")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 20
Mechanisms of Neurotoxicity of Organophosphate Pesticides and Their Relation to Neurological Disorders
Authors Chen Y, Yang Z, Nian B, Yu C , Maimaiti D, Chai M, Yang X, Zang X , Xu D
Received 24 May 2024
Accepted for publication 12 November 2024
Published 21 November 2024 Volume 2024:20 Pages 2237—2254
DOI https://doi.org/10.2147/NDT.S479757
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Yixin Chen,1,* Zhuo Yang,2,* Bin Nian,3 Chenglin Yu,4 Dilimulat Maimaiti,5 Min Chai,1 Xinran Yang,1 Xiuxian Zang,1 Dahai Xu1
1Department of Emergency Medicine, The First Hospital of Jilin University, Changchun, Jilin, 130000, People’s Republic of China; 2Department of Emergency Intensive Care Unit of Jilin University, Changchun, Jilin, 130000, People’s Republic of China; 3Department of Ultrasonography, Yanbian University Hospital, Yanji, Jilin, People’s Republic of China; 4Department of Emergency Medicine, Yanbian University Hospital, Yanji, Jilin, 133000, People’s Republic of China; 5Department of Emergency Medicine, Seventh Affiliated Hospital of Xinjiang Medical University, Urumqi, Xinjiang, 830054, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dahai Xu; Xiuxian Zang, Department of Emergency Medicine, The First Hospital of Jilin University, No. 1 Xinmin Street, Chaoyang District, Changchun, Jilin, 130000, People’s Republic of China, Tel +8615043032548 ; +8613596097922, Email [email protected]; [email protected]
Abstract: Organophosphates (OPs) refers to a diverse group of phosphorus-containing organic compounds; they are widely used all over the world and have had an important beneficial impact on industrial and agricultural production and control of vector transmission. Exposure to OPs of different toxicities (high, moderate, slight, and low toxicity) can all have negative consequences on the nervous system, such as nausea, vomiting, muscle tremors, and convulsions. In severe cases, it can lead to respiratory failure or even death. Notably, OPs induce neuropathy in the nervous system through specific interactions with nicotinic or muscarinic receptors, phosphorylating acetylcholinesterase, or neuropathic target esterases. This review summarizes the possible toxicological mechanisms and their interplay underlying OP pesticide poisoning, including cholinesterase inhibition and non-cholinesterase mechanisms. It outlines the possible links between OP pesticide poisoning and neurological disorders, such as dementia, neurodevelopmental diseases, and Parkinson’s disease. Additionally, it explores OP interactions’ potential therapeutic implications that may help mitigate the deleterious impact of OPs on the nervous system.
Keywords: organophosphate pesticides, neurotoxicity, acetylcholinesterase, neurodegenerative diseases
Introduction
From an epidemiologic perspective, the current situation of organophosphate (OP) poisoning is critical. Since the late 1930s, several OPs have been produced and sold as insecticides for use in various dosages all over the world, with their use peaking in the 1970s.1 OPs account for 38% of the globally used pesticides because of their high efficacy.2 The application of OP insecticides has increased in a wide range of occupational and non-occupational uses in the last six decades.3 The incidence of organophosphorus poisoning (OPP) is increasing yearly worldwide, particularly in rural areas.3 Internationally, OPP is among the predominant causes of poisoning-related deaths (intentional and unintentional) because of easy availability of OPs and their poorly regulated usage. According to WHO, as early as a decade ago, millions of people are poisoned by OPs globally each year, mostly in underdeveloped nations, with about 200,000 deaths recorded annually.4 Between 2018 and 2021, Asia, Africa, and the European Union were the top three in pesticide consumption, with the European Union having the highest consumption at 4.50×108 metric tons (MT), followed by Asia and Africa.5 This consumption is staggering, particularly for organophosphorus pesticides, which pose a significant threat to human health and the environment. Over 40,000 military personnel are thought to be exposed to carbamate-containing pesticides or OP-based acetylcholinesterase inhibitors.6 Humans are exposed to OP pesticides in various ways, such as gastrointestinal and respiratory exposure, skin absorption, and non-dietary ingestion of OP pesticide-containing dust.7 Living in pesticide-sprayed houses, occupational exposure, and indirect contact with occupationally exposed persons8 are also common exposure pathways. Therefore, an in-depth understanding of the mechanisms of OPs is crucial.
We conducted a comprehensive literature search on PubMed, Web of Science, Google Scholar, and Medline for references related to “organophosphorus pesticides”, “neurotoxicity”, and other related terms and applied logical operators (AND/OR/NOT) to filter relevant references, and consequently, we identified 3,145 articles. To ensure the comprehensiveness of the references, we also considered the articles cited in some of the studies. We only considered articles from the past 20 years, with a greater priority on including those from the past 10 years, and after careful selection, we ultimately obtained 235 papers. Acetylcholinesterase (AChE) inhibitors (eg detoxadine and atropine) and other regimens have been widely used in the treatment of acute OP poisoning. However, owing to a lack of understanding of the underlying mechanisms by which OP causes neurological damage, there is currently no accurate treatment strategy for neurological damage caused by long-term repeated exposure to OP and lower levels of OP. Therefore, in this paper, we focus on the neurological mechanisms of OP poisoning and its treatments, which include various aspects of axonal transport defects, oxidative stress, neuroinflammation, autoimmune effects, neurotrophins, microbes, RNA, and genes. Furthermore, we focus on the neurodegenerative pathologies caused by its exposure in humans, such as Parkinson’s disease (PD), epilepsy, and Alzheimer’s disease (AD).
Chemical Properties of OPs
Organophosphates (OPs) are a group of phosphorus-containing organic compounds. Although the skeleton and properties of the functional groups involved are quite different, OPs are usually associated with phosphoric acid derivatives, particularly phosphate esters containing organic parts. Therefore, the structure of OP compounds usually contains a phosphorus atom linked to an oxygen atom, and one or more of these can be further replaced with sulfur or nitrogen. The central phosphorus atom has two side groups, namely R1 and R2 (which are alkoxy groups), and a leaving group (X), which is nucleophilically substituted by the oxygen of serine esterase enzymes. The inhibition rate of OP pesticides on serine esterase or, in other words, their neurotoxicity potential depends on the hydrolytic propensity of the leaving group. Among the leaving groups, the hydrolysis tendency of fluorine is the highest and that of alkyl and aryl groups is the least.9
According to the consensus of clinical experts on the diagnosis and treatment of OPP in China, OPs are organophosphate esters or sulfated phosphate esters, and according to the substituents on the basic chemical structure, OPs are divided into seven categories: phosphate esters, phosphorothioate, dithiophosphate, phosphonate, fluorophosphate, amidophosphate/diamidophosphate, and pyrophosphate. The differences in their chemical structures make their physicochemical properties different, and the current varieties have amounted to more than 100 species. The toxicity of OPs is divided into four categories according to the orally consumed single dose that could be lethal to the body (LD50)—extremely toxic: LD50, <10 mg/kg (eg, methomyl, endosulfan, and parathion); highly toxic: LD50, 10–100 mg/kg (eg, methyl parathion, methamidophos, oxytetracycline, and dichlorvos); poisoning: LD50, 100–1000 mg/kg [ethephon, trichlorfon, diazinon, and chlorpyrifos (CPF)]; and low toxicity: LD50, 1000–5000 mg/kg (malathion, phoxim, and chlorothion).
Clinical Manifestations of OP Poisoning
Depending on the time of onset of symptoms and dose of consumption, OPs can lead to cholinergic crisis, intermediate syndrome, OP-induced delayed polyneuropathy, neuropsychiatric disorders, and multiple organ damage. High levels of OP exposure can lead to death within minutes due to apnea.4 Besides the potentially lethal nature of this cholinergic overstimulation, OPP can also trigger neurodegenerative processes. However, unlike the acute phase of poisoning, wherein symptoms appear within minutes, neurodegenerative changes are chronic and occur much more slowly. OPs induce neuropathy and affect neurobehavior in humans by phosphorylating acetylcholinesterase or neuropathic target esterases or by selectively attaching to nicotinic or muscarinic receptors. In human patients and animal models of OPP,10 the first neurodegenerative changes have been noted days, weeks, and even months after poisoning.
OPP also reportedly causes opsoclonus–myoclonus syndrome, which can manifest as myoclonus and involuntary, conjugate sweeping eye movements,11 with strabismic ocular clonus–myoclonus occurring only rarely. Subacute exposure to OPs leads to neurotoxicity during development in neonates. A study including almost 400 singleton term infants12 explored the connection between newborn neurobehavior and intrauterine and early postnatal OP exposure by measuring six metabolites of organophosphorus pesticides in maternal urine; the findings revealed that OP exposure adversely affected neonatal reflexes. Many OPs have been shown to induce breast tumors in experimental models;13 however, their tumorigenic effects in humans have not been clearly demonstrated.
Mechanisms of Neurotoxicity in OPs
Mechanisms of Cholinesterase Inhibition
OPs inhibit various forms of cholinesterase, leading to peripheral and central hyperactivity of cholinergic activity, which is the main and widely recognized mechanism of acute toxicity. The cholinesterase family comprises 13 isoenzymes. The liver synthesizes 11 of these, which are distributed in all tissues and called “nonspecific” isoenzymes (pseudocholinesterases or butyrylcholinesterases) and can be released into the serum.14 In addition, only OPs containing the “P = O” bond are powerful AChE inhibitors, and a compound with a “P = S” group must be activated to a “P = O” group before it can function.15
The main target of OPs is AChE, and its influence in humans is mainly through the phosphorylation of the hydroxyl group of serine residues and irreversible inhibition of the active site of cholinesterase; this leads to the loss of cholinesterase’s ability to catabolize acetylcholine, resulting in increased accumulation of acetylcholine, overactivation of acetylcholine receptors, and sustained impulses of the cholinergic nerves in the body. These effects produce a series of muscarinic-like symptoms (M-symptoms), nicotinic-like symptoms (N-symptoms), and central nervous system symptoms, with severe cases often dying of respiratory failure.16
Chronic OP exposure may trigger cholinergic anti-inflammatory pathways by downregulating cholinergic receptors, leading to the inhibition of T-cell activity and susceptibility to cancer.17 Malaoxon, a metabolite of malathion, can bind to the active site of acetylcholinesterase, resulting in irreversible inhibition of the enzyme and consequently preventing the breakdown of acetylcholine to accumulate in the synaptic cleft, thus leading to the blockade neural signal transmission and nervous system dysfunction.18 Some researchers used pregnant guinea pigs as model animals; they were given subcutaneous injection of 20-mg/kg malathion or vehicle daily from the 53rd to the 63th day of pregnancy. The offspring of malathion exposed mice showed significant spatial learning deficits and memory impairment,19 providing the first direct evidence for a link between in utero exposure to low doses of malathion and cognitive deficits. In addition, the toxicity study of malathion using zebrafish brain cells revealed increased inhibition of brain cell viability with increasing malathion concentration,20 which also suggests that the toxicity of malathion to zebrafish brain cells is closely related to its inhibitory effect on AChE.
Some researchers have found the role of RNAs in the regulation of cholinergic mechanisms to be crucial, having a significant impact on controlling the expression of associated proteins and gene activity.21 At the neurochemical level, OPs change the RNA regulators of cholinergic signaling at different levels, and noncoding RNAs play a key role in the regulation of genes that encode proteins associated with cholinergic mechanisms. Notably, OP exposure during pregnancy may disrupt the placental gene network, thus leading to downstream changes in placental function22 and ultimately affecting the neurodevelopment of the developing fetus. The interactions of RNAs and genes with the mechanisms of OPs need further investigation.
Non-Cholinesterase Mechanisms
As mentioned previously, acetylcholinesterase inhibition plays a key role in OP toxicity. However, the typical cholinesterase mechanism alone cannot explain all clinical manifestations resulting from OP exposure. Preclinical studies have shown that OP-induced toxicity can occur in animals even when the concentration of OP is below the threshold for AChE inhibition. It is hypothesized that OP toxicity can occur in the absence of cholinergic signs and that it is not necessarily dependent on AChE and other mechanisms may also contribute to the neurotoxicity caused by OP.23 Therefore, in recent years, non-cholinergic mechanisms related to OPs have been increasingly studied. Table 1 indicates different mechanisms of OP-induced neurotoxicity.
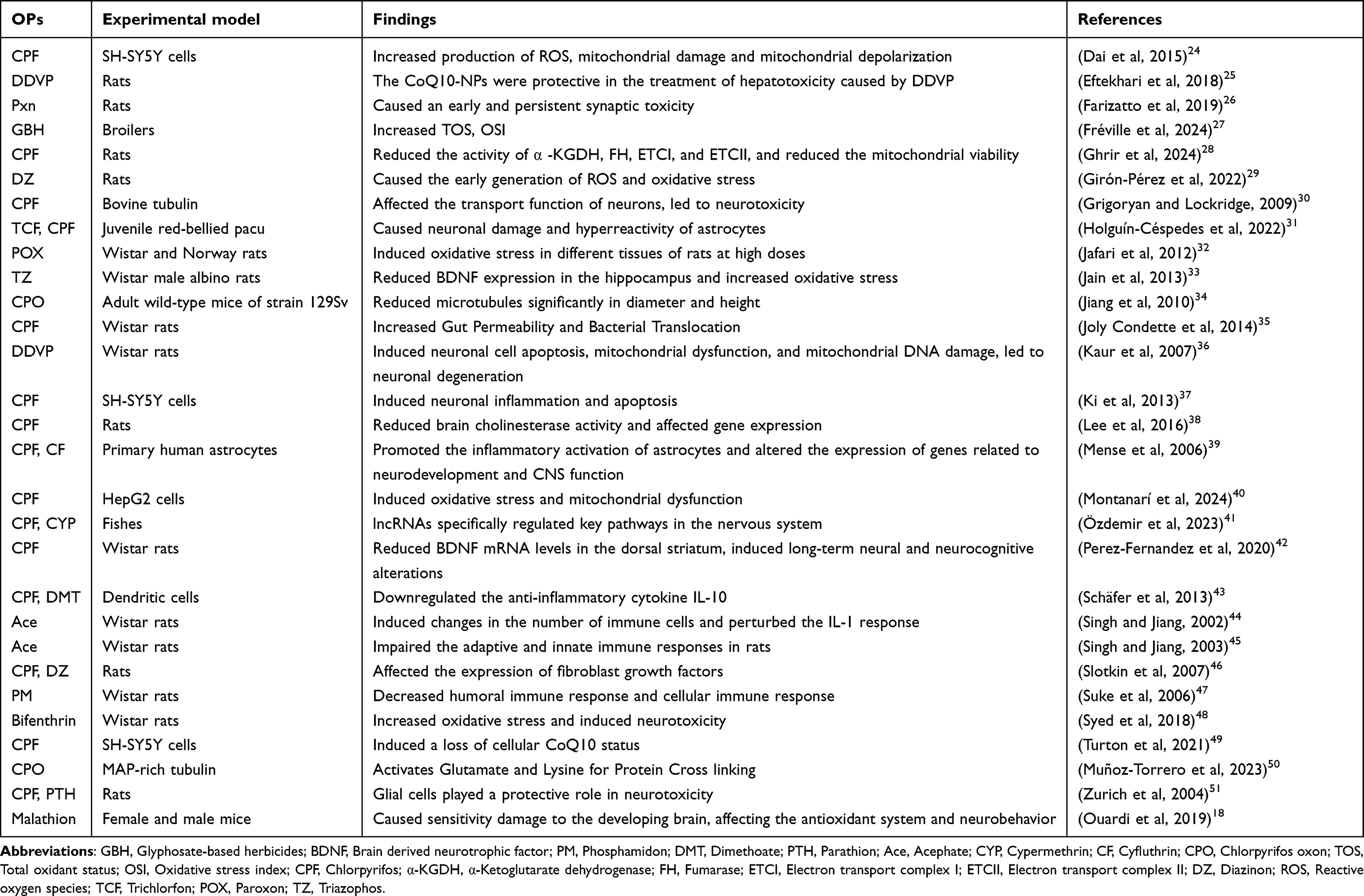 |
Table 1 Different Mechanisms of OP-Induced Neurotoxicity |
Long-term neurologic symptoms occur in the context of prodromal symptoms of acute toxicity but not necessarily in the context of cholinesterase inhibition. OPs may affect some other targets besides acetylcholinesterase, such as various proteins, receptors, and enzymes, and new action sites are still being studied. Non-cholinesterase mechanisms include OP-related axon transport defects, oxidative stress, and neuroinflammatory and autoimmune effects. In addition, OP toxicity in humans has been linked to mitochondrial dynamics and neurotrophins. These non-cholinesterase mechanisms may be interconnected. Non-cholinesterase enzymes may be affected in both acute high-level exposures and repeated low-level exposures. It is possible to use some of these targets therapeutically.
Axonal Transport Impairment
Axonal transport is responsible for transporting lipids, mitochondria, mRNA, enzymes, receptor proteins, synaptic vesicles, growth factors, and macromolecules52 to the cell body of the neuron via the axoplasm. Early studies have shown that OPs inhibit rapid paracrine axonal transport and accumulate in tubular vesicle profiles within degenerating anterior axons, which is a pathology consistent with membrane transport arrest.53 Further experiments show that the hypothesis that OPs negatively affect the driving protein is of great significance.23 Microtubules are important structural (cytoskeletal) components in neurons and are also the substrate along which the motility protein moves. Microtubules are aggregates of microtubule protein dimers that are polarized and arranged in specialized (dense parallel) arrays within the cell.
Cis-axonal transport is carried out by kinesins and is mainly seen in organelles with membrane structures, such as the mitochondria, protruding vesicles, and secretory granules. The motor domain of the head of the kinesin has a cross-bridge with ATPase activity and binds to microtubule-binding proteins on microtubules; the tail, which comprises light chains, connects to the organelle being transported. When one head of the kinesin binds to a microtubule, its ATPase activity is activated; next, the cross-bridge breaks down ATP and becomes energized, and the neck of the kinesin twists, and then, the other head binds to the next site on the microtubule.54 This process alternates, and substances (organelles) are then transported to the axon terminals along the microtubules, and the microtubules simultaneously continue to form toward the end of the axon terminals (positive end, formative end) and disintegrate at their end toward the cytosol. Reverse axonal transport is mainly seen for substances that are phagocytosed at axon terminals and transported backward to the neuronal cytosol in the presence of dynamin proteins, thus affecting neuronal activity and survival (Figure 1).
 |
Figure 1 Schematic diagram of the synaptic mechanism. Cis-axonal transport occurs with the assistance of kinesin. Microtubules are the main cytoskeleton of neurons and are formed by the polymerization of tubulin. Kinesin moves along these microtubules. OPs can disrupt the function of kinesin and the polymerization of tubulin, causing defects in axonal transport (Created in BioRender. Yx, C. (2024) https://BioRender.com/p58s105). |
OPs can lead to protein reactions that disrupt the motor structural domains of microtubule motor proteins and polymerization of microtubule proteins, thus leading to defects in axonal transport. Using atomic force microscopy, it has been confirmed that CPF oxon disrupt microtubule protein polymerization,30 and using mass spectrometry,34 it has been further demonstrated that covalent binding of CPF metabolites to microtubule proteins may be responsible for disrupting microtubule protein polymerization. Recent studies have shown the cross-linking effect of CPF on MAP-rich microtubule proteins. OP-glutamate adducts were indeed present in CPF-treated proteins. This upholds the notion that the interaction between glutamate and lysine is triggered by OP adducts on glutamate. This suggests that organophosphorus-induced formation of high-molecular-weight protein aggregates could be the missing link in the disruption of cellular function.50 In addition, OPs can bind to tyrosine in the kinesin motor structural domain.
Neurodegenerative diseases are also affected by synaptotoxicity. The relationship between synaptotoxicity and OP toxicity has recently been investigated from a new perspective, wherein biomarkers for presynaptic and postsynaptic connections gradually decreased in cultures of hippocampus slices subjected to paraoxon (POX) treatment.26 If these observations can be confirmed in an in vivo model, they could suggest a new mechanism by which acute OP exposure causes neurological disorders.
Some researchers believe that there will be new therapeutic strategies for OPP that incorporate our understanding of the characteristics of axon transport. In their experiments, Naughton et al identified kinesin separation, decreased microtubule outgrowth, and decreased tubulin acetylation as specific mechanisms underlying axonal transport damage, and this led to the development of new therapeutic strategies.55
Oxidative Stress
It is currently believed that one of the main mechanisms of OP toxicity is disruption of the balance between oxidative and antioxidant components in neurons and the resulting oxidative stress,56 which is accompanied by glutathione depletion and increased lipid peroxidation and occurs in vivo both externally and internally.57
Among various organs of POX-treated rats, oxidative stress reportedly affects the brain first.32 Because of glucose depletion and the relatively weak antioxidative effect in the brain, oxidative stress can greatly affect the brain.58
OP exposure causes oxidative stress by producing reactive oxygen species (ROS), thus stimulating the mitogen-activated protein kinase (MAPK) signaling pathway in tissues, which plays a significant role in gene expression regulation in all cells. It receives extracellular signals in the form of mitosis or genotoxicity and responds to cytokines. In mammals, these serine/threonine kinases comprise three subfamilies, namely extracellular kinases (ERKs), c-Jun amino-terminal kinases (JNKs), and p38-MAPKs.41 These are key players in intracellular signaling37 and are involved in intracellular signaling regulation via lipid peroxidation, protein oxidation, and apoptosis, consequently leading to further tissue damage. However, different OPs and cell exposures can activate different MAPK signaling pathways, which can induce different cellular responses.59 OPs may lead to apoptosis by blocking the regulation of the MAPK signaling pathway (Figure 2). The toxic effects of OPs are significantly impacted by the activation of MAPKs in the target organs. Based on the pathway analysis and collation of the literature, we constructed a schematic showing the potential molecular mechanisms that may be induced by OP exposure.59
 |
Figure 2 OPs cause mitochondrial dysfunction, cell apoptosis, and inflammatory responses by blocking signal pathways (Created in BioRender. Yx, C. (2024) https://BioRender.com/l34w839). Abbreviations: OP, organophosphate pesticide; ERK, Extracellular signal-regulated protein kinase; MAPK, Mitogen-activated protein kinases; MSK1/2, Mitogen- and stress-activated protein kinases 1 and 2; ATF2, Activating transcription factor 2; Cyt, Cytochrome; CREB, cAMP response element; NF-κB, Nuclear transcription factor kappa-β; IL-6, Interleukin-6; IL-1β, Interleukin-1β; cox2, Cyclooxygenase-2; TNF-α, Tumor necrosis factor alpha; JNK, c-Jun NH2-terminal kinase; ASK1, Apoptosis-signal-regulating kinase 1; ROS, Reactive oxygen species; Trx, Thioredoxin; DrP1, Dynamin-related protein 1. |
Notably, POX causes oxidative stress and ROS generation in a dose-dependent manner.60 Oxidative stress can alter the metabolism and important functions of cells by damaging the DNA, RNA, and proteins involved in defense mechanisms.61 Wang et al were the first to explore the association of genes encoding miRNA with telomere length under long-term low-dose omethoate exposure; they showed that telomere length was prolonged in peripheral blood leukocytes of workers exposed to omethoate, which may be attributed to chromosomal damage caused by omethoate.62 In adult rats, chronic low-dose exposure to dichlorvos increases the oxidative stress and induces apoptotic neurodegeneration by elevating Ca+ levels in the mitochondria and impairing the function of mitochondrial complexes I, III, and IV.36
Nrf2 is a key transcription factor involved in the oxidative stress response; it can regulate the expression of multiple genes encoding antioxidant and detoxification proteins,63 and it is involved in CPF-related neurotoxicity. In a previous study, CPF exposure reportedly increased the amount of Nrf2 protein by 44%,40 thus significantly increasing its expression level and showing antioxidant effects. Activation of Nrf2 may help mitigate the effects of mitochondrial dysfunction and oxidative stress.
Recent studies have found that antioxidants may have protective effects against oxidative damage caused by OPs in the nervous system. Animal experiments have found that vitamins E and C protect cells from damage caused by harmful oxygen free radicals64 and promote humoral and cellular immune responses, thus reducing the oxidative stress index.27 Melatonin showed significant cytoprotective effects in the in vitro cell model SH-SY 5Y,65 suggesting that it may have therapeutic potential for oxidative stress in the nervous system.
Ghrir et al evaluated oxidative stress through quantitative determination of biological molecular oxidative damage, and the results showed that grape seed extract effectively reduced the oxidation effect caused by OP and maintained it at a normal level. In addition, some OPs like CPF can also act via different signaling pathways, and future work should focus on inhibiting the role of different pathways in OP-induced neurological symptoms.28 Recent studies have shown that the organic selenium compound diphenyl diselenide can exert neuroprotective effects in rats by eliminating oxidative inflammatory stress and activating caspase-3 and that it has anti-tumor, antioxidant, anti-inflammatory, and anti-mutational effects. At present, its effects on chemotherapy-induced neurotoxicity have been confirmed by experiments, and as the neurotoxicity caused by OP has a similar underlying mechanism, we can reasonably speculate diphenyl diselenide to have a similar effect on OPP, and we hope that this can be further clarified by relevant studies in the future.66
Mitochondrial Dysfunction
Mitochondria play a significant role in energy regulation, cellular metabolism, cell proliferation, and anti-inflammatory processes.67 Animal studies have shown that exposure to dichlorvos causes significant changes in the morphology of neuronal and glial cell mitochondria, including chromatin condensation and mitochondrial swelling. Dichlorvos exposure increases mitochondrial ROS production, decreases the activity of complex I and IV in the respiratory chain, and reduces ATP production, thus resulting in α-synuclein aggregation and neuronal death. This also plays a crucial role in the pathogenesis of Parkinson’s disease.68 Concomitant mitochondrial respiration defects and increased ROS generation lead to the release of cytochrome c into the cytosol to initiate the apoptotic cascade.24 In addition, control experiments using rats have revealed that exposure to OPs significantly reduces the activity of NADH dehydrogenase and ATPase.69 NADH dehydrogenase is a part of the mitochondrial electron transport chain, and ATPase is a complex on the mitochondrial membrane responsible for synthesizing ATP. The reduced activity of these enzymes indicates that the function of the mitochondrial respiratory chain has been inhibited.
Notably, OPs disrupt calcium homeostasis and inhibit cellular respiration enzymes, affecting cellular respiration, ATP generation, and ROS production, consequently inducing apoptosis.57 They trigger cell death through the release of mitochondrial cytochrome c and subsequent activation of caspases, leading to DNA damage.
Coenzyme Q10 (CoQ10) is a fat-soluble substance also known as ubiquinone or coenzyme Q. It acts as a proton–electron carrier in the cellular mitochondrial electron transport chain, participating in the production of energy, that is, the synthesis of ATP.70 CoQ10 plays a crucial role in maintaining mitochondrial function. Turton et al exposed SH-SY 5Y cells to different OPs, including dichlorvos, methyl parathion, and CPF, and then detected the levels of CoQ10 in the cells using high-pressure liquid chromatography. The experimental results showed that the levels of CoQ10 decreased by 72%, 62%, and 43% after treatment with dichlorvos, methyl parathion, and CPF, respectively.49 This indicates that OPs significantly reduce CoQ10 levels, thus affecting mitochondrial function.
Syed et al elucidated that bifenthrin impairs the affinity of cellular proteins and can increase the protein carbonyl levels in brain regions, thus showing that such pesticides can lead to excessive ROS production by the mitochondrial respiratory chain.48 This can further result in oxidative stress. The mitochondrial membrane potential is the fundamental control unit for cellular electrical activity and several biological processes, such as ATP production. Studies have shown that 24-h exposure to CPF resulted in a decrease in mitochondrial membrane potential ranging from 10% (at 70 μM) to 20% (at 17.5 μM). In addition, the production of mitochondrial superoxide anions and lipid peroxidation significantly increased, particularly at a CPF concentration of 70 μM, which showed an increase in the production of MDA (malondialdehyde) by 11%.40 These findings suggest that CPF may cause cytotoxicity by inducing oxidative stress and mitochondrial dysfunction.
CoQ10 has potential therapeutic effects in the treatment of OP poisoning; however, higher doses may be required.49 Through the evaluation of cell viability, ROS levels, and other indicators in experimental rats, researchers found that CoQ10 nanoparticles have a protective effect against oxidative stress induced by dichlorvos,25 and more in-depth study in this regard is required in the future. Antioxidants like rosmarinic acid enhance mitochondrial glutathione levels by stimulating the PI3K/Akt/Nrf2 pathway,71 thus regulating mitochondrial activity.
Neuroinflammation
Studies have suggested that inflammatory activation of astrocytes is an important mechanism underlying neurotoxicity caused by OPs like CPF and cyhalothrin. Astrocytes are the most numerous and functionally complex cells in the brain; they can exert a trophic impact on neuronal growth, development, survival, and functional maintenance through various neurotrophic factors secreted by them. Cyfluthrin and CPF increase the critically important inflammatory mediator interleukin 6, which induces astrocyte proliferation, and they also increase glial fibrillary acidic protein, a marker of inflammatory astrocyte activation.39 Parathion induces astrocyte proliferation at relatively low concentrations, and subtoxic doses of CPF inhibit neural protrusion growth, neuronal cell replication and differentiation, glial cell replication, gliogenesis, and glioma differentiation and disrupt glial cell development in vivo.51 OPs upregulate the expression of proinflammatory mediators and inflammatory activation of astrocytes or astrocyte cell dysfunction. OPs affect cell viability,72 and their induced developmental neurotoxicity can be modeled in vitro using RNA-seq to mimic human neurodevelopmental processes, which include proliferation of neural progenitor cells, neuronal and neuroglial differentiation, neurite extension, synaptogenesis, and synaptic transmission, the details of which need to be refined.73 Some animal studies have shown that compared to the brains of animals not exposed to OPs, those of animals subchronically exposed to OPs for 25 days show phenotypic alterations in astrocytes, which are characterized by shortening, hypertrophy, and amoeboid morphology of cytoplasmic processes and detectable astrocyte hyperreactivity.31
Notably, OP compounds affect the release of inflammatory factors (eg, IL-1β and IL-8), and IL-10 is reduced in OP-exposed dendritic cells. Protein kinases like the Akt family or ERK, which are essential for cell survival and proliferation, are inhibited by OPs.43 Furthermore, repeated exposure to low levels of acephate can reportedly induce an inflammatory response in astrocytes39 and increase inflammatory cytokines.45 To date, however, no evident functional connection has been identified between OP-associated neurobehavioral deficits and inflammatory response.74
Some researchers have suggested that in OP-induced neuroinflammation, microglia regulate the appearance of proinflammatory cytokines that damage neurons and cause neurodegenerative changes, and thus, focusing on therapies that inactivate microglia and suppress the inflammatory response could work against the disease to some extent.21,75
Autoimmunity
Innate immunity is the first line of defense against antigens and comprises various effector molecules, such as natural antibodies, growth inhibitors, interferons, and protein inhibitors, as well as mast cells, macrophages, and neutrophils. Highly differentiated cells, such as a proliferating population of lymphocytes including B cells, T cells, and natural killer (NK) cells, as well as immunoglobulin and major tissue compatibility complex systems are the components of adaptive autoimmunity.
Notably, OPs have the ability to influence both in vivo and in vitro immune responses. They exert this effect via the generation of autoantibodies, generation of IL-2, T-cell proliferation, CD5 cell reduction, CD26 cell increase, and antibody production.76 Furthermore, their influence on immune response is also attributable to their modification of Th1 and Th2 cytokine profiles, suppression of NK cells and lymphokine-activated killer (LAK) cells, and the activity of cytotoxic T lymphocytes. Immune cells express cholinergic components in large quantities, and OPs can alter lymphocyte cholinergic signaling by inhibiting AChE.77 Studies have shown that OPs like acetamiprid inhibit the number of immune cells in the blood, thus compromising body’s immunity. Acetamiprid exposure enhances the acute phase response and may downregulate the immune system by modulating the IL-1 response by altering glucocorticoid concentrations in mammals.44 IL-1 can stimulate the central nervous system and behavior; therefore, OP-induced dysregulation of the IL-1 response may also lead to behavioral abnormalities.78 Exposure to OPs negatively affects both innate and adaptive immunity, preventing host defense against foreign substances and promoting dysregulation of some humoral and cellular processes, such as cytokine expression, phagocytosis, antibody production, cell proliferation, and cell differentiation.79 Studies have shown that active metabolites of OPs interact with nicotinic receptors, G protein-coupled receptors (GPCR), and interleukin receptors (ILR), thus modifying the expression of genes and signaling pathways in both intrinsic and adaptive immune system cells to regulate the processes of phagocytosis, lymphocyte proliferation, cellular senescence, cell death, complementation, antibody production, antigen presentation, and other aspects.77 OP exposure can also diminish the complement system. However, the immunotoxic effects of long-term exposure to OPs remain inadequately studied.
Studies have shown that exposure to OPs like malathion and phosphamidon can trigger macrophage migration and can directly lead to reduced nitrite production. Lipopolysaccharide (LPS)-induced tumor necrosis factor alpha (TNF-α) release from macrophages is reduced, and TNF-α and inducible nitric oxide synthase levels are reduced as well.45,47 In addition, it has been suggested that diazinon may affect the immune system through premature oxidative stress induction.29
Neurotrophins
Neurotrophic factors are classically defined as a class of protein or peptide molecules which are produced by effector tissues innervated by nerves (eg, muscle) and glial cells (primarily astrocytes) and are essential for neuronal growth and survival. They play a critical role in neuronal genesis, neuronal migration, neuronal differentiation, neuronal apoptosis, brain assembly, and recovery from neuronal injury.80 In recent years, owing to the continuous discovery of new neurotrophic factors, there is accumulating information on their sources and the types of tissues and cells they regulate. It has become difficult to clearly distinguish between the trophic effect of nerves on effector tissues and the regulation of neurons by neurotrophic factors.81
Researchers treated chickens with the nerve agents diisopropylfluorophosphate or cyclic phenyl saligenin phosphate, and after 3 days, cells receiving tissue extracts from chickens began to elongate and extend protrusions compared to controls that did not receive OPs; conversely, the morphology of cells not receiving extracts from chickens remained unaffected or was minimally affected. Pope et al elucidated the factors produced in situ after OP exposure and found them to be neurotrophic factors, which also led them to coin the term “neurotrophic factor”.82 Local expression of mRNA encoding fibroblast growth factor and its receptors in the forebrain and brain stem was examined, and organophosphorus exposure was found to reduce neurotrophic factor expression in the forebrain and brain stem of newborn rats. Specifically, both CPF and diazinons significantly inhibited the expression of forebrain Fgf20 and brainstem Fgf2, increased the expression of brainstem Fgfr4, and caused a small amount of Fgf22 deletion, indicating that organophosphorus causes damage to and leads to poor outcomes for developing neurons.46 In addition, it has been demonstrated in recent years that OPs may act by targeting neurotrophic factors and their receptors (eg, Ntf3, Ntrk1, Ntrk2, Ngfr, Fgf2, Fgf11, Fgf22, and Fgfr4) to produce neurological effects.
It has been shown that low doses of CPF reduce the level of brain-derived neurotrophic factors’ mRNA expression in the male rats’ dorsal striatum, resulting in long-term changes to the nervous system, particularly inattention and impulsive behavior.42 Transcriptomic and epigenetic evidence suggests that neurotrophin-mediated pathways are activated after repeated exposure to CPF in rats.38 Chronic exposure to triazophos was found to significantly reduce mRNA expression and protein levels of brain-derived neurotrophic factor (BDNF) in the rat hippocampus. BDNF has been implicated in neuronal growth, differentiation, and survival, and reduced hippocampal BDNF expression may contribute to the decline in acquired memory retention and impaired cognitive functioning in rats.33
Microorganisms
Although a substantial amount of research and experimentation has been done on OPs, few studies have assessed the influence of OPs on the nervous system through the action of the microbiota. The microbiota is essential for the maintenance of human health. The human body is colonized by a wide range of bacteria and microorganisms, which are mainly found in the gastrointestinal tract; however, their effects are not limited to the gastrointestinal tract. They can also influence other organs like the brain through the microbiome–gut–brain axis, playing roles in metabolic, immune, and host homeostasis. Studies have found the disruption of the gut microbiome in the gut–brain axis to be associated with neurological disorders caused by environmental chemicals.83 Studies have shown that the microbiota can be temporarily altered or modified by OPs, thus affecting metabolic and neurotoxic outcomes.84
Studies have shown that although communication between the gut flora and the central nervous system is not very strong,85 the gut flora has an important role in bidirectional interactions between the gut and the nervous system; this communication contributes to modulating brain physiology and influencing the neuroendocrine system for aspects related to stress response, anxiety, and memory function through various mechanisms.86 This suggests that modulating the microbiota may be a viable approach to treating OP-induced CNS disorders.
Using animal models, some researchers have found that chronic exposure to OPs like CPF leads to gut microbial dysbiosis and that this exposure can alter the intestinal epithelium’s morphology, resulting in bacterial displacement and increased intestinal permeability, particularly in developmental stages of organs.35 The researchers examined the effects of CPF on the gut microbiota in mice. Notably, CPF does not directly damage the bacteria but rather disrupts bacterial metabolism, leading to changes in bacterial metabolic pathways, such as amino acids, glucose metabolism, and glycolysis. OPs may inhibit the amino acid metabolism pathway, reduce the intracellular amino acid content, and adversely affect the intestinal function.87 Kopjar et al found that low (0.01 mg/mL), medium (0.1 mg/mL), and high doses (1 mg/mL) of OP exposure caused significant changes in gut flora and bacterial metabolic levels, which significantly altered the structure and function of the gut microbial community. Their results suggest for the first time that DNA damage and DNA instability in host cells88 may occur in the gut microbiota. The dysbiosis of gut microbes changes the permeability of the gut, allowing the passage of harmful substances and consequently activating the immune system to release cytokines and chemokines, which can regulate the nervous system and lead to functional abnormalities.89 Dai et al found that glyphosate exposure reduces the antioxidant capacity and increase the serum lipid and proinflammatory factors, whereas hawthorn leaf flavonoid can improve the intestinal flora balance, enhance intestinal immunity, reduce inflammation, improve nutrient absorption, and inhibit harmful bacteria growth while alleviating the negative effects of glyphosate, improving the antioxidant capacity, and enhancing the intestinal barrier function.90 Therefore, dysregulation of the microbiota and the related products should be considered when discussing the neurological damage caused by OPs.
The Link Between OP and Neurodegenerative Diseases
Neurotoxicity has been defined as deleterious changes in the structure and function of the central and peripheral nervous system.91 Long-term neuropsychiatric consequences, such as deficits in signal detection, information processing, memory, sustained attention, flexibility of thought, and depression, have been reported in patients with acute OP poisoning. Similarly, there is significant evidence that occupational exposure and accidental, low-dose consumption of OP residues via food and drinking water can also cause neurotoxicity. Long-term opioid exposure may lead to anomalies in neurobehavioral functioning, presenting as issues with working and visual memory, psychomotor speed, executive function, and visuospatial ability.92 Chronic occupational exposure to OPs has been linked to cognitive consequences.
Considerable evidence from animal studies shows that both prenatal and early postnatal exposure to OPs (at dose ranges not associated with acute toxicity) can lead to multiple lasting cognitive impairments that become more evident with age.
Epilepsy
A class of organophosphorus substances called neurotoxic gases inhibits the cholinesterase enzyme, which breaks down acetylcholine. OPs and nerve agents are potent neurotoxic compounds that cause seizures, status epilepticus, brain injury, or death93 When these medications are administered to humans, they can cause cholinergic toxicity, tremors, seizures, hypersecretion, coma, and even death. We think that OPs cause seizures via the enhanced glutamate release from presynaptic terminals.94 Organophosphorus can induce the accumulation of acetylcholine at synaptic endings and activation of M1 muscarinic receptor, thus inhibiting the KCNQ2/3 potassium channel. A recent study established an animal model of OP intoxication and assessed the impact of OPs on excitatory α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-mediated glutamatergic neurotransmission in the hippocampus using membrane-clamp electrophysiologic techniques. These findings imply that organophosphates improve glutamatergic transmission by increasing neurotransmitter release. At least partially, this action is mediated by M1 muscarinic receptors. This impact is mediated by muscarinic receptors, which block certain KCNQ2/3 potassium channels involved in M-currents.95 One medication that works well against OP-induced seizures is flupirtine, which is an open channel medication.95
Muscarinic activation modulates glutamatergic transmission. Widely expressed in the brain, muscarinic acetylcholine receptors are the GPCRs expressed in the central nervous system. The major muscarinic acetylcholine receptor in the striatum, the thalamus, hippocampal regions, and the cerebral cortex is the M1 subtype.96 Activation of muscarinic receptors enhances the release of glutamate at the presynaptic level from penetrating fibers to dentate granule cells. Voltage-gated calcium channel-mediated calcium influx mediates muscarinic effects.97
Over a prolonged period, the accumulation of ACh caused by the OP-mediated inhibition of AChE leads to overstimulation of nicotinic or muscarinic receptors, which activates the glutamatergic system and leads to seizures. Brain injury and neuritis may result from cholinergic anti-inflammatory response blockade and seizures caused by OPs.98 Recent studies have shown that organophosphorus poisoning can cause damage to the blood-brain barrier, and loss of the integrity of the blood-brain barrier may contribute to the eclampsia process.99 In addition, OP-induced seizures are reportedly associated with increased oxidative stress.100
Alzheimer’s Disease
Notably, AD often begins in the cholinergic system101 and leads to progressive cognitive deterioration. The cholinergic hypothesis of AD suggests that dysfunction of neurons in the ACh-producing brain contributes to the cognitive decline associated with AD,102 and therefore, cholinesterase inhibitors (ChEIs) are one of the few treatments for AD caused by OP intoxication. Recent studies from the United States, the United Kingdom, and continental Europe have shown increased risk of dementia in individuals taking anticholinergic drugs over a long term.103 AChE promotes the development of β-amyloid fibers in the central nervous system, which plays a role in the etiology of AD. Therefore, AChE inhibitors have been introduced in the palliative treatment of patients with AD to restore central cholinergic function and prevent β-amyloid fibril deposition.104
In addition, impaired axonal transport is thought to underlie the pathology of AD.105 The pathogenesis of AD may depend on the reduced activity and different polymorphisms of the paraoxonase-1 (PON1) enzyme, which accumulates in the body as a result of long-term exposure to OP pesticides, and the reduction of PON1 activity leads to an increase in activity against OP pesticides, which consequently causes symptoms of dementia, memory impairment, and other AD symptoms.106–108
As previously mentioned, modification of OPs can disrupt several sites of microtubule function, such as polymerization of microtubule proteins, leading to defects in axonal transport.23 In addition, Tau is a microtubule-associated protein whose misfolding, hyperphosphorylation, loss of normal function, and enhanced toxic function are associated with a variety of neurodegenerative diseases, including AD109 in various neurologic degenerative diseases.
In addition, RNA metabolism-related brain damage has been observed in cholinergic neurons of the Alzheimer’s brain, and microRNAs and tRNA fragments have been found to act as regulators of ACh production and cholinergic activity in brain regions and cell types with different cholinergic activity. This mechanism may provide guidance for the treatment of secondary diseases.101
One study evaluated the relationship between elderly people’s risk of developing AD and occupational exposure to OP using Cox proportional risk survival analysis.110 The authors concluded that exposure to OP pesticides may have long-term adverse effects on the central nervous system, which may increase the risk of developing AD later in life.
Slow-binding inhibitors of acetylcholinesterase are reportedly promising agents for the management of neurological diseases like Alzheimer’s dementia.111 However, evidence supporting the association between OP pesticide exposure and AD in adults is limited by there not being enough published studies in this regard.91
Parkinson’s Disease
After AD, Parkinson’s disease (PD) is the second most common neurological illness, and its prevalence is expected to double by 2030.112 It is characterized by the gradual degeneration of nigrostriatal dopaminergic neurons, which is accompanied by the degeneration of nerve endings in the striatum along with marked neuroinflammation and immune dysfunction. Once the disappearance of dopaminergic cells reaches about 80%, clinical signs like bradykinesia, muscular tonus, resting tremor, and stiffness of the neck, trunk, and limbs occur. Even if some abnormalities in certain genes are linked to the inherited form of PD, the vast majority of PD cases are sporadic. PD is now recognized as a multi-system disease, with neuroinflammation being implicated in its pathogenesis. Currently, there are multiple lines of evidence suggesting an association between environmental stressors (eg, viral and bacterial exposures, pesticide exposure, and alterations in diet and gut microbiota) and an increased risk of PD, with exposure to pesticides like organophosphorus pesticides being recognized as environmental factors associated with increased risk of PD.
Dhillon et al conducted a case-control study of PD in East Texas residents113 and identified an association between pesticide exposure and the risk of developing PD. The use of CPF was associated with a greater than twofold rise in PD risk, and the incidence of PD was 90% higher in pesticide-exposed individuals than in those who were not exposed to pesticides. In 2009, Gatto et al applied for the first time a semi-quantitative method to estimate pesticide exposure in well water, and among the 26 pesticides included in the study, the risk of developing PD was higher in people drinking well water contaminated with OP pesticides, and this risk showed an increasing trend with the increase in the number of pesticide types in well water. Water-soluble organophosphorus pesticides are likely to increase the risk of PD. This particularly holds true for CPF and diazinon, with their exposure being higher in residents having well water exposure than in those without well water exposure, resulting in 70%–90% increased relative risk of developing PD in the former.114 In a study by Chuang et al, to determine the rate of new-onset PD, 45,594 patients were observed for a maximum period of 12 years, and Poisson regression modeling was used to identify PD predictors. Using a Kaplan–Meier plot, the cumulative incidence of PD in the two groups was estimated. In the multivariate model, compared to control patients, those with OP poisoning showed 1.36-fold higher incidence of PD during the study period. According to this retrospective investigation, OP poisoning may be a separate risk factor for PD.115
Parkinson disease protein 7 (PARK7, DJ-1) controls and interacts with paraoxophosphorase-2, which is an enzyme that has increased activity during oxidative stress response and is crucial for neuronal survival during oxidative stress. DJ-1 guards neurons from oxidative stress in PD models both in vitro and in vivo. The DJ-1 protein is believed to mitigate the effects of the oxidative stress caused by environmental pollutants like OP insecticides and has a neuroprotective function in preventing PD. Paraoxonase-2 (PON2) successfully alleviated the heightened vulnerability to oxidative stress caused by DJ-1 deficiency.116 In addition, RNA metabolism-related brain damage was observed in PD brains from both cholinergic-deficient animals and human donors.117
Discussion
As mentioned earlier, the use of OPs as insecticides is common in agriculture and near human settlements. Considering their deleterious health effects on human beings, governments have raised environmental concerns associated with their use. According to scientific evidence, unintentional or direct long-term exposure to various OPs is a major public health threat as it can lead to chronic neurological diseases like epilepsy and AD. Therefore, it is critical to understand the mechanisms that lead to the toxicity of OPs to find new and more effective treatments to minimize and hopefully eliminate the health hazards associated with OP use. A large body of data currently indicates that the cholinesterase mechanism of OPs alone cannot explain all neurological manifestations caused by their use, particularly long-term neuropsychiatric symptoms. Some of the long-lasting impacts of OPs could be attributed to their interactions with processes like axonal transport, oxidative stress, neuroinflammation, microbial regulation, neurotrophic factor support, and mitochondrial function (Figure 3).
 |
Figure 3 The summary of all the different forms of toxicity caused OPs (Created in BioRender. Yx, C. (2024) https://BioRender.com/j98s954). |
The mechanisms of OPs discussed in this review may function as a foundation for the development of future therapeutic approaches. There are still many questions to be answered, such as whether some of the non-cholinesterase-related targets and therapeutic strategies identified in vitro and in animal studies are also applicable in vivo and whether there are more drugs that can exert therapeutic effects for the targets identified so far. In addition, there are few studies on the molecular mechanism of OPs related to RNA and microorganisms. Most published works describe oxidative stress and other aspects of OPs, and it is hoped that more studies will support the RNA, gene and microbial aspects in the future. We hope that through the induction and summary of the mechanism of organophosphorus pesticide neurotoxicity, we can contribute to the new research and experiments in this field and propose new therapeutic strategies and programs for these targets and their interactions.
Abbreviations
OP, Organophosphate; OPP, organophosphorus poisoning; PD, Parkinson’s disease; AD, Alzheimer’s disease; CPF, chlorpyrifos; AChE, acetylcholinesterase; POX, paraoxon; ROS, reactive oxygen species; MAPK, mitogen-activated protein kinase; ERK, extracellular kinase; JNK, c-Jun amino-terminal kinase; NK, natural killer; LAK, lymphokine-activated killer; GPCR, G protein-coupled receptors; ILR, interleukin receptor; BDNF, brain-derived neurotrophic factor; ChEI, cholinesterase inhibitor.
Acknowledgments
This study was supported by Special Projects for Health in Jilin Province (2008SCZWSZX-050). We thank Medjaden Inc. for its assistance in the preparation of this manuscript.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Costa LG. Organophosphorus Compounds at 80: some Old and New Issues. Toxicol Sci. 2018;162(1):24–35. doi:10.1093/toxsci/kfx266
2. Millán D, Tapia RA, Pavez P. Efficient Nucleophilic Degradation of an Organophosphorus Pesticide “Diazinon” Mediated by Green Solvents and Microwave Heating. Front Chem. 2018;6:669. doi:10.3389/fchem.2018.00669
3. Patel V, Ramasundarahettige C, Vijayakumar L, et al. Suicide mortality in India: a nationally representative survey. Lancet. 2012;379(9834):2343–2351. doi:10.1016/S0140-6736(12)60606-0
4. Hulse EJ, Davies JO, Simpson AJ, Sciuto AM, Eddleston M. Respiratory complications of organophosphorus nerve agent and insecticide poisoning. Implications for respiratory and critical care. Am J Respir Crit Care Med. 2014;190(12):1342–1354. doi:10.1164/rccm.201406-1150CI
5. Kiran PS, Mandal P, Jain M, Ghosal PS, Gupta AK. A comprehensive review on the treatment of pesticide-contaminated wastewater with special emphasis on organophosphate pesticides using constructed wetlands. J Environ Manage. 2024;368:122163. doi:10.1016/j.jenvman.2024.122163
6. Friedman A, Janulewicz Lloyd PA, Carlson J, et al. Preliminary Findings from the Gulf War Women’s Cohort: reproductive and Children’s Health Outcomes among Women Veterans. Int J Environ Res Public Health. 2022;19(14):8483. doi:10.3390/ijerph19148483
7. Muñoz-Quezada MT, Lucero BA, Barr DB, et al. Neurodevelopmental effects in children associated with exposure to organophosphate pesticides: a systematic review. Neurotoxicology. 2013;39:158–168. doi:10.1016/j.neuro.2013.09.003
8. Curl CL, Fenske RA, Kissel JC, et al. Evaluation of take-home organophosphorus pesticide exposure among agricultural workers and their children. Environ Health Perspect. 2002;110(12):A787–792. doi:10.1289/ehp.021100787
9. Ganie SY, Javaid D, Hajam YA, Reshi MS. Mechanisms and treatment strategies of organophosphate pesticide induced neurotoxicity in humans: a critical appraisal. Toxicology. 2022;472:153181. doi:10.1016/j.tox.2022.153181
10. Kadar T, Shapira S, Cohen G, Sahar R, Alkalay D, Raveh L. Sarin-induced neuropathology in rats. Hum Exp Toxicol. 1995;14(3):252–259. doi:10.1177/096032719501400304
11. Haridas A, Ravi P. Opsoclonus-myoclonus syndrome caused by organophosphate poisoning. Pract Neurol. 2023;23(3):243–245. doi:10.1136/pn-2022-003612
12. Young JG, Eskenazi B, Gladstone EA, et al. Association between in utero organophosphate pesticide exposure and abnormal reflexes in neonates. Neurotoxicology. 2005;26(2):199–209. doi:10.1016/j.neuro.2004.10.004
13. Ventura C, Zappia CD, Lasagna M, et al. Effects of the pesticide chlorpyrifos on breast cancer disease. Implication of epigenetic mechanisms. J Steroid Biochem Mol Biol. 2019;186:96–104. doi:10.1016/j.jsbmb.2018.09.021
14. Ramadori GP. Organophosphorus Poisoning: acute Respiratory Distress Syndrome (ARDS) and Cardiac Failure as Cause of Death in Hospitalized Patients. Int J Mol Sci. 2023;24(7):6658. doi:10.3390/ijms24076658
15. Jokanović M. Biotransformation of organophosphorus compounds. Toxicology. 2001;166(3):139–160. doi:10.1016/S0300-483X(01)00463-2
16. Gupta RC. Brain regional heterogeneity and toxicological mechanisms of organophosphates and carbamates. Toxicol Mech Methods. 2004;14(3):103–143. doi:10.1080/15376520490429175
17. Banks CN, Lein PJ. A review of experimental evidence linking neurotoxic organophosphorus compounds and inflammation. Neurotoxicology. 2012;33(3):575–584. doi:10.1016/j.neuro.2012.02.002
18. Ouardi FZ, Anarghou H, Malqui H, et al. Gestational and Lactational Exposure to Malathion Affects Antioxidant Status and Neurobehavior in Mice Pups and Offspring. J Mol Neurosci. 2019;69(1):17–27. doi:10.1007/s12031-018-1252-6
19. Lumsden EW, McCowan L, Pescrille JD, et al. Learning and memory retention deficits in prepubertal Guinea pigs prenatally exposed to low levels of the organophosphorus insecticide malathion. Neurotoxicol Teratol. 2020;81:106914. doi:10.1016/j.ntt.2020.106914
20. Liu Y, Xu Y, Yuan B, et al. Bioaccumulation mediated by water solubility leads to differences in the acute toxicity of organophosphorus insecticides to zebrafish (Danio rerio). Ecotoxicology. 2024;33(7):750–761. doi:10.1007/s10646-024-02775-7
21. Kovarik Z, Moshitzky G, Maček Hrvat N, Soreq H. Recent advances in cholinergic mechanisms as reactions to toxicity, stress, and neuroimmune insults. J Neurochem. 2024;168(4):355–369. doi:10.1111/jnc.15887
22. Li Q, Lesseur C, Srirangam P, et al. Associations between prenatal organophosphate pesticide exposure and placental gene networks. Environ Res. 2023;224:115490. doi:10.1016/j.envres.2023.115490
23. Terry Jr AV. Functional consequences of repeated organophosphate exposure: potential non-cholinergic mechanisms. Pharmacol Ther. 2012;134(3):355–365. doi:10.1016/j.pharmthera.2012.03.001
24. Dai H, Deng Y, Zhang J, et al. PINK1/Parkin-mediated mitophagy alleviates chlorpyrifos-induced apoptosis in SH-SY5Y cells. Toxicology. 2015;334:72–80. doi:10.1016/j.tox.2015.06.003
25. Eftekhari A, Ahmadian E, Azami A, Johari-Ahar M, Eghbal MA. Protective effects of coenzyme Q10 nanoparticles on dichlorvos-induced hepatotoxicity and mitochondrial/lysosomal injury. Environ Toxicol. 2018;33(2):167–177. doi:10.1002/tox.22505
26. Farizatto KLG, Almeida MF, Long RT, Bahr BA. Early Synaptic Alterations and Selective Adhesion Signaling in Hippocampal Dendritic Zones Following Organophosphate Exposure. Sci Rep. 2019;9(1):6532. doi:10.1038/s41598-019-42934-z
27. Fréville M, Bernardi O, Ramé C, Froment P, Dupont J. Vitamin E alleviates glyphosate-based herbicide-induced progesterone secretion inhibition and oxidative stress increase in chicken primary granulosa cells. Poult Sci. 2024;103(11):104194. doi:10.1016/j.psj.2024.104194
28. Ghrir S, Ben Abbes W, Chourabi A, et al. Grape seed extract prevents chlorpyrifos-induced toxicity in rat liver through the modulation of Phase I detoxification pathway. Environ Sci Pollut Res Int. 2024;31(12):18566–18578. doi:10.1007/s11356-024-32201-8
29. Girón-Pérez MI, Mary VS, Rubinstein HR, Toledo-Ibarra GA, Theumer MG. Diazinon toxicity in hepatic and spleen mononuclear cells is associated to early induction of oxidative stress. Int J Environ Health Res. 2022;32(10):2309–2323. doi:10.1080/09603123.2021.1962814
30. Grigoryan H, Lockridge O. Nanoimages show disruption of tubulin polymerization by chlorpyrifos oxon: implications for neurotoxicity. Toxicol Appl Pharmacol. 2009;240(2):143–148. doi:10.1016/j.taap.2009.07.015
31. H-CG K, Céspedes-Rubio ÁE, Rondón-Barragán IS. First study on response of astrocytes in alevines of red-bellied pacu (Piaractus brachypomus) to subchronic exposure to chlorpyrifos and trichlorfon. Veterinary World. 2022;15(7):1676–1683. doi:10.14202/vetworld.2022.1676-1683
32. Jafari M, Salehi M, Asgari A, et al. Effects of paraoxon on serum biochemical parameters and oxidative stress induction in various tissues of Wistar and Norway rats. Environ Toxicol Pharmacol. 2012;34(3):876–887. doi:10.1016/j.etap.2012.08.011
33. Jain S, Banerjee BD, Ahmed RS, Arora VK, Mediratta PK. Possible role of oxidative stress and brain derived neurotrophic factor in triazophos induced cognitive impairment in rats. Neurochem Res. 2013;38(10):2136–2147. doi:10.1007/s11064-013-1122-0
34. Jiang W, Duysen EG, Hansen H, Shlyakhtenko L, Schopfer LM, Lockridge O. Mice treated with chlorpyrifos or chlorpyrifos oxon have organophosphorylated tubulin in the brain and disrupted microtubule structures, suggesting a role for tubulin in neurotoxicity associated with exposure to organophosphorus agents. Toxicol Sci. 2010;115(1):183–193. doi:10.1093/toxsci/kfq032
35. Joly Condette C, Khorsi-Cauet H, Morlière P, et al. Increased gut permeability and bacterial translocation after chronic chlorpyrifos exposure in rats. PLoS One. 2014;9(7):e102217. doi:10.1371/journal.pone.0102217
36. Kaur P, Radotra B, Minz RW, Gill KD. Impaired mitochondrial energy metabolism and neuronal apoptotic cell death after chronic dichlorvos (OP) exposure in rat brain. Neurotoxicology. 2007;28(6):1208–1219. doi:10.1016/j.neuro.2007.08.001
37. Ki YW, Park JH, Lee JE, Shin IC, Koh HC. JNK and p38 MAPK regulate oxidative stress and the inflammatory response in chlorpyrifos-induced apoptosis. Toxicol Lett. 2013;218(3):235–245. doi:10.1016/j.toxlet.2013.02.003
38. Lee YS, Lewis JA, Ippolito DL, et al. Repeated exposure to neurotoxic levels of chlorpyrifos alters hippocampal expression of neurotrophins and neuropeptides. Toxicology. 2016;340:53–62. doi:10.1016/j.tox.2016.01.001
39. Mense SM, Sengupta A, Lan C, et al. The common insecticides cyfluthrin and chlorpyrifos alter the expression of a subset of genes with diverse functions in primary human astrocytes. Toxicol Sci. 2006;93(1):125–135. doi:10.1093/toxsci/kfl046
40. Montanarí C, Franco-Campos F, Taroncher M, Rodríguez-Carrasco Y, Zingales V, Ruiz MJ. Chlorpyrifos induces cytotoxicity via oxidative stress and mitochondrial dysfunction in HepG2 cells. Food Chem Toxicol. 2024;192:114933. doi:10.1016/j.fct.2024.114933
41. Özdemir S, Aydın Ş, Laçin BB, Arslan H. Identification and characterization of long non-coding RNA (lncRNA) in cypermethrin and chlorpyrifos exposed zebrafish (Danio rerio) brain. Chemosphere. 2023;344:140324. doi:10.1016/j.chemosphere.2023.140324
42. Perez-Fernandez C, Morales-Navas M, Guardia-Escote L, Colomina MT, Giménez E, Sánchez-Santed F. Postnatal exposure to low doses of Chlorpyrifos induces long-term effects on 5C-SRTT learning and performance, cholinergic and GABAergic systems and BDNF expression. Exp Neurol. 2020;330:113356. doi:10.1016/j.expneurol.2020.113356
43. Schäfer M, Koppe F, Stenger B, et al. Influence of organophosphate poisoning on human dendritic cells. Chem Biol Interact. 2013;206(3):472–478. doi:10.1016/j.cbi.2013.08.011
44. Singh AK, Jiang Y. Immunotoxicity of acute acephate exposure in control or IL-1-challenged rats: correlation between the immune cell composition and corticosteroid concentration in blood. J Appl Toxicol. 2002;22(5):279–291. doi:10.1002/jat.852
45. Singh AK, Jiang Y. Lipopolysaccharide (LPS) induced activation of the immune system in control rats and rats chronically exposed to a low level of the organothiophosphate insecticide, acephate. Toxicol Ind Health. 2003;19(2–6):93–108. doi:10.1191/0748233703th181oa
46. Slotkin TA, Seidler FJ, Fumagalli F. Exposure to organophosphates reduces the expression of neurotrophic factors in neonatal rat brain regions: similarities and differences in the effects of chlorpyrifos and diazinon on the fibroblast growth factor superfamily. Environ Health Perspect. 2007;115(6):909–916. doi:10.1289/ehp.9901
47. Suke SG, Ahmed RS, Tripathi AK, Chakraborti A, Banerjee BD. Immunotoxicity of phosphamidon following subchronic exposure in albino rats. Indian J Exp Biol. 2006;44(4):316–320.
48. Syed F, Awasthi KK, Chandravanshi LP, et al. Bifenthrin-induced neurotoxicity in rats: involvement of oxidative stress. Toxicol Res (Camb). 2018;7(1):48–58. doi:10.1039/C7TX00205J
49. Turton N, Heaton RA, Ismail F, et al. The Effect of Organophosphate Exposure on Neuronal Cell Coenzyme Q(10) Status. Neurochem Res. 2021;46(1):131–139. doi:10.1007/s11064-020-03033-y
50. Muñoz-Torrero D, Schopfer LM, Lockridge O. Chlorpyrifos Oxon Activates Glutamate and Lysine for Protein Cross-linking. Chem Res Toxicol. 2023;36(1):112–121. doi:10.1021/acs.chemrestox.2c00333
51. Zurich MG, Honegger P, Schilter B, Costa LG, Monnet-Tschudi F. Involvement of glial cells in the neurotoxicity of parathion and chlorpyrifos. Toxicol Appl Pharmacol. 2004;201(2):97–104. doi:10.1016/j.taap.2004.05.003
52. Duncan JE, Goldstein LS. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006;2(9):e124. doi:10.1371/journal.pgen.0020124
53. Chretien M, Patey G, Souyri F, Droz B. ‘Acrylamide-induced’ neuropathy and impairment of axonal transport of proteins. II. Abnormal accumulations of smooth endoplasmic reticulum as sites of focal retention of fast transported proteins. Electron microscope radioautographic study. Brain Res. 1981;205(1):15–28. doi:10.1016/0006-8993(81)90716-2
54. Maeder CI, Shen K, Hoogenraad CC. Axon and dendritic trafficking. Current Opinion in Neurobiology. 2014;27:165–170. doi:10.1016/j.conb.2014.03.015
55. Naughton SX, Beck WD, Wei Z, Wu G, Terry Jr AV. Multifunctional compounds lithium chloride and methylene Blue attenuate the negative effects of diisopropylfluorophosphate on axonal transport in rat cortical neurons. Toxicology. 2020;431:152379. doi:10.1016/j.tox.2020.152379
56. Teleanu DM, Niculescu AG, Lungu II, et al. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int J Mol Sci. 2022;23(11):5938. doi:10.3390/ijms23115938
57. Pearson JN, Patel M. The role of oxidative stress in organophosphate and nerve agent toxicity. Ann N Y Acad Sci. 2016;1378(1):17–24. doi:10.1111/nyas.13115
58. Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222(3):236–245. doi:10.1046/j.1525-1373.1999.d01-140.x
59. Farkhondeh T, Mehrpour O, Buhrmann C, Pourbagher-Shahri AM, Shakibaei M, Samarghandian S. Organophosphorus Compounds and MAPK Signaling Pathways. Int J Mol Sci. 2020;21(12):4258. doi:10.3390/ijms21124258
60. Sobolev VE, Sokolova MO, Jenkins RO, Goncharov NV. Molecular Mechanisms of Acute Organophosphate Nephrotoxicity. Int J Mol Sci. 2022;23(16):8855. doi:10.3390/ijms23168855
61. Ledda C, Cannizzaro E, Cinà D, et al. Oxidative stress and DNA damage in agricultural workers after exposure to pesticides. J Occup Med Toxicol. 2021;16(1):1. doi:10.1186/s12995-020-00290-z
62. Wang W, Zhang H, Duan X, et al. Association of genetic polymorphisms of miR-145 gene with telomere length in omethoate-exposed workers. Ecotoxicol Environ Saf. 2019;172:82–88. doi:10.1016/j.ecoenv.2019.01.023
63. Zhao MW, Yang P, Zhao LL. Chlorpyrifos activates cell pyroptosis and increases susceptibility on oxidative stress-induced toxicity by miR-181/SIRT1/PGC-1α/Nrf2 signaling pathway in human neuroblastoma SH-SY5Y cells: implication for association between chlorpyrifos and Parkinson’s disease. Environ Toxicol. 2019;34(6):699–707. doi:10.1002/tox.22736
64. Tahmasebi K, Jafari M, Heydari J, et al. Tissues toxicity attenuation by vitamin E on oxidative damage induced by diazinon. Environ Anal Health Toxicol. 2022;37(4):e2022036–2022030. doi:10.5620/eaht.2022036
65. Mallamaci R, Barbarossa A, Carrieri A, Meleleo D, Carocci A. Evaluation of the Potential Cytoprotective Effect of Melatonin in Comparison with Vitamin E and Trolox against Cd(2+)-Induced Toxicity in SH-SY5Y, HCT 116, and HepG2 Cell Lines. Int J Mol Sci. 2024;25(15):8055. doi:10.3390/ijms25158055
66. Da-Silva OF, Adelowo AR, Babalola AA, et al. Diphenyl Diselenide Through Reduction of Inflammation, Oxidative Injury and Caspase-3 Activation Abates Doxorubicin-Induced Neurotoxicity in Rats. Neurochem Res. 2024;49(4):1076–1092. doi:10.1007/s11064-023-04098-1
67. Boskabady M, Marefati N, Farkhondeh T, Shakeri F, Farshbaf A, Boskabady MH. The effect of environmental lead exposure on human health and the contribution of inflammatory mechanisms, a review. Environ Int. 2018;120:404–420. doi:10.1016/j.envint.2018.08.013
68. Binukumar BK, Gupta N, Bal A, Gill KD. Protection of dichlorvos induced oxidative stress and nigrostriatal neuronal death by chronic coenzyme Q10 pretreatment. Toxicol Appl Pharmacol. 2011;256(1):73–82. doi:10.1016/j.taap.2011.07.015
69. Taha MAI, Badawy MEI, Abdel-Razik RK, Younis HM, Abo-El-Saad MM. Mitochondrial dysfunction and oxidative stress in liver of male albino rats after exposing to sub-chronic intoxication of chlorpyrifos, cypermethrin, and imidacloprid. Pestic Biochem Physiol. 2021;178:104938. doi:10.1016/j.pestbp.2021.104938
70. Sangwan M, Chaudhary H, Mehan S, et al. Effect of mitochondrial coenzyme-Q10 precursor solanesol in gentamicin-induced experimental nephrotoxicity: evidence from restoration of ETC-complexes and histopathological alterations. Pharmacol Res Perspect. 2024;12(5):e70022. doi:10.1002/prp2.70022
71. de Oliveira MR, Ferreira GC, Schuck PF. Protective effect of carnosic acid against paraquat-induced redox impairment and mitochondrial dysfunction in SH-SY5Y cells: role for PI3K/Akt/Nrf2 pathway. Toxicol In Vitro. 2016;32:41–54. doi:10.1016/j.tiv.2015.12.005
72. Pistollato F, de Gyves EM, Carpi D, et al. Assessment of developmental neurotoxicity induced by chemical mixtures using an adverse outcome pathway concept. Environ Health. 2020;19(1):23. doi:10.1186/s12940-020-00578-x
73. De leeuw VC, van Oostrom CTM, Wackers PFK, et al. Neuronal differentiation pathways and compound-induced developmental neurotoxicity in the human neural progenitor cell test (hNPT) revealed by RNA-seq. Chemosphere. 2022;304:135298. doi:10.1016/j.chemosphere.2022.135298
74. Rohlman DS, Anger WK, Lein PJ. Correlating neurobehavioral performance with biomarkers of organophosphorous pesticide exposure. Neurotoxicology. 2011;32(2):268–276. doi:10.1016/j.neuro.2010.12.008
75. Zhong X, Wu J, Ke W, et al. Neonatal exposure to organophosphorus flame retardant TDCPP elicits neurotoxicity in mouse hippocampus via microglia-mediated inflammation in vivo and in vitro. Arch Toxicol. 2020;94(2):541–552. doi:10.1007/s00204-019-02635-y
76. Corsini E, Sokooti M, Galli CL, Moretto A, Colosio C. Pesticide induced immunotoxicity in humans: a comprehensive review of the existing evidence. Toxicology. 2013;307:123–135. doi:10.1016/j.tox.2012.10.009
77. Mokarizadeh A, Faryabi MR, Rezvanfar MA, Abdollahi M. A comprehensive review of pesticides and the immune dysregulation: mechanisms, evidence and consequences. Toxicol Mech Methods. 2015;25(4):258–278. doi:10.3109/15376516.2015.1020182
78. Berkenbosch F, Rey AD, Besedovsky HO, de Goeij DEC. Neuroendocrine, sympathetic and metabolic responses induced by interleukin-1. Neuroendocrinology. 1989;50(5):570–576. doi:10.1159/000125283
79. Bernal-González KG, Covantes-Rosales CE, Camacho-Pérez MR, et al. Organophosphate-Pesticide-Mediated Immune Response Modulation in Invertebrates and Vertebrates. Int J Mol Sci. 2023;24(6):5360. doi:10.3390/ijms24065360
80. Ghaffarzadegan R, Akhondzadeh S, Nikasa Z, et al. New Insights into Contradictory Changes in Brain-Derived Neurotrophic Factor (BDNF) in Rodent Models of Posttraumatic Stress Disorder (PTSD). Neurochem Res. 2024;49(12):3226–3243. doi:10.1007/s11064-024-04242-5
81. Liberona A, Jones N, Zúñiga K, et al. Brain-Derived Neurotrophic Factor (BDNF) as a Predictor of Treatment Response in Schizophrenia and Bipolar Disorder: a Systematic Review. Int J Mol Sci. 2024;25(20):11204. doi:10.3390/ijms252011204
82. Pope C, diLorenzo K, Ehrich M. Possible involvement of a neurotrophic factor during the early stages of organophosphate-induced delayed neurotoxicity. Toxicol Lett. 1995;75(1–3):111–117. doi:10.1016/0378-4274(94)03167-6
83. Giambò F, Costa C, Teodoro M, Fenga C. Role-Playing Between Environmental Pollutants and Human Gut Microbiota: a Complex Bidirectional Interaction. Front Med (Lausanne). 2022;9:810397. doi:10.3389/fmed.2022.810397
84. Roman P, Cardona D, Sempere L, Carvajal F. Microbiota and organophosphates. Neurotoxicology. 2019;75:200–208. doi:10.1016/j.neuro.2019.09.013
85. Borre YE, Moloney RD, Clarke G. The Impact of Microbiota on Brain and Behavior: mechanisms & Therapeutic Potential[M]//Lyte M. In: Cryan JF, editor. Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease. Vol. 817. New York, NY: Springer; 2014:373–403.
86. Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28(2):203–209.
87. Nichols RG, Rimal B, Hao F, Peters JM, Davenport ER, Patterson AD. Chlorpyrifos modulates the mouse gut microbiota and metabolic activity. Environ Int. 2024;192:109022. doi:10.1016/j.envint.2024.109022
88. Kopjar N, Žunec S, Mendaš G, et al. Evaluation of chlorpyrifos toxicity through a 28-day study: cholinesterase activity, oxidative stress responses, parent compound/metabolite levels, and primary DNA damage in blood and brain tissue of adult male Wistar rats. Chem Biol Interact. 2018;279:51–63. doi:10.1016/j.cbi.2017.10.029
89. He X, Yang Y, Zhou S, et al. Alterations in microbiota-metabolism-circRNA crosstalk in autism spectrum disorder-like behaviours caused by maternal exposure to glyphosate-based herbicides in mice. Ecotoxicol Environ Saf. 2024;285:117060. doi:10.1016/j.ecoenv.2024.117060
90. Dai H, Wang J, Li Y, Lv Z. Hawthorn-leaf flavonoid alleviate intestinal health and microbial dysbiosis problems induced by glyphosate. Ecotoxicol Environ Saf. 2024;284:116901. doi:10.1016/j.ecoenv.2024.116901
91. Jokanović M, Oleksak P, Kuca K. Multiple neurological effects associated with exposure to organophosphorus pesticides in man. Toxicology. 2023;484:153407. doi:10.1016/j.tox.2022.153407
92. Muñoz-Quezada MT, Lucero BA, Iglesias VP, et al. Chronic exposure to organophosphate (OP) pesticides and neuropsychological functioning in farm workers: a review. Int J Occup Environ Health. 2016;22(1):68–79. doi:10.1080/10773525.2015.1123848
93. Reddy DS, Singh T, Ramakrishnan S, Huber M, Wu X. Neuroprotectant Activity of Novel Water-Soluble Synthetic Neurosteroids on Organophosphate Intoxication and Status Epilepticus-Induced Long-Term Neurological Dysfunction, Neurodegeneration, and Neuroinflammation. J Pharmacol Exp Ther. 2024;388(2):399–415. doi:10.1124/jpet.123.001819
94. Kozhemyakin M, Rajasekaran K, Kapur J. Central cholinesterase inhibition enhances glutamatergic synaptic transmission. J Neurophysiol. 2010;103(4):1748–1757. doi:10.1152/jn.00949.2009
95. Williamson J, Singh T, Kapur J. Neurobiology of organophosphate-induced seizures. Epilepsy Behav. 2019;101(Pt B):106426. doi:10.1016/j.yebeh.2019.07.027
96. Bernheim L, Mathie A, Hille B. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci U S A. 1992;89(20):9544–9548. doi:10.1073/pnas.89.20.9544
97. Fu L, Luo Y, Niu L, et al. M(1)/M(4) receptors as potential therapeutic treatments for schizophrenia: a comprehensive study. Bioorg Med Chem. 2024;105:117728. doi:10.1016/j.bmc.2024.117728
98. Aroniadou-Anderjaska V, Figueiredo TH, Apland JP, Braga MF. Targeting the glutamatergic system to counteract organophosphate poisoning: a novel therapeutic strategy. Neurobiol Dis. 2020;133:104406. doi:10.1016/j.nbd.2019.02.017
99. Bernardino PN, Luo AS, Andrew PM, et al. Evidence Implicating Blood-Brain Barrier Impairment in the Pathogenesis of Acquired Epilepsy following Acute Organophosphate Intoxication. J Pharmacol Exp Ther. 2024;388(2):301–312. doi:10.1124/jpet.123.001836
100. Gupta RC, Milatovic D, Dettbarn WD. Depletion of energy metabolites following acetylcholinesterase inhibitor-induced status epilepticus: protection by antioxidants. Neurotoxicology. 2001;22(2):271–282. doi:10.1016/S0161-813X(01)00013-4
101. Shulman D, Dubnov S, Zorbaz T, et al. Sex-specific declines in cholinergic-targeting tRNA fragments in the nucleus accumbens in Alzheimer’s disease. Alzheimers Dement. 2023;19(11):5159–5172. doi:10.1002/alz.13095
102. Bartus RT, Dean RL, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408–414. doi:10.1126/science.7046051
103. Coupland CAC, Moore M, Hippisley-Cox J. Association of Anticholinergic Drug Exposure With Increased Occurrence of Dementia-Reply. JAMA Intern Med. 2019;179(12):1730–1731. doi:10.1001/jamainternmed.2019.4908
104. Silman I, Sussman JL. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr Opin Pharmacol. 2005;5(3):293–302. doi:10.1016/j.coph.2005.01.014
105. Stokin GB, Goldstein LS. Axonal transport and Alzheimer’s disease. Annu Rev Biochem. 2006;75(1):607–627. doi:10.1146/annurev.biochem.75.103004.142637
106. Saeidi M, Shakeri R, Marjani A, Khajeniazi S. Alzheimer’s Disease and Paraoxonase 1 (PON1) Gene Polymorphisms. Open Biochem J. 2017;11(1):47–55. doi:10.2174/1874091X01711010047
107. Wehr H, Bednarska-Makaruk M, Graban A, et al. Paraoxonase activity and dementia. J Neurol Sci. 2009;283(1–2):107–108. doi:10.1016/j.jns.2009.02.317
108. Yan D, Zhang Y, Liu L, Yan H. Pesticide exposure and risk of Alzheimer’s disease: a systematic review and meta-analysis. Sci Rep. 2016;6(1):32222. doi:10.1038/srep32222
109. Khan SS, Bloom GS. Tau: the Center of a Signaling Nexus in Alzheimer’s Disease. Front Neurosci. 2016;10:31. doi:10.3389/fnins.2016.00031
110. Hayden KM, Norton MC, Darcey D, et al. Occupational exposure to pesticides increases the risk of incident AD: the Cache County study. Neurology. 2010;74(19):1524–1530. doi:10.1212/WNL.0b013e3181dd4423
111. Lushchekina SV, Masson P. Slow-binding inhibitors of acetylcholinesterase of medical interest. Neuropharmacology. 2020;177:108236. doi:10.1016/j.neuropharm.2020.108236
112. Schapira AH. Recent developments in biomarkers in Parkinson disease. Curr Opin Neurol. 2013;26(4):395–400. doi:10.1097/WCO.0b013e3283633741
113. Dhillon AS, Tarbutton GL, Levin JL, et al. Pesticide/environmental exposures and Parkinson’s disease in East Texas. J Agromedicine. 2008;13(1):37–48. doi:10.1080/10599240801986215
114. Gatto NM, Cockburn M, Bronstein J, Manthripragada AD, Ritz B. Well-water consumption and Parkinson’s disease in rural California. Environ Health Perspect. 2009;117(12):1912–1918. doi:10.1289/ehp.0900852
115. Chuang CS, Su HL, Lin CL. Risk of Parkinson disease after organophosphate or carbamate poisoning. Acta Neurologica Scandinavica. 2017;136(2):129–137. doi:10.1111/ane.12707
116. Parsanejad M, Bourquard N, Qu D, et al. DJ-1 interacts with and regulates paraoxonase-2, an enzyme critical for neuronal survival in response to oxidative stress. PLoS One. 2014;9(9):e106601. doi:10.1371/journal.pone.0106601
117. Paldor I, Madrer N, Vaknine Treidel S, Shulman D, Greenberg DS, Soreq H. Cerebrospinal fluid and blood profiles of transfer RNA fragments show age, sex, and Parkinson’s disease-related changes. J Neurochem. 2023;164(5):671–683. doi:10.1111/jnc.15723
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.