")
Back to Journals » Cancer Management and Research » Volume 16
Pediatric Adrenocortical Carcinoma: The Nuts and Bolts of Diagnosis and Treatment and Avenues for Future Discovery
Authors O'Neill AF, Ribeiro RC, Pinto EM , Clay MR, Zambetti GP, Orr BA, Weldon CB, Rodriguez-Galindo C
Received 5 January 2024
Accepted for publication 26 June 2024
Published 7 September 2024 Volume 2024:16 Pages 1141—1153
DOI https://doi.org/10.2147/CMAR.S348725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Allison F O’Neill,1 Raul C Ribeiro,2 Emilia M Pinto,3 Michael R Clay,4 Gerard P Zambetti,3 Brent A Orr,3 Christopher B Weldon,5 Carlos Rodriguez-Galindo2,6
1Department of Pediatric Oncology, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Boston, MA, USA; 2Department of Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA; 3Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA; 4Department of Pathology, Children’s Hospital Colorado, Denver, CO, USA; 5Department of Surgery, Boston Children’s Hospital, Boston, MA, USA; 6Department of Global Pediatric Medicine, St. Jude Children’s Research Hospital, Memphis, TN, USA
Correspondence: Allison F O’Neill, Department of Pediatric Oncology, Dana-Farber Cancer Institute, 450 Brookline Avenue – DA3111, Boston, MA, 02215, USA, Email [email protected]
Abstract: Adrenocortical tumors (ACTs) are infrequent neoplasms in children and adolescents and are typically associated with clinical symptoms reflective of androgen overproduction. Pediatric ACTs typically occur in the context of a germline TP53 mutation, can be cured when diagnosed at an early stage, but are difficult to treat when advanced or associated with concurrent TP53 and ATRX alterations. Recent work has demonstrated DNA methylation patterns suggestive of prognostic significance. While current treatment standards rely heavily upon surgical resection, chemotherapy, and hormonal modulation, small cohort studies suggest promise for multi-tyrosine kinases targeting anti-angiogenic pathways or immunomodulatory therapies. Future work will focus on novel risk stratification algorithms and combination therapies intended to mitigate toxicity for patients with perceived low-risk disease while intensifying therapy or accelerating discoveries aimed at improving survival for patients with difficult-to-treat disease.
Keywords: adrenocortical, pediatric, management, discovery, carcinoma, tumors
Introduction
Adrenocortical tumors (ACTs) are rare neoplasms that span the spectrum from benign to malignant behavior with malignant tumors generally referred to as either adrenocortical carcinomas (ACCs) or malignant ACTs: the focus of this manuscript. Per standard convention, the term ACT will be utilized for the remainder of the manuscript to refer to the malignant entity. Malignant ACTs can affect children of any age from birth to adolescence and young adulthood. A recently concluded Children’s Oncology Group trial (COG ARAR0332) has provided unique insights into tumor biology, guides current standards of care, and has served as a valuable tumor repository for discovery. Treatment for relapsed or refractory disease is often extrapolated from adult literature with variable therapeutic success. Recent discoveries in the genomic and immunophenotype of ACTs may guide an evolution in risk stratification and the treatment algorithm of newly diagnosed or recurrent disease. Herein we will outline the epidemiology, clinical presentation, surgical and oncologic management of these tumors, and predict future directions based on recent preclinical and translational developments.
Epidemiology
Malignant ACTs occur with a frequency of approximately 0.2–0.3 cases per million children per year in the United States and account for approximately 0.2% of all pediatric malignancies.1,2 Pediatric ACTs occur with a bimodal incidence in children less than 5 years and greater than 10 years of age with a female predominance in both groups.3 In the south of Brazil, there is a 10–15-fold increase in pediatric ACT diagnoses, particularly in younger children, due to the high prevalence of the TP53 p.R337H founder variant. In contrast to the majority of pediatric malignancies, pediatric ACTs, even apart from those in the Brazilian cohort, are typically linked to a cancer predisposition syndrome.4,5
Germline Associations
Pediatric ACTs are most often associated with Li-Fraumeni Syndrome6,7 and the more broadly defined Li-Fraumeni spectrum. Additional genetic disorders related to ACTs include Beckwith-Wiedemann, Lynch syndrome, and Multiple Endocrine Neoplasia (MEN) Type 1.8 The Li-Fraumeni spectrum is associated with germline TP53 mutations, with variable penetrance and latency, that predispose carriers to diverse tumor types that span early childhood to late adulthood.9 TP53 is located on chromosome 17p13, and its main function is to regulate the cell cycle, apoptosis, DNA repair, metabolism, and angiogenesis in response to DNA damage and other cellular stressors.10,11 The prevalence of germline TP53 mutations in patients with sporadic ACT varies by age group and is low (3–6%) in adults,12 but significantly higher (50–80%) in children.13,14 Of note, the types of germline TP53 mutations in children with ACT appear to differ from those typically found in Li Fraumeni Syndrome (LFS)-core cancer carriers, as many pediatric ACTs harbor hypomorphic TP53 variants encoding a partially functional p53 protein. The excess of pediatric ACTs associated with hypomorphic TP53 alleles suggests that the adrenal cortex is particularly susceptible to reduced TP53 tumor suppressor activity during embryogenesis and developmental tissue remodeling of the adrenal gland. Therefore, pediatric ACTs can serve as a sentinel tumor for families within the LFS spectrum warranting genetic testing for TP53 variants in the proband and other family members.
An increased incidence of ACTs is also observed within the context of Beckwith-Wiedemann syndrome (BWS), which is associated with tissue overgrowth and tumor predisposition.15 The classical form of BWS includes patients with pre- and post-natal overgrowth, visceromegaly, macroglossia, neonatal hypoglycemia, hemihypertrophy, and cancer susceptibility. Phenotypic variability among individuals and tissue mosaicism extends the classification [Beckwith-Wiedemann syndrome spectrum (BWSp)] to include patients with isolated lateralized overgrowth and those with suggestive features (large for gestational age, facial nevus simplex, polyhydramnios, transient hypoglycemia, nephromegaly, hepatomegaly, umbilical hernia, and tumors) that do not otherwise fit the classical clinical criteria of BWS.16 The molecular events associated with BWSp are complex and result from abnormal regulation of imprinting centers located on chromosome 11p15 that control the expression of IGF2, CDKN1C, KCNQ1, and H19. IGF2 is a paternally expressed fetal growth factor, whereas the cell cycle inhibitor CDKN1C (p57), potassium channel protein KCNQ1, and noncoding H19 transcripts are expressed from the maternal allele.17 BWSp associated tumors (Wilms tumor, neuroblastoma, hepatoblastoma, and rhabdomyosarcoma), including adrenocortical tumors, are dependent on chromosome 11p15 alterations (paternal uniparental disomy, mutations, duplication, or deletions) that usually occur during early embryonic development.18 However, germline genetic and epigenetic alterations at chromosome 11p15 have also been observed in pediatric patients with ACT without classic clinical features of BWS.19,20
Clinical Presentation
Symptoms and Work-Up
Both benign and malignant ACTs can secrete excess androgens and approximately 80% of children with malignant ACTs have functional tumors.21 Therefore, clinical signs associated with the hyperproduction of androgens (virilization) or cortisol (Cushing syndrome) should alert the physician to the possibility of an ACT. Cursory laboratory assessments should include serum electrolytes (sodium, potassium), fasting blood glucose, cortisol, adrenocorticotropic hormone (ACTH), adrenal androgens (DHEA, DHEA sulfate (DHEAS), androstenedione, testosterone), and 17-hydroxyprogesterone. In most cases, an initially obtained abdominal ultrasound will reveal an adrenal mass.
Subsequent evaluation of a child with a presumptive diagnosis of ACT is based on features relevant to surgical decision-making. Magnetic resonance imaging (MRI) and computed tomography (CT) of the abdomen and pelvis can detect invasion of adjacent organs and lymph nodes as well as vascular tumor extension. Distant metastases, usually in the liver, lungs, kidneys, or bones, are further evaluated by 18-fluorodeoxyglucose positron emission tomography (PET) integrated with MRI or CT. Conventional radiologic modalities must assess potential metastasis in those anatomic areas if PET-MRI/CT is unavailable.
Needle biopsy of the primary tumor should be avoided as this can cause rupture of the tumor, resulting in intraperitoneal dissemination. If clinical observations (virilization or signs and symptoms of Cushing syndrome), laboratory test results (elevated androgens and/or cortisol), and imaging findings (calcifications and necrosis) strongly support the diagnosis of an ACT, but complete resection of the tumor is not possible, it is reasonable to initiate empiric preoperative chemotherapy before the definitive surgery. In patients with metastatic disease, a biopsy of the metastatic lesion might have fewer adverse consequences than a needle biopsy of the primary tumor.
Histopathology
The histopathologic classification of pediatric adrenocortical tumors is a complex process, combining both gross and microscopic features. Unlike in adult disease, the Weiss and Modified Weiss criteria are not recommended for classification in the pediatric population, as they tend to overestimate the risk of malignant behavior.22–24 Instead, the Wieneke Criteria represent the most commonly utilized system for risk stratification in the pediatric population, and have been established as a reliable means of differentiating between adrenocortical adenomas and carcinomas (Table 1).25
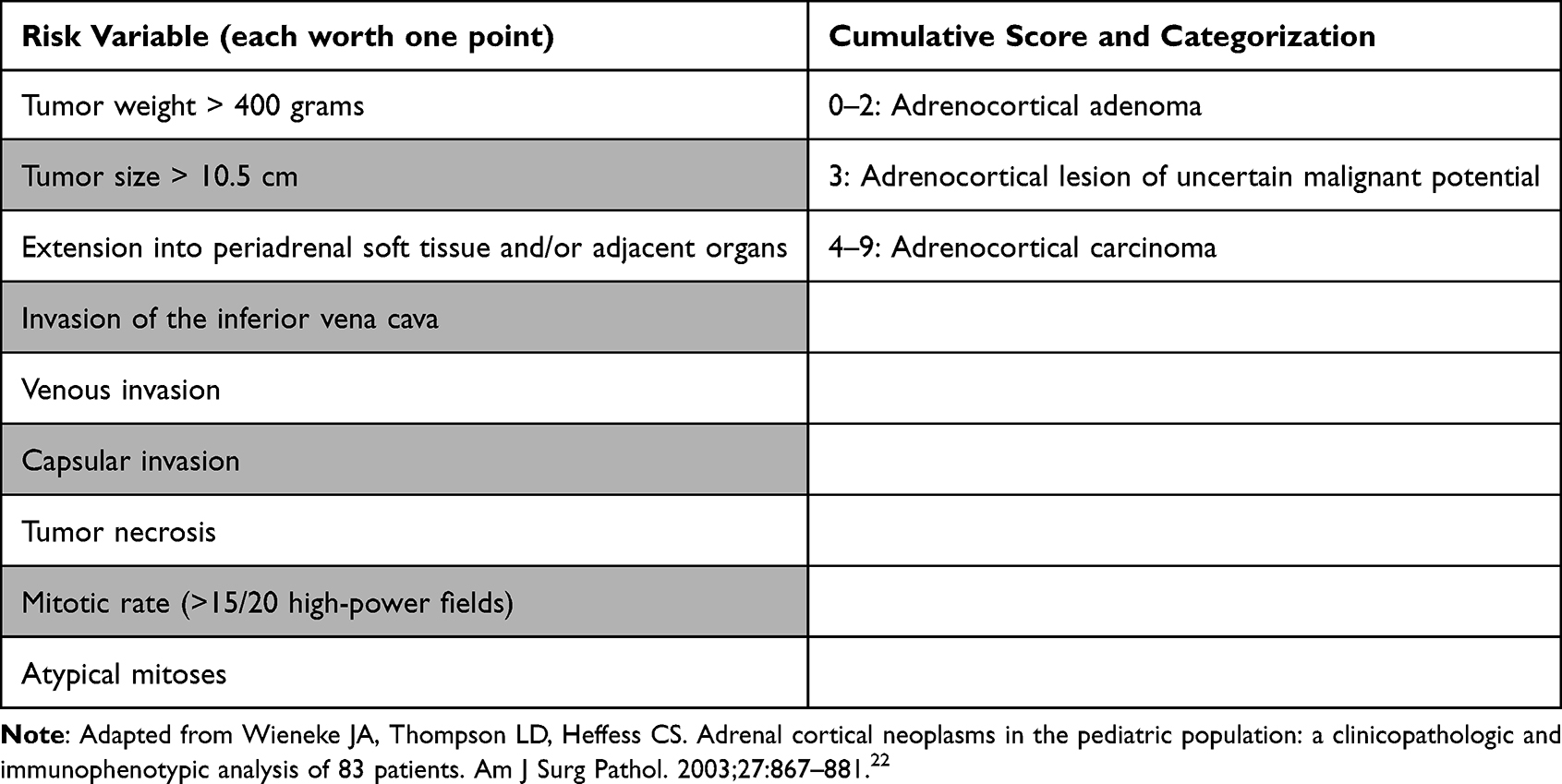 |
Table 1 Wieneke Classification Criteria of Pediatric Adrenocortical Neoplasias |
The Wieneke system is unique in that it includes the addition of the gross clinical variables, tumor size, and weight to the classification model. This system also intentionally uses a slightly different point allocation system, putting less weight on microscopic variables when compared with the Modified Weiss model. Familiarity with the Wieneke model has grown, and the proposed criteria have now been thoroughly vetted and are considered user-friendly by general surgical pathologists.26
Several localized microscopic variables are evaluated including tumor necrosis, elevated mitotic rate, and atypical mitoses (Figure 1). In addition, evidence of aggressive local growth is captured by review of capsular invasion, venous invasion, direct invasion of the inferior vena cava, and generalized extension into the periadrenal soft tissue. Immunohistochemical staining plays a key role in the diagnosis of adrenocortical neoplasia (Figure 2). Studies can be divided into those that correlate with cell of origin (expected positive: synaptophysin, SF-1, Inhibin, Melan-A, Cytokeratin Cam 5.2. Expected negative: chromogranin), those that provide prognostic data (Ki-67)27–30 and those that correlate with molecular alterations (p53, beta-catenin, MSH2, MLH1, MSH6, PMS2).
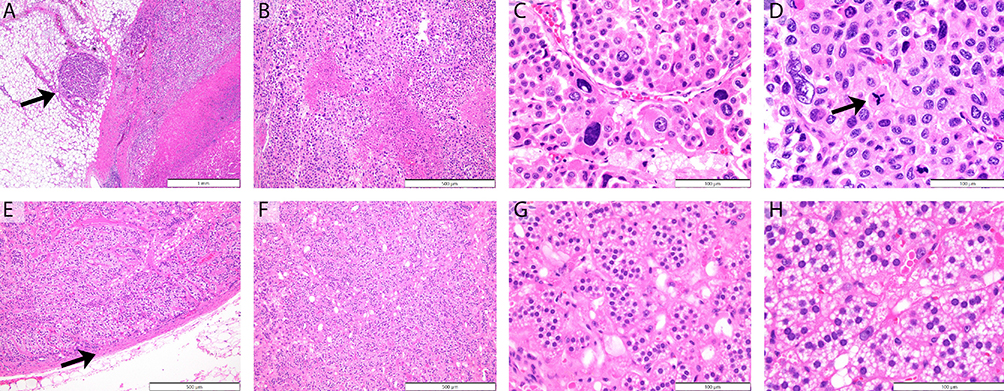 |
Figure 1 Histologic features of malignant adrenocortical tumors (ACTs) and adrenocortical adenoma (ACAs). ACTs often display irregular capsular surfaces, with soft tissue extension (A). Coagulative tumor cell necrosis (B), prominent cell to cell variability (C), and increased mitotic activity with atypical mitotic divisional forms (D) are frequently identified. In contrast, ACAs have intact encapsulated borders (E), an absence of necrosis (F), cellular monotony (G), and rare to absent mitotic activity (H). (A) 4x HE, ACT with irregular capsular invasion with soft tissue nodule (arrow). (B) 10x HE, ACT with coagulative necrosis. (C) 40x HE, ACT with nuclear atypia and irregular nested growth. (D) 60x HE, ACT with atypical mitosis (arrow) and single cell necrosis. (E) 10x HE, ACA with smooth capsular border (arrow). (F) 10x HE, ACA with monotonous cellularity. (G) 40x HE, ACA with regular nested growth and no evidence of necrosis. (H) 60x HE, ACA with cellular uniformity and absent mitotic division. |
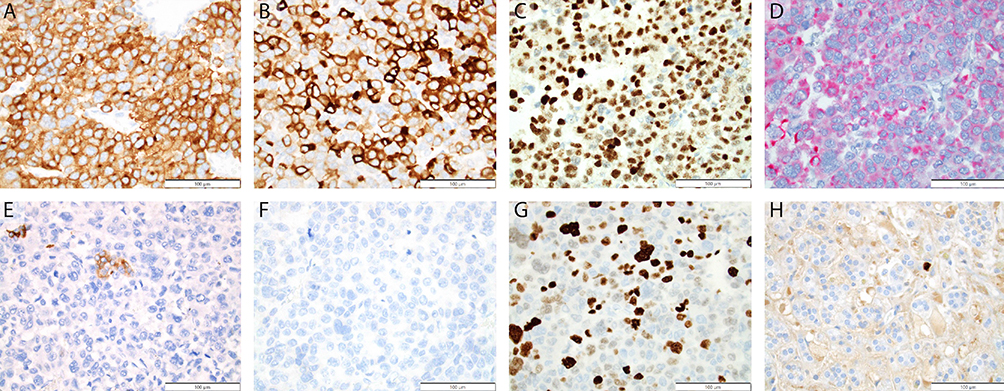 |
Figure 2 Immunohistochemical findings in ACT. Adrenocortical neoplasms are reliably positive for Synaptophysin (A), Inhibin (B), SF1 (C), and Melan A (D) in most instances. These tumors may show weak or focal staining in higher grade lesions, particularly with cytokeratin expression. Cytokeratin Cam 5.2 (E), is often at least focally positive. In contrast to medullary lesions, Chromogranin (F) is reliably negative. Ki-67 staining shows an elevated proliferative index in ACTs (G), when compared to adrenocortical adenoma (H). (A) 40x Synaptophysin – positive. (B) 40x Inhibin – positive. (C) 40x SF1 – positive. (D) 40x Melan-A Red – Positive. (E) 40x Cytokeratin Cam 5.2, Positive, focal. (F) 40x Chromogranin – Negative. (G) 40x Ki-67 in ACC – 30-40%. (H) 40x Ki-67 in ACA – <5%. |
Taken together, determining whether a neoplasm is of adrenocortical origin is often quite straightforward. Most lesions are also easily classified as either having features of pure benign adenoma or malignant carcinoma. The difficulty lies in those tumors with intermediate grade and features. In such cases, collaborative discussions in a multidisciplinary tumor board environment are often necessary particularly given therapeutic implications to diagnostic accuracy.
Historical Context
Prior to the 1980s, there were few organized efforts to meaningfully source data regarding these rare tumors creating a primary barrier to uniform diagnostic and therapeutic approach. Since then, there have been several international efforts aimed at sourcing aggregate data to ascertain prognostic factors and guide therapy for pediatric patients with ACTs. With the recognition that single-disease, prospective registries have the potential to generate data on the natural history, histology, treatment response, and outcomes of rare tumors, one of the first organized attempts to collect data was the International Adrenocortical Tumor Registry (IPACTR), launched by St. Jude’s Children’s Research Hospital in response to a cluster of cases of ACT in South Brazil and with the goal to integrate clinical data with laboratory investigation.31
The objective of the first version of the St. Jude IPACTR registry was to perform a retrospective demographic and clinical analysis of cases admitted to St. Jude or to Brazilian hospitals. Approximately 80% of the registered patients were from institutions in southern Brazil, and data analysis showed that tumor size (weight <100 g) and age (<4 years) were each independently associated with prognosis in patients with completely resected tumors. Patients with metastatic disease had dismal outcomes.21 The discovery that the registered patients from Brazil carried a founder TP53 variant raised questions about whether one could generalize these findings to patients with wild-type TP53 ACT.32,33 The second version of the IPACTR aimed to prospectively collect demographic and medical information, including any family history of cancer and data from yearly follow-ups. Limited (targeted) gene sequencing was performed for clinical use, and tumor tissue and blood were stored in a biorepository for research studies.
Since then, other collaborative efforts have contributed to our understanding of pediatric ACTs. Cecchetto et al analyzed data from Germany, France, Poland and Italy as part of an initiative led by the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT). In this 82 patient cohort, progression-free survival and overall survival were demonstrated to be most heavily impacted by the presence of distant metastases and tumor volume >200 cm3.34
The findings that age, tumor size, and even the presence of metastatic disease at diagnosis in patients with wild-type TP53 are not independently associated with prognosis have contributed greatly to the field.19,35 IPACTR data formed the basis for development of the Children’s Oncology Group Phase III trial ARAR0332, which enrolled patients at all participating COG sites and in Brazil. This trial has valuably contributed to our current accepted medical and surgical standards in treating ACTs.
Current Treatment Standards
Pediatric ACT management is complex and requires a multidisciplinary approach. Careful consideration from the perspectives of surgery, oncology, radiology, and endocrinology is critical. Because ACTs can progress relatively quickly, the time between initial suspicion of the diagnosis and definitive treatment should be as brief as possible.
Surgery
Surgery is indispensable for the successful treatment of pediatric ACT. Although pediatric ACT is partially chemosensitive and radiosensitive, neither chemotherapy nor radiotherapy can eradicate ACT without surgery. Therefore, the main goal of surgery is complete removal of the tumor including all or part of the involved adrenal gland without tumor capsule violation resulting in spoilage of the retroperitoneum or peritoneum (ie, spill).
The decision on whether to use an open or a laparoscopic surgical approach for local control depends on the experience of the surgeon and the tumor characteristics. The minimally invasive technique is appropriate for small tumors (<5 cm in their largest diameter) that are restricted to the adrenal gland and have benign radiographic characteristics. Experts strongly recommend that laparoscopic resection be performed at centers with experienced surgeons and a high case volume.36 In the absence of this skill set, minimally invasive approaches (laparoscopic and/or robotic) to the primary tumor are traditionally avoided so as not to increase the risk of spill and upstaging.36 This approach is supported by data from the GPOH-MET 97 non-randomized, single-arm study which demonstrated an increased rate of operative complications and a poorer overall survival in patients with tumors >300 mL in volume. Pre-operative biopsy and, unsurprisingly, tumor rupture were also associated with a poorer overall survival.37
When an open surgical procedure is considered more appropriate, this is performed via an abdominal incision (laparotomy) or a thoracoabdominal incision. A thoracoabdominal approach is preferred to decrease the risk of rupture, bleeding, and spillage. This approach is also recommended for small tumors that are densely adherent to the liver capsule. The risk of tumor rupture and spillage is high, even with relatively small tumors, because of the thin, friable pseudocapsule and the gelatinous tumor contents resulting from ongoing necrosis and calcification. Spillage has been associated with poor prognosis and upstaging of patients from stage II to stage III. Because many children with ACT have contralateral adrenal suppression (either due to tumor-mediated steroid production with suppression of endogenous ACTH or intentional adrenal suppression as part of systemic therapy), patients must receive corticosteroids with glucocorticoid and mineralocorticoid activity before, during, and after surgery.
Assessment of the surrounding area for occult disease is also mandated, including sampling suspicious lesions in the adjacent soft tissues and/or removing the ipsilateral draining lymph node basins. Resection of frank loco-regional (or even distant) metastatic deposits, in a select cadre of patients, can also be considered in facilitating the conduct of an operation (en-bloc procedure) and/or to reduce tumor burden and apparent endocrinopathies from hormone overproduction in metabolically active cases.38
An area of active study surrounds investigation of the role and extent of retroperitoneal lymph node dissection (RPLND) in this disease. There are published data in adult patients with stage I–III localized adrenocortical carcinoma, suggesting a survival benefit following pursuit of locoregional lymphadenectomy.39 The recently published COG trial sought to study the role of an RPLND in pediatric ACT, but the data are mixed with no clear benefit demonstrated in patients with Stage II disease.40 It is known that the draining lymph node basins for the adrenal glands are wide and the identification of precise draining lymph nodes is difficult despite use of adjuvants (indocyanine green) effective in other pediatric malignancies.41 Hence, better studies are warranted to determine the utility of this maneuver and to more adequately define the nodal basin. Tumor extension to blood vessels represents another surgical challenge in pediatric ACTs. In some cases, cardiac bypass may be needed to complete resection, especially when the thrombus reaches the right atrium.
Chemotherapy
Until 1960, surgery was the only available treatment modality for patients with ACTs. After mitotane was found to be effective in treating advanced ACC, the National Cancer Institute sponsored the production and distribution of this drug. In July 1970, the FDA approved mitotane for treating inoperable, functional, or nonfunctional ACTs. Since then, no other medication has been approved for treating adults or children with ACTs. In 1980, Tattersall et al reported that four adults with advanced ACTs had objective responses to cisplatin.42 Subsequently, several small retrospective series were reported in which patients with advanced disease were treated with a combination of mitotane and cisplatin with other agents, including etoposide, doxorubicin, cyclophosphamide, 5-fluorouracil, streptozotocin, and gemcitabine. A prospective randomized Phase III study of mitotane plus etoposide, doxorubicin, and cisplatin (EDP) vs streptozotocin in 304 patients aged >18 years with advanced adrenocortical carcinoma (tumor stage III or IV) showed that response and progression-free survival rates were significantly better with mitotane plus EDP than with mitotane plus streptozotocin. However, there was no significant difference in overall survival (greater than 70% of the patients died within 2 years of study entry).43 The available evidence indicates that patients with inoperable or metastatic ACT have dismal outcomes when treated with conventional chemotherapy regimens.
In adjuvant settings, the role of chemotherapy with mitotane alone or combined with cisplatin and etoposide has been investigated in adults. A recent prospective randomized study investigated mitotane vs surveillance in patients with a perceived low-to-intermediate risk of recurrence (ie, with completely resected tumors, stage I–III, and Ki-67 ≤10%). Patients who refused the randomization agreed to provide follow-up information via the European Network for the Study of Adrenal Tumors (ENSAT). The 5-year recurrence-free survival was 79% in the mitotane group and 75% in the surveillance group. Based on these results, the study group concluded that mitotane is not indicated for adults with a low-to-intermediate risk of recurrence.44 An ongoing randomized study is comparing mitotane alone with mitotane plus cisplatin and etoposide for patients with a high risk of relapse.
A pediatric focused, single-arm, prospective registry study of multi-agent chemotherapy ± mitotane was first conducted in Germany (GPOH-MET 97). Patients enrolled received chemotherapy with vincristine, ifosfamide, adriamycin, carboplatin and etoposide. Of the analyzed cohort, 34 patients (56.6%) received chemotherapy across various settings (neoadjuvant, adjuvant, salvage) while 32 patients (53.3%) received mitotane. Patients receiving mitotane for >6 months with levels consistently >14 mg/l achieved significantly better survival. EFS and OS were reported at 43.3% and 64.8%, respectively, but treatment conclusions, apart from the noted efficacy of combination chemotherapy + mitotane in a subset of patients, are difficult to derive given heterogeneity in treatment approach.45
The Children’s Oncology Group (COG) prospectively investigated the efficacy of a mitotane and cisplatin–based regimen for advanced childhood ACT.40 The ARAR0332 study adapted this treatment to the tumor stage according to the COG criteria. Patients with stage I tumors were treated with surgery alone; patients with stage II tumors underwent surgery of the primary tumor and RPLND; and patients with stage III or IV tumors were treated with surgery of the primary tumor, RPLND, and eight courses of EDP plus mitotane. Of 24 patients with stage I tumors, three had events, two experienced tumor relapse, and one developed acute lymphoblastic leukemia. Progression of the primary tumor was noted in seven of 15 patients with stage II tumors, four of 24 patients with stage III and 11 of 14 patients with stage IV tumors. The 5-year EFS estimates for stages I, II, III, and IV were 86.2%, 53.3%, 81%, and 7.1%, respectively (Figure 3). As mentioned previously, results did not confirm the benefit of RPLND for patients with stage II disease. Patients with stage II disease (tumors > 100g or 200cm3) who underwent resection of the primary tumor and RPLND had a surprisingly poor prognosis; whether this was due to variability in surgical approach to RPLND, omission of chemotherapy, biological factors, or interference with local lymphoid tumor immune surveillance is not known. Patients with stage III disease benefited from adjuvant chemotherapy. The outcomes of patients with metastatic disease were dismal. Notably, a feasibility analysis concluded that the chemotherapeutic regimen was not feasible, with approximately one-third of patients requiring significant dose reductions, suggesting that modifications are necessary to improve its tolerability.
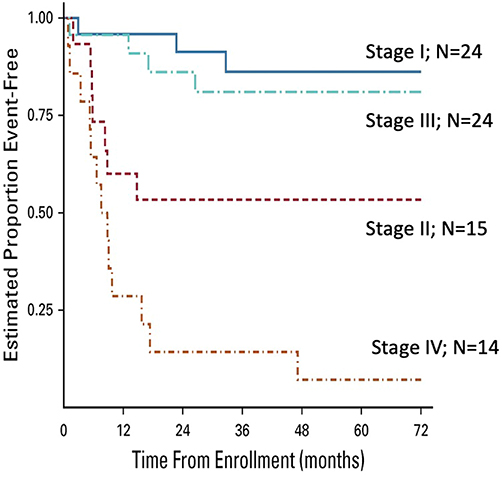 |
Figure 3 Event-free survival probabilities for 77 patients enrolled on COG ARAR0332. Notes: Reproduced with permission from Carlos Rodriguez-Galindo, Mark D Krailo, Emilia M Pinto, Farzana Pashankar, Christopher B Weldon, Li Huang, Eliana M Caran, John Hicks, M Beth McCarville, David Malkin, Jonathan D Wasserman, Antonio G de Oliveira Filho, Michael P LaQuaglia, Deborah A Ward, Gerard Zambetti, Maria J Mastellaro, Alberto S Pappo, Raul C Ribeiro. Treatment of Pediatric Adrenocortical Carcinoma With Surgery, Retroperitoneal Lymph Node Dissection, and Chemotherapy: The Children’s Oncology Group ARAR0332 Protocol. JCO. 2021 Aug 1;39(22):2463-2473. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8462560/.40 |
The role of mitotane in pediatric patients has been further explored via a questionnaire to pediatric oncology providers administered through the European Network for the Study of Adrenal Tumors Pediatric Working Group (ENSAT-PACT) and the International Consortium of Pediatric Adrenocortical Tumors (ICPACT). Results from this questionnaire concluded, in alignment with early adult data, that mitotane should be reserved for the treatment of patients with stage III and IV disease and incomplete resection or tumor spillage. Further recommendations were included in this consensus statement regarding dosing, serum levels, duration of therapy, and management of associated adrenal insufficiency.46
Radiotherapy
The evidence for the effectiveness of radiotherapy in pediatric ACT is weak. In adults with ACT, retrospective uncontrolled studies have yielded results suggestive of response and efficacy. In a retrospective analysis of 80 adults with advanced ACT in whom a total of 132 tumoral lesions were irradiated with different radiation doses, six lesions (4.5%) had a complete response, 52 (39%) had a partial response, 14 (10.4%) showed progression, and 60 (45.5%) remained stable. Survival among the 63 patients treated with curative intent was superior to patients treated with palliative intent.47 No prospective, randomized, controlled study has compared radiotherapy with observation or mitotane for adults and children with ACT in the adjuvant setting. Retrospective analyses of adult patients with ACT with resected primary tumors and a high risk of relapse revealed that local recurrence rates in irradiated patients were lower and survival was higher than in those receiving mitotane alone.48 In a recent retrospective literature survey of 1181 pediatric cases of ACT, it was reported that 80 of the patients were treated with radiotherapy. Radiotherapy was used with curative intent in 32 of these patients, for palliation in nine, and with unknown intent in the remaining 39 patients. The authors of the report could not determine the potential benefit of radiotherapy because of a lack of information on the outcomes.49 Local radiation might be an alternative to systemic chemotherapy for patients who do not carry germline TP53 variants and who have microscopic residual disease.
Summary and Future Directions
There is an urgent need to improve the management of pediatric ACT, particularly for those patients with metastatic disease. Conventional chemotherapy with mitotane is ineffective for these patients and is associated with severe toxicity; therefore, patients with stage IV disease should be considered for clinical trials investigating novel approaches. Patients with localized or regional disease can benefit from chemotherapy, but the current risk stratification is inadequate to distinguish patients at high risk of relapse. Histopathologic and genomic data might improve the current stage classification, and this information could be integrated into the management of the tumor. For example, tumor size correlates strongly with the occurrence of tumor relapse and progression in cases with germline TP53 mutations,14 whereas tumor size has no prognostic implications for patients with constitutional paternal uniparental disomy of chromosome 11p15.20
Therapies Under Investigation
As immunotherapeutics demonstrate promise across adult malignancies, they have likewise been trialed in adult patients with advanced or relapsed/refractory ACT. In one of the earliest series published, Habra et al studied single-agent pembrolizumab in a small cohort of 16 adult patients with advanced disease. Of the 14 response-evaluable patients, two achieved a partial response, seven had stable disease (lasting >4 months for six of the seven patients), and five progressed (14% objective response).50 Raj et al reported on 39 adult patients with advanced ACC who received pembrolizumab 200mg q3 weeks. The objective response rate in these patients was 23%, the disease control rate 52%, and the median progression-free survival 2.1 months, failing to meet the goal median duration of response set forth in the study. Two of six patients with MSI-high/MMR-D tumors responded.51 A similar experience was reported in a small cohort of patients treated with nivolumab.52 Avelumab was studied in 50 adult patients with relapsed/refractory ACT, 50% of whom received avelumab in combination with mitotane; 42% had stable disease as the best response and a median progression-free survival of 2.6 months. While PD-L1 expression trended with response, expression was not statistically linked to response.53 Cixutumumab has been studied in concert with mitotane as has cytotoxin interleukin-13 conjugated to the truncated, mutated form of Pseudomonas aeruginosa exotoxin, both with disappointing results.54–56
While immunotherapy approaches using chimeric antigen receptor (CAR) T cells can be effective in treating hematological malignancies,57 they have met less success in treating solid tumors, with the identification of tumor-specific antigens representing a key obstacle.58 Autologous CAR-T cells targeting B7-H3 (CD276) across solid tumors, ACTs included, are currently under study (NCT04897321). The possibility of combining strategies, including CAR-T cells with components targeting the steroidogenic pathway or synergizing with chemotherapeutic components, warrants further investigation.
The study of tyrosine kinase inhibitors in ACTs has been limited in scope. A retrospective series of eight heavily pre-treated adults with refractory ACT demonstrated that the combination of pembrolizumab and lenvatinib, a predominant VEGF-targeted multi-kinase inhibitor, yielded a partial response in two patients, stable disease in one patient, and progressive disease in five patients.59 Cabozantinib, a preferential inhibitor of cMET signaling shown to be relevant preclinically for viability and proliferation in ACTs60 has demonstrated a 19% partial response rate in a multi-institutional case series and more recently a 7.2-month PFS advantage in an adult Phase II trial.61,62 On the basis of these findings, efficacy of cabozantinib with or without checkpoint inhibition is the objective of study in a forthcoming clinical trial to enroll adults and pediatric patients >12 years of age with advanced or relapsed/refractory ACC. This trial may be compelling for pediatric patients given that in a basket trial of single-agent pembrolizumab, 8 of 136 patients with solid tumors had a partial response, two of whom had a diagnosis of ACT.63
The relatively unsuccessful outcomes for immunotherapy-based trials to date may be related to an excess of adrenal steroid hormones that are postulated to be immunosuppressive to circulating and tumor infiltrating immune cells.64 Insights into the immunophenotype of pediatric ACTs have been gleaned from analyses of tumor tissue collected from the IPACTR cohort of pediatric patients with germline TP53 mutations and from specimens collected for correlative studies on COG ARAR0332. Pediatric adrenocortical carcinomas express significantly lower levels of MHC class II mRNA compared to the normal adrenal cortex and adenomas as shown by microarray profiling.65 Follow-up studies combining microarray and IHC analyses have demonstrated that the MHC class II antigens detected in pediatric ACTs are primarily expressed by tumor-infiltrating immune cells, which are at basal levels in the normal cortex, most abundant in adenomas and lowest in carcinomas, especially those with worse prognosis.66 Conversely, MHC II expression has been shown to be higher in lower stage-, improved PFS-tumors, suggesting a potential role for CD4+ T-cell immune surveillance in disease control.66 A statistically significant increase in tumor infiltrating CD8+ cytotoxic T-lymphocytes has been demonstrated in tumors of patients with younger age and lower stage disease.67 Additional work is underway to pursue multiplex immunohistochemistry on samples from the COG ARAR0332 cohort to further define the immunophenotype of pediatric tumors across stages and prognostic categories.
Discovery
Genomic Landscape
Pediatric adrenocortical tumors are very heterogeneous at a molecular level. The genetic landscape of pediatric ACTs includes important somatic events (Figure 4). Copy neutral loss of heterozygosity (cn-LOH) selecting against maternal chromosome 11 resulting in the overexpression of IGF2 and concomitant downregulation of CDKN1C and H19 is observed in nearly all cases of pediatric ACTs (>90%).14,19 Moreover, genomic analyses of pediatric ACTs have identified cn-LOH of chromosome 11 as a potential driver that occurs early during tumorigenesis in both adenomas and carcinomas and is not related to prognosis. Widespread somatic 9q copy number gains and 4q34 copy number loss, as well as acquired mutations in CTNNB1 (Wnt signaling pathway) and ATRX (ATPase/helicase involved in chromosome remodeling and segregation) are also recurrently observed in childhood ACT. Notably, tumors harboring concurrent variants in both TP53 and ATRX, are associated with particularly poor outcome.14,40 Unfortunately, neither variant is clinically actionable.
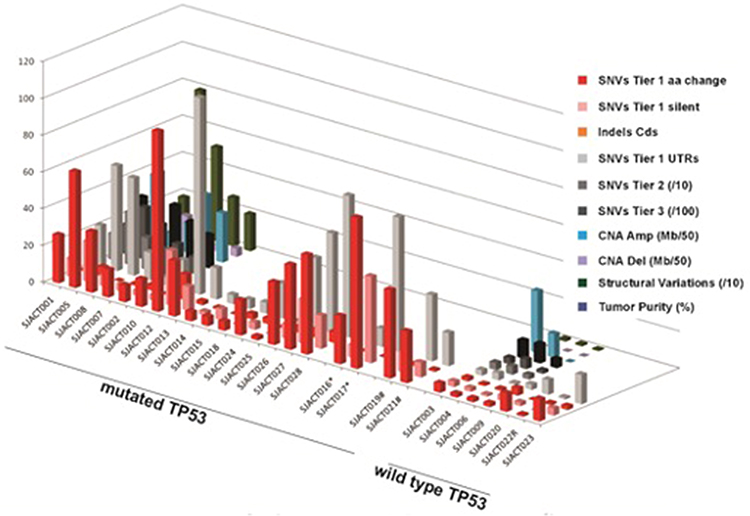 |
Figure 4 Molecular characterization of pediatric adrenocortical tumors. Whole genome and whole exome sequencing reveals massive chromosomal copy number alterations and additional molecular events in pediatric adrenocortical tumors associated with germline TP53 variants compared to tumors with wild-type TP53 sequence, which retain rather quiet genomes. |
DNA Methylation
DNA methylation at cytosines within CpG dinucleotides is critical for tissue specification during development and regulation of gene expression in cells.68 CpG sites are enriched within CpG islands located in promoter and enhancer regions in the genome, where the methylation state regulates the binding and activity of transcription factors.69,70 Similar to normal tissues, tumors also have stereotypic DNA methylation patterns.71 These patterns are believed to represent an interaction of signals derived from the tumor’s cell of origin and tumor-specific driver abnormalities.68 By matching tumor-specific methylation patterns to clinicopathologic and sequencing data, these signatures have been exploited to identify both novel tumor types and molecular subtypes within established histologically defined tumor entities.72 The most common assay for evaluating genome-wide DNA methylation across the genome is the Illumina Infinium BeadChip Array (450K or EPIC) due to its relatively robust performance on both fresh/frozen and formalin-fixed paraffin-embedded (FFPE) tissues.72 The high fidelity on FFPE tissue makes DNA methylation profiling amenable to use for clinical diagnostics, and comprehensive supervised classification models have been established for both brain tumors and sarcomas.73,74
DNA methylation profiling has revealed molecular heterogeneity in ACTs, both in adult and pediatric cohorts.35,75 These molecular differences likely have clinical implications, as global hypomethylation and a CpG-island methylator phenotype (CIMP) have been associated with aggressive behavior in adult ACTs.76,77 In pediatric ACTs, two subtypes have been identified (pACT A1 and A2) that differ in tumor characteristics, age of onset, hormonal symptoms, and prognosis.35,78 While DNA methylation groups have also been defined in adult ACTs, the pediatric classes appear distinct from their adult counterparts.35 pACT-A1 tumors have significantly inferior clinical outcomes compared to A2 tumors.76,77 In fact, DNA methylation class remains an independent predictor of outcome in pediatric ACTs, even after adjusting for other clinical variables.35,78
The underlying molecular differences driving the pACT molecular subtypes are currently unknown. pACT-A1 and A2 tumors cannot be entirely distinguished by other molecular abnormalities, such as germline and somatic mutations or copy number changes. Germline and somatic TP53 mutations are found in both A1 and A2 tumors.35,78 In fact, the founder TP53 p.R337H variant distributes in both A1 and A2 groups.78 CTNNB1 mutations are enriched in the A1 group but are also not exclusive to that group or uniformly detected.35 Diverse WNT-activating mechanisms have been described in ACTs and are associated with aggressive behavior.77,79 Additional study is necessary to determine if these alternative mechanisms are also active in pediatric tumors, and whether WNT activation is a primary driver of aggressive behavior in pACT-A1 tumors. Among other highly differentially methylated genes in the pACT-A1 and -A2 groups are genes in pathways implicated in adrenocortical tumor biology, including ADM, CACNA1H, CDKN1B, CLDN1, GNAS, IGF2, and PRKCA.35 Further evaluation of the class-specific activity of these genes and pathways may add additional insight into underlying biological differences between pACT methylation classes. Validation of these methylation findings is currently underway from tumor specimens collected on the COG ARAR0332 trial with cross-cutting analyses planned to determine whether these methylation findings correlate with multiplex immunohistochemical findings.
Future Directions
Several knowledge gaps have critical implications for managing pediatric ACTs and pose challenges that need to be addressed, including developing optimal treatment for children with advanced-stage disease, resolving the heterogeneity of cases currently classified as stage II, innovating mechanisms of disease staging and risk stratification taking into account germline and somatic genomic alterations, integrating methylation profiling into pediatric ACT classification, understanding the tumor immune milieu, and forging international collaborations (Figure 5). The degree to which each of these factors impact prognosis and/or will contribute to protocol design, risk stratification, and the assignment of therapy remains unknown. As novel therapies are discovered, trials bridging the adult and pediatric populations will provide unique insight as to the influence of developmental age, germline status, immunologic function, and disease biology on patient outcomes. As with other rare tumors, adrenocortical neoplasms have the capacity to serve as an ideal paradigm for collaboration between clinicians, basic and translational researchers, and adult and pediatric providers.
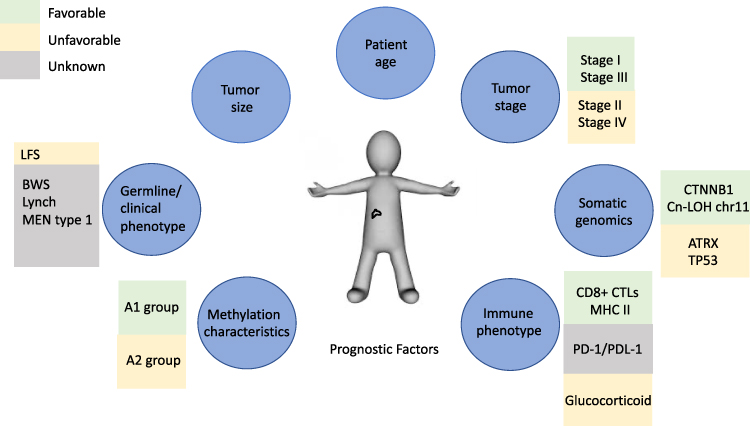 |
Figure 5 Figure depicting prognostic factors for ACTs guiding potential future risk stratification and treatment assignment (green = favorable, yellow = unfavorable, gray = unknown). |
Disclosure
The authors report no conflicts of interest in this work.
References
1. Varley JM, McGown G, Thorncroft M, et al. Are there low-penetrance TP53 Alleles? evidence from childhood adrenocortical tumors. Am J Hum Genet. 1999;65:995–1006. doi:10.1086/302575
2. Wooten MD, King DK. Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer. 1993;72:3145–3155. doi:10.1002/1097-0142(19931201)72:11<3145::AID-CNCR2820721105>3.0.CO;2-N
3. Ilanchezhian M, Varghese DG, Glod JW, et al. Pediatric adrenocortical carcinoma. Front Endocrinol. 2022;13(961650). doi:10.3389/fendo.2022.961650
4. Jouinot A, Bertherat J. Diseases predisposing to adrenocortical malignancy (Li-Fraumeni syndrome, Beckwith-Wiedemann syndrome, and Carney complex). Exp. 2019;(Suppl 111):149–169.
5. Miele E, Di Giannatale A, Crocoli A, et al. Clinical, genetic, and prognostic features of adrenocortical tumors in children: A 10-year single-center experience. Front Oncol. 2020;10(554388). doi:10.3389/fonc.2020.554388
6. Li FP, Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71:747–752. doi:10.7326/0003-4819-71-4-747
7. Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi:10.1126/science.1978757
8. Pinto EM, Zambetti GP, Rodriguez-Galindo C. Pediatric adrenocortical tumours. Best Pract Res Clin Endocrinol Metab. 2020;34(101448):101448. doi:10.1016/j.beem.2020.101448
9. Mai PL, Best AF, Peters JA, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the national cancer institute Li-Fraumeni syndrome cohort. Cancer. 2016;122:3673–3681. doi:10.1002/cncr.30248
10. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi:10.1038/35042675
11. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170:1062–1078. doi:10.1016/j.cell.2017.08.028
12. Raymond VM, Else T, Everett JN, et al. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98:E119–E125. doi:10.1210/jc.2012-2198
13. Wasserman JD, Novokmet A, Eichler-Jonsson C, et al. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children’s oncology group study. J Clin Oncol. 2015;33:602–609. doi:10.1200/JCO.2013.52.6863
14. Pinto EM, Chen X, Easton J, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun. 2015;6(6302). doi:10.1038/ncomms7302
15. Duffy KA, Cielo CM, Cohen JL, et al. Characterization of the Beckwith-Wiedemann spectrum: diagnosis and management. Am J Med Genet C Semin Med Genet. 2019;181:693–708. doi:10.1002/ajmg.c.31740
16. Brioude F, Kalish JM, Mussa A, et al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14:229–249. doi:10.1038/nrendo.2017.166
17. Smith AC, Choufani S, Ferreira JC, et al. Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatr Res. 2007;61:43R–47R. doi:10.1203/pdr.0b013e3180457660
18. Maas SM, Vansenne F, Kadouch DJ, et al. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A. 2016;170:2248–2260. doi:10.1002/ajmg.a.37801
19. Pinto EM, Rodriguez-Galindo C, Pounds SB, et al. Identification of clinical and biologic correlates associated with outcome in children with adrenocortical tumors without germline tp53 mutations: A St Jude adrenocortical tumor registry and children’s oncology group study. J Clin Oncol. 2017;35:3956–3963. doi:10.1200/JCO.2017.74.2460
20. Pinto EM, Rodriguez-Galindo C, Lam CG, et al. adrenocortical tumors in children with constitutive chromosome 11p15 paternal uniparental disomy: implications for diagnosis and treatment. Front Endocrinol. 2021;12(756523). doi:10.3389/fendo.2021.756523
21. Michalkiewicz E, Sandrini R, Figueiredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the international pediatric adrenocortical tumor registry. J Clin Oncol. 2004;22:838–845. doi:10.1200/JCO.2004.08.085
22. Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol. 2003;27:867–881. doi:10.1097/00000478-200307000-00001
23. Weiss LM. Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am J Surg Pathol. 1984;8:163–170. doi:10.1097/00000478-198403000-00001
24. Aubert S, Wacrenier A, Leroy X, et al. Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol. 2002;26:1612–1619. doi:10.1097/00000478-200212000-00009
25. Das S, Sengupta M, Islam N, et al. Weineke criteria, Ki-67 index and p53 status to study pediatric adrenocortical tumors: is there a correlation? J Pediatr Surg. 2016;51:1795–1800. doi:10.1016/j.jpedsurg.2016.07.014
26. Chatterjee G, DasGupta S, Mukherjee G, et al. Usefulness of Wieneke criteria in assessing morphologic characteristics of adrenocortical tumors in children. Pediatr Surg Int. 2015;31:563–571. doi:10.1007/s00383-015-3708-x
27. Beuschlein F, Weigel J, Saeger W, et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab. 2015;100:841–849. doi:10.1210/jc.2014-3182
28. Zhang F, Zhang F, Liu Z, et al. Prognostic role of Ki-67 in Adrenocortical carcinoma after primary resection: A retrospective mono-institutional study. Adv Ther. 2019;36:2756–2768. doi:10.1007/s12325-019-01050-0
29. Martins-Filho SN, Almeida MQ, Soares I, et al. Clinical impact of pathological features including the Ki-67 labeling index on diagnosis and prognosis of adult and pediatric adrenocortical tumors. Endocr Pathol. 2021;32:288–300. doi:10.1007/s12022-020-09654-x
30. Picard C, Orbach D, Carton M, et al. Revisiting the role of the pathological grading in pediatric adrenal cortical tumors: results from a national cohort study with pathological review. Mod Pathol. 2019;32:546–559. doi:10.1038/s41379-018-0174-8
31. Ribeiro RC, Pinto EM, Zambetti GP, et al. The international pediatric adrenocortical tumor registry initiative: contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol Cell Endocrinol. 2012;351:37–43. doi:10.1016/j.mce.2011.10.015
32. Pinto EM, Billerbeck AE, Villares MC, et al. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq Bras Endocrinol Metabol. 2004;48:647–650. doi:10.1590/S0004-27302004000500009
33. Ribeiro RC, Sandrini F, Figueiredo B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001;98:9330–9335. doi:10.1073/pnas.161479898
34. Cecchetto G, Ganarin A, Bien E, et al. Outcome and prognostic factors in high-risk childhood adrenocortical carcinomas: a report from the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT). Pediatr Blood Cancer 64. 2017. doi:10.1002/pbc.26368
35. Clay MR, Pinto EM, Cline C, et al. DNA methylation profiling reveals prognostically significant groups in pediatric adrenocortical tumors: a report from the international pediatric adrenocortical tumor registry. JCO Precis Oncol. 2019;3. doi:10.1200/PO.19.00163
36. Abib SCV, Weldon CB. Management of adrenal tumors in pediatric patients. Surg Oncol Clin N Am. 2021;30:275–290. doi:10.1016/j.soc.2020.11.012
37. Hubertus J, Boxberger N, Redlich A, et al. Surgical aspects in the treatment of adrenocortical carcinomas in children: data of the GPOH-MET 97 trial. Klin Padiatr. 2012;224:143–147. doi:10.1055/s-0032-1304627
38. Kemp CD, Ripley RT, Mathur A, et al. Pulmonary resection for metastatic adrenocortical carcinoma: The national cancer institute experience. Ann Thorac Surg. 2011;92:1195–1200. doi:10.1016/j.athoracsur.2011.05.013
39. Hendricks A, Muller S, Fassnacht M, et al. Impact of lymphadenectomy on the oncologic outcome of patients with adrenocortical carcinoma-a systematic review and meta-analysis. Cancers (Basel). 2022;14:291. doi:10.3390/cancers14020291
40. Rodriguez-Galindo C, Krailo MD, Pinto EM, et al. Treatment of pediatric adrenocortical carcinoma with surgery, retroperitoneal lymph node dissection, and chemotherapy: the children’s oncology group arar0332 protocol. J Clin Oncol. 2021;39:2463–2473. doi:10.1200/JCO.20.02871
41. Abdelhafeez A, Talbot L, Murphy AJ, et al. Indocyanine green-guided pediatric tumor resection: approach, utility, and challenges. Front Pediatr. 2021;9(689612). doi:10.3389/fped.2021.689612
42. Tattersall MH, Lander H, Bain B, et al. Cis-platinum treatment of metastatic adrenal carcinoma. Med J Aust. 1980;1:419–421. doi:10.5694/j.1326-5377.1980.tb134997.x
43. Fassnacht M, Terzolo M, Allolio B, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012;366:2189–2197. doi:10.1056/NEJMoa1200966
44. Terzolo M, Fassnacht M, Perotti P, et al. Adjuvant mitotane versus surveillance in low-grade, localised adrenocortical carcinoma (ADIUVO): an international, multicentre, open-label, randomised, Phase 3 trial and observational study. Lancet Diabetes Endocrinol. 2023;11:720–730. doi:10.1016/S2213-8587(23)00193-6
45. Redlich A, Boxberger N, Strugala D, et al. Systemic treatment of adrenocortical carcinoma in children: data from the German GPOH-MET 97 trial. Klin Padiatr. 2012;224:366–371. doi:10.1055/s-0032-1327579
46. Riedmeier M, Antonini SRR, Brandalise S, et al. International consensus on mitotane treatment in pediatric patients with adrenal cortical tumors: indications, therapy, and management of adverse effects. Eur J Endocrinol. 2024;190:G15–G24. doi:10.1093/ejendo/lvae038
47. Kimpel O, Schindler P, Schmidt-Pennington L, et al. Efficacy and safety of radiation therapy in advanced adrenocortical carcinoma. Br J Cancer. 2023;128:586–593. doi:10.1038/s41416-022-02082-0
48. Gharzai LA, Green MD, Griffith KA, et al. Adjuvant radiation improves recurrence-free survival and overall survival in adrenocortical carcinoma. J Clin Endocrinol Metab. 2019;104:3743–3750. doi:10.1210/jc.2019-00029
49. Wiegering V, Riedmeier M, Thompson LDR, et al. Radiotherapy for pediatric adrenocortical carcinoma - Review of the literature. Clin Transl Radiat Oncol. 2022;35:56–63. doi:10.1016/j.ctro.2022.05.003
50. Habra MA, Stephen B, Campbell M, et al. Phase II clinical trial of pembrolizumab efficacy and safety in advanced adrenocortical carcinoma. J Immunother Cancer. 2019;7(253). doi:10.1186/s40425-019-0722-x
51. Raj N, Zheng Y, Kelly V, et al. PD-1 Blockade in Advanced Adrenocortical Carcinoma. J Clin Oncol. 2020;38:71–80. doi:10.1200/JCO.19.01586
52. Carneiro BA, Konda B, Costa RB, et al. Nivolumab in Metastatic Adrenocortical Carcinoma: results of a Phase 2 Trial. J Clin Endocrinol Metab. 2019;104(12):6193–6200. doi:10.1210/jc.2019-00600
53. Le Tourneau C, Hoimes C, Zarwan C, et al. Avelumab in patients with previously treated metastatic adrenocortical carcinoma: Phase 1b results from the JAVELIN solid tumor trial. J Immunother Cancer. 2018;6(111). doi:10.1186/s40425-018-0424-9
54. Haluska P, Worden F, Olmos D, et al. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Canc Chemother Pharm. 2010;65:765–773. doi:10.1007/s00280-009-1083-9
55. Lerario AM, Worden FP, Ramm CA, et al. The combination of insulin-like growth factor receptor 1 (IGF1R) antibody cixutumumab and mitotane as a first-line therapy for patients with recurrent/metastatic adrenocortical carcinoma: a multi-institutional NCI-sponsored trial. Horm Canc. 2014;5:232–239. doi:10.1007/s12672-014-0182-1
56. Jain M, Zhang L, He M, et al. Interleukin-13 receptor alpha2 is a novel therapeutic target for human adrenocortical carcinoma. Cancer. 2012;118:5698–5708. doi:10.1002/cncr.27629
57. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–448. doi:10.1056/NEJMoa1709866
58. Marofi F, Motavalli R, Safonov VA, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12(81). doi:10.1186/s13287-020-02128-1
59. Bedrose S, Miller KC, Altameemi L, et al. Combined lenvatinib and pembrolizumab as salvage therapy in advanced adrenal cortical carcinoma. J Immunother Canc. 2020;8. doi:10.1136/jitc-2020-001009
60. Phan LM, Fuentes-Mattei E, Wu W, et al. Hepatocyte Growth Factor/cMET Pathway Activation Enhances Cancer Hallmarks in Adrenocortical Carcinoma. Cancer Res. 2015;75:4131–4142. doi:10.1158/0008-5472.CAN-14-3707
61. Kroiss M, Megerle F, Kurlbaum M, et al. Objective response and prolonged disease control of advanced adrenocortical carcinoma with cabozantinib. J Clin Endocrinol Metab. 2020;105:1461–1468. doi:10.1210/clinem/dgz318
62. Campbell MT JC, Long JP, Varghese J, et al. 1MO An open-label, phase II trial of cabozantinib for advanced adrenocortical carcinoma. ESMO Abstract. 2022;33.
63. Geoerger B, Kang HJ, Yalon-Oren M, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020;21:121–133. doi:10.1016/S1470-2045(19)30671-0
64. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. 2017;17:233–247. doi:10.1038/nri.2017.1
65. West AN, Neale GA, Pounds S, et al. Gene expression profiling of childhood adrenocortical tumors. Cancer Res. 2007;67:600–608. doi:10.1158/0008-5472.CAN-06-3767
66. Pinto EM, Rodriguez-Galindo C, Choi JK, et al. Prognostic significance of major histocompatibility complex class ii expression in pediatric adrenocortical tumors: A St. Jude and children’s oncology group study. Clin Cancer Res. 2016;22:6247–6255. doi:10.1158/1078-0432.CCR-15-2738
67. Parise IZS, Parise GA, Noronha L, et al. The Prognostic Role of CD8(+) T Lymphocytes in Childhood Adrenocortical Carcinomas Compared to Ki-67, PD-1, PD-L1, and the Weiss Score. Cancer. 2019;11:1730. doi:10.3390/cancers11111730
68. Luo C, Hajkova P, Ecker JR. Dynamic DNA methylation: in the right place at the right time. Science. 2018;361:1336–1340. doi:10.1126/science.aat6806
69. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412–1417. doi:10.1073/pnas.0510310103
70. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:1062–1078. doi:10.1038/nrg3230
71. Feinberg AP, Vogelstein B. Vogelstein B: Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi:10.1038/301089a0
72. Hovestadt V, Remke M, Kool M, et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013;125:913–916. doi:10.1007/s00401-013-1126-5
73. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–474. doi:10.1038/nature26000
74. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12(498). doi:10.1038/s41467-020-20603-4
75. Zheng S, Cherniack AD, Dewal N, et al. Comprehensive pan-genomic characterization of adrenocortical Carcinoma. Cancer Cell. 2016;29:723–736. doi:10.1016/j.ccell.2016.04.002
76. Rechache NS, Wang Y, Stevenson HS, et al. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J Clin Endocrinol Metab. 2012;97:E1004–13. doi:10.1210/jc.2011-3298
77. Barreau O, Assie G, Wilmot-Roussel H, et al. Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J Clin Endocrinol Metab. 2013:98
78. Bueno AC, da Silva RMP, Stecchini MF, et al. DNA methylation is a comprehensive marker for pediatric adrenocortical tumors. Endocr Relat Cancer. 2022;29:599–613.
79. Lerario AM, Moraitis A, Hammer GD. Genetics and epigenetics of adrenocortical tumors. Mol Cell Endocrinol. 2014;386:67–84. doi:10.1016/j.mce.2013.10.028
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.