")
Back to Journals » Drug Design, Development and Therapy » Volume 19
LncRNA MALAT1/Calpain-1 Axis in ATO Induced hERG Channel Deficiency
Authors Yan C, Li Y, Li X, Li Y, Zhang Y, Sun J, Ding Q, Zhao X, Li B
Received 29 November 2024
Accepted for publication 21 February 2025
Published 28 February 2025 Volume 2025:19 Pages 1475—1487
DOI https://doi.org/10.2147/DDDT.S502776
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Tamer Ibrahim
Caichuan Yan,* Yuexin Li,* Xiaoxu Li, Yang Li, Yuhao Zhang, Jinyang Sun, Qirui Ding, Xin Zhao, Baoxin Li
Department of Pharmacology, College of Pharmacy, Harbin Medical University, Harbin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Baoxin Li; Xin Zhao, Department of Pharmacology, College of Pharmacy, Harbin Medical University, Xuefu Road 194, Harbin, Heilongjiang, 150081, People’s Republic of China, Email [email protected]; [email protected]
Background: KCNH2 encodes the hERG potassium channel, which is associated with drug-induced long QT syndrome. Arsenic trioxide (ATO) is an effective therapeutic agent for acute promyelocytic leukemia; however, its long-term use can lead to cardiotoxicity, particularly in cases of acquired long QT syndrome (acLQTS), which may result in torsade de pointes (TdP). Therefore, it is essential to comprehend the mechanisms behind acLQTS and to develop effective preventive and therapeutic strategies.
Aim: This study sought to investigate the role and molecular mechanism of MALAT1 in ATO-induced acLQTS. Furthermore, it sought to identify pharmacological agents that could mitigate the cardiotoxic effects of ATO and establish viable intervention targets for the prevention and management of acLQTS.
Methods: First, we employed gene chip arrays to identify target long noncoding RNAs (lncRNAs). Subsequently, we performed quantitative qRT-PCR and RNA-binding protein immunoprecipitation (RIP) to assess lncRNA levels. Next, we utilized Western blotting for protein expression analysis, and finally, we conducted whole-cell patch-clamp recordings to evaluate hERG currents.
Results: Our results revealed a significant upregulation of lncRNA MALAT1 expression in HEK293-hERG cells treated with ATO. Mechanistically, MALAT1 interacts with calpain-1, inhibiting its ubiquitin-mediated degradation and enhancing the cleavage activity of calpain-1 on the hERG channel. FEX and TAN were found to mitigate the effects of ATO on the MALAT1/calpain-1 pathway, ultimately restoring hERG protein levels.
Conclusion: This study demonstrated that ATO-induced enhancement of calpain-1 and reduction of hERG may be linked to the aberrant overexpression of lncRNA MALAT1. Tanshinone IIA and fexofenadine restored the hERG protein levels potentially by decreasing MALAT1 expression and counteracting ATO’s effects on the MALAT1/calpain-1 pathway. Collectively, our research uncovers a previously unreported regulatory mechanism underlying ATO-induced acLQTS. Moreover, it identifies potential molecular targets and intervention strategies for acLQTS therapy.
Keywords: hERG, MALAT1, calpain-1, ATO, LQTS
Introduction
The rapidly activating delayed rectifier potassium-selective ion channel (IKr) is critical for the third repolarization phase of cardiac action potential duration (APD).1 The hERG (or KCNH2) gene encodes the pore-forming subunit of IKr, which is an important off-target for drugs that induce cardiotoxicity. Mutations in the hERG gene, direct blockage of the hERG channel by drugs, or disordered synthesis/degradation of the hERG protein under pathological conditions can prolong APD, which is known as long QT syndrome (LQTS), which results in arrhythmia or sudden death.
Unintended off-target effects of cardiotoxic drugs due to their interactions with hERG have led to the withdrawal of large amounts of drugs such as terfenadine, cisapride, and droperidol.2–4 Arsenic trioxide (ATO) is a cardiotoxic drug that can prolong QT interval by targeting the hERG channel.5 Based on the annals of recorded history, arsenic has been used to treat various ailments including ulcers, plagues and malaria for more than 2400 years.6 Research enthusiasm for ATO as a chemotherapy agent was rekindled after its identification as an effective therapeutic agent for acute promyelocytic leukemia (APL).7 However, numerous studies have reported the cardiotoxicity (including QT prolongation) of the therapeutic dose of ATO, which largely limit its clinical application.8–10 To the best of our knowledge, ATO-induced cardiotoxicity frequently manifests as a deficiency of the hERG channel, and the underlying mechanisms include abnormal expression of microRNAs, disordered trafficking of hERG protein, or aberrant signal pathways, etc.10–12 In the present study, we discovered an alternative mechanism of ATO-induced hERG channel defects, which might help to better understand drug-induced complex biological networks and to explore new approaches to eliminate the undesigned off-target effects of cardiotoxic drugs.
It’s widely accepted that long non-coding RNAs (lncRNAs) regulate cellular activities at multiple levels via their RNA forms.13,14 These regulatory mechanisms include transcriptional regulation, post-transcriptional control, and post-translational modifications.15,16 LncRNAs can bind to microRNAs (miRNAs) or RNA-binding proteins, regulating gene expression and protein levels either through the mechanism of competing endogenous RNAs or by directly binding to specific sites on proteases.17,18 MALAT1 (Metastasis Associated Lung Adenocarcinoma Transcript 1), the first lncRNA identified in non-small cell lung cancer, promotes the expression of NLRP3 inflammasome in the heart undergoing ischemia-reperfusion injury by sequestering miR-133. It is a potential regulator of autophagy during myocardial I/R injury. MALAT1 plays a crucial regulatory role in numerous cardiovascular diseases; however, there is no report in the literature regarding its participation in cardiac arrhythmias.19
The abnormally elevated expression of MALAT1 in neonatal rat cardiomyocytes following ATO administration was screened by the gene chip, and it was further confirmed by an in vitro qRT-PCR assay that MALAT1 was significantly increased in the presence of ATO in HEK293-hERG cells. Considering that lncRNAs can directly bind to their downstream target proteins and thereby change their function or stability, we attempted to identify MALAT1-binding proteins associated with hERG channel deficiency. Interestingly, using the molecular docking software catRAPID, we found a possible binding association between the calcium-activated calpain-1 protein and MALAT1. It’s reported that calpain-1 is a calcium-dependent protease that facilitates hERG channel degradation.20 Thus, we focused on the research between MALAT1/calpain-1 and found that the binding of calpain-1 with MALAT1 inhibited the ubiquitination degradation of calpain-1 protein, which eventually enhanced the cleavage of hERG protein mediated by elevated calpain-1.
Materials and Methods
Neonatal Rat Cardiomyocytes
First, the chest of Sprague Dawley rats of one to two-day-old was opened and the heart was removed. Then, the cardiac muscle tissues were cut into small pieces. The chopped heart tissue was transferred into 0.1% trypsin digestion solution and shaken at room temperature to separate the cardiomyocytes into a single-cell suspension. The cells were then placed in a 37°C incubator for 1.5 hours to remove fibroblasts that adhered quickly. Subsequently, the cells were cultured in a medium containing 20 μmol/L BrdU, 1% penicillin-streptomycin and 10% fetal bovine serum (FBS; Bioind, Israel) at 37°C. After 48 h, neonatal rat cardiomyocytes were prepared for subsequent experiments. All animal experiments were conducted in accordance with ethical standards and national guidelines, and were approved by the Ethics Committee of Harbin Medical University (IRB3020622, Harbin, China).
Gene Chip Assay
After the neonatal rat cardiomyocytes were incubated for 24 h in 3 μM ATO, the total RNA of the cells (along with the blank control cells) was extracted and the samples were sent to KangChen Bio-tech (Shanghai, China) for lncRNA gene chip analysis.
Bioinformatics Analysis
CatRAPID (http://service.tartaglialab.com/page/catrapid_group) was used to predict the binding sites between MALAT1 and calpain-1.
Cell Culture
Human embryonic kidney 293 cells stably expressing the hERG channel (hERG-HEK293 cells) were donated by the Montreal Heart Institute (donated). hERG-HEK293 cells were maintained in Dulbecco’s Modified Eagle’s medium (DMEM; VivaCell, Shanghai, China) containing 10% fetal bovine serum (FBS; Bioind, Israel) and 400 μg/mL gentamycin (G418; Thermo Fisher Scientific, USA).
The use of hERG-HEK293 cells was also approved by the Ethics Committee of Harbin Medical University (IRB3020622, Harbin, China).
Cellular Electrophysiology
The hERG current was recorded using the whole-cell patch clamp technique by V-Clamp mode. The current was amplified and digitized at 37°C using Axopatch 700B amplifier and Digidata 1440B converter. PClamp 10.6 (Axon Instruments) was used for electrophysiological recording and analysis of hERG current. All data were normalized to the values of the control group at +40 mV. The analysis was performed using the experimental protocol described in the literature.21
Cell Transfection
For knockdown of lncRNA MALAT1, si-MALAT1 (5’-GATCCATAATCGGTTTCAA-3’) and si-NC (5’-UUCUCCGAACGUGUCACGUTT-3,’ negative control) were obtained from GenePharma Co., Ltd. (Shanghai, China). For the knockdown of calpain-1, shRNA1, shRNA2, shRNA3, and sh-NC (negative control) were purchased from GenePharma Co., Ltd. (Shanghai, China). All oligonucleotides or plasmids were transfected into the cells using Lipofectamine 2000 (Invitrogen) for 24 h. After 24 h of transfection, qRT-PCR was performed to confirm transfection efficiency.
Quantitative Real Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated and extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and transcribed into complementary DNA (cDNA) using a cDNA Reverse Transcription Kit (Invitrogen). GAPDH was used as an internal control. The primers used in the reverse transcription were listed below: The primers used in the qRT-PCR were listed below: GAPDH, forward 5’-AAGAAGGTGGTGAAGCAGGC-3’, reverse 5’-TCCACCACCCTGTTGCTGTA −3’; MALAT1, forward 5’-CAGTGGGGAACTCTGACTCG-3’, reverse 5’-GTGCCTGGTGCTCTCTTACC-3’; calpain-1, forward 5’-AGCTCATCATCACCCGCTAC-3’, reverse 5’-CCTCATGCAAACATGGTCAG-3’.
Western Blot
Cells were lysed using RIPA buffer (Beyotime Biotechnology, Shanghai, China) supplemented with 1% protease inhibitors (Roche Applied Science, Indianapolis, IN, USA), and protein concentrations were measured using NanoDrop 2000 (Thermo Fisher, USA). Proteins were then mixed with a loading buffer (Beyotime Biotechnology, China) and denatured in water at 100°C for 8 min. The SDS-PAGE was used to separate the different molecular weight proteins. Next, the proteins on the gel were electrotransferred to nitrocellulose membranes. The nitrocellulose membranes were blocked with 5% nonfat milk in PBS and immunoblotted with the corresponding primary antibodies at 4°C overnight. On the second day, the membranes were scanned using Odyssey CLx (Li-COR, Lincoln, NE, USA), followed by incubation with secondary antibodies (Li-COR, Lincoln, NE, USA). Odyssey v1.2 software was used to measure the intensity of each band. β-actin was used for normalization.
Co-Immunoprecipitation (Co-IP)
Briefly, whole-cell protein (0.5 mg) was incubated with 2 μg calpain-1 antibody or ubiquitin antibody overnight at 4°C on a 360° shaker. On the second day, the immune complexes were mixed with 50 μL of ProteinA/G plus beads for additional 24h at 4°C on a 360° shaker. Subsequently, the beads were washed with ice-cold PBST buffer and the precipitate was mixed with the loading buffer. Finally, the supernatants were collected for Western blot analysis, following centrifugation at 1500 rpm for 5 min.
RNA Binding Protein Immunoprecipitation
An RNA-binding protein immunoprecipitation assay was performed using a Magna RIP RNA-Binding Protein Immunoprecipitation Kit (#17-700, Merck, MA, USA) according to the manufacturer’s instructions. Briefly, hERG-HEK293 cells were lysed in lysis buffer and protein A/G magnetic beads were incubated with calpain-1 antibody (sc-271313, Santa Cruz Biotechnology, USA) or anti-IgG antibody (sc-69786, Santa Cruz Biotechnology, USA) as a negative control. Afterwards, the cell lysate suspension was mixed with the bead-antibody complex and incubated at 4° for 6 h. Subsequently, protease K was added to the immunoprecipitation complex to remove proteins. Pure RNA precipitation was used to detect the amount of MALAT1 in different groups using reverse transcription quantitative polymerase chain reaction (qRT-PCR).
Molecular Docking
Molecular docking was used to investigate the binding modes of TAN to MALAT1 and calpain-1 to MALAT1. The most stable conformations of TAN and MALAT1, calpain-1 and MALAT1 were calculated using Autodock vina 2.1.6 and Discovery Studio, respectively. HEX 8.0 was applied to analyse the interaction types and binding energies.22 Finally, interactions were visualised and mapped by Pymol 3.1.
Statistical Analysis
Data were processed using GraphPad Prism 5.0, and presented as mean±standard error of the mean (SEM). One-way ANOVA with Bonferroni’s post-hoc test was performed to identify differences among three or more groups. The Student’s t-test was used to assess the differences between the two groups. Statistical significance was set at P < 0.05.
Results
The Forced Expression of MALAT1 in the Presence of ATO
First, the differential expression profiles of lncRNAs in the presence or absence of ATO were obtained using a gene chip (based on the total RNA), as shown in Figure 1A (volcano map) and Figure 1B (heat map). Firstly, the unnormalized raw lncRNA and mRNA data were converted to log2 values. Then we screened several lncRNAs (MALAT1, MIAT, and TINCR) related to cardiac disease (genes with an expression fold change >2 and FDR-adjusted p < 0.05 were considered statistically significant) for further verification by qRT-PCR (Figure 1C). We focused our research on MALAT1, a conserved lncRNA (conservation of approximately 70%) reported to be associated with heart arrhythmia.23 However, how MALAT governs cardiomyocyte activity remains unknown. Calpain-1 is a protein which has been found to regulate the expression of hERG channels.20 Thus, we first attempted to identify the proteins of interest to which MALAT1 may directly bind, and we used Discovery Studio 4.0 to predict the possible binding between MALAT1 and calpain-1. As depicted in Figure 1D, there was a mutual binding relationship between MALAT1 and calpain-1 protein, and with a binding score of −85.009 (the greater the absolute value, the higher the possibility of combination). Furthermore, Western blotting was performed to determine the protein expression levels of calpain-1 and hERG at different concentrations of ATO (1μM, 3μM, 5μM). The results showed that ATO significantly promotes the expression levels of calpain-1 while inhibiting the expression level of hERG (Figure 1E-G). Collectively, the above evidence suggests that hERG channel deficiency in the presence of ATO may be attributed to the abnormal MALAT1 and calpain-1 expression levels.
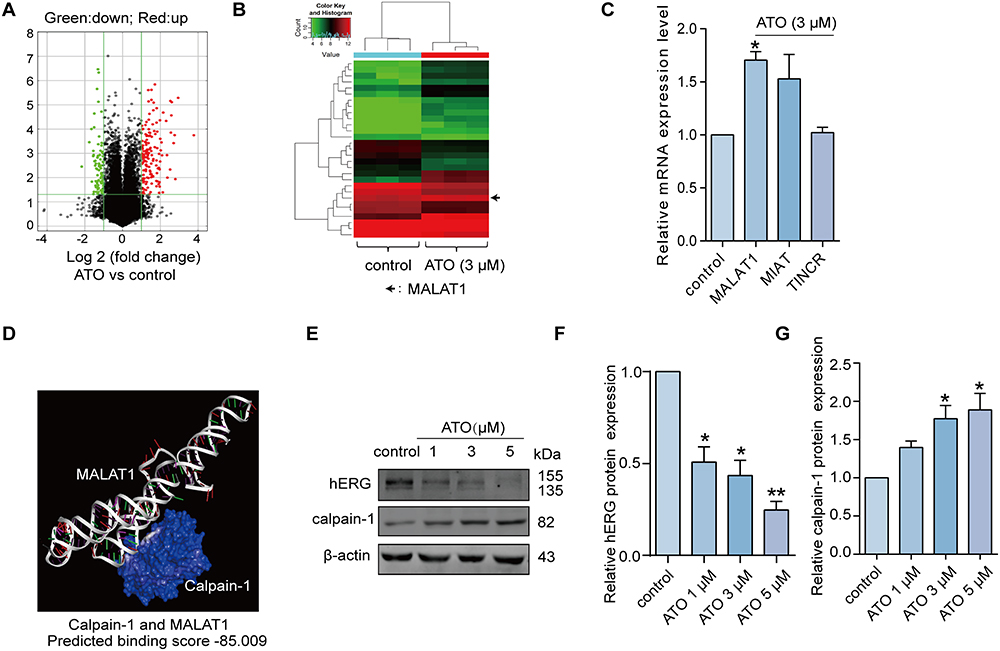 |
Figure 1 The forced expression of MALAT1 in the presence of ATO. (A) The volcano diagram represented the differentially expressed lncRNA profile in the presence or absence of 3μM ATO. Red denoted significantly up-regulated genes, and green denoted significantly down-regulated genes. Genes with an expression fold change >2 and FDR-adjusted p < 0.05 were considered statistically significant. (B) The heat map illustrated the differentially expressed lncRNAs, including MALAT1, in the presence or absence of 3μM ATO. Each column represented a sample, and each row represented a gene. Red indicated high relative expression and green indicated low relative expression. (C) The expression of lncRNAs (MALAT1, MIAT, TINCR) under the presence or absence of 3μM ATO determined by qRT-PCR. n=5, *p < 0.05 versus control. (D) The binding relationship between MALAT1 and calpain-1 protein. The most stable conformations of calpain-1 and MALAT1 were calculated using Discovery Studio, and then HEX 8.0 was applied to analyse the interaction types and binding energies. (E-G) The protein expression levels of hERG and calpain-1 in the presence or absence of ATO, and the statistical histogram. n=5, *p < 0.05, **p < 0.01 versus control. |
Knockdown of MALAT1 Reverses the ATO Induced hERG Channel Defects
To determine whether the aberrant increase in MALAT expression exerts an impact on the calpain-1/hERG axis, we first knocked down MALAT1 by transfecting si-MALAT1 in the HEK293-hERG cell line. Knockdown efficiency was determined using qRT-PCR (Figure 2A). As expected, MALAT1 knockdown decreased the protein expression level of calpain-1 markedly compared to the control group, whereas the negative control group showed no significant change. Correspondingly, the protein expression of hERG was negatively correlated with that of calpain-1 (Figure 2B and C). Based on the above evidence, we can preliminarily speculate that the upregulated MALAT1 play a key role in the ATO-induced hERG deficiency, and knockdown of MALAT1 may theoretically reverse the cell phenotypic changes caused by ATO. Interestingly, the experimental results were consistent with our expectations; knockdown of MALAT1 significantly reversed the increase in calpain-1 and the decrease in hERG induced by ATO (Figure 2D and E). Figure 2F shows the representative traces of the hERG current recorded by the patch clamp, and Figure 2G shows the statistical results of the hERG tail current (right part of the hERG-current traces). These findings indicate that ATO inhibited hERG current and knockdown of MALAT1 rescued the ATO-induced reduction in hERG current.
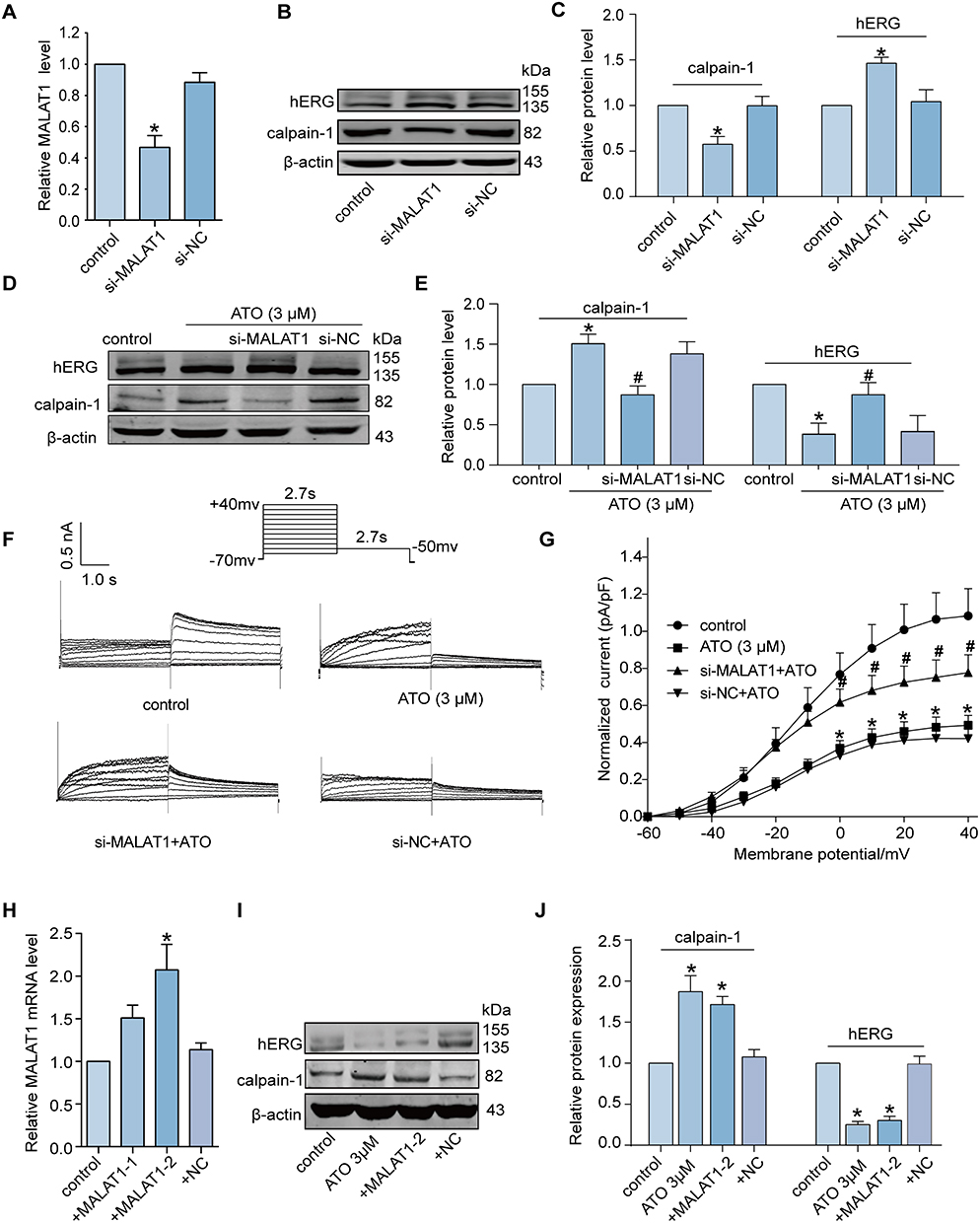 |
Figure 2 MALAT1 regulates the expression level of calpain-1 and hERG channel under ATO. (A-C) The knockdown efficiency of MALAT1 was determined by qRT-PCR and Western blot. n=5, *p < 0.05 versus control. (D-E) Knockdown of MALAT1 reversed the decrease in hERG protein and increase in calpain-1 protein in the administration of ATO. n=5, *p < 0.05 versus control, #p < 0.05 versus ATO. (F) The representative traces of hERG current recorded by patch clamp. Knockdown of MALAT1 reversed the decrease in hERG current of ATO. The resting membrane potential was −70 mV. Currents were generated by applying depolarizing voltage steps ranging from −70 mV to +40 mV. Tail currents were measured upon returning to −50 mV. (G) The statistical results of hERG tail current. I–V relationships show ATO inhibits hERG currents in hERG-HEK293 cells, and knockdown of MALAT1 rescued the ATO-induced reduction in hERG current. n=9, *p < 0.05 versus control, #p < 0.05 versus ATO. (H) The overexpression efficiency of MALAT1 determined by qRT-PCR. n=5, *p < 0.05 versus control. (I-J) The expression levels of calpain-1 and hERG proteins in the cell transfected with pRP [ncRNA]-MALAT1-2. n=5, *p < 0.05 versus control. |
Overexpression of MALAT1 Leads to Disorder of Cell Signaling Pathway Similar to ATO
To mimic the cell signaling pathway disorder induced by ATO, we overexpressed MALAT1 in normal HEK293-hERG cells and examined whether similar phenotypes might occur. First, we screened plasmids with higher MALAT1 overexpression efficiency (pRP [ncRNA]-MALAT1-2) by qRT-PCR (Figure 2H). Subsequently, Western blotting was used to detect the expression of calpain-1 and hERG proteins in cells transfected with pRP [ncRNA]-MALAT1-2. As shown in Figure 2I and J, overexpression of MALAT1 significantly increased the expression level of calpain-1 and inhibited the expression of hERG. This effect exhibited a similar trend to that observed in cells incubated with ATO.
Silencing of Calpain-1 Attenuates the ATO-Induced hERG Channel Defects
Considering that calpain-1 plays a pivotal role in the degradation of hERG channels, we used Co-IP to determine the direct binding relationship between calpain-1 and hERG channels. As shown in Figure 3A, calpain-1/hERG complexes were isolated by immunoprecipitation with anti-hERG or anti-calpain-1 antibodies. The results demonstrated that calpain-1 directly binds to hERG. Additionally, we knocked down calpain-1 expression to verify the regulatory relationship between calpain-1 and hERG. We then applied calpain-1 shRNA (sh-1, sh-2, and sh-3) to determine whether silencing calpain-1 could alleviate the ATO-induced hERG channel deficiency. Figure 3B shows the knockdown efficiency of calpain-1 shRNA. As shown in Figure 3C and D, ATO treatment induced a significant increase in calpain-1 and a decrease in hERG channels in HEK293-hERG cells. However, this effect was mitigated when calpain-1 was silenced (transfection of calpain-1 shRNA-3).
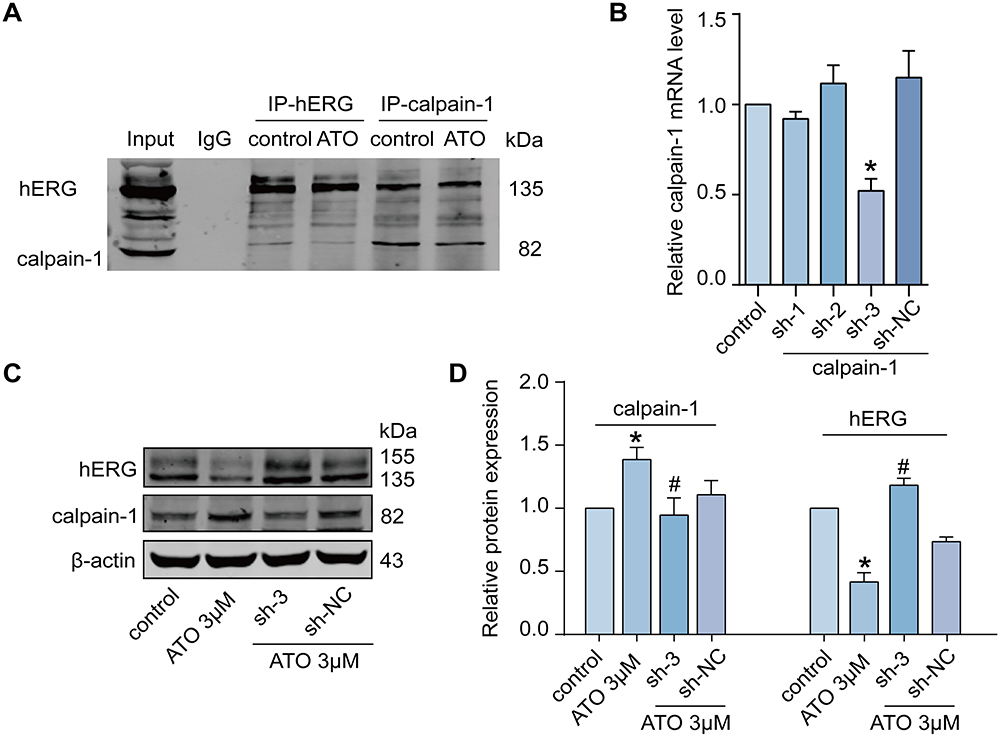 |
Figure 3 Silencing of calpain-1 attenuates the ATO-induced hERG channel defect. (A) Co-IP analyzed the direct binding relationship between calpain-1 and hERG. An anti-hERG antibody and anti-calpain-1 antibody were used to immunoprecipitate and isolate hERG-calpain-1 complexes, respectively. n=5. (B) The knockdown efficiency of calpain-1 shRNA. n=5, *p < 0.05 versus control. (C and D) Knockdown of calpain-1 reversed the protein expression of calpain-1 and hERG in the presence of ATO. n=5, *p < 0.05 versus control; #p < 0.05 versus ATO+sh-NC. |
MALAT1 Inhibits the Ubiquitination Degradation of Calpain-1
MALAT1 and calpain-1 are abnormally elevated under ATO conditions, but the mechanism by which MALAT1 governs the expression of calpain-1 remains unknown. Considering that MALAT1 could directly bind to calpain-1 protein in certain regions, as predicted by Discovery Studio 4.0, we wondered whether MALAT1 can inhibit the degradation process of calpain-1. To elucidate the underlying mechanism, RIP and Co-IP assays were performed. Figure 4A and B present the data from the catRAPID database, which presents the potential binding sites between MALAT1 and calpain-1. The results of RIP (Figure 4C) further confirmed the direct binding between MALAT1 and calpain-1. Considering that proteins have multiple degradation pathways, including the ubiquitin- proteasome, lysosomal, and caspase pathways, we initially sought to identify the degradation pathways of calpain-1. Figure 4D shows that in the presence of the proteasome inhibitor (MG132), the expression level of calpain-1 was significantly elevated compared to that in the control group, which indicates that calpain-1 was degraded, at least partially, through the ubiquitin-proteasome pathway. Subsequently, we performed co-IP to evaluate ubiquitination-dependent degradation of calpain-1 under different conditions. As illustrated in Figure 4E, compared to the control group, ATO greatly reduced the ubiquitination level of calpain-1 (the area of IB: Ub), and this effect was largely reversed by MALAT1 knockdown (using si-MALAT1), rather than by si-NC. Taken together, these data strongly imply that MALAT1 might competitively bind to the ubiquitination modification site of calpain-1 or cover its ubiquitination modification site. As a consequence, the ubiquitination process of calpain-1 is impeded, ultimately leading to the up-regulation of calpain-1 expression.
 |
Figure 4 MALAT1 inhibits the ubiquitination degradation of calpain-1. (A) The data from catRAPID database showed the potential binding sites between MALAT1 and calpain-1. The x-axis in represented the RNA sequence, and the y-axis represented the interaction score. Interaction Profile represented the interaction score (y-axis) of the protein along the RNA sequence (x-axis), giving information about the segments of RNA that are most likely to be bound by the protein. (B) The data from catRAPID database showed the predicted interactions between MALAT1 and calpain-1. The x-axis represented the RNA sequence and the y-axis represented the protein sequence. The shade of red markers on the heatmap indicated the interaction scores for individual amino acid and nucleotide pairs. (C) The RIP assay determined the direct binding status between MALAT1 and calpain-1 by qRT-PCR. n=5, *p < 0.05 versus IgG. (D) The protein expression level of calpain-1 in the administration of MG132. n=5, *p < 0.05 versus control. (E) Co-IP analyzed the ubiquitination-dependent degradation level of calpain-1. Whole-cell lysates were immunoprecipitated (IP) by anti-calpain-1 antibody and immunoblotted (IB) by both anti-calpain-1 antibody and anti- ubiquitination antibody, n=5. |
Reversal of ATO-Induced LQTS by Fexofenadine (FEX) and Tanshinone IIA (TAN)
Based on the literature and our previous experimental work, we used Hex8.0 software to analyze the binding between certain drugs (which can effectively reverse drug-induced acLQTS) and MALAT1, and predict the possibility of the drugs binding to MALAT1 and the binding fraction of the drugs to MALAT1. Tanshinone IIA (TAN) and fexofenadine (FEX) exhibit varying degrees of binding affinity to MALAT1. The prediction scores are displayed in Table 1, with higher absolute values of the scores representing greater binding affinity. The binding mode and interaction of TAN with MALAT1 is shown in Figure 5A. To validate these findings, qRT-PCR was performed on hERG-HEK293 cells. The results presented in Figure 5B revealed that FEX and TAN significantly reversed MALAT1 expression. Additionally, Figure 5C and D show a significant reversal in calpain-1 and hERG protein expression with TAN and ATO co-incubation, when compared to the treatment with ATO alone. We also confirmed changes in calpain-1 and hERG protein expression following coincubation with FEX and ATO. The results depicted in Figure 5E and F suggest that FEX also had an analogous effect of reversing the expression of calpain-1 and hERG proteins. Collectively, these findings suggest that both Tanshinone IIA (TAN) and fexofenadine (FEX) have the potential to restore the levels of hERG protein. They achieve this by downregulating the expression of MALAT1 and mitigating the impact of ATO on the MALAT1/calpain-1 signaling pathway. This underlying mechanism may provide a molecular rationale for the use of TAN and FEX in the treatment of LQTS.
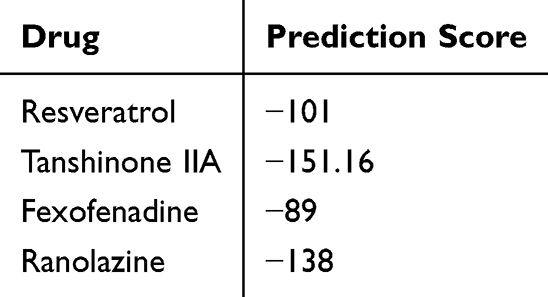 |
Table 1 The Binding Score Between Different Drugs and MALAT1 |
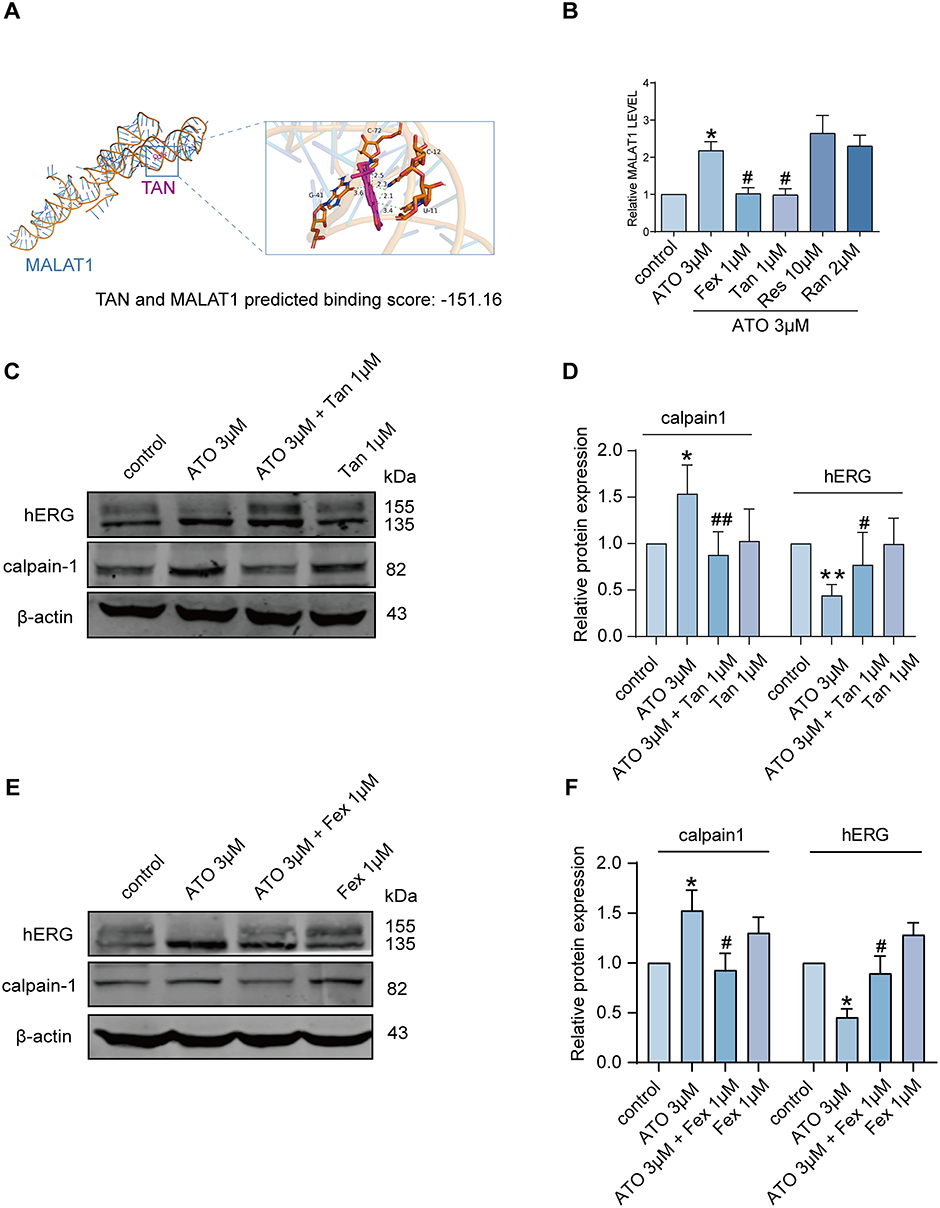 |
Figure 5 TAN and FEX may reverse the hERG channel defect caused by ATO by inhibiting LncRNA MALAT1/calpain-1 axis. (A) Predicted binding scores for TAN and MALAT1. The most stable conformations of TAN and MALAT1 were calculated using Autodock vina 2.1.6, and then HEX 8.0 was applied to analyse the interaction types and binding energies. TAN forms hydrogen bonds with multiple bases on the nucleic acid. (B) Fex and TAN significantly inhibited ATO-induced elevation of MALAT1, while Res and Ran had no effect on the elevated expression of MALAT1. *p < 0.05 versus control, #p < 0.05 versus ATO, n = 5. (C and D) Protein expression levels of hERG and calpain-1 in the presence of ATO and TAN alone or in combination. Statistical graphs of hERG and calpain-1 protein expression levels. *p < 0.05, **p < 0.01 versus control; #p < 0.05, ##p < 0.01 versus ATO, n = 5. (E and F) Protein expression levels of hERG and calpain-1 in the presence of ATO and FEX either alone or in combination. Statistical graphs of hERG and calpain-1 protein expression levels. *p < 0.05 versus control; #p < 0.05, versus ATO, n = 5. Abbreviations: Fex, fexofenadine; Tan, tanshinone IIA; Res, resveratrol; Ran, ranolazine. |
Discussion
In the present study, we provide evidence that in the presence of ATO, abnormally elevated lncRNA MALAT1 enhances the expression of calpain-1, which facilitates the degradation of the hERG potassium channel and reduces the hERG current. Mechanistically, MALAT1 directly binds to calpain-1 to impede the ubiquitination degradation of calpain-1, thereby enhancing the cleavage of hERG by the calcium protease calpain-1. The 200–900nt sequence fragment of MALAT1 is probably responsible for its interaction with calpain-1. These findings reveal the possible molecular basis of the lncRNA MALAT1/calpain-1 axis in ATO-induced hERG channel deficiency, and suggest the potential clinical application of lncRNA MALAT1 intervention for drug-associated QT prolongation.
Emerging evidence has shown that ncRNAs (non-coding RNAs) play pivotal roles in transcriptional and post-transcriptional cellular activity. Therefore, abnormally expressed non-coding RNA often serves as an indicator of the intracellular disorder status. In addition to ceRNAs (competing endogenous RNAs) that have been widely reported, lncRNAs participate in governing cellular functions, including chromatin remodeling, transcriptional regulation and protein degradation.24–26 Here, we demonstrated that lncRNA MALAT1 acts as a scaffold to bind to calpain-1 and stabilize calpain-1 protein, which inhibits calpain-1’s ubiquitination degradation. Consequently, forced expression of calpain-1 enhances the cleavage and further degradation of hERG channels. Silencing MALAT1 or calpain-1 greatly abrogates the hERG channel deficiency caused by ATO, suggesting that the MALAT1/calpain-1 axis is the key pathway involved in the regulation of hERG potassium channels.
Currently, treatment strategies for LQTS include pharmacological interventions, surgery, and cardioverter defibrillators. Notably, drugs such as fexofenadine, ranolazine, resveratrol, and tanshinone IIA exhibit potential therapeutic or rescue effects on acquired LQTS and associated arrhythmias. To investigate whether these drugs affect MALAT1 expression, we screened a series of substances and found that tanshinone IIA and fexofenadine could effectively and significantly suppress the up-regulation of MALAT1 induced by arsenic trioxide (ATO). In contrast, resveratrol and ranolazine failed to reverse the elevated levels of MALAT1. The molecular docking prediction showed that the binding scores of TAN and FEX to MALAT1 were much higher than those of resveratrol and ranolazine, suggesting that TAN and FEX are likely to rescue ATO-induced defects in hERG channels by directly binding to and inhibiting the function of MALAT1. Our experimental results revealed that both TAN and FEX were able to downregulate the expression of MALAT1. Moreover, they counteracted the effects of ATO on the MALAT1-calpain-1 pathway. Nevertheless, the underlying mechanisms and specific targets of these drugs in reversing the associated effects still await in-depth exploration.
Similarly, a limitation of this study lies in the lack of in vivo experimental data. Assuming that we obtained results consistent with those of in vitro experiments in subsequent animal experiments, this will greatly advance the clinical application prospects of lncRNA MALAT1 in cardiovascular diseases.
In summary, the study indicated that the ATO-induced enhancement of calpain-1 and reduction of hERG might result from the aberrant over-expression of lncRNA MALAT1. It was confirmed that Tanshinone IIA and Fexofenadine could restore the hERG protein level by reducing the expression of MALAT1, reversing the effect of ATO on the MALAT1/calpain-1 pathway, thus providing an experimental basis for the application of these drugs in the treatment of LQTS. Figure 6 shows a diagram of the main mechanism of the study.
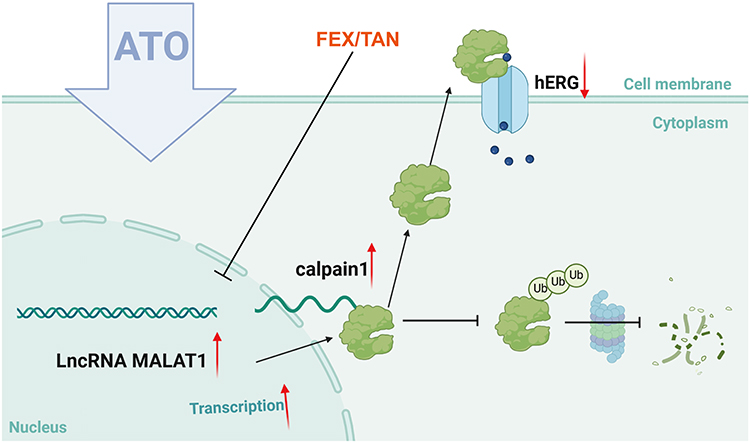 |
Figure 6 Mechanism of ATO-induced hERG deficiency via MALAT1/calpain-1 axis. The expression levels of lncRNA MALAT1 were markedly elevated in HEK293-hERG cells treated with ATO. Mechanistically, MALAT1 associates with calpain-1, inhibiting its ubiquitin-dependent degradation, which enhances the cleavage of hERG channels by calpain-1. FEX and TAN mitigated the impact of ATO on the MALAT1/calpain-1 pathway, ultimately reinstating the hERG protein levels. |
Future research should be further expanded in several aspects. Firstly, it is necessary to thoroughly explore the specific mechanisms of action and targets of TAN and FEX as reversal drugs, clarifying their detailed intracellular processes and potential regulatory pathways. Secondly, in vivo experiments are urgently needed to verify the in vitro research results using animal models, and to evaluate the effectiveness and safety of MALAT1 as a therapeutic target in vivo. In addition, considering the important role of ncRNAs, the mechanisms of other ncRNAs in cardiovascular diseases and their relationships with MALAT1 and other pathogenic factors should be further explored. Meanwhile, attempts should be made to explore combination therapy strategies based on the MALAT1/calpain-1 axis, to evaluate their synergistic therapeutic effects, and to provide more comprehensive and effective solutions for the prevention and treatment of cardiovascular diseases.
Data Sharing Statement
All data are available upon reasonable request.
Funding
This study was financially supported by the National Natural Science Foundation of China (grant numbers 81673636 and 82274029) and the Joint Guidance Project of the Heilongjiang Natural Science Foundation (grant number LH2022H014).
Disclosure
The authors declare no competing interests in this work.
References
1. Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–469. doi:10.1038/nature04710
2. Hishigaki H, Kuhara S. hERGAPDbase: a database documenting hERG channel inhibitory potentials and APD-prolongation activities of chemical compounds. Database. 2011;2011:bar017.
3. Kalyaanamoorthy S, Barakat KH. Development of Safe Drugs: the hERG Challenge. Med Res Rev. 2018;38(2):525–555. doi:10.1002/med.21445
4. Villoutreix BO, Taboureau O. Computational investigations of hERG channel blockers: new insights and current predictive models. Adv Drug Deliv Rev. 2015;86:72–82.
5. Zhang Y, Dong Z, Jin L, et al. Arsenic trioxide-induced hERG K(+) channel deficiency can be rescued by matrine and oxymatrine through up-regulating transcription factor Sp1 expression. Biochem Pharmacol. 2013;85(1):59–68. doi:10.1016/j.bcp.2012.09.002
6. Waxman S, Anderson KC. History of the development of arsenic derivatives in cancer therapy. Oncologist. 2001;6(Suppl 2):3–10. doi:10.1634/theoncologist.6-suppl_2-3
7. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111(5):2505–2515. doi:10.1182/blood-2007-07-102798
8. Haybar H, Shahrabi S, Rezaeeyan H, Jodat H, Saki N. Strategies to inhibit arsenic trioxide-induced cardiotoxicity in acute promyelocytic leukemia. J Cell Physiol. 2019;234(9):14500–14506. doi:10.1002/jcp.28292
9. Dennis A, Wang L, Wan X, Ficker E. hERG channel trafficking: novel targets in drug-induced long QT syndrome. Biochem Soc Trans. 2007;35(Pt 5):1060–1063. doi:10.1042/BST0351060
10. Yan M, Feng L, Shi Y, et al. Mechanism of As2O3-induced action potential prolongation and using hiPS-CMs to evaluate the rescue efficacy of Drugs with different rescue mechanism. Toxicol Sci. 2017;158(2):379–390. doi:10.1093/toxsci/kfx098
11. Vineetha VP, Raghu KG. An overview on arsenic trioxide-induced cardiotoxicity. Cardiovasc Toxicol. 2019;19(2):105–119. doi:10.1007/s12012-018-09504-7
12. Zhao X, Shi YQ, Yan CC, et al. Up-regulation of miR-21 and miR-23a contributes to As2 O3 -induced hERG channel deficiency. Basic Clin Pharmacol Toxicol. 2015;116(6):516–523. doi:10.1111/bcpt.12348
13. Zhang Y, Du W, Yang BJP. Long non-coding RNAs as new regulators of cardiac electrophysiology and arrhythmias: molecular mechanisms, therapeutic implications and challenges. Pharmacol Therap 2019;203:107389. doi:10.1016/j.pharmthera.2019.06.011
14. Bridges M, Daulagala A, AJTJocb K. LNCcation: lncRNA localization and function. J Cell Biol. 2021;220(2). doi:10.1083/jcb.202009045
15. Zhang Y, Sun L, Xuan L, et al. Long non-coding RNA CCRR controls cardiac conduction via regulating intercellular coupling. Nat Commun. 2018;9(1):4176. doi:10.1038/s41467-018-06637-9
16. Fang Y, Xu Y, Wang R, et al. Recent advances on the roles of LncRNAs in cardiovascular disease. J Cellular Molecular Med. 2020;24(21):12246–12257. doi:10.1111/jcmm.15880
17. Bai X, Yang C, Jiao L, et al. LncRNA MIAT impairs cardiac contractile function by acting on mitochondrial translocator protein TSPO in a mouse model of myocardial infarction. Sign Trans Targ Therapy. 2021;6(1):172. doi:10.1038/s41392-021-00538-y
18. Yang C, Zhang Y, Yang BJC. CMLS mls. MIAT, a potent CVD-promoting lncRNA. Cellular and Molecular Life Sci. 2021;79(1):43. doi:10.1007/s00018-021-04046-8
19. Jiao L, Li M, Shao Y, et al. lncRNA-ZFAS1 induces mitochondria-mediated apoptosis by causing cytosolic Ca overload in myocardial infarction mice model. Cell Death Dis. 2019;10(12):942. doi:10.1038/s41419-019-2136-6
20. Lamothe SM, Song W, Guo J, et al. Hypoxia reduces mature hERG channels through calpain up-regulation. FASEB J. 2017;31(11):5068–5077. doi:10.1096/fj.201700255R
21. Zhan G, Wang F, Ding YQ, et al. Rutaecarpine targets hERG channels and participates in regulating electrophysiological properties leading to ventricular arrhythmia. J Cell Mol Med. 2021;25(11):4938–4949.
22. Kharb S, Yadav S, Singh A, Sarkar A, Tomar R. Molecular docking and physicochemical studies of 1,3-benzodioxole tagged Dacarbazine derivatives as an anticancer agent. Artif Cells Nanomed Biotechnol. 2023;51(1):520–530. doi:10.1080/21691401.2023.2253470
23. Zhu P, Yang M, Ren H, et al. Long noncoding RNA MALAT1 downregulates cardiac transient outward potassium current by regulating miR-200c/HMGB1 pathway. J Cell Biochem. 2018;119(12):10239–10249. doi:10.1002/jcb.27366
24. Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145(2):178–181. doi:10.1016/j.cell.2011.03.014
25. Su W, Wang L, Zhao H, et al. LINC00857 interacting with YBX1 to regulate apoptosis and autophagy via MET and phosphor-AMPKa signaling. Mol Ther Nucleic Acids. 2020;22:1164–1175. doi:10.1016/j.omtn.2020.10.025
26. Mathy NW, Burleigh O, Kochvar A, et al. A novel long intergenic non-coding RNA, Nostrill, regulates iNOS gene transcription and neurotoxicity in microglia. J Neuroinflammation. 2021;18(1):16. doi:10.1186/s12974-020-02051-5
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.