")
Back to Journals » International Journal of Nanomedicine » Volume 19
Ischemic-Preconditioning Induced Serum Exosomal miR-133a-3p Improved Post-Myocardial Infarction Repair via Targeting LTBP1 and PPP2CA
Authors Yang N , Hou YB , Cui TH, Yu JM , He SF, Zhu HJ
Received 8 February 2024
Accepted for publication 31 July 2024
Published 4 September 2024 Volume 2024:19 Pages 9035—9053
DOI https://doi.org/10.2147/IJN.S463477
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Na Yang,1,2,* Yong-Bo Hou,2,3,* Tian-Hao Cui,2 Jun-Ma Yu,2 Shu-Fang He,4,5 Hai-Juan Zhu1
1Department of Anesthesiology, Maternal and Child Medical Center of Anhui Medical University, Hefei, Anhui, People’s Republic of China; 2Department of Anesthesiology, The Third Affiliated Hospital of Anhui Medical University (The First People’s Hospital of Hefei), Hefei, Anhui, People’s Republic of China; 3Department of Anesthesiology, Wannan Medical College, Wuhu, Anhui, People’s Republic of China; 4Department of Anesthesiology and Perioperative Medicine, The Second Affiliated Hospital of Anhui Medical University, Hefei, People’s Republic of China; 5Key Laboratory of Anesthesiology and Perioperative Medicine of Anhui Higher Education Institutes, Anhui Medical University, Hefei, Anhui, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shu-Fang He; Hai-Juan Zhu, Email [email protected]; [email protected]
Background: Ischemic preconditioning-induced serum exosomes (IPC-exo) protected rat heart against myocardial ischemia/reperfusion injury. However, whether IPC-exo regulate replacement fibrosis after myocardial infarction (MI) and the underlying mechanisms remain unclear. MicroRNAs (miRs) are important cargos of exosomes and play an essential role in cardioprotection. We aim to investigate whether IPC-exo regulate post-MI replacement fibrosis by transferring cardioprotective miRs and its action mechanism.
Methods: Exosomes obtained from serum of adult rats in control (Con-exo) and IPC groups were identified and analyzed, subsequently intracardially injected into MI rats following ligation. Their miRs profiles were identified using high-throughput miR sequencing to identify target miRs for bioinformatics analysis. Luciferase reporter assays confirmed target genes of selected miRs. IPC-exo transfected with selected miRs antagomir or NC were intracardially administered to MI rats post-ligation. Cardiac function and degree of replacement fibrosis were detected 4 weeks post-MI.
Results: IPC-exo exerted cardioprotective effects against excessive replacement fibrosis. MiR sequencing and RT-qPCR identified miR-133a-3p as most significantly different between IPC-exo and Con-exo. MiR-133a-3p directly targeted latent transforming growth factor beta binding protein 1 (LTBP1) and protein phosphatase 2, catalytic subunit, alpha isozyme (PPP2CA). KEGG analysis showed that transforming growth factor-β (TGF-β) was one of the most enriched signaling pathways with miR-133a-3p. Comparing to injection of IPC-exo transfected with miR-133a-3p antagomir NC, injecting IPC-exo transfected with miR-133a-3p antagomir abolished protective effects of IPC-exo on declining excessive replacement fibrosis and cardiac function enhancement, while increasing the messenger RNA and protein expression of LTBP1, PPP2CA, and TGF-β 1in MI rats.
Conclusion: IPC-exo inhibit excessive replacement fibrosis and improve cardiac function post-MI by transferring miR-133a-3p, the mechanism is associated with directly targeting LTBP1 and PPP2CA, and indirectly regulating TGF-β pathway in rats. Our finding provides potential therapeutic effect of IPC-induced exosomal miR-133a-3p for cardiac repair.
Keywords: ischemic preconditioning, myocardial infarction, Exosomes, MicroRNAs, cardiac function, myocardial fibrosis
Graphical Abstract:

Introduction
Myocardial infarction (MI) refers to the process of cardiomyocyte death and myocardial dysfunction caused by persistent ischemia and hypoxia in the blood flow to the heart. Cardiac dysfunction following MI is a leading cause of worldwide mortality and morbidity.1 During the early repair phase after MI, the activation and differentiation of replacement myofibroblasts, as well as excessive deposition of extracellular matrix (ECM) in the cardiac interstitium, are integral to the repairing process resulting from the loss of cardiomyocytes. Although they maintain the integrity of cardiac structure, in the pathophysiological context of persistent myocardial infarction, long-term replacement fibrosis gradually becomes decompensated fibrosis, which results in increased myocardial stiffness, systolic dysfunction, ventricular remodeling, and ultimately heart failure.2 Inhibiting long-term replacement fibrosis caused by cardiac myofibroblast activation can be a potential treatment strategy for heart failure post-MI.
Ischemic preconditioning (IPC) has been shown to possess potent cardioprotective effects. Emerging evidence confirms that the therapeutic effects of IPC on cardiac repair are mediated through their paracrine mechanism, particularly exosomes, especially in specific pathological conditions.3,4 However, the efficacy of IPC-induced cardioprotection may be compromised and diminished by postinfarct ventricular remodeling or chronic heart failure.5,6 Conversely, a recent study reveals that plasma exosomes induced by IPC can restore the protective effect against myocardial ischemia/reperfusion (I/R) injury in post-infarcted failing hearts.3 This finding strengthens the evidence supporting exosomes as crucial effectors of IPC-mediated cardioprotection. Previous investigations have demonstrated that plasma exosomes derived from remote ischemic preconditioning (RIPC) exhibit protective effects against I/R injury in cardiomyocytes.7–9Moreover, they may also offer distinct benefits in enhancing cardiac function and angiogenesis in rats following an infarction.10 More recently, it was demonstrated that serum exosomes obtained from IPC rats possess protective effects against I/R injury.11 However, the potential therapeutic effects of IPC-induced serum exosomes on post-MI cardiac repair, such as inhibiting overactive replacement fibrosis and heart failure, have yet to be investigated.
Exosomes are natural, cell-free systems that contain a diverse range of biological molecules, including proteins, lipids, and nucleic acids. The unique characteristics of exosomes protect their internal contents from degradation by cellular processes.12,13 Due to their ability to be secreted and taken up by neighboring or distant cells, exosomes have garnered significant attention as a primary medium for intercellular signal transfer mechanisms.14 Various studies have demonstrated the potential of exosomes to act as carriers for transferring genetic information, such as messenger RNA (mRNA) and microRNA (miR), for the treatment of cardiovascular diseases in animal models.9,15–17 For example, exosomes from the mesenchymal stem cells of adipose tissue have been effective in reducing cardiac fibrosis following myocardial infarction, aiding in heart repair by delivering miR-205.17 Furthermore, exosomes generated by the physical stimulus of shock wave therapy have shown a decrease in fibrosis during the treatment of ischemic myocardium, primarily through the transportation of miR-19a-3p.18
MiRs are short, non-encoding RNAs, generally 20–24 nucleotides long, playing pivotal roles in regulating cellular functions by blocking the translation and facilitating the breakdown of specific mRNAs.19 Previous research has highlighted miRs’ involvement in the cardioprotective effects derived from morphine and hypoxic preconditioning.20,21 The significance of exosomal miRs in heart repair is well-documented.8,9,16 Yet, the capacity of miRs within serum exosomes from IPC rats to mediate excessive replacement fibrosis during cardiac repair following MI remains to be fully understood. This study focuses on investigating the effectiveness of IPC-induced serum exosomes in inhibiting long-term replacement fibrosis during cardiac repair and uncovering the miR-mediated molecular mechanisms in rats post-MI.
Materials and Methods
Animals
Adult male Sprague-Dawley (SD) rats (250–280 g) were sourced from the Experimental Animal Center of Anhui Medical University (China). These rats had free access to standard rodent food and water, housed in a room where the temperature was kept at a steady 22–24°C. All research activities adhered to the principles of the Care and Use of Laboratory Animals and received approval from the Institutional Animal Care and Use Committee at Anhui Medical University (approval number: LLSC20210983). For the experimental procedures, the rats were sedated with a 3% pentobarbital solution, administered intraperitoneally at a dose of 50 mg/kg.
Animal Model of IPC
Following the administration of anesthesia, IPC was carried out by occluding the left anterior descending coronary artery (LAD) approximately 3 mm below the left atrium using a 6–0 nylon thread. This procedure involved three cycles of 5 minutes of ischemia alternated with 5 minutes of reperfusion. The induction of myocardial ischemia was verified by the pallor of the myocardial tissue beneath the suture and towards the heart’s apex, coupled with S-T segment elevation observed in electrocardiograms, which were recorded using the BL-420S biological signal acquisition and processing system (Taimeng, China). After this procedure, blood samples were drawn from the left ventricle using a sterile intravenous needle attached to a coagulation tube. Rats in the control group did not receive any processing, blood was taken in the same manner but without the ischemic preconditioning step.
Isolation of Exosomes and Free-Exosomes Serum
Exosome isolation was performed through a sequence of differential centrifugation steps at 4°C as follows: Initially, whole blood collected in coagulation tubes underwent a centrifugation at 3000×g for 15 min to separate serum. This serum was then centrifuged again at 12000×g for 20 min to eliminate cellular debris. Afterward, the serum was passed through a 0.22 μM pore size filter (Millex®-GP filter, Millipore, catalog number: SLGP033RB). This filtration was followed by two rounds of ultracentrifugation at 110000×g for 70 min each, using a Type 100 Ti rotor (Beckman, Germany), to precipitate the exosomes. The sediment after this process was identified as isolated exosomes, and the supernatant from the initial ultracentrifugation step was labeled as serum devoid of exosomes. The exosomes derived from 3 mL of serum were resuspended in 200 µL of Phosphate-buffered saline (PBS) and then frozen at −80°C for subsequent analysis. Similarly, the serum without exosomes was preserved at −80°C for additional studies.
Identification of Exosomes
The morphology and size of the exosomes were examined using a transmission electron microscope (TEM, FEI, Tecnai G2 Spirit BioTwin) and Nanoparticle Tracking Analysis (NanoFCM, Nottingham, UK). Moreover, the presence of specific biomarker proteins CD9, CD63, and HSP70, which are indicative of exosomes, was confirmed through Western blot analysis. The protein concentrations of the exosomes were determined using the BCA protein assay kit (BCA protein assay kit, Beyotime, catalog number: P0009).
Exosome Labeling and Internalization
To assess the uptake of exosomes in both in vivo and in vitro settings, fluorescent dye PKH26 was used to label the exosomes. The labeling procedure was performed utilizing the Red Fluorescent Cell Linker Kit (PKH26 Red Fluorescent Cell Linker Mini Kit, Sigma, catalog number: MINI26). Briefly, 100 μL of exosomes were combined with 200 µL of Diluent C in a clean centrifuge tube. In a separate centrifuge tube, 200 µL of Diluent C and 4 µL of PKH26 were mixed. The contents of the latter tube were promptly added to the former tube and thoroughly mixed for 1 min. The resulting mixture was incubated at room temperature for 10 min.
To halt the reaction, an equal volume of exosome-free serum was added, and the resulting mixture was centrifuged at 110000×g for 70 min to eliminate any residual unbound dye. In the in vitro experiment, H9c2 cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China), pre-labeled exosomes were introduced into the H9c2 cell culture medium and incubated for 1, 6, and 24 h, respectively. Subsequently, the H9c2 cells were thoroughly washed five times with PBS, fixed with 4% paraformaldehyde, and stained with 4’,6-diamino-2-phenylindoles (DAPI) (Antifade Mounting Medium with DAPI, Beyotime, catalog number: P0131) under dark conditions. The samples were then observed using confocal microscopy (Leica, Germany). For the in vivo experiment, pre-labeled exosomes were intramyocardially injected, and after 24 h, the heart was processed in a similar manner. The co-localization of exosomes and cardiomyocyte nuclei was examined using a fluorescence microscope.
MI Model Establishment
Prior to the surgical procedure, male SD rats were anesthetized with an intraperitoneal injection of 3% pentobarbital sodium (50 mg/kg) and mechanically ventilated. The LAD was ligated 3 mm below the left atrium using a 6–0 nylon suture. Immediately after the ligation, exosomes (50 µg in 25 µL PBS), free-exosome serum (50 µg in 25 µL PBS), or 25 µL PBS were administered intracardially at three sites surrounding the infarct area. Rats in the sham group underwent identical procedures without ligation, and 25 µL PBS was injected instead. Serum samples were harvested from the tail vein of the rats 7 and 28 days after MI to measure relevant cytokines. Transthoracic two-dimensional M-mode echocardiography at the papillary muscle level was implemented under isoflurane anesthesia to assess cardiac function 28 days after MI.8 Subsequently, the rats were euthanized for subsequent experiments.
Enzyme-Linked Immunosorbent Assay (ELISA)
Serum was obtained from blood samples collected from the tail veins of the rats. The levels of interleukin (IL)-1β (Rat IL-1β ELISA KIT, catalog number: CRE0006-096), tumor necrosis factor (TNF)-α (Rat TNF-α ELISA KIT, catalog number: CRE0003-048), and IL-6 (Rat IL-6 ELISA KIT, catalog number: CRE0005-096) were measured in each serum sample using ELISA kits (4A Biotech, Beijing). An ELISA kit (Rat N-Terminal Pro-Brain Natriuretic Peptide (NT-proBNP) ELISA Kit, Elabscience, catalog number: E-EL-R3023) was employed to assess the levels of NT-proBNP in the serum samples.
Histopathological Analysis
Following the sacrifice of rats, the heart tissues were collected for further analysis. The tissues were then fixed in a solution of 4% paraformaldehyde for one day. Following fixation, then dehydrated with gradient alcohol, and thin sections measuring 5 µm were obtained from each segment through paraffin embedding. Subsequently, these sections were subjected to staining using the Masson trichrome (Masson) and hematoxylin-eosin (HE) stains. This allowed for the evaluation of fibrosis and collagen area, as well as the analysis of tissue morphology. The stained sections were captured and observed using a fluorescence microscope (Zeiss, Germany). The fibrosis extent was measured by ImageJ software, and the ratio of the fibrosis area to the total cardiac tissue region is defined as the percentage of the fibrosis area.
RNA Isolation and Real-Time Reverse Transcription-PCR (RT-qPCR)
RNA from both heart tissue and exosomes were isolated utilizing Trizol reagent (Invitrogen, catalog number: 15596018CN). This RNA was subsequently converted into cDNA templates via reverse transcription using the miR First Strand cDNA Synthesis (Tailing Reaction) kit (catalog number: B532451) from Sangon Biotech, Shanghai, with 1 µg of RNA as the starting material. The synthesized cDNA served as the template for the following RT-qPCR assays, conducted using 2X SG Fast qPCR Master Mix (catalog number: B639271) provided by BBI Life Science, Shanghai. Primers specific to each gene studied are detailed in Supplementary Table 1. Gene expression was quantified utilizing the 2−ΔΔCt method, employing GAPDH and U6 as the normalization controls for mRNA and miR analyses, respectively.
Western Blot Analysis
Proteins were extracted from both exosomes and fresh heart tissue using a lysis buffer (RIPA Lysis Buffer, Beyotime, catalog number: P0013B) provided by Beyotime, China, and their concentrations were assessed using the BCA protein assay kit from the same company. Then, 20–40 µg of these proteins were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (SDS-PAGE Gel Quick Preparation Kit, Beyotime, catalog number: P0012AC) for separation. The separated proteins were then transferred onto polyvinylidene difluoride (PVDF) membranes (Immobilon-PSQ Transfer Membrane, Millipore, catalog number: ISEQ00010).
Blocking of the membranes was performed using Protein Free Rapid Blocking Buffer (Yamei, catalog number: PS108P) for 30 min, followed by an overnight incubation at 4°C with primary antibodies. Subsequent to three TBST washes, membranes were exposed to a secondary antibody (1:10,000) (Goat Anti-Rabbit IgG H&L, Abcam, catalog number: ab205718) for 1 h at ambient temperature. Detection of protein bands was achieved with electrochemiluminescence (ECL) reagent (SuperSignalTM West Femto Maximum sensitive substrate, ThermoFisher, catalog number: 34096), and their integral optical density was quantified utilizing Image-Pro Plus 6.0 software. The primary antibodies applied in the Western blot analysis are listed in Supplementary Table 2.
High-Throughput miR Sequencing for Exosomes
Total RNA was extracted from serum exosomes of rats subjected to IPC or control conditions using TRIzol. Subsequently, the extracted RNA was used to prepare and sequence a miR library utilizing an Illumina HiSeq 2500 platform (Ribobio, China). Bioinformatics analysis, including cluster analysis, was conducted to investigate the differential expression of miRs between IPC-exo and Con-exo. The miRs were identified based on a p-value < 0.05, with established thresholds for up- and downregulated genes. The miR sequencing data can be accessed in the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/) through the accession number GSE272122. To predict target genes for rno-miR-133a-3p, TargetScan (http://www.targetscan.org/), miRDB (http://www.mirdb.org/), and miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html) databases were utilized. Genes identified by all three databases were selected for further experimentation.
Dual-Luciferase Reporter Assay
Luciferase reporter vectors (LTBP1-wt/LTBP1-mut and PPP2CA-wt/PPP2CA-mut) were constructed by Asia Biotechnology (Shanghai, China). HEK293T cells were then seeded in a 24-well plate at a density of 5×104 cells/well and incubated for 24 h. Following that, the cells were transfected with various vectors along with either miR-133a-3p mimics or negative control (NC) using Lipofectamine 2000 Reagent (Invitrogen, catalog number: 11668500) when they reached 70% confluency. After a 72-hour transfection, the cells were harvested and lysed. The miR level was assessed using chemiluminescence with the Dual-Luciferase Reporter Assay System (Promega) upon introducing detection reagents into the medium.
Exo-FectTM Loading of IPC-Exo
IPC-induced serum exosomes were loaded with miR-133a-3p antagomir or antagomir NC (Sangon Biotech, Shanghai) using Exo-FectTM (Exo-FectTM Exosome Transfection Kit, System Biosciences, catalog number: EXFT20A-1).22
To prepare the samples, 10 µL of Exo-effect Solution was combined with 20 pmol of either miR-133a-3p antagomir or NC, along with 50 µL of purified exosomes and 70µL of sterile PBS. This mixture was incubated at 37°C for 10 min before the centrifuge tube was placed on ice. Next, 30 µL of the ExoQuick-TC reagent provided in the kit was added, and the sample was gently mixed by inverting it 6 times. The transfected exosome sample was then kept on ice for 30 min and centrifuged at 13,000 rpm for 3 min at 4°C. The transfected exosomes were suspended in 30 µL of PBS for subsequent in vivo experiments. For in vivo studies, the final samples were composed of 20 pmol of miR-133a-3p antagomir or NC per 50µg of exosomes in a total volume of 30 µL.
Statistical Analysis
The data are expressed as the mean ± standard error of the mean (SEM) and percentages. GraphPad 8.0 (GraphPad Software, USA) was adopted for data analysis. Unpaired two-tailed t-tests were performed to compare between two groups. For comparisons involving three or more groups, one-way ANOVA followed by a Bonferroni’s post-test was applied. A p < 0.05 was deemed statistically significant for all comparisons.
Results
Characterization of Serum Exosomes and Exosomal miRs Analysis
The IPC rat model was established following the IPC protocol, as evidenced by S-T segment elevation and pathological Q waves observed in electrocardiograms (Supplementary Figure S1A), and subsequently, exosomes were isolated from both IPC and control rats (Figure 1A). The characterization of serum exosomes from control rats (Con-exo) and IPC rats (IPC-exo) was performed using TEM and nanoparticle tracking analysis. TEM (Figure 1B) revealed cup-shaped double-layered particles with diameters ranging from 30 to 150 nm, obtained through ultracentrifugation. Nanoparticle tracking analysis (Figure 1C) confirmed a single peak in the volume distribution profile for both Con-exo and IPC-exo, with diameters ranging from 30 to 150 nm. No significant differences were observed in particle distribution between Con-exo and IPC-exo (Figure 1D). Interestingly, the protein quantification assay showed that IPC induced an increase in the protein content of serum exosomes (Figure 1E). Western blotting analysis demonstrated the expression of exosome-specific protein markers HSP70, CD63, and CD9 in these particles (Figure 1F). IPC-exo exhibited higher expression of these protein markers compared to Con-exo (Figure 1G). Overall, these findings confirm that IPC leads to increased production of serum exosomes, consistent with previous reports.3
Figure 1 Continued. Figure 1 Characterization of serum exosomes and miR-133a-3p was upregulated in IPC exosomes. (A) The process flow of how two types of serum exosomes are obtained. (B) Representative electron micrograph of exosomes. Scale bar: 100 nm. (C) A nanoparticle trafficking system (NTA) was used to analyze the sizes of the IPC-exo or Con-exo. The results indicated particle sizes of both IPC-exo and Con-exo ranged between 30 and 150nm. (D) Mean size of IPC-exo and Con-exo (n = 3). (E) The protein concentration of exosomes was measured by BCA protein assay (n = 6). (F) Representative images of exosomes-associated marker proteins HSP70, CD63, and CD9 in IPC-exo and Con-exo. (G) The data of exosomal marker protein expression level in IPC-exo and Con-exo (n = 4). (H) Heat map based on high-throughput miR sequencing representing the 40 identified miRs with the most significant abundance differences between control exosomes and IPC exosomes (p < 0.05, n=3). (I) Volcano plot showing log2 (fold change) on x-axis and -log10 (p value) on y-axis. (J) Specific fold changes of 40 identified miRs with the most significant abundance differences (p < 0.05) between control exosomes and IPC exosomes (n=3). (K) MiR-133a-3p expression level in control and IPC exosomes determined by RT-qPCR (n=6). Abbreviations: IPC, ischemic preconditioning; IPC-exo, exosomes isolated from IPC rat serum; Con-exo, exosomes isolated from control rat serum; miR, microRNA. Notes: *p<0.05, **p<0.01, ****p<0.0001 and nsp>0.05. Data are presented as the mean ± SEM.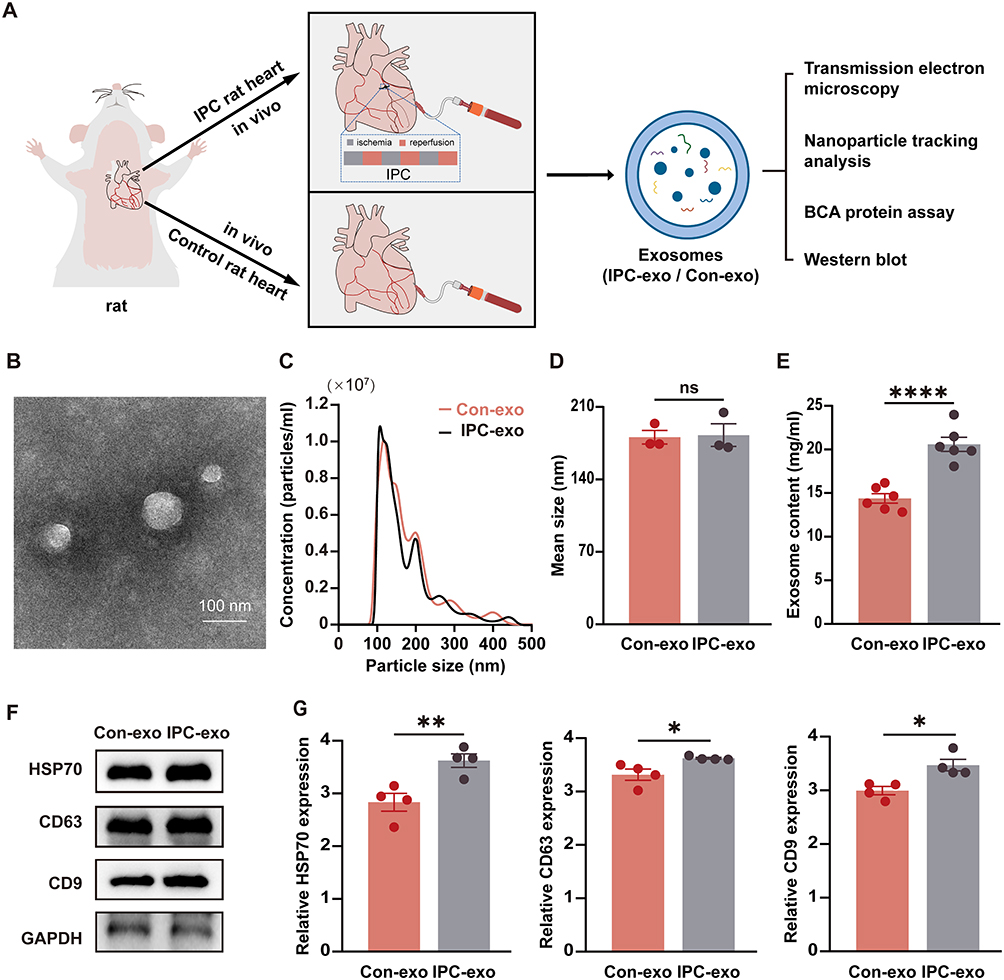

To evaluate the impact of IPC on the expression of serum exosomal miRs, we conducted miR sequencing (miR-seq) to examine the miR profiles of both Con-exo and IPC-exo. Figure 1H demonstrates the supervised clustering of the 40 identified miRs with the most significant differences (P < 0.05) in abundance between IPC-exo and Con-exo. These 40 miRs consist of 23 upregulated and 17 downregulated miRs, as shown in Figure 1I. Additionally, Figure 1J presents the fold changes of these 40 miRs, with miR-133a-3p exhibiting a fold change > 2. According to the results of miR-seq, we selected five miRs (miR-206-3p, miR-18a-5p, miR-133a-5p, miR-133a-3p, miR-1224) with significant differential expression based on their fold change and p value. Among these five miRs, further RT-qPCR validation revealed that miR-133a-3p exhibited the most significant difference in expression between IPC-exo and Con-exo (Figure 1K and supplementary Figure S1B).
Internalization of Serum Exosomes
To investigate the uptake of serum exosomes by myocardial cells, we performed intramyocardial injection of a fluorescent dye PKH26-labelled Con-exo and IPC-exo. After 24 h, we observed colocalization of PKH26-labelled Con-exo and IPC-exo with cardiomyocytes, indicating an efficient absorption of exosomes by heart tissue (Supplementary Figure S2A). Furthermore, we added PKH26-labelled Con-exo and IPC-exo to the culture medium of H9c2 cells for 1 h, 6 h, and 24 h. Through confocal microscopy analysis, we observed the presence of small PKH26-labelled Con-exo and IPC-exo localized around the nucleus within 1 h. The H9c2 cells displayed effective uptake of Con-exo and IPC-exo after 6 h, with a substantial amount of exosomes being absorbed within 24 h (Supplementary Figure S2B). These findings suggested that serum exosomes could be internalized by heart tissue in vivo and H9c2 cells in vitro in a time-dependent manner.
IPC-Exo Demonstrated Cardioprotective Effects Against MI in vivo
To investigate the cardioprotective effects of IPC-exo in vivo, we established a MI rat model with ST segment elevation observed in electrocardiograms (Supplementary Figure S3A). Subsequently, we intracardially injected 50 µg of Con-exo, IPC-exo, or free- exosomes serum from control (Con-s) or IPC rats (IPC-s) resuspended in 25 µL of PBS, as well as 25 µL of PBS alone after the ligation of the LAD (Figure 2A). After 4 weeks, we assessed cardiac function using echocardiography. The results revealed a significant decrease in left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS) (Figure 2B-C), and increase in left ventricular internal diameter at end systole (LVIDs) and left ventricular internal diameter at end diastole (LVIDd) (Supplementary Figure 3B) in the MI group relative to the sham group. Treatment with IPC-exo enhanced cardiac function compared to MI rats treated with PBS (LVEF: 65.93±2.05% versus 40.08±1.52%, p < 0.0001; LVFS: 36.94±1.72% versus 20.36±0.83%, p < 0.0001) or Con-exo (LVEF: 65.93±2.05% versus 46.00±1.63%, p < 0.0001; LVFS: 36.94±1.72% versus 22.67±1.24%, p < 0.0001) (Figure 2B-C). Furthermore, in accordance with these findings, rats treated with IPC-exo exhibited an amelioration in the severity of heart failure, demonstrated by a decrease in NT-proBNP levels in the serum after 28 days of MI (Figure 2D). ELISA was also utilized to quantify the levels of serum IL-6, IL-1β, and TNF-α at both 7 and 28 days post-MI. The results uncovered that IPC-exo reduced the levels of pro-inflammatory indicators on the 7th day post-surgery, and subsequently inhibited the prolonged inflammatory response (Supplementary Figure S3C). Moreover, the results obtained from HE staining revealed a disordered arrangement of cardiomyocytes and an enlarged gap between cells in the MI group. In contrast, treatment with IPC-exo showed an improvement in the morphological alterations observed in MI rats (Figure 2E).
Figure 2 Continued. Figure 2 IPC-induced serum exosomes protect against myocardial infarction in vivo. Exosomes, free-exosome serum, or PBS were injected into the ischemia heart tissue after the myocardial infarction model was established. (A) Schematic diagram and flowchart illustrating the experimental protocols in vivo. (B) Representative echocardiographic images of rats in each group exposed to myocardial infarction for 28 days. (C) Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) of rats in each group (n=6). (D) ELISA assay of the N-Terminal Pro-Brain Natriuretic Peptide (NT-proBNP) in rat serum 28 days post-MI (n = 6). (E) The representative images of histology (HE staining) of transverse heart sections and local magnified images of the selected infarct border zone tissue from different groups rats. Red scale bar: 1 mm; black scale bar: 50 µm. (F) Representative images of Masson trichrome (Masson staining) of transverse heart sections and local magnified images of the selected infarct border zone tissue from different groups rats. Red scale bar: 1 mm; black scale bar: 50 µm. (G) The data of fibrosis area in rat transverse heart sections exposed to different treatments (n=5). (H) Representative images of fibrosis-related and apoptosis-related proteins in the infarct border zone of heart tissue in response to different treatments. Relative proteins α-SMA (I), CTGF (J), Bax/Bcl-2 (K) expression measured by Western blot analysis in the infarct border zone of heart tissue in response to different treatments (n=4). Abbreviations: IPC, ischemic preconditioning; IPC-exo, exosomes isolated from IPC rat serum; Con-exo, exosomes isolated from control rat serum; MI, myocardial infarction; Con-s, free-exosomes serum from control rats; IPC-s, free-exosomes serum from IPC rats; PBS, phosphate buffered solution; α-SMA, alpha-smooth muscle actin; CTGF, connective tissue growth factor; Bax, BCL2-associated X; Bcl-2, B-cell lymphoma-2. Notes: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Data are presented as the mean ± SEM and percentages.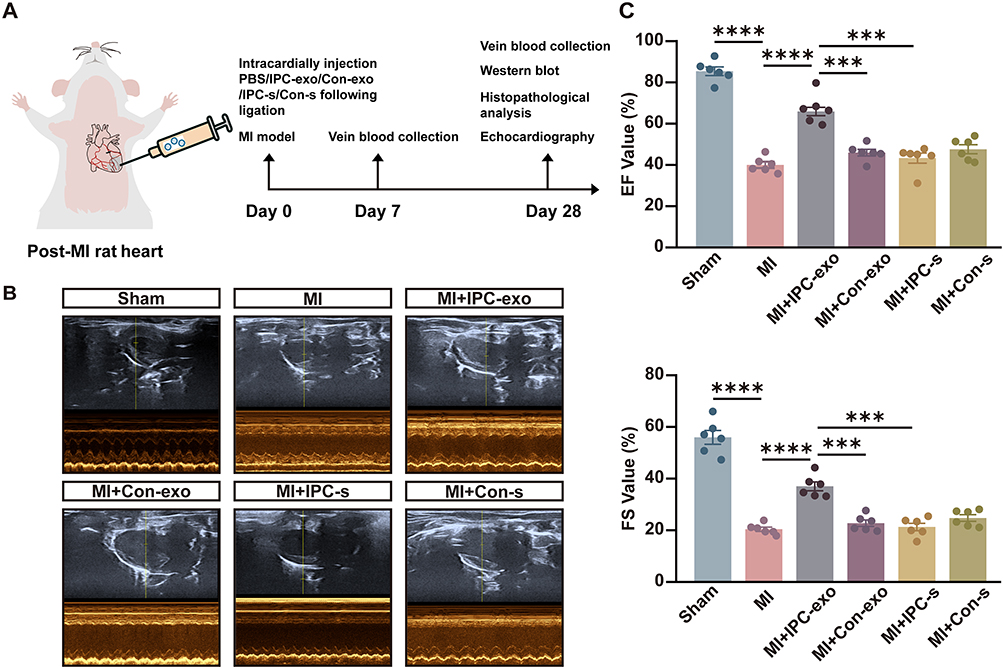

We further assessed myocardial replacement fibrosis in these groups using Masson’s staining. As shown in Figure 2F-G, rats in the MI group demonstrated significant interstitial collagen deposition. Importantly, IPC-exo therapy led to a suppression in collagen density (MI+IPC-exo versus MI: 7.74±0.71% versus 17.31±1.87%, p < 0.05; MI+IPC-exo versus MI+Con-exo: 7.74±0.71% versus 13.84±1.23%, p < 0.05). Additionally, the expression levels of alpha-smooth muscle actin (α-SMA) and connective tissue growth factor (CTGF) proteins in the infarct border zone consistently decreased in MI rats treated with IPC-exo. Conversely, neither Con-exo nor free-exosome serum treatment resulted in a reduction in fibrosis proteins α-SMA and CTGF in MI rats (Figure 2H-J).
Next, we investigated the effect of exosomes on apoptosis in the infarct border zone of the MI rat heart. We evaluated the expression of the apoptosis-associated proteins B-cell lymphoma-2 (Bcl-2) and BCL2-associated X (Bax) through Western blot analysis 28 days post-surgery. The Bax/Bcl-2 ratio was significantly declined in MI rats treated with IPC-exo (Figure 2H and K). In conclusion, these findings provide evidence of the therapeutic potential of serum exosomes isolated from IPC rats for the treatment of cardiac repair following MI. This potential is achieved through suppression of persistent inflammatory stimuli and apoptosis, reduction of replacement fibrosis and cardiac remodeling, and the improvement of cardiac function.
MiR-133a-3p Was Involved in the Myocardial Protection of IPC-Exo
In our in vivo experiments, we observed a noteworthy increase in the expression of miR-133a-3p in the border infarcted myocardial tissue following the administration of IPC-exo post-MI, as depicted in Figure 3A.
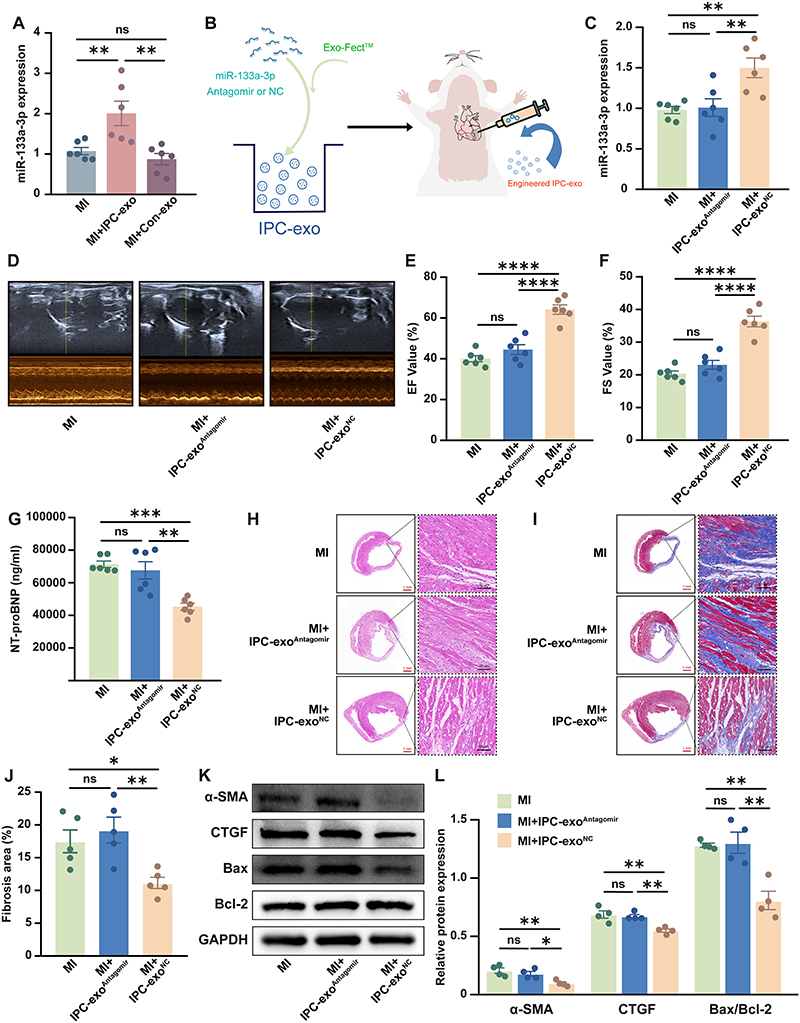 |
Figure 3 MiR-133a-3p derived from IPC-exo mediated cardioprotection. (A) MiR-133a-3p expression level in the infarct border zone of rat heart tissues treated with different methods was detected by RT-qPCR (n=6). (B) Schematic diagram illustrating that miR-133a-3p antagomir or negative control (NC) were transfected into IPC exosomes. Engineered exosomes and PBS were injected into the ischemia myocardium infarct border zone of rat heart tissues after the myocardial infarction model was established. (C) MiR-133a-3p antagomir or NC transfected into IPC exosomes alerted the miR-133a-3p expression level in heart tissues (n=6). (D) Representative echocardiographic images of rats in each group exposed to myocardial infarction for 28 days. (E) The improvement of left ventricular ejection fraction (LVEF) by IPC exosomes was blunted when miR-133a-3p antagomir was transfected into IPC-exo (n=6). (F) Left ventricular fractional shortening (LVFS) of rats was decreased in the MI+IPC-exoAntagomir group compared with the MI+IPC-exoNC group (n=6). (G) ELISA assay of the N-Terminal Pro-Brain Natriuretic Peptide (NT-proBNP) in rat serum 28 days post-MI (n=6). (H) The representative images of histology (HE staining) and (I) Masson trichrome (Masson staining) of transverse heart sections and local magnified images of the selected infarct border zone tissue from different groups rats. Red scale bar: 1 mm; black scale bar: 50 µm. (J) The data of fibrosis area in rat transverse heart sections exposed to different treatments (n=5). (K) Representative images of fibrosis-related and apoptosis-related proteins in the infarct border zone of heart tissue in response to different treatments. (L) Relative α-SMA, CTGF, Bax/Bcl-2 expression measured by Western blot analysis in the infarct border zone of heart tissue in response to different treatments (n=4). Abbreviations: IPC, ischemic preconditioning; IPC-exo, exosomes isolated from IPC rat serum; Con-exo, exosomes isolated from control rat serum; MI, myocardial infarction; MI+IPC-exoAntagomir, MI rat injected with miR-133a-3p antagomir transfected IPC-exo; MI+IPC-exoNC, MI rat injected with miR-133a-3p antagomir NC transfected IPC-exo; miR, microRNA; PBS, phosphate buffered solution; α-SMA, alpha-smooth muscle actin; CTGF, connective tissue growth factor; Bax, BCL2-Associated X; Bcl-2, B-cell lymphoma-2. Notes: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001and nsp>0.05. Data are presented as the mean ± SEM and percentages. |
To further confirm the involvement of miR-133a-3p in mediating the cardioprotective effects of IPC-exo and to gain a deeper understanding of the role of exosomal miR-133a-3p in vivo, we transfected miR-133a-3p antagomir or an NC into IPC-exo. Subsequently, we performed intracardiac injection of IPC-exo transfected with miR-133a-3p antagomir or NC in rats post-MI (Figure 3B). The RT-qPCR assay demonstrated that treatment with IPC-exo transfected with miR-133a-3p antagomir inhibited the elevation of miR-133a-3p expression in the infarct border zone of the rats’ hearts, while IPC-exo transfected with NC restored the increased expression of miR-133a-3p (Figure 3C). Furthermore, our findings demonstrated that the introduction of miR-133a-3p antagomir into IPC-exo significantly attenuated the protective effects of IPC-exo. As depicted in Figure 3D-F, the improvement in cardiac function facilitated by IPC-exo was nullified by the inhibition of miR-133a-3p in IPC-exo (MI+IPC-exoNC versus MI+IPC-exoAntagomir: LVEF, 64.21±2.30% versus 44.65±2.40%, p < 0.0001; LVFS, 36.26±1.64% versus 23.08±1.36%, p < 0.0001). LVIDs and LVIDd of three groups were presented in Supplementary Figure S4A. Coincidentally, the reduced serum NT-proBNP index, indicative of heart failure, observed in MI rats treated with IPC-exo, was significantly blunted by the transfection of miR-133a-3p antagomir into IPC-exo (Figure 3G). In comparison to the IPC-exo transfected NC group, elevated levels of pro-inflammatory indicators were observed in the rat serum 7 days post-surgery as a result of miR-133a-3p depletion in IPC-exo (Supplementary Figure S4B). The repair of damaged heart tissue and cardiomyocytes in rats treated with IPC-exo was diminished by the administration of miR-133a-3p antagomir (Figure 3H). Additionally, the administration of miR-133a-3p antagomir resulted in the elimination of reduced fibrosis area observed in MI rats treated with IPC-exo (MI+IPC-exoNC versus MI+IPC-exoAntagomir: 11.16±0.85% versus 19.23±1.97%, p < 0.01) (Figure 3I and J). Similarly, consistent alterations in the expression of both α-SMA and CTGF proteins were observed (Figure 3K and L). Finally, IPC-exo treatment led to a reduction in myocardial apoptosis, which was reversed by the addition of miR-133a-3p antagomir (Figure 3K and L). The collective findings from our in vivo results unfold that the depletion of miR-133a-3p diminishes the cardioprotective effects of IPC-exo following MI. MiR-133a-3p plays an essential role in the cardioprotective effects conferred by IPC-exo.
The Exploration of Potential Signaling Pathways and Target Genes Involved in Cardioprotection of Exosomal miR-133a-3p
To further elucidate the underlying mechanism responsible for the cardioprotective effect induced by IPC-exo, we conducted target gene prediction and analysis of the KEGG for the upregulated miR-133a-3p. Figure 4A showed significant enrichment of signaling pathways as analyzed by KEGG analysis (P<0.001). Among the numerous enriched signaling pathways, the transforming growth factor-β (TGF-β) signaling pathway stood out as one of the most prominent. Through the utilization of three bioinformatics platforms for concurrent prediction, we identified latent transforming growth factor beta binding protein 1 (LTBP1) and protein phosphatase 2, catalytic subunit, alpha isozyme (PPP2CA) as highly enriched genes associated with the TGF-β signaling pathway (Figure 4B and C).
Figure 4 Continued. Figure 4 The potential signaling pathway and target genes involved in cardioprotection of exosomal miR-133a-3p. (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses showed the top 30 significantly enriched signaling pathways of miR-133a-3p. (B) The relationship between predicted TGF-β signaling pathway with different identified miRs. (C) Common genes involved in the TGF-β signaling pathway and targets of miR-133a-3p. (D) The predicted binding sites and sequences of miR-133a-3p and the 3’UTR of the LTBP1 and PPP2CA genes according to the Targetscan database. (E) Relative luciferase activity in HEK293T cells transfected with luciferase reporter vectors carrying wild-type or mutant binding site of miR-133a-3p in 3ʹUTR of LTBP1 and miR-133a-3p mimic and negative control (n=3). (F) Relative luciferase activity in HEK293T cells transfected with luciferase reporter vectors carrying wild-type or mutant binding site of miR-133a-3p in 3ʹUTR of PPP2CA and miR-133a-3p mimic and negative control (n=3). Abbreviations: miR, microRNA; LTBP1, latent transforming growth factor beta binding protein 1; PPP2CA, protein phosphatase 2, catalytic subunit, alpha isozyme; TGF-β, transforming growth factor-β. Notes: **p<0.01 and nsp>0.05. Data are presented as the mean ± SEM.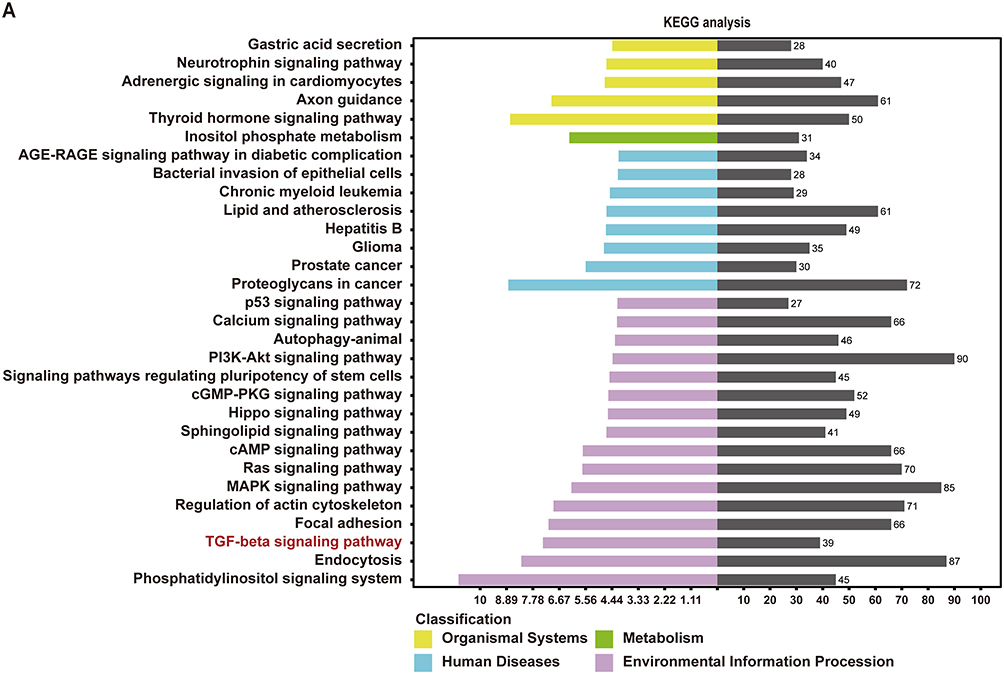

According to the prediction results from TargetScan, both LTBP1 and PPP2CA were found to have binding sites within the 3’UTR region of miR-133a-3p (Figure 4D). To confirm the interaction between LTBP1 and PPP2CA with miR-133a-3p, luciferase reporter assays were conducted. Luciferase vectors were generated with either the wild-type or mutant 3’-UTR sequences of LTBP1 and PPP2CA, as demonstrated in Figure 4E and F. Cells transfected with miR-133a-3p mimic and LTBP1 3’UTR-wt vectors or PPP2CA 3’UTR-wt vectors exhibited a significant decrease in luciferase activity. Conversely, the relative luciferase activity of cells transfected with the mutant vectors was not affected by the miR-133a-3p mimic, indicating the specificity of the repression. These results indicate that LTBP1 and PPP2CA are validated as the target mRNA hosts of miR-133a-3p.
Exosomal miR-133a-3p Promoted Post-MI Repair by Targeting LTBP1 and PPP2CA, and Suppressing TGF-β Signaling
In our in vivo experiments, we observed a reduction in LTBP1 mRNA and protein expression in the infarct border zone of myocardial tissue treated with IPC-exo following infarction (Figure 5A-C). Conversely, this treatment resulted in an accumulation of miR-133a-3p in the heart tissue, as previously mentioned (Figure 3A). Similar changes were observed in PPP2CA, TGF-β1, and TGF-β2, with decreased levels of mRNA and protein (Figure 5A-C). Furthermore, comparing to rats in MI group, rats in MI+IPC-exoNC group had significant decreased mRNA and protein levels of LTBP1, PPP2CA, and TGF-β1 and also TGF-β2 mRNA; however, comparing with MI+IPC-exoNC group, rats in the MI+IPC-exoAntagomir group had significant increased mRNA and protein levels of LTBP1, PPP2CA, and TGF-β1, as well as TGF-β2 mRNA. Among three groups, TGF-β2 protein expression level did not reach statistical significance (Figure 5D-F). The inconsistency observed in TGF-β2 mRNA and protein expression in the MI+IPC-exoNC group may be attributed to various factors influencing post-transcriptional and translational modifications.
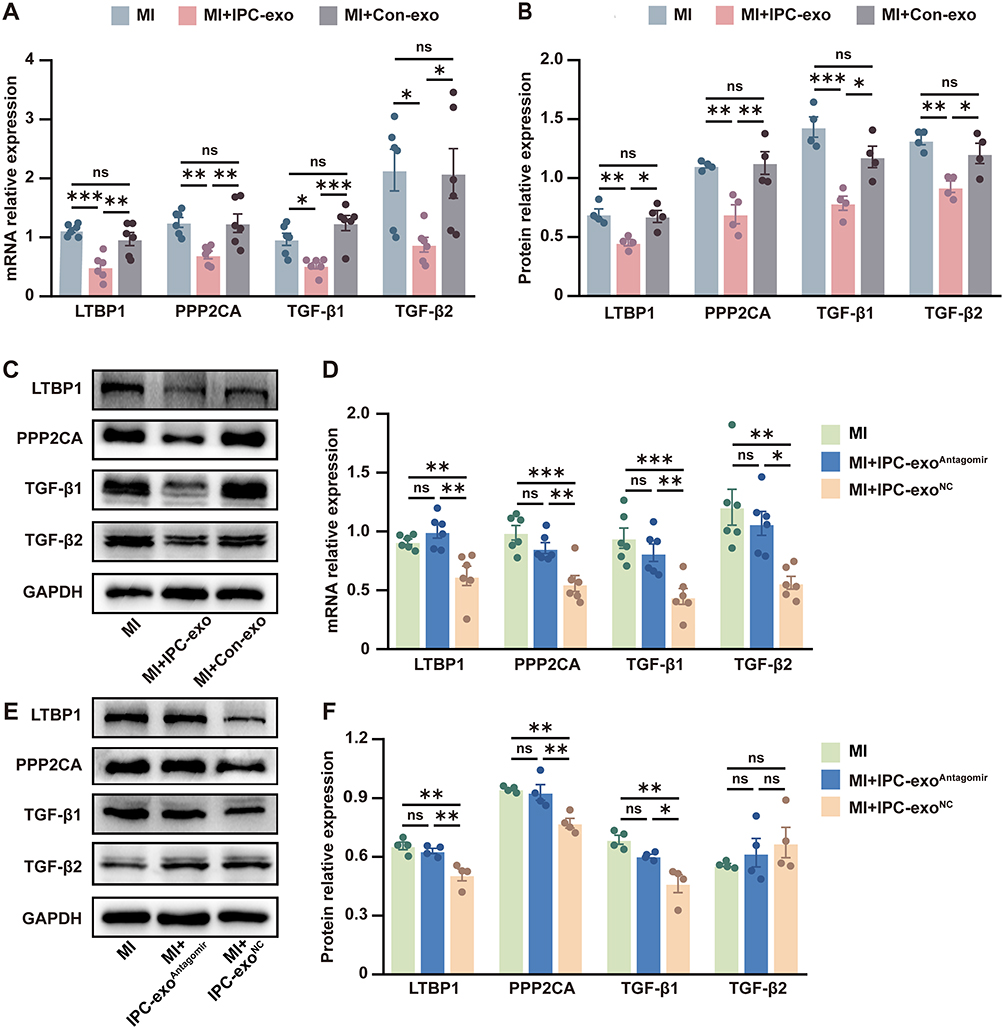 |
Figure 5 Exosomal miR-133a-3p promoted post-MI repair which is associated with directly targeting LTBP1 and PPP2CA, and indirectly suppressing TGF-β signaling pathway. (A) mRNA expression of LTBP1, PPP2CA, TGF-β1 and TGF-β2 in the infarct border zone of heart tissues treated with different exosomes and PBS (n=6). (B) Protein expression levels of LTBP1, PPP2CA, TGF-β1 and TGF-β2 in the infarct border zone of heart tissues treated with different exosomes and PBS (n=4). (C) The representative images of the Western blot in the infarct border zone of heart tissues treated with different exosomes and PBS. (D) Regulation of LTBP1, PPP2CA, TGF-β1 and TGF-β2 mRNA expression levels by transfecting miR-133a-3p antagomir or negative control into IPC-exo (n=6). (E) Representative images of the Western blot in heart tissues injected with engineered exosomes or PBS. (F) Protein expression of LTBP1, PPP2CA, and TGF-β1 in heart tissue were regulated by exosomal miR-133a-3p (n = 4). Abbreviations: IPC, ischemic preconditioning; IPC-exo, exosomes isolated from IPC rat serum; Con-exo, exosomes isolated from control rat serum; MI, myocardial infarction; MI+IPC-exoAntagomir, MI rat injected with miR-133a-3p antagomir transfected IPC-exo; MI+IPC-exoNC, MI rat injected with miR-133a-3p antagomir NC transfected IPC-exo; PBS, phosphate buffered solution; mRNA, message RNA; LTBP1, latent transforming growth factor beta binding protein 1; PPP2CA, protein phosphatase 2, catalytic subunit, alpha isozyme; TGF-β, transforming growth factor-β. Notes: *p<0.05, **p<0.01, ***p<0.001 and nsp>0.05. Data are presented as the mean ± SEM. |
Discussion
Our data demonstrated that IPC-exo suppressed excessive replacement fibrosis to promote cardiac repair post-MI. Cardioprotective effects of IPC-exo were mediated by transferring miR-133a-3p to ischemic myocardium. The mediation of IPC-exo’s protective effects by miR-133a-3p is related with direct targeting of LTBP1 and PPP2CA, and indirect suppression of TGF-β signaling pathway.
The process of cardiac repair following MI involves a complex sequence of events. It begins with intense inflammation and immune cell infiltration, ultimately leading to cardiac replacement fibrosis.23 However, overactive fibrosis in infarcted patients may lead to adverse remodeling, potentially triggering the pathogenesis of heart failure.23 The reason is that initial activation of replacement myofibroblasts facilitates collagen deposition around dead cardiomyocytes, offering structural stability to the heart chamber. While chronic excessive collagen accumulation can cause ventricular wall stiffening, impair systolic function, and diminish overall cardiac performance.2 Therapeutic modulation of replacement fibrosis could offer potential for averting heart failure post-MI.24
Clinical application of cardioprotective IPC has been hindered due to its invasive nature.25 Exosome therapy, with its low immunogenicity and minimal tumorigenicity, is a promising avenue for cardiac repair,26 using the bloodstream to transport cell signaling molecules.27 Studies demonstrated that exosomes induced by RIPC or IPC exhibited cardioprotective effects against myocardial I/R injury in rats.3,7,11 In our study, we initially discovered that IPC stimulation caused changes in miRs within serum exosomes, while serum exosomes could be internalized and taken up by cardiomyocytes, which also suggests that exosomes may have the potential to deliver protective substances to ischemic myocardium. Furthermore, our animal experiments demonstrated that IPC-exo promoted cardiac repair by mitigating sustained inflammatory stimuli, suppressing excessive replacement fibrosis and preserving heart function in rats following MI.
Exosomes transport genetic material to facilitate cell-to-cell communication and various biological processes.28 Among the cargos carried by exosomes, miRs play a prominent role and are considered crucial in regulating cardiac function.16 In the present study, miR-133a-3p was identified as one of the most significant upregulated miRs by IPC-exo. Relevant reports indicated that miR-133a levels were reduced in the myocardial tissue of both humans and animals afflicted with MI.29,30 Additionally, miR-133a-3p is associated with cardiovascular diseases29 and involved in modulating the fibrotic process within the myocardium,26,31 overexpression of miR-133a-3p curbs keloid fibroblast fibrosis by inhibiting the TGF-β/Smad2 pathway32 and is encapsulated in exosomes from engineered mesenchymal stem cells, which facilitate myocardial repair in rats post-acute infarction.33 Further, we tested the idea if miR-133a-3p mediates protective effect of IPC-exo, and we observed that miR-133a-3p antagomir transfection of IPC-exo reversed the elevation of miR-133a-3p expression level in myocardial tissue by IPC-exo injection. Of note, transfection of miR-133a-3p antagomir into IPC-exo also greatly attenuated the myocardial protective effect of IPC-exo against excessive replacement fibrosis post-MI. These data suggest that IPC-exo inhibit excessive replacement fibrosis to exert protective effects post-MI, at least in part, by transferring miR-133a-3p.
KEGG assays results revealed significant enrichment of 30 signaling pathways (P<0.001). We aim to identify the pro-fibrotic signaling pathway and TGF-β signaling pathway stood out among the highly enriched pathways as a typical pro-fibrotic signaling route, frequently cited in the literature concerning myocardial fibrosis.34–37 MiR-133a-3p was also evidenced to direct target transforming growth factor beta receptor 1 (TGFBR1)38 and have regulatory effect on the TGF-β/Smad2 pathway.32 In our study, exosomal miR-133a-3p suppressed the mRNA and protein expression levels of TGF-β1 post-MI. Thus, TGF-β signaling pathway chosen for elucidating the underlying molecular mechanisms. To further explore the relationship between the anti-fibrotic effects of miR-133–3p and the TGF-β signaling pathway, two high-confidence mRNA targets of miR-133a-3p, namely LTBP1 and PPP2CA, were selected for further experimentation. Our data revealed that miR-133a-3p directly targeted LTBP1 and PPP2CA (Figure 4E-F), and IPC-exo did inhibit the expression levels of LTBP1 and PPP2CA by transferring miR-133a-3p in MI rats.
As a target gene of miR-133a-3p, PPP2CA functions as the catalytic subunit (36-kDa) of protein phosphatase 2A (PP2A),39,40 which play an important role in heart disease.41 Within canonical pro-fibrotic TGF-β signaling cascade, PP2A serves as a downstream effector of TGF-β1 signaling.42 Overexpression of PPP2CA has been linked to fibrosis in cardiomyocytes and detrimental cardiac remodeling in both transgenic and myocardial infarction mouse models,39,43 indicating that PPP2CA was a pro-fibrotic substance. Accordingly, direct inhibition of PPP2CA by miR-133a-3p may be correlated with an indirect suppression on TGF-β.
Previous studies illustrated that LTBP1 plays a key role in the TGF-β pathway.44 Elevated levels of secreted LTBP1 enhance the formation and extracellular deposition of TGF-β complexes.45 Additionally, this increase bolsters matrix binding post-MI, thereby amplifying the profibrotic effects of TGF-β.46 In natural killer /T-cell lymphoma, inhibition of LTBP1 inactivates the TGF-β/Smad pathway, LTBP1 is recognized as a crucial positive regulator of TGF-β1, and the LTBP1/TGF-β1 signaling axis has been well-established.47 Therefore, miR-133a-3p may exert its influence on the TGF-β signaling pathway by repressing LTBP1.
Collectively, our data indicate that IPC-exo mitigate excessive replacement fibrosis, thereby therapeutically modulating cardiac repair in rats following MI. This effect is potentially achieved by transferring miR-133a-3p to directly target PPP2CA and LTBP1, and to indirectly regulate the TGF-β signaling pathway. Our study identifies a potent therapeutic substance for targeted delivery of engineered extracellular vesicle systems, which has the potential to mitigate the progression of ventricular remodeling after MI and promote cardiac repair by ensuring its controlled release in the target myocardium.48 The precise delivery of IPC-exo to cardiac tissue presents a promising therapeutic approach for patients with chronic MI who have missed the critical period for reperfusion, potentially improving their clinical outcomes.
Our study has several limitations. Firstly, miR-133a-3p within IPC-exo is not necessarily the only miR that influences post-MI repair in rats. Other miRs contained in IPC-exo potentially synergize to play important roles in promoting cardiac repair. While the miR-mediated mechanism is known to engage multiple targets and pathways, our investigation has only identified two candidate molecules so far, other target genes involved in cardioprotection should be explored. Intriguingly, miR-133, when upregulated in the myocytes of those with heart failure, advances the hyperphosphorylation of ryanodine receptors by inhibiting the activity of PP2A, consequently elevating the risk of arrhythmias.49 Moreover, miR-133a-3p has been confirmed to induce atrial myocardial repolarization defects and to modulate ventricular electrophysiology by directly targeting and modulating the expression of PPP2CA.50 However, we have not monitored the impact of arrhythmia in rats in this study. Future studies should investigate the impact of IPC-induced exosomal miR-133a-3p on electrophysiological parameters and ventricular arrhythmia. Lastly, our results lack direct evidence of LTBP1 and PPP2CA regulating the TGF-β signaling pathway, experiments would be conducted to provide direct evidence of LTBP1 and PPP2CA’s regulatory role in the TGF-β signaling pathway in our future study.
Conclusion
In conclusion, IPC-exo transfer miR-133a-3p to ischemic myocardium to promote cardiac repair by inhibiting excessive replacement fibrosis post-MI, and mechanism may be related to the direct targeting of LTBP1 and PPP2CA, and indirect regulation of TGF-β signaling pathway.
Acknowledgments
We acknowledge the Research Center of the Second Affiliated Hospital of Anhui Medical University for providing the instruments.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by National Natural Science Foundation of China (grant number 82100315), Key Laboratory of Anesthesiology and Perioperative Medicine of Anhui Higher Education Institutes, Anhui Medical University, Key Project of Open Subject (grant number MZKF202001) and Anhui Medical University research fund project (grant number 2021xkj232).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Roth GA, Mensah GA, Johnson CO, et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: update From the GBD 2019 Study. J Am Coll Cardiol. 2020;76(25):2982–3021. doi:10.1016/j.jacc.2020.11.010
2. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2021;117(6):1450–1488.
3. Luo Z, Hu X, Wu C, et al. Plasma exosomes generated by ischaemic preconditioning are cardioprotective in a rat heart failure model. Br J Anaesth. 2023;130(1):29–38.
4. Lassen TR, Just J, Hjortbak MV, et al. Cardioprotection by remote ischemic conditioning is transferable by plasma and mediated by extracellular vesicles. Basic Res Cardiol. 2021;116(1):16.
5. Seeger JP, Benda NM, Riksen NP, et al. Heart failure is associated with exaggerated endothelial ischaemia-reperfusion injury and attenuated effect of ischaemic preconditioning. Eur J Prev Cardiol. 2016;23(1):33–40.
6. Miki T, Miura T, Tsuchida A, et al. Cardioprotective mechanism of ischemic preconditioning is impaired by postinfarct ventricular remodeling through angiotensin II type 1 receptor activation. Circulation. 2000;102(4):458–463.
7. Cheng XF, He ST, Zhong GQ, et al. Exosomal HSP90 induced by remote ischemic preconditioning alleviates myocardial ischemic/reperfusion injury by inhibiting complement activation and inflammation. BMC Cardiovasc Disord. 2023;23(1):58.
8. Minghua W, Zhijian G, Chahua H, et al. Plasma exosomes induced by remote ischaemic preconditioning attenuate myocardial ischaemia/reperfusion injury by transferring miR-24. Cell Death Dis. 2018;9(3):320.
9. Li D, Zhao Y, Zhang C, et al. Plasma Exosomes at the Late Phase of Remote Ischemic Pre-conditioning Attenuate Myocardial Ischemia-Reperfusion Injury Through Transferring miR-126a-3p. Front Cardiovasc Med. 2021;8:736226.
10. Chen Q, Huang M, Wu J, et al. Exosomes isolated from the plasma of remote ischemic conditioning rats improved cardiac function and angiogenesis after myocardial infarction through targeting Hsp70. Aging. 2020;12(4):3682–3693. doi:10.18632/aging.102837
11. Zhang J, Zhang X. Ischaemic preconditioning-induced serum exosomes protect against myocardial ischaemia/reperfusion injury in rats by activating the PI3K/AKT signalling pathway. Cell Biochem Funct. 2021;39(2):287–295. doi:10.1002/cbf.3578
12. Krylova SV, Feng D. The Machinery of Exosomes: biogenesis, Release, and Uptake. Int J Mol Sci. 2023;24(2):1337. doi:10.3390/ijms24021337
13. Kim KU, Kim WH, Jeong CH, et al. More than Nutrition: therapeutic Potential of Breast Milk-Derived Exosomes in Cancer. Int J Mol Sci. 2020;21(19):7327. doi:10.3390/ijms21197327
14. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977. doi:10.1126/science.aau6977
15. Ke X, Liao Z, Luo X, et al. Endothelial colony-forming cell-derived exosomal miR-21-5p regulates autophagic flux to promote vascular endothelial repair by inhibiting SIPL1A2 in atherosclerosis. Cell Commun Signal. 2022;20(1):30. doi:10.1186/s12964-022-00828-0
16. Liao Z, Chen Y, Duan C, et al. Cardiac telocytes inhibit cardiac microvascular endothelial cell apoptosis through exosomal miRNA-21-5p-targeted cdip1 silencing to improve angiogenesis following myocardial infarction. Theranostics. 2021;11(1):268–291. doi:10.7150/thno.47021
17. Wang T, Li T, Niu X, et al. ADSC-derived exosomes attenuate myocardial infarction injury by promoting miR-205-mediated cardiac angiogenesis. Biol Direct. 2023;18(1):6. doi:10.1186/s13062-023-00361-1
18. Gollmann-Tepeköylü C, Pölzl L, Graber M, et al. miR-19a-3p containing exosomes improve function of ischaemic myocardium upon shock wave therapy. Cardiovasc Res. 2020;116(6):1226–1236. doi:10.1093/cvr/cvz209
19. Xie L, Zhao H, Wang Y, et al. Exosomal shuttled miR-424-5p from ischemic preconditioned microglia mediates cerebral endothelial cell injury through negatively regulation of FGF2/STAT3 pathway. Exp Neurol. 2020;333:113411. doi:10.1016/j.expneurol.2020.113411
20. He SF, Zhu HJ, Han ZY, et al. MicroRNA-133b-5p Is Involved in Cardioprotection of Morphine Preconditioning in Rat Cardiomyocytes by Targeting Fas. Can J Cardiol. 2016;32(8):996–1007.
21. Pan YL, Han ZY, He SF, et al. miR‑133b‑5p contributes to hypoxic preconditioning‑mediated cardioprotection by inhibiting the activation of caspase‑8 and caspase-3 in cardiomyocytes. Mol Med Rep. 2018;17(5):7097–7104.
22. Esteves M, Abreu R, Fernandes H, et al. MicroRNA-124-3p-enriched small extracellular vesicles as a therapeutic approach for Parkinson’s disease. Mol Ther. 2022;30(10):3176–3192.
23. Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: from Inflammation to Fibrosis. Circ Res. 2016;119(1):91–112.
24. Wang X, Gaur M, Mounzih K, et al. Inhibition of galectin-3 post-infarction impedes progressive fibrosis by regulating inflammatory profibrotic cascades. Cardiovasc Res. 2023;119(15):2536–2549.
25. He SF, Jin SY, Wu H, et al. Morphine preconditioning confers cardioprotection in doxorubicin-induced failing rat hearts via ERK/GSK-3β pathway independent of PI3K/Akt. Toxicol Appl Pharmacol. 2015;288(3):349–358.
26. Zhu W, Sun L, Zhao P, et al. Macrophage migration inhibitory factor facilitates the therapeutic efficacy of mesenchymal stem cells derived exosomes in acute myocardial infarction through upregulating miR-133a-3p. J Nanobiotechnology. 2021;19(1):61.
27. Yu D, Li Y, Wang M, et al. Exosomes as a new frontier of cancer liquid biopsy. Mol Cancer. 2022;21(1):56.
28. Wang C, Li Z, Liu Y, et al. Exosomes in atherosclerosis: performers, bystanders, biomarkers, and therapeutic targets. Theranostics. 2021;11(8):3996–4010.
29. Bian Y, Pang P, Li X, et al. CircHelz activates NLRP3 inflammasome to promote myocardial injury by sponging miR-133a-3p in mouse ischemic heart. J Mol Cell Cardiol. 2021;158:128–139.
30. Boštjančič E, Brandner T, Zidar N, et al. Down-regulation of miR-133a/b in patients with myocardial infarction correlates with the presence of ventricular fibrillation. Biomed Pharmacother. 2018;99:65–71.
31. Yao L, Zhou B, You L, et al. LncRNA MIAT/miR-133a-3p axis regulates atrial fibrillation and atrial fibrillation-induced myocardial fibrosis. Mol Biol Rep. 2020;47(4):2605–2617.
32. Huang Y, Wang Y, Lin L, et al. Overexpression of miR-133a-3p inhibits fibrosis and proliferation of keloid fibroblasts by regulating IRF5 to inhibit the TGF-β/Smad2 pathway. Mol Cell Probes. 2020;52:101563.
33. Sun L, Zhu WW, Zhang J, et al. Exosomes derived from miR-133a-3p engineered mesenchymal stem cells promote myocardial repair in rats after acute myocardial infarction. Zhonghua Xin Xue Guan Bing Za Zhi. 2024;52(1):72–78.
34. Frangogiannis NG. Transforming growth factor-β in myocardial disease. Nat Rev Cardiol. 2022;19(7):435–455.
35. Ko T, Nomura S, Yamada S, et al. Cardiac fibroblasts regulate the development of heart failure via Htra3-TGF-β-IGFBP7 axis. Nat Commun. 2022;13(1):3275.
36. Cheng X, Wang L, Wen X, et al. TNAP is a novel regulator of cardiac fibrosis after myocardial infarction by mediating TGF-β/Smads and ERK1/2 signaling pathways. EBioMedicine. 2021;67:103370.
37. Tian J, Zhang M, Suo M, et al. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J Cell Mol Med. 2021;25(16):7642–7659.
38. Jiang L, Shi X, Liu Y, et al. LncRNA LINC00847 Accelerates Melanoma Progression by Regulating MiR-133a-3p/TGFBR1 Axis. Comb Chem High Throughput Screen. 2024;27(8):1231–1241.
39. Gergs U, Boknik P, Buchwalow I, et al. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J Biol Chem. 2004;279(39):40827–40834.
40. Xie F, Li F, Li R, et al. Inhibition of PP2A enhances the osteogenic differentiation of human aortic valvular interstitial cells via ERK and p38 MAPK pathways. Life Sci. 2020;257:118086.
41. Zhu D, Liu S, Huang K, et al. Intrapericardial Exosome Therapy Dampens Cardiac Injury via Activating Foxo3. Circ Res. 2022;131(10):e135–e150.
42. Guo Y, Deng Y, Li X, et al. Glutaminolysis Was Induced by TGF-β1 through PP2Ac Regulated Raf-MEK-ERK Signaling in Endothelial Cells. PLoS One. 2016;11(9):e0162658.
43. Hoehn M, Zhang Y, Xu J, et al. Overexpression of protein phosphatase 2A in a murine model of chronic myocardial infarction leads to increased adverse remodeling but restores the regulation of β-catenin by glycogen synthase kinase 3β. Int J Cardiol. 2015;183:39–46.
44. Lockhart-Cairns MP, Cain SA, Dajani R, et al. Latent TGFβ complexes are transglutaminase cross-linked to fibrillin to facilitate TGFβ activation. Matrix Biol. 2022;107:24–39.
45. Taipale J, Miyazono K, Heldin CH, et al. Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein. J Cell Biol. 1994;124(1–2):171–181.
46. Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74(2):184–195.
47. Lin R, Li X, Wu S, et al. Suppression of latent transforming growth factor-β (TGF-β)-binding protein 1 (LTBP1) inhibits natural killer/ T cell lymphoma progression by inactivating the TGF-β/Smad and p38(MAPK) pathways. Exp Cell Res. 2021;407(1):18.
48. Pezzana C, Agnely F, Bochot A, et al. Extracellular Vesicles and Biomaterial Design: new Therapies for Cardiac Repair. Trends Mol Med. 2021;27(3):231–247.
49. Belevych AE, Sansom SE, Terentyeva R, et al. MicroRNA-1 and −133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One. 2011;6(12):e28324.
50. Kuzmin VS, Ivanova AD, Filatova TS, et al. Micro-RNA 133a-3p induces repolarization abnormalities in atrial myocardium and modulates ventricular electrophysiology affecting I(Ca,L) and Ito currents. Eur J Pharmacol. 2021;908:174369.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.