")
Back to Journals » Infection and Drug Resistance » Volume 18
Pan-Genome-Wide Association Study Identifies Genetic Factors Associated with the Pathogenicity of Invasive Serotype 19F Streptococcus Pneumoniae
Authors Shi X, Patil S , Yi Q, Liu Z , Wang H, Zhu C, Chen Y, Zheng Y , Dong S , Bao Y
Received 4 March 2025
Accepted for publication 20 May 2025
Published 12 June 2025 Volume 2025:18 Pages 2963—2975
DOI https://doi.org/10.2147/IDR.S519795
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Xing Shi,1,* Sandip Patil,2,* Qiuwei Yi,1,* Zihao Liu,1 Heping Wang,1 Chunqing Zhu,1 Yunsheng Chen,3 Yuejie Zheng,1 Shaowei Dong,2 Yanmin Bao1
1Department of Respiratory Medicine, Shenzhen Children’s Hospital, Shenzhen, Guangdong, People’s Republic of China; 2Department of Haematology and Oncology, Shenzhen Children’s Hospital, Shenzhen, Guangdong, People’s Republic of China; 3Department of Clinical Microbiology Laboratory, Shenzhen Children’s Hospital, Shenzhen, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanmin Bao, Email [email protected] Shaowei Dong, Email [email protected]
Background: Streptococcus pneumoniae is a common respiratory pathogen that poses significant health concerns in children, particularly serotype 19F strains that demonstrate high level of invasiveness in China. To investigate the genetic variations associated with high invasiveness of serotype 19F S. pneumoniae strains isolated from children in Shenzhen.
Methods: We compared the genomic profiles of 42 invasive and 162 noninvasive strains from children’s respiratory tracts and employed pan-genome-wide association methods to elucidate the origins of genetic variation.
Results: Significant gene presence variability was observed between invasive and noninvasive strains, suggesting a genetic basis for their pathogenicity differences. Invasive 19F strains demonstrated enhanced adhesion in co-culture experiments with human epithelial cells, with adhesion abilities correlating with the presence of specific genes. Despite high non-susceptibility to common antibiotics across all strains, no significant differences in antimicrobial susceptibility patterns were found between invasive and noninvasive groups.
Conclusion: Although genomic differences within serotype 19F were relatively minor, invasive and noninvasive strains exhibited significant differences in adherence and invasiveness in the host microenvironment. While the underlying regulatory mechanisms remain uncertain, genetic differences play a crucial role in determining the invasiveness of S. pneumoniae serotype 19F strains in children.
Keywords: pneumococcus, serotype 19F, invasive strains, pan-GWAS, genetic variation
Introduction
Streptococcus pneumoniae is one of the most prevalent conditionally pathogenic bacteria in pediatric populations. The carriage rate of S. pneumoniae among children in China are generally high, with studies reporting prevalence rates ranging from 21.4% to 30.4%, indicating its widespread presence.1,2 This bacterium primarily colonizes the mucosal surfaces of the upper respiratory tract in humans as a common commensal organism. However, when it invades the lower respiratory tract, as well as the bloodstream and other organs, it can become a pathogen responsible for a range of respiratory and invasive diseases, including pneumonia, meningitis, and bacteremia. In 2015, pneumococcal disease caused an estimated 294,000 deaths in children under five years of age around the world.3 The high morbidity and mortality rates associated with pneumococcal infections underscore its role as a leading cause of death in young children. Among the various serotypes, 19F exhibited the highest prevalence and invasiveness in Chinese pediatric populations, persisting both prior to and subsequent to the implementation of the PCV13 (13-valent pneumococcal conjugate vaccine) vaccination.2,4–10 Although PCV13 was introduced in China in 2016, it has not been included in the national immunization program, leading to limited adoption. A multicenter prospective study on pediatric IPDs (invasive pneumococcal diseases) conducted from 2019 to 2021 found that the PCV13 vaccine covers 85.1% of serotypes, yet serotype 19F still accounted for 24.2% of cases.11 Moreover, in cities like Shenzhen where PCV13 vaccination rates approximate 50%, the respiratory colonization rate of serotype 19F continue to be the highest observed.6 Despite the effectiveness of PCV13 in decreasing the overall S. pneumoniae infection rate, serotype 19F maintains the highest rate of invasiveness in China, attributable to its high colonization rate in the respiratory tract, significant antibiotic resistance, and superior capacity to evade vaccine-induced immunity.2,12,13 Colonization in respiratory tract by S. pneumoniae is a critical prerequisite to the development of invasive pneumococcal diseases.14 The primary characteristic of colonization is the adherence of S. pneumoniae to host cells and tissues, which disrupts mucosal innate and adaptive immunity. This disruption facilitates the pathogen’s persistence and potential transition from a commensal organism to an invasive pathogen.15 The adhesive capabilities of various serotypes critically influence colonization rates, thereby modulating the incidence of infections and the occurrence of invasive diseases.16 And the infection process is influenced by individual immunity, viral infections, as well as the pathogenicity of S. pneumoniae in a complex interplay that determines the severity and outcome of the disease.17,18 Ultimately, certain serotypes, such as 1, 3, 19A, and 19F, can lead to severe clinical outcomes and high morbidity.19,20 Despite the clinical importance of serotype 19F, the influence of genome-wide variation on the pathogenicity of 19F strains, particularly in the post-PCV era, has not been thoroughly investigated. Notably, there remains an insufficient understanding how the same serotype 19F strains can exhibit diverse colonization and invasion phenotypes. To address this gap, it is essential to investigate the adhesive and invasives capacities and associated genetic factors among 19F S. pneumoniae isolates from different infection sites. This study examines 19F isolates from sterile body fluid and samples from children’s respiratory tract collected in Shenzhen Children’s Hospital to explore the underlying genetic factors prone to invasiveness. By comparing pan-genomic differences and assessing adhesion capabilities between the two 19F groups from different sources, we found that the invasive 19F strains demonstrated superior adaptability within the human epithelial cell environment and was closely related to their genomic composition. However, deeper insights into these adaptive mechanisms will necessitate further molecular and omics study.
Materials and Methods
Isolation of Clinical Pneumococcal Strains
Clinical strains of S. pneumoniae were isolated from respiratory tracts, blood, and cerebrospinal fluid (CSF) using standard microbiological techniques. To ensure patient privacy throughout our study, all clinical samples were de-identified before analysis. We assigned unique artificial codes to each patient and their corresponding samples. We have checked that each pneumococcal strain in our analysis represented a distinct patient case, thereby preventing statistical bias from multiple sampling of genetically identical isolates. S. pneumoniae cultures were obtained following previously described methods.6 Strains were preserved in brain heart infusion (BHI) broth with 40% glycerol broth medium at −80°C for further analysis.
Bacterial and Lung Epithelial Co-Culture
Co-culture experiments were conducted by inoculating S. pneumoniae strains with human lung epithelial cells. Initially, A549 cells (ATCC CCL-185) were grown in T75 culture flasks and passaged at a 1:3 ratio to maintain optimal growth. Simultaneously, S. pneumoniae is revived from glycerol-stored stocks and cultured overnight on blood agar plates at 37°C and 5% CO2. Selected colonies are transferred into Todd-Hewitt broth (THB) medium to ensure optimal growth. The bacterial cultures are then placed in a 96-well plate, each well containing 200 µL of medium, and incubated while monitoring the optical density at 600 nm until it reaches approximately 0.1, equivalent to about 5×107 CFU/mL, which is suitable for further experiments. For co-culturing, these S. pneumoniae cultures are added to antibiotic-free A549 cells seeded at about 5×106 cells per flask at a multiplicity of infection (MOI) of 10. This setup allows bacterial adhesion over 1h period. After incubation, non-adherent and loosely attached bacteria are washed away with phosphate-buffered saline (PBS). The epithelial cells are then treated with trypsin to detach them from the flask surface, followed by centrifugation to remove the trypsin. The resulting cell pellet is resuspended in 200 µL of 3% FBS in PBS to stabilize the cells. This suspension is serially diluted and spread onto blood agar plates for bacterial colony growth. By counting these colonies, adherent bacterial were quantitatively evaluated.
Whole Genome Sequencing and Analysis
Genomic DNA was extracted from bacterial cultures using a cell lysis solution, ribonuclease (RNase) solution, and proteinase solution to remove contaminants. Whole genome sequencing (WGS) was performed using an Illumina Novaseq6000 from Novogene Co., Ltd. Raw reads were filtered using the Trimmomatic v0.36.21 Filtered reads were assembled de novo using SPAdes v3.11,22 with contigs less than 500 bp being removed. For molecular serotyping, raw sequencing data were processed using seroBA v1.0.2,23 employing default settings and the advised k-mer size of 71. Detection of virulence factors (VFs) was achieved through a BLAST search against the virulence factor database (VFDB, http://www.mgc.ac.cn/VFs/). Genes displaying a nucleotide identity of over 95% were classified as VFs.
Pan-Genomic Analysis
Pan-genomic analysis was conducted using Roary v3.11.2 to determine the core and accessory genomes of the S. pneumoniae isolates.24 The pan-genome-wide association studies (pan-GWAS) based on the gene presence and absence table were analyzed using Scoary after correcting for covariates such as age, sex.25 To control for the false discoveries, Bonferroni-adjusted method was used to perform multiple testing for p-values that accounts for correlation. Pyseer was utilized to correlate gene presence with co-culture results, adjusting for the same covariates.20 Significant genes were defined as Bonferroni_p < 0.01 for Scoary results, and lrt-pvalue < 0.001 for Pyseer results. Venn diagrams were used to visualize the gene overlap between the groups, providing insights into shared genetic factors across different conditions or populations.
Random Forest Modeling
Feature selection for distinguishing invasive from respiratory strains was performed using the Boruta algorithm,26 which iteratively compares the importance of pan-GWAS features against the shadow features using statistical tests. Features that consistently prove more significant than their shadows was deemed relevant, while those do not are rejected. This process allows Boruta to capture both strongly and weakly relevant features, making it particularly useful in high-dimensional datasets where many features may have subtle but important relationships with the target variable. For Boruta implementation, we used a RandomForestClassifier base estimator with balanced class weights and maximum depth of 5, setting alpha=0.05 as the significance threshold. After Boruta identified relevant features, we ranked them according to their importance scores derived from the random forest model. The top 10 features were then selected for subsequent model construction. For the classification model, we used a RandomForeastClassifier with 100 trees implemented with scikit-learn v1.5.2.27 The predictive accuracy of the RandomForest classifier was evaluated using stratified 5-fold cross-validation with shuffling, together with Receiver Operating Characteristic (ROC) curve analysis.
Statistical Power Analysis and Sample Size Estimation
To estimate the statistical power for detecting an effect size of 0.5 between two groups, we employed a two-sample t-test power analysis using the statsmodels package at a significance level of 0.05. Additionally, a Monte Carlo simulation approach was implemented to estimate the power of the Mann–Whitney U-test under the same sample sizes and effect size, by generating 10,000 simulated datasets and calculating the proportion of tests rejecting the null hypothesis at the 0.05 significance level. The resulting power values from the parametric t-test and nonparametric Mann–Whitney U-test were 0.819 and 0.808, respectively. These results demonstrate that our sample size is adequate for detecting a medium effect size with sufficient statistical power.
Statistical Analysis
Statistical analyses were conducted using various tests appropriate for the data types involved. Normality of continuous data was assessed using the Shapiro–Wilk test. The chi-square test was utilized for analyzing categorical data, while Fisher’s exact test was employed for small sample sizes less than 5. To compare means of continuous variables, the Student’s t-test was applied. For non-parametric data, the Mann–Whitney U-test was used.
Results
Comparative Clinical Characteristics of Invasive and Noninvasive Pneumococcal Strains
In our study, we first conducted a comparative analysis of the clinical characteristics between 42 invasive and 162 noninvasive pneumococcal strains isolated from children’s respiratory tracts in Shenzhen Children’s Hospital. Of the noninvasive strains, 153 samples had complete clinical records documenting sex, age, and inflammatory biomarkers. The 42 invasive isolates were predominantly collected from blood (34 samples) and cerebrospinal fluid (CSF) (3 samples). The 153 documented noninvasive samples consisted of 135 sputum specimens and 18 bronchoalveolar lavage fluid (BALF) samples. The primary disease types of 42 invasive pneumococcal diseases (IPDs) included sepsis (n=37) and meningitis (n=7), while the main types of noninvasive isolate cases were bronchopneumonia (n=67), sepsis (n=21), acute bronchitis (n=15), and protracted bacterial bronchitis (n=6) (Table 1). The age and sex distribution did not significantly differ between the two groups. Invasive isolate cases have significantly higher results compared to respiratory isolate cases across inflammatory indicators, such as white blood cell (WBC) count (median 19.57 vs 10.90; p<0.001), neutrophil percentage (median 76.65 vs 47.80; p<0.001), C‐reactive protein (CRP) (median 69.90 vs 7.88; p<0.001), and procalcitonin (PCT) (median 2.45 vs 0.35; p<0.001) (Figure 1A–D). Overall, it is not surprising that invasive 19F infected cases are associated with a significantly stronger inflammatory response compared to noninvasive cases. However, this highlights the necessity of distinguishing between invasive and noninvasive 19F strains and understanding the molecular mechanisms underlying invasiveness particularly given their similar genetic backgrounds.
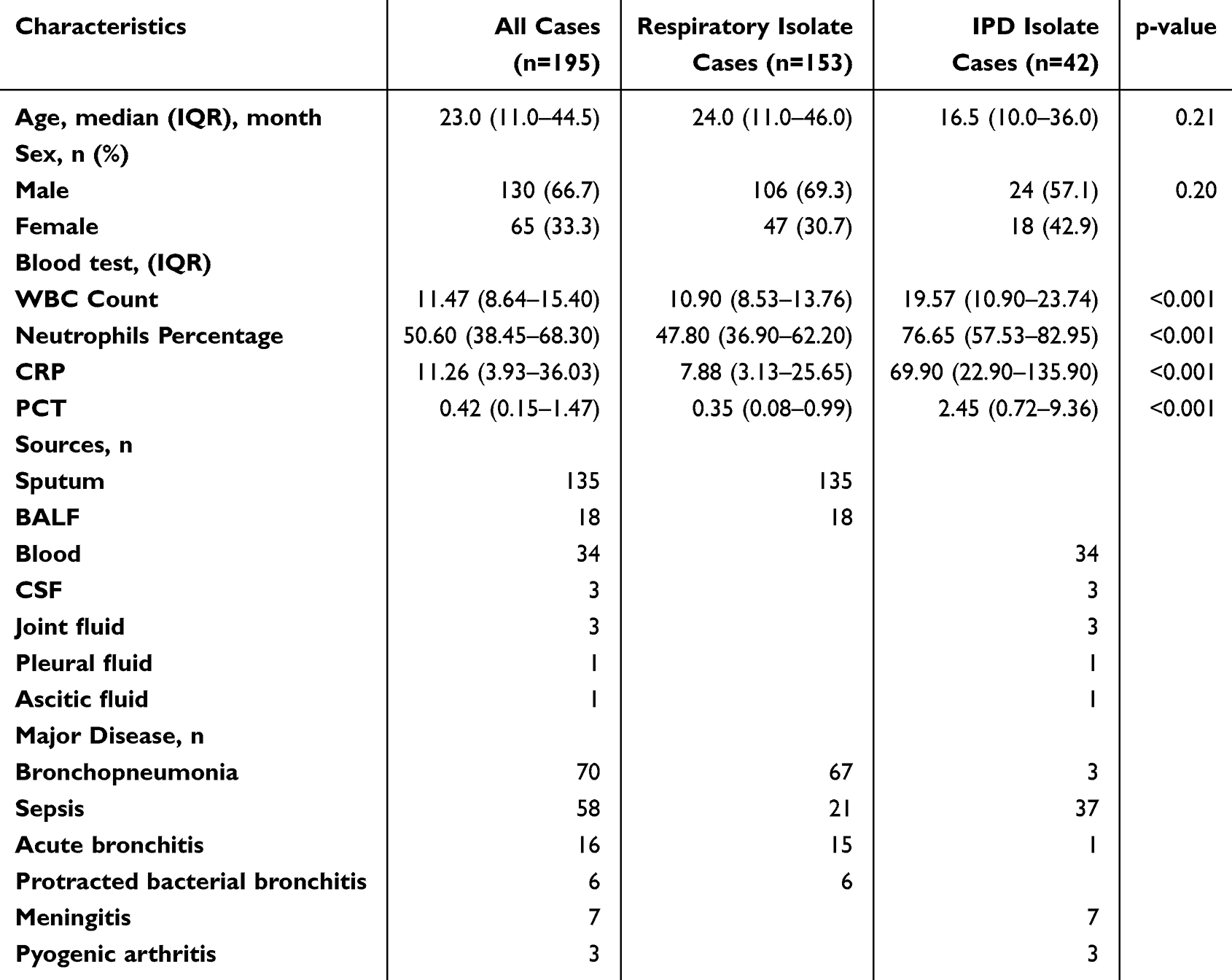 |
Table 1 Clinical Characteristics |
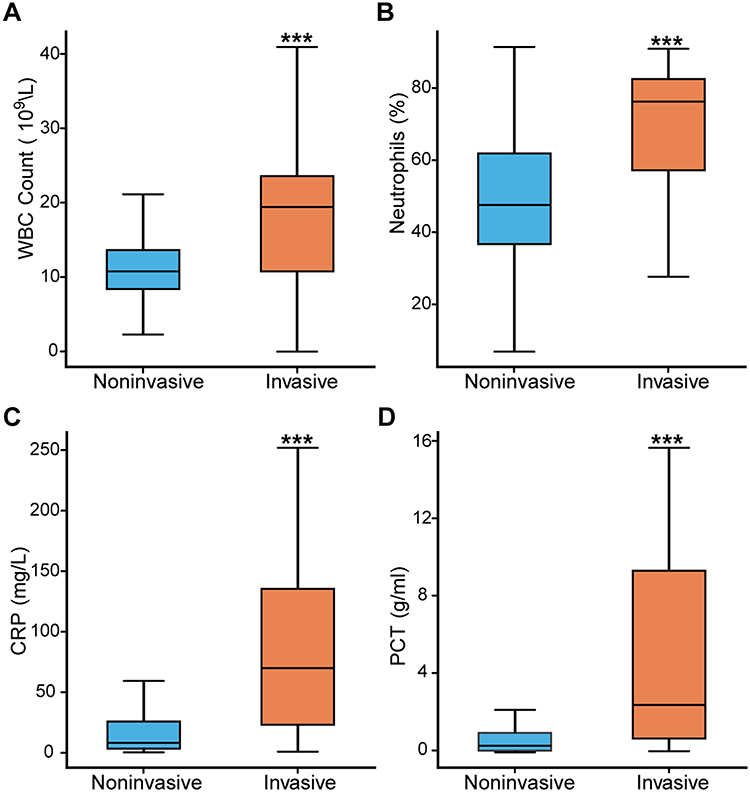 |
Figure 1 The inflammatory biomarkers comparison between respiratory isolated and invasive cases. (A) White blood cell (WBC) count distribution between respiratory isolated and invasive groups, with significantly higher WBC counts observed in the invasive group.(B) Neutrophil percentage distribution, indicating a significantly higher neutrophil percentage in invasive cases compared to respiratory isolated cases.(C) C-reactive protein (CRP) levels, showing significantly elevated CRP levels in invasive cases compared to respiratory isolated cases.(D) Procalcitonin (PCT) levels, demonstrating significantly higher PCT levels in invasive cases compared to respiratory isolated cases. ***, p < 0.001. |
Antimicrobial Susceptibility Testing (AST) Results
The AST results for Erythromycin (ERY), Penicillin (PEN), ceftriaxone (CRO), vancomycin (VAN), and levofloxacin (LVX) between noninvasive and invasive 19F strains revealed no significant differences. These AST results indicate that both noninvasive and invasive strains show high levels of non-susceptibility to CRO (75.68% vs 61.04%), ERY (100% vs 99.34%) and PEN (100% vs 97.40%), with no significant difference between the groups. And all isolates demonstrated complete susceptibility to vancomycin (VAN) and levofloxacin (LVX) (Table S1).
Virulence Factor (VFs) Analysis
We conducted VFs gene annotation by aligning genes with the VFDB databases using BLASTN, and subsequently conducted a statistical analysis using the chi-square test. The analysis revealed no statistically significant differences in the presence of these genes between noninvasive and invasive 19F strains (Table S2). Genes such as srtC-1/srtB, rrgA, rrgB, cbpD, cps4A, lytA, and cps4D exhibited an odds ratio around 1.00, indicating no preferential association with either the noninvasive or invasive phenotypes (p ranging from 0.15 to 1). The absence of significant differences in classical virulence genes between invasive and noninvasive strains highlights the complexity of pneumococcal pathogenicity, suggesting that its virulence potential is modulated by additional genetic elements. Beyond the mere presence or absence of these well-characterized virulence genes, factors such as genome plasticity, epigenetic regulatory mechanisms, niche-specific gene expression patterns, and metabolic adaptations likely play crucial roles in determining pneumococcal invasiveness.
Pan-Genome Analysis for 19F Strains
To facilitate global gene comparisons, we conducted pan-genome annotation using the Roary pipeline. The pan-genome analysis of the 19F strains revealed that total 3864 genes across all strains, in which 1649 genes were identified as core genes (present in 99% to 100% strains), 202 genes were soft-core genes (present in 95% to 99% strains), 223 genes were shell genes (present in 15% to 95% strains), and 1790 genes were cloud genes (present in less than 15% strains) (Figure 2A). The number of conserved genes stabilizes as the number of genomes increases, while the total number of genes continues to rise, reflecting the high diversity of the accessory genome among the 19F strains (Figure 2B). We conducted Scoary analysis to investigate the presence or absence of specific genes in 19F strains associated with noninvasive and invasive infections. This involved statistically assessing gene distribution patterns across these two groups to identify unique potential genetic markers. The analysis identified 37 genes with significant differences between noninvasive and invasive 19F strains (Table S3). These genes include various hypothetical proteins and transposases, such as those from the ISL3, IS5, and IS630 families, indicating potential roles in genetic mobility and variability. And the sensitivity and specificity values varied, with top5 genes specific to invasive isolates, ie group_196, group_3471, group_5, group_7, and group_377, showing high odds ratios (96.84 to 177.10), specificity (98.77 to 99.38) and sensitivity (40.48 to 54.76), suggesting strong associations with either invasive or noninvasive strains (Table S3). These results highlight specific genetic elements that could contribute to the pathogenicity and distinct characteristics of invasive versus noninvasive 19F strains.
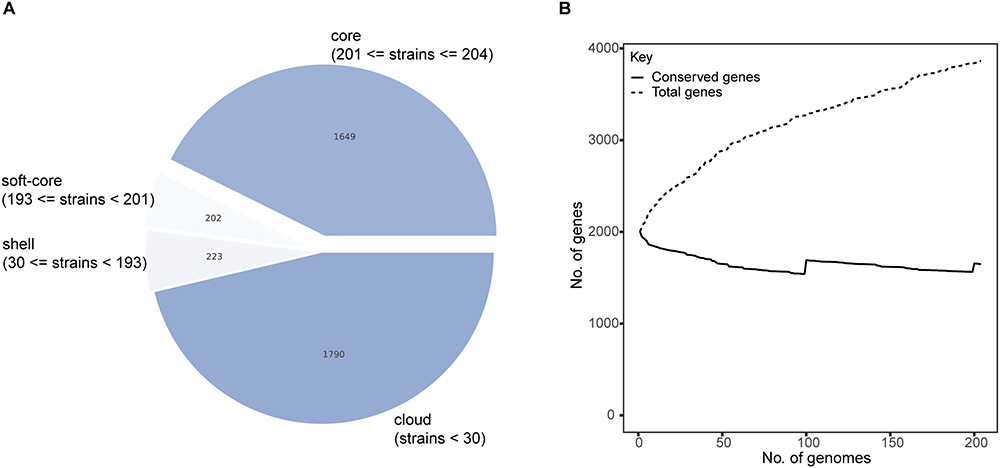 |
Figure 2 Pan-genome and phylogenetic analysis of noninvasive and invasive 19F isolates. (A) Pie chart representing the distribution of genes within the pan-genome of 19F isolates, categorized into core genes, soft-core genes, shell genes, and cloud genes. (B) Pan-genome accumulation curve showing the total number of genes and the number of conserved genes as the number of genomes increases. The curve indicates a steady increase in the total number of genes with additional genomes, while the number of conserved genes plateaus, reflecting the pan-genome’s open nature. |
Enhanced Adhesion of Invasive 19F Strains in Human Epithelial Cell Co-Culture
In our co-culture experiment, we observed that invasive 19F strains exhibited significant adhesion capabilities than noninvasive 19F strains in human respiratory epithelial A549 cells co-culture. CFU counts revealed a much higher bacterial load in invasive group during the exponential growth phase (median 3200 vs 400, p-value < 0.001, Mann–Whitney U-test) (Figure 3A). These findings suggest that invasive 19F strains can colonize and invade epithelial cells more effectively, potentially contributing to its pathogenicity.
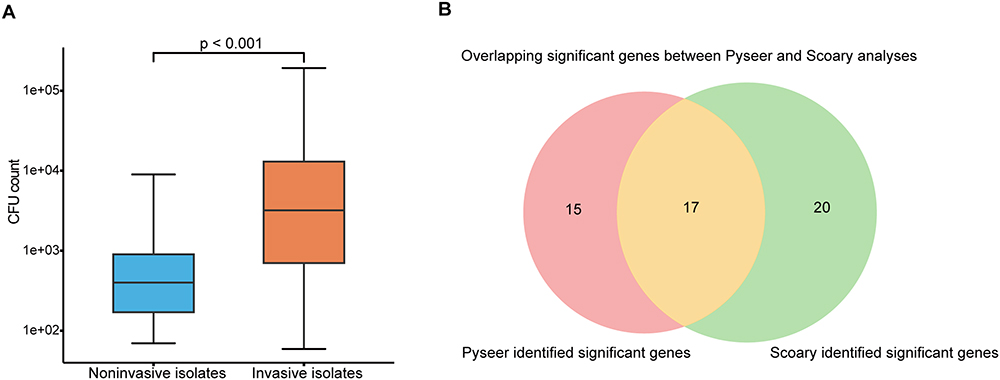 |
Figure 3 Adhesion and invasion associated genes analysis. (A) Box plot comparing the CFU values of human epithelial cell coculture results between respiratory isolates and invasive isolates. The invasive isolates show significantly higher values than respiratory isolates, with a statistical significance of p < 0.001. (B) Venn diagram showing the overlap of significant genes identified by Pyseer GWAS and Scoary analyses. A total of 15 genes are uniquely significant in GWAS, 20 genes are uniquely significant in Scoary, and 17 genes are found to be significant by both methods, highlighting the convergence between these two analytical approaches. |
Significant Overlap Between Gene Variability and Adhesion Correlation Highlights Key Pathogenic Factors
The analysis using Scoary and Pyseer identified 17 overlapping genes (Table 2). These genes were found to be shared between the 32 genes revealed by Scoary as differentially present in invasive versus non-invasive groups and the 37 genes identified by Pyseer as correlating with the co-culture results of pneumococcus and A549 cells (Figure 3B). Genes identified exclusively by either Scoary or Pyseer exhibited lower sensitivity, whereas those detected by both tools demonstrated higher sensitivity. This result indicates that genetic variations between groups, leading to differences in adhesion within the lung epithelial microenvironment, are likely significant triggers of invasiveness. For instance, genes encoding HTH-type transcriptional regulator sarX was enriched in invasive group and significantly associated with increased adhesion metrics.
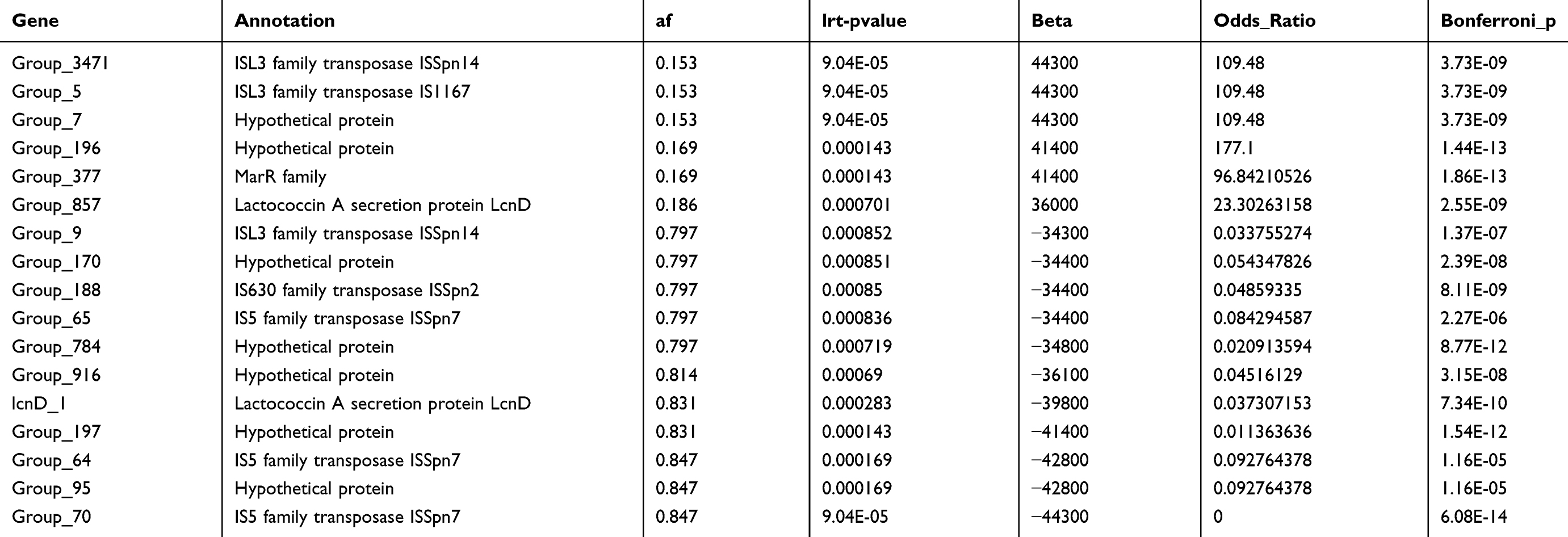 |
Table 2 Gene Presence/Absence and Pan-GWAS of Coculture Results |
The genetic differences between the two groups are closely linked to their adhesion abilities, highlighting the importance of these genes in bacterial pathogenicity. This insight provides a valuable foundation for further research into the genetic determinants of bacterial virulence and their potential as targets for medical intervention.
Development of a Predictive Model Using Gene Data to Identify Invasive 19F Strains
By analyzing genetic features, we developed a predictive model to distinguish invasive 19F strains based on the presence and absence of specific genes. When building such a model, we initially employ the Boruta algorithm for feature filtering to identify the most relevant variables for our analysis. This method works by creating shadow features and systematically evaluating whether the real variables have more statistical significance than these shadows. By doing so, we can effectively distinguish between essential and redundant features, ensuring that our model includes only those variables that truly contribute to its predictive accuracy (Figure 4A). The model demonstrated high performance and the area under the receiver operating characteristic curve (AUROC) was 0.96, indicating excellent discriminative ability (Figure 4B). The most important genes for the model were those encoding transposases, such as IS630 family transposase ISSpn2 and ISL3 family transposase ISSpn14, which were significantly correlated with the bacteria’s adhesion abilities. In future, biological validation through experiments is necessary to confirm the functional importance of key genes. Overall, the predictive model effectively identifies invasive strains, providing valuable insights into the genetic determinants of bacterial virulence and potential targets for medical intervention.
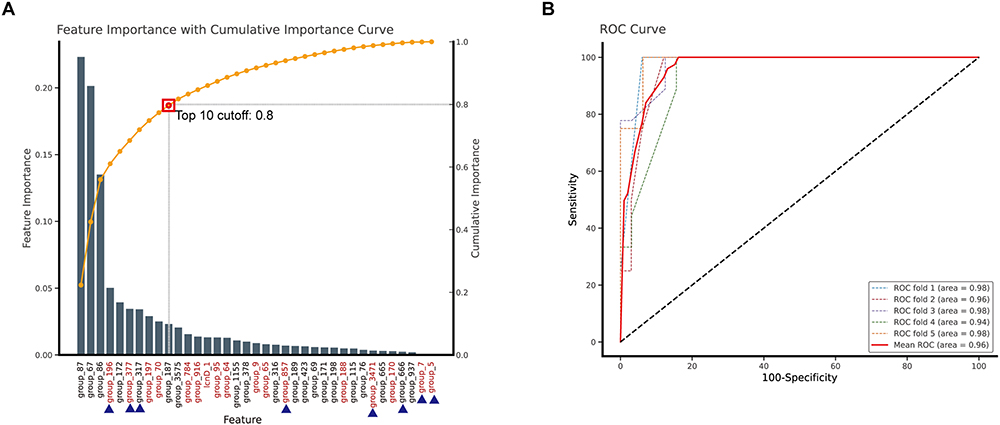 |
Figure 4 Random forest model for invasive isolate distinguishes. (A) The sorted feature importance from the Random Forest model used to differentiate invasive isolates. The brown dashed line indicates the cumulative importance values for features, indicating the top 10 features’ cumulative importance value is above this threshold 0.8. Genes highlighted in red indicate significance in both co-culture and invasive phenotype-associated genes, while genes labelled with “■” have an odds ratio (OR) greater than 1. (B) The ROC curve analysis for discrimination between noninvasive and invasive isolates. This result displays the performance of a Random Forest machine learning model using the top 10 combined features to discriminate between noninvasive and invasive isolates. The model’s performance was validated through over 5-fold cross-validation, wherein the dataset was partitioned into five equal subsets, with four used for training and one for testing. The ROC curve plots sensitivity (true positive rate) on the y-axis against 100-specificity (false negative rate). The colored dashed lines represent the performance of individual validation folds, which demonstrated the consistent classification performance across different data subsets. The solid red line displays the mean ROC curve with an AUC of 0.96. The black dashed line with a 45-degree slope represents the line of no discrimination (AUC=0.5). The consistently high AUC values (0.94–0.98) across all validation folds suggest that the Random Forest model-based ROC analysis provides a reliable discrimination between invasive and noninvasive isolates. |
Discussion
In our previous research, we found that although the PCV13 vaccination coverage in Shenzhen has been close to 50% in 2021 and 2022, serotype 19F remains the most prevalent in colonization in respiratory tract in hospitalized children, reaching to 23% in all pneumococcal positive cases.6 Various clinical studies also revealed that 19F has the highest invasiveness rate in China.4–10 In this study, we found that compared to 19F isolates from the lower respiratory tract, invasive 19F exhibits stronger adhesion capabilities in the human epithelial co-culture microenvironment, which is closely associated with the genetic variations unique to invasive 19F. This indicates that, despite not forming a stable evolutionary lineage, the same serotype may exhibit a phenotype with a marked tendency towards aggressiveness, which is closely correlated with considerable genetic variability. These discoveries expand our understanding of the mechanisms behind the invasiveness of 19F, providing a basis for future research on mechanisms behind invasiveness.
Invasive occurrences significantly compromise the survival outcomes and therapeutic prognosis of pediatric patients. Interpreting invasive strains’ genomic and co-culture data helps us further understand the sources of invasiveness variability within the same serotype and the molecular mechanisms of high invasiveness. The diagnosis of pneumococcal disease can be achieved through culturing the pathogen and using molecular diagnostics for serotyping to evaluate strain virulence further. However, in clinical settings, the same serotype can exhibit different phenotypes, which are closely related to patient immunity, co-infection with influenza viruses, among other factors, but whether these are related to the strain characteristics themselves merits further exploration. While numerous studies report genomic differences between invasive pneumococci and carriage strains, the serotype distribution between these groups introduces variability, with most differences likely still originating from inter-serotype variations, making it challenging to pinpoint invasive mechanisms in the same serotype.28–30 Reports on comparative studies between invasive and carriage strains of serotype 3 and 19A, have find significant genomic differences.31,32 Serotype 19F, highly invasive in Shenzhen, is also reported to be highly invasive in other countries and regions,4,20 and has been identified as high virulence and resistance serotype, with virulence genes facilitating adhesion, invasion, and immune evasion.33 This serotype serves as an optimal serotype for studying the sources of invasiveness within the same serotype category.
In our study, we conducted a pan-genomic comparison analysis of the genomes from invasive and noninvasive strains, identifying a tendency for each group to carry different gene sequences. Due to sample limitations, there was a significant difference in time period between the two groups, which may lead to significant branches in the evolutionary tree. In this analysis, we use Scoary and Pyseer to perform the pan-GWAS analysis, which both control for biases introduced by clonal sampling and evolutionary transitions. Even after adjusting for population structure, we still identified significant differences in certain genes. Most of these genes were associated with transposase which improve strain adaptation and genetic diversity.34
To confirm how the differential genes identified influence the invasiveness of the strains, we evaluated the adhesion and invasion capabilities of both groups in co-culture with lung epithelial cells, and conducted a correlation analysis. Ultimately, we found that over a certain percentage of the invasive strain specific genes and the strains’ adhesion capabilities associated genes, including the ISL3 family transposase and MarR family genes. The finding shows that the ability of pneumococci to adhere to and invade epithelial cells is a prerequisite for invasive infection. The gene group_377 was annotated by the 3D-homology alignment tool HHblits,35 and the top three best hits were MarR family, virulence regulator SarX, and virulence regulator SlyA. The virulence regulator SarX and SlyA are belong to or share homolog with MarR family of transcriptional regulators.36,37 SarX has been identified as a crucial regulator of biofilm formation in Staphylococcus aureus by upregulating the transcription of the icaADBC operon and promoting the production of polysaccharide intercellular adhesin (PIA).36,38,39 Meanwhile, SlyA serves as an essential transcriptional regulator that increases the virulence of Salmonella by inducing the expression of cytolysin A, which aids in bacterial invasion.40 ISL3 is belong to DDE transposases that which is a major group of transposase enzymes and play crucial roles in genome dynamics. For instance, in Streptococcus agalactiae, DDE transposable elements have been shown to significantly enhance virulence and adaptability by promoting the expression of virulence genes and facilitating genomic rearrangements, enabling the bacterium to colonize various host environments effectively.41 Additionally, the power of transposase activity is evident in Escherichia coli, where a single transposon insertion upstream of the yrfF can lead to transition from commensal strain to pathogenic strain. This insertion enhances the bacterium’s ability to survive and evade the immune response of macrophages, highlighting the profound impact of DDE transposases on microbial pathogenicity and adaptation.42 In previous studies, researchers also found that serotype 14 isolated from blood also present different invasive capability in mouse infection, and genetic variations including many transposase gene such as IS1515 and IS1380-Spn1. Another study focus on the genetic variations of invasive and carriage 19A in China found that the different gene include many IS5 family transposases.32 However, in a study of serotype 1 found that the hyper-invasiveness of serotype 1 strains is likely an intrinsic trait rather than being associated with specific genetic variations, suggesting that all strains of this serotype may share similar pathogenic capabilities despite their different clinical outcomes.43 These differences in the results may be due to differences in the genetic background and competitive advantage of the different serotypes, and highlights the necessity of ongoing surveillance and in-depth research into pneumococcal serotypes to enhance clinical management and vaccine development. Based on these research findings, we believe that the identification of transposases as significant markers of pneumococcus invasiveness has important clinical implications. Transposon insertions can directly facilitate adaptive changes during infection processes, providing a mechanistic foundation for developing targeted anti-transposase interventions. However, further systematic investigation of their regulatory mechanisms and activation conditions remains essential. Additionally, more precise, efficient, and convenient transposase classification and detection methodologies must be developed to strengthen our collective ability to anticipate, prevent, and effectively respond to IPD threats. Establishing comprehensive surveillance systems to monitor transposase and transposon prevalence and distribution patterns in pneumococcal populations would significantly enhance our predictive capabilities regarding emergent pathogenic strains, ultimately improving global pneumococcal disease management strategies.
Limitations
In this study, the results may not be universally applicable. (1) Our strains were isolated from Shenzhen and the analysis was restricted to comparing isolates from respiratory tracts and invasive diseases. The results cannot fully capture the genetic diversity of strains isolated from other body sites. (2) Our study population showed a gender imbalance (around 66% male and 33% female), which may affect the generalizability of findings across genders, particularly if there are gender-specific host-pathogen interactions that influence invasiveness. (3) Our analysis was limited to the 19F serotype, which has the highest proportion of invasiveness, while other serotypes require further study. (4) Despite about half of the differential genes we identified are related to adhesion capabilities, their underlying molecular mechanisms still need further experimental validation, which will be a focus of our next research. To address the gender imbalance, we implemented statistical corrections through pan-genome analyses. And to strengthen our findings, we compared our results with published studies from different geographical regions and serotypes.
Conclusion
Our pan-GWAS analyses identified mobile genetic elements, particularly transposases, as key contributors to the invasiveness of pneumococcal serotype 19F. These findings not only enhance our understanding of the genetic factors driving pneumococcal pathogenicity but also open new avenues for developing more precise diagnostic tools and targeted therapeutic strategies. Future research focused on the functional characterization of these elements will be essential to fully elucidate their mechanistic role in invasiveness and their potential as targets for intervention.
Data Sharing Statement
All data supporting this study has been included in the manuscript and/or the supplementary materials.
Ethics Statement
The study was approved by the Shenzhen Children’s Hospital Institutional Ethics Committee (202200302).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The study was supported by Shenzhen Fundamental Research Program (JCYJ20180228175330567, JCYJ20210324115607021, JCYJ20220530152800001).
Disclosure
Xing Shi, Sandip Patil, Qiuwei Yi are co-first authors for this study. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wang J, Qiu L, Bai S, et al. Prevalence and serotype distribution of nasopharyngeal carriage of Streptococcus pneumoniae among healthy children under 5 years of age in Hainan Province, China. Infect Dis Poverty. 2024;13(7). doi:10.1186/s40249-024-01175-7
2. Wang L, Fu J, Liang Z, Chen J. Prevalence and serotype distribution of nasopharyngeal carriage of Streptococcus pneumoniae in China: a meta-analysis. BMC Infect Dis. 2017;17:765. doi:10.1186/s12879-017-2816-8
3. Wahl B, O’Brien KL, Greenbaum A, et al. Burden of Streptococcus pneumoniae and Haemophilus influenzae type b disease in children in the era of conjugate vaccines: global, regional, and national estimates for 2000–15. Lancet Glob Health. 2018;6:e744–e757. doi:10.1016/S2214-109X(18)30247-X
4. Huang L-D, Yang M-J, Huang -Y-Y, et al. Molecular characterization of predominant serotypes, drug resistance, and virulence genes of Streptococcus pneumoniae isolates from East China. Front Microbiol. 2022;13:892364. doi:10.3389/fmicb.2022.892364
5. Wang X, Cong Z, Huang W, Li C. Molecular characterization of Streptococcus pneumoniae isolated from pediatric patients in Shanghai, China. Pediat Pulmonol. 2020;55:2135–2141. doi:10.1002/ppul.24877
6. Shi X, Patil S, Wang Q, et al. Prevalence and resistance characteristics of multidrug-resistant Streptococcus pneumoniae isolated from the respiratory tracts of hospitalized children in Shenzhen, China. Front Cell Infect Microbiol. 2024;13. doi:10.3389/fcimb.2023.1332472
7. Chen H, Liu C. Molecular epidemiology of Streptococcus pneumoniae isosslated from children with community-acquired pneumonia under 5 years in Chengdu, China. Epidemiol Infect. 2022;151:e2. doi:10.1017/S0950268822001881
8. Ma M, Yuan M, Li M, et al. Serotype distribution and characteristics of the minimum inhibitory concentrations of Streptococcus pneumoniae isolated from pediatric patients in Kunming, China. Curr Microbiol. 2021;78:954–960. doi:10.1007/s00284-021-02365-4
9. Fu J, Yi R, Jiang Y, et al. Serotype distribution and antimicrobial resistance of Streptococcus pneumoniae causing invasive diseases in China: a meta-analysis. BMC Pediatric. 2019;19:424. doi:10.1186/s12887-019-1722-1
10. Guo M-Y, Shi X-H, Gao W, et al. The dynamic change of serotype distribution and antimicrobial resistance of pneumococcal isolates since PCV13 administration and COVID-19 control in Urumqi, China. Front Cell Infect Microbiol. 2023;13. doi:10.3389/fcimb.2023.1110652
11. Ning X, Li L, Liu J, et al. Invasive pneumococcal diseases in Chinese children: a multicentre hospital-based active surveillance from 2019 to 2021. Emerg Microbes Infect. 2024;13(1):2332670. doi:10.1080/22221751.2024.2332670
12. Xiaofei L, Yudan LI, Qinghui C, et al. Effectiveness of 13-valent pneumococcal conjugate vaccine against vaccine-serotype community acquired pneumococcal diseases among children in China: a test-negative case-control study. Vaccine. 2024;42(6):1275–1282. doi:10.1016/j.vaccine.2024.01.068
13. Liang Z, Fu J, Li L, et al. Molecular epidemiology of Streptococcus pneumoniae isolated from pediatric community-acquired pneumonia in pre-conjugate vaccine era in Western China. Ann Clin Microbiol Antimicrob. 2021;20(4). doi:10.1186/s12941-020-00410-x
14. Bogaert D, Groot RD, Hermans PWM. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis. 2004;4:144–154. doi:10.1016/S1473-3099(04)00938-7
15. Weiser JN, Ferreira DM, Paton JC. Streptococcus pneumoniae: transmission, colonization and invasion. Nat Rev Microbiol. 2018;16:355–367. doi:10.1038/s41579-018-0001-8
16. Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6:288–301. doi:10.1038/nrmicro1871
17. Brooks LRK, Mias GI. Streptococcus pneumoniae’s virulence and host immunity: aging, diagnostics, and prevention. Front Immunol. 2018;9:1366. doi:10.3389/fimmu.2018.01366
18. McCullers JA. Insights into the Interaction between Influenza virus and Pneumococcus. Clin Microbiol Rev. 2006;19:571–582. doi:10.1128/CMR.00058-05
19. Song JY, Nahm MH, Moseley MA. Clinical implications of Pneumococcal Serotypes: invasive disease potential, clinical presentations, and antibiotic resistance. J Korean Med Sci. 2013;28:4–15. doi:10.3346/jkms.2013.28.1.4
20. Müller A, Kleynhans J, de Gouveia L, et al. Streptococcus pneumoniae Serotypes associated with death, South Africa, 2012–2018. Emerg Infect Dis J. 2022;28(1):166–179. doi:10.3201/eid2801.210956
21. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi:10.1093/bioinformatics/btu170
22. Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A. Using SPAdes De Novo assembler. Curr Protoc Bioinform. 2020;70(1). doi:10.1002/cpbi.102
23. Epping L, van Tonder AJ, Gladstone RA, et al. SeroBA: rapid high-throughput serotyping of Streptococcus pneumoniae from whole genome sequence data. Microbial Genomics. 2018;4:e000186. doi:10.1099/mgen.0.000186
24. Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi:10.1093/bioinformatics/btv421
25. Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016;17:238. doi:10.1186/s13059-016-1108-8
26. Kursa MB, Rudnicki WR. Feature Selection with the Boruta Package. J Stat Softw. 2010;36:1–13. doi:10.18637/jss.v036.i11
27. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit-learn: machine learning in python. J Mach Learn Res.2011;12:2825–2830.
28. Obolski U, Gori A, Lourenço J, et al. Identifying genes associated with invasive disease in S. pneumoniae by applying a machine learning approach to whole genome sequence typing data. Sci Rep. 2019;9:4049. doi:10.1038/s41598-019-40346-7
29. Mohale T, Wolter N, Allam M, et al. Genomic differences among carriage and invasive nontypeable pneumococci circulating in South Africa. Microb Genom. 2019;5:e000299. doi:10.1099/mgen.0.000299
30. Obolski U, Gori A, Lourenço J, et al. Identifying Streptococcus pneumoniae genes associated with invasive disease using pangenome-based whole genome sequence typing. bioRxiv. 2018;2018:314666. doi:10.1101/314666
31. Cleary DW, Lo SW, Kumar N, et al. Comparative genomic epidemiology of serotype 3 IPD and carriage isolates from Southampton, UK between 2005 and 2017. Microb Genom. 2023;9:mgen000945. doi:10.1099/mgen.0.000945
32. Li T, Huang J, Yang S, et al. Pan-genome-wide association study of serotype 19A Pneumococci identifies disease-associated genes. Microbiol Spectr. 2023;11:e04073–22. doi:10.1128/spectrum.04073-22
33. Liu L, Wang Y, Ge L, et al. Integrated genomic analysis of antibiotic resistance and virulence determinants in invasive strains of Streptococcus pneumoniae. Front Cell Infect Microbiol. 2023;13. doi:10.3389/fcimb.2023.1238693
34. Oggioni MR, Claverys J-P. Repeated extragenic sequences in prokaryotic genomes: a proposal for the origin and dynamics of the RUP element in Streptococcus pneumoniae. Microbiology. 1999;145:2647–2653. doi:10.1099/00221287-145-10-2647
35. Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–248. doi:10.1093/nar/gki408
36. Hao Z, Guo Y, Rao L, et al. Deletion of SarX decreases biofilm formation of Staphylococcus aureus in a Polysaccharide Intercellular Adhesin (PIA)-dependent manner by downregulating spa. Infect Drug Resist. 2021;14:2241. doi:10.2147/IDR.S305650
37. Ellison DW, Miller VL. Regulation of virulence by members of the MarR/SlyA family. Curr Opin Microbiol. 2006;9:153–159. doi:10.1016/j.mib.2006.02.003
38. Rowe SE, Mahon V, Smith SG, O’Gara JP. A novel role for SarX in Staphylococcus epidermidis biofilm regulation. Microbiology. 2011;157:1042–1049. doi:10.1099/mic.0.046581-0
39. Cue D, Lei MG, Lee CY. Activation of sarX by Rbf is required for biofilm formation and icaADBC expression in Staphylococcus aureus. J Bacteriol. 2013;195:1515–1524. doi:10.1128/JB.00012-13
40. Krone L, Mahankali S, Geiger T. Cytolysin A is an intracellularly induced and secreted cytotoxin of typhoidal Salmonella. Nat Commun. 2024;15:8414. doi:10.1038/s41467-024-52745-0
41. Fléchard M, Gilot P. Physiological impact of transposable elements encoding DDE transposases in the environmental adaptation of Streptococcus agalactiae. Microbiology. 2014;160:1298–1315. doi:10.1099/mic.0.077628-0
42. Proença JT, Barral DC, Gordo I. Commensal-to-pathogen transition: one-single transposon insertion results in two pathoadaptive traits in Escherichia coli -macrophage interaction. Sci Rep. 2017;7:4504. doi:10.1038/s41598-017-04081-1
43. Chaguza C, Ebruke C, Senghore M, et al. Comparative genomics of disease and carriage serotype 1 Pneumococci. Genome Biol Evol. 2022;14:evac052. doi:10.1093/gbe/evac052
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.