")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 21
Obstacles to Early Diagnosis of Gaucher Disease
Authors Nishimura S, Ma C, Sidransky E, Ryan E
Received 24 October 2024
Accepted for publication 11 January 2025
Published 25 January 2025 Volume 2025:21 Pages 93—101
DOI https://doi.org/10.2147/TCRM.S388266
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Samantha Nishimura,* Charis Ma,* Ellen Sidransky, Emory Ryan
National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA
*These authors contributed equally to this work
Correspondence: Ellen Sidransky, National Human Genome Research Institute, National Institutes of Health, Building 35A -1E623 Convent Drive, MSC 3708, Bethesda, MD, 20892-3708, USA, Tel +1-301-451-0901, Fax +301-402-6438, Email [email protected]
Abstract: Gaucher disease (GD) is a rare lysosomal storage disorder resulting from a deficiency of the lysosomal enzyme glucocerebrosidase caused by biallelic variants in the GBA1 gene. Patients may present with a wide spectrum of disease manifestations, including hepatosplenomegaly, thrombocytopenia, bone manifestations, and in the case of GD types 2 and 3, neurodegeneration, cognitive delay, and/or oculomotor abnormalities. While there is no treatment for neuronopathic GD, non-neuronopathic manifestations can be efficiently managed with enzyme replacement therapy or substrate reduction therapy. However, many patients with GD experience a lengthy diagnostic odyssey, which can negatively affect their access to care and clinical outcomes. The cause of this diagnostic delay is multifaceted. Since genotype/phenotype correlations in GD are not always clear, it is difficult to predict the presence, severity, and onset of clinical manifestations. This heterogeneity, combined with the molecular complexity of the GBA1 locus, low disease prevalence, and limited knowledge of GD among providers serves as a barrier to early diagnosis of GD. In this review, we discuss such obstacles and challenges, considerations, and future steps toward improving the diagnostic journey for patients with GD.
Keywords: Gaucher disease, newborn screening, diagnosis
Introduction
Rare diseases are not rare. Collectively, there are 7,000–10,000 identified rare diseases that in total affect 1 in 10 people in the US,1 with a worldwide prevalence of 3.5–5.9%. This is similar to the incidence of Alzheimer disease in those over 65 (1 in 9), as well as the rate of diabetes in the US (1 in 10).2 However, less than 2.4% of rare diseases have a treatment,3 and physicians tend to be less aware of these than of drugs for more common disorders. Gaucher disease (GD) is a lysosomal storage disorder resulting from decreased glucocerebrosidase levels due to biallelic pathogenic variants in the gene GBA1, with an overall prevalence of 1 in 10,000–60,000 worldwide. GD manifests with a spectrum of features, with overlap between the three classically defined types: non-neuronopathic Gaucher disease type 1 (GD1, OMIM 230800), acute neuronopathic Gaucher disease type 2 (GD2, OMIM 230900), and chronic neuronopathic Gaucher disease type 3 (GD3, OMIM 231000).4–6 Patients may present at any time between birth to old age, with a myriad of manifestations including splenomegaly, bone pain, fractures, thrombocytopenia, anemia, fatigue, and in more severe forms, cognitive delay, seizures, scoliosis/kyphosis, and neurodegeneration. Due to the heterogenous nature of the disorder, relatively low prevalence, variation of presentation, and low provider familiarity, patients may experience lengthy diagnostic delays as well as extraneous testing and procedures.7,8 This delay may have long term sequelae, both for the patient and the caregivers,9,10 and earlier diagnosis is essential for improving patient outcomes. In this paper we explore some barriers to early diagnosis as well as current attempts to ameliorate them.
Barriers to Diagnosis
Molecular Complexity
Many factors, such as non-specific biomarkers with limited disease correlation and inaccurate genotyping, contribute to delays in diagnosing all three types of GD. Currently, the use of sequencing to identify GBA1 variants, coupled with evaluation of GCase activity, remains the diagnostic gold standard. However, technical intricacies in sequencing and measuring enzymatic activity can make diagnosis difficult. Obtaining an accurate GBA1 genotype can be challenging due to the existence of the highly homologous GBA1 pseudogene (GBAP1).11 Located only 16 kb downstream from the GBA1 gene, GBAP1 increases the risk for recombination and gene conversion events, resulting in complex, pathogenic recombinant alleles. These can be categorized into nonreciprocal events, when a portion of GBA1 is replaced by the homologous sequence in GBAP1, and reciprocal events, when a segment of GBA1 is fused with a segment of GBAP1 to establish a novel allele (Figure 1). Despite multiple decades of study, the gene regulation, expression, and structure at the GBA1/GBAP1 locus remains poorly understood, especially within different cell types.12,13 Prior to the availability of next generation sequencing (NGS), identification of GBA1 variants relied on the use of PCR-based genotyping panels with limited numbers of pathogenic variants, and Sanger sequencing, which requires avoiding specific primers that may lead to incorrect genotyping and failure to correctly diagnose GD.14 The use of selected panels can miss up to 15–20% of pathogenic variants.15 NGS can be a powerful tool to analyze large target regions, enabling the discovery of new, rare variants. However, despite its many advantages, WGS can introduce errors during library preparation and data analysis, for example, by failing to specifically amplify the functional gene.16,17 Both long and short read NGS often cannot reliably detect recombinant alleles. Recent efforts to enhance mapping accuracy include a software tool called Gauchian. This platform recognizes most reciprocal recombinations, but can miss rare gene conversion events.18 Further advancements are necessary to consistently ensure accurate GBA1 genotyping.
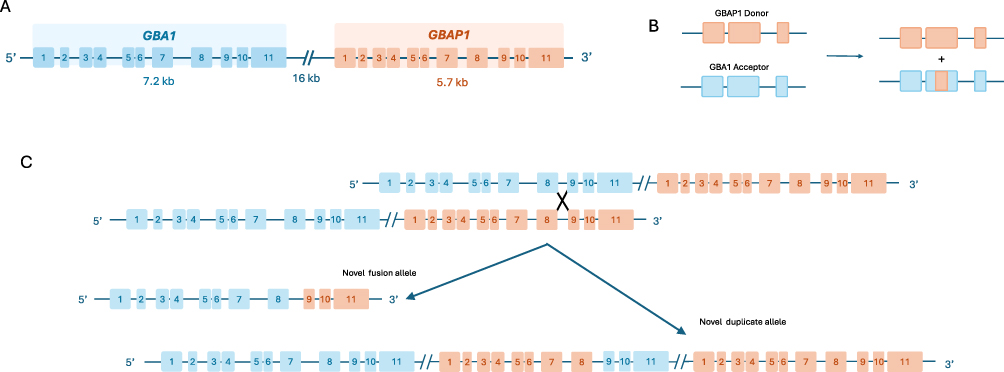 |
Figure 1 Recombination and gene conversion events between GBA1 and GBAP1. (A) The GBA1 gene has a highly homologous pseudogene, GBAP1, located ~16 kb downstream from the functional gene. (B) An example of an allelic gene conversion, a type of nonreciprocal recombination event where a portion of GBA1 is replaced by the homologous sequence in GBAP1. (C) An example of a reciprocal recombination event, where a crossover leads to a novel fusion allele and a novel duplicate allele.Adapted from Tayebi N, Stubblefield BK, Park JK, et al. Reciprocal and nonreciprocal recombination at the glucocerebrosidase gene region: implications for complexity in Gaucher disease. Am J Hum Genet. 2003;72(3):519–534. Copyright 2003, with permission from Elsevier”.11 |
There are also shortcomings when using biomarkers to screen for GD. Chitotriosidase was historically used to monitor disease progression in patients with GD but was found to be an unreliable diagnostic tool, as about 40% of the general population carries at least one CHIT1 variant, leading to absent chitotriosidase activity in 10%, and raising concerns for false negatives.19,20 Furthermore, chitotriosidase is a non-specific biomarker and can be elevated in other lysosomal storage disorders such as Niemann Pick disease21 and some gangliosidoses.22 Other biomarkers used such as CCL18,23 angiotensin-converting enzyme (ACE), and TRAP5b are also not specific for GD.24
Recently, glucosylsphingosine (lyso-Gb1) has emerged as a more promising GD-specific biomarker. Unlike previously identified biomarkers not directly involved in GD pathology, lyso-Gb1 is a direct metabolite of glucosylceramide. Elevated levels were first detected in the brains of patients with neuronopathic GD. Since then, plasma lyso-Gb1 levels have been found to correlate with genotype and disease burden, as well as clinical GD symptoms, including hepatosplenomegaly.20,25,26 Subsequently, plasma lyso-Gb1 has been evaluated as a useful tool to monitor disease severity and patients’ response to therapy. However, studies have demonstrated that lyso-Gb1 levels can fluctuate in patients, and there is not yet a consensus on a reliable cut-off level to distinguish patients with GD from carriers and healthy controls.27–29 It is also important to note that as a biomarker, lyso-Gb1 has limitations. An 8-year longitudinal study has found that lyso-Gb1 levels do not consistently correlate with GD severity, concluding that, contrary to previous study results, the relationship between the marker and disease progression is not linear.30 Another study also reported a patient affected with GD who did not have elevated levels of lyso-Gb1, raising a concern for false negative results.31 Further research is needed to identify specific lyso-Gb1 cut-off levels that can inform GD diagnosis and treatment timelines.
Clinical Ambiguity
Lengthy Diagnostic Odyssey
It is well established that GD presents with a variety of symptoms. The patient’s initial workup may be non-specific and include anemia, thrombocytopenia, hepatosplenomegaly, or lymphadenopathy, leading to an unclear clinical picture and difficult diagnosis.32 For example, hematological abnormalities and lymphadenopathy can be caused by various childhood leukemias, lymphomas, or viral pathogens. The signs and symptoms of GD can mimic those of rarer inborn errors of metabolism, other lysosomal storage disorders, inborn errors of immunity, and autoimmune disorders.33 Also, access to accurate genetic testing is not universal, creating further delays and difficulties for providers seeking to diagnose patients with a rare disease.34 Performing accurate genotyping of all GD patients may ultimately improve our understanding of genotype/phenotype correlations in this disease.
As mentioned, difficulty in diagnosis is often exacerbated by lack of knowledge among providers regarding GD. Patients are often misdiagnosed or referred to multiple different specialists, most often hematologist-oncologists, before receiving a diagnosis.35,36 Among hematologists, there remains a lack of awareness of GD, as a study across seven countries found that only 20% of 406 surveyed hematologist oncologists initially considered GD in a patient presenting with common GD manifestations such as anemia, thrombocytopenia, hepatomegaly, splenomegaly, and bone pain, instead focusing on leukemia, lymphoma, and multiple myeloma.9,37 In addition to emotional distress over a prolonged diagnostic journey, patients may undergo unnecessary invasive procedures or receive inappropriate treatment.
Unclear Management Guidelines
While enzyme replacement therapy and substrate reduction therapy are effective at managing non-neurological manifestations of Gaucher disease, there is a lack of universal standards of care or established management goals. Expert consensus opinions have converged on a few general principles of treatment such as normalizing hematological parameters, reducing organomegaly, and maintaining quality of life and participation in work or school.38,39 Most diagnostic guidelines recommend genetic testing confirmed with enzyme activity assays.40 However, the clinical utility of available biomarkers, optimal dosing, and criteria for initiation of treatment are still debated. Limited genotype/phenotype correlations make it difficult to predict which patients may experience the greatest symptom burden and benefit most from treatment.41,42 Clearer management guidelines and disease surveillance throughout the entire spectrum of GD would help to better quantify the benefits of early treatment initiation, particularly in patients who show few disease manfestations.
Public Policy Considerations
Newborn Screening in the US
Newborn screening (NBS) in the United States is a public health initiative developed and funded at the state level, with wide variation in both screening implementation and disease inclusion. The Recommended Uniform Screening Panel (RUSP) serves as a nationwide guideline that communicates which conditions are appropriate for NBS. The RUSP currently does not include GD or mandate its adoption.43 In addition, the timeline from inclusion to implementation can be lengthy and irregular. Since the current RUSP’s official inception in 2010, eight new disorders have been added, including MPS I, MPS II, and Pompe disease.44 However, state implementation of those guidelines can be slow and inconsistent; for example, MPS I was approved in 2015 and added to the RUSP in 2016, but currently, only 34 states conduct or plan to conduct NBS for MPS I.45
Gaucher disease faces its own specific hurdles to widespread screening. The benefits of adding GD to the RUSP and state-specific newborn screening programs have been debated given unclear genotype/phenotype correlation, variable age of symptom onset, unreliable biomarkers, and the lack of effective treatment for its neuronopathic manifestations46 (Figure 2). Thus, opinions regarding the utility of NBS are varied. Results from one study exploring genetic healthcare providers’ attitudes towards population-wide NBS for lysosomal storage disorders indicated that Fabry and GD were considered lower priority than infantile Pompe disease and MPS I, for example, due to a perceived later age of onset.47 However, a Delphi consensus group of GD experts supported NBS for GD since an earlier disease onset conveys a high risk of morbidity and there is often a lengthy diagnostic odyssey.39 Patients themselves lack a consensus opinion on whether it would be preferable to expand NBS efforts48 due to the lack of a definitive cure for GD, the observation that many patients may have a low symptom burden, and lack of evidence for any long-term effects of mild patients going untreated in childhood.49,50 In light of these debates and subsequent lack of inclusion in the RUSP, GD is currently part of the newborn screens in only six states – Illinois, Missouri, New Jersey, New Mexico, Oregon, and Tennessee – in addition to the ongoing ScreenPlus pilot NBS program in New York City.51
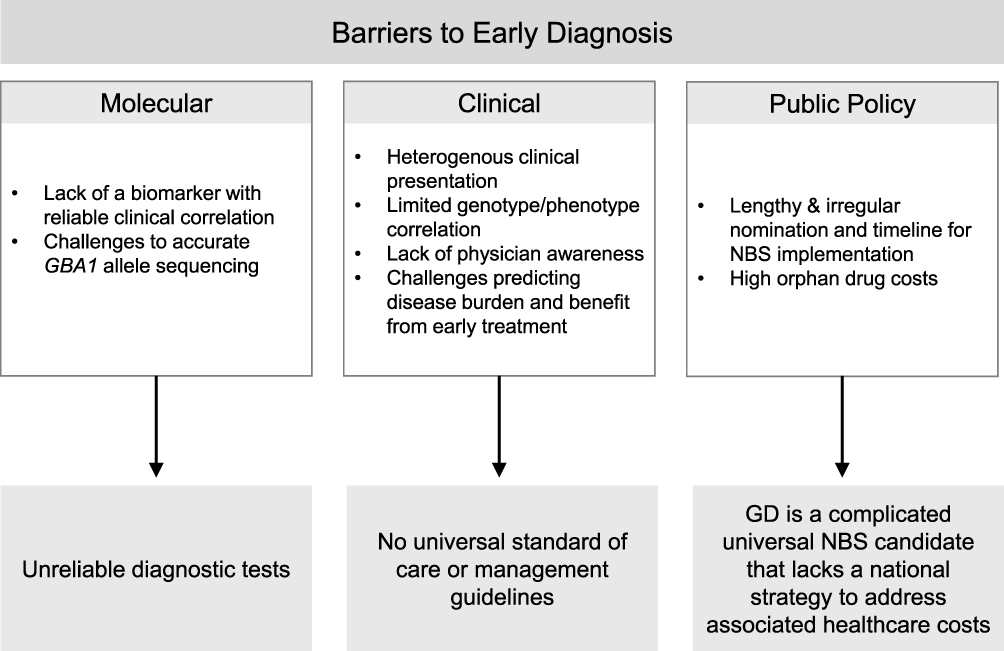 |
Figure 2 Medical and institutional barriers to early diagnosis. Abbreviations: GD, Gaucher disease; NBS, Newborn Screening. |
Economic Considerations
The Orphan Drug Act of 1983 changed the landscape of drug development in the rare disease space through research incentives. As of 2022, there have been 1035 therapeutic approvals for drug indications that received orphan status.52 Nevertheless, having a rare disease is expensive both before and after diagnosis. A study of Pompe disease found that the cost of disease was $176,000 in the year of diagnosis and $371,000 after, with a cost of $46,000 to $87,000 per year of delayed diagnosis.53 Even though newer technologies and diagnostic strategies, such as early exome and genomic studies, often reduce the time to diagnosis, associated costs, and caregiver burden,54,55 they still may not reduce overall disease-associated expense due to treatment costs.56 In GD, ERT is estimated to cost between $240,000 and $750,000 per patient per year depending on specific drug, patient weight, and associated costs of drug administration, rendering it inaccessible to patients in many regions of the world.57 Orphan drugs in total are estimated to generate $185 billion dollars in revenue this year, occupying a full fifth of drug expenditures58 with a price increase of up to 44% over their non-orphan drug similars in the last 15 years.52 Development costs and price justification for new therapies are difficult to assess, as drug developers also benefit from patient groups and other government funded agencies and make use of basic science and preclinical work by publicly funded institutions.59,60
Future Steps
Harnessing the AI Advantage
The numerous biological, political, and logistical hurdles faced by patients with GD highlight the need for improved diagnostic tools, wider access to care, and a better understanding of the effect of genotype on clinical manifestations. The larger rare disease community currently uses several tools to address these shortfalls through data collection and analysis and by facilitating disease diagnosis.
Specialized search engines are one such tool. FindZebra.com curates articles from various databases, including the Online Mendelian Inheritance in Man (OMIM), Orphanet, National Organization for Rare Disorders (NORD), and more. Recently, a new online search tool, deep.findzebra.com, was piloted, which mines PubMed case data to identify rare phenotypes of rare diseases. This deep learning model has been trialed for Fabry disease and GD. Validation by medical experts showed that the tool extracted clinically relevant articles more often for Fabry disease than for GD, perhaps due to the limited genotype/phenotype correlation in GD and changing therapeutic guidelines. However, this tool still represents an important step in taking computational approaches to aid in making a more rapid diagnosis of rare diseases.61
Artificial intelligence applied to electronic health records has the potential to recognize rare disease diagnoses by identifying previously undiagnosed patients and validating diagnostic scoring systems. Certain rare disease algorithms trained on EHR data from US patients have been shown to be 10–20-fold more efficient at identifying patients with GD than previous clinical diagnostic algorithms.62 Such approaches still require that patients receive confirmatory diagnostic testing, but identifying high-risk patients early can help to shorten the lengthy diagnostic odyssey. Current scoring systems such as the Gaucher Earlier Diagnosis Consensus (GED-C) developed by GD specialists can be validated and applied wide-scale to EHR data to identify potential undiagnosed GD cases.7,63–65
In addition to collating online resources to give physicians and patients better access to specialized and accurate information on GD, the use of artificial intelligence to evaluate facial phenotypes provides another promising diagnostic tool. The DeepGestalt algorithm and its subsequent tools such as GestaltMatcher and D-Score are leaders in this “next-gen phenotyping” space.66 The Face2Gene platform, powered by DeepGestalt, has already been applied to compare photos of healthy controls and patients with GD2 or GD3, yielding a mean accuracy of 76.6%, compared to 37.7% for random comparison.67
Public Policy
Public policy first defined the rare disease space more than four decades ago. With the advent of new technologies, the field has expanded, with a large increase in the number of identified rare diseases, as well as an improved understanding of their pathogenesis, enabling new treatment modalities. With new knowledge, new challenges have arisen that would benefit from updated policy at the national level. The rising cost of new therapies is one of those areas. The mean cost for single use treatments such as gene therapy was $2,643,487 for approved therapies from 2016 to 2023, often with limited clinical evidence in support of FDA approval.68 Several strategies have been proposed to address these current and future shortcomings, such as re-defining the definition of rare disease in the orphan drug space, adopting outcome-based risk sharing, and multistate purchasing initiatives.59,68–70 No unified strategy to address the economic considerations of rare diseases has yet emerged.
NBS varies throughout the nation, but its implementation is guided by common principles. These include the availability of screening tests that can identify the disorder shortly after birth, when disease manifestations may still be subclinical, with appropriate sensitivity and specificity. In addition, there must be a demonstrable benefit to early detection, intervention, and treatment.71 Historically, the ability to accurately screen for GD and benefits to widespread screening have been debated. However, improved enzyme assays, sequencing capabilities, and the advent of ERT and SRT that treat non-neuronopathic GD symptoms are promising developments. While the field still lacks effective treatment for the neuronopathic manifestations of GD, new and emerging therapies may necessitate updates to universal screening guidelines for GD and the RUSP nomination process. A recent survey of NBS experts supported the idea that the current NBS nomination and RUSP process required modernization. They posited possible solutions that included 1) revising and improving timeliness of RUSP review; 2) creating mechanisms to offer screening for conditions in addition to the RUSP, for example, through public-private partnerships to support expanded NBS; 3) accelerating and expanding data collection to inform policy and implementation; 4) helping states expedite comprehensive implementation of screening for new disorders; and 5) evaluating emerging methods of screening and their consequences.72
Conclusion
Obstacles to early diagnosis of GD stem from its phenotypic heterogeneity and overlap in manifestations with other disorders. The dearth of reliable biomarkers, wide spectrum of disease manifestations, and limited genotype/phenotype correlation lead to inadequate provider knowledge about this rare disease. Defining appropriate diagnostic algorithms, increasing access to accurate genetic testing, and extending current research on standards of care may help to rectify this problem. Once diagnosed, the lack of consensus on management guidelines in asymptomatic individuals further complicates the utility of universal NBS, as the benefits of early initiation of treatment in asymptomatic non-neuronopathic patients are unclear. The high cost of available therapies is another consideration, as well as the lack of brain penetrant therapies for those affected with the most severe forms of GD. Thus, GD remains a nuanced addition to nationwide screening guidelines. A few of these challenges can be addressed through computational efforts, such as AI, that can identify previously undiagnosed patients, validate current GD symptom scoring systems, and enhance our understanding of GD’s varied clinical manifestations. In addition, public policy efforts to expand funding and facilitate public-private stakeholder partnerships can help to address the current obstacles to diagnosis at a nationwide level.
Author Contributions
All authors made a significant contribution to this work reported, including in the conception, study design, execution, acquisition of data, analysis and interpretation. Each took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by the Intramural Research Programs of the National Human Genome Research Institute and the National Institutes of Health.
Disclosure
The authors report no conflicts of interest related to this work.
References
1. Haendel M, Vasilevsky N, Unni D, et al. How many rare diseases are there? Nat Rev Drug Discov. 2020;19(2):77–78. doi:10.1038/d41573-019-00180-y
2. Sun H, Saeedi P, Karuranga S, et al. IDF diabetes atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabet Res Clin Pract. 2022;183:109119. doi:10.1016/j.diabres.2021.109119
3. Ferreira CR. The burden of rare diseases. Am J Med Genet A. 2019;179(6):885–892. doi:10.1002/ajmg.a.61124
4. Daykin EC, Ryan E, Sidransky E. Diagnosing neuronopathic Gaucher disease: new considerations and challenges in assigning Gaucher phenotypes. Mol Gene Metabol. 2021;132(2):49–58. doi:10.1016/j.ymgme.2021.01.002
5. Roshan Lal T, Seehra GK, Steward AM, et al. The natural history of type 2 Gaucher disease in the 21st century: a retrospective study. Neurology. 2020;95(15):e2119–e2130. doi:10.1212/WNL.0000000000010605
6. Mistry PK, Lopez G, Schiffmann R, Barton NW, Weinreb NJ, Sidransky E. Gaucher disease: progress and ongoing challenges. Mol Gene Metabol. 2017;120(1–2):8–21. doi:10.1016/j.ymgme.2016.11.006
7. Mehta A, Kuter DJ, Salek SS, et al. Presenting signs and patient co-variables in Gaucher disease: outcome of the Gaucher Earlier Diagnosis Consensus (GED-C) Delphi initiative. Intern Med J. 2019;49(5):578–591. doi:10.1111/imj.14156
8. Gleason AM, D’Souza A, Ryan E, et al. The D409H variant in GBA1: challenges in predicting the Gaucher phenotype in the newborn screening era. Am J Med Genet A. 2023;191(7):1783–1791. doi:10.1002/ajmg.a.63202
9. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82(8):697–701. doi:10.1002/ajh.20908
10. Koto Y, Narita A, Noto S, et al. Burden of caregivers of patients with neuronopathic and non-neuronopathic Gaucher disease in Japan: a survey-based study. Mol Gene Metabol Rep. 2023;36:100994. doi:10.1016/j.ymgmr.2023.100994
11. Tayebi N, Stubblefield BK, Park JK, et al. Reciprocal and nonreciprocal recombination at the glucocerebrosidase gene region: implications for complexity in Gaucher disease. Am J Hum Genet. 2003;72(3):519–534. doi:10.1086/367850
12. Miyoshi K, Hagita H, Horiguchi T, Tanimura A, Noma T. Redefining GBA gene structure unveils the ability of Cap-independent, IRES-dependent gene regulation. Commun Biol. 2022;5(1):639. doi:10.1038/s42003-022-03577-5
13. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567–583. doi:10.1002/humu.20676
14. Woo EG, Tayebi N, Sidransky E. Next-generation sequencing analysis of GBA1: the challenge of detecting complex recombinant alleles. Front Genet. 2021;12:684067. doi:10.3389/fgene.2021.684067
15. Ruskey JA, Greenbaum L, Ronciere L, et al. Increased yield of full GBA sequencing in Ashkenazi Jews with Parkinson’s disease. Eur J Med Genet. 2019;62(1):65–69. doi:10.1016/j.ejmg.2018.05.005
16. Gorostidi A, Marti-Masso JF, Bergareche A, Rodriguez-Oroz MC, Lopez de Munain A, Ruiz-Martinez J. Genetic mutation analysis of Parkinson’s disease patients using multigene next-generation sequencing panels. Mol Diagn Ther. 2016;20:481–491. doi:10.1007/s40291-016-0216-1
17. den Heijer JM, Cullen VC, Quadri M, et al. A large-scale full GBA1 gene screening in Parkinson’s disease in the Netherlands. Mov Disord. 2020;35:1667–1674. doi:10.1002/mds.28112
18. Toffoli M, Chen X, Sedlazeck FJ, et al. Comprehensive short and long read sequencing analysis for the Gaucher and Parkinson’s disease-associated GBA gene. Commun Biol. 2022;5(1):670. doi:10.1038/s42003-022-03610-7
19. Giraldo P, Lopez de Frutos L, Cebolla JJ. Biomarker combination is necessary for the assessment of Gaucher disease? Ann Transl Med. 2018;6(Suppl 1):S81. doi:10.21037/atm.2018.10.69
20. Murugesan V, Chuang WL, Liu J, et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am J Hematol. 2016;91(11):1082–1089. doi:10.1002/ajh.24491
21. De Castro-Oros I, Irun P, Cebolla JJ, et al. Assessment of plasma chitotriosidase activity, CCL18/PARC concentration and NP-C suspicion index in the diagnosis of Niemann-Pick disease type C: a prospective observational study. J Transl Med. 2017;15(1):43. doi:10.1186/s12967-017-1146-3
22. Kim S, Whitley CB, Jarnes JR. Chitotriosidase as a biomarker for gangliosidoses. Mol Gene Metabol Rep. 2021;29:100803. doi:10.1016/j.ymgmr.2021.100803
23. Brinkman J, Wijburg FA, Hollak CE, et al. Plasma chitotriosidase and CCL18: early biochemical surrogate markers in type B Niemann-Pick disease. J Inherit Metab Dis. 2005;28(1):13–20. doi:10.1007/s10545-005-4416-9
24. Aerts JM, Kallemeijn WW, Wegdam W, et al. Biomarkers in the diagnosis of lysosomal storage disorders: proteins, lipids, and inhibodies. J Inherit Metab Dis. 2011;34(3):605–619. doi:10.1007/s10545-011-9308-6
25. Dekker N, van Dussen L, Hollak CE, et al. Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood. 2011;118(16):e118–27. doi:10.1182/blood-2011-05-352971
26. Rolfs A, Giese AK, Grittner U, et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS One. 2013;8(11):e79732. doi:10.1371/journal.pone.0079732
27. Cozma C, Cullufi P, Kramp G, et al. Treatment efficiency in Gaucher patients can reliably be monitored by quantification of Lyso-Gb1 concentrations in dried blood spots. Int J mol Sci. 2020;21(13). doi:10.3390/ijms21134577
28. Hurvitz N, Dinur T, Becker-Cohen M, et al. Glucosylsphingosine (lyso-Gb1) as a biomarker for monitoring treated and untreated children with Gaucher disease. Int J mol Sci. 2019;20(12):3033. doi:10.3390/ijms20123033
29. Giuffrida G, Markovic U, Condorelli A, et al. Glucosylsphingosine (Lyso-Gb1) as a reliable biomarker in Gaucher disease: a narrative review. Orphanet J Rare Dis. 2023;18(1):27. doi:10.1186/s13023-023-02623-7
30. Dubiela P, Szymanska-Rozek P, Hasinski P, et al. Long- and short-term glucosphingosine (lyso-Gb1) dynamics in gaucher patients undergoing enzyme replacement therapy. Biomolecules. 2024;14(7):842. doi:10.3390/biom14070842
31. Menkovic I, Boutin M, Alayoubi A, Mercier FE, Rivard GE, Auray-Blais C. Identification of a reliable biomarker profile for the diagnosis of gaucher disease type 1 patients using a mass spectrometry-based metabolomic approach. Int J mol Sci. 2020;21(21):7869. doi:10.3390/ijms21217869
32. Elstein D, Belmatoug N, Bembi B, et al. Twelve years of the Gaucher Outcomes Survey (GOS): insights, achievements, and lessons learned from a global patient registry. J Clin Med. 2024;13(12):3588. doi:10.3390/jcm13123588
33. Costagliola G, De Marco E, Massei F, et al. The etiologic landscape of lymphoproliferation in childhood: proposal for a diagnostic approach exploring from infections to inborn errors of immunity and metabolic diseases. Ther Clin Risk Manag. 2024;20:261–274. doi:10.2147/TCRM.S462996
34. Thorpe E, Williams T, Shaw C, et al. The impact of clinical genome sequencing in a global population with suspected rare genetic disease. Am J Hum Genet. 2024;111(7):1271–1281. doi:10.1016/j.ajhg.2024.05.006
35. Mengel E, Gaedeke J, Gothe H, et al. The patient journey of patients with Fabry disease, Gaucher disease and Mucopolysaccharidosis type II: a German-wide telephone survey. PLoS One. 2020;15(12):e0244279. doi:10.1371/journal.pone.0244279
36. Mehta A, Belmatoug N, Bembi B, et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol Gene Metabol. 2017;122:122–129. doi:10.1016/j.ymgme.2017.08.002
37. Thomas AS, Mehta AB, Hughes DA. Diagnosing Gaucher disease: an on-going need for increased awareness amongst haematologists. Blood Cells mol Dis. 2013;50(3):212–217. doi:10.1016/j.bcmd.2012.11.004
38. Biegstraaten M, Cox TM, Belmatoug N, et al. Management goals for type 1 Gaucher disease: an expert consensus document from the European working group on Gaucher disease. Blood Cells mol Dis. 2018;68(Supplement C):203–208. doi:10.1016/j.bcmd.2016.10.008
39. Kishnani PS, Al-Hertani W, Balwani M, et al. Screening, patient identification, evaluation, and treatment in patients with Gaucher disease: results from a Delphi consensus. Mol Gene Metabol. 2022;135(2):154–162. doi:10.1016/j.ymgme.2021.12.009
40. Dardis A, Michelakakis H, Rozenfeld P, et al. Patient centered guidelines for the laboratory diagnosis of Gaucher disease type 1. Orphanet J Rare Dis. 2022;17(1):442. doi:10.1186/s13023-022-02573-6
41. Hassan S, Lopez G, Stubblefield BK, Tayebi N, Sidransky E. Alleles with more than one mutation can complicate genotype/phenotype studies in Mendelian disorders: lessons from Gaucher disease. Mol Gene Metabol. 2018;125(1–2):1–3. doi:10.1016/j.ymgme.2018.06.013
42. Ryan E, Seehra GK, Sidransky E. Mutations, modifiers and epigenetics in Gaucher disease: blurred boundaries between simple and complex disorders. Mol Gene Metabol. 2019;128(1–2):10–13. doi:10.1016/j.ymgme.2019.08.006
43. McCandless SE, Wright EJ. Mandatory newborn screening in the United States: history, current status, and existential challenges. Birth Defects Res. 2020;112(4):350–366. doi:10.1002/bdr2.1653
44. Kemper AR, Green NS, Calonge N, et al. Decision-making process for conditions nominated to the recommended uniform screening panel: statement of the US Department of Health and Human Services Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children. Genet Med. 2014;16(2):183–187. doi:10.1038/gim.2013.98
45. Ellinwood NM. Newborn screening and the recommended uniform screening panel: optimal submissions and suggested improvements based on an advocacy organization’s decade-long experience. Am J Med Genet C Semin Med Genet. 2022;190(2):156–161. doi:10.1002/ajmg.c.32001
46. Wasserstein MP, Orsini JJ, Goldenberg A, et al. The future of newborn screening for lysosomal disorders. Neurosci lett. 2021;760:136080. doi:10.1016/j.neulet.2021.136080
47. Lisi EC, McCandless SE. Newborn screening for lysosomal storage disorders: views of genetic healthcare providers. J Genet Couns. 2016;25(2):373–384. doi:10.1007/s10897-015-9879-8
48. DeLuca JM. Public attitudes toward expanded newborn screening. J Pediatr Nurs. 2018;38:e19–e23. doi:10.1016/j.pedn.2017.10.002
49. Lisi EC, Ali N. Opinions of adults affected with later-onset lysosomal storage diseases regarding newborn screening: a qualitative study. J Genet Couns. 2021;30(6):1544–1558. doi:10.1002/jgc4.1421
50. Lisi EC, Gillespie S, Laney D, Ali N. Patients’ perspectives on newborn screening for later-onset lysosomal storage diseases. Mol Gene Metabol. 2016;119(1–2):109–114. doi:10.1016/j.ymgme.2016.07.009
51. Ryan E, Jong T, Sidranksy E. Newborn screening in Gaucher disease: a bright and complicated future. OBM Gene. 2022;6(3):1–21. doi:10.21926/obm.genet.2203165
52. Michaeli T, Jurges H, Michaeli DT. FDA approval, clinical trial evidence, efficacy, epidemiology, and price for non-orphan and ultra-rare, rare, and common orphan cancer drug indications: cross sectional analysis. BMJ. 2023;381:e073242. doi:10.1136/bmj-2022-073242
53. The Lewin Group I. The cost of delayed diagnosis in rare disease: a health economic study. 2023. Available from: https://everylifefoundation.org/delayed-diagnosis-study/#:~:text=On%20average%2C%20the%20diagnostic%20odyssey,benefit%20from%20recent%20medical%20advancements.
54. Srivastava S, Love-Nichols JA, Dies KA, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21(11):2413–2421. doi:10.1038/s41436-019-0554-6
55. Klau J, Abou Jamra R, Radtke M, et al. Exome first approach to reduce diagnostic costs and time - retrospective analysis of 111 individuals with rare neurodevelopmental disorders. Eur J Hum Genet. 2022;30(1):117–125. doi:10.1038/s41431-021-00981-z
56. Weymann D, Buckell J, Fahr P, et al. Health care costs after genome-wide sequencing for children with rare diseases in England and Canada. JAMA Netw Open. 2024;7(7):e2420842. doi:10.1001/jamanetworkopen.2024.20842
57. Farahbakhshian S, Inocencio TJ, Poorman G, Wright E, Pathak RR, Bullano M. The budget impact of enzyme replacement therapy in type 1 Gaucher disease in the United States. J Med Econ. 2022;25(1):755–761. doi:10.1080/13696998.2022.2082200
58. Senior M. The new report is out: are orphan drugs losing their sparkle? 2024. Available from: https://www.evaluate.com/thought-leadership/orphan-drug-report-2024/.
59. Alonso Ruiz A, Large K, Moon S, Vieira M. Pharmaceutical policy and innovation for rare diseases: a narrative review. F1000Res. 2023;12:211. doi:10.12688/f1000research.130809.2
60. Kesselheim AS, Tan YT, Avorn J. The roles of academia, rare diseases, and repurposing in the development of the most transformative drugs. Health Aff. 2015;34(2):286–293. doi:10.1377/hlthaff.2014.1038
61. Lievin V, Hansen JM, Lund A, et al. FindZebra online search delving into rare disease case reports using natural language processing. PLOS Digit Health. 2023;2(6):e0000269. doi:10.1371/journal.pdig.0000269
62. Wilson A, Chiorean A, Aguiar M, et al. Development of a rare disease algorithm to identify persons at risk of Gaucher disease using electronic health records in the United States. Orphanet J Rare Dis. 2023;18(1):280. doi:10.1186/s13023-023-02868-2
63. Savolainen MJ, Karlsson A, Rohkimainen S, et al. The Gaucher earlier diagnosis consensus point-scoring system (GED-C PSS): evaluation of a prototype in Finnish Gaucher disease patients and feasibility of screening retrospective electronic health record data for the recognition of potential undiagnosed patients in Finland. Mol Gene Metabol Rep. 2021;27:100725. doi:10.1016/j.ymgmr.2021.100725
64. Pehrsson M, Heikkinen H, Wartiovaara-Kautto U, et al. Screening for potential undiagnosed Gaucher disease patients: utilisation of the Gaucher earlier diagnosis consensus point-scoring system (GED-C PSS) in conjunction with electronic health record data, tissue specimens, and small nucleotide polymorphism (SNP) genotype data available in Finnish biobanks. Mol Gene Metabol Rep. 2022;33:100911. doi:10.1016/j.ymgmr.2022.100911
65. Revel-Vilk S, Shalev V, Gill A, et al. Assessing the diagnostic utility of the Gaucher Earlier Diagnosis Consensus (GED-C) scoring system using real-world data. Orphanet J Rare Dis. 2024;19(1):71. doi:10.3390/ijms20123033
66. Reiter AMV, Pantel JT, Danyel M, Horn D, Ott CE, Mensah MA. Validation of 3 computer-aided facial phenotyping tools (DeepGestalt, GestaltMatcher, and D-Score): comparative diagnostic accuracy study. J Med Internet Res. 2024;26:e42904. doi:10.2196/42904
67. Daykin E, Fleischer N, Abdelwahab M, et al. Investigation of a dysmorphic facial phenotype in patients with Gaucher disease types 2 and 3. Mol Gene Metabol. 2021;134(3):274–280. doi:10.1016/j.ymgme.2021.09.008
68. Odouard IC, Ballreich J, Lee B, Socal MP. Clinical evidence supporting FDA approval of gene and RNA therapies for rare inherited conditions. Paediatr Drugs. 2024;26:741–752. doi:10.1007/s40272-024-00645-7
69. Odouard IC, Ballreich J, Socal MP. Medicaid spending and utilization of gene and RNA therapies for rare inherited conditions. Health Aff Sch. 2024;2(5):qxae051. doi:10.1093/haschl/qxae051
70. Ng QX, Ong C, Chan KE, et al. Comparative policy analysis of national rare disease funding policies in Australia, Singapore, South Korea, the United Kingdom and the United States: a scoping review. Health Econ Rev. 2024;14(1):42. doi:10.1186/s13561-024-00519-1
71. American College of Medical Genetics Newborn Screening Expert G. Newborn screening: toward a uniform screening panel and system—executive summary. Pediatrics. 2006;117(5 Pt 2):S296–307. doi:10.1542/peds.2005-2633I
72. Bailey DB, Porter KA, Andrews SM, Raspa M, Gwaltney AY, Peay HL. Expert evaluation of strategies to modernize newborn screening in the United States. JAMA Netw Open. 2021;4(12):e2140998. doi:10.1001/jamanetworkopen.2021.40998
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.