")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Metabolism and Excretion of 8-O-Acetylharpagide in Rats and Identification of Its Potential Anti-Breast Cancer Active Metabolites
Authors Zhao X , Su S, Zhou J, Gao J, Tang X, Wen B
Received 22 July 2024
Accepted for publication 28 March 2025
Published 10 April 2025 Volume 2025:19 Pages 2795—2815
DOI https://doi.org/10.2147/DDDT.S487898
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Muzammal Hussain
Xinyu Zhao, Sijia Su, Jingna Zhou, Junfeng Gao, Xu Tang, Binyu Wen
Dongfang Hospital, Beijing University of Chinese Medicine, Beijing, 100078, People’s Republic of China
Correspondence: Binyu Wen, Dongfang Hospital, Beijing University of Chinese Medicine, Beijing, 100078, People’s Republic of China, Email [email protected]
Background: Ajuga decumbens, a traditional Chinese medicine, possesses anti-breast cancer effects. Its main component, 8-O-acetylharpagide, exhibits potential anticancer activity; however, the active metabolites and mechanisms underlying its effects remain unclear.
Methods: The metabolism and excretion of 8-O-acetylharpagide in rats were investigated using ultra-high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry analysis of bile, urine, and feces. Active metabolites were identified and evaluated using network pharmacology, molecular docking, and Western blotting assays.
Results: A total of 21 metabolites were identified, with demethylation, hydrolysis, and glucuronidation being the primary metabolic pathways. M3 and M5 were identified as key metabolites, showing strong binding affinity to cancer-related targets, such as AKT1, MMP9, and STAT3. M5 displayed strong pharmacokinetic properties, including better lipid solubility and reduced polarity. Network pharmacology analysis indicated that these metabolites exert anticancer effects by modulating the PI3K/AKT signaling pathway. In vivo experiments demonstrated that oral administration of 8-O-acetylharpagide significantly inhibited the proliferation of 4T1 tumor tissues by suppressing the expression of the AKT/NF-κB/MMP9 signaling axis. This may be related to inhibition of the expression of the AKT/NF-κB/MMP9 signaling axis in 4T1 tumor tissues after metabolism of 8-O-acetylharpagide to M5 and M3.
Conclusion: As a prodrug, 8-O-acetylharpagide is metabolized to M5, which may subsequently exert an anti-breast cancer effect through downregulation of the AKT/NF-κB/MMP9 signaling axis. This study provides a theoretical basis for the clinical application of Ajuga decumbens in breast cancer therapy.
Keywords: 8-O-acetylharpagide, breast cancer, ultra-high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry, metabolite, network pharmacology, AKT/NF-κB/MMP9 signaling pathway
Graphical Abstract:

Introduction
Ajuga decumbens is a traditional Chinese medicine in China, mainly in the south of Qinling Mountains. It has a long history of folk medicinal use in China. It is mainly used for treating lung-heat cough, sore throat, bruises, carbuncle boils, and sores.1–4 In Traditional Chinese Medicine Oncology, Yu recorded that this medicine is commonly used in the treatment of breast and lung cancer.5 The iridoid glycosides extracted from Ajuga decumbens were found to have anti-breast cancer effects. The mechanisms underlying these effects were related to inhibition of the activation of extracellular signal-regulated kinase 1/2/mitogen-activated protein kinase (ERK1/2/MAPK) and phosphatidylinositol 3 kinase/protein kinase B (PI3K/AKT) signaling pathways, as well as inhibition of the expression of matrix metalloproteinase 9 (MMP9) and MMP2 in breast cancer tissues.6–8The PI3K/AKT signaling pathway plays a crucial role in promoting breast cancer progression by regulating cell survival, proliferation, and metabolism.9 Matrix metalloproteinases (MMPs), particularly MMP9 and MMP2, facilitate tumor invasion and metastasis by degrading the extracellular matrix and basement membranes.10,11
As the primary iridoid glycoside in Ajuga decumbens, 8-O-acetylharpagide constitutes approximately 35% of the iridoid glycosides in this plant.12 It has been reported that 8-O-acetylharpagide has vasoconstrictive, antibacterial, antiviral, analgesic, antiprotozoal, hepatoprotective, and renoprotective activities.13–18 Moreover, 8-O-acetylharpagide could exert anti-inflammatory effects by inhibiting leukocyte adhesion and migration. Additionally, it could significantly inhibit endothelial cell migration to alleviate the progression of chronic inflammation.19 8-O-acetylharpagide regulates the related biomarkers senescence-associated-β-galactosidase and p53 in old human dermal fibroblasts, leading to inhibition of cellular senescence.20 It is also a potentially promising anticancer agent that exerts Epstein–Barr Virus Early Antigen-induced inhibitory effects on 7.12-dimethylbenzo[a]-anthracene and 12-O-tetradecanoylphorbol-13-acetate-induced skin cancers, and inhibits lung cancer.21,22 Furthermore, our latest study found that 8-O-acetylharpagide significantly inhibits the growth of breast cancer. The mechanism underlying the anti-breast cancer effect of 8-O-acetylharpagide may be related to the regulation of primary bile acid biosynthesis and arachidonic acid metabolism, as well as the modulation of the abundance of Akkermansia and Firmicutes.23 Most studies on traditional Chinese medicine and its components for breast cancer treatment focus on pharmacological activities and mechanisms. However, research on their pharmacokinetics remains insufficient. It is well established that pharmacokinetics plays a crucial role in elucidating the mechanism of action of these drugs. For 8-O-acetylharpagide, pharmacokinetic research has revealed that its absolute bioavailability is only 7.7%.24 This low bioavailability implied a low exposure in vivo,25 suggesting that 8-O-acetylharpagide does not exert its anti-breast cancer effect in the form of a prototype drug. Therefore, the aim of this study was to provide insights into the mechanisms underlying the anti-breast cancer effects of 8-O-acetylharpagide by elucidating the metabolic pathways and identifying active metabolites.
In this study, 8-O-acetylharpagide metabolites were identified in rat plasma, bile, urine, feces, and in vitro intestinal flora. Subsequently, the content of 8-O-acetylharpagide in bile, urine and faces of rats after gavage of 8-O-acetylharpagide was quantified. Network pharmacological analyses and molecular docking were performed to identify potential active metabolites of 8-O-acetylharpagide for the treatment of breast cancer and elucidate the underlying pharmacological mechanisms. Finally, the effects of 8-O-acetylharpagide on the expression of related proteins in breast cancer tumors were determined by Western blotting analysis.
Materials and Methods
Chemicals and Reagents
8-O-acetylharpagide (99.52%; Chemical Abstracts Service number: 6926–14-3) was obtained from Chengdu Must Bio-technology Co., Ltd. (Chengdu, China). Fine bore polythene tubing (diameter: 0.58 mm, length: 30 m) was purchased from Smiths Medical International Ltd. (Kent, UK). Gifu Anaerobic Medium (GAM), vitamin K1, and hemin were purchased from Qingdao Hope Bio Co., Ltd (Qingdao, China). Biological saline was purchased from Shijiazhuang NO.4 Pharmaceutical Co., Ltd (Shijiazhuang, China). Ultra-pure water was acquired from Watsons (Beijing, China). Acetonitrile, methanol, and formic acid (liquid chromatography–mass spectrometry grade) were obtained from Thermo Fisher Scientific (Shanghai, China).
Metabolites of 8-O-Acetylharpagide in Rat Plasma, Bile, Urine and Feces
Animal Handling and Ethical Considerations
Healthy female Sprague–Dawley rats (specific pathogen-free grade, weight: 200–220 g) were obtained from Charles River (Beijing, China; Certificate No. SCXK (Jing) 2021–0011). Animals were placed in an animal environment under the following conditions: 12-h light/dark cycle, 22°C, 50–60% humidity, and free access to water and feed. The experiments were conducted in strict agreement with the protocols approved by the Laboratory Animal Ethics Committee of Dongfang Hospital, Beijing University of Chinese Medicine (Beijing, China; protocol code: DFYY2023B32R).
Sample Collection and Pretreatment
The 48 rats were adaptively fed for 7 days and fasted for 12 h prior to the experiment. Subsequently, they were randomly divided into four groups for plasma, bile, urine, and feces collection. Each group had two subgroups, namely control and 8-O-acetylharpagide treatment (10 mg/kg). Blood, bile, urine, and feces samples were collected at various time points following oral administration of the drug.
Plasma samples were collected at 0.5, 1, 1.5, 2, 2.5, 3, 4, and 8 h post gavage. Blood was collected in heparin tubes, centrifuged, and stored at −80°C. For bile collection,26 rats underwent choledochal intubation under isoflurane anesthesia, and bile was collected at 30-min intervals for up to 12 h. Urine and feces were collected in metabolic cages for 24–48 h post dosing.
All biological matrices underwent similar pretreatment procedures, with minor differences based on sample type.
Pooled plasma samples were mixed with methanol (3× volume), vortexed, and centrifuged. The supernatant was evaporated and reconstituted in methanol for analysis.
Pooled bile samples were mixed with methanol, vortexed, and centrifuged. The supernatant was evaporated, redissolved, and analyzed.
Pooled urine samples were mixed with methanol and an internal standard (IS) solution (lidocaine), vortexed, and centrifuged. The supernatant was used for analysis.
Fecal samples were homogenized, sonicated, and centrifuged. The supernatant was treated similarly to urine for analysis.
Metabolites of 8-O-Acetylharpagide in Intestinal Flora
To prepare 0.1% vitamin K1 solution, vitamin K1 (0.01 g) was dissolved in anhydrous ethanol (10 mL), vortexed to for full dissolution, and filtered through a 0.22-µm organic membrane to obtain sterile 0.1% vitamin K1 solution.
For the preparation of hemin chloride (5 mg/mL) solution, hemin chloride (50 mg) was dissolved in 0.1 mL sodium hydroxide solution (1 mol/L); after adding ultrapure water to reach a volume of 10 mL, the mixture was autoclaved at 121°C for 15 min, and subsequently cooled to room temperature for use.
GAM was prepared as follows. Initially, GAM (4.9 g) (HB8518-1; Qingdao Hope Bio-Technology Co., Ltd.) was dissolved in water (100 mL), autoclaved at 121°C for 15 min, and cooled to room temperature. Next, sterile 0.1% vitamin k1 solution and hemoglobin chloride (5 mg/mL) solution (0.1 mL each) were added, and the mixture was mixed for use.
Saline (8 mL) was added to fresh feces (2 g) obtained from healthy female rats, and the mixture was vortexed, homogenized, and centrifuged (37°C, 2000 r/min, 10 min). Subsequently, the supernatant (2 mL) was added to GAM (18 mL), and the mixture was vortexed, and placed into an anaerobic incubator (C31; Mitsubishi Gas Chemical Co., Inc., Tokyo, Japan) at 37°C for 24 h of constant temperature oscillation to obtain the intestinal flora culture solution (two samples). One of the samples was autoclaved at 121°C for 25 min (sterile control group), whereas the other was not autoclaved (untreated group). In both groups, 8-O-acetylharpagide (dissolved in ultra-pure water, 2.5 mg/mL) (2 mL) was added, and the samples were placed in an anaerobic incubator, incubated at constant temperature (37°C) and vibration. Samples (600 μL each) were obtained at 0, 2, 4, and 24 h (three copies were obtained at each time point). Cold methanol solution (600 μL) was added to each sample, and the mixtures were vortexed. After centrifugation (12,000 r/min, 10 min, 4°C), the supernatant (1 mL) of each sample was obtained and concentrated to dryness by nitrogen blowing at 37°C. Prior to UPLC-Q-TOF-MS/MS analysis, each sample was reconstituted with 50% methanol water (150 μL), and supernatant (100 μL) was obtained by centrifugation (13,000 r/min, 10 min, 4°C).
Analytical Conditions
Chromatographic evaluations were conducted on a Waters Acquity UPLC system (Waters Corporation, Milford, MA, USA) equipped with a binary solvent delivery system, an autosampler, and an ACQUITY UPLC® HSS T3 column (100 × 2.1 mm, 1.8 μm; Waters Corporation). The mobile phase consisted of water containing 0.1% formic acid (A) and methanol (B) at a flow rate of 0.4 mL/min. The elution gradient was programmed as follows: 0–2 min, 0% B; 2–22 min, 0–95% B; 22–27 min, 95% B; 27–27.1 min, 95–0% B; 27.1–30 min, 0% B. The temperature of the autosampling chamber was 15°C, and the temperature of the column was 40°C. The injection volume was 5.0 μL.
Mass spectra were obtained using Waters Synapt G2 (Waters Corporation). Both positive and negative ion detection modes were used in this analysis. Capillary voltage was 3.0 kV, while the sampling cone voltage was 15 V. The source temperature was 120°C, and the desolvation temperature was 500°C, with a nitrogen gas flow rate of 800 L/h. The collision energy of the ramp ranged 10–80 V. High-resolution quality data were recorded using Waters MSE acquisition mode. The collision energy of the ramps ranged 10–80 V. The data were centered from 100 to 1500 Da for all samples with a scan time of 0.2 s over an analysis time of 30 min. LockSprayTM was employed for the validation of the mass precision. A lock mass solution of leucine enkephalin (556.2771 and 554.2615 m/z for positive and negative electrospray modes, respectively) was infused through a LockSprayTM probe at a flow rate of 10 µL/min at 10 s intervals to correct any mass drift during analysis.
Strategy for Systems Analysis of Metabolites
UNIFI 1.8.2 (Waters Corporation) software was employed for data processing. The Binary Compare function was used for the identification of effective metabolites. Evaluated metabolites did not exist in the equivalent control sample or existed at low ion intensities. The relative intensity threshold was set at 3 or 5, and metabolites that fulfilled the underlined criteria could be evaluated. Common and predictable metabolites were subsequently determined by extracted ion chromatography. The neutral loss filtering function was utilized to search for two-phase metabolites. For example, in the UNIFI software, the parameters could be set at 176.0321 to search for possible glucuronic acid conjugates. Post processing, a neutral loss could be set in the method or identified. Mass fragment was used to determine or characterize the detected structures of the metabolites. The spectral interpretation function of UNIFI was the main function used to analyze secondary fragmentation of parent components. This function can be used for rapid verification of the fragmentation path whether reasonable.
Quantification of 8-O-Acetylharpagide in Rat Bile, Urine, and Feces
Animal Handling and Ethical Considerations
Healthy female Sprague–Dawley rats (specific pathogen-free grade, weight: 250±30 g) were obtained from Charles River (Certificate No. SCXK (Ji) 2016–006). The adaptive feeding and animal ethics were consistent with those described in the above experiment.
Sample Collection and Pretreatment
The experimental setup for the quantitative excretion assay was similar to the one used for metabolite identification, with the following key differences: for bile sample collection, 8-O-acetylharpagide was administered at 12 mg/kg for 4 h; for urine and fecal sample collection, 8-O-acetylharpagide was administered at 10.5 mg/kg for 48 h.
The pretreatment steps for bile, urine, and fecal sample collection were the same as those performed for metabolite identification, except that IS was not added to the samples in this assay.
Analytical Conditions
The analysis was performed using an Agilent 1200 series high-performance liquid chromatograph equipped with an autosampler and a quadruple gradient pump, as well as an atmospheric pressure ionization 4000QTRAP mass spectrometer (Applied Biosystems, Inc., USA) equipped with an electrospray ionization source and Analysis 1.4.2 data processing system.
The chromatographic separation was performed on a Waters Atlantis dC18 column (2.1×150 mm, 5 µm). The mobile phase consisted of aqueous solution containing 0.1% formic acid (A) and acetonitrile solution containing 0.1% formic acid (B) at a flow rate of 0.3 mL/min with the following gradient elution pattern: 0–2 min, 10% B, 2–3 min, 10–20% B, 3–7 min, 20–100% B, and 7–10 min, 10% B. The mass spectrometry was performed in the positive ion mode. The mass spectrometry was performed in the multiple reaction monitoring mode, and the samples were analyzed by monitoring the ionic excursions 429.00→369.00 m/z (8-O-acetylharpagide) and 235.12→86.00 m/z (IS: lidocaine). The capillary voltage was 1.5 kV, the ion source temperature was 130°C, the drying gas temperature was 350°C, the drying gas flow rate was 76 L/h, the detector voltage was 650 V, and the collision gas pressure was 3.13×10−3 mbar.
Method Validations
The specificity of the method was determined by measuring the levels of interfering components from six independent sources in blank biological matrices to exclude the retention time of analytes and IS from any potential interference.
The blank samples were diluted to a series of mass concentrations of 0.02, 0.1, 0.2, 0.4, 0.6, 0.8, 0.9, 1.0 μg/mL (bile); 0.02, 0.05, 0.1, 0.2, 0.5, 1.0, 2.0, 5.0 μg/mL (urine); and 0.004, 0.01, 0.02, 0.04, 0.1, 0.2, 0.4, 1.0 μg/mL (feces). Calibration curves were constructed using a weighted least squares linear regression analysis of the drug concentrations versus peak area ratios of 8-O-acetylharpagide/IS.
Network Pharmacology Analysis
Target Genes Related to 8-O-Acetylharpagide and Metabolites
Simplified Molecular Input Line Entry System (SMILES) strings were obtained directly through PubChem (https://pubchem.ncbi.nlm.nih.gov/) or after recognizing structures based on StoneMIND Collector. The obtained SMILES strings were imported into SwissTargetPrediction (http://www.swisstargetprediction.ch) and SEA (https://sea.bkslab.org/) databases.
Potential Target Genes of Breast Cancer
Information on breast cancer target genes was obtained from the GeneCard database (http://www.genecards.org/), Therapeutic Target Database (TTD; http://db.idrblab.net/ttd/), and Online Mendelian Inheritance in the Man® (OMIM) database (https://omim.org/). The keyword “breast cancer” was used, and the GeneCard database had a relevance score >5.27 The Search Tool for the Retrieval of Interacting Genes (STRING) database (https://cn.string-db.org/) was used for gene name conversion of target proteins.
Construction of Networks
Association of target genes of 8-O-acetylharpagide and its metabolites with breast cancer was investigated using the Venny 2.1 website (https://bioinfogp.cnb.csic.es/tools/venny/index.html). Protein–protein interaction (PPI) analysis was performed using the functional protein association network (STRING), and the core targets were screened according to a degree ≥5.28 Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of core target genes was conducted in the Database for Annotation, Visualization, and Integrated Discovery (DAVID) database (https://david.ncifcrf.gov/), and potential pathways for 8-O-acetylharpagide and its metabolites were determined.
Compound-target-disease networks and compound-target-pathway networks were constructed using Cytoscape (ver. 3.7.2). Nodes in the Comparative Toxicogenomics Database (CTD) network represent interactions between compounds and target genes, and compound target pathway (CTP) indicates interactions between compounds, target genes, and pathways.
Molecular Docking
The aim of this study was to determine the interactions between metabolites and their protein targets, as well as to explore their binding modes. Therefore, six compounds and six core proteins were selected for molecular docking validation. The three-dimensional (3D) structures of 8-O-acetylharpagide and its metabolites were downloaded from PubChem or drawn by ChemDraw and Chem 3D software. Docked proteins were selected from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) database (https://www.rcsb.org/), UniProt database (https://www.uniprot.org), and AlphaFold database (https://alphafold.ebi.ac.uk) with the following selection criteria: “Homo sapiens” proteins; refinement resolution ≤2.5 A; complete conformational sequence, including small molecule information in the structural complex; and crystallization pH close to the normal human physiological range. Component-target molecule docking was performed using AutoDock Tools (ver. 1.5.6). The visualization of docking results was realized using PyMOL software.
Western Blotting Analysis
The experiments on animals were approved by the Laboratory Animal Ethics Committee of the Dongfang Hospital Beijing University of Chinese Medicine (protocol code: DFYY2023B33M). Female Balb-C mice (age: 4 weeks; weight: 22±2 g) were purchased from Beijing Vital River Laboratory Animal Technology Co. (animal acquisition license: SCXK (JING) 2021-0006). Mouse modeling of breast cancer was performed by subcutaneously injecting 4T1 cells into the back of each animal.7,23 Appearance of nodular tumors with a diameter measuring 2–4 mm within 1 week denoted successful modeling. Thereafter, mice with breast cancer were divided into four groups: control group (treated with 0.1 mL saline once daily: n=5); low-dose 8-O-acetylharpagide group (75 mg/kg once daily, n=5); high-dose 8-O-acetylharpagide group (150 mg/kg once daily, n=5); and positive drug group (intraperitoneal cyclophosphamide, 60 mg/kg, once weekly, n=5). After 4 weeks, the mice were anesthetized with isoflurane inhalation (induced 4%, maintenance 1.5–2.0% at room temperature) in an animal anesthesia machine (RWD, Shenzhen, China), and tumor tissues were obtained for Western blotting analysis.
Breast cancer tumor tissues were treated on ice with radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate), which contains sodium orthovanadate, sodium fluoride, ethylenediaminetetraacetic acid, leupeptin, and other inhibitors (Beyotime, Shanghai, China). The lysates were centrifuged at 12,000 rpm for 15 min, and the proteins were quantified using the BCA Protein Assay Kit (MDL, Beijing, China). Total proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. The primary antibodies used in the analyses were AKT (1:1000; Biodragon, China), p-AKT (1:1000; proteintech, China), NF-κB p65 (1:1000; proteintech, China), MMP9 (1:1000; Biodragon, China), and β-actin (1:3,000; Affinity, China). The secondary antibody was a goat anti-rabbit conjugated to horseradish peroxidase (1:10,000, MDL). Western blotting results were detected and analyzed using Image Studio Lite.
Statistical Analysis
GraphPad Prism 8 (GraphPad Software, San Diego, CA, USA) was used to conduct statistical analyses. The data were presented as the mean ± standard error of the mean. Comparisons between two groups were analyzed by t-test, while analysis between multiple groups was performed using one-way analysis of variance. P-values < 0.05 indicate statistically significant differences.
Results
Metabolite Identification
The identification of metabolites of 8-O-acetylharpagide was carried out using UNIFI software. Binary comparison method was used to eliminate the interference of blank matrix. The compounds detected only in drug-containing samples were considered metabolites, and the identification was based on binary comparisons, characteristic fragments, and retention time comparisons with their prototypes. Metabolic product identification was based on the ion fragmentation pattern of 8-O-acetylharpagide (Supplementary Figures S1 and S2). A total of 21 metabolites were screened in plasma (n=3), urine (n=15), feces (n=15), bile (n=10), and intestinal flora (n=2), and the chemical structures of nine of these were determined (Supplementary Table S1). Metabolic reactions included hydrolysis, dehydration, reduction, methylation, and glucuronidation (Figure 1). The extracted ion chromatograms and MS/MS spectra of the above metabolites are shown in Supplementary Figure S3. All identified 8-O-acetylharpagide metabolites are listed in Table 1.
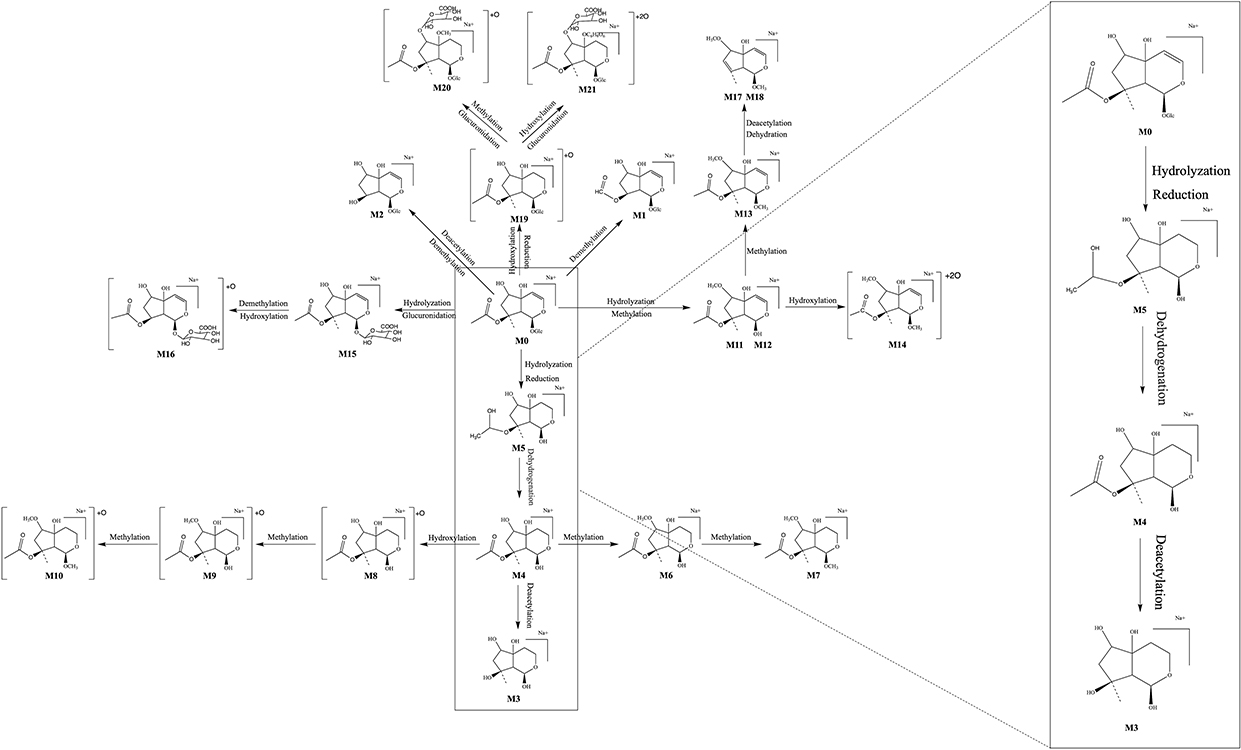 |
Figure 1 Metabolic profile and proposed metabolic pathways of 8-O-acetylharpagide in vitro and in vivo. |
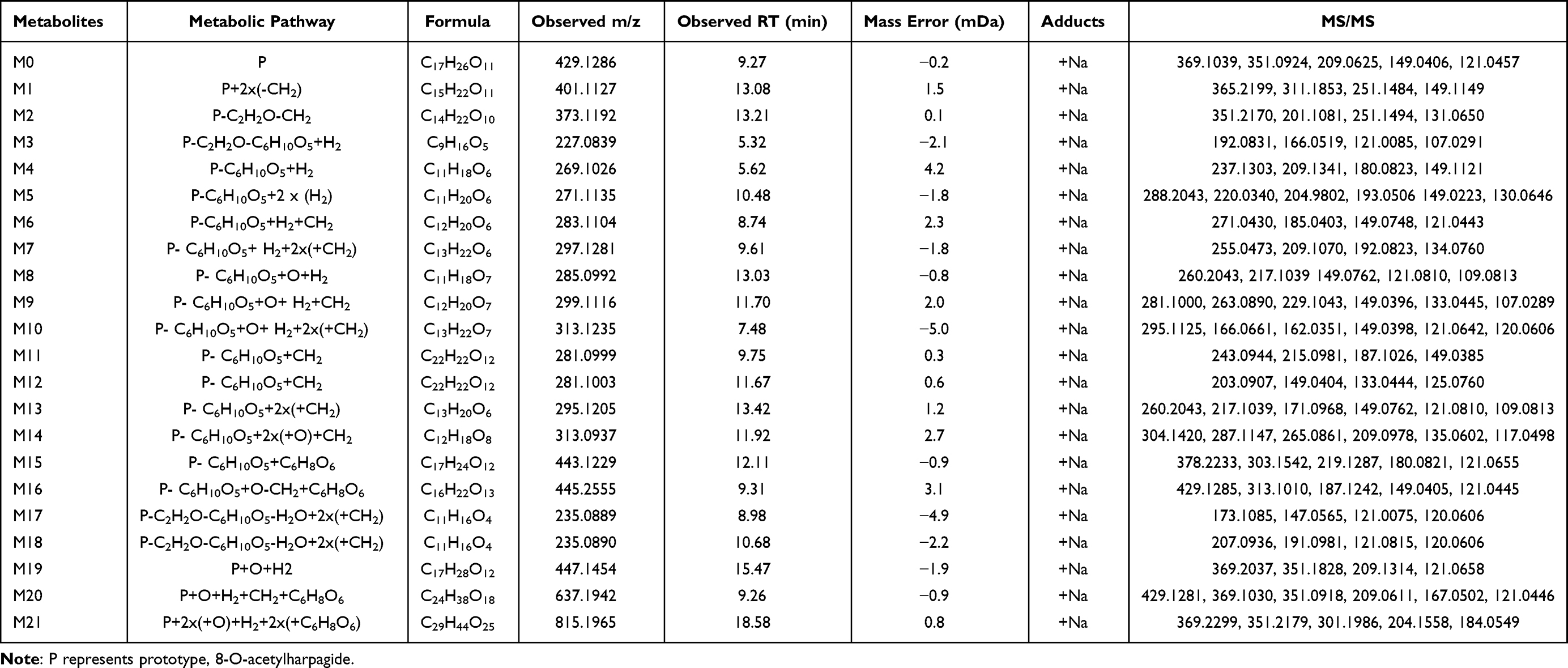 |
Table 1 The Analytical Profile of 8-O-Acetylharpagide Metabolites Identified by UPLC-Q-TOF-MS/MS |
One such metabolite, M1, eluted at 13.08 min. The quasimolecular ion peak [M+Na]+ of M1 was observed at 401.1127 m/z in the positive ion mass spectrum, suggesting a molecular formula of C15H22O11. The MS/MS mass spectrum of M1 showed a product ion at 149.1149 m/z [M+Na-Glu-HCOOH-CH3CHO]+, mirroring the product ion of 8-O-acetylharpagide at 149.1286. This correlation, alongside the molecular formula speculation, preliminarily identified M1 as a demethylation metabolite of 8-O-acetylharpagide.
The retention time of the metabolite M2 was 13.21 min. The cationic first-order mass spectrum showed the quasimolecular ion peak [M+Na]+ at 373.1192 m/z, predicting a molecular formula of C14H22O10. The secondary mass spectrum showed the fragmentation ion at 201.1081 m/z [M+Na-H2O-Glu-CH3CHO]+. The molecular weight was 56 Da and 28 Da lower than that 8-of O-acetylharpagide and M1, respectively. Combined with the predicted molecular formula, this evidence indicated that M2 is a demethylation and deacetylation metabolite of 8-O-acetylharpagide.
The excimer ion peak [M+Na]+ of metabolite M3 was at 227.0839 m/z, eluting at 5.32 min. The molecular formula was predicted to be C9H16O5. The secondary fragment ion was at 121.0085 m/z [M+Na-H2O-H2-CH3CHO-CH2-CO]+. Based on the above data and in conjunction with the literature, it was speculated that M3 is the dihydrogen-harpagenin, the deacetylation, hydrolysis and reduction product of 8-O-acetylharpagide.29
Metabolite M4, eluting at 5.62 min, showed an excimer ion peak [M+Na]+ at 269.1026 m/z in the positive ion mass spectrum. Its predicted molecular formula was C11H18O6. The secondary mass spectrum featured product ions at 149.1121 m/z [M+Na-CH3COOH-H2-CH3CHO-CH2]+ and 209.1341 m/z [M+Na-H2-CH3CHO-CH2]+, aligning with the product ions of 8-O-acetylharpagide at 149 and 209. M4, lighter by C6H8O5 compared with 8-O-acetylharpagide and 42 Da heavier than M3, is presumed to be a hydrolyzation and reduction metabolite.
Metabolite M5, eluting at 10.48 min, showed an excimer ion peak [M+Na]+ at 271.1135 m/z in the positive ion mass spectrum. Its predicted molecular formula was C11H20O6. The secondary mass spectrum featured product ions at 149.0223 m/z [M+Na-CH3COOH-2H2-CH3CHO-CH2]+ aligned with the product ions of 8-O-acetylharpagide at 149. M5 is 2 Da heavier than M4; thus, it is presumed to be a reduction metabolite of M4.
The metabolite M6 excimer ion peak [M+Na]+ was at 283.1104 m/z and eluted at 8.74 min. The molecular formula was predicted to be C12H20O6. The secondary fragment ions were at 121.0443 m/z [M+Na-CH3COOH-H2-CH3CHO-2CH2-CO]+ and 149.0748 m/z [M+Na-CH3COOH-H2-CH3CHO-2CH2]+. The molecular weight of M6 was 14 Da higher than that of M4. Combined with the predicted molecular formula, these findings indicate that M6 is a methylation metabolite of M4. According to the steric hindrance of the methyl substituent, methylation is most likely to occur at the 6-position hydroxyl group.30
The quasimolecular ion peak [M+Na]+ of metabolite M7 was at 297.1281 m/z with a retention time of 9.61 min. Its molecular formula was predicted to be C13H22O6. The secondary fragmentation ion was at 209.1070 m/z [M+Na-H2-CH3CHO-3CH2]+. It was 14 Da and 28 Da heavier than metabolites M6 and M4, respectively. Hence, it was determined that M7 is a methylation product of M6. Based on the steric hindrance of the methyl substituent, it is possible to determine where methylation is most likely to occur.30
The excimer ion peak [M+Na]+ of metabolite M8 was at 285.0992 m/z; it weighed 16 Da more than M4 and eluted at 13.03 min. Its predicted molecular formula was C11H18O7. The major secondary fragment ions were at 149.0762 m/z [M+Na-CH3COOH-H2O-CH3CHO-CH2]+ and 121.0810 m/z [M+Na-CH3COOH-H2O-CH3CHO-CH2-CO]+. Therefore, it is speculated that M8 is the hydroxylation product of M4. However, the concrete hydroxylation site could not be confirmed.31
The metabolite M9 was eluted at 11.70 min, and the molecular ion peak [M+Na]+ was at 299.1116 m/z, indicating that it weighed 16 Da and 14 Da more than M6 and M8, respectively. The predicted molecular formula of M9 was C12H20O7, and the characteristic secondary fragment ion was at 149.0396 m/z [M+Na-CH3COOH-H2O-CH3CHO-2CH2]+. Therefore, it was predicted that M9 is the hydroxylation product of M6 or the methylation product of M8.
Metabolite M10, eluting at 7.48 min, displayed an excimer ion peak [M+Na]+ at 313.1235 m/z. Its predicted molecular formula was C13H22O7, and it was 14 Da, 28 Da, and 16 Da heavier than M9, M8, and M7, respectively. This suggests that M10 is a methylation product of M9. The mass spectrum revealed characteristic fragment ions at 121.0642 m/z [M+Na-CH3COOH-H2O-CH3CHO-3CH2-CO]+ and 149.0398 m/z [M+Na-CH3COOH-H2O-CH3CHO-3CH2]+.
Metabolite M11–M12 is an isomer with quasimolecular ion peaks [M+Na]+ at 281.0999 and 281.1003 m/z with retention times of 9.75 and 11.67 min, respectively. The molecular formula was predicted to be C22H22O12.The characteristic secondary fragment ion was at 149.0385 and 149.0404 m/z [M+Na-CH3COOH-CH3CHO-2CH2]+, respectively. It weighed 2 Da less than the metabolite M6, and it could be preliminarily determined that M11–M12 were the dehydrogenation products of M6.
The retention time of metabolite M13 was 13.42 min. The primary mass spectrum showed the quasimolecular ion peak [M+Na]+ to be 295.1205 m/z. The molecular formula was predicted to be C13H20O6. Fragment ions at 121.0810 m/z [M+Na-CH3COOH-CH3CHO-3CH2-CO]+ and 149.0762 m/z [M+Na-CH3COOH-CH3CHO-3CH2]+ were present. M13 weighed 14 Da more than M11–M12 and was presumed to be a methylation product of M11–M12.
The metabolite M14 eluted at 11.92 min with an excimer ion peak [M+Na]+ at 313.0937 m/z. Its molecular formula was predicted to be C12H18O8. The characteristic fragmentation ion was at 209.0978 m/z [M+Na-2O-CH3CHO-2CH2]+ generated by the secondary mass spectrum. Its molecular weight was 32 Da higher than those of M11–M12 and may contain two additional oxygen. Therefore, it was presumed to be a hydroxylation product of M11–M12.
The retention time of metabolite M15 was 12.11 min. The cation first-order mass spectra showed that the excimer ion peak [M+Na]+ was at 443.1229 m/z. The molecular formula was predicted to be C17H24O12. The fragment ion was at 121.0655 m/z [M+Na-C6H8O6-CH3CHO-CH2-CO]+. Combined with the molecular formula, this evidence suggested that M15 was a hydrolyzation and glucuronidation metabolite.
The metabolite M16 excimer ion peak [M+Na]+ was at 445.2555 m/z and eluted at 9.31 min. The molecular formula was expected to be C16H22O13. Its molecular weight was 2 Da higher than that of M15. Thus, it was presumed that M16 was a demethylation and hydroxylation product of M15. Secondary mass spectrometry generated characteristic fragment ions at 121.0445 m/z [M+Na-C6H8O6-O-CH3CHO-CO]+ and 149.0405 m/z [M+Na-C6H8O6-O-CH3CHO]+.
Metabolites M17–M18 are isomers with quasimolecular ion peaks [M+Na]+ at 235.0889 and 235.0890 m/z with retention times of 8.98 and 10.68 min, respectively. The molecular formula was predicted to be C11H16O4.The characteristic secondary fragment ion was at 121.0075 and 121.0815 m/z [M+Na-CH3CHO-3CH2]+. It weighed 60 Da less than the metabolite M13. In combination with the structural formula, this evidence indicated that M17–M18 were deacetylation and dehydration products of M13.
The metabolite M19 had a quasimolecular ion peak [M+Na]+ at 447.1454 m/z and eluted at 15.47 min. The molecular formula was predicted to be C17H28O12. Its molecular weight was 18 Da higher than that of 8-O-acetylharpagide; hence, it was presumed that M19 was a product of reduction and hydroxylation of 8-O-acetylharpagide. Secondary mass spectra generated characteristic fragment ions at 369.2037 m/z [M+Na-CH3COOH-H2O]+, 351.1828 m/z [M+Na-CH3COOH-2H2O]+, 209.1314 m/z [M+Na-Glc-H2O-CH3CHO-CH2]+, and 121.0658 m/z [M+Na-Glc-CH3COOH-H2O-CH3CHO-CH2-CO]+.
The metabolite M20 had a quasimolecular ion peak [M+Na]+ at 637.1942 m/z and eluted at 9.26 min. The molecular formula was predicted to be C24H38O18. Its molecular weight was 208 Da higher than that of 8-O-acetylharpagide. Thus, it was presumed that M20 was a product of reduction, methylation, hydroxylation and glucuronidation of 8-O-acetylharpagide. Secondary mass spectra generated characteristic fragment ions at 369.1030 m/z [M+Na–CH3COOH–H2O–C6H8O6–CH2]+, 351.0918 m/z [M+Na–CH3COOH–2H2O–C6H8O6–CH2]+, 209.0611 m/z [M+Na–Glc–H2O–C6H8O6–CH3CHO–2CH2]+, 149.0396 [M+Na–Glc–CH3COOH–H2O–C6H8O6–CH3CHO–2CH2]+, and 121.0446 m/z [M+Na–Glc–CH3COOH–H2O–C6H8O6–CH3CHO–2CH2–CO]+. The hydroxyl group at positions 5 and 6 of aglycones can be glucosylated, and the steric hindrance of the hydroxyl group at position 6 is the least; hence, hydroxyl substitution may occur at position 6.30
The metabolite M21 excimer ion peak [M+Na]+ was at 815.1965 m/z and eluted at 18.58 min. The molecular formula was expected to be C29H44O25. Its molecular weight was 368 Da higher than that of M19. Therefore, it was presumed that M21 was a glucuronidation and hydroxylation product of M19. Secondary mass spectrometry generated characteristic fragment ions at 369.2299 m/z [M+Na–CH3COOH–2O–H2–2C6H8O6]+ and 351.2179 m/z [M+Na–CH3COOH–H20-2O-H2-2C6H8O6]+.
Optimization of Mass Spectrometry and Chromatography and Method Validation
The conditions for chromatography and mass spectrometry were optimized for the sensitive and efficient analysis of 8-O-acetylharpagide and lidocaine. Gradient elution and 0.1% formic acid improved sensitivity, and the absence of interference from endogenous substances in urine or fecal samples was confirmed after sample treatment (Supplementary Figure S4).
The typical calibration curve equations and linear ranges for 8-O-acetylharpagide in bile, urine, and feces are shown in Supplementary Table S2. The results showed a good correlation between the peak area ratios and the concentrations of 8-O-acetylharpagide over the linear range.
Excretion Study
The cumulative excretion results of the analytes in rat bile, urine, and feces after oral administration of 8-O-acetylharpagide are displayed in Figure 2. The total recovery of 8-O-acetylharpagide in bile was 0.84% over 4 h compared with the administered dose. The lower recovery of 8-O-acetylharpagide in bile indicated that few untransformed forms were excreted in bile; this observation might be due to the first-pass effect. The cumulative urinary and fecal excretion of 8-O-acetylharpagide stabilized after 24 h. The excretion data for 8-O-acetylharpagide in urine and feces indicate that <1.5% of the administered dose was excreted in unconverted form. In addition, the fecal excretion was less compared with the urinary excretion.
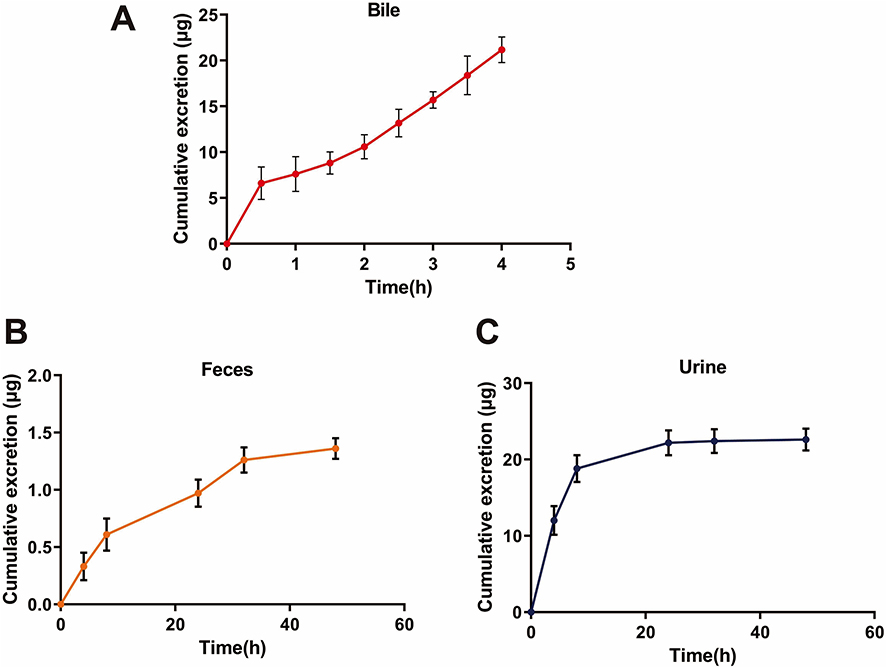 |
Figure 2 Cumulative excretion of 8-O-acetylharpagide in the bile of rats after oral administration of a single dose of 12 mL/kg 8-O-acetylharpagide (A). Fecal (B) and urinary (C) cumulative excretion of 8-O-acetylharpagide in rats following oral administration at a single dose of 10.5 mL/kg 8-O-acetylharpagide (n = 5). |
Network Pharmacology Analysis
Prediction of Compound- and Breast Cancer-Related Targets
Five active metabolites (ie, M1, M2, M3, M5, and M15) of 8-O-acetylharpagide were identified by SwissTargetPrediction and SEA. Moreover, a total of 280 relevant targets for 8-O-acetylharpagide and its five active metabolites were evaluated. Furthermore, 5020 genes related to breast cancer were screened from OMIM, GeneCards, and TTD databases. The 312 identified targets included fibroblast growth factor 1 (FGF1), FGF2, vascular endothelial growth factor A (VEGFA), MMP family, and heat shock protein 90 alpha family class A member 1 (HSP90AA1),10,32–35 which are closely related to breast cancer. This result suggests that 8-O-acetylharpagide and its metabolites exert anti-breast cancer effects through various mechanisms. Using VENNY 2.1 software to create the drug-disease overlapping target Venn diagram, a total of 188 overlapping targets were identified (Figure 3E). To preliminarily analyze the medicinal value of these six compounds in the treatment of breast cancer, a CTD network was constructed using Cytoscape v3.7.2 software (Figure 3A). The network showed 219 nodes, including six compounds, 213 target genes, and 465 compound-target gene interaction edges (Supplementary Table S3). All compounds had an interaction degree >15; this finding implied that these compounds might regulate multiple targets to exert different therapeutic effects.
 |
Figure 3 (A) The CTD network was built by the compounds (green) and potential targets (blue); (B) The PPI network of 184 potential targets; (C) CTP network was built by the compounds (yellow), potential targets (blue), and pathways (green); (D) Enriched KEGG pathways analysis of breast cancer potential targets; (E) Venn diagram of the shared genes of 8-O-acetylharpagide and its active metabolites targets with breast cancer. |
PPI Analysis
To elucidate the relationship between genes and identify more valuable targets for further analysis, a PPI network consisting of 184 nodes and 3614 edges was established (Figure 3B; Supplementary Table S4). The 178 genes with a degree ≥5 were selected for KEGG analysis.
KEGG Enrichment Analysis and CTP Network Establishment
KEGG enrichment analysis was performed using DAVID on 178 targets selected by PPI. The top 20 pathways were selected for further analysis as shown in Supplementary Table S5. In order to visualize the relationship between the six compounds, 20 pathways, and 123 targets from the 20 pathways, a CTP network diagram was constructed using Cytoscape 3.9.1 (Figure 3C; Supplementary Table S6). As shown in Figure 3D, those pathways were mainly related to metabolism (n=2), disease (n=10, of which seven were directly related to cancer), environmental information processing (n=5), cellular processes (n=1), and organismal systems (n=2).
M5 as a Key Active Metabolite
Molecular docking between the top six breast cancer-related targets (glyceraldehyde-3-phosphate dehydrogenase [GAPDH, PDB ID: IU8F]; AKT serine/threonine kinase 1 [AKT1, PDB ID: 3O96]; epidermal growth factor receptor [EGFR, PDB ID: 1M17]; signal transducer and activator of transcription 3 [STAT3, PDB ID: 5AX3]; caspase 3 [CASP3, PDB ID: 6X8I]; and MMP9 [PDB ID: 1GKC]) selected based on the PPI network and 8-O-acetylharpagide and its active metabolites were performed to assess protein-ligand binding potential. The docking combinations between 8-O-acetylharpagide and its metabolites and targets are shown in Table 2. Binding energy <0 Kcal/mol denoted docking between the component and the protein, while binding energy <−1.2 Kcal/mol indicated satisfactory docking.36
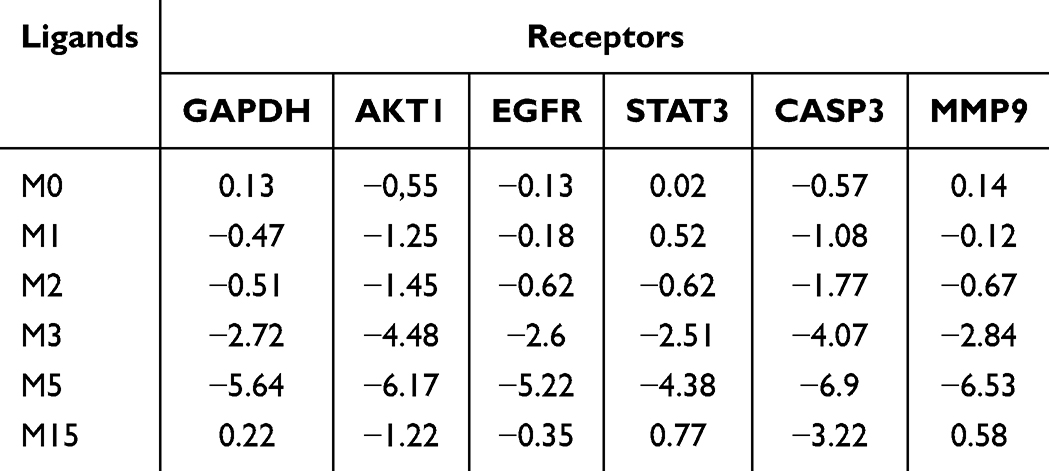 |
Table 2 Docking Binding Energy of Potential Active Metabolites with Key Targets (Kcal/Mol) |
Based on the docking results, 8-O-acetylharpagide did not exhibit significant binding affinity with GAPDH, STAT3, or MMP9 and showed low affinity for AKT1 and CASP3. Among its metabolites, M3 and M5 demonstrated the strongest binding with multiple targets. Specifically, M5 showed favorable docking with GAPDH, AKT1, EGFR, STAT3, CASP3, and MMP9, with binding energies of −5.64 kcal/mol, −6.17 kcal/mol, −5.22 kcal/mol, −4.38 kcal/mol, −6.9 kcal/mol, and −6.53 kcal/mol, respectively. Similarly, M3 exhibited strong interactions with GAPDH, AKT1, EGFR, STAT3, CASP3, and MMP9, with binding energies <−1.2 kcal/mol for all. The 3D docking models of compounds with binding energies <−1.2 kcal/mol are shown in Figure 4.
 |
Figure 4 Molecular docking model 3D diagram. (A) M1 binds to AKT1. (B) M2 binds to AKT1 (a) and CASP3 (b). (C) M3 binds to AKT1 (c), CASP3 (d), GAPDH (e), MMP9 (f), STAT3 (g) and EGFR (h). (D) M5 binds to AKT1 (i), CASP3 (j), GAPDH (k), MMP9 (l), STAT3 (m) and EGFR (n). (E) M15 binds to AKT1 (o) and CASP3 (p). |
Effect of 8-O-Acetylharpagide on the AKT/NF-κB/MMP9 (AKT/Nuclear Factor-κB/MMP9) Signaling Axis in Mouse Breast Tumors
Based on the in silico findings, the effects of 8-O-acetylharpagide treatment on the AKT/NF-κB/MMP9 signaling axis were evaluated using Western blotting analysis (Figure 5A). As shown in Figure 5B–F, the protein levels of MMP9 and AKT were significantly decreased in the tumors of the high-dose 8-O-acetylharpagide group compared with the model group. Notably, the levels of phosphorylated AKT (p-AKT) and NF-κB (p65) were also markedly reduced, indicating a suppression of the AKT/NF-κB pathway. In addition, the p-AKT/AKT ratio was significantly lower in the 8-O-acetylharpagide-treated group, further supporting the inhibitory effect of 8-O-acetylharpagide on the AKT signaling. These results suggest that 8-O-acetylharpagide may exert its anti-breast cancer effects through modulation of the AKT/NF-κB/MMP9 signaling axis, providing valuable insights into its mechanism of action.
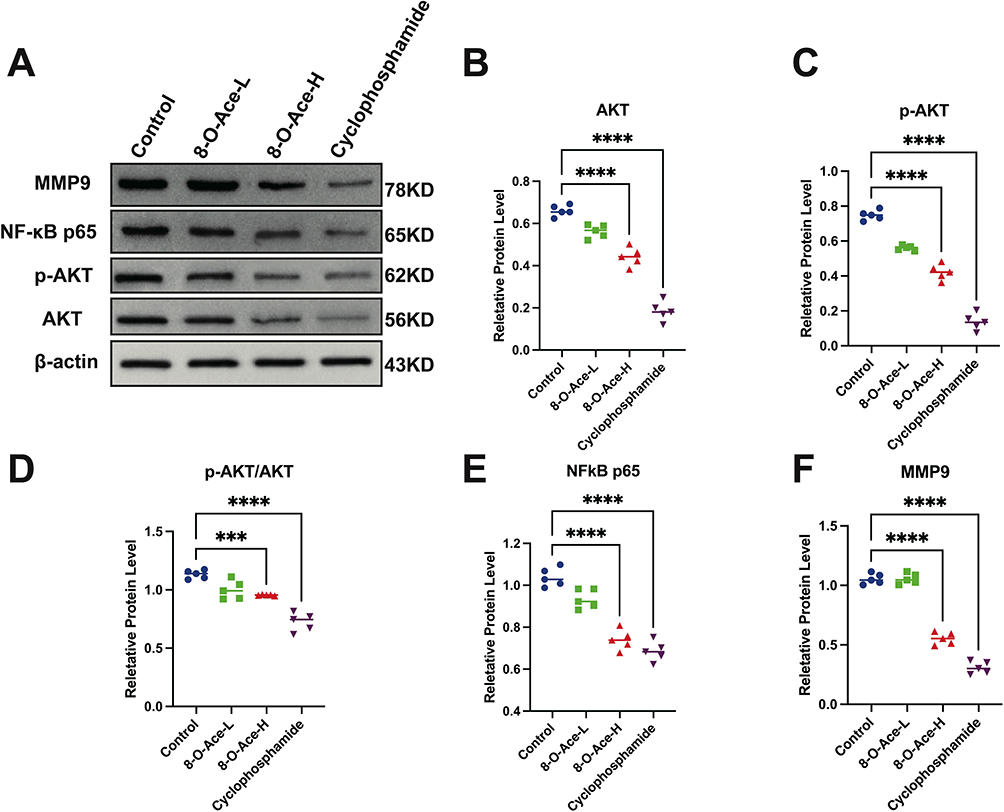 |
Figure 5 Changes in AKT, p-AKT, NF-κB (p65) and MMP9 levels in breast cancer tissues of different groups of mice (n = 5). (A) Representative Western blot images of the AKT, p-AKT, NF-κB (p65) and MMP9 levels; (B–F) Quantification of the AKT, p-AKT, p-AKT/AKT, NF-κB (p65) and MMP9 levels. **** P < 0.0001; *** P < 0.001. |
Discussion
Ajuga decumbens, the primary constituent of the Xiaoai Jiedu Formula, plays a crucial role in its therapeutic efficacy. When combined with fluorouracil, epirubicin, and cyclophosphamide chemotherapy, this formula significantly enhances clinical outcomes in patients with breast cancer by improving treatment efficacy, boosting immune function, and alleviating clinical symptoms.37,38 Additionally, it mitigates chemotherapy-induced adverse effects, such as gastrointestinal discomfort and bone marrow suppression,39 making it a valuable adjunctive therapy in comprehensive cancer treatment. Notably, 8-O-acetylharpagide is the principal bioactive component of Ajuga decumbens6–8 and exhibits promising anticancer potential.22,23,40 Given that the pharmacological activity of traditional Chinese medicine is closely linked to its chemical constituents, and that metabolic transformation can generate active metabolites with enhanced pharmacological effects,41 understanding the metabolic fate of 8-O-acetylharpagide is critical. Investigating the in vivo processes and identifying the active metabolites of 8-O-acetylharpagide will provide essential insights into its mechanism of action, potentially uncovering novel therapeutic agents and optimizing its clinical application.
As the primary metabolic organ, the liver is rich in enzymes, particularly cytochrome P450 enzymes, which play a crucial role in drug biotransformation.42,43 Additionally, the intestine serves as an important site for drug metabolism, with the gut microbiota significantly influencing drug absorption, metabolism, and toxicity.44,45 Oral administration of 8-O-acetylharpagide in a breast cancer mouse model resulted in tumor growth inhibition.23 However, in a pharmacokinetic study, Shen et al demonstrated that its absolute bioavailability was only 7.7%.24 This finding suggested that its anti-breast cancer effects may not be mediated by the parent compound. Therefore, the objective of this study was to characterize the metabolites of 8-O-acetylharpagide in rat plasma, bile, feces, and in vitro intestinal flora. The results showed that 8-O-acetylharpagide may be hydrolyzed and reduced by intestinal microbiota and hepatic enzymes and metabolized to metabolites M3 and M5. Previous studies have shown that iridoid glycosides can be hydrolyzed by gut microbial β-glucosidases into their active aglycones, which are subsequently absorbed to exert pharmacological effects.46,47 For example, Zhi-zi-chi Decoction components geniposide and genipin-1-gentiobioside could be metabolized to genipin, a known antidepressant metabolite.48 Additionally, the reduction reactions mediated by the gut microbiota play an essential role, potentially influencing drug absorption and therapeutic efficacy. Koppel et al49 reported that gut bacterial reductases could convert intestinally poorly absorbed berberine into the absorbable form dihydroberberine. Next, dihydroberberine is absorbed by the gut, oxidized back to berberine in intestinal tissues, and subsequently enters the bloodstream to exert its pharmacological effects. These findings align with our results, highlighting the importance of metabolic transformation in drug efficacy. This study emphasizes the significant role of metabolism, particularly the involvement of the hepatic and intestinal microbiota in the bioactivation of 8-O-acetylharpagide. The evidence suggests that the therapeutic effects of 8-O-acetylharpagide may be mediated by its metabolites rather than the parent compound itself. Therefore, it is critical to understand these metabolic pathways to optimize the efficacy of 8-O-acetylharpagide and other similar compounds, especially in drug development and personalized medicine.
The excretion profile of 8-O-acetylharpagide further supports the notion that extensive metabolism occurs before elimination. In this study, only 0.84% of the administered 8-O-acetylharpagide was recovered in bile over a 4-h period, and <1.5% of the dose was excreted in urine and feces in its unchanged form within 24 h. These findings indicate that 8-O-acetylharpagide undergoes rapid and extensive metabolic transformation in vivo, with minimal direct excretion. This phenomenon is consistent with observations from previous studies on structurally similar iridoid glycosides, which have also shown low recovery of the parent compound in excreta due to significant hepatic and microbial metabolism before elimination.50 The presence of multiple metabolites in feces and bile suggests that enterohepatic circulation and gut microbial metabolism play a key role in the metabolic fate of 8-O-acetylharpagide (Supplementary Figure S5), influencing its pharmacokinetic behavior and overall bioavailability.51 These results emphasize the necessity to further investigate the metabolic pathways and active metabolites of 8-O-acetylharpagide to better understand its pharmacological effects.
Network pharmacology, a powerful tool for explaining complex interactions from a systemic perspective, has been widely used to predict the main active components and possible targets in Chinese medicine.52 In this study, our network pharmacology analysis revealed 20 relevant pathways. Among the acquired signaling pathways, hypoxia inducible factor-1 (HIF-1) was a major regulator of breast cancer progression,53 and modulation of HIF-1 inhibited breast cancer growth.54 The PI3K/AKT pathway plays an important role in the development and progression of breast cancer, and activation of the PI3K/AKT signaling pathway reduces apoptosis, stimulates cell growth, and increases cell proliferation.55 Similarly, iridoids in Ajuga decumbens could inhibit the breast cancer stem cell invasion by suppressing the PI3K/AKT and ERK1/2 MAPK signaling pathways.6,8 These findings support the results of our KEGG pathway enrichment analysis. The data also suggested that 8-O-acetylharpagide and its metabolites might be the active components of iridoids in Ajuga decumbens exerting anti-breast cancer effects.
Molecular docking simulations were performed to explore the binding interactions between 8-O-acetylharpagide and its five active metabolites with six key targets. The docking results indicated that none of the binding energies for 8-O-acetylharpagide to the key targets were <−1.2 kcal/mol. This finding suggested that 8-O-acetylharpagide itself may not be the primary compound responsible for its anti-breast cancer effects. In contrast, metabolites M3 (a hydrolysis, reduction, and deacetylation product) and M5 (a hydrolysis and reduction product) exhibited stronger binding affinities to the six breast cancer-related targets. Notably, M3 and M5 showed significant binding to MMP9, aligning with previous evidence demonstrating that Ajuga decumbens extract inhibits breast cancer metastasis by downregulating MMP9 expression.7 High expression of MMP9 in tumor or stromal cells is correlated with poor prognosis in breast cancer.56–58 Studies have shown that MMP9 overexpression in tumors is associated with lymph node metastasis and predicts shorter overall survival in patients.59 Additionally, MMP9 promotes tumor growth and angiogenesis through interactions with vascular endothelial growth factor/vascular endothelial growth factor receptor 2 (VEGF/VEGFR2) receptors,60 enhancing the invasiveness of breast cancer cells and facilitating lung metastasis.61,62 These findings suggest that M3 and M5 inhibit breast cancer progression by suppressing MMP9 expression.
SwissADME63 analysis revealed that among 8-O-acetylharpagide and its metabolites, only M3 and M5 demonstrated high gastrointestinal absorption and bioavailability, with a bioavailability score of 0.55, indicating promising drug-like properties (Supplementary Table S7). The iLOGP value of M5 (1.55) was higher than that of M3 (1.04), suggesting that M5 possesses better lipid solubility,63 which may enhance its absorption. Moreover, the retention time of M5 (10.48 min) was longer than that of M3 (5.32 min), further supporting the idea that M5 is less polar. This characteristic may enhance its ability to permeate the cell membrane and exert more potent effects on breast cancer cells. The fluid-mosaic model of cell membranes explains that nonpolar compounds preferentially diffuse through the phospholipid bilayer, leading to enhanced bioavailability.64 Therefore, M5 appears to be the main active metabolite of 8-O-acetylharpagide, and its pharmacological effects warrant further investigation.
Building upon the molecular docking and SwissADME findings, Western blotting analysis was conducted to further elucidate the potential therapeutic mechanisms of 8-O-acetylharpagide in breast cancer. The results showed that after treatment with 8-O-acetylharpagide, key proteins involved in tumor progression, including AKT and MMP9, were significantly downregulated. Notably, while the parent compound exhibited minimal direct target binding (<−1.2 kcal/mol), its in vivo efficacy likely stems from metabolic activation to M5, a hypothesis supported by SwissADME predictions of excellent druggability for M5. The reduction in p-AKT and NF-κB (p65) levels, along with a decrease in the p-AKT/AKT ratio, suggest that 8-O-acetylharpagide inhibits the activation of the AKT/NF-κB signaling axis, which is critical for cell proliferation, survival, and metastasis in breast cancer. Activation of AKT upregulates NF-κB (p65), which in turn induces the expression of MMP9, a key protease involved in extracellular matrix degradation and cancer cell invasion.65,66 The suppression of both AKT and NF-κB signaling, along with the downregulation of MMP9 expression, likely inhibit breast cancer cell invasion and metastasis. The inhibition of MMP9, a critical factor in tumor metastasis, by 8-O-acetylharpagide and its metabolites may therefore contribute to its anti-breast cancer effects. These findings are consistent with those of previous studies, which have shown that inhibition of the AKT/NF-κB/MMP9 signaling axis suppresses tumor growth and metastasis in breast cancer.67,68 Taken together, these results provide strong evidence that 8-O-acetylharpagide, potentially through its metabolites, exerts anticancer effects by modulating the AKT/NF-κB/MMP9 signaling axis. These findings highlight the potential of 8-O-acetylharpagide and its metabolites as promising therapeutic agents for targeting the AKT/NF-κB/MMP9 signaling axis in breast cancer treatment. This evidence offers a novel strategy for inhibiting tumor progression and metastasis.
In this study, we identified the metabolic products of 8-O-acetylharpagide and, through network pharmacology, molecular docking, and experimental validation, revealed potential anticancer metabolites involved in its anti-breast cancer effects. However, this study had several limitations. Firstly, we did not directly validate the metabolite M5 in experimental models; hence, its specific therapeutic effects remain to be confirmed. Secondly, the relationship between 8-O-acetylharpagide and its metabolites, particularly M5, in terms of metabolic conversion requires further exploration. Future research should focus on the potential development of M5 or similar metabolites as therapeutic agents, including formulating new treatments, devising targeted strategies, or investigating their synergistic effects with existing therapies. Additionally, a deeper understanding of the active ingredients in other herbs from the Xiaoai Jiedu Formula, as well as isolating monomeric compounds to create formulations that reflect the multi-component, multi-pathway nature of traditional Chinese medicine, may enhance treatment efficacy. Lastly, the lack of direct clinical validation in humans limits the generalizability of these findings. Future studies should aim to evaluate the safety and clinical relevance of the identified metabolites, ensuring their potential for widespread therapeutic use.
Conclusions
In summary, this research identified M5 as a key metabolite of 8-O-acetylharpagide. The results showed that oral administration of 8-O-acetylharpagide exerts anti-breast cancer effects by inhibiting the AKT/NF-κB/MMP9 signaling axis. Furthermore, this effect may be achieved after 8-O-acetylharpagide is metabolized to M5. This provides a foundation for developing M5 as a novel therapeutic agent and integrating traditional Chinese medicine into modern cancer treatment. However, further validation in human models is needed to confirm its therapeutic potential, and strategies to improve the bioavailability of 8-O-acetylharpagide should be explored. Future studies should focus on optimizing drug delivery systems and evaluating the efficacy of M5 in advanced breast cancer models to facilitate its clinical translation.
Abbreviations
MMP9, matrix metalloproteinase 9; PI3K/AKT, phosphatidylinositol 3 kinase/protein kinase B; IS, internal standard; HPLC, high performance liquid chromatograph; ESI, electrospray ionization source; MRM, multiple reaction monitoring; GAM, Gifu Anaerobic Medium; TTD, Therapeutic Target Database; OMIM, Online Mendelian Inheritance in the Man®; PPI, Protein–Protein Interaction; CTP, compound-target-pathway.
Ethics Statement
The animal study protocol was approved by the Laboratory Animal Ethics Committee of Dongfang Hospital, Beijing University of Chinese Medicine (protocol code DFYY2023B32R and DFYY2023B33M).
Data Sharing Statement
The data that support the findings of this study are available on request from the corresponding authors.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Beijing Science and Technology Development Fund Project of Traditional Chinese Medicine (No. JJ-2020-39) and National High Level Chinese Medicine Hospital Clinical Research Funding (No. DFGZRB-2024GJRC018).
Disclosure
The authors report there are no competing interests to declare.
References
1. Wu D. Analysis of the medicinal value of Ajuga decumbens. Fujian J Animal Husbandry Veterinary Med. 1997;1997(01):32–33+26.
2. Tang J. The Integrated Active Filter of Ajuga Decumbens ─anti-Inflammatory and Regulating Blood Lipid. North Sichuan Medical College; 2017.
3. Peng W. Progress in the study of chemical composition and pharmacological effects of Ajuga decumbens Thunb. J Pract Tradit Chin Med. 2012;28(04):322–323.
4. Ma J. National Compendium of Chinese Herbal Medicine, third edition. J Tradit Chin Med Management. 2014;(04):594.
5. RY. Traditional Chinese Medicine (TCM) (Oncology). Beijing: Science Press; 1983.
6. Peng B, Han J, He R, Xu Q, Li J. Intervention Effect of Ajuga decumbens Iridoids on Characteristics of Breast Cancer Stem Cells via ERK MAPK and PI3K/Akt Signaling Pathways. Chin J Exp Traditional Med Formulae. 2017;23(18):94–99.
7. Peng B, He R, Xu Q, Gao J, Lu Y, Li J. Correlation between antimetastatic action of Ajuga decumbens and expression of MMPs and TIMPs. Zhongguo Zhong Yao Za Zhi. 2011;36(24):3511–3514.
8. Peng B, Yang Y, Wang J, et al. Inhibitory Effect of Ajuga decumbens Iridoids on the Metastasis of Triple Negative Breast Cancer and Its Related Mechanism. Chin Pharm J. 2017;52(21):1903–1908.
9. Miricescu D, Totan A, Stanescu II S, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: from Molecular Landscape to Clinical Aspects. Int J mol Sci. 2020;22(1):173. doi:10.3390/ijms22010173
10. Dong H, Diao H, Zhao Y, et al. Overexpression of matrix metalloproteinase-9 in breast cancer cell lines remarkably increases the cell malignancy largely via activation of transforming growth factor beta/SMAD signalling. Cell Prolif. 2019;52(5):e12633. doi:10.1111/cpr.12633
11. Raeeszadeh-Sarmazdeh M, Do LD, Hritz BG. Metalloproteinases and Their Inhibitors: potential for the Development of New Therapeutics. Cells. 2020;9(5):145. doi:10.3390/cells9051313
12. Wen HR, Li P, Li P, et al. Pharmacokinetics of 8-O-acetylharpagide and harpagide after oral administration of Ajuga decumbens Thunb extract in rats. J Ethnopharmacol. 2013;147(2):503–508. doi:10.1016/j.jep.2013.03.048
13. Breschi MC, Martinotti E, Catalano S, Flamini G, Morelli I, Pagni AM. Vasoconstrictor activity of 8-O-acetylharpagide from Ajuga reptans. J Nat Prod. 1992;55(8):1145–1148. doi:10.1021/np50086a019
14. Ganaie HA, Ali MN, Ganai BA, Meraj M, Ahmad M. Antibacterial activity of 14, 15-dihydroajugapitin and 8-o-acetylharpagide isolated from Ajuga bracteosa Wall ex. Benth against human pathogenic bacteria. Microb Pathog. 2017;103:114–118. doi:10.1016/j.micpath.2016.12.017
15. Tundis R, Loizzo MR, Menichini F, Statti GA, Menichini F. Biological and pharmacological activities of iridoids: recent developments. Mini Rev Med Chem Apr. 2008;8(4):399–420. doi:10.2174/138955708783955926
16. Usmanov D, Azamatov A, Baykuziyev T, Yusupova U, Rasulev B. Chemical constituents, anti-inflammatory and analgesic activities of iridoids preparation from Phlomoides labiosa bunge. Nat Prod Res. 2023;37(10):1709–1713. doi:10.1080/14786419.2022.2104274
17. Atay I, Kirmizibekmez H, Kaiser M, Akaydin G, Yesilada E, Tasdemir D. Evaluation of in vitro antiprotozoal activity of Ajuga laxmannii and its secondary metabolites. Pharm Biol. 2016;54(9):1808–1814. doi:10.3109/13880209.2015.1129542
18. Abdel-Kader MS, Alqasoumi SI. In Vivo Hepatoprotective and Nephroprotective Activity of Acylated Iridoid Glycosides from Scrophularia hepericifolia. Biology. 2021;10(2). doi:10.3390/biology10020145
19. You Y, Wang J, Tong Y, et al. Anti-inflammatory effect of acetylharpagide demonstrated by its influence on leukocyte adhesion and transmigration in endothelial cells under controlled shear stress. Clin Hemorheol Microcirc. 2014;56(3):205–217. doi:10.3233/ch-131704
20. Hsu WH, Lin BZ, Leu JD, et al. Involvement of 8-O-acetylharpagide for Ajuga taiwanensis mediated suppression of senescent phenotypes in human dermal fibroblasts. Sci Rep. 2020;10(1):19731. doi:10.1038/s41598-020-76797-6
21. Takasaki M, Tokuda H, Nishino H, Konoshima T. Cancer chemopreventive agents (antitumor-promoters) from Ajuga decumbens. J Nat Prod. 1999;62(7):972–975. doi:10.1021/np990033w
22. Konoshima T, Takasaki M, Tokuda H, Nishino H. Cancer chemopreventive activity of an iridoid glycoside, 8-acetylharpagide, from Ajuga decumbens. Cancer Lett. 2000;157(1):87–92. doi:10.1016/s0304-3835(00)00479-1
23. Qian J, Zhao X, Yuan S, et al. Metabolome-microbiome insights into therapeutic impact of 8-O-acetylharpagide against breast cancer in a murine model. Biomed Chromatogr. 2024:e5880. doi:10.1002/bmc.5880.
24. Shen X, Chen C, Wen C, et al. A newly developed UPLC-MS/MS method for simultaneous quantitative analysis of ajuforrestin A, ajuforrestin B, ajugamacrin and 8-O-acetylharpagide derived from Ajuga plants in mice blood and the in vivo pharmacokinetics. Drug Dev Ind Pharm. 2024;2024:1–9. doi:10.1080/03639045.2024.2328731
25. Dong Y, Yang N, Li X, et al. Research progress on mechanism of effective components of poorly absorbable oral Chinese materia medica. Chin Tradit Herbal Drugs. 2020;51(03):769–779.
26. Liu H, Sun H, Lu D, et al. Identification of glucuronidation and biliary excretion as the main mechanisms for gossypol clearance: in vivo and in vitro evidence. Xenobiotica. 2014;44(8):696–707. doi:10.3109/00498254.2014.891780
27. Zhang Z, Liu J, Liu Y, Shi D, He Y, Zhao P. Virtual screening of the multi-gene regulatory molecular mechanism of Si-Wu-tang against non-triple-negative breast cancer based on network pharmacology combined with experimental validation. J Ethnopharmacol. 2021;269:113696. doi:10.1016/j.jep.2020.113696
28. Huang X, Jia M, Liu Y, et al. Identification of bicyclol metabolites in rat plasma, urine and feces by UPLC-Q-TOF-MS/MS and evaluation of the efficacy and safety of these metabolites based on network pharmacology and molecular docking combined with toxicity prediction. J Pharm Biomed Anal. 2022;220:114947. doi:10.1016/j.jpba.2022.114947
29. Yosioka I, Sugawara T, Yoshikawa K, Kitagawa I. Soil Bacterial Hydrolysis leading to Genuine Aglycone. VII. On Monoterpenoid Glucosides of Scrophularia buergeriana MIQ. and Paeonia albiflora PALLAS. Chem Pharm Bull. 1972;20(11):2418–2421. doi:10.1248/cpb.20.2418
30. Liu ZX, Wang F, Z. J, Liu GX, Shang MY, Cai SQ. Identification and Analysis of Harpagide Metabolites in Rats in vivo. China Pharm. 2017;28(10):1310–1315.
31. Zhao M, Tao J, Du L, Jiang S, Qian D, Duan J. UPLC-Q-TOF/MS-Based Metabolic Profiling Comparison of Two Major Bioactive Components and Their Metabolites in Normal and CKD Rat Plasma, Urine and Feces Following Oral Administration of Fructus Corni Extract. J Chromatogr Sci. 2017;55(8):857–865. doi:10.1093/chromsci/bmx046
32. Castillo-Castrejon M, Sankofi BM, Murguia SJ, et al. FGF1 supports glycolytic metabolism through the estrogen receptor in endocrine-resistant and obesity-associated breast cancer. Breast Cancer Res. 2023;25(1):99. doi:10.1186/s13058-023-01699-0
33. Hosaka K, Yang Y, Seki T, et al. Therapeutic paradigm of dual targeting VEGF and PDGF for effectively treating FGF-2 off-target tumors. Nat Commun. 2020;11(1):3704. doi:10.1038/s41467-020-17525-6
34. Palazon A, Tyrakis PA, Macias D, et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell. 2017;32(5):669–683.e5. doi:10.1016/j.ccell.2017.10.003
35. Zhang Y, Zhao G, Yu L, et al. Heat-shock protein 90α protects NME1 against degradation and suppresses metastasis of breast cancer. Br J Cancer. 2023;129(10):1679–1691. doi:10.1038/s41416-023-02435-3
36. Li H, Xiao J, Li X, Huang Q, Liu Q, Zhang Q. Mechanism of morusin on breast cancer via network pharmacology and in vitro experiments. Medicine. 2023;102(28):e34300. doi:10.1097/md.0000000000034300
37. Pan J, Cheng H. Clinical Efficacy of Xiaoai Jiedu Formula Combined with FEC Chemotherapy on Breast Cancer Patients. Chin J Exp Traditional Med Formulae. 2019;25(06):95–100.
38. Pan J, Li J. Improvement of prognosis of breast cancer by Xiaoai Jiedu formula combined with FEC chemotherapy. Doctor. 2023;8(19):69–72.
39. Pan J, Cheng H. Clinical observation of XiaoaiJiedu Formula improving side effects of chemotherapy for breast cancer. China Modern Doctor. 2018;56(34):110–113.
40. Zhou X, Yin Q, Huang R, Jian B, Cai X. 8-O-Acetylharpagide induces apoptosis of colon cancer HCT116 cells through Wnt signaling pathway. Nat Prod Res Dev. 2022;34(06):996–1004+1020. doi:10.16333/j.1001-6880.2022.6.012
41. Liu Y, Yang L. Early metabolism evaluation making traditional Chinese medicine effective and safe therapeutics. J Zhejiang Univ Sci B. 2006;7(2):99–106. doi:10.1631/jzus.2006.B0099
42. Almazroo OA, Miah MK, Venkataramanan R. Drug Metabolism in the Liver. Clin Liver Dis. 2017;21(1):1–20. doi:10.1016/j.cld.2016.08.001
43. Zhao M, Ma J, Li M, et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int J mol Sci. 2021;22(23):12808. doi:10.3390/ijms222312808
44. Noh K, Kang YR, Nepal MR, et al. Impact of gut microbiota on drug metabolism: an update for safe and effective use of drugs. Arch Pharm Res. 2017;40(12):1345–1355. doi:10.1007/s12272-017-0986-y
45. Li H, He J, Jia W. The influence of gut microbiota on drug metabolism and toxicity. Expert Opin Drug Metab Toxicol. 2016;12(1):31–40. doi:10.1517/17425255.2016.1121234
46. Yao Y, Ma X, Li T, et al. Quantification of isoflavone glycosides and aglycones in rat plasma by LC-MS/MS: troubleshooting of interference from food and its application to pharmacokinetic study of Semen Sojae Praeparatum extract. J Pharm Biomed Anal. 2018;161:444–454. doi:10.1016/j.jpba.2018.09.011
47. Tan XB, Wei YJ, Jia XB, Liu WB. Disposition of geniposide and genipin via intestinal absorption barrier. J Chin Pharmal Sci. 2013;48(15):1289–1293.
48. Gao FY, Chen XF, Cui LX, et al. Gut microbiota mediates the pharmacokinetics of Zhi-zi-chi decoction for the personalized treatment of depression. J Ethnopharmacol. 2023;302(Pt B):115934. doi:10.1016/j.jep.2022.115934
49. Feng R, Shou JW, Zhao ZX, et al. Transforming berberine into its intestine-absorbable form by the gut microbiota. Sci Rep. 2015;5(1):12155. doi:10.1038/srep12155
50. Jeong SH, Jang JH, Cho HY, Lee YB. Simultaneous determination of three iridoid glycosides of Rehmannia glutinosa in rat biological samples using a validated hydrophilic interaction-UHPLC-MS/MS method in pharmacokinetic and in vitro studies. J Sep Sci. 2020;43(22):4148–4161. doi:10.1002/jssc.202000809
51. Tsunoda SM, Gonzales C, Jarmusch AK, Momper JD, Ma JD. Contribution of the Gut Microbiome to Drug Disposition, Pharmacokinetic and Pharmacodynamic Variability. Clin Pharmacokinet. 2021;60(8):971–984. doi:10.1007/s40262-021-01032-y
52. Zhang R, Zhu X, Bai H, Ning K. Network Pharmacology Databases for Traditional Chinese Medicine: review and Assessment. Front Pharmacol. 2019;10:123. doi:10.3389/fphar.2019.00123
53. Wicks EE, Semenza GL. Hypoxia-inducible factors: cancer progression and clinical translation. J Clin Invest. 2022;132(11):1. doi:10.1172/jci159839
54. Zheng Z, Xu T, Liu Z, et al. Cryptolepine suppresses breast adenocarcinoma via inhibition of HIF-1 mediated glycolysis. Biomed Pharmacother. 2022;153:113319. doi:10.1016/j.biopha.2022.113319
55. Soltani A, Torki S, Ghahfarokhi MS, Jami MS, Ghatrehsamani M. Targeting the phosphoinositide 3-kinase/AKT pathways by small molecules and natural compounds as a therapeutic approach for breast cancer cells. mol Biol Rep. 2019;46(5):4809–4816. doi:10.1007/s11033-019-04929-x
56. Joseph C, Alsaleem M, Orah N, et al. Elevated MMP9 expression in breast cancer is a predictor of shorter patient survival. Breast Cancer Res Treat. 2020;182(2):267–282. doi:10.1007/s10549-020-05670-x
57. Zhao S, Ma W, Zhang M, et al. High expression of CD147 and MMP-9 is correlated with poor prognosis of triple-negative breast cancer (TNBC) patients. Med Oncol. 2013;30(1):335. doi:10.1007/s12032-012-0335-4
58. Mylona E, Nomikos A, Magkou C, et al. The clinicopathological and prognostic significance of membrane type 1 matrix metalloproteinase (MT1-MMP) and MMP-9 according to their localization in invasive breast carcinoma. Histopathology. 2007;50(3):338–347. doi:10.1111/j.1365-2559.2007.02615.x
59. Jiang H, Li H. Prognostic values of tumoral MMP2 and MMP9 overexpression in breast cancer: a systematic review and meta-analysis. BMC Cancer. 2021;21(1):149. doi:10.1186/s12885-021-07860-2
60. Mira E, Lacalle RA, Buesa JM, et al. Secreted MMP9 promotes angiogenesis more efficiently than constitutive active MMP9 bound to the tumor cell surface. J Cell Sci. 2004;117(Pt 9):1847–1857. doi:10.1242/jcs.01035
61. Wang M, Chen J, Zhao S, et al. Atrazine promotes breast cancer development by suppressing immune function and upregulating MMP expression. Ecotoxicol Environ Saf. 2023;253:114691. doi:10.1016/j.ecoenv.2023.114691
62. Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget. 2014;5(9):2736–2749. doi:10.18632/oncotarget.1932
63. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi:10.1038/srep42717
64. Schultz SG. Membrane Transport, General Concepts. In: Lennarz WJ, Lane MD, editors. Encyclopedia of Biological Chemistry.
65. Zhang R, Nag SA, Wang W. Targeting the NFκB Signaling Pathways for Breast Cancer Prevention and Therapy. Curr Med Chem. 2015;22(2):264–289. doi:10.2174/0929867321666141106124315
66. Ko HS, Lee HJ, Kim SH, Lee EO. Piceatannol suppresses breast cancer cell invasion through the inhibition of MMP-9: involvement of PI3K/AKT and NF-κB pathways. J Agric Food Chem. 2012;60(16):4083–4089. doi:10.1021/jf205171g
67. Tan H, Zhang M, Xu L, Zhang X, Zhao Y. Gypensapogenin H suppresses tumor growth and cell migration in triple-negative breast cancer by regulating PI3K/AKT/NF-κB/MMP-9 signaling pathway. Bioorg Chem. 2022;126:105913. doi:10.1016/j.bioorg.2022.105913
68. Kuo HP, Chuang TC, Tsai SC, et al. Berberine, an isoquinoline alkaloid, inhibits the metastatic potential of breast cancer cells via Akt pathway modulation. J Agric Food Chem. 2012;60(38):9649–9658. doi:10.1021/jf302832n
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.