figure 2A and Table 1).
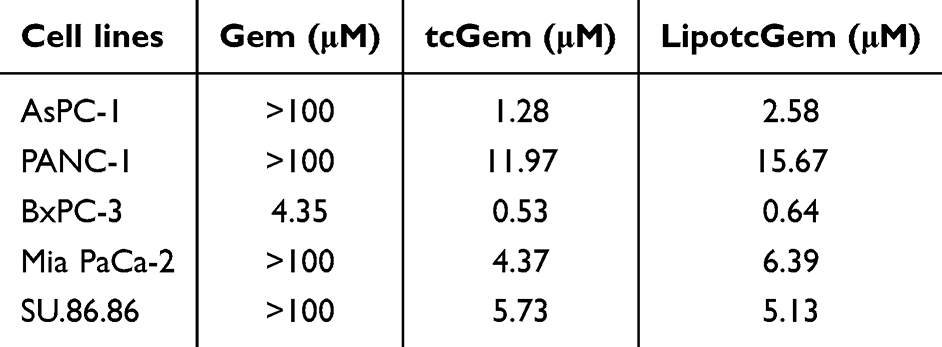 |
Table 1 The IC50 Values of Gem, tcGem and LipotcGem After 48 h Incubation |
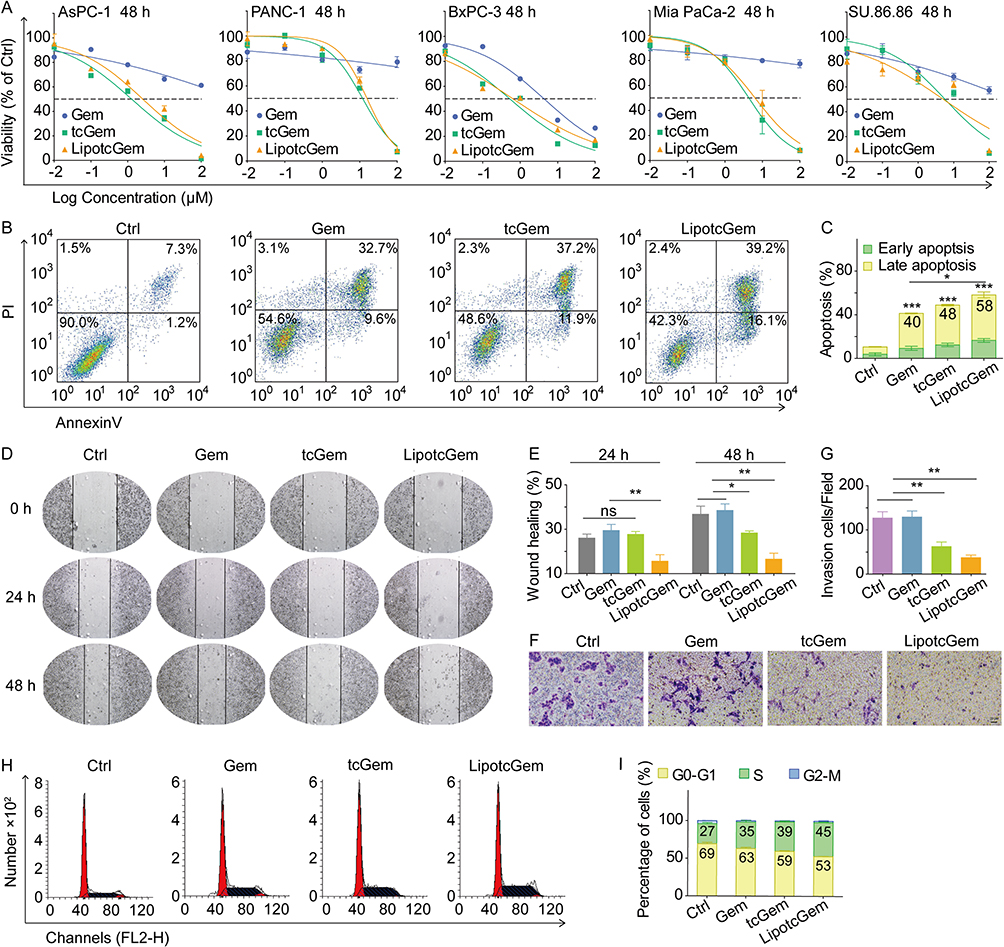 |
Figure 2 Antitumor activity in vitro. (A) Cytotoxicity of Gem, tcGem, and LipotcGem on AsPC-1, PANC-1, BxPC-3, Mia PaCa-2, and SU.86.86 cell lines after 48 h of incubation at a series of concentrations of 0.01, 0.1, 1, 10 and 100 μM. (B) Cellular apoptosis of AsPC-1 cells after 48 h treatment with 10 μM Gem, tcGem, or LipotcGem using a flow cytometer. (C) Histograms of percentages of early and late apoptosis. (D and E) The representative phase-contrast photomicrographs (100 ×) and histograms of wound healing (%) of initial width of AsPC-1 cells grown in presence of 10 μM Gem, tcGem or LipotcGem at 0, 24 and 48 h after wound creation. (F and G) The images and histograms of invading cells on the lower surface of transwell chamber after 48 h treatment with 1 μM Gem, tcGem or LipotcGem. (H and I) The images and histograms of cell cycle analysis of AsPC-1 cells after 24 h treatment with 10 μM Gem, tcGem, or LipotcGem. Data were presented as mean ± SD (n = 3). *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference. |
Notably, the IC50 of Gem is higher than that reported in some publications. We observed the increased Gem IC50 can primarily be attributed to the increased cell density, which plays a crucial role in determining the Gem IC50 value.
For AsPC-1 cells, after 48 h of incubation, the Gem IC50 value increased to over 100 μM when the cell density reached 2 × 104 cells per well. While, this value decreased to 11.78 μM when the cell density was reduced to 5 × 103 cells per well (Figure S7). Similarly, in Mia PaCa-2 cells, the IC50 value of Gem decreased to 73.53 μM when the cell density was reduced to 3 × 103 cells per well after a 72-hour treatment (Figure S8). For PANC-1 cells, cell viability remained above 50% even at lower cell densities and with extended exposure time (Figure S9). Overall, in the three pancreatic cancer cell lines, the IC50 value of Gem showed a decline when the cell density decreased and the treatment duration was prolonged. The decreased Gem IC50 is similar to that reported in other publications.42–48
The IC50 values of LipotcGem also exhibited an increasing trend with rising cell density. Notably, at a high concentration of 100 μM, LipotcGem showed the capacity to effectively eliminate nearly all tumor cells (Figure S10). Given Gem’s limited efficacy in eradicating tumor cells, LipotcGem displayed a significant ability to eliminate these cells, highlighting its remarkable anti-tumor properties.
Enhanced Cell Apoptosis
The percentages of early apoptotic and late apoptotic cells induced by Gem, tcGem, and LipotcGem were 40%, 48%, and 58%, respectively, indicating that LipotcGem significantly induced cellular apoptosis in AsPC-1 cells (Figure 2B and C). LipotcGem significantly up-regulated the pro-apoptotic protein ratio of cleaved caspase-3/caspase-3 compared to Gem (Figure S11), suggesting that LipotcGem induced cell apoptosis through the caspase 3-mediated pathway.
Inhibited Cell Migration
The migratory property of AsPC-1 cells was assessed using the wound healing assay after treatment with Gem, tcGem and LipotcGem, respectively. The results exhibited that the percentage of wound healing in LipotcGem group was significantly lower than that of Gem after 24 and 48 h incubation (Figure 2D and E), indicating that LipotcGem could significantly inhibit AsPC-1 cells migration compared to Gem.
Suppressed Cell Invasion
The invasion ability of AsPC-1 cells was investigated using the transwell chamber by quantifying the number of stained cells that migrated to the lower surface of the chamber. The counts were 127 ± 25, 129 ± 27, 62 ± 20, 37 ± 12 cells per field in Control, Gem, tcGem, and LipotcGem groups, respectively (Figure 2F and G), indicating that LipotcGem significantly suppressed AsPC-1 cells migration and invasion compared to Gem.
It was reported that, only after 24 h treatment, Gem dose-dependently (1–10 M) upregulated the expression of stemness-associated genes and proteins as well as increased fractions of cancer stem cells phenotype in pancreatic cancer cells, accompanied by increased cell migration, chemoresistance, and tumorigenesis.49,50 Furthermore, the authors also observed the attenuated inductive effect of cancer stem cells and stemness-associated molecules with a high concentration of Gem. They assumed that an adequate concentration of Gem would suppress its capability to promote stem cell induction.49 LipotcGem significantly increased the intracellular accumulation, which may be an explanation of the increased ability to suppress AsPC-1 cells migration and invasion.
Increased Cell Cycle Arrest
Gem is a cell cycle specific drug that increases the proportion of cells in the S phase.51–53 In this test, the percentages of cells in different phases of the cell cycle were analyzed using flow cytometry. Figure 2H and I showed that the cell cycle arrest was consistent between LipotcGem and Gem, with a 10% increase in the proportion of cells in the S-phase (45% vs 35%).
Improved Uptake and Mechanism of LipotcGem
Cellular Uptake in a Concentration- and Time-Dependent Manner
DiI-Liposome was employed instead of LipotcGem. The results showed that the cellular uptake of DiI-Liposome was in a concentration- and time-dependent manner via fluorescence microscopy observation (Figure 3A and B) and flow cytometer analysis (Figure 3C–F).
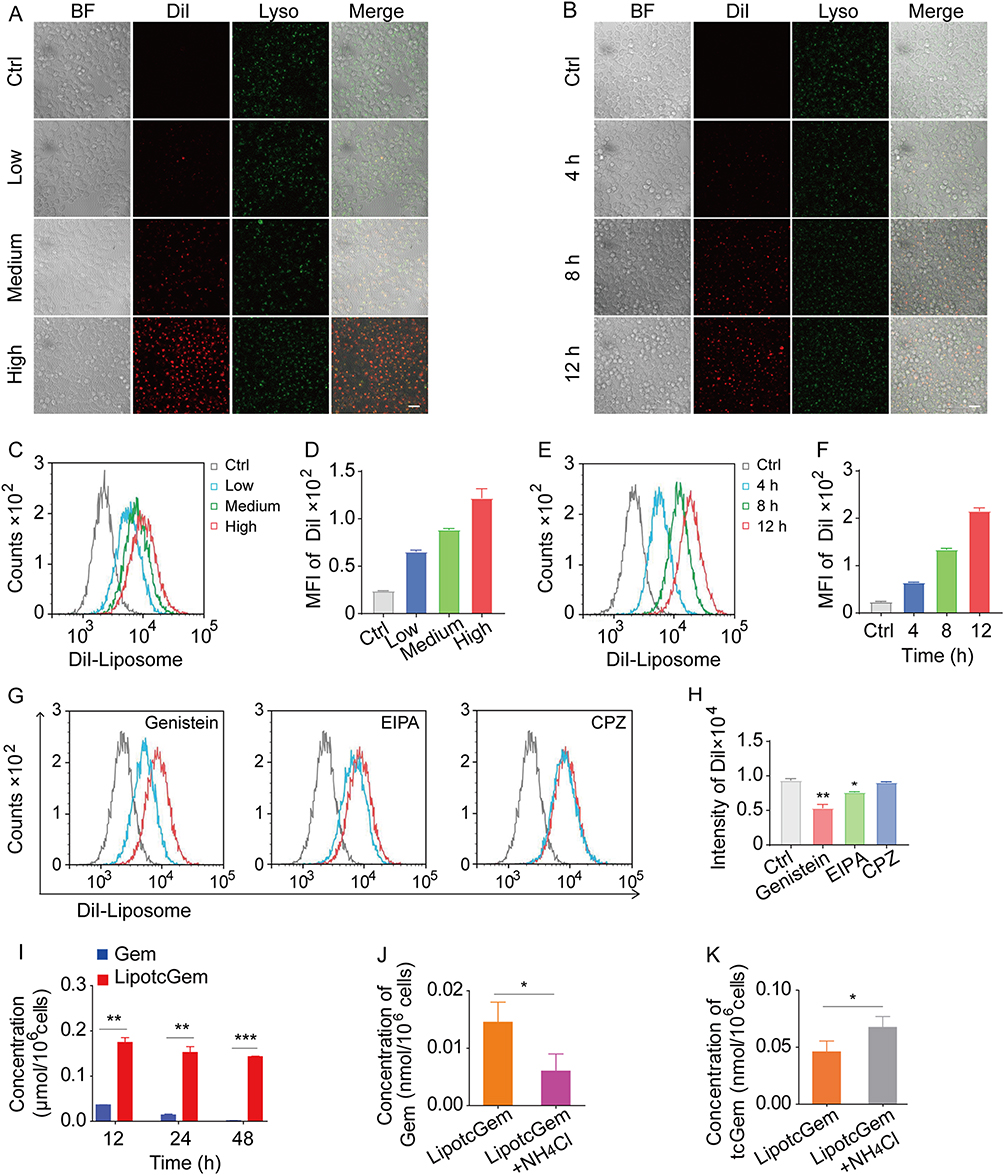 |
Figure 3 Cellular uptake and endocytic mechanism of DiI labeled liposome (DiI-Liposome). (A and B) CLMS images of AsPC-1 cells treated with DiI-Liposome for 4 h or for 4, 8, 12 h at 37°C. Red signals represented DiI-Liposome, green spots represented lysosomes. Scale bar, 20 μm. (C–F) Intracellular mean fluorescent intensity (MFI) after incubation with DiI-Liposome for 4 h or for 4, 8, 12 h at 37°C measured by a flow cytometer. (G and H) Endocytic mechanism of DiI-Liposome in the presence of genistein, EIPA or CPZ for 4 h incubation at 37°C by a flow cytometer. (I) Intracellular accumulation from Gem or LipotcGem treated cells detected by LC-MS/MS. (J and K) Intracellular accumulation of Gem and tcGem from LipotcGem in the presence of 30 mmol/L NH4Cl or not. Data were presented as mean ± SD (n = 3). *p<0.05, **p<0.01, ***p<0.001. |
Endocytosis
Endocytosis is the primary way for nanomedicines to enter cells, including micropinocytosis, clathrin- and caveolae-mediated pathways. Figure 3G and H indicated that the endocytosis of DiI-Liposome was highly significantly inhibited by Genistein and amiloride (EIPA), which are inhibitors of caveolae-dependent endocytosis and micropinocytosis, respectively. While it was not influenced by chlorpromazine (CPZ), an inhibitor of clathrin-dependent endocytosis, suggesting that endocytosis was primarily mediated by caveolae with a minor contribution from micropinocytosis.
Subcellular Location
Figure 3A and B exhibited a fine colocalization between DiI-Liposome and LysoTracker Green, a lysosomal marker, indicating that LipotcGem was transferred to lysosomes after endocytosis, where the acidic environment might play an important role in the release and biotransformation of Gem.54
Intracellular Accumulation
After 48 h of incubation, the intracellular accumulation of Gem and tcGem from LipotcGem was approximately 100 times higher than that of Gem (0.15 vs 0.0015 nmol/106 cells) (Figure 3I).
Intracellular Release
After weakening the acidic environment of lysosomes by treating cells with NH4Cl,55 the intracellular Gem from LipotcGem decreased by ~ 60%, while the intracellular remained tcGem increased by around 30% compared to cells in normal lysosomal environment (Figure 3J and K), suggesting that lysosomes played a key role in the hydrolysis of tcGem into Gem in LipotcGem treated cells.
We speculated that the remarkably increased intracellular accumulation may result from the modification at the 4-amino position by directly linking a myristyl group, protecting Gem from enzymatic deactivation,56 and the benefit from liposomal vesicles, facilitating greater intracellular uptake due to the ability to penetrate the cell membrane by fusion or endocytosis.26,57 Further, the significantly increased intracellular accumulation of Gem and tcGem from LipotcGem is primarily responsible for the substantial improvement in anti-tumor efficiency. Conversely, the lower intracellular accumulation of Gem was predominantly limited by active transportation by nucleoside transporters and enzymatic deactivation by deoxycytidine deaminase.11
Improved Antitumor Efficacy in vivo and Neglected Systematic Toxicity
In the subsequent in vivo assessments of antitumor efficacy, PK and tissue distribution, free tcGem was failed to be set as a control due to its poor solubility in water. The solubility of tcGem could not meet the requirements for intravenous administration, even at the maximal injection volume, with 5% DMSO being the universally allowable concentration for in vivo studies.
The AsPC-1 subcutaneous and AsPC-1-luc orthotopic xenograft BALB/c nude mouse models were used in the test, with Gem chosen as the positive control.
In the subcutaneous model, the tumor volume profiles showed that LipotcGem significantly retarded tumor growth without causing noticeable body weight loss compared to Gem throughout the treatment (Figure 4A and B). The tumor inhibition rate (TIR) of LipotcGem was notably higher than that of Gem. For tumor volume, LipotcGem reached a TIR of 70% compared to 50% for Gem (Figure 4A). For tumor weight, the TIR was 69% for LipotcGem versus 49% for Gem on Day 30 (Figure 4C–E). Following cessation of treatment up to Day 30, the tumors in LipotcGem group exhibited the slowest growth rate, followed by Gem group, while those in Control group grew the fastest, suggesting that LipotcGem displayed a long-acting and delayed effect. No significant weight loss was observed among mice treated with LipotcGem, Gem, or Control (Figure 4B). H&E staining was utilized to examine the histological features of the heart, liver, spleen, lung, kidney, and pancreas. The results showed no visible pathological damage in LipotcGem group (Figure 4F), indicating that LipotcGem did not induce additional systemic toxicity, such as hepatotoxicity or nephrotoxicity. As for tumor tissues, the necrotic areas in the LipotcGem treated group were larger than that in Gem and Control group. In Control group, many necrotic regions were also present, however, the regions were small and scattered among proliferative tumor cells. Conversely, in LipotcGem treated group, the necrotic areas formed large clusters with almost no proliferative cells in the middle.
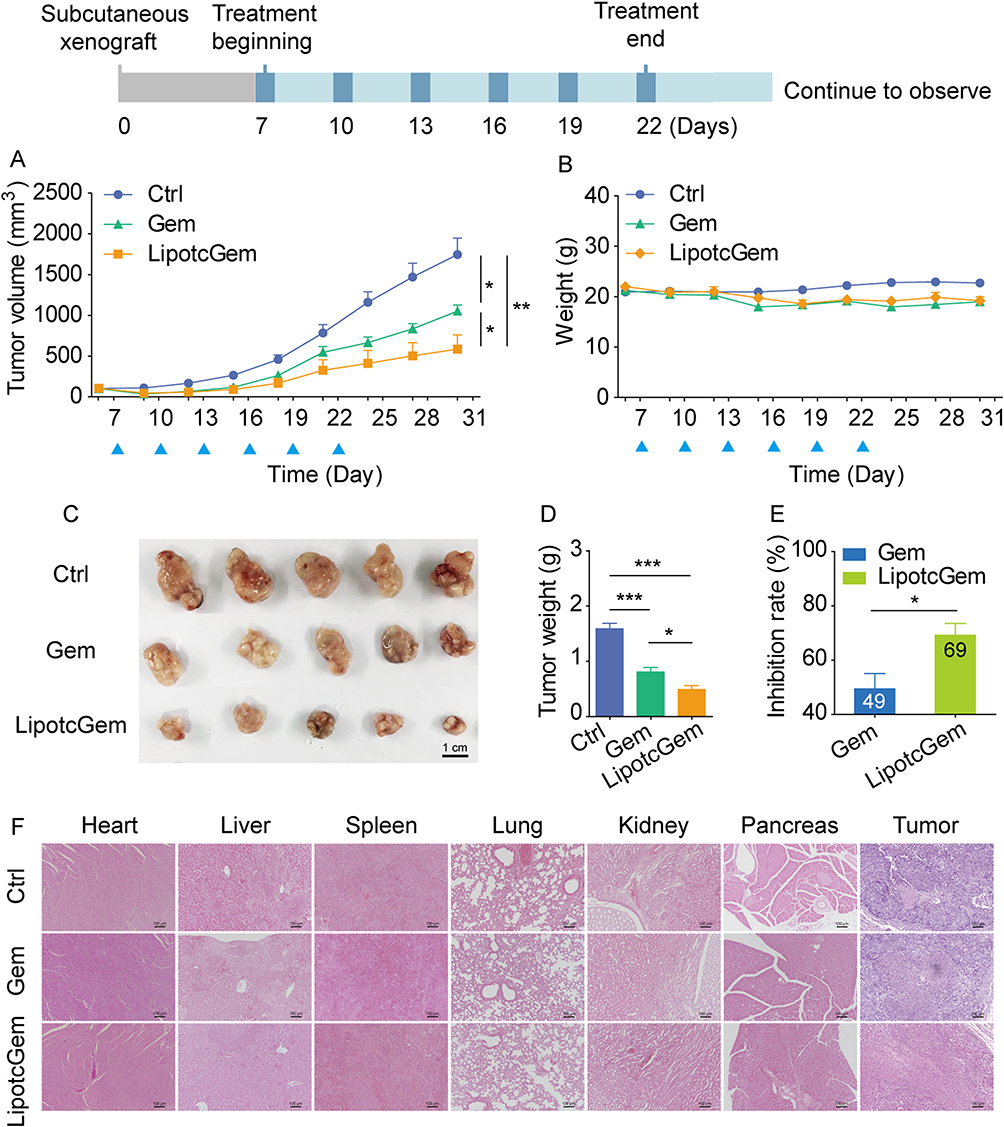 |
Figure 4 In vivo antitumor efficiency evaluation in AsPC-1 subcutaneous xenograft BALB/c nude mouse model. (A) Curves of tumor volume and (B) body weight of tumor bearing mice throughout the treatment of Control (5% glucose), Gem (10 mg/Kg) or LipotcGem (5 mg/Kg) administration (Q3D × 6). The blue triangles present the time of administration. (C) Picture of tumors, (D) histograms of tumor weight and (E) TIR (%) on Day 30 or one week after the last administration. (F) Representative histological images of the heart, liver, spleen, lung, kidney, pancreas and tumor from the mice in each treatment group. Scale bar, 100 μm. Data were presented as mean ± SD (n = 5). *p<0.05, **p<0.01, ***p<0.001. |
In the orthotopic model, the results revealed that the intensity of bioluminescent signals was significantly reduced in LipotcGem group compared to those in the Gem group (Figure 5A). On Day 27 (5 days after the last administration), the radiance in LipotcGem group was only about 1.5% of that in Gem group. The TIR of LipotcGem was approximately 90%, far exceeding the 50% TIR of Gem (Figure 5A, B and E). The survival rate (%) of mice treated with LipotcGem was extended by 15 days compared to Control group, and by 9 days compared to Gem-treated mice (Figure 5D), suggesting that LipotcGem showed excellent antitumor activity in vivo, consistent with the findings from the subcutaneous model.
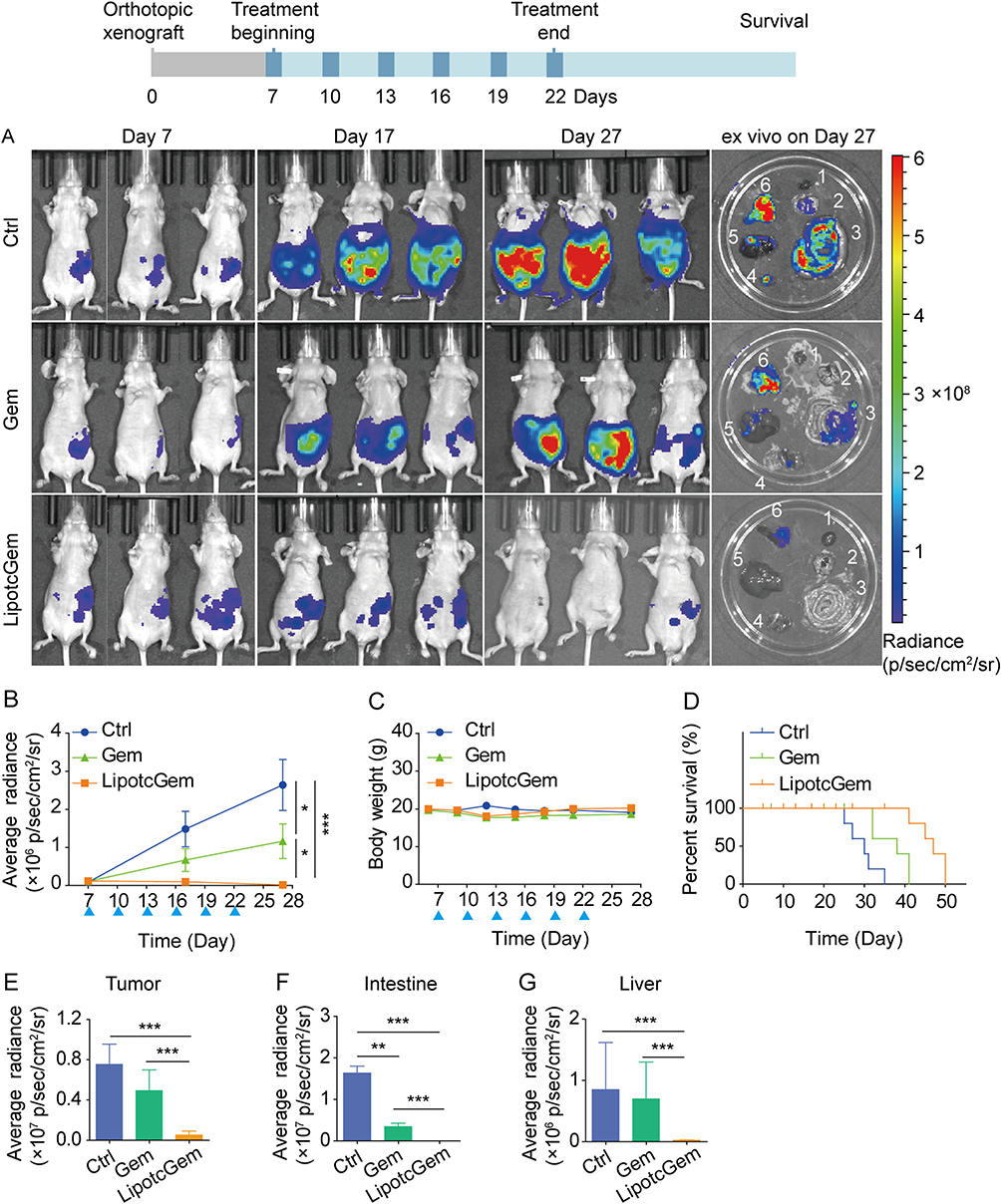 |
Figure 5 In vivo antitumor efficiency evaluation in AsPC-1-luc orthotopic xenograft BALB/c nude mouse model. (A) Images of in vivo imaging at Day 7, Day 17 and Day 27 after inoculation. After the last scanning, the tissues were collected and scanned ex vivo. 1, heart; 2, lung; 3 intestine, 4 kidney; 5, liver; 6, tumor and spleen. (B) Curves of average radiance, (C) body weight and (D) percent survival (%) of mice in Control (5% glucose), or Gem (10 mg/kg) or LipotcGem (5 mg/kg) groups (the blue triangles present the time of administration). Histograms of average radiance signal in (E) tumors, (F) intestine, (G) liver on Day 27. Data were presented as mean ± SD (n = 3). *p<0.05, **p<0.01, ***p<0.001. |
Semi-quantitative analysis of bioluminescent signals indicated that the metastatic burden was nearly undetectable in LipotcGem group, moderate in the Gem group, and heavily spread to the liver and intestine in Control group, suggesting that LipotcGem could forcefully inhibit tumor metastasis (Figure 5A, E, F and G), consistent with the above results regarding tumor migration and invasion in vitro. Throughout the entire treatment course, there was no significant reduction in body weight in LipotcGem group (Figure 5C). Given that the major adverse effects of Gem are myelosuppression with thrombocytopenia and anemia,10,58–60 hematologic and biochemical analysis were performed. The results showed that LipotcGem did not cause myelosuppression, suggesting LipotcGem had a good safety. The concentrations of lactate dehydrogenase, alanine aminotransferase and aspartate aminotransferase were significantly increased in Ctrl group, indicating a severe tumor burden and abnormal liver function caused by tumor metastasis. In contrast, these parameters in LipotcGem group were similar to those in the healthy group and far superior to those in Gem group, suggesting that LipotcGem exhibited better antitumor metastasis ability than Gem (Figure 6).
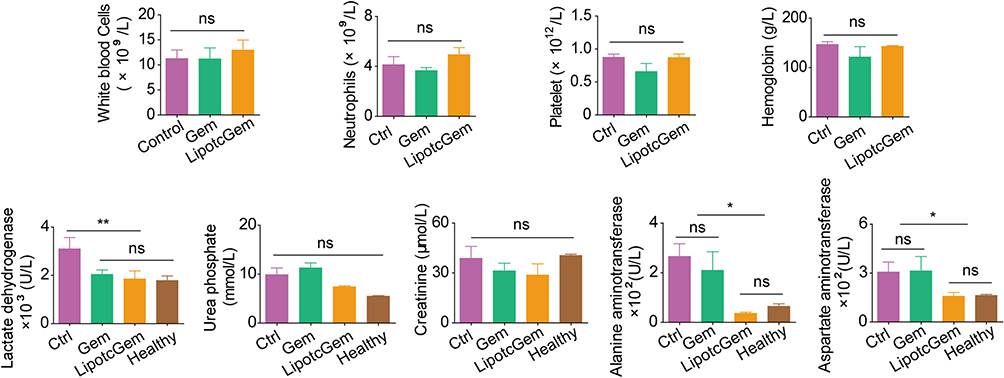 |
Figure 6 Histograms of hematological and biochemical evaluation of mice bearing AsPC-1-luc orthotopic xenograft after the treatment with Gem or LipotcGem. Data were presented as mean ± SD (n = 3). *p<0.05, **p<0.01. ns, no significant difference. |
Pk
An equimolar dose of 38 μmol/kg of Gem or LipotcGem were intravenously administered to healthy BALB/c mice via tail vein. The PK parameters are listed in Table 2 using a non-compartmental method. Compared to Gem, LipotcGem showed significantly prolonged T1/2 (3.5-fold, 5.23 vs 1.46 h), increased Cmax (2.7-fold, 498.90 vs.183.07 μmol/L), and AUC 0–48h (8-fold, 441.66 vs 55.59 h×μmol/L) (Figure 7A), suggesting LipotcGem could potentially improve antitumor efficacy.
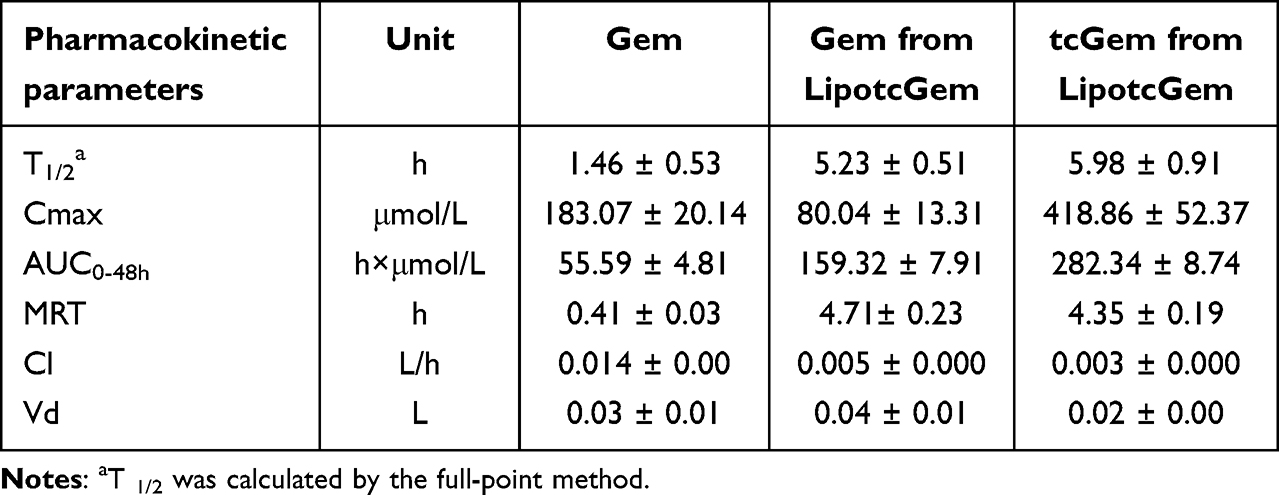 |
Table 2 The PK Parameters of Gem and LipotcGem in BALB/c Mice (Mean ± SD, n = 6) |
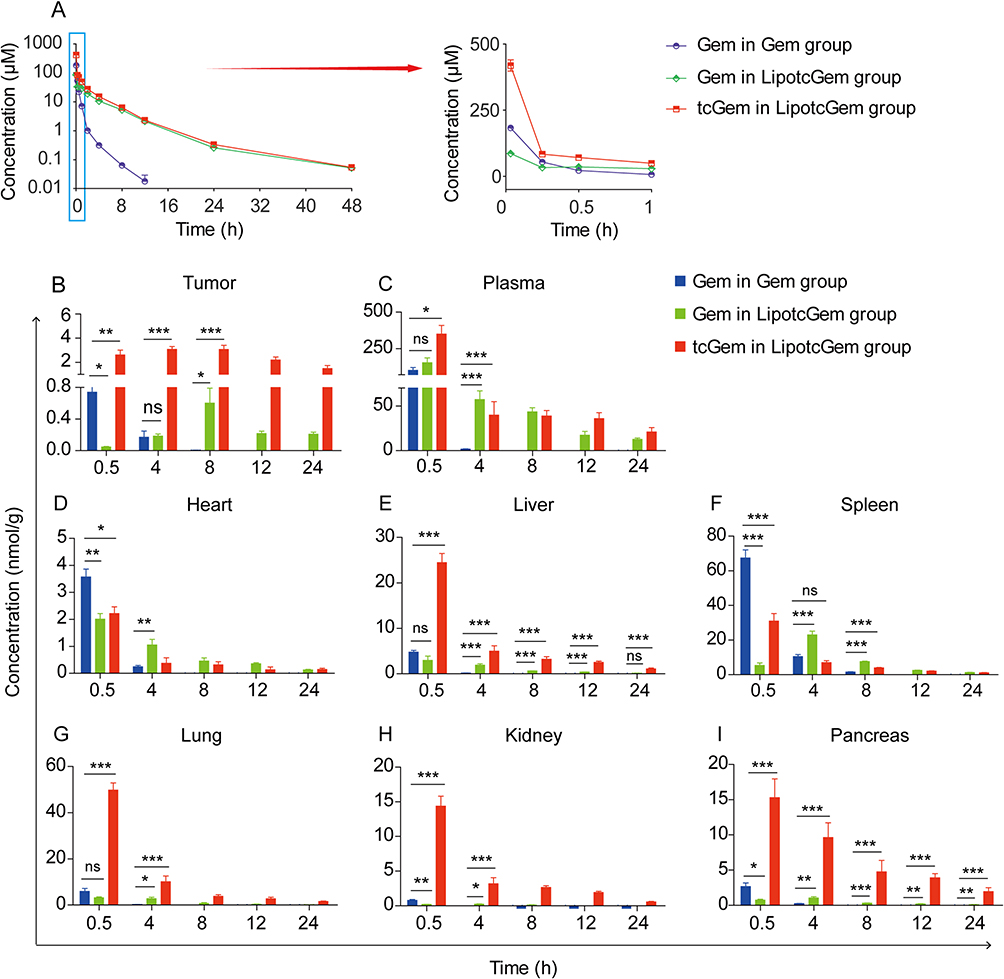 |
Figure 7 PK profiles and histograms of tissue distribution of Gem and LipotcGem. (A) Curves depicting the mean plasma concentration-time of Gem and LipotcGem in healthy BALB/c mice injected via tail vein at an equimolar dose of 38 μmol/kg of Gem or LipotcGem. Tissue distribution of Gem and LipotcGem in the (B) tumor, (C) plasma, (D) heart, (E) liver, (F) spleen, (G) lung, (H) kidney, (I) pancreas at 0.5, 4, 8, 12 and 24 h in AsPC-1 tumor-bearing mice. Data were presented as mean ± SD (n = 6). *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference. |
Tissue Distribution
Gem and LipotcGem, at an equimolar dose of 38 μmol/kg, were intravenously administered to BALB/c nude mice bearing AsPC-1 xenografts, respectively. In tumor tissues and plasma (Figure 7B and C), the Gem concentration in Gem group decreased sharply over time and become undetectable beyond 8 h. While the concentrations of both Gem and tcGem from LipotcGem group increased continuously within the first 8 h and peaked 3.0 μmol/g at 8 h, which was 750 times higher than that of Gem (0.004 μmol/g) in tumors (Figure 7B), further explaining the better antitumor efficacy of LipotcGem than Gem.
The accumulation of LipotcGem in the heart and spleen was maintained at a low concentration, in contrast with the significantly higher concentration of Gem at 0.5 h, indicating that LipotcGem imposed a weaker burden than Gem on these organs. The reduced burden of LipotcGem in the heart was especially important, as Gem was reported to be associated with cardiotoxicity.61–63 LipotcGem exhibited higher accumulation in liver, lung, kidney and pancreas, while positively, the concentration of tcGem in LipotcGem group in these organs decreased sharply, suggesting a potential re-distribution of LipotcGem from these organs to the tumor (Figure 7D–I).
Conclusion
A novel Gem derivative (tcGem) was designed and synthesized, and the tcGem-loaded liposomes have also been successfully prepared. The optimized LipotcGem was characterized as suitable particle size, excellent EE and DL. Compared to Gem, LipotcGem exhibited better antitumor efficacy in vitro and in vivo, as evidenced by its enhanced ability to inhibit tumor cell growth, induce cell apoptosis, show a higher tumor inhibition rate, suppress tumor metastasis, and prolong survival, due to the higher and longer drug exposure. Additionally, the endocytosis of LipotcGem was mainly mediated by caveolae, and then processed in the lysosomes, where some of tcGem was released and hydrolyzed into Gem. These findings provide insight into the reason that Gem acylation can significantly improve antitumor efficacy and offer a potentially effective strategy for pancreatic cancer treatment.
Acknowledgments
This work was supported by the Fundamental Research Funds for the Central Universities (3332023049), the CAMS Innovation Fund for Medical Sciences (2021-I2M-1-030, 2021-I2M-1-070 and 2021-I2M-1-026) and the NSFC 81903477.
Disclosure
Dr Dan Wang reports a patent Anti-tumor lipid composition licensed to CN112245391. Miss Yan Li reports a patent Anti-tumor lipid composition issued to CN202011145564.6. Prof. Dr. Apeng Wang reports a patent Anti-tumor compound licensed to CN202011145601.3. Professor Mingliang Liu reports a patent Anti-tumor compound licensed to CN202011145601.3, a patent Anti-tumor lipid composition issued to CN202011145564.6. Professor Guimin Xia reports a patent Anti-tumor compound licensed to CN202011145601.3, a patent Anti-tumor compound composition issued to CN202011145564.6. The authors report no other conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Wagle NS, et al. Cancer statistics, 2023. CA Cancer J Clin. 2023;3(1):17–48. doi:10.3322/caac.21763
2. Halbrook CJ, Lyssiotis CA, Di Magliano MP, et al. Pancreatic cancer: advances and challenges. Cell. 2023;186(8):1729–1754. doi:10.1016/j.cell.2023.02.014
3. Christenson ES, Jaffee E, Azad NS. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: a bright future. Lancet Oncol. 2020;21(3):e135–e145. doi:10.1016/S1470-2045(19)30795-8
4. Stoop TF, Theijse RT, Seelen LWF, et al. Preoperative chemotherapy, radiotherapy and surgical decision-making in patients with borderline resectable and locally advanced pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2024;21(2):101–124. doi:10.1038/s41575-023-00856-2
5. Mizrahi JD, Surana R, Valle JW, et al. Pancreatic cancer. Lancet. 2020;395(10242):2008–2020. doi:10.1016/S0140-6736(20)30974-0
6. Wood LD, Canto MI, Jaffee EM, et al. Pancreatic Cancer: pathogenesis, screening, diagnosis, and treatment. Gastroenterology. 2022;163(2):386–402.e1. doi:10.1053/j.gastro.2022.03.056
7. Hosein AN, Dougan SK, Aguirre AJ, et al. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat Cancer. 2022;3(3):272–286. doi:10.1038/s43018-022-00349-2
8. Conroy T, Castan F, Lopez A, et al. Five-Year outcomes of FOLFIRINOX vs gemcitabine as adjuvant therapy for pancreatic cancer: a randomized clinical trial. JAMA Oncol. 2022;8(11):1571–1578. doi:10.1001/jamaoncol.2022.3829
9. Kunzmann V, Siveke JT, Algül H, et al. Nab-paclitaxel plus gemcitabine versus nab-paclitaxel plus gemcitabine followed by FOLFIRINOX induction chemotherapy in locally advanced pancreatic cancer (NEOLAP-AIO-PAK-0113): a multicentre, randomised, Phase 2 trial. Lancet Gastroenterol Hepatol. 2021;6(2):128–138. doi:10.1016/S2468-1253(20)30330-7
10. Wainberg ZA, Melisi D, Macarulla T, et al. NALIRIFOX versus nab-paclitaxel and gemcitabine in treatment-naive patients with metastatic pancreatic ductal adenocarcinoma (NAPOLI 3): a randomised, open-label, Phase 3 trial. Lancet. 2023;402(10409):1272–1281. doi:10.1016/S0140-6736(23)01366-1
11. Mini E, Nobili S, Caciagli B, et al. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17 Suppl 5:v7–12. doi:10.1093/annonc/mdj941
12. de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014;741:8–16. doi:10.1016/j.ejphar.2014.07.041
13. Bouffard DY, Laliberté J, Momparler RL. Kinetic studies on 2’,2’-difluorodeoxycytidine (Gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochem Pharmacol. 1993;45(9):1857–1861. doi:10.1016/0006-2952(93)90444-2
14. Shelton J, Lu X, Hollenbaugh JA, et al. Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem Rev. 2016;116(23):14379–14455. doi:10.1021/acs.chemrev.6b00209
15. Birhanu G, Javar HA, Seyedjafari E, et al. Nanotechnology for delivery of gemcitabine to treat pancreatic cancer. Biomed Pharmacother. 2017;88:635–643. doi:10.1016/j.biopha.2017.01.071
16. Han H, Li S, Zhong Y, et al. Emerging pro-drug and nano-drug strategies for gemcitabine-based cancer therapy. Asian J Pharm Sci. 2022;17(1):35–52. doi:10.1016/j.ajps.2021.06.001
17. Tarannum M, Vivero-Escoto JL. Nanoparticle-based therapeutic strategies targeting major clinical challenges in pancreatic cancer treatment. Adv Drug Deliv Rev. 2022;187:114357. doi:10.1016/j.addr.2022.114357
18. Yu X, Di Y, Xie C, et al. An in vitro and in vivo study of gemcitabine-loaded albumin nanoparticles in a pancreatic cancer cell line. Int J Nanomed. 2015;10:6825–6834. doi:10.2147/IJN.S93835
19. Guo Z, Wang F, Di Y, et al. Antitumor effect of gemcitabine-loaded albumin nanoparticle on gemcitabine-resistant pancreatic cancer induced by low hENT1 expression. Int J Nanomed. 2018;13:4869–4880. doi:10.2147/IJN.S166769
20. Wang Y, Fan W, Dai X, et al. Enhanced tumor delivery of gemcitabine via PEG-DSPE/TPGS mixed micelles. Mol Pharm. 2014;11(4):1140–1150. doi:10.1021/mp4005904
21. Tam YT, Huang C, Poellmann M, et al. Stereocomplex prodrugs of Oligo(lactic acid) n Gemcitabine in Poly(ethylene glycol)-block -poly(d, l -lactic acid) micelles for improved physical stability and enhanced antitumor efficacy. ACS Nano. 2018;12(7):7406–7414. doi:10.1021/acsnano.8b04205
22. Oliveira C, Neves NM, Reis RL, et al. Gemcitabine delivered by fucoidan/chitosan nanoparticles presents increased toxicity over human breast cancer cells. Nanomedicine. 2018;13(16):2037–2050. doi:10.2217/nnm-2018-0004
23. Wang G, Wu B, Li Q, et al. Active transportation of liposome enhances tumor accumulation, penetration, and therapeutic efficacy. Small. 2020;16(44):e2004172. doi:10.1002/smll.202004172
24. Matsumoto T, Komori T, Yoshino Y, et al. A Liposomal Gemcitabine, FF-10832, Improves plasma stability, tumor targeting, and antitumor efficacy of gemcitabine in pancreatic cancer xenograft models. Pharma Res. 2021;38(6):1093–1106. doi:10.1007/s11095-021-03045-5
25. Paolino D, Cosco D, Racanicchi L, et al. Gemcitabine-loaded PEGylated unilamellar liposomes vs GEMZAR: biodistribution, pharmacokinetic features and in vivo antitumor activity. J Control Release. 2010;144(2):144–150. doi:10.1016/j.jconrel.2010.02.021
26. Federico C, Morittu VM, Britti D, et al. Gemcitabine-loaded liposomes: rationale, potentialities and future perspectives. Int J Nanomed. 2012;7:5423–5436. doi:10.2147/IJN.S34025
27. Tamam H, Park J, Gadalla HH, et al. Development of liposomal gemcitabine with high drug loading capacity. Mol Pharm. 2019;16(7):2858–2871. doi:10.1021/acs.molpharmaceut.8b01284
28. Cai H, Wang R, Guo X, et al. Combining gemcitabine-loaded macrophage-like nanoparticles and erlotinib for pancreatic cancer therapy. Mol Pharm. 2021;18(7):2495–2506. doi:10.1021/acs.molpharmaceut.0c01225
29. He B, Wang C, Wang F, et al. Differentiation therapy for murine myelofibrosis model with MLN8237 loaded low-density lipoproteins. J Control Release. 2023;356:554–566. doi:10.1016/j.jconrel.2023.03.024
30. Nejad ZK, Khandar AA, Khatamian M, et al. Investigating of the anticancer activity of salen/salophen metal complexes based on graphene quantum dots: induction of apoptosis as part of biological activity. Int J Pharm. 2023;642:123092. doi:10.1016/j.ijpharm.2023.123092
31. Wang Z, Liu Z, Wang S, et al. Implantation of hydrogel-liposome nanoplatform inhibits glioblastoma relapse by inducing ferroptosis. Asian J Pharm Sci. 2023;18(3):100800. doi:10.1016/j.ajps.2023.100800
32. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–333. doi:10.1038/nprot.2007.30
33. Cai H, Dai X, Guo X, et al. Ataxia telangiectasia mutated inhibitor-loaded copper sulfide nanoparticles for low-temperature photothermal therapy of hepatocellular carcinoma. Acta Biomater. 2021;127:276–286. doi:10.1016/j.actbio.2021.03.051
34. Liu X, Xu J, Shen B, et al. USP33 promotes pancreatic cancer malignant phenotype through the regulation of TGFBR2/TGFβ signaling pathway. Cell Death Dis. 2023;14(6):362. doi:10.1038/s41419-023-05871-4
35. Qiu W, Su GH. Development of orthotopic pancreatic tumor mouse models. Methods Mol Biol. 2013;980:215–223. doi:10.1007/978-1-62703-287-2_11
36. Jiang YJ, Lee CL, Wang Q, et al. Establishment of an orthotopic pancreatic cancer mouse model: cells suspended and injected in Matrigel. World J Gastroenterol. 2014;20(28):9476–9485. doi:10.3748/wjg.v20.i28.9476
37. Thompson BR, Shi J, Zhu HJ, et al. Pharmacokinetics of gemcitabine and its amino acid ester prodrug following intravenous and oral administrations in mice. Biochem Pharmacol. 2020;180:114127. doi:10.1016/j.bcp.2020.114127
38. Deer EL, González-Hernández J, Coursen JD, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39(4):425–435. doi:10.1097/MPA.0b013e3181c15963
39. Yao H, Song W, Cao R, et al. An EGFR/HER2-targeted conjugate sensitizes gemcitabine-sensitive and resistant pancreatic cancer through different SMAD4-mediated mechanisms. Nat Commun. 2022;13(1):5506. doi:10.1038/s41467-022-33037-x
40. Mukhopadhyay S, Goswami D, Adiseshaiah PP, et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-Driven pancreatic cancers. Cancer Res. 2020;80(8):1630–1643. doi:10.1158/0008-5472.CAN-19-1363
41. Kim D, Lee S, Na K. Immune stimulating antibody-photosensitizer conjugates via Fc-mediated dendritic cell phagocytosis and phototriggered immunogenic cell death for KRAS-mutated pancreatic cancer treatment. Small. 2021;17(10):e2006650. doi:10.1002/smll.202006650
42. Palam LR, Gore J, Craven KE, et al. Integrated stress response is critical for gemcitabine resistance in pancreatic ductal adenocarcinoma. Cell Death Dis. 2015;6(10):e1913. doi:10.1038/cddis.2015.264
43. Zhang Z, Han H, Rong Y, et al. Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J of Exp Clin Cancer Res. 2018;37:291. doi:10.1186/s13046-018-0972-3
44. Zhao G, Cui J, Zhang JG, et al. SIRT1 RNAi knockdown induces apoptosis and senescence, inhibits invasion and enhances chemosensitivity in pancreatic cancer cells. Gene Ther. 2011;18(9):920–928. doi:10.1038/gt.2011.81
45. Zhao H, Wu S, Li H, et al. ROS/KRAS/AMPK signaling contributes to Gemcitabine-induced stem-like cell properties in pancreatic cancer. Mol Ther Oncolytics. 2019;14:299–312. doi:10.1016/j.omto.2019.07.005
46. Santos VS, Vieira GM, Ruckert MT, et al. Atypical phosphatase DUSP11 inhibition promotes nc886 expression and potentiates gemcitabine-mediated cell death through NF-kB modulation. Cancer Gene Ther. 2024;31(9):1402–1411. doi:10.1038/s41417-024-00804-5
47. Elia A, Henry-Grant R, Adiseshiah C, et al. Implication of 4E-BP1 protein dephosphorylation and accumulation in pancreatic cancer cell death induced by combined gemcitabine and TRAIL. Cell Death Dis. 2017;8(12):3204. doi:10.1038/s41419-017-0001-z
48. Affram KO, Smith T, Ofori E, et al. Cytotoxic effects of gemcitabine-loaded solid lipid nanoparticles in pancreatic cancer cells. J Drug Delivery Sci Technol. 2020;55:101374. doi:10.1016/j.jddst.2019.101374
49. Zhang Z, Duan Q, Zhao H, et al. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade. Cancer Lett. 2016;382(1):53–63. doi:10.1016/j.canlet.2016.08.023
50. Quint K, Tonigold M, Di Fazio P, et al. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. Int J Oncol. 2012;41(6):2093–2102. doi:10.3892/ijo.2012.1648
51. Warren NJH, Eastman A. Inhibition of checkpoint kinase 1 following gemcitabine-mediated S phase arrest results in CDC7- and CDK2-dependent replication catastrophe. J Biol Chem. 2019;294(6):1763–1778. doi:10.1016/j.jconrel.2013.03.033
52. Miao X, Koch G, Ait-Oudhia S, et al. Pharmacodynamic modeling of cell cycle effects for gemcitabine and trabectedin combinations in pancreatic cancer cells. Front Pharmacol. 2016;7:421. doi:10.3389/fphar.2016.00421
53. Ono H, Basson MD, Ito H. PTK6 potentiates gemcitabine-induced apoptosis by prolonging S-phase and enhancing DNA damage in pancreatic cancer. Mol Cancer Res. 2015;13(8):1174–1184. doi:10.1158/1541-7786.MCR-15-0034
54. Wonganan P, Lansakara-P DSP, Zhu S, et al. Just getting into cells is not enough: mechanisms underlying 4-(N)-stearoyl gemcitabine solid lipid nanoparticle’s ability to overcome gemcitabine resistance caused by RRM1 overexpression. J Control Release. 2013;169(1–2):17–27. doi:10.1016/j.jconrel.2013.03.033
55. Wang L, Cai J, Zhao X, et al. Palmitoylation prevents sustained inflammation by limiting NLRP3 inflammasome activation through chaperone-mediated autophagy. Mol Cell. 2023;83(2):281–297.e10. doi:10.1016/j.molcel.2022.12.002
56. Immordino ML, Brusa P, Rocco F, et al. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100(3):331–346. doi:10.1016/j.jconrel.2004.09.001
57. Celia C, Malara N, Terracciano R, et al. Liposomal delivery improves the growth-inhibitory and apoptotic activity of low doses of gemcitabine in multiple myeloma cancer cells. Nanomedicine. 2008;4(2):155–166. doi:10.1016/j.nano.2008.02.003
58. Abbruzzese JL, Grunewald R, Weeks EA, et al. A Phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol. 1991;9(3):491–498. doi:10.1200/JCO.1991.9.3.491
59. Storniolo AM, Allerheiligen SR, Pearce HL. Preclinical, pharmacologic, and phase I studies of gemcitabine. Semin Oncol. 1997;24(2 Suppl 7).
60. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. doi:10.1056/NEJMoa1011923
61. Hilmi M, Ederhy S, Waintraub X, et al. Cardiotoxicity associated with gemcitabine: literature review and a pharmacovigilance study. Pharmaceuticals. 2020;13(10):325. doi:10.3390/ph13100325
62. Alam S, Illo C, Ma YT, et al. Gemcitabine-induced cardiotoxicity in patients receiving adjuvant chemotherapy for pancreatic cancer: a case series. Case Rep Oncol. 2018;11(1):221–227. doi:10.1159/000488139
63. Mohebali D, Matos J, Chang JD, et al. Gemcitabine induced cardiomyopathy: a case of multiple hit cardiotoxicity. ESC Heart Fail. 2017;4(1):71–74. doi:10.1002/ehf2.12113
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.