")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Metabolomic Analysis, Antiproliferative, Anti-Migratory, and Anti-Invasive Potential of Amlodipine in Lung Cancer Cells
Authors Alqudah MAY , Yaseen MM, Alzoubi KH , Al-Husein BA , Bardaweel SK, Abuhelwa AY, Semreen AM, Zenati RA, El-Awady R, Shara M, Bustanji Y, Soares NC, Abu-Gharbieh E, Ramadan WS, Semreen MH
Received 27 June 2024
Accepted for publication 6 February 2025
Published 19 February 2025 Volume 2025:19 Pages 1215—1229
DOI https://doi.org/10.2147/DDDT.S484561
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Mohammad AY Alqudah,1,2 Mahmoud M Yaseen,3 Karem H Alzoubi,1,2 Belal A Al-Husein,2 Sanaa K Bardaweel,4 Ahmad Y Abuhelwa,1 Ahlam M Semreen,5 Ruba A Zenati,5 Raafat El-Awady,1,5 Mohd Shara,1 Yasser Bustanji,5– 7 Nelson C Soares,8– 10 Eman Abu-Gharbieh,5– 7 Wafaa S Ramadan,5 Mohammad H Semreen5,8
1Department of Pharmacy Practice and Pharmacotherapeutics, University of Sharjah, Sharjah, United Arab Emirates; 2Department of Clinical Pharmacy, Jordan University of Science and Technology, Irbid, Jordan; 3Department of Medical Laboratory Sciences, Jordan University of Science and Technology, Irbid, Jordan; 4Department of Pharmaceutical Sciences, School of Pharmacy, the University of Jordan, Amman, Jordan; 5Research Institute of Medical and Health Sciences, University of Sharjah, Sharjah, United Arab Emirates; 6Department of Clinical Sciences, University of Sharjah, Sharjah, United Arab Emirates; 7Department of Biopharmaceutics and Clinical Pharmacy, the University of Jordan, Amman, Jordan; 8Department of Medicinal Chemistry, University of Sharjah, Sharjah, United Arab Emirates; 9Center for Applied and Translational Genomics (CATG), Mohammed Bin Rashid University Medicine and Health Sciences (MBRU), Dubai Health, Dubai, United Arab Emirates; 10College of Medicine, Mohammed Bin Rashid University of Medicine and Health Sciences (MBRU), Dubai Health, Dubai, United Arab Emirates
Correspondence: Mohammad AY Alqudah, Department of Pharmacy Practice and Pharmacotherapeutics, University of Sharjah, Sharjah, United Arab Emirates, Tel +962 2 7201000, Fax +962 2 7095123, Email [email protected]
Background and Objective: Lung cancer stands as the leading cause of cancer-related fatalities worldwide. While chemotherapy remains a crucial treatment option for managing lung cancer in both early-stage and advanced cases, it is accompanied by significant drawbacks, including severe side effects and the development of chemoresistance. Overcoming chemoresistance represents a considerable challenge in lung cancer treatment. Amlodipine cytotoxicity was previously demonstrated and could make lung cancer cells more susceptible to chemotherapies. This research aims to examine the metabolomics changes that may occur due to amlodipine’s anticancer effects on non-small cell lung cancer (NSCLC) cells.
Methods: Amlodipine’s effects on A549 and H1299 NSCLC were evaluated using a colorimetric MTT assay, a scratch wound-healing assay and Matrigel invasion chambers to measure cell viability, cell migration and cell invasion. Ultra-high-performance liquid chromatography-electrospray ionization quadrupole time-of-flight mass spectrometry (UHPLC-ESI-QTOF-MS) was used for the untargeted metabolomics investigation.
Results: Our study revealed that amlodipine significantly reduced proliferation of cancer cells in a dose-dependent fashion with IC50 values of 23 and 25.66 μM in A549 and H1299 cells, respectively. Furthermore, amlodipine reduced the invasiveness and migration of cancer cells. Metabolomics analysis revealed distinct metabolites to be significantly dysregulated (Citramalic acid, L-Proline, dGMP, L-Glutamic acid, Niacinamide, and L-Acetylcarnitine) in amlodipine-treated cells.
Conclusion: The present study illustrates the anticancer effects of amlodipine on lung cancer proliferation, migration, and invasion in vitro and enhance our understanding of how amlodipine exerts its anticancer potential by casting light on these mechanisms.
Keywords: Lung cancer, amlodipine, cytotoxicity, cell migration, cell invasion, metabolomics
Introduction
Globally, lung cancer ranks as the second most prevalent cancer across both genders among all types of cancer.1 In the United States, it was estimated that there were 235,760 new cases of lung cancer and 131,880 deaths related to the disease in 2021.2 Non-small cell lung cancer (NSCLC) forms approximately 80% of all lung cancer cases.3,4 NSCLC is further categorized into three major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large-cell carcinoma.5,6 Due to recent advancements in genomic techniques, all patients with adenocarcinoma histology must be tested for actionable oncogenes such as mutant epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase-1 (ALK-1), B-Raf proto-oncogene (BRAF), among other several targetable mutations.6 Interestingly, these targetable mutations are rare among patients with squamous and large cell histology.5,6
Molecular targeted therapies such as EGFR tyrosine kinase inhibitors (EGFR-TKIs) and ALK inhibitors are currently the preferred treatment options for tumors with driver mutations.6 On the other hand, chemotherapy remains the primary treatment approach for other subtypes.5,6 Platinum-based regimens, in combination with gemcitabine, paclitaxel, or pemetrexed, are specifically favored as the initial chemotherapy treatments.5,6 Despite the development of new therapies, resistance to chemotherapy has remained a significant challenge.5,7 Furthermore, combination chemotherapies often lead to additional adverse effects and impose a greater economic burden on the healthcare system.8 Therefore, it is crucial to emphasize the significance of ongoing research in discovering new therapeutics or exploring the repurposing of medications currently used to treat non-cancer-related conditions. Particularly, focus should be given to drugs that demonstrate potential anti-cancer effects while exhibiting fewer side effects than current chemotherapeutic agents.9 Repurposing such drugs offers distinct advantages over developing entirely new anti-cancer medications, saving money, time, and effort.
The calcium ions (Ca2+) function as a second messenger within cells, regulating various cellular activities such as gene transcription, cell proliferation, cell migration, and cell death.10 Maintaining intracellular Ca2+ homeostasis is crucial under normal physiological conditions. However, in cancer settings, the balance of intracellular Ca2+ is disrupted or altered. This dysregulation has been linked to various activities are contributing to tumor formation and disease progression, including tumor initiation, angiogenesis, apoptosis resistance, progression, and metastasis.10–12 Consequently, targeting Ca2+, either directly or indirectly, holds therapeutic potential in cancer.
Calcium channel blockers (CCBs), a well-known class of drugs primarily used for hypertension treatment, have demonstrated anti-tumor effects in multiple cancer types.13–17 For instance, verapamil, a non-dihydropyridine CCB, has shown the ability to reverse multidrug resistance (MDR) in vitro, enhance the accumulation of chemotherapy drugs, and sensitize chemoresistant cancer cells to autophagy-induced cell death and apoptosis in various cancer types, including lung cancer.18–22 While some clinical trials have indicated that verapamil could improve outcomes in NSCLC,23 cardiotoxicity has limited its effectiveness.24 Consequently, researchers shifted their focus to dihydropyridine CCBs. Several studies have highlighted the anticancer potential of dihydropyridine CCBs in different types of cancer. For instance, nifedipine inhibited the in vitro growth of prostate and colorectal cancer cells but still, it has no effect in lung cancer.22 Conversely, nifedipine could stimulate the proliferation and migration of breast cancer cells.25,26 Other dihydropyridine CCBs, such as nicardipine, lercanidipine, and amlodipine, have exhibited cytotoxic effects when combined with proteasome inhibitors in breast and liver cancer cells.27,28 In vitro and in vivo studies on human epidermoid cancer cells have demonstrated the ability of several dihydropyridine CCBs, including amlodipine, nicardipine, and nimodipine, to inhibit cancer cell growth.29
In patients with unresectable histologically proven pancreatic ductal adenocarcinoma, patients prescribed dihydropyridine CCBs such as amlodipine and nifedipine demonstrated significantly improved overall survival.30 Amlodipine has also been shown to induce cell cycle arrest and suppress cell growth in colorectal cancer cells in vitro.31 In addition, amlodipine is a powerful inhibitor of programmed death-ligand 1 (PD-L1) production in cancer cells, which may increase tumor-specific T cell cytotoxicity.9 Interestingly, amlodipine, when combined with doxorubicin, has also shown promise in reducing multidrug resistance in leukemia.32
We previously showed that amlodipine is cytotoxic and could sensitize lung cancer cells to chemotherapies.33 This work aims to investigate the in vitro effects of amlodipine on the suppression of cancer cell proliferation, migration, and invasion in NSCLC cells using the A549 and H1299 cell lines with a specific focus on analyzing potential metabolomic alterations in the A549 cell line as it is the most common model used in previous lung cancer studies.
Materials and Methods
Cell Lines, Cell Culture and Drug Treatments
A549 and H1299 human lung adenocarcinoma cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained in Roswell Park Memorial Institute (RPMI-1640) media supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 0.1 mg/mL streptomycin in a humidified atmosphere with 5% CO2 at 37°C. Drug treatments were purchased from Tocris Bioscience (Bristol, UK). Amlodipine was dissolved in dimethyl sulfoxide (DMSO) to produce 10, 50 and 100 mm DMSO stock concentrations. Of note, to minimize the cytotoxic effects of DMSO, the final concentration has never exceeded 0.1% in all treatment groups. Furthermore, we treated cells of each cell line used in this study All experiments were carried out with a sample size of 5 in a quadruplicate manner with DMSO as a positive control.
Cell Viability Assay
To assess the effect of amlodipine on the viability of lung cancer cells, cell viability was measured by the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay as described previously.34,35 MTT was purchased from Sigma Aldrich (St Louis, MO, USA). Briefly, cells were seeded in a 96-well plate at 1×104 cells per well density. After 24 hours, cells were treated with amlodipine at concentrations of 10, 15, 20, 30, 40 µM (4 replicates/group) or vehicle for 48 hours.
Following a 48-hour incubation period, 10 µL of MTT solution was added to each well, reaching a final concentration of 0.5 mg/mL, and then incubated for an additional 4 hours at 37 °C. Afterward, DMSO was added to solubilize the formazan crystals. The absorbance was measured at 490 nm on a microplate reader (BioTech, Winooski, VT, USA). The absorbance values were standardized, and results were presented as a percentage of viable cells normalized to vehicle-treated cells according to the following equations:


Scratch Wound-Healing Assay
An in vitro scratch wound-healing assay was used to determine the impact of amlodipine on the directionality of NSCLC cell migration in two dimensions at concentrations of 10, 15 and 20 µM. These concentrations were selected because they are below the IC50, aiming to observe a significant influence on migration while minimizing impacts on cell viability and avoiding excessive cytotoxicity.34 In brief, A549 and H1299 cell lines were seeded until confluence in sterile flat-bottom 6-well plates (4 replicates per group). Then, cells were synchronized using serum-free media for 6 hours. Afterward, a wound was done in the confluent monolayer of lung cancer cells using sterile 200 µL pipette tips. A serum-free media containing the final amlodipine concentrations was added to the scratched cells and maintained until the wounds became completely closed. The anti-migratory effect of amlodipine was normalized to the width of control cells. Finally, we measured the distance traveled by A549 and H1299 after measuring the wound width at 48 h and subtracting it from the wound width at the start of treatment (ie, time zero) using NIH-Image J software.
Cell Invasion Assay
To assess the anti-invasive effect of amlodipine treatment in A549 and H1299 lung cancer cells, we used Corning BioCoat Matrigel Invasion Chambers (Corning Inc., Acton, MA, USA) as previously described.36 Following trypsinization, cells were counted, centrifuged, and resuspended into serum-free media (SFM) with drug treatments at concentrations of 10, 15 and 20 µM. Subsequently, 5 x 104 cells/well were introduced into the top chambers pre-coated with basement membrane extract. Bottom chambers were supplemented with 10% serum-containing media. After a 48-hour incubation at 37°C, the invading cells were fixed with cold ethanol for 5 minutes and stained with 0.1% crystal violet solution in 20% ethanol for 30 minutes. Non-invading cells were swabbed off from the upper membrane surface. Stained cells were PBS-washed, air-dried, and quantified in 4 random fields per sample using NIH (National Institutes of Health) ImageJ software. Results were expressed as average values ± SE relative to the control invading cells.
NSCLC A549 Cell Harvest and Lysis for Metabolomics Analyses
Approximately 2×106 NSCLC A549 cells were seeded per replicate to perform metabolomic analysis in separate T25 flasks. The cells were treated individually with either amlodipine (IC50 values of 23 µM) or a vehicle solution (DMSO) as a negative control for 24 hours. IC50 concentration of amlodipine ensures that the metabolomics analysis is conducted at a point where cellular responses are maximally sensitive to the drug. The decision to conduct metabolomic analysis after 24 hours of amlodipine treatment was deliberate, considering that prolonged treatment durations could induce cell death or provoke additional stress responses, potentially introducing complexity to the cellular metabolic profile. Maintaining a consistent cells number per flask ensured that any observed effects were not influenced by variable cell quantities. After the treatment period, the cells and buffer samples were transferred to Eppendorf tubes and centrifuged at 14000 rpm for 5 minutes. The buffer was discarded, and the cell pellets were preserved for further analysis. Each sample was supplemented with 400 µL of lysis buffer and allowed to rest for 10 minutes. Subsequently, the samples were transferred to 10 mL tubes and subjected to vortexing for 2–4 minutes, followed by sonication using a COPLEY probe-sonicator (QSONICA SONICATOR, USA) for 30 seconds at a 30% amplifier setting in an ice bath. The sonicated samples were then returned to Eppendorf tubes and centrifuged at 14000 rpm for 5 minutes. The resulting supernatant was carefully transferred to another Eppendorf tube, and a mixture of 400 µL of methanol and 300 µL of chloroform was added. After thorough vortexing for 30 seconds, the samples underwent a subsequent centrifugation step at 14000 rpm for 5 minutes. This step facilitated the separation of two distinct layers. The upper layer containing the interest metabolites was selectively collected from each sample and transferred to glass vials. An additional 400 µL of methanol was added, and the mixture was vortexed and centrifuged. The remaining supernatant was combined with the previously collected upper layers in the glass vials, while the excess liquid was evaporated to dryness. The dried metabolomics samples were resuspended in 200 µL of a solution containing 0.1% formic acid in water. Finally, these resuspended samples were injected into an UHPLC system coupled to a quadrupole-time-of-flight mass spectrometer (Q-TOF, enabling comprehensive metabolite characterization.
Ultra-High‐performance Liquid Chromatography-Tandem Mass Spectrometry (HPLC–MS/MS)
Metabolite analysis was conducted using an Elute UHPLC system coupled with a Q-TOF Mass Spectrometer (Bruker, Bremen, Germany) as described previously.37 The Elute HPG 1300 pumps, Elute Autosampler (Bruker, Bremen, Germany), and Hamilton® Intensity Solo 2 C18 column (100 mm × 2.1 mm, 1.8 m beads) were utilized for reversed-phase chromatography. The separation solvents consisted of 0.1% formic acid (FA) in LC-grade water (solvent A) and 0.1% FA in acetonitrile (solvent B). The LC-QTOF-MS system was maintained at a consistent column temperature of 35 °C. Five biological replicates were analysed, with each sample extract subjected to duplicate injections at a standardized volume of 10 μL. This protocol was designed to control for technical variability and enhance the precision of the measurements, ensuring robust and reproducible metabolomic data across all experimental conditions A 30-minute gradient elution was employed, starting with 1% acetonitrile for 2 minutes, followed by a ramp to 99% acetonitrile over 15 minutes. The elution was then held at 99% acetonitrile for 3 minutes before re-equilibrating with 1% acetonitrile for 10 minutes. The flow rate was set at 0.25 mL/min for 20 minutes, increased to 0.35 mL/min for 8.3 minutes, and returned to 0.25 mL/min for 1.7 minutes. The ESI source conditions for each injection were as follows: a drying gas flow rate of 10.0 L/min at 220 °C, a capillary voltage of 4500 V, an end plate offset of 500 V, and a nebulizer pressure of 2.2 bar. In metabolomics analysis, MS2 acquisition involved varying the collision energy between 100–250% of 20 eV, with an end plate offset of 500 volts. Auto MS scans for sodium formate were conducted from 0 to 0.3 minutes, followed by auto MS/MS scans for fragmentation from 0.3 to 30 minutes. Both acquisition sections were performed in positive mode at 12 hz. The scan range encompassed 20 to 1300 m/z, with a precursor ion width of 0.5 and a precursor number of 3. The cycle time was set at 0.5 s, and the threshold was set to 1000 counts. An active exclusion process was implemented after three spectra, and the exclusion was lifted after 0.2 minutes.
Metabolomics Data Processing and Analysis
The data obtained from the metabolite analysis was processed using licensed copy of MetaboScape® 4.0 software (Bruker, Bremen, Germany). In the T-ReX 2D/3D workflow, molecular feature detection settings were applied: a minimum intensity threshold of 1000 counts and a maximum peak duration of seven spectra. Mass recalibration was performed within a retention time range of 0 to 0.3 minutes.38 Only features detected in at least six of the 24 samples per cell type were considered. The MS/MS import method involved averaging, and bucketing parameters were set with a retention duration of 0.3 to 25 minutes and a mass range of 50 to 1000 m/z. Metabolite identification was accomplished by comparing combined MS/MS, precursor m/z values, and isotope pattern scores with the human metabolome database (HMDB) 4.0. The annotation quality score (AQ score) was used to select the best matching feature when multiple features matched a particular database entry. Furthermore, the metabolite data was saved as CSV files for further analysis and integrated into the comprehensive metabolomics platform MetaboAnalyst 5.0 software (https://www.metaboanalyst.ca) (Accessed: June 22, 2023). Two-tailed independent Student’s t-tests were conducted for each medication to identify significantly different metabolites compared to DMSO with a significance threshold set at p < 0.05. False discovery rate (FDR) correction was applied to address multiple hypothesis testing and eliminate false positives. A volcano plot was generated to visualize the statistical significance and fold change of cellular metabolite alteration for each condition. Enrichment analysis and Principal Component Analysis (PCA) were performed using MetaboAnalyst (version 5.0, http://www.metaboanalyst.ca) to compare the two groups and gain further insights.
Statistical Analyses
Statistical analyses were performed using licensed copy of GraphPad Prism® (version 9.1.1; GraphPad software, San Diego, California, USA). One-way Analysis of variance (ANOVA) test and Tukey’s multiple comparison test were used to detect statistical significance. Half maximal (50%) inhibitory concentration (IC50) values were obtained by applying nonlinear regression curve fit analysis. P<0.05 was considered statistically significant. For all experiments, results were expressed as mean ± SEM.
Results
The Cytotoxic Effect of Amlodipine on NSCLC Cells
The effect of in vitro amlodipine treatment on the viability of two NSCLC cell lines is illustrated in Figure 1. Amlodipine treatment showed a reduction in A549 and H1299 cell viability in a dose-dependent manner. Treatment of A549 with an amlodipine dose range of 15–40 µM resulted in significant inhibition of cell growth compared to control cells. The same pattern was observed in H1299, 15–40 µM amlodipine significantly inhibited H1299 cell growth compared to control cells. The IC50 values for amlodipine treatment were 23 and 25.66 μM in A549 and H1299 cells, respectively.
 |
Figure 1 Antiproliferative effect of amlodipine on A549 and H1299 cell lines. (A) Cell viability of A549 cells treated with increasing concentrations of amlodipine. (B) Cell viability of H1299 cells treated with increasing concentrations of amlodipine. Each bar represents mean ± SEM of cell viability (%) in each treatment group. * indicates P < 0.05 and ** indicates P < 0.01. compared with control vehicle-treated group. DMSO, dimethyl sulfoxide. |
Amlodipine suppressed the migration of NSCLC cells
The effect of increasing concentrations of amlodipine on the migration of NSCLC cancer cells, namely A549 and H1299 cell lines, is shown in Figure 2. Almost complete closure of inflicted wounds in A549 and H1299 cells was observed after 48 h of incubation. Treatment with 10–20 µM amlodipine concentrations has resulted in a dose-dependent inhibition of cell motility after 48 h in culture, as shown in Figure 2.
 |
Figure 2 Effect of amlodipine on A549 and H1299 cells migration. (A) Representative images of cell migration and (B) quantitative analysis of cell migration percentage by A549 cells following treatment with amlodipine (10, 15 or 20 µM) compared with the control treated cells. (C) Quantitative analysis of cell migration percentage and (D) representative microscopic images for cell migration by H1299 cells treated with amlodipine (10, 15 or 20 µM) compared with the control treated cells. * indicates P < 0.05 and ** indicates P < 0.01. compared with control vehicle-treated group. DMSO, dimethyl sulfoxide. |
Amlodipine Suppressed the Invasion of NSCLC Cells
The effect of various concentrations of amlodipine on the invasiveness of NSCLC cancer cells, namely A549 and H1299 cell lines, is shown in Figure 3. Amlodipine treatment in concentrations of 10–20 µM significantly suppressed the invasiveness of A549 and H1299 cells compared to vehicle-treated control groups, as illustrated in Figure 3.
 |
Figure 3 Effect of amlodipine on A549 and H1299 cells invasion. (A) Representative images of cell invasion and (B) quantitative analysis of cell invasion percentage by A549 cells following treatment with amlodipine (10, 15 or 20 µM) compared with the control treated cells. (C) Quantitative analysis of cell invasion percentage and (D) representative microscopic images for cell invasion by H1299 cells treated with amlodipine (10, 15 or 20 µM) compared with the control treated cells. * indicates P < 0.05 and ** indicates P < 0.01. compared with control vehicle-treated group. DMSO, dimethyl sulfoxide. |
Metabolomics of Amlodipine NSCLC A549 Treated Cells
NSCLC A549 cells were treated with vehicle (DMSO) or amlodipine (23 µM). The MTT assay revealed that the application of amlodipine (23 µM) suppressed the growth of A549 cells. The observed reduction in cell proliferation, accompanied by the induction of cell death, following treatment with amlodipine compound 48 hours later, indicated the cytotoxic effects of these compounds on the target cells. Consequently, we opted to delve deeper into the changes occurring in the metabolome after 24 hours of treatment. This investigation aimed to identify the initial events triggered by amlodipine that led to cell death in the treated cells.
We conducted untargeted LC-MS/MS-based metabolomics analysis to compare the metabolome of control cells and cells treated with amlodipine. The experiments were designed using five biological replicates to account for intrinsic biological variation across independent samples, thereby enhancing the robustness and statistical validity of the data. Each metabolite extract derived from these biological replicates was subjected to duplicate injections for LC-QTOF-MS analysis, which allowed for the mitigation of potential technical variability arising from instrumental fluctuations. This approach ensured both biological and technical accuracy, resulting in 12 distinct samples and a total of 24 LC-QTOF-MS analyses. Following filtration, we confidently assigned 127 metabolites to the HMDB 4.0 library with a significance level of p < 0.05. Upon conducting Principal Component Analysis (PCA), the results (as depicted in Figure 4) showed a partial overlap between the group of cells treated with amlodipine and the control group. This observation suggests that there are some differences between the two groups, although some similarities are also present. Moreover, through further metabolomics analysis, the comparison between control and amlodipine-treated cells revealed significant changes in the abundance of six metabolites with a fold change cutoff value of 1.5 (see Table 1, and Figure 5). Among these metabolites, five (citramalic acid, L-proline, 2’-Deoxyguanosine 5’-monophosphate, niacinamide, and L-acetylcarnitine) exhibited an increase, while one (L-glutamic acid) showed a decrease following treatment (Supplementary Figure 1). Subsequently, we conducted a functional enrichment analysis of the metabolites that significantly changed (p-value < 0.05) due to amlodipine treatment. We used the Small Molecule Pathway Database (SMPDB) and MetaboAnalyst 5.0 for this analysis. The pathway enrichment analysis revealed that the differentially abundant metabolites were enriched in pathways such as nicotinate and nicotinamide metabolism, arginine and proline metabolism, and purine metabolism (as illustrated in Figure 6).
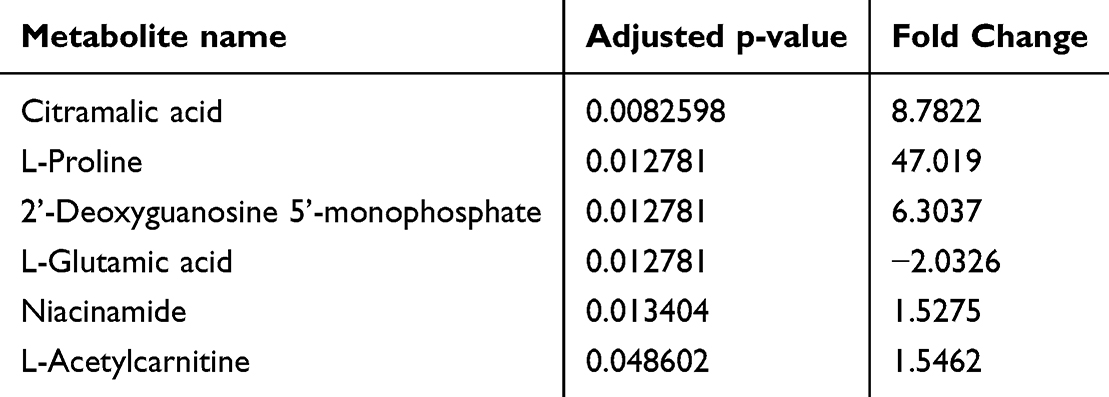 |
Table 1 Significantly Altered Metabolites Between Amlodipine Treated Cells and Controls |
 |
Figure 4 Principal Component (PC) Analysis presenting the correlation between amlodipine treated cells compared to control in A549 cells. |
 |
Figure 5 Volcano plot presenting the abundance of altered metabolites between the two groups. Red color indicates that the metabolite is increased in treated A549 cells. Blue color indicates that the metabolite is decreased in treated A549 cells. |
 |
Figure 6 Enriched pathways of significantly altered metabolites in A549 cells treated with amlodipine. |
Discussion
Lung cancer, specifically NSCLC, continues to be a significant worldwide health concern, representing a major cause of cancer-related fatalities. Repositioning/repurposing is one of the promising treatment approaches in cancer research to overcome chemoresistance.39 We previously showed that amlodipine is cytotoxic and could sensitize lung cancer cells to chemotherapies.33 In this study, we assessed other potential anti-cancer effects of amlodipine in NSCLC cells. Our results showed that amlodipine, when used as a monotherapy, is cytotoxic to lung cancer cells with squamous or non-squamous histology with wild-type EGFR and ALK. This study shed light on the potential of amlodipine as an anti-migratory and anti-invasive agent in the context of cancer. The findings revealed significant changes in metabolomics, offering new perspectives and potential targets for cancer therapy.
The effects of amlodipine on cell viability of NSCLC cells are in parallel with previous studies in which dihydropyridine CCBs have been shown to possess an anti-proliferative role in several human cancers such as prostate, lung, breast, liver, and colorectal cancers.27,28,31,40,41 Amlodipine treatment showed a significant reduction of lung cancer cell viability in a dose-dependent manner. The growth inhibition effect was significantly started at moderate amlodipine concentration (15 µM) and maintained to the maximum effect at 40 µM. Although the IC50 value for A549 in our conditions was higher than to what was previously reported by Fu et al (23 vs 9.641 μM),35 these cytotoxic effects of amlodipine were more promising in A549 cells compared to H1299 cells, the latter being p53 defective and thus interpret this difference.42 The differences in IC50 values between the two studies can also be attributed to variations in experimental conditions, including: cell seeding counts (4 x 103 vs 1×104 cells/well), MTT concentration (5 vs 0.5 mg/mL), as well as potential variances in the passage number of cell lines. Our data showed that amlodipine treatment could suppress NSCLC cell survival, demonstrating its anti-proliferative effects. Previous studies have shown several mechanisms by which amlodipine could inhibit tumor progression in vitro and in vivo.29,35 These mechanisms are mainly represented by the inhibition of cell cycle at G0/G1 phase via modulation of cell cycle-related proteins such as cyclin D1, p-Rb, p27, and p21.29,31,35 In addition, amlodipine and lercanidipine have been shown to inhibit PD-L1 expression in H1299 cells.9,43 Moreover, a study reported that verapamil could sensitize resistant H1299 sublines to chemotherapies.44 Further, other studies have shown that amlodipine could prevent the transformation and tumorigenesis of lung epithelial cells.45 Interestingly, recent studies have reported that amlodipine suppresses lung cell proliferation by inhibiting major altered, pro-oncogenic pathways in lung cancer such as the PI3K/Akt and Raf/MEK/ERK through the inhibition of epidermal growth factor receptor (EGFR) phosphorylation.29,35,46
Cancer cell migration and invasion are critical factors for cancer progression and metastasis.47 In the present study, the anti- migratory and the ant-invasive effects of amlodipine in NSCLC cells were significant in a concentration-dependent manner. The inhibitory effect of amlodipine on cell invasion was slightly more observable at 10 and 15 µM in the H1299 cell line, whereas the inhibitory effect at 20 µM was notably more prominent in A549 cell line. Previous studies have implicated amlodipine and other CCBs in cancer cell migration and invasion.28,35,48 For instance, amlodipine has been shown to diminish A549 cell migration via the inhibition of EGFR-mediated PI3K/Akt and Raf/ERK pathways.35 Additionally, the anti-invasive effects of amlodipine in breast cancer were accompanied by downregulation in p-ERK1/2 and integrin β1 protein expression.28 Moreover, nicardipine inhibitory effects on breast cancer cell migration has been shown to be mediated via matrix metalloproteinase-9 reduction.48 Further, EGFR mutation has been shown to be the main driver of H1299 cell invasion via ERK-dependent activation of CXCL12-CXCR4 signaling pathways.49 Together, these studies strongly support the in vitro anti-migratory and anti-invasive effects of amlodipine in lung cancer cells via modulating major, altered signaling pathways.
For the metabolomics analyses, the alteration observed in the nicotinate and nicotinamide metabolism pathway after treating lung cancer cells A549 with amlodipine suggests that amlodipine may have an impact on the metabolism of nicotinate and nicotinamide, which are forms of vitamin B3 (niacin). This pathway involves various enzymes and reactions responsible for synthesizing, breaking down, and converting nicotinamide.50 While amlodipine is primarily known for its effects on calcium channels, it can also have off-target effects on other cellular processes.35 Notably, niacinamide, a key metabolite in this pathway, plays a vital role in maintaining NAD+/NADH homeostasis, which regulates cellular energy production and oxidative stress response. The increase in niacinamide observed in amlodipine-treated cells suggests potential disruption of NAD+ synthesis, which may impair ATP production via glycolysis and oxidative phosphorylation. This pathway is crucial in cancer cells, where elevated NAD+ levels support rapid proliferation and DNA repair. Amlodipine’s impact may thus inhibit tumor growth by reducing the availability of NAD+ and enhancing oxidative stress.51
Moreover, the alteration observed in arginine and proline metabolism after treating A549 cells with amlodipine suggests that this drug may impact the metabolism of arginine and proline, which are amino acids involved in various cellular processes. Also, arginine and proline metabolism pathway involve enzymes and reactions responsible for the synthesis, breakdown, and conversion of arginine and proline.52 These amino acids play crucial roles in protein synthesis, cell signaling, immune function, and nitric oxide production, among other important functions.53 Increased L-proline levels observed in amlodipine-treated cells may indicate heightened oxidative stress, as proline metabolism generates reactive oxygen species. Furthermore, arginine metabolism contributes to immune evasion in cancer by suppressing T-cell function through nitric oxide signaling. Amlodipine, as a calcium channel blocker,54 may disrupt these pathways by altering intracellular calcium levels, which can affect the activity of enzymes involved in amino acid metabolism, including nitric oxide synthase. Such disruptions could reverse immune suppression and impair tumor progression by modulating the metabolic and signaling pathways critical for cancer cell survival.55
Furthermore, the alteration observed in the purine metabolism after treating lung cancer cells A549 with amlodipine suggests that this medication may have an impact on the metabolism of purines, which are essential components of DNA, RNA, and various cellular processes.56 This pathway involves a series of enzymatic reactions responsible for the synthesis, breakdown, and interconversion of purines, such as adenine and guanine.57 These purine nucleotides are fundamental building blocks for the synthesis of DNA and RNA, as well as energy carriers (eg, ATP, GTP) and important signaling molecules (eg, cAMP).56 Amlodipine indirectly influences purine metabolism through its impact on cellular energy metabolism by affecting intracellular calcium levels, which can modulate ATP production and energy metabolism.58 Changes in cellular energy status can impact the availability and utilization of purine nucleotides, potentially altering the purine metabolism.59 Amlodipine might affect gene expression or enzyme activity, either directly or indirectly through signaling pathways. It could modulate the activity of specific enzymes involved in purine metabolism, leading to alterations in the pathway. In accordance with our results, decreased glutamic acid abundance was observed in amlodipine treated A549 cells. Glutamic acid, an amino acid vital for various metabolic pathways including purine metabolism, plays a crucial role in purine synthesis. Glutamic acid provides nitrogen atoms that are integral to the formation of the purine ring structure. This incorporation of nitrogen atoms is a critical step in the biosynthesis of adenine and guanine, the two major types of purines.60 A reduction in glutamic acid can lead to impaired purine metabolism. This impairment hinders the synthesis of nucleotides, thereby disrupting DNA and RNA synthesis. Consequently, this disruption can result in decreased cell proliferation.
Additionally, the alteration observed in the malate-aspartate shuttle pathway after treating lung cancer cells A549 with amlodipine suggests that this medication may impact this specific metabolic pathway involved in the transfer of reducing equivalents between cellular compartments.61 The malate-aspartate shuttle is an essential pathway involved in transferring reducing equivalents, specifically in the form of electrons, from the cytoplasm to the mitochondria. This shuttle relies on the conversion of malate to oxaloacetate in the cytoplasm, followed by the transport of oxaloacetate into the mitochondria, where it is converted back to malate.62 This process generates NADH, which can be used in the mitochondrial electron transport chain to produce ATP. Amlodipine indirectly influences this pathway by impacting cellular metabolism or energy balance. One potential explanation is that amlodipine may alter the redox balance or mitochondrial function, leading to changes in the activity or expression of enzymes involved in the malate-aspartate shuttle. These alterations can affect the flow of reducing equivalents, ultimately influencing cellular energy production and metabolism. Furthermore, amlodipine might modulate gene expression or enzyme activity directly or indirectly, through signaling pathways. It could impact the expression or activity of enzymes involved in the malate-aspartate shuttle, resulting in alterations in the pathway.
Overall, the changes observed in the nicotinate and nicotinamide, arginine and proline, purine, and malate-aspartate shuttle pathways upon treatment of lung cancer cells A549 with amlodipine suggest that this medication affects cellular metabolism and potentially influences various metabolic processes within the cells. However, it is important to acknowledge that the exact mechanisms and implications of these changes in the context of lung cancer and amlodipine treatment are not well understood. These findings indicate that amlodipine might have unintended effects on cellular metabolism, which could have positive and negative consequences. Further investigation is necessary to explore the downstream effects of these pathway alterations and their specific roles in lung cancer biology. Understanding the functional significance of these changes can provide valuable insights into potential therapeutic targets or mechanisms of action of amlodipine in lung cancer treatment. Also, it is essential to interpret these findings within the broader scope of lung cancer research, considering other factors that can influence cellular metabolism and tumor biology. Additional studies, including genetic analyses and in vivo experiments, will help elucidate the implications of these pathway alterations and their potential impact on lung cancer progression and response to treatment. This study has a few limitations. First, it lacks a positive control in the cell viability experiments. Second, further investigations are needed to identify the exact molecular protein targets mediating the anticancer effects of amlodipine, whether through inhibition of calcium ion channels or other signal transduction pathways. Third, the metabolomics study was conducted on only one cell line. Finally, this study does not address the time-effect relationship of amlodipine’s actions. Future in vitro and/or in vivo studies could incorporate time-course analyses, autophagy markers and the role of reactive oxygen species to provide additional insights into the progression and duration of the observed anticancer effects.
Conclusion
In conclusion, our study revealed the ability of amlodipine treatment to inhibit the growth, cell migration, and invasiveness of NSCLC cells with significant metabolomics changes. However, the effect of amlodipine in enhancing the sensitization of NSCLC to chemotherapy remains to be determined in future investigations.
Data Sharing Statement
The metabolomics data of this study have been deposited in the Metabolomics Workbench under the study ID ST002755.
Acknowledgments
We are grateful to the Deanship of Research at Jordan University of Science and Technology to fund this project.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Deanship of Research at Jordan University of Science and Technology (Receiver: MAYA; Grant# 81/2016). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors have declared that no competing interests exist.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries.n/a(n/a).
2. Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics. CA: a Cancer Journal for Clinicians. 2021;71(1):7–33. doi:10.3322/caac.21654
3. Gazdar AF. Should we continue to use the term non-small-cell lung cancer? Ann Oncol. 2010;21(Suppl 7):vii225–vii229. doi:10.1093/annonc/mdq372
4. Field RW, Smith BJ, Platz CE, et al. Lung cancer histologic type in the surveillance, epidemiology, and end results registry versus independent review. JNCI J National Cancer Inst. 2004;96(14):1105–1107. doi:10.1093/jnci/djh189
5. Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4(1):36–54. doi:10.3978/j.issn.2218-6751.2014.05.01
6. Ettinger DS, Wood DE, Aisner DL, et al. Non-small cell lung cancer, version 3.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2022;20(5):497–530. doi:10.6004/jnccn.2022.0025
7. Kim ES. Chemotherapy resistance in lung cancer. advances in experimental medicine and biology. Advances in Experimental Medicine and Biology. 2016;893:189–209. doi:10.1007/978-3-319-24223-1_10
8. Pearce A, Haas M, Viney R, et al. Incidence and severity of self-reported chemotherapy side effects in routine care: a prospective cohort study. PLoS One. 2017;12(10):e0184360. doi:10.1371/journal.pone.0184360
9. Li C, Yao H, Wang H, et al. Repurposing screen identifies Amlodipine as an inducer of PD-L1 degradation and antitumor immunity. Oncogene. 2021;40(6):1128–1146. doi:10.1038/s41388-020-01592-6
10. Cui C, Merritt R, Fu L, et al. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7(1):3–17. doi:10.1016/j.apsb.2016.11.001
11. Martinez-Delgado G, Felix R. Emerging role of CaV1.2 channels in proliferation and migration in distinct cancer cell lines. Oncology. 2017;93(1):1–10. doi:10.1159/000464293
12. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11(8):609–618. doi:10.1038/nrc3105
13. Shchepotin IB, Soldatenkov V, Buras RR, et al. Apoptosis of human primary and metastatic colon adenocarcinoma cell lines in vitro induced by 5-fluorouracil, verapamil, and hyperthermia. Anticancer Res. 1994;14(3A):1027–1031.
14. Woods N, Trevino J, Coppola D, et al. Fendiline inhibits proliferation and invasion of pancreatic cancer cells by interfering with ADAM10 activation and beta-catenin signaling. Oncotarget. 2015;6(34):35931–35948. doi:10.18632/oncotarget.5933
15. Taylor JM, Simpson RU. Inhibition of cancer cell growth by calcium channel antagonists in the athymic mouse. Cancer Res. 1992;52(9):2413–2418.
16. Uehara H, Nakaizumi A, Baba M, et al. Inhibition by verapamil of hepatocarcinogenesis induced by N-nitrosomorpholine in Sprague-Dawley rats. Br J Cancer. 1993;68(1):37–40. doi:10.1038/bjc.1993.283
17. Sato K, Ishizuka J, Cooper CW, et al. Inhibitory effect of calcium channel blockers on growth of pancreatic cancer cells. Pancreas. 1994;9(2):193–202. doi:10.1097/00006676-199403000-00009
18. Inaba M, Kobayashi H, Sakurai Y, et al. Active efflux of daunorubicin and Adriamycin in sensitive and resistant sublines of P388 leukemia. Cancer Res. 1979;39(6 Pt 1):2200–2203.
19. Harker WG, Bauer D, Etiz BB, et al. Verapamil-mediated sensitization of doxorubicin-selected pleiotropic resistance in human sarcoma cells: selectivity for drugs which produce DNA scission. Cancer Res. 1986;46(5):2369–2373.
20. Feng MR, Liebert M, Wedemeyer G, et al. Effect of verapamil on the uptake and efflux of etoposide (VP16) in both sensitive and resistant cancer cells. Selective Cancer Therapeutics. 1991;7(2):75–83. doi:10.1089/sct.1991.7.75
21. Roepe PD. Analysis of the steady-state and initial rate of doxorubicin efflux from a series of multidrug-resistant cells expressing different levels of P-glycoprotein. Biochemistry. 1992;31(50):12555–12564. doi:10.1021/bi00165a003
22. Wong B-S, Chiu L-Y, Tu D-G, et al. Anticancer effects of antihypertensive L-type calcium channel blockers on chemoresistant lung cancer cells via autophagy and apoptosis. Cancer Manag Res. 2020;12:1913–1927. doi:10.2147/CMAR.S228718
23. Millward MJ, Cantwell BM, Munro NC, et al. Oral verapamil with chemotherapy for advanced non-small cell lung cancer: a randomised study. Br J Cancer. 1993;67(5):1031–1035. doi:10.1038/bjc.1993.189
24. Ozols RF, Cunnion RE, Klecker RW Jr, et al. Verapamil and Adriamycin in the treatment of drug-resistant ovarian cancer patients. J Clin Oncol. 1987;5(4):641–647. doi:10.1200/JCO.1987.5.4.641
25. Guo D-Q, Zhang H, Tan S-J, et al. Nifedipine promotes the proliferation and migration of breast cancer cells. PLoS One. 2014;9(12):e113649. doi:10.1371/journal.pone.0113649
26. Zhao T, Guo D, Gu Y, et al. Nifedipine stimulates proliferation and migration of different breast cancer cells by distinct pathways. Mol Med Rep. 2017;16(2):2259–2263. doi:10.3892/mmr.2017.6818
27. Lee AR, Seo MJ, Kim J, et al. Lercanidipine synergistically enhances bortezomib cytotoxicity in cancer cells via enhanced endoplasmic reticulum stress and mitochondrial Ca(2+) overload. Int J mol Sci. 2019;20(24):6112. doi:10.3390/ijms20246112
28. Alqudah MAY, Al-Samman R, Azaizeh M, et al. Amlodipine inhibits proliferation, invasion, and colony formation of breast cancer cells. Biomed Rep. 2022;16(6):50. doi:10.3892/br.2022.1533
29. Yoshida J, Ishibashi T, Nishio M. Antitumor effects of amlodipine, a Ca2+ channel blocker, on human epidermoid carcinoma A431 cells in vitro and in vivo. Eur J Pharmacol. 2004;492(2–3):103–112. doi:10.1016/j.ejphar.2004.04.006
30. Tingle SJ, Severs GR, Moir JAG, et al. Calcium channel blockers in pancreatic cancer: increased overall survival in a retrospective cohort study. Anticancer Drugs. 2020;31(7):737–741. doi:10.1097/CAD.0000000000000947
31. Alqudah MA, Alrababah BA, Mhaidat NMJJJo PS, Mhaidat NM. Amlodipine inhibits cell proliferation and induces cell cycle arrest in colorectal cancer cells. OncoTargets and Therapy. 2017;10(3):4869–4883. doi:10.2147/OTT.S148604
32. Ji BS, He L, Liu GQ. Reversal of p-glycoprotein-mediated multidrug resistance by CJX1, an amlodipine derivative, in doxorubicin-resistant human myelogenous leukemia (K562/DOX) cells. Life Sci. 2005;77(18):2221–2232. doi:10.1016/j.lfs.2004.12.050
33. Alqudah MAY, Al-Samman R, Alzoubi KH. The interactive effect of amlodipine and chemotherapeutic agents in lung cancer cells. Inf Med Unlocked. 2022;32:101066. doi:10.1016/j.imu.2022.101066
34. Alqudah MA, Agarwal S, Al-Keilani MS, et al. NOTCH3 is a prognostic factor that promotes glioma cell proliferation, migration and invasion via activation of CCND1 and EGFR. PLoS One. 2013;8(10):e77299. doi:10.1371/journal.pone.0077299
35. Fu B, Dou X, Zou M, et al. Anticancer effects of amlodipine alone or in combination with gefitinib in non-small cell lung cancer. Front Pharmacol. 2022;13:902305. doi:10.3389/fphar.2022.902305
36. Ayoub NM, Al-Shami KM, Alqudah MA, et al. Crizotinib, a MET inhibitor, inhibits growth, migration, and invasion of breast cancer cells in vitro and synergizes with chemotherapeutic agents. Onco Targets Ther. 2017;10:4869–4883.
37. Semreen AM, Alsoud LO, El-Huneidi W, et al. Metabolomics analysis revealed significant metabolic changes in brain cancer cells treated with paclitaxel and/or etoposide. Int J mol Sci. 2022;23(22):13940. doi:10.3390/ijms232213940
38. Zenati RA, Giddey AD, Al-Hroub HM, et al. Evaluation of two simultaneous metabolomic and proteomic extraction protocols assessed by ultra-high-performance liquid chromatography tandem mass spectrometry. International Journal of Molecular Sciences. 2023;24(2):1354. doi:10.3390/ijms24021354
39. Alqudah MAY, Mansour HT, Mhaidat N. Simvastatin enhances irinotecan-induced apoptosis in prostate cancer via inhibition of MCL-1. Saudi Pharm J. 2018;26(2):191–197. doi:10.1016/j.jsps.2017.12.012
40. Chen R, Zeng X, Zhang R, et al. Cav1.3 channel α1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers11This work was partially supported by grants from DoD PCRP program (W81XWH-09-1-0455) and KUMC Valk Foundation to Dr Benyi Li, and grants from China Natural Science Foundation to Dr Benyi Li (NSFC #81172427) and Dr Jun Yang (NSFC #81101927). This project was also supported by the “Chutian Scholar” program funded by Hubei Province of China dedicated to China Three Gorges University. J Urol Oncol. 2014;32(5):524–536.
41. Wu L, Lin W, Liao Q, et al. Calcium channel blocker nifedipine suppresses colorectal cancer progression and immune escape by preventing NFAT2 Nuclear Translocation. Cell Rep. 2020;33(4):108327. doi:10.1016/j.celrep.2020.108327
42. Yusein-Myashkova S, Stoykov I, Gospodinov A, et al. The repair capacity of lung cancer cell lines A549 and H1299 depends on HMGB1 expression level and the p53 status. J Biochem. 2016;160(1):37–47. doi:10.1093/jb/mvw012
43. Pan X, Li R, Guo H, et al. Dihydropyridine calcium channel blockers suppress the transcription of PD-L1 by Inhibiting the Activation of STAT1. Front Pharmacol. 2020;11:539261. doi:10.3389/fphar.2020.539261
44. Chiu LY, Ko JL, Lee YJ, et al. L-type calcium channel blockers reverse docetaxel and vincristine-induced multidrug resistance independent of ABCB1 expression in human lung cancer cell lines. Toxicol Lett. 2010;192(3):408–418. doi:10.1016/j.toxlet.2009.11.018
45. Boo H-J, Min H-Y, Jang H-J, et al. The tobacco-specific carcinogen-operated calcium channel promotes lung tumorigenesis via IGF2 exocytosis in lung epithelial cells. Nat Commun. 2016;7(1):12961. doi:10.1038/ncomms12961
46. Yoshida J, Ishibashi T, Yang M, et al. Amlodipine, a Ca2+ channel blocker, suppresses phosphorylation of epidermal growth factor receptor in human epidermoid carcinoma A431 cells. Life Sci. 2010;86(3–4):124–132. doi:10.1016/j.lfs.2009.11.014
47. Luanpitpong S, Talbott SJ, Rojanasakul Y, et al. Regulation of lung cancer cell migration and invasion by reactive oxygen species and caveolin-1. J Biol Chem. 2010;285(50):38832–38840. doi:10.1074/jbc.M110.124958
48. Chen YC, Chen JH, Tsai CF, et al. Nicardipine inhibits breast cancer migration via Nrf2/HO-1 axis and matrix metalloproteinase-9 regulation. Front Pharmacol. 2021;12:710978. doi:10.3389/fphar.2021.710978
49. Tsai MF, Chang TH, Wu SG, et al. EGFR-L858R mutant enhances lung adenocarcinoma cell invasive ability and promotes malignant pleural effusion formation through activation of the CXCL12-CXCR4 pathway. Sci Rep. 2015;5(1):13574. doi:10.1038/srep13574
50. Chand T, Savitri B. Vitamin B3, niacin. Industrial Biotechnology of Vitamins, Biopigments, and Antioxidants. 2016;41–65.
51. Surjana D, Halliday GM, Damian DL. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J Nucleic Acids. 2010;2010(1). doi:10.4061/2010/157591
52. Thompson JF. 10 - Arginine Synthesis, Proline Synthesis, and Related Processes. In: Miflin BJ, editor. Amino Acids and Derivatives: Elsevier. 1980;375–402.
53. Wu G, Morris SMJBJ Jr. Arginine metabolism: nitric oxide and beyond. The Biochemical Journal. 1998;336(1):1–17. doi:10.1042/bj3360001
54. Zhang X, Thjc H. Amlodipine releases nitric oxide from canine coronary microvessels: an unexpected mechanism of action of a calcium channel–blocking agent. Circulation. 1998;97(6):576–580. doi:10.1161/01.cir.97.6.576
55. Conigrave AD, Simpson SJ. Calcium-Sensing Receptors Mediate Amino Acid Signals From the Liver to Islet Alpha Cells. J Clin Endocrinol Metab. 2023;108(9):e893–e894.
56. FBJTJon R. The biochemistry and physiology of nucleotides. The Journal of Nutrition. 1994;124(suppl_1):124S–127S. doi:10.1093/jn/124.suppl_1.124S
57. Fan TW, Bruntz RC, Yang Y, et al. De novo synthesis of serine and glycine fuels purine nucleotide biosynthesis in human lung cancer tissues. The Journal of Biological Chemistry. 2019;294(36):13464–13477. doi:10.1074/jbc.RA119.008743
58. Batova S, DeWever J, Godfraind T, et al. The calcium channel blocker amlodipine promotes the unclamping of eNOS from caveolin in endothelial cells. Cardiovascular Research. 2006;71(3):478–485. doi:10.1016/j.cardiores.2006.04.013
59. Moffatt BA, Ashihara H. Purine and pyrimidine nucleotide synthesis and metabolism. Arabidopsis Book. 2002;1:e0018.
60. Zhang Y, Morar M, Ealick SEJC, et al. Structural biology of the purine biosynthetic pathway. Cellular and Molecular Life Sciences: CMLS. 2008;65(23):3699–3724. doi:10.1007/s00018-008-8295-8
61. Lu M, Zhou L, Stanley WC, et al. Role of the malate–aspartate shuttle on the metabolic response to myocardial ischemia. Journal of Theoretical Biology. 2008;254(2):466–475. doi:10.1016/j.jtbi.2008.05.033
62. Borst P. The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life. 2020;72(11):2241–2259.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Clinical Utility and Application of Liquid Biopsy Genotyping in Lung Cancer: A Comprehensive Review
Nigro MC, Marchese PV, Deiana C, Casadio C, Galvani L, Di Federico A, De Giglio A
Lung Cancer: Targets and Therapy 2023, 14:11-25
Published Date: 3 February 2023