")
Back to Journals » International Journal of Nanomedicine » Volume 20
Liver-Targeting Nanoparticles GA-MSe@AR Treat NAFLD Through Dual Lipid-Lowering and Antioxidant Efficacy
Authors Lei S , Wu Q, Zhang B, Lu M, Xia Y, Li N
Received 5 December 2024
Accepted for publication 4 April 2025
Published 18 April 2025 Volume 2025:20 Pages 5017—5037
DOI https://doi.org/10.2147/IJN.S510577
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lijie Grace Zhang
Sheng Lei,1 Qiang Wu,1 Bin Zhang,1 Minqiang Lu,1 Yu Xia,1,2 Ning Li1
1Department of HBP Surgery, The Second Affiliated Hospital, School of Medicine, South China University of Technology, Guangzhou, 510180, People’s Republic of China; 2Department of Gastroenterology and Hepatology, Guangzhou Digestive Disease Center, The Second Affiliated Hospital, School of Medicine, South China University of Technology, Guangzhou, 510180, People’s Republic of China
Correspondence: Ning Li, Department of HBP Surgery, The Second Affiliated Hospital, School of Medicine, South China University of Technology, Guangzhou, 510180, People’s Republic of China, Email [email protected] Yu Xia, Department of Gastroenterology and Hepatology, Guangzhou Digestive Disease Center, The Second Affiliated Hospital, School of Medicine, South China University of Technology, Guangzhou, 510180, People’s Republic of China, Email [email protected]
Background and Purpose: Non-alcoholic fatty liver disease (NAFLD) is prevalent worldwide and lacks effective treatments. Arctiin (AR), a natural product, has shown promise for NAFLD therapy, due to its antioxidant, anti-inflammatory, and inhibition adipogenesis properties. However, its therapeutic efficacy is hindered by low water solubility, poor bioavailability, and inadequate liver targeting. In this study, selenium-based antioxidant nanoparticles were developed to load and deliver AR to the liver for synergistic AR and selenium effective treatment of NAFLD.
Methods: The therapeutic potential of AR was analyzed by network pharmacology. GA-MSe@AR was synthesized by encapsulating AR within galactose-modified mesoporous selenium nanoparticles (GA-MSe) for liver-specific targeting. The nanoparticle size, chemical structure, and elemental composition were explored. The toxicity, cellular uptake, lysosomal escape, and AR release efficiency of GA-MSe@AR were investigated by in vitro experiments. The liver targeting ability of GA-MSe@AR was evaluated through live imaging. The lipid-lowering and antioxidant activities of GA-MSe@AR were assessed in both in vitro and in vivo NAFLD models. Additionally, its effects on inflammation and pancreatic function were analyzed in vivo.
Results: Network pharmacology analysis revealed AR may against NAFLD through regulating metabolism, inflammation, and oxidative stress. GA-MSe@AR exhibited low toxicity, efficient cellular uptake, remarkable lysosomal escape ability, and high AR release efficiency in vitro. In both in vitro and in vivo NAFLD models, GA-MSe@AR demonstrated more pronounced lipid-lowering and antioxidant properties than AR and GA-MSe. Additionally, GA-MSe@AR effectively targeted the liver, resulting in a greater decrease in blood glucose, lipids, ALT, AST levels, and reduction liver inflammation, as well as improved pancreatic function in high-fat diet (HFD)-fed mice compared to AR alone.
Conclusion: The GA-specific modification enhanced liver-targeted accumulation of the selenium-based nanoparticles, enabling precise targeted delivery of AR. GA-MSe@AR demonstrated superior lipid-lowering efficacy and antioxidant activity in a NAFLD mice model. These findings collectively establish GA-MSe@AR as a promising therapeutic candidate for NAFLD treatment.
Keywords: arctiin, selenium nanoparticles, lipid deposition, oxidative stress, network pharmacology
Graphical Abstract:

Introduction
Non-alcoholic fatty liver disease (NAFLD) has become increasingly prevalent due to modern lifestyle changes, affecting approximately 25% of the global adult population and representing the most common chronic liver disease-a major public health concern.1 Treatment options remain limited, primarily due to the complex pathogenesis of the condition.2 Excessive lipid accumulation in the liver is recognized as the primary driver of NAFLD development and progression.3 This lipid overload leads to the overproduction of reactive oxygen species (ROS), which causes hepatocyte damage and liver tissue injury.4 Additionally, elevated ROS levels trigger liver inflammation, further exacerbating tissue damage.5 This inflammatory response, in turn, amplifies ROS production, creating a cycle that intensifies liver inflammation and injury, ultimately accelerating NAFLD progression.6 Thus, targeting both ROS generation and liver inflammation concurrently presents a promising approach for the prevention and treatment of NAFLD.
In recent decades, natural products have gained significant attention as potential treatments for NAFLD, owing to their diverse bioactivities, therapeutic potential, and relatively low side effect profiles.7,8 Especially arctiin (AR), the primary active component extracted from traditional Chinese medicine Arctium lappa L., with low toxicity and few side effects. In addition, modern pharmacological studies have shown that AR exhibited remarkable antioxidant,9 anti-inflammatory,10 metabolic regulatory,11 and immune-boosting properties.12 These properties make AR a promising natural compound for NAFLD treatment. Network pharmacology analyses have further confirmed the therapeutic potential of AR against NAFLD. Despite AR showing promising potential for the treatment of NAFLD, its efficacy has not been fully evaluated due to challenges such as low water solubility, poor bioavailability and insufficient hepatic targeting.13 Consequently, innovative strategies are needed to address these limitations to enhance the therapeutic efficacy of AR for NAFLD, facilitating its application into clinical practice.
Nanoparticles have emerged as promising drug carriers due to their high bioavailability, substantial drug-loading capacity, prolonged circulation time, extended half-life, and ability to deliver drugs to specific organs or lesions.14–17 Among these, selenium-based nanoparticles (Se NPs) have demonstrated unique advantages for NAFLD treatment.18 Selenium, the primary metabolite of Se NPs, is an essential trace element that supports human health.19 Moderate selenium supplementation promotes the synthesis of selenium-dependent antioxidant enzymes in the liver, such as glutathione peroxidase (GPx) and selenocysteine lyase (SCLY), thereby enhancing the antioxidant capacity of the liver to mitigate NAFLD.20,21 Furthermore, selenium supplementation may attenuate NAFLD by improving the hepatic injury and insulin resistance.22 Additionally, Se NPs can be synthesized under environmentally friendly conditions in aqueous solutions at room temperature, further supporting their utility as carriers for loading AR.23 Therefore, selenium-based nanoparticles (Se NPs) are ideal drug carriers for the treatment of NAFLD, due to their antioxidant properties and metabolic modulation capabilities. To enhance the AR loading ability of Se NPs, mesoporous selenium nanoparticles (MSe NPs) were developed, offering enhanced drug-loading capacity.24 While MSe@AR nanoparticles exhibit high AR loading, their liver-targeting capability remains suboptimal. To address this issue, galactose (GA) was identified as a promising liver-targeting ligand.25 GA exhibits selectivity and high affinity binding to the asialoglycoprotein receptor (ASGPR), a receptor that is specifically expressed on hepatocytes with minimal expression in non-hepatocytes.26 Upon GA binds to ASGPR with high affinity, ASGPR initiates internalization via clathrin-mediated endocytosis. Thus, this endocytosis mechanism not only enhances the bioavailability of hydrophobic drugs but also facilitates targeted drug delivery to the liver. Therefore, by conjugating GA to the surface of MSe@AR, functionalized GA-MSe@AR nanoparticles were developed, enabling targeted delivery of AR to the liver.
In this study, the therapeutic potential of AR for NAFLD was validated. Based on this, a novel liver-targeted functional selenium nanoparticle GA-MSe@AR was successfully developed as a therapeutic agent for NAFLD by combining the antioxidant properties of selenium and the therapeutic effects of AR. As illustrated in Scheme 1, MSe was prepared to enable effective loading of AR, resulting in the formulation of MSe@AR. Subsequently, galactose (GA) was conjugated to the surface of MSe@AR to construct liver-targeted nanoparticles, GA-MSe@AR. The antioxidant and lipid-lowering properties of GA-MSe@AR were investigated in NAFLD cells models. The liver-targeting ability of GA-MSe@AR was investigated using a live imaging system. The effect of GA-MSe@AR in reduced hepatic steatosis, oxidative stress, and improved pancreatic function were evaluated in a high-fat diet (HFD) mouse model.
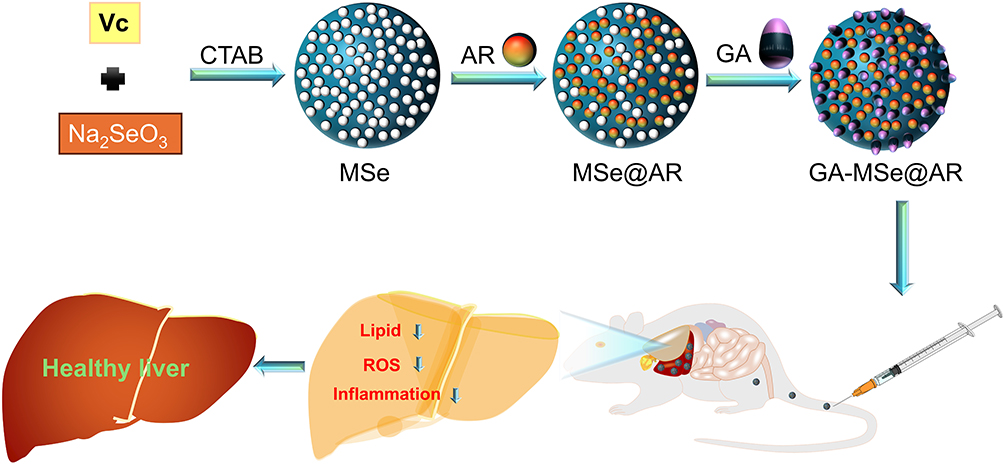 |
Scheme 1 Development of GA-MSe@AR nanoparticles and their therapeutic effect on non-alcoholic fatty liver disease (NAFLD). |
Materials and Methods
Reagents
Cetyltrimethyl ammonium bromide (CTAB), NaOH, Na2SeO3, L-ascorbic acid, and galactose were sourced from Sigma Aldrich Co. (MO, USA). Fetal bovine serum (FBS) and Dulbecco’s Modified Eagle Medium (DMEM)/high glucose medium were obtained from Gibco (MA, USA). Arctiin, palmitic acid (PA), and oleic acid (OA) were purchased from Macklin (Shanghai, China). Hoechst 33342, dialysis bags, DCFH-DA, and Cell Counting Kit-8 (CCK8) were gained from Solarbio Technology Co., Ltd. (Beijing, China). The assay kits for aspartate aminotransferase (AST), triglycerides (TG), alanine aminotransferase (ALT), and total cholesterol (TC) were provided by Nanjing Jiancheng Bioengineering Research Institute (Nanjing, China). Additionally, BODIPY 493/503, LysoTracker Red, JC-1 staining kit, GSH/GSSG assay kit, and Malondialdehyde (MDA) test kit were procured from Beyotime Biotechnology (Shanghai, China). Oil Red O dye solution was obtained from M&C Gene Technology Ltd. (Beijing, China). For protein analysis, BCA Protein Concentration Assay Kit, RIPA lysis buffer, phenylmethanesulfonyl fluoride (PMSF), horseradish peroxidase (HRP)-linked secondary antibodies, anti-F4/80, anti-β-actin, anti-p-PI3K, anti-p-AKT, and anti-IGF1 were sourced from Servicebio Biotechnology (Wuhan, China). Antibodies specific to SREBP-1c, FAS, PPAR-γ, NOX4, SOD, and CAT were purchased from Proteintech (Wuhan, China).
Network Pharmacology
The metabolites present in Arctium lappa L. were identified using the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP; https://old.tcmsp-e.com/tcmsp.php).27 Compounds with drug-likeness (DL) scores ≥ 0.18 and oral bioavailability (OB) ≥ 30% were selected as potential active ingredients. Subsequently, the targets of AR were obtained from the Swiss Target Prediction database (http://www.swisstargetprediction.ch/),28 ChEMBL database (https://www.ebi.ac.uk/chembl/),29 and the PharmMapper database (http://www.lilab-ecust.cn/pharmmapper/).30 “Non-alcoholic Fatty Liver Disease” was used as a keyword to search the OMIM database (https://www.omim),31 GeneCards (https://www.gene-(cards.org/),32 and TTD database (http://bidd.nus.edu.sg/group/cjttd/)33 for NAFLD-related targets. To ensure consistency, all target names were standardized using the UniProt database (https://www.uniprot.org/).34 Shared targets between NAFLD and AR were identified using a Venn diagram generated through the bioinformatics tool (https://bioinfogp.cnb.csic.es/). A protein-protein interaction (PPI) network was constructed using the STRING database (http://string-db.org/)35 and visualized in Cytoscape version 3.10.2. Additionally, Cytoscape was used to create a drug-target-disease network. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the DAVID database (https://david.ncifcrf.gov/)36 to explore the biological functions and pathways associated with the identified targets.
Molecular Docking
Molecular docking studies were performed to evaluate the binding affinity between AR and potential targets. X-ray crystallographic data for the proteins were obtained from the Protein Data Bank (PDB, https://www.rcsb.org/).37 The CB-DOCK2 (https://cadd.labshare.cn/cb-dock2/php/index.php)38 was used for molecular docking, and the binding affinity was assessed using the Vina score. To visualize the docking results, PyMOL (version 2.5.5) and Discovery Studio 2024 software were employed.
Preparation of GA-MSe@AR
A solution of CTAB (4 mg) was added to sodium hydroxide (NaOH, 0.2 mol/L) and stirred thoroughly until the temperature reached 80°C. Next, 2 mL of L-ascorbic acid (VC, 20 mM) and 2 mL of sodium selenite (Na₂SeO₃, 5 mM) were added to the mixture, which was stirred for 2 hours at pH 8.0. The solution was centrifuged at 8000 rpm for 5 minutes and washed three times each with water and ethanol. The resulting specimen was dispersed in 30 mL of ethanol, stirred at 800 rpm, and refluxed at 80°C for 24 hours to remove CTAB, yielding MSe. Then, the MSe solution was mixed with 2 mL of AR (4 mM) solution and stirred for 1 hour to prepare MSe@AR. Following this, the MSe@AR solution was mixed with 10 mg of GA and stirred at 800 rpm, pH 7.8, and 37°C for 2 hours. Free AR and GA were then removed through dialysis (3.5 kDa MWCO) with ultrapure water for 36 hours at 4°C. The dialyzed mixture was centrifuged at 8000 rpm for 10 minutes, washed three times with water, and freeze-dried for 12 hours to obtain GA-MSe@AR. The amount of AR in the GA-MSe@AR was calculated using high-performance liquid chromatography (Thermo Fisher Scientific, MA, USA).
For the preparation of Coumarin 6- or Sulfo-Cyanine 3 (Cy3)-labeled GA-MSe@AR, a procedure similar to GA-MSe@AR synthesis was followed. Briefly, 5 μg of either Coumarin 6 or Cy3 was added to the GA-MSe@AR solution and stirred at 800 rpm for 2 hours. Dialysis was then performed using a dialysis bag with a 3.5 kDa cutoff filled with ultrapure water to remove free coumarin 6 or Cy3; subsequently, the solution was centrifuged, washed, and freeze-dried to obtain the final Coumarin 6- or Cy3-labeled GA-MSe@AR.
Characterization of GA-MSe@AR
Transmission Electron Microscopy (TEM) and Energy-Dispersive X-Ray (EDX) Analysis
The GA-MSe@AR samples were diluted and placed on a copper grid covered with nitrocellulose. Phosphotungstic acid was used to perform negative staining, after which the samples were left to dry. The morphology and elemental composition of GA-MSe@AR were analysed by standard bright field and energy dispersive X-ray of TEM (HT7700, Hitachi, Japan).
Fourier Transform Infrared (FTIR) Spectroscopy
In the analysis of functional groups, the samples (MSe, GA-MSe, GA, AR, and GA-MSe@AR) were compressed into potassium bromide (KBr) pellets. Thereafter, the pellets were examined using an FTIR spectrometer (Thermo Fisher, USA) with a wavelength ranging from 500 to 4000 cm−1 at 4 cm−1 resolution for 128 scans.
Dynamic Light Scattering (DLS) Analysis
DLS studies were used to measure the size distribution of GA-MSe@AR using Zetasizer Nano PRO instruments (Malvern Panalytical, UK). The instrument contains a 4 mW He-Ne laser operating at a wavelength of 632.8 nm with 175° detection. Samples were dissolved in pure water, loaded into measuring tubes and the measurements were repeated three times at 25°C.
Cells Culture
The human and mouse normal hepatocyte cell line (LO2 cells and AML12 cells) were purchased from American Type Culture Collection (ATCC, Manassas, USA). All the cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 1% streptomycin and penicillin at 37 °C in 5% CO2.
Cellular Uptake of GA-MSe@AR
To evaluate cellular uptake, LO2 and AML12 cells (2 × 105) were cultured in 24-well plates and incubated with coumarin 6-labeled GA-MSe@AR (5 μg/mL) for varying durations (1, 2, and 4 hours). After incubation, the nuclei of the cells were stained with Hoechst 33342 (10 μM) for 10 minutes. Fluorescence microscopy (Leica Microsystems, Wetzlar, Germany) was then employed to observe the internalization of GA-MSe@AR.
Lysosomal Escape of GA-MSe@AR
To examine the escape process of GA-MSe@AR from lysosomes, LO2 cells and AML12 cells were treated with coumarin 6-labeled GA-MSe@AR (5 μg/mL) for 0.25, 0.5, 1, 2, and 4 hours in 12-well plates. Cells were then stained with Lysotracker Red to label lysosomes (75 nM) and Hoechst 33342 (10 μM) to label nuclei. The location of GA-MSe@AR and lysosomes was visualized by fluorescence microscopy (Leica Microsystems, Wetzlar, Germany).
Lipid Deposition Assay
Lipid deposition in LO2 and AML12 cells was evaluated using Oil Red O and BODIPY fluorescence staining. Cellular lipid accumulation was induced with free fatty acids (FFA, palmitate/oleate = 1/2, 1 mM), while untreated cells served as negative controls. After induction, cells were treated overnight with GA-MSe@AR, GA-MSe, or AR (each at 5 μg/mL) and fixed with 4% paraformaldehyde. The fixed cells were stained with BODIPY 493/503 (10 µM) for 30 minutes or Oil Red O for 15 minutes. Lipid droplets were visualized using fluorescence microscopy (Leica Microsystems, Wetzlar, Germany) for BODIPY staining and light microscopy (Olympus Corporation, Tokyo, Japan) for Oil Red O staining.
DCFH-DA Staining for ROS
ROS generation in LO2 cells and AML12 cells was induced by treatment with PA (0.5 mM). The cells were then treated separately with GA-MSe@AR, GA-MSe, or AR (each at 5 μg/mL). Following this, the cells were stained with DCFH-DA (10 μM) for 10 minutes. ROS production was visualized using fluorescence microscopy (Leica Microsystems, Wetzlar, Germany) by detecting the green fluorescence emitted from DCFH-DA, which indicates ROS generation.
Mitochondrial Membrane Potential (MMP) Measurement
MMP was assessed using the JC-1 staining kit. LO2 and AML12 cells (5 × 10⁵ per well) were cultured overnight in 12-well plates and exposed to 0.5 mM palmitic acid (PA) alone or in combination with 5 μg/mL of GA-MSe@AR, GA-MSe, or AR for 24 hours. The cells were then stained with JC-1 and observed under fluorescence microscopy (Leica Microsystems, Wetzlar, Germany).
Mice and Intervention
Four-week-old male C57BL/6J mice, housed in a controlled environment (22–24°C, 60% humidity) with a 12-hour light-dark cycle, were acclimated for 2 weeks. The mice were then randomly assigned to five groups (n = 6). The mice received either a low-fat diet (LFD, 10% kcal fat) with saline, or a high-fat diet (HFD, 60% kcal fat) supplemented with saline, GA-MSe, AR, or GA-MSe@AR (all at 5 mg/kg, administered twice weekly, i.v). Weekly body weights were recorded. Following 16 weeks of treatment, the mice were fasted overnight, anesthetized, and then euthanized. Blood samples were collected via cardiac puncture, and the livers were excised, separated by immersion in 4% paraformaldehyde, and snap-frozen in liquid nitrogen. All procedures were conducted following the Guide for the Care and Use of Experimental Animals, as approved by the Ethics Committee of Guangdong Medical Laboratory Animal Center (Certificate No. D202408-10).
In vivo Biodistribution of GA-MSe@AR
The C57BL/6J mice were injected with a single dose of Sulfo-Cyanine 3 (Cy3)-labeled GA-MSe@AR (5 mg/kg) via the tail vein. Untreated mice served as controls. After 15, 30, and 60 minutes of injection, the organs including the heart, liver, spleen, lungs, and kidneys were harvested and placed in cell culture dishes. These organs were then imaged using the IVIS Spectrum in vivo imaging system (PerkinElmer, Boston, USA).
Determination of Oxidative Stress
The levels of glutathione (GSH) and malondialdehyde (MDA) were measured as indicators of antioxidants and oxidative products, respectively, in the liver of mice. Liver tissues were homogenized in RIPA lysate (Servicebio, Wuhan, China) after harvesting and washing. The supernatant was collected by centrifugation at 12,000 rpm for 15 minutes. The concentrations of GSH and MDA were then determined using commercial assay kits (Beyotime, Shanghai, China) according to the manufacturer’s instructions.
Biochemical Analysis
Serum was collected from mouse blood samples by centrifugation at 3000 rpm for 20 minutes. The concentrations of AST, ALT, TC, and TG were measured using commercial assay kits, following the protocols provided by Nanjing Jiancheng Bioengineering Research Institute (Nanjing, China).
Insulin Tolerance Tests (ITT) and Glucose Tolerance Tests (GTT)
The ITT and GTT experiments were conducted to assess insulin and glucose tolerance. After a 6-hour fast, mice received intraperitoneal injections of either insulin (0.75 U/kg) or glucose (1.5 g/kg). Blood glucose levels were measured at 0, 15, 30, 60, 90, and 120 minutes post-injection using a blood glucometer (CVS Advanced Health) via tail snipping. The area under the curve (AUC) for both ITT and GTT was calculated using Prism GraphPad 8 software.
Histological Staining and Oil Red O Staining
For histological analysis, liver samples were fixed in neutral formalin, embedded in paraffin, and sectioned into 6 μm slices. The paraffin sections were deparaffinized, rehydrated through graded ethanol/water solutions, and stained with hematoxylin and eosin (H&E). Alternatively, liver specimens were fixed with 4% paraformaldehyde, dehydrated in 20% sucrose, embedded in optimal cutting temperature (OCT) compound, and cryosectioned at 7 μm. These cryosections were stained with Oil Red O after immersion in 60% isopropanol for 10 minutes, followed by hematoxylin counterstaining of the nuclei.
Immunofluorescence Staining
Liver sections were prepared according to a standard procedure and subsequently blocked in a 10% bovine serum albumin (BSA) solution (Solarbio Life Science, Beijing, China). The sections were then coated with anti-F4/80 primary antibodies (Servicebio, Wuhan, China) and incubated at 4°C overnight. After removing the unbound primary antibody, the sections were incubated at room temperature for one hour with a secondary antibody conjugated to FITC (Servicebio, Wuhan, China). The slides were sealed with an anti-fluorescence quenching solution containing DAPI (Servicebio, Wuhan, China) and images were captured using fluorescence microscopy (Leica Microsystems, Wetzlar, Germany).
Western Blotting
Liver tissues were lysed using RIPA buffer (Servicebio, China) to obtain protein samples. Equal amounts of protein were separated by SDS-PAGE (Servicebio, China) and transferred to PVDF membranes (Millipore, USA). The membranes were blocked with 5% skim milk and incubated overnight at 4°C with the following primary antibodies: anti-FASN, anti-PPARγ, anti-SREBP1c, anti-SOD2, anti-CAT, anti-NOX4, anti-IGF1, anti-p-AKT, and anti-p-PI3K (all from Proteintech, China, and Servicebio, China). β-actin (Servicebio, China) was used as the endogenous control. The membranes were then incubated with a secondary antibody conjugated to HRP (Servicebio, China) for one hour. Immunoreactive bands were visualized using the ChemiDoc™ imaging system (Bio-Rad, CA, USA).
Statistical Analysis
The data are presented as mean ± SD and were analyzed using Prism GraphPad 8 (San Diego, USA). ANOVA, followed by Tukey’s test, was adopted to compare the experimental groups. Statistical significance was defined as *p < 0.05, **p < 0.01, or ***p < 0.001.
Results and Discussion
Bioinformatic Analysis Between AR and NAFLD
Arctium lappa L., a traditional Chinese medicine (TCM), possesses metabolic regulatory, hepatoprotective, and anti-inflammatory properties,39 making it a potential candidate for the treatment of NAFLD. AR, identified as the main active component through the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform, exhibited the highest drug-likeness (0.84) (Table 1). The therapeutic potential of AR in NAFLD treatment was further explored through network pharmacology. A total of 258 AR-related targets were retrieved from online databases based on the chemical structure of AR (Figure 1A). In parallel, 4997 disease-associated targets related to NAFLD were identified. A total of 55 shared targets between AR and NAFLD were then analyzed through Protein-Protein Interaction (PPI) network analysis using the STRING database (Figure 1B and C). Based on the network’s degree, betweenness, and closeness centrality metrics (Table S1), the following hub targets were identified as key players in AR’s therapeutic effects on NAFLD: HSP90AA1, EGFR, GSK3β, IGF1, and CDC42 (Figure 1D). GO enrichment analysis showed that AR may mitigate NAFLD through negative regulation of the apoptotic process, inflammatory response, and involvement in ATP binding (Figure 1E). KEGG enrichment analysis revealed that AR predominantly influences the metabolic pathways, the PI3K-Akt signaling pathway, the FoxO signaling pathway, the MAPK signaling pathway, and lipid and atherosclerosis (Figure 1F). These pathways are well-known for their roles in regulating metabolism, inflammation, and oxidative stress in NAFLD.40 Collectively, these findings suggest that AR holds significant potential as a small molecule therapeutic for NAFLD.
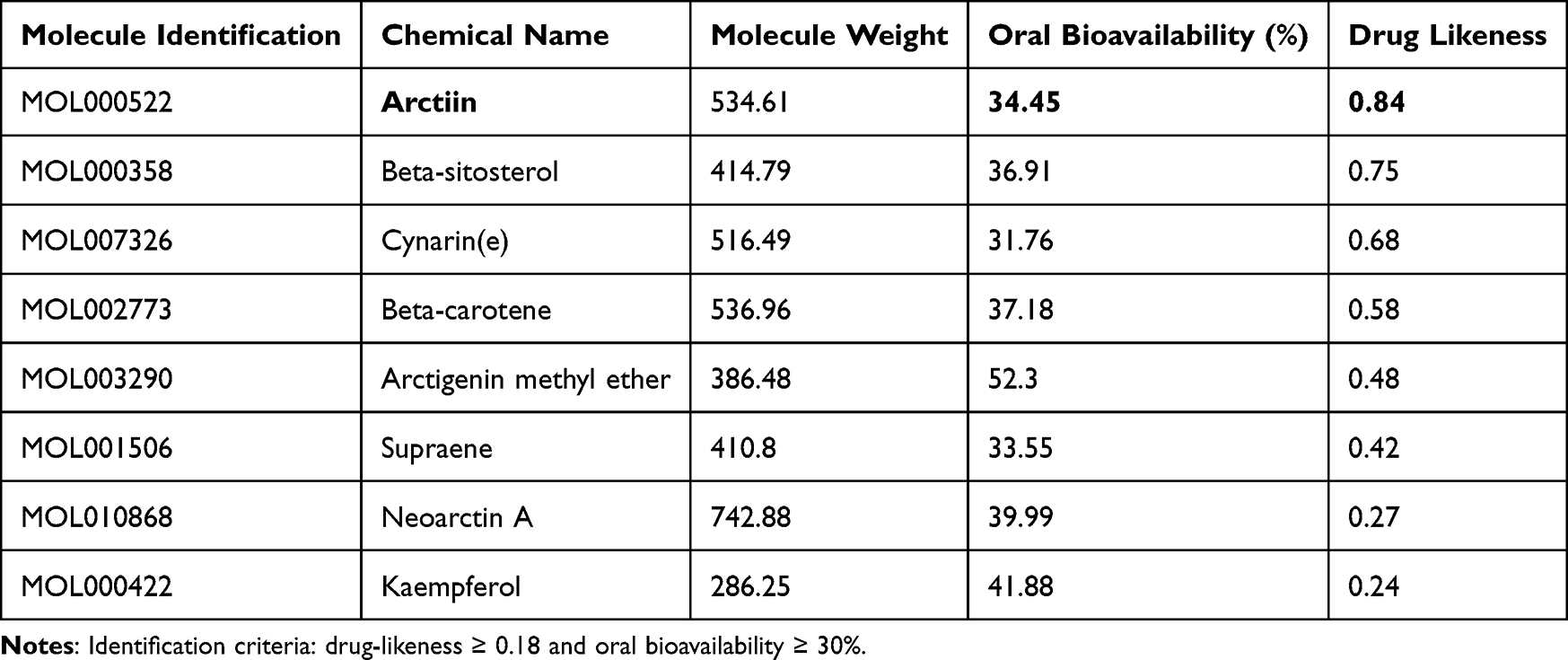 |
Table 1 The Top 8 Active Ingredients of Arctium lappa L |
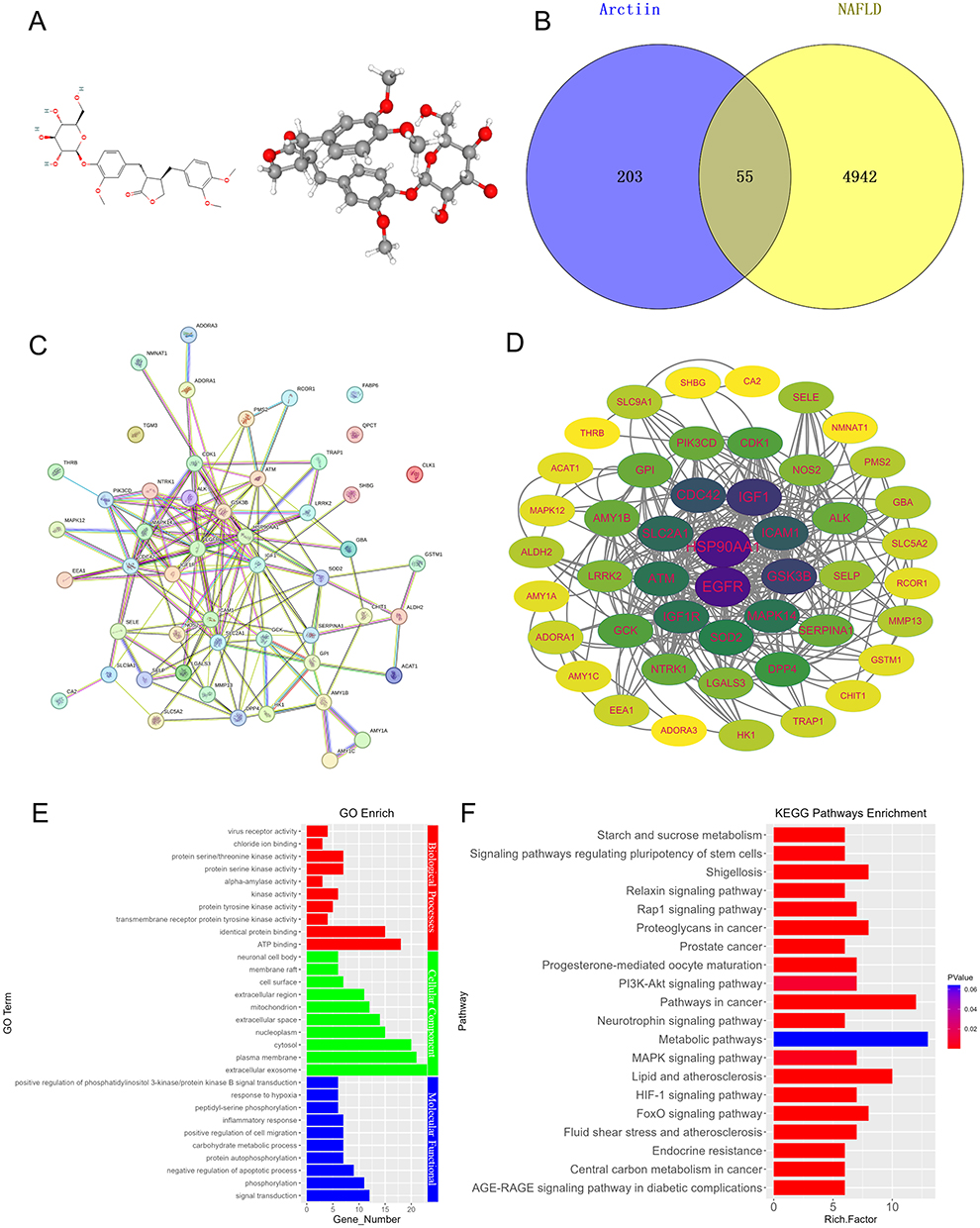 |
Figure 1 Network pharmacological analysis of arctiin (AR) and non-alcoholic fatty liver disease (NAFLD). (A) 2D and 3D chemical structures of AR were obtained from the PubChem database. (B) Shared targets of AR and NAFLD summarized by Venn diagram. (C) Protein-protein interaction (PPI) network conducted by shared targets. (D) Hub target genes of AR and NAFLD identified using Cytoscape. (E) Gene Ontology analysis (GO) of AR targets related to NAFLD, revealing significant enrichments in biological process, molecular function, and cellular components. (F) Kyoto Encyclopedia of Genes and Genomes analysis (KEGG) for the potential pathway of AR treat NAFLD. |
Preparation and Characterization of GA-MSe@AR
Despite the promising potential of AR for the treatment of NAFLD, its therapeutic efficacy is limited by low water solubility, poor bioavailability, and non-liver-targeting specificity. To enhance the therapeutic efficacy of AR, MSe NPs with numerous microporous structures and optimal particle size were synthesized using CTAB template method. AR was subsequently loaded onto the surface of the MSe NPs to fabricate MSe@AR. To enable liver-targeted delivery, the surface of MSe@AR was modified with GA, resulting in the construction of GA-MSe@AR. This modification leverages the specific binding interaction between GA and ASGPR, which is highly expressed in hepatocytes.
The successful synthesis of GA-MSe@AR was confirmed through various characterization techniques. TEM revealed a spherical morphology (Figure 2A). Particle size analysis showed that GA-MSe@AR had a diameter range of 20–40 nm, with an average diameter of approximately 30 nm (Figure 2B). The EDX analysis identified the elemental composition of GA-MSe@AR, displaying classical selenium signal peaks from MSe NPs, along with carbon and oxygen peaks derived from GA and/or AR (Figure 2C). This confirmed the successful loading of GA and AR onto the surface of MSe NPs. Furthermore, FTIR spectroscopy was used to characterize the chemical structure of GA-MSe@AR. The FTIR spectrum (Figure 2D) revealed peaks similar to those of MSe, a typical lactone bond peak at 1765 cm−1 from AR, and hydroxyl peaks between 3650 cm−1 and 3150 cm−1 from GA, further confirming the successful functionalization of MSe with GA and AR.
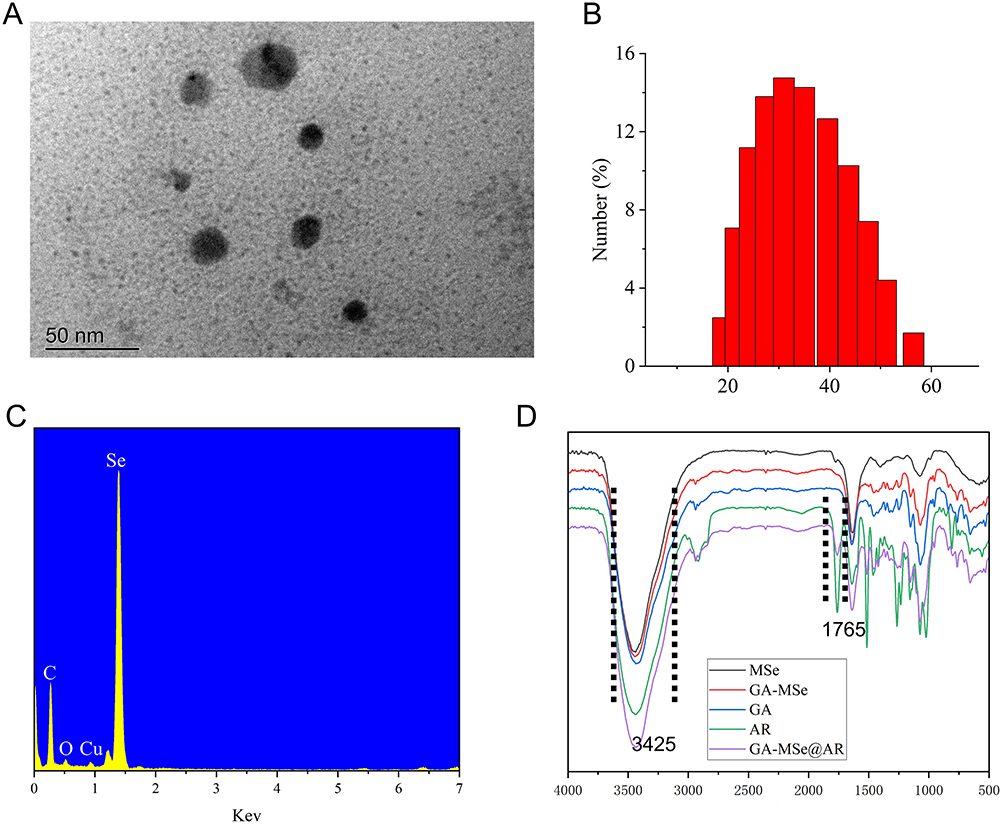 |
Figure 2 Characterization of GA-MSe@AR. (A) Representative transmission electron microscopy (TEM) image of GA-MSe@AR, scale bar = 50 nm. (B) Size distribution of GA-MSe@AR. (C) The elemental analysis of GA-MSe@AR by Energy-dispersive X-ray (EDX). (D) The chemical structure of GA-MSe@AR was characterized by Fourier transform infrared (FTIR). Notes: MSeNPs (Mesoporous selenium nanoparticles), GA (galactose), AR (arctiin), and GA-MSe@AR (GA-modified MSeNPs loaded with AR). |
Toxicity of GA-MSe@AR
The biosafety of nanoparticles is a critical consideration for their therapeutic application. To evaluate the cytotoxicity of GA-MSe@AR, a CCK-8 assay was performed in vitro. As shown in Figure S1, GA-MSe@AR exhibited minimal impact on hepatocyte viability at concentrations below 400 ng/mL in LO2 cells and 800 ng/mL in AML12 cells. Even at the highest tested concentration of 1600 ng/mL, the majority of hepatocytes in both cell lines remained viable. These findings suggest that GA-MSe@AR possesses low cytotoxicity, supporting its potential as a safe therapeutic agent for NAFLD treatment.
Cellular Uptake of GA-MSe@AR
Effective cellular uptake of GA-MSe@AR is critical for its biological effects. To visualize this process, coumarin 6-labeled GA-MSe@AR was employed. As shown in Figure S2, a faint green fluorescence (coumarin 6-labeled GA-MSe@AR) was observed in both LO2 and AML12 cells after 1 hour of co-incubation, indicating the initial uptake of GA-MSe@AR. The fluorescence intensity increased notably after 2 hours, demonstrating enhanced cellular internalization. By 4 hours, GA-MSe@AR had permeated significantly throughout the cytoplasmic region. These results demonstrate that GA-MSe@AR is taken up effectively by cells in a time-dependent manner.
Furthermore, to verify whether GA-MSe@AR targets the liver by binding to ASGPR on hepatocytes through GA, we evaluated the uptake efficiency of GA-MSe@AR in AML12 cells (with high ASGPR expression) and DU145 cells (with low ASGPR expression). As shown in Figure S3, AML12 cells exhibited significantly higher uptake rates and greater amounts of GA-MSe@AR compared to DU145 cells in 1–4 hours of co-incubation. The superior uptake efficiency of GA-MSe@AR in hepatocytes is likely due to the binding of the GA ligand to ASGPR on the hepatocyte surface, which promotes the uptake of GA-MSe@AR by hepatocytes. Collectively, these findings validate the ASGPR-dependent targeting strategy of GA-MSe@AR, establishing its potential for selective hepatocyte delivery and subsequent therapeutic applications in liver-targeted therapy.
AR Release Efficiency from GA-MSe@AR
Following cellular uptake, the release of AR from GA-MSe@AR is essential for its biological activity. To evaluate the release efficiency of AR under different pH conditions (pH 5.4 versus 7.4), high-performance liquid chromatography (HPLC) was employed. GA-MSe@AR slowly released 45% of AR after 16 hours of incubation at pH 7.4 (mimicking the physiological environment) (Figure S4). In contrast, at pH 5.4 (the analogous lysosomal environment), AR was rapidly released, reaching about 60% within the first 1 hour and 70% cumulative release after 16 hours. The results revealed a notably higher release efficiency of AR at pH 5.4 (70%) compared to pH 7.4 (45%) (**p < 0.01), suggesting that the acidic environment facilitates the release of AR from the GA-MSe@AR. This acid-dependent release of GA-MSe@AR may be attributed to the occurrence of an acid hydrolysis reaction within an acidic environment, which subsequently facilitated the dissociation of AR from GA-MSe@AR.
Lysosomal Escape Capacity of GA-MSe@AR
Upon entering the cell, GA-MSe@AR initially accumulates in the lysosomes before escaping to the cytoplasm. To observe the lysosomal escape dynamics of GA-MSe@AR in LO2 and AML12 cells, fluorescence microscopy was employed. In LO2 cells, a brief 0.25-hour incubation with coumarin 6-labeled GA-MSe@AR (green fluorescence) showed limited uptake and colocalization with lysosomes (red fluorescence) (Figure 3A). After 0.5 hours of incubation, an increase in GA-MSe@AR accumulation within lysosomes was noted, with the green and red fluorescence largely overlapping. Notably, GA-MSe@AR escaped from lysosomes after 1 hour of incubation, marked by the emergence of non-overlapping green fluorescence. Furthermore, the lysosomal escape effect became more pronounced after 2 and 4 hours of incubation, evidenced by the green fluorescence dispersed throughout the cytoplasm. Additionally, a similar lysosomal escape process of GA-MSe@AR was also observed in AML12 cells (Figure 3B), further demonstrating that GA-MSe@AR could efficiently escape from the lysosomal region into the cytoplasm. This lysosomal escape capability enhances the potential of GA-MSe@AR to exert its biological effects within hepatocytes.
 |
Figure 3 Lysosomal escape of GA-MSe@AR. The lysosomes were labeled with LysoTracker (75 nM, red fluorescence), and nuclei were stained with Hoechst 33342 (10 µM, blue fluorescence). Subsequently, coumarin 6-labeled GA-MSe@AR (5 μg/mL, green fluorescence) was co-cultured with LO2 cells (A) and AML12 cells (B) for 0.25, 0.5, 1, 2, and 4 hours. Scale bar = 50 µm. |
GA-MSe@AR Inhibits Cellular Lipid Deposition in Cells
Excessive lipid accumulation in hepatocytes leads to NAFLD. To evaluate the lipid level-lowering effects of GA-MSe@AR, FFA (palmitate/oleate =1/2)-induced lipid deposition models were established in LO2 and AML12 cells. After FFA treatment for 24 hours, abundant lipid droplets (red droplets) were observed in both LO2 cells (Figure 4A) and AML12 cells (Figure 4B) by Oil Red O staining, in comparison to the control group. Subsequent 24-hour treatments with GA-MSe, AR, or GA-MSe@AR demonstrated a dose-dependent reduction in lipid accumulation. Notably, the GA-MSe@AR group exhibited the most pronounced decrease in lipid droplets, outperforming the groups treated with GA-MSe or AR alone. To further investigate the lipid-lowering effect of GA-MSe@AR, BODIPY fluorescence dye was used. Consistent with the Oil Red O staining results, GA-MSe@AR demonstrated the most pronounced reduction in lipid droplets (green fluorescence) in both LO2 cells and AML12 cells, followed by AR and GA-MSe (Figure 4C and D). These results indicate that GA-MSe@AR effectively reduces lipid accumulation in hepatocytes, probably through synergistic action of GA-MSe and AR.
 |
Figure 4 Lipid-lowering effect of GA-MSe@AR in vitro. Lipid deposition cell models were established by exposing LO2 cells and AML12 cells to FFA (1 mM). The cells were subsequently incubated with GA-MSe@AR, GA-MSe, or AR (each at 5 μg/mL) overnight. Untreated cells were served as controls. Lipid deposition in LO2 cells (A) and AML12 cells (B) was detected using Oil Red O staining and captured using a light microscope, scale bar = 100 μm. In addition, lipid droplets in LO2 cells (C) and AML12 cells (D) were visualized using BODIPY fluorescent staining (10 μM, green fluorescence), nuclei were stained with Hoechst 33342 (10 μM, blue fluorescence), scale bar = 50 μm. |
GA-MSe@AR Reduced Oxidative Stress in Cells
Abnormal lipid deposition in hepatocytes initiates a cascade of oxidative stress, which causes hepatocyte damage and exacerbates the progression of NAFLD. To evaluate the antioxidant potential of GA-MSe@AR, oxidative stress cell models were established by exposing LO2 cells and AML12 cells to PA. DCFH-DA was then employed to measure ROS levels, reflecting the oxidative stress status of cells. As depicted in Figure 5A and B, the PA treatment significantly increased ROS levels (green fluorescence) in both LO2 cells and AML12 cells. Subsequently, the ROS levels were slightly reduced by GA-MSe treatment, probably due to the selenium element promoting the synthesis of the selenium-dependent enzyme selenocysteine lyase (SCLY), which is an antioxidant enzyme. Furthermore, AR treatment significantly reduced ROS levels. As anticipated, GA-MSe@AR resulted in the most pronounced ROS reduction, possibly due to the combined antioxidant activity of AR and GA-MSe.
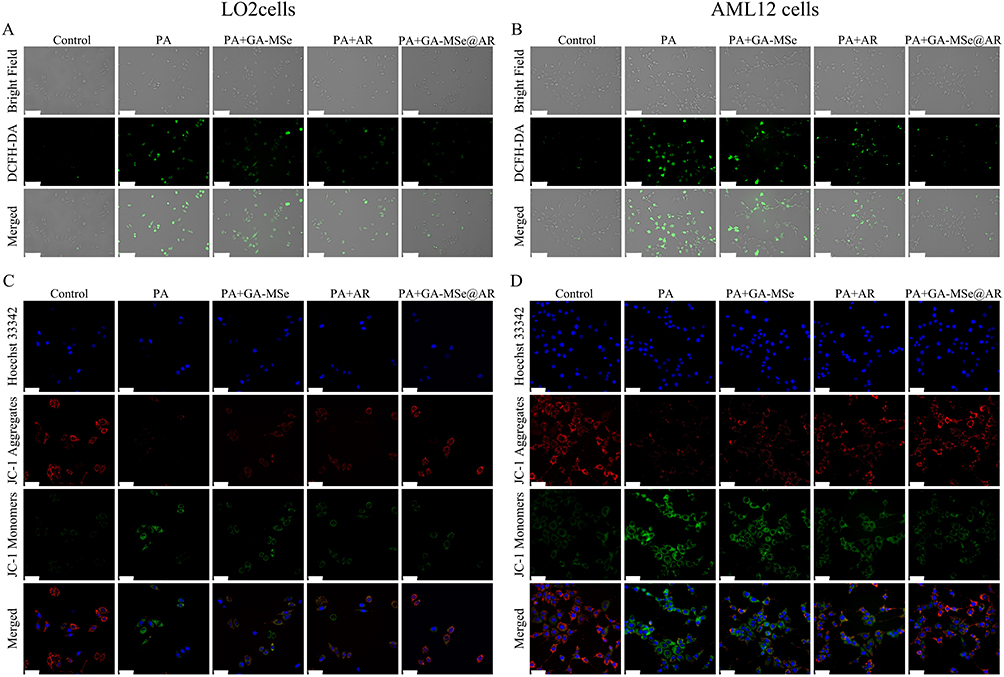 |
Figure 5 GA-MSe@AR reduced ROS production and protected against mitochondrial damage in cells. Oxidative stress cell models were established by exposing LO2 cells and AML12 cells to 0.5 mM PA. Subsequently, the cells were treated with 5 μg/mL GA-MSe@AR, GA-MSe, or AR for 24 hours. Untreated cells served as controls. ROS levels were assessed using DCFH-DA fluorescence staining in LO2 cells (A) and AML12 cells (B). Scale bar = 100 μm. MMP changes in LO2 cells (C) and AML12 cells (D) were detected to monitor mitochondrial status using JC-1 fluorescence staining. Scale bar = 50 μm. |
Given that mitochondria are primary sites of ROS production and play a critical role in cellular oxidative stress responses,41 MMP was measured as an indicator of mitochondrial health. MMP was assessed using JC-1 staining, with red fluorescence (JC-1 aggregates) indicating healthy mitochondria and green fluorescence (JC-1 monomers) signifying damaged mitochondria. Compared to control cells, PA-treated LO2 (Figure 5C) and AML12 cells (Figure 5D) displayed increased green fluorescence and diminished red fluorescence, indicating mitochondrial dysfunction. However, treatments with GA-MSe, AR, and GA-MSe@AR mitigated mitochondrial damage to varying extents, with GA-MSe@AR exhibiting the strongest protective effect. These findings suggest that GA-MSe@AR not only reduces ROS production but also protects mitochondrial integrity, highlighting its potent antioxidant properties.
In vivo Biodistribution of GA-MSe@AR
Although GA-MSe@AR has demonstrated potent lipid-lowering and antioxidant properties in vitro, its ability to specifically target the liver for effective NAFLD treatment remains uncertain. To evaluate its liver-targeting capability, sulfo-cyanine 3 (Cy3)-labeled GA-MSe@AR (5 mg/kg) was injected into mice, and fluorescence signals were visualized using a live imaging system. As depicted in Figure 6, no fluorescent signals were observed in any organ of the control group. However, after 15 minutes of GA-MSe@AR injection, a distinct red fluorescence appeared exclusively in the livers of the mice, indicating initial accumulation of GA-MSe@AR in the liver. At 30 and 60 minutes post-injection, the fluorescence signal in the liver became even more intense, suggesting a substantial accumulation of GA-MSe@AR in this organ. Additionally, a faint fluorescence was observed in the kidneys, possibly due to the metabolism and excretion of GA-MSe@AR by the kidneys, while no fluorescence was observed in other organs. These findings indicate that GA-MSe@AR predominantly accumulates in the liver, likely due to the specific binding between GA and ASGPR. This targeted delivery property underscores the potential of GA-MSe@AR for in vivo treatment of NAFLD.
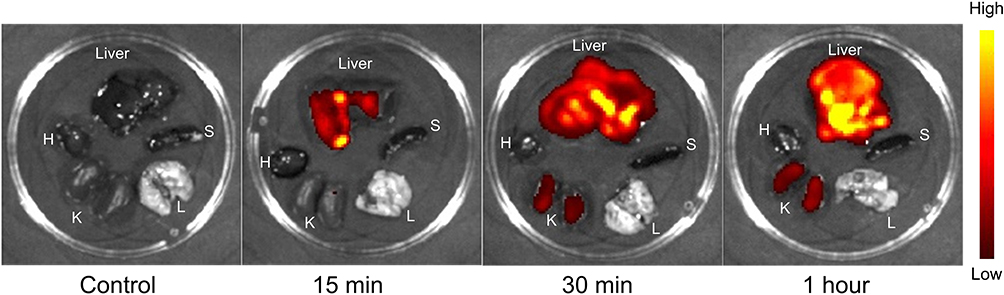 |
Figure 6 In vivo biodistribution of GA-MSe@AR. Mice were injected with 5 mg/kg of sulfo-cyanine 3 (Cy3)-labeled GA-MSe@AR via the tail vein. Untreated mice were used as controls. The mice were euthanized at 15, 30, and 60 minutes post-injection, then the hearts (H), livers, spleens (S), lungs (L), and kidneys (K) were harvested immediately for biodistribution analysis of GA-MSe@AR using a living imaging system. |
GA-MSe@AR Suppressed Body Weight Gain and Decreased the Levels of Biomarkers in Mice
To further evaluate the potential of GA-MSe@AR against NAFLD in vivo, an NAFLD mouse model was established using an HFD, while control mice were fed an LFD. As described in Figure 7A, the mice were injected with GA-MSe, AR, or GA-MSe@AR at 5 mg/kg via the tail vein twice weekly for 16 weeks. An equal volume of saline was injected into the control mice following the same schedule. Compared to the LFD group, the body weight of mice in the HFD group was rapidly increased (Figure 7B). This HFD-induced weight gain was significantly inhibited by GA-MSe@AR, followed by AR and GA-MSe.
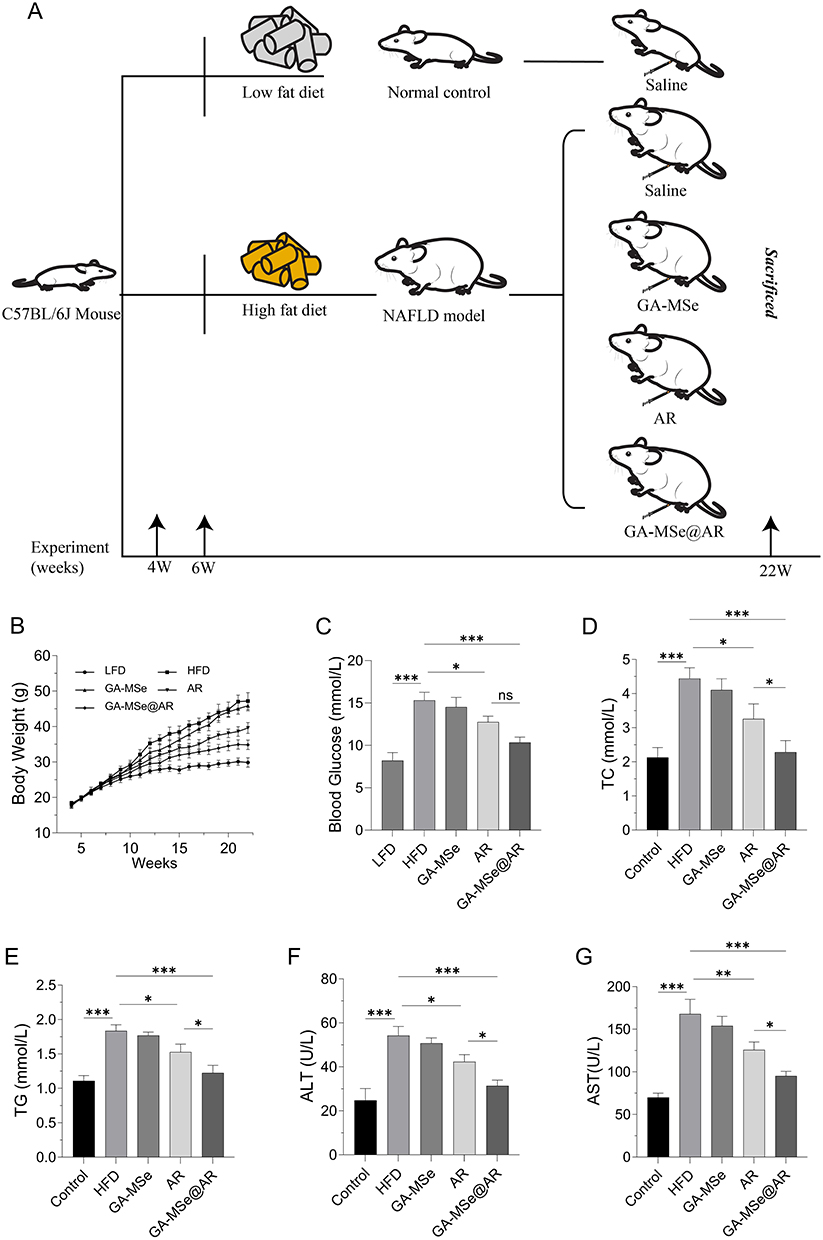 |
Figure 7 GA-MSe@AR reduced body weight and blood biochemical parameters in mice. (A) Scheme of the mouse experiment. C57BL/6J mice were randomly divided into five groups (n = 6). The mice received either a low-fat diet (LFD, 10% kcal fat) with saline, or a high-fat diet (HFD, 60% kcal fat) supplemented with saline, GA-MSe, AR, or GA-MSe@AR (all at 5 mg/kg, administered twice weekly, i.v.) for 16 weeks. (B) Body weight changes of mice in each group during the experiment. Blood glucose (C), serum total cholesterol (TC) (D), and serum triglycerides (TG) (E) were measured to evaluate the metabolic status of the mice. Liver injury markers alanine aminotransferase (ALT) (F) and aspartate aminotransferase (AST) (G) were measured to assess the extent of hepatocyte injury in each group of mice. *p<0.05, **p<0.01, ***p<0.001. |
Furthermore, blood glucose, serum TC, and TG levels were measured to reflect the metabolic status in different groups of mice. As shown in Figure 7C–E, the blood glucose levels (***p<0.001), TC (***p<0.001), and TG (***p<0.001) in the GA-MSe@AR group were significantly reduced compared to the HFD group, followed by AR and GA-MSe group, indicating that GA-MSe@AR possesses superior hypoglycemic and lipid-lowering properties compared to AR and GA-MSe. In addition, the impact of GA-MSe@AR on hepatocyte damage was evaluated. Liver damage markers, ALT and AST, were significantly elevated in HFD-fed mice compared to the LFD group (Figure 7F and G). However, treatment with GA-MSe@AR markedly reduced ALT (***p<0.001) and AST (***p<0.001) levels, while the AR and GA-MSe groups exhibited lesser reductions. These results suggest that GA-MSe@AR offers superior hepatocyte protection compared to AR and GA-MSe, likely through its potent blood glucose- and lipid-lowering effects.
GA-MSe@AR Reduced Hepatic Lipid Deposition and Improved Pancreatic Function in Mice
To further evaluate the lipid-lowering effects of GA-MSe@AR, liver sections from treated mice were subjected to hematoxylin and eosin (H&E) staining. In the control group, liver lobules were well-organized, with minimal vacuole formation (Figure 8A). Conversely, the HFD group exhibited substantial hepatic lipid deposition, as indicated by the presence of extensive white vacuoles and disrupted liver lobule structures. Notably, the GA-MSe, AR, and GA-MSe@AR groups showed a gradual reduction in lipid deposition, as shown by the number and size of white vacuoles. Furthermore, Oil Red O staining was performed to more accurately and visually analyze lipid deposition in mice liver. The LFD group showed almost no lipid accumulation (Figure 8B), whereas abundant lipid accumulation (red dots) was observed in the HFD group. In contrast, a marked decrease in lipid accumulation was observed in GA-MSe, AR, and GA-MSe@AR treatment groups compared to the HFD group, with GA-MSe@AR exhibiting the strongest lipid reduction effect. These results indicated that GA-MSe@AR exhibited a significantly greater lipid-lowering effect than AR and GA-MSe.
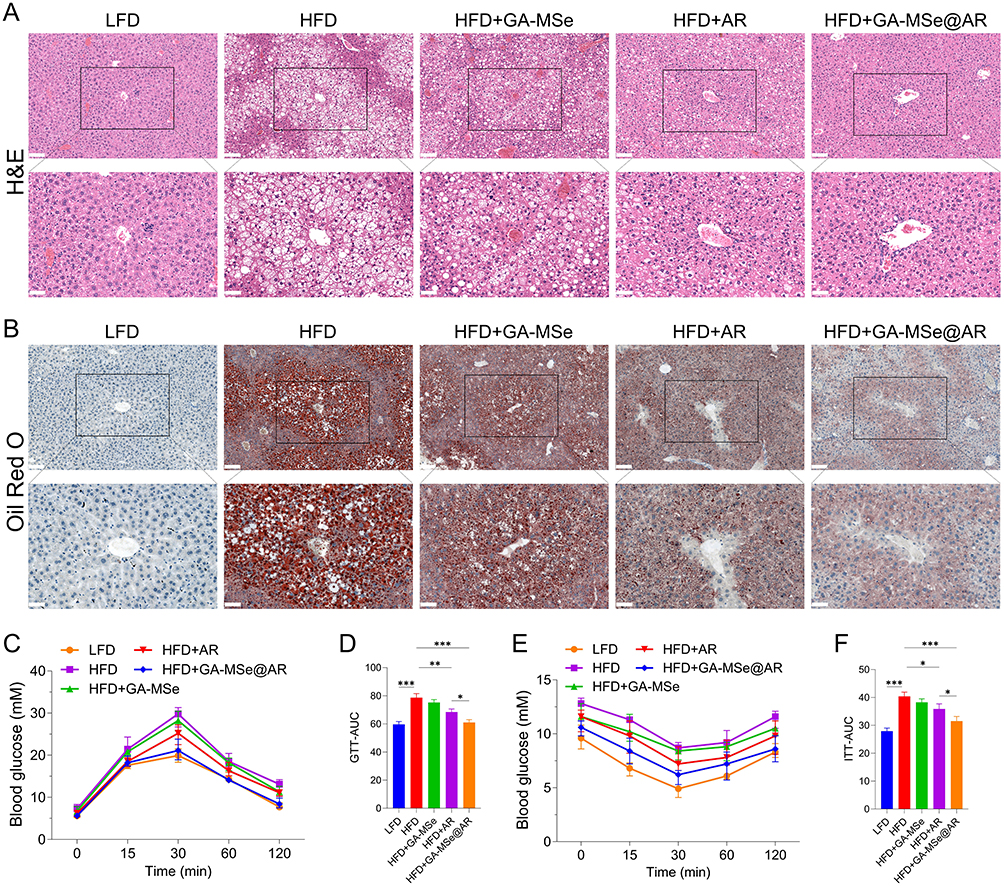 |
Figure 8 GA-MSe@AR reduced hepatic lipid deposition and improved pancreatic function in mice. H&E (A) and Oil red O (B) staining of mouse liver were performed to monitor lipid deposition, scale bar = 100 μm and 50 μm. The mouse glucose tolerance test (GTT) (C) and the corresponding area under the curve (AUC) (D) in each group, and the mouse insulin tolerance test (ITT) (E) and the corresponding AUC (F) were performed to reflect the mouse pancreatic function in different groups. *p<0.05, **p<0.01, ***p<0.001. |
In addition, pancreatic function plays a pivotal role in mediating lipid deposition in vivo.42 Therefore, the GTT and ITT experiments were conducted to monitor pancreatic function in mice. Notably, the continuous HFD diet impaired glucose tolerance (Figure 8C and D) and reduced insulin sensitivity (Figure 8E and F) in mice. As expected, GA-MSe@AR reversed HFD-induced pancreatic dysfunction, thereby ameliorating insulin resistance in HFD-fed mice to a greater extent than AR and GA-MSe. These findings suggest that GA-MSe@AR reduces hepatic lipid deposition by improving pancreatic function in vivo.
GA-MSe@AR Exhibited Antioxidant and Anti-Inflammatory Effect in Mice
Increased oxidative stress damages hepatocytes and accelerates the progression of NAFLD.5 To assess the oxidative stress status, hepatic levels of MDA and glutathione disulfide (GSSG) (oxidative stress markers) were measured, along with GSH levels and the GSH/GSSG ratio (antioxidant indicators). In the HFD group, hepatic MDA and GSSG levels were significantly elevated (Figure 9A and B), while GSH levels and the GSH/GSSG ratio were markedly reduced (Figure 9C and D) compared to the LFD group. As anticipated, GA-MSe@AR significantly reduced the hepatic oxidative stress levels and increased the antioxidative levels, followed by AR and GA-MSe. The potent antioxidant activity of GA-MSe@AR was probably due to the combined antioxidant properties of GA-MSe and AR.
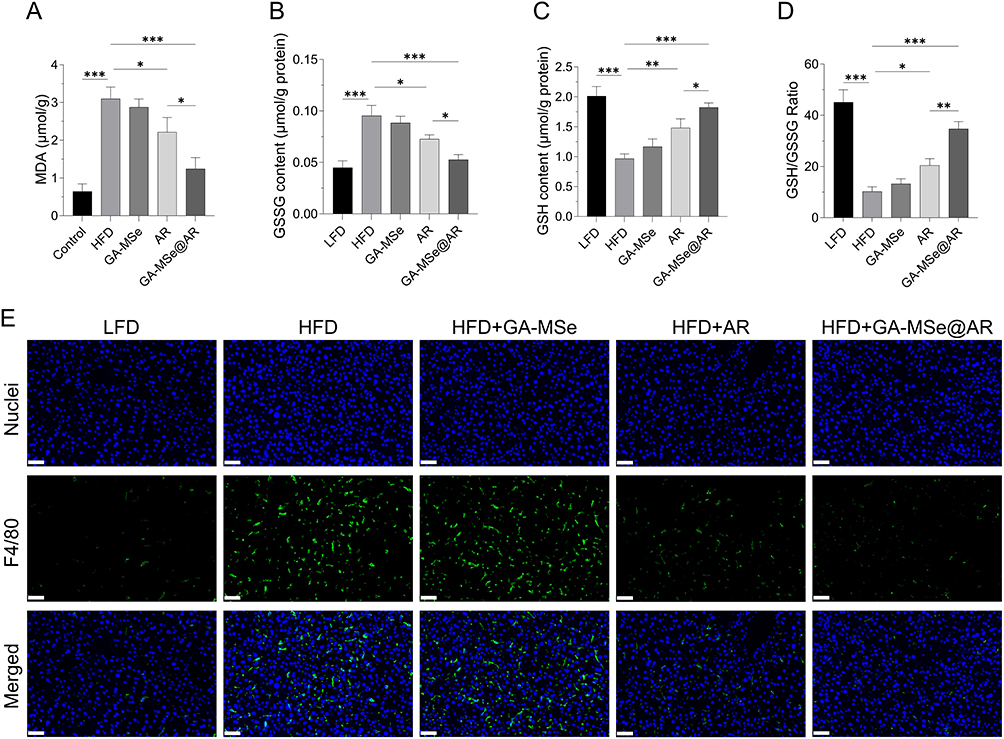 |
Figure 9 GA-MSe@AR exhibited antioxidant and anti-inflammatory effects in mice. The oxidative stress markers, malondialdehyde (MDA) (A) and glutathione disulfide (GSSG) (B), alongside antioxidant indicators glutathione (GSH) (C) and GSH/GSSG ratio (D), were measured to assess the oxidative stress status of the mice liver. *p<0.05, **p<0.01, ***p<0.001. (E) Inflammatory status of the liver was monitored by F4/80 immunofluorescence staining, with nuclei and F4/80 stained blue and green, respectively. Scale bar = 50 μm. |
Oxidative stress is also known to initiate and exacerbate inflammation, further damaging liver tissue and worsening NAFLD.6 To evaluate the inflammatory status, the mouse macrophage marker F4/80 was used for immunofluorescence staining. The HFD group exhibited severe liver inflammation, evidenced by increased macrophage fluorescence intensity and number (Figure 9E) compared to the LFD group. Treatment with GA-MSe, AR, and GA-MSe@AR progressively decreased liver inflammation, with GA-MSe@AR showing the most pronounced anti-inflammatory effect. These findings suggest that GA-MSe@AR exerts hepatoprotective effects by mitigating hepatic oxidative stress and inflammation, demonstrating its potential as a superior therapeutic agent for NAFLD.
GA-MSe@AR Regulated Adipogenesis- and Oxidative Stress-Related Proteins in Mice Liver
To investigate how GA-MSe@AR exhibits lipid-lowering and antioxidant properties, protein levels relative to adipogenesis and oxidative stress in mice liver were examined by Western blotting analysis. In comparison to the LFD group, key adipogenesis proteins, including the fatty acid synthase (FASN), peroxisome proliferator-activated receptor-γ (PPAR-γ), and sterol regulatory element binding protein-1c (SREBP-1c), were markedly elevated in the HFD group (Figure 10A–D). In contrast, GA-MSe@AR significantly downregulated the expression levels of those adipogenesis proteins. Concurrently, the protein levels of the antioxidant enzymes, superoxide dismutase 2 (SOD2) and catalase (CAT), were significantly reduced in the HFD group compared to the LFD group. This reduction was accompanied by elevated expression of NADPH oxidase 4 (NOX4), a key marker of oxidative stress (Figure 10E–H). Remarkably, these alterations were effectively reversed by GA-MSe@AR intervention. These findings suggest that GA-MSe@AR exerts lipid-lowering and antioxidant effects by inhibiting adipogenesis protein expression, suppressing oxidative stress markers, and upregulating antioxidant protein levels.
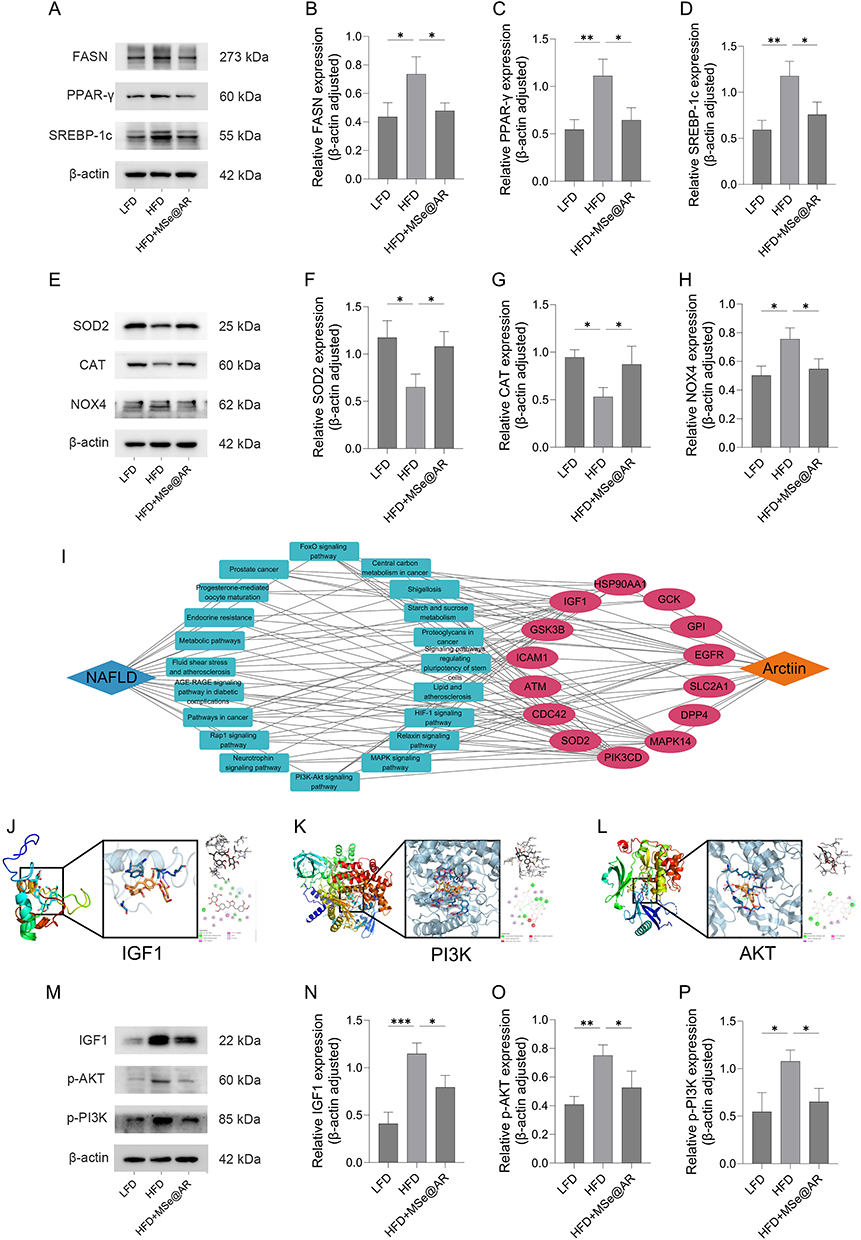 |
Figure 10 GA-MSe@AR inhibited the IGF1/PI3K/Akt pathway. (A–D). Western blotting analysis of the fatty acid synthase (FASN), peroxisome proliferator-activated receptor-γ (PPARγ), and sterol regulatory element binding protein-1c (SREBP-1c) proteins and their quantitative measurements in mice liver. (E–H). Expression of superoxide dismutase 2 (SOD2), catalase (CAT), and NADPH Oxidase 4 (NOX4) in the liver of the mice. (I). Drug-target-pathway-disease networks were constructed to explore the potential mechanism of AR in the treatment of NAFLD. The binding capability of AR with insulin-like growth factor 1 (IGF1) (J), phosphatidylinositol-3-kinase (PI3K) (K), and protein kinase B (Akt) (L) was investigated by molecular docking. (M–P). The protein expression of IGF1, p-PI3K, and p-Akt in the liver of the mice. *p < 0.05, **p < 0.01, ***p < 0.001. |
GA-MSe@AR Inhibited IGF1/PI3K/AKT Pathway
To explore the underlying mechanism of GA-MSe@AR in treating NAFLD, drug-target-pathway-disease (DTPD) networks were constructed. Analysis of the DTPD network (Figure 10I) suggested that AR may treat NAFLD by modulating insulin-like growth factor 1 (IGF1) to regulate the PI3K-Akt pathway. Additionally, IGF1 was identified as a promising target for NAFLD therapy,43 and the PI3K-Akt pathway has been reported to regulate lipid synthesis44 and oxidative stress.45 Furthermore, molecular docking revealed that AR could strongly bind to IGF1 (Figure 10J), PI3K (Figure 10K), and Akt (Figure 10L) (Table S2). Thus, we inferred that GA-MSe@AR could reduce lipid deposition and oxidation through the IGF1/PI3K/Akt pathway. Consistent with the predicted result, the IGF1, p-PI3K, and p-Akt protein expression in the liver of HFD-fed mice were significantly increased compared to LFD-fed mice. Treatment with GA-MSe@AR reversed these changes (Figure 10M–P). These results demonstrate that GA-MSe@AR exerts its dual lipid-lowering and antioxidant activity by inhibiting the IGF1-PI3K-Akt signaling pathway, and thus represents a promising approach for the treatment of NAFLD.
Conclusion
In this study, AR was identified as a potential therapeutic natural product for NAFLD, owing to its antioxidant, anti-inflammatory, and metabolic regulatory properties. To improve the therapeutic efficacy of AR, a novel liver-targeting selenium nanoparticle GA-MSe@AR was successfully synthesized for delivery AR to treat NAFLD. GA-MSe@AR demonstrated low toxicity, acid-dependent release, good lysosomal escape ability, marked cellular uptake in vitro. Living imaging analysis revealed that GA-MSe@AR showed liver-targeting ability in mice. GA-MSe@AR exhibited more pronounced antioxidant and lipid-lowering activity than AR alone both in vitro and in vivo. This was paralleled by a more effective reduction in the blood biochemical index, hepatic inflammation, and an improvement in pancreatic function in HFD mice. Furthermore, GA-MSe@AR reduced the protein expression of adipogenesis and oxidative stress by inhibiting the IGF1/PI3K/Akt pathway. In conclusion, GA-MSe@AR emerges as a promising multi-targeted therapeutic agent for NAFLD, leveraging the combination of selenium and AR with targeted delivery to the liver. This innovative selenium-based nanotherapeutic agent exhibits potent antioxidant, lipid-lowering, anti-inflammatory, and insulin resistance reducing properties. Notably, even GA-MSe demonstrates mild anti-NAFLD effects, offering a novel perspective for advancing drug development strategies in NAFLD.
Abbreviations
NAFLD, non-alcoholic fatty liver disease; AR, arctiin; MSe, the mesoporous selenium nanoparticles; GA, galactose; GA-MSe, GA modified MSe; GA-MSe@AR, GA modified MSe coated AR; ROS, reactive oxygen species; ASGPR, the asialoglycoprotein receptor; CTAB, cetyltrimethyl ammonium bromide; TEM, transmission electron microscope; DLS, dynamic light scattering; EDX, energy dispersive X-ray spectroscopy; FTIR, Fourier transform infrared spectroscopy; OTC, ornithine carbamyltransferase; BSA, bovine serum albumin; FBS, fetal bovine serum; DCFH-DA, 2′,7′-dichlorodihydrofluorescein diacetate; AST, aspartate aminotransferase; ALT, alanine aminotransferase; TC, total cholesterol; TG, triglycerides; RIPA, radio immunoprecipitation assay buffer; PMSF, phenylmethanesulfonyl fluoride; BCA, bicinchoninic acid; MDA, malondialdehyde; GSH/GSSG, glutathione/glutathione disulfide; HFD, high-fat diet; LFD, low-fat diet; GTT, glucose tolerance test; ITT, insulin tolerance test; SREBP-1c, sterol regulatory element-binding protein-1c; FAS, fatty acid synthase; PPAR-γ, peroxisome proliferator-activated receptor gamma; NOX4, NADPH oxidase 4; SOD2, superoxide dismutase 2; CAT, catalase; F4/80, a macrophage marker; p-PI3K, phosphorylated phosphatidylinositol 3-kinase; p-AKT, phosphorylated AKT; IGF1, insulin-like growth factor 1.
Acknowledgments
This research was funded by Guangzhou Science and Technology Program (2023A03J0952) and the Guangdong Basic and Applied Basic Research Foundation (2021A1515110457).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Riazi K, Azhari H, Charette JH, et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2022;7(9):851–861. doi:10.1016/s2468-1253(22)00165-0
2. Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. Lancet. 2021;397(10290):2212–2224. doi:10.1016/s0140-6736(20)32511-3
3. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell mol Life Sci. 2018;75(18):3313–3327. doi:10.1007/s00018-018-2860-6
4. Scorletti E, Carr RM. A new perspective on NAFLD: focusing on lipid droplets. J Hepatol. 2022;76(4):934–945. doi:10.1016/j.jhep.2021.11.009
5. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020;152:116–141. doi:10.1016/j.freeradbiomed.2020.02.025
6. Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwälder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. 2022;77(4):1136–1160. doi:10.1016/j.jhep.2022.06.012
7. Guo X, Yin X, Liu Z, Wang J. Non-Alcoholic Fatty Liver Disease (NAFLD) pathogenesis and natural products for prevention and treatment. Int J mol Sci. 2022;23(24):15489. doi:10.3390/ijms232415489
8. Wang L, Yan Y, Wu L, Peng J. Natural products in non-alcoholic fatty liver disease (NAFLD): novel lead discovery for drug development. Pharmacol Res. 2023;196:106925. doi:10.1016/j.phrs.2023.106925
9. Fierascu RC, Georgiev MI, Fierascu I, et al. Mitodepressive, antioxidant, antifungal and anti-inflammatory effects of wild-growing Romanian native Arctium lappa L. (Asteraceae) and veronica persica poiret (Plantaginaceae). Food Chem Toxicol. 2018;111:44–52. doi:10.1016/j.fct.2017.11.008
10. Zhang X, Zhang N, Kan J, et al. Anti-inflammatory activity of alkali-soluble polysaccharides from Arctium lappa L. and its effect on gut microbiota of mice with inflammation. Int J Biol Macromol. 2020;154:773–787. doi:10.1016/j.ijbiomac.2020.03.111
11. Yosri N, Alsharif SM, Xiao J, et al. Arctium lappa (Burdock): insights from ethnopharmacology potential, chemical constituents, clinical studies, pharmacological utility and nanomedicine. Biomed Pharmacother. 2023;158:114104. doi:10.1016/j.biopha.2022.114104
12. Ma K, Sheng W, Gao R, et al. Ethanolic extract of root from Arctium lappa L ameliorates obesity and hepatic steatosis in rats by regulating the AMPK/ACC/CPT-1 pathway. J Food Biochem. 2022;46(12):e14455. doi:10.1111/jfbc.14455
13. Gao Q, Yang M, Zuo Z. Overview of the anti-inflammatory effects, pharmacokinetic properties and clinical efficacies of arctigenin and arctiin from Arctium lappa L. Acta Pharmacol Sin. 2018;39(5):787–801. doi:10.1038/aps.2018.32
14. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
15. Lv Y, Li W, Liao W, et al. Nano-drug delivery systems based on natural products. Int J Nanomed. 2024;19:541–569. doi:10.2147/ijn.S443692
16. Lu J, Zeng Y, Zhong H, et al. Dual-stimuli-responsive gut microbiota-targeting nitidine chloride-CS/PT-NPs improved metabolic status in NAFLD. Int J Nanomed. 2024;19:2409–2428. doi:10.2147/ijn.S452194
17. Fan N, Zhao J, Zhao W, et al. Celastrol-loaded lactosylated albumin nanoparticles attenuate hepatic steatosis in non-alcoholic fatty liver disease. J Control Release. 2022;347:44–54. doi:10.1016/j.jconrel.2022.04.034
18. Khurana A, Tekula S, Saifi MA, Venkatesh P, Godugu C. Therapeutic applications of selenium nanoparticles. Biomed Pharmacother. 2019;111:802–812. doi:10.1016/j.biopha.2018.12.146
19. Xia Y, Xiao M, Zhao M, et al. Doxorubicin-loaded functionalized selenium nanoparticles for enhanced antitumor efficacy in cervical carcinoma therapy. Mater Sci Eng C Mater Biol Appl. 2020;106:110100. doi:10.1016/j.msec.2019.110100
20. Xia Y, Tang G, Guo M, et al. Silencing KLK12 expression via RGDfC-decorated selenium nanoparticles for the treatment of colorectal cancer in vitro and in vivo. Mater Sci Eng C Mater Biol Appl. 2020;110:110594. doi:10.1016/j.msec.2019.110594
21. Tran PA, O’Brien-Simpson N, Palmer JA, et al. Selenium nanoparticles as anti-infective implant coatings for trauma orthopedics against methicillin-resistant Staphylococcus aureus and epidermidis: in vitro and in vivo assessment. Int J Nanomed. 2019;14:4613–4624. doi:10.2147/ijn.S197737
22. Xu L, Lu Y, Wang N, Feng Y. The role and mechanisms of selenium supplementation on fatty liver-associated disorder. Antioxidants. 2022;11(5):922. doi:10.3390/antiox11050922
23. Mikhailova EO. Selenium Nanoparticles: green Synthesis and Biomedical Application. Molecules. 2023;28(24):8125. doi:10.3390/molecules28248125
24. Xia Y, Guo M, Xu T, et al. siRNA-loaded selenium nanoparticle modified with hyaluronic acid for enhanced hepatocellular carcinoma therapy. Int J Nanomed. 2018;13:1539–1552. doi:10.2147/ijn.S157519
25. Xia Y, Zhong J, Zhao M, et al. Galactose-modified selenium nanoparticles for targeted delivery of doxorubicin to hepatocellular carcinoma. Drug Deliv. 2019;26(1):1–11. doi:10.1080/10717544.2018.1556359
26. D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting - strategies and applications. J Control Release. 2015;203:126–139. doi:10.1016/j.jconrel.2015.02.022
27. Ru J, Li P, Wang J, et al. TCMSP: a database of systems pharmacology for drug discovery from herbal medicines. J Cheminform. 2014;6:13. doi:10.1186/1758-2946-6-13
28. Daina A, Michielin O, Zoete V. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019;47(W1):W357–w364. doi:10.1093/nar/gkz382
29. Zdrazil B, Felix E, Hunter F, et al. The ChEMBL database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 2024;52(D1):D1180–d1192. doi:10.1093/nar/gkad1004
30. Wang X, Shen Y, Wang S, et al. PharmMapper 2017 update: a web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017;45(W1):W356–w360. doi:10.1093/nar/gkx374
31. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–98. doi:10.1093/nar/gku1205
32. Safran M, Rosen N, Twik M, et al. The GeneCards Suite. In: Abugessaisa I, Kasukawa T, editors. Practical Guide to Life Science Databases. Springer Nature Singapore; 2021;27–56. doi:10.1007/978-981-16-5812-9_2
33. Zhu F, Han B, Kumar P, et al. Update of TTD: therapeutic target database. Nucleic Acids Res. 2010;38:D787–91. doi:10.1093/nar/gkp1014
34. UniProt, Consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 2023;51(D1):D523–d531. doi:10.1093/nar/gkac1052
35. von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31(1):258–261. doi:10.1093/nar/gkg034
36. Sherman BT, Hao M, Qiu J, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022;50(W1):W216–w221. doi:10.1093/nar/gkac194
37. Burley SK, Bhikadiya C, Bi C, et al. RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023;51(D1):D488–d508. doi:10.1093/nar/gkac1077
38. Liu Y, Yang X, Gan J, Chen S, Xiao ZX, Cao Y. CB-Dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022;50(W1):W159–w164. doi:10.1093/nar/gkac394
39. Guo H, Cui BD, Gong M, et al. An ethanolic extract of Arctium lappa L. leaves ameliorates experimental atherosclerosis by modulating lipid metabolism and inflammatory responses through PI3K/Akt and NF-κB singnaling pathways. J Ethnopharmacol. 2024;325:117768. doi:10.1016/j.jep.2024.117768
40. Tacke F, Puengel T, Loomba R, Friedman SL. An integrated view of anti-inflammatory and antifibrotic targets for the treatment of NASH. J Hepatol. 2023;79(2):552–566. doi:10.1016/j.jhep.2023.03.038
41. Mansouri A, Gattolliat CH, Asselah T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology. 2018;155(3):629–647. doi:10.1053/j.gastro.2018.06.083
42. watt MJ, Miotto PM, De Nardo W, Montgomery MK. The liver as an endocrine organ-linking NAFLD and insulin resistance. Endocr Rev. 2019;40(5):1367–1393. doi:10.1210/er.2019-00034
43. Dichtel LE, Cordoba-Chacon J, Kineman RD. Growth hormone and insulin-like growth factor 1 regulation of nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2022;107(7):1812–1824. doi:10.1210/clinem/dgac088
44. Savova MS, Mihaylova LV, Tews D, Wabitsch M, Georgiev MI. Targeting PI3K/AKT signaling pathway in obesity. Biomed Pharmacother. 2023;159:114244. doi:10.1016/j.biopha.2023.114244
45. Alshehade S, Alshawsh MA, Murugaiyah V, et al. The role of protein kinases as key drivers of metabolic dysfunction-associated fatty liver disease progression: new insights and future directions. Life Sci. 2022;305:120732. doi:10.1016/j.lfs.2022.120732
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.