")
Back to Journals » Journal of Pain Research » Volume 18
Myeloid Differentiation Primary Response Protein 88: An Important Therapeutic Target for Chronic Pain
Authors Liang H , Fu L , Li Z, Liu Z
Received 8 October 2024
Accepted for publication 25 February 2025
Published 4 March 2025 Volume 2025:18 Pages 1061—1069
DOI https://doi.org/10.2147/JPR.S487685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Hanlin Liang,1 Linbin Fu,1 Zheng Li,1,2,* Zhiheng Liu1,*
1Department of Anesthesiology, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital, Shenzhen, People’s Republic of China; 2Department of Anesthesiology, Department of Anesthesiology and Pain Medicine, Hubei Key Laboratory of Geriatric Anesthesia and Perioperative Brain Health, and Wuhan Clinical Research Center for Geriatric Anesthesia, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zheng Li, Email [email protected] Zhiheng Liu, Email [email protected]
Abstract: Chronic pain is a major cause of suffering. This interferes with daily functioning and is often accompanied by distress. However, current therapeutic strategies for chronic pain are unsatisfactory because of poor understanding of its mechanisms. Therefore, more comprehensive therapeutic targets must be identified to improve the quality of life of these patients. Myeloid differentiation primary response protein 88 (MyD88) is an adaptor protein of the toll-like receptor (TLR) and interleukin-1 receptor (IL-1R) families. Recently, overexpression of MyD88 in the spinal and dorsal root ganglia was observed in multiple pain models, which also revealed that MyD88 plays an important role in the development and maintenance of chronic pain. In this review, we summarized the roles and mechanisms of MyD88 in the progression of different pain models, including chemotherapy-induced peripheral neuropathy (CIPN), diabetic neuropathic pain (DNP), spinal nerve ligation (SNL), chronic constriction injury (CCI), spinal cord injury (SCI) and inflammatory pain.
Keywords: MyD88, chronic pain, neuropathic pain, inflammatory pain
Introduction
Physiological pain plays an important role in the protection against nociceptive stimulation. However, chronic pain compromises quality of life. Major pathological phenomena of chronic pain include allodynia (pain due to a stimulus that does not normally provoke pain), hyperalgesia (increased pain from a stimulus that normally provokes pain) and spontaneous pain (pain felt without apparent external stimulus).1 Chronic pain has multiple mechanisms that can exacerbate and maintain pain, including central sensitization and peripheral sensitization.2,3 Several studies had illustrated that many pathological processes are characterized by chronic pain, such as chemotherapy-induced peripheral neuropathy (CIPN), diabetic neuropathic pain (DNP), spinal nerve ligation (SNL), chronic constriction injury (CCI), spinal cord injury (SCI) and inflammatory pain. However, the mechanisms identified by previous researchers in these pain models have not achieved good clinical conversion. Therefore, there is an urgent need to understand the mechanisms underlying the development and maintenance of pain during therapeutic treatment.
Damage to the nervous system causes the release of cytokines, chemokines and pro-inflammatory mediators, which activate resident immune and glial cells and attract circulating leukocytes to the site of injury and throughout the neural pain axis (including the dorsal root ganglia, dorsal horn spinal cord and supraspinal brain areas). A lot of immune cells participate in neuroinflammation including neutrophils, macrophages, dendritic cells, mast cells, T cells, antibody-producing B cells and microglia. These cells release inflammatory mediators, such as tumour necrosis factor, diverse interleukins, reactive oxygen and nitrogen species, bradykinin, growth factors, and prostaglandins, which increase neuronal excitability and suppress inhibitory pathways. The result of these changes is sensitization of the somatosensory signaling pathway and development of neuropathic pain symptoms.4
Myeloid differentiation primary response protein 88 (MyD88) was discovered in 1990 and was upregulated in the murine leukemic myeloblast cell line M1D+ during IL-6-induced cellular differentiation.5 MyD88 is mainly composed of an N-terminal death domain (DD), an intermediate domain (INT), and a C-terminal toll-interleukin-1 receptor (TIR) domain. Moreover, the INT domain of MyD88 links DD and TIR.6 As downstream of the toll-like receptor (TLR) and interleukin-1 (IL-1) receptor family members, MyD88 is a typical adaptor in the inflammatory signaling pathway. Activation of IRAK family kinases leads to a variety of functional outputs, including the activation of nuclear factor-kappa B (NFkB), mitogen-activated protein kinases, and activator protein 1, making MyD88 a central node of inflammatory pathways. TLR2 and TLR4 recruit MyD88 indirectly by bridging the adapter MAL (TIR-domain-containing adaptor protein), whereas other TLRs recruit it directly.7,8 MyD88 plays a central role in connecting the IL-1 receptor (IL-1R) or TLR family members with IL-1R-associated kinase (IRAK). Subsequently, it leads to the activation of nuclear factor-kappa B (NF-κB), activator protein-1 (AP-1), and interferon regulatory factors (IRFs) (Figure 1).9 MyD88 signaling can lead to distinct outputs depending on the context, usually leading to pro-inflammatory cytokine or type I IFN production. Distinct pathways downstream of IRAK family members regulate these outputs, and the outcome of signaling can be influenced by the cell type and location of signal initiation.6 Recently, increasing evidence has been reported on the role of MyD88 in pain processing. A deeper understanding of the mechanism of MyD88 in different pain models will pave the way for future studies of MyD88 and its role in weakening or regulating pain. Therefore, the role of MyD88 in different pain models is worth investigating.
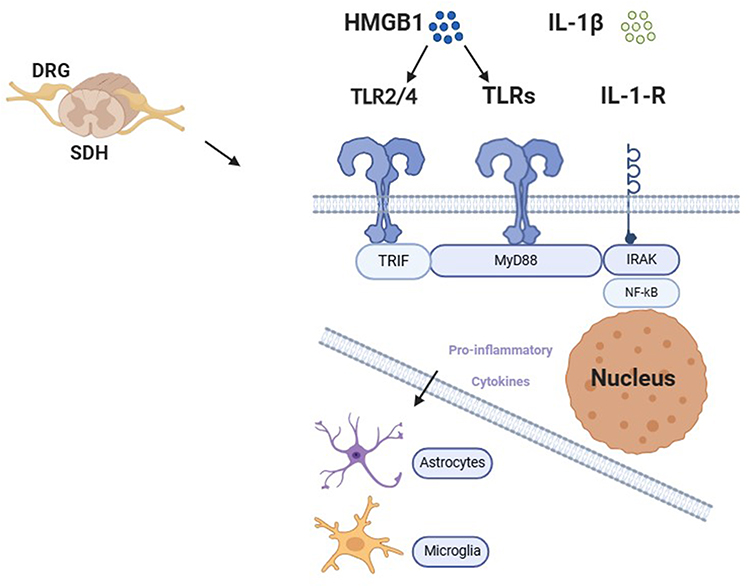 |
Figure 1 Schematic illustration demonstrates MyD88 signaling pathways of chronic pain. The binding of HMGB1 and IL-1β to their receptors (TLR2/4 and IL-1R, respectively) activates MyD88 in the DRG and SDH, which phosphorylate NF-κB. Phosphorylated NF-κB subsequently enter the nucleus to regulate the expression of proinflammation cytokine. Which regulates the expression of certain cytokines and activates glial cells. All these signaling events consequently result in central and peripheral sensitizations that produce chronic pain. |
MyD88 and Chronic Peripheral Neuropathic Pain
Chronic peripheral neuropathic pain is chronic pain result from a lesion or disease of the peripheral somatosensory nervous system.10 Population prevalence of neuropathic pain is estimated between 6.9% and 10%.11 According to the different causes that affect the peripheral or central nervous system, neuropathic pain can be divided into peripheral neuropathic pain and chronic central neuropathic pain.10 Painful peripheral neuropathies, post-herpetic neuralgia and traumatic nerve injury are classical etiologies of peripheral neuropathic pain.12 Insights into the mechanism of how MyD88 acts in the occurrence and development of neuropathic pain will be a new therapeutic strategy.
MyD88 and Chemotherapy-Induced Peripheral Neuropathy
Chemotherapy-induced peripheral neuropathy (CIPN) is the most common adverse effect associated with chemotherapy. The prevalence rate can reach 30–40% after chemotherapy.13 CIPN is characterized by pungent pain, numbness, severe burning pain, and electric shock-like pain.14 These symptoms after paclitaxel treatment can be explained by ectopic spontaneous activity (SA) in distal nerve endings15 and primary sensory neurons.16
At present, the mechanism causing CIPN mainly focuses on nuclear and mitochondrial DNA damage, destabilization of microtubule polymers and stabilization of microtubule polymers.17 Therefore, it is worth exploring the role of MyD88 in CIPN.
In a rat model of paclitaxel-induced peripheral neuropathy, the expression of MyD88 in dorsal root ganglion (DRG) reached the peak at days 3 and 7 and decreased gradually at days 14 and 28.18 Compared to wild-type (WT) mice, MyD88-conditional knockout (CKO) mice reduced paw withdrawal frequency at days 3, 7, and 10, with no difference at days 14, 21, or 28 after paclitaxel treatment. Furthermore, Li et al19 found that intrathecal administration of MyD88 homodimerization inhibitory peptide (MIP) significantly increased the paw withdrawal threshold by one hour and reached its maximal effect by 3 h in a model of paclitaxel-induced peripheral neuropathic pain. Park et al20 revealed that MyD88−/− mice treated with cisplatin had significantly increased withdrawal thresholds on days 3, 10, and 17. The overall trend of paw withdrawal threshold increased and returned to baseline after day 17. Likewise, Woller et al21 MyD88−/− male mice showed slightly reduced tactile allodynia during cisplatin administration, which did not change after discontinuing cisplatin treatment.21 Overall, the results listed above indicated the important role of MyD88 in the development of paclitaxel- or cisplatin-induced peripheral neuropathic pain.
A study found that MyD88 is required for toll-like receptor-4 signal transduction,7 and Illias et al22 found that cotreatment with the TLR4 antagonist lipopolysaccharide derived from Rhodobacter sphaeroides (LPS-RS) could reduce the increased expression of MyD88 in the DRG and prevent the development of mechanical hypersensitivity induced by oxaliplatin treatment. Similarly, Chen et al23 revealed that TLR4 and MyD88 levels were increased in L4-6 DRGs after intraperitoneal paclitaxel administration in a rat model. Regular administration of Shaoyao Gancao decoction (SGD) reversed the overexpression of TLR4 and MyD88, indicating a protective role of SGD in relieving paclitaxel-induced peripheral neuropathy by inhibiting the high expression of TLR4-MyD88 signaling. Furthermore, Li et al24 also noted that electroacupuncture (EA) alleviated pain hypersensitivity by suppressing TLR4 and its downstream signaling molecule MyD88 overexpression in L4-L6 DRGs neuron in a rat model of paclitaxel-induced neuropathic pain. These results indicate that TLR4 can regulate the expression of MyD88 in the DRG, and TLR4-MyD88 signaling play an important role in the development of paclitaxel- or cisplatin-induced peripheral neuropathic pain (Figure 2).
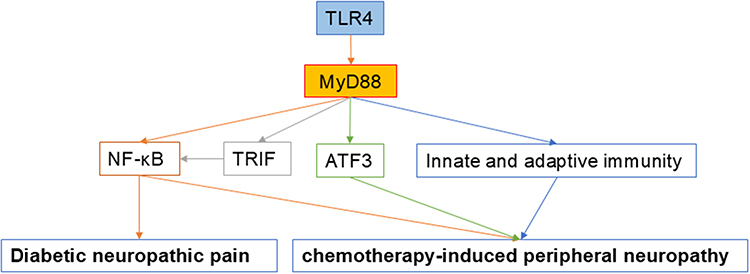 |
Figure 2 Schematic representation of the potential mechanism by which MyD88 are involved in diabetic neuropathic pain and chemotherapy-induced peripheral neuropathy. Abbreviations: TLR, toll-like receptor; MyD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor-kappa B; TRIF, TIR domain-containing adaptor inducing IFN-β; ATF3, activating transcription factor 3. |
MyD88 and Diabetic Neuropathic Pain
Diabetic neuropathic pain (DNP) can also be described as a painful numbness, burning, or a stabbing sensation from distal to proximal, known as stocking-glove neuropathy.25 At present, controlling blood sugar is a universally accepted treatment strategy that prevents the development of DNP. First-line drugs for DNP relief in clinical settings include anticonvulsants (pregabalin and gabapentin), antidepressants, and opioids.26 However, pain relief remains unsatisfactory for many patients.
Animal treatment with streptozotocin (STZ) intraperitoneal injection is one common DNP model, the blood glucose of rats is larger than 16.7 mmol/L 3 days later and the mechanical withdrawal threshold and thermal withdrawal latency all decreased in rats after 3 days after STZ injection.27 The results of immunohistochemistry and Western blot showed that the expression of TLR4, MyD88, and NF-κB were upregulated in spinal dorsal horn of DNP model.28 What’s more, the activated microglia and astrocytes promoting the release of pro-inflammatory cytokines to induce and maintain DNP. Intraperitoneal injection of duloxetine in DNP rats could alleviate the behavioral outcomes and decrease the expression of TLR4, MyD88, and NF-κB. However, the author did not prove that whether modulating MyD88 by duloxetine directly. This result indicated the potential role of MyD88 in the development of DNP.
In another study, Liu et al29 found that intrathecal injection of MyD88 homodimerization inhibitory peptide (MIP) dramatically increased paw withdrawal threshold (PWT) and paw withdrawal thermal latency (PWTL) in DNP rats. Moreover, this study found that intrathecal injection of baclofen (a GABAB receptor agonist) can also increase PWT and PWTL in DNP rats, whereas saclofen (a GABAB receptor antagonist) had the opposite effect on behavior compared to the DNP group. Furthermore, baclofen downregulated the expression of TLR4, MyD88, and NF-κB in the DNP model. These results illustrate that activation of GABAB receptors in the spinal cord downregulated the expression of factors in the TLR4/Myd88/NF-κB signaling pathway and alleviated neuronal inflammation, which is closely related to the analgesic effects of GABAB receptor agonists (Figure 2).
The TLR4/Myd88/NF-κB signaling pathway and downstream inflammatory factors, including IL-1 and TNF-α, play a key role in the process of neuropathic pain.30,31 TLR is G protein-coupled receptors and can bind to the HMGB1 in cytoplasm, which expression level is rised in diabetes.32,33
MyD88 and Chronic Constriction Injury (CCI)-Induced Neuropathic Pain
In a rat model of CCI, immunohistochemistry assay showed that MyD88 increased in DRG neuron 10 days after CCI, and intrathecal injection MIP from postoperative day (POD) 3 to POD 5 alleviated neuropathic pain and lasted from POD 7 to POD 14.34 Moreover, Liu et al35 found that the expression of MyD88 increased in spinal dorsal horn (SDH) and DRG. The levels of phosphorylated NF-κB p65 and total NF-κB p65 significantly increased in the SDH after CCI. Moreover, upregulation was observed in the expression of extracellular signal-regulated protein kinase 1/2 (ERK1 and ERK2), a pathway that participates in TLR-mediated pro-inflammatory responses. Intrathecal injection of the MyD88 inhibitor MIP suppressed CCI-induced pain and decreased the expression of MyD88 in the DRG and SDH. Moreover, MIP downregulated the phospho-NF-κB p65 and ERK protein levels. In addition, cotreatment with frankincense and myrrh alleviated thermal and mechanical hypersensitivity in a CCI mouse model. Western blotting results showed a reduction in TLR4, MyD88, and p-p65 expression after frankincense and myrrh treatment.34 These results confirm that the MyD88/NF-κB and MyD88/ERK signaling pathways are activated within days after CCI.35
Intrathecal injection of exogenous recombinant TNF-α-stimulated gene 6 (TSG-6) effectively attenuated CCI-induced neuropathic pain. TSG-6 protein, released by bone marrow mesenchymal stem cells, alleviated neuropathic pain via inhibiting the TLR2/MyD88/NF-ΚB signaling and downregulated the expression of pro-inflammatory cytokines, such as IL-1β, interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) of the spinal microglia in CCI rats.36 Furthermore, Wang et al37 explained that botulinum toxin type A (BTX-A) alleviates CCI-induced neuropathic pain via decreasing the secretion of pro-inflammatory factors from microglia by inhibition of TLR2/MyD88 signaling. In a mouse model of chronic constriction injury of the distal infraorbital nerve (dIoN-CCI), Chen et al38 illustrated that dIonN-CCI surgery remarkably increased TLR2 and MyD88 expression in the ipsilateral trigeminal nucleus caudalis (TNC). And TLR2 deficiency mice decreased bilateral mechanical pain and the expression of MyD88 in the TNC of dIonN-CCI mice. TLR2/MyD88 signaling participates in the development of CCI-induced neuropathic pain.
The nucleotide-binding domain-like receptor protein 3 (NLRP3) inflammasome is a cytoplasmic multi-protein complex. Selective inhibition of NLRP3 alleviates neuropathic pain.39 Moreover, Huang et al indicated that miR-185-5p alleviated CCI-induced neuropathic pain by suppressing NLRP3 inflammasome through targeting MyD88 and CXCR4 in BV2 microglia.40 However, the specific function of MyD88 and NLRP3 in NP were unproven. These suggested the regulatory role of MyD88 on NLRP3 inflammasome activation. Together, these results confirmed that MyD88 in the DRG and SDH promotes the development of CCI-induced neuropathic pain (Figure 3).
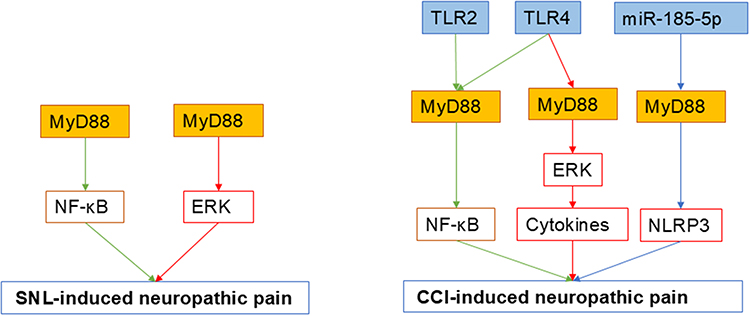 |
Figure 3 Schematic representation of the potential mechanism by which MyD88 are involved in SNI-induced neuropathic pain and CCI-induced neuropathic pain. Abbreviations: MyD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor-kappa B; ERK, extracellular signal-regulated protein kinase; SNL, spinal nerve ligation; TLR, toll-like receptor; NLRP3, nucleotide-binding domain-like receptor protein 3; CCI, chronic constriction injury. |
MyD88 and Spinal Nerve Ligation (SNL) and Spared Nerve Injury (SNI)-Induced Pain
In a rat model of SNI, Chen et al41 reported that MyD88 and its downstream pro-inflammatory pathways were highly expressed in the ligated ipsilateral spinal dorsal horn (iSDH) compared to the control group. Downgrading the expression of these pro-inflammatory pathways regulates neuroinflammation in the central nervous system and relieves neuropathic pain induced by SNI. In addition, researchers found that the acetylation level of MyD88 was increased after treatment with ACY-1215 (HDAC6 inhibitor), accompanied by an improvement in neuropathic pain induced by SNI. This suggests a role for MyD88 acetylation in the development of SNI-induced neuropathic pain via pro-inflammatory pathways. Similarly, mice deficient in MyD88 showed an approximately 50% reduction in withdrawal thresholds and reduced ipsilateral Iba-1, a marker of active microglia, after L5 spinal nerve ligation.42 These implied an important role in SNL and SNI-induced neuropathic pain (Figure 3).
MyD88 and Chronic Central Neuropathic Pain
Chronic central neuropathic pain is chronic pain resulting from a lesion or disease of the central somatosensory nervous system. The most common etiologies of central neuropathic pain are spinal cord injury (SCI), brain injury and stroke.10
MyD88 and Spinal Cord Injury (SCI)-Induced Neuropathic Pain
SCI is a neurogenic damage and pathological state that causes major motor, sensory, and autonomic dysfunctions43 with a prevalence rate of neuropathic pain were 53% after spinal cord injury.44 Recently, mountains of studies have highlighted the important role of MyD88 in SCI-induced neuropathic pain.
In an adult male rat model of SCI-induced neuropathic pain, interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), downstream of TLR4/MyD88/NF-κB, were found to participate in the inflammatory pathway and development of neuropathic pain. Treatment with TAK-242, a TLR4 inhibitor, suppressed the expression of TLR4 in microglia and of these two inflammatory factors.45 This result suggests that TLR4/MyD88/NF-κB regulates the inflammatory pathway, resulting in SCI-induced neuropathic pain. In addition, Wang et al46 found that salvianolic acid B (SalB) inhibited neuropathic pain by suppressing the TLR4/MyD88 pathway and downstream TNF-α and substance P, indicating that MyD88-mediated inflammation could participate in SCI-induced neuropathic pain.
In addition, Fan et al47 revealed that M1 microglia/macrophages could induce necroptosis of astrocytes partially by TLR/MyD88 signaling and that inhibiting astrocytic necroptosis could be beneficial for preventing secondary SCI. In another study, Rong et al48 illustrated that disturbance of the gut microbiota activates the TLR4/MyD88 pathway, which could promote apoptosis and cause nerve damage by regulating the expression of pro-inflammatory factors. These results indicated that MyD88 plays an important role in SCI development through multiple mechanisms (Figure 4).
 |
Figure 4 Schematic representation of the potential mechanism by which MyD88 are involved in SCI-induced neuropathic pain and inflammatory pain. Abbreviations: TLR, toll-like receptor; MyD88, myeloid differentiation primary response protein 88; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; SCI, spinal cord injury; HMGB1, high mobility group box 1; NF-κB, nuclear factor-kappa B. |
MyD88 and Inflammatory Pain
Inflammation pain often results from the high excitability of peripheral nociceptive sensory fibers to inflammatory mediators.49 Persistent contact with pro-inflammatory mediators sensitizes peripheral pain-sensing neurons, leading to chronic inflammatory pain. Skin, joints, and gut are particularly susceptible to the development of inflammatory pain.50 The C-terminal tail region of high mobility group box 1 (HMGB1) binding to TLR5 initiates the activation of NF-kB signaling pathway in a MyD88-dependent manner, leading pro-inflammatory cytokine production and pain enhancement in vivo.51
Endometriosis is an estrogen-dependent disease with endometrial tissue occurring outside the uterine cavity.52 Its estimated affect 10% of reproductive age women.53 In a rat model of endometriosis, Su et al54 revealed that HMGB1-TLR4-MyD88 signaling pathway in the DRG and SDH might be involved in endometriosis-related hyperpathia. Intrathecal application of LPS-RS-Ultra (LRU) and MIP alleviated mechanical pain by blocking TLR4 and MyD88 expression. Daily oral administration of an inhibitor of IL-1R-associated kinase 4 (IRAK4), a downstream signaling molecule of MyD88, significantly inhibits epithelial cell proliferation and cystic lesion growth. Endometriotic lesion volume was almost completely suppressed in MyD88−/− mice.52
Intra-articular (ia) injection of lipopolysaccharide (LPS) can induce inflammatory hyperalgesia in joints. The mechanical nociceptive threshold was reduced in a dose- and time-dependent manner.55 And LPS-induced joint mechanical hyperalgesia was abolished in TLR4−/−and MyD88−/− mice, respectively.55 This result indicated that the TLR4/MyD88 signaling pathway is involved in the mechanism of inflammatory joint pain. Recently, Cai et al56 illustrated that the overexpression of fat mass and obesity-associated gene (FTO) could reduce cell apoptosis and inhibit inflammation in the synovial fluid. Inhibitory effect of FTO on LPS-induced cell injury through the miR-515-5p/TLR4/MyD88/NF-κB axis. In another rabbit model of knee osteoarthritis (KOA), Xu et al57 indicated that Zhuifeng Tonggu (ZFTG) capsules reduced chondrocyte inflammation and apoptosis by suppressing the expression of TLR2, TLR4, and MyD88. Moreover, with the modulation of miR-665 and circRNF121, MyD88 expression is altered in an OA model.58
In addition, in a model of inflammatory pain treated with LPS paw injection, WT mice produced mechanical hyperalgesia while this performance was absent in TLR4 mutant and MyD88 null mice.59 Bexarotene was found to play anti-inflammatory and analgesic roles in systemic inflammatory response model triggered by LPS intraperitoneal injection by suppression of TLR4/MyD88/TAK1/NF-κB/COX-2 pathway.60 Qin et al61 found that the level of MyD88 mRNA and protein were increased in a dose- and time-dependent manner in intervertebral disc (IVD) degeneration model, in which IVD nucleus pulposus cells treated with LPS. These results from various models suggest that the activation of the MyD88-dependent signaling pathway plays an important role in inflammatory pain (Figure 4).
Conclusion
By reviewing current evidence, we discuss the relationship between MyD88 and chronic pain. These studies provide robust evidence that MyD88 plays a vital role in the development of chronic pain such as CIPN, DNP, neuropathic pain, inflammatory pain, and MIP by various mechanisms. Treatment with the MyD88 homodimerization inhibitory peptide MIP attenuated mechanical allodynia and thermal hyperalgesia caused by chronic pain, indicating that inhibitors of MyD88 may be beneficial therapeutic tools for chronic pain. Moreover, the development of upstream of MyD88 signaling pathway (such as TLR4) inhibitors/modulators also show effective therapeutic effects for the treatment of chronic pain. These findings provide a convincing theoretical basis for the development of drugs targeting chronic pain. However, improved MyD88 inhibitors with fewer side effects should be explored in the future.
Funding
This study was supported by the National Natural Science Foundation of China (no. 82101308), Guangdong Basic and Applied Basic Research Nature Science Foundation (2023A1515011649), and the Sanming Project of Medicine in Shenzhen (no. SZSM20221107), and Shenzhen High-level Hospital Construction Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen S-P, Zhou Y-Q, Liu D-Q, et al. PI3K/Akt pathway: a potential therapeutic target for chronic pain. Curr Pharm Des. 2017;23(12):1860–1868. doi:10.2174/1381612823666170210150147
2. Walk D, Poliak-Tunis M. Chronic pain management. Med Clin North Am. 2016;100(1):1–16. doi:10.1016/j.mcna.2015.09.005
3. Li C, Kim HJ, Back SK, Na HS. Common and discrete mechanisms underlying chronic pain and itch: peripheral and central sensitization. Pflügers Archiv Eur J Physiol. 2021;473(10):1603–1615. doi:10.1007/s00424-021-02599-y
4. Fiore NT, Debs SR, Hayes JP, Duffy SS, Moalem-Taylor G. Pain-resolving immune mechanisms in neuropathic pain. Nat Rev Neurol. 2023;19(4):199–220. doi:10.1038/s41582-023-00777-3
5. Lord KA, Hoffman-Liebermann B, Liebermann DA. Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene. 1990;5:1095–1097.
6. Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000Prime Rep. 2014;6. doi:10.12703/P6-97
7. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413(6851):78–83. doi:10.1038/35092578
8. Santos-Sierra S, Deshmukh SD, Kalnitski J, et al. Mal connects TLR2 to PI3Kinase activation and phagocyte polarization. EMBO J. 2009;28(14):2018–2027. doi:10.1038/emboj.2009.158
9. Chen L, Zheng L, Chen P, Liang G. Myeloid differentiation primary response protein 88 (MyD88): the central hub of TLR/IL-1R signaling. J Med Chem. 2020;63(22):13316–13329. doi:10.1021/acs.jmedchem.0c00884
10. Scholz J, Finnerup NB, Attal N, et al. The IASP classification of chronic pain for ICD-11: chronic neuropathic pain. Pain. 2019;160(1):53–59. doi:10.1097/j.pain.0000000000001365
11. van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain. 2014;155(4):654–662. doi:10.1016/j.pain.2013.11.013
12. Bouhassira D. Neuropathic pain: definition, assessment and epidemiology. Rev Neurol. 2019;175(1–2):16–25. doi:10.1016/j.neurol.2018.09.016
13. Pike CT, Birnbaum HG, Muehlenbein CE, Pohl GM, Natale RB. Healthcare costs and workloss burden of patients with chemotherapy-associated peripheral neuropathy in breast, ovarian, head and neck, and nonsmall cell lung cancer. Chemother Res Pract. 2012;2012:1–10. doi:10.1155/2012/913848
14. Zajączkowska R, Kocot-Kępska M, Leppert W, Wrzosek A, Mika J, Wordliczek J. Mechanisms of chemotherapy-induced peripheral neuropathy. Int J Mol Sci. 2019;20(6):1451. doi:10.3390/ijms20061451
15. Xiao WH, Bennett GJ. Chemotherapy-evoked neuropathic pain: abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-l-carnitine. Pain. 2008;135(3):262–270. doi:10.1016/j.pain.2007.06.001
16. Zhang H, Dougherty PM. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology. 2014;120(6):1463–1475. doi:10.1097/ALN.0000000000000176
17. Staff NP, Grisold A, Grisold W, Windebank AJ. Chemotherapy-induced peripheral neuropathy: a current review. Ann Neurol. 2017;81(6):772–781. doi:10.1002/ana.24951
18. Liu XJ, Zhang Y, Liu T, et al. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res. 2014;24(11):1374–1377. doi:10.1038/cr.2014.106
19. Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2014;15(7):712–725. doi:10.1016/j.jpain.2014.04.001
20. Park HJ, Stokes JA, Corr M, Yaksh TL. Toll-like receptor signaling regulates cisplatin-induced mechanical allodynia in mice. Cancer Chemother Pharmacol. 2013;73(1):25–34. doi:10.1007/s00280-013-2304-9
21. Woller SA, Corr M, Yaksh TL. Differences in cisplatin-induced mechanical allodynia in male and female mice. Eur J Pain. 2015;19(10):1476–1485. doi:10.1002/ejp.679
22. Illias AM, Yu KJ, Hwang SH, et al. Dorsal root ganglion toll-like receptor 4 signaling contributes to oxaliplatin-induced peripheral neuropathy. Pain. 2022;163(5):923–935. doi:10.1097/j.pain.0000000000002454
23. Chen Y, Lu R, Wang Y, Gan P. Shaoyao gancao decoction ameliorates paclitaxel-induced peripheral neuropathy via suppressing TRPV1 and TLR4 signaling expression in rats. Drug Des Devel Ther. 2022;16:2067–2081. doi:10.2147/DDDT.S357638
24. Li Y, Yin C, Li X, et al. Electroacupuncture alleviates paclitaxel-induced peripheral neuropathic pain in rats via suppressing TLR4 signaling and TRPV1 upregulation in sensory neurons. Int J Mol Sci. 2019;20(23):5917. doi:10.3390/ijms20235917
25. Feldman EL, Nave K-A, Jensen TS, Bennett DLH. New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron. 2017;93(6):1296–1313. doi:10.1016/j.neuron.2017.02.005
26. Schreiber AK. Diabetic neuropathic pain: physiopathology and treatment. World J Diabetes. 2015;6(3):432. doi:10.4239/wjd.v6.i3.432
27. Furman BL. Streptozotocin-induced diabetic models in mice and rats. Curr Protoc. 2021;1(4):e78. doi:10.1002/cpz1.78
28. Zhou DM, Zhuang Y, Chen WJ, Li W, Miao B. Effects of duloxetine on the toll-like receptor 4 signaling pathway in spinal dorsal horn in a rat model of diabetic neuropathic pain. Pain Med. 2018;19(3):580–588. doi:10.1093/pm/pnx125
29. Liu P, Yuan HB, Zhao S, et al. Activation of GABA(B) receptor suppresses diabetic neuropathic pain through toll-like receptor 4 signaling pathway in the spinal dorsal horn. Mediators Inflamm. 2018;2018:6016272. doi:10.1155/2018/6016272
30. Wu FX, Bian JJ, Miao XR, et al. Intrathecal siRNA against toll-like receptor 4 reduces nociception in a rat model of neuropathic pain. Int J Med Sci. 2010;7(5):251–259. doi:10.7150/ijms.7.251
31. Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59(2):242–255. doi:10.1002/glia.21094
32. Wang Y, Zhong J, Zhang X, et al. The role of HMGB1 in the pathogenesis of type 2 diabetes. J Diabetes Res. 2016;2016:2543268. doi:10.1155/2016/2543268
33. Magna M, Pisetsky DS. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med. 2014;20(1):138–146. doi:10.2119/molmed.2013.00164
34. Liao Y, Guo C, Wen A, et al. Frankincense-Myrrh treatment alleviates neuropathic pain via the inhibition of neuroglia activation mediated by the TLR4/MyD88 pathway and TRPV1 signaling. Phytomedicine. 2023;108:154540. doi:10.1016/j.phymed.2022.154540
35. Liu F, Wang Z, Qiu Y, et al. Suppression of MyD88-dependent signaling alleviates neuropathic pain induced by peripheral nerve injury in the rat. J Neuroinflammation. 2017;14(1). doi:10.1186/s12974-017-0822-9
36. Yang H, Wu L, Deng H, et al. Anti-inflammatory protein TSG-6 secreted by bone marrow mesenchymal stem cells attenuates neuropathic pain by inhibiting the TLR2/MyD88/NF-kappaB signaling pathway in spinal microglia. J Neuroinflammation. 2020;17(1):154. doi:10.1186/s12974-020-1731-x
37. Wang X, Tian S, Wang H, et al. Botulinum toxin type A alleviates neuropathic pain and suppresses inflammatory cytokines release from microglia by targeting TLR2/MyD88 and SNAP23. Cell Biosci. 2020;10(1):141. doi:10.1186/s13578-020-00501-4
38. Chen WJ, Niu JQ, Chen YT, et al. Unilateral facial injection of Botulinum neurotoxin A attenuates bilateral trigeminal neuropathic pain and anxiety-like behaviors through inhibition of TLR2-mediated neuroinflammation in mice. J Headache Pain. 2021;22(1):38. doi:10.1186/s10194-021-01254-2
39. Zhang A, Wang K, Ding L, et al. Bay11-7082 attenuates neuropathic pain via inhibition of nuclear factor-kappa B and nucleotide-binding domain-like receptor protein 3 inflammasome activation in dorsal root ganglions in a rat model of lumbar disc herniation. J Pain Res. 2017;10:375–382. doi:10.2147/JPR.S119820
40. Huang A, Ji L, Huang Y, Yu Q, Li Y. miR-185-5p alleviates CCI-induced neuropathic pain by repressing NLRP3 inflammasome through dual targeting MyD88 and CXCR4. Int Immunopharmacol. 2022;104:108508. doi:10.1016/j.intimp.2021.108508
41. Chen C, Liu A, Lu Q, et al. HDAC6 inhibitor ACY-1215 improves neuropathic pain and its comorbidities in rats of peripheral nerve injury by regulating neuroinflammation. Chem Biol Interact. 2022;353:109803. doi:10.1016/j.cbi.2022.109803
42. Stokes JA, Cheung J, Eddinger K, Corr M, Yaksh TL. Toll-like receptor signaling adapter proteins govern spread of neuropathic pain and recovery following nerve injury in male mice. J Neuroinflammation. 2013;10:148.
43. Anjum A, Yazid MD, Fauzi Daud M, et al. Spinal cord injury: pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int J Mol Sci. 2020;21(20):7533. doi:10.3390/ijms21207533
44. Burke D, Fullen BM, Stokes D, Lennon O. Neuropathic pain prevalence following spinal cord injury: a systematic review and meta-analysis. Eur J Pain. 2017;21(1):29–44. doi:10.1002/ejp.905
45. Li Z, Bai H, Zhang R, et al. Systematic analysis of critical genes and pathways identified a signature of neuropathic pain after spinal cord injury. Eur J Neurosci. 2022;56(2):3991–4008. doi:10.1111/ejn.15693
46. Wang Y, Xu X, Hu P, Jia N, Ji S, Yuan H. Effect of toll-like receptor 4/Myeloid differentiation factor 88 inhibition by Salvianolic Acid B on neuropathic pain after spinal cord injury in mice. World Neurosurg. 2019;132:e529–e534. doi:10.1016/j.wneu.2019.08.086
47. Fan H, Zhang K, Shan L, et al. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol Neurodegener. 2016;11:14. doi:10.1186/s13024-016-0081-8
48. Rong Z, Huang Y, Cai H, et al. Gut microbiota disorders promote inflammation and aggravate spinal cord injury through the TLR4/MyD88 signaling pathway. Front Nutr. 2021;8:702659. doi:10.3389/fnut.2021.702659
49. Linley JE, Rose K, Ooi L, Gamper N. Understanding inflammatory pain: ion channels contributing to acute and chronic nociception. Pflügers Archiv Eur J Physiol. 2010;459(5):657–669. doi:10.1007/s00424-010-0784-6
50. Muley MM, Krustev E, McDougall JJ. Preclinical assessment of inflammatory pain. CNS Neurosci Ther. 2016;22(2):88–101. doi:10.1111/cns.12486
51. Das N, Dewan V, Grace Peter M, et al. HMGB1 activates proinflammatory signaling via TLR5 leading to allodynia. Cell Rep. 2016;17(4):1128–1140. doi:10.1016/j.celrep.2016.09.076
52. Kato T, Yasuda K, Matsushita K, et al. Interleukin-1/-33 signaling pathways as therapeutic targets for endometriosis. Front Immunol. 2019;10:2021. doi:10.3389/fimmu.2019.02021
53. Zondervan KT, Longo DL, Becker CM, Missmer SA. Endometriosis. N Engl J Med. 2020;382(13):1244–1256. doi:10.1056/NEJMra1810764
54. Su W, Cui H, Wu D, et al. Suppression of TLR4-MyD88 signaling pathway attenuated chronic mechanical pain in a rat model of endometriosis. J Neuroinflammation. 2021;18(1):65. doi:10.1186/s12974-020-02066-y
55. Guerrero AT, Pinto LG, Cunha FQ, et al. Mechanisms underlying the hyperalgesic responses triggered by joint activation of TLR4. Pharmacol Rep. 2016;68(6):1293–1300. doi:10.1016/j.pharep.2016.08.006
56. Cai D, Zhang J, Yang J, Lv Q, Zhong C. Overexpression of FTO alleviates osteoarthritis by regulating the processing of miR-515-5p and the TLR4/MyD88/NF-kappaB axis. Int Immunopharmacol. 2023;114:109524. doi:10.1016/j.intimp.2022.109524
57. Xu X, Li N, Wu Y, et al. Zhuifeng tougu capsules inhibit the TLR4/MyD88/NF-kappaB signaling pathway and alleviate knee osteoarthritis: in vitro and in vivo experiments. Front Pharmacol. 2022;13:951860. doi:10.3389/fphar.2022.951860
58. Wang T, Hao Z, Liu C, et al. LEF1 mediates osteoarthritis progression through circRNF121/miR-665/MYD88 axis via NF-small ka, CyrillicB signaling pathway. Cell Death Dis. 2020;11(7):598. doi:10.1038/s41419-020-02769-3
59. Wang T, Calil IL, Zarpelon AC, et al. Lipopolysaccharide induces inflammatory hyperalgesia triggering a TLR4/MyD88-dependent cytokine cascade in the mice paw. PLoS One. 2014;9(3).
60. Senol SP, Temiz-Resitoglu M, Guden DS, Sari AN, Sahan-Firat S, Tunctan B. Suppression of TLR4/MyD88/TAK1/NF-kappaB/COX-2 signaling pathway in the central nervous system by bexarotene, a selective RXR agonist, prevents hyperalgesia in the lipopolysaccharide-induced pain mouse model. Neurochem Res. 2021;46(3):624–637. doi:10.1007/s11064-020-03197-7
61. Qin C, Zhang B, Zhang L, et al. MyD88-dependent Toll-like receptor 4 signal pathway in intervertebral disc degeneration. Exp Ther Med. 2016;12(2):611–618. doi:10.3892/etm.2016.3425
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.