")
Back to Journals » International Journal of Nanomedicine » Volume 19
PEGylated β-Cell-Targeting Exosomes from Mesenchymal Stem Cells Improve β Cell Function and Quantity by Suppressing NRF2-Mediated Ferroptosis
Authors Xia L , Yang M, Zang N, Song J, Chen J, Hu H, Wang K , Xiang Y, Yang J, Wang L , Zou Y, Lv X, Hou X , Chen L
Received 20 February 2024
Accepted for publication 27 August 2024
Published 14 September 2024 Volume 2024:19 Pages 9575—9596
DOI https://doi.org/10.2147/IJN.S459077
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. RDK Misra
Longqing Xia,1 Mengmeng Yang,1 Nan Zang,1– 4 Jia Song,1– 4 Jun Chen,1– 4 Huiqing Hu,1 Kewei Wang,1 Yingyue Xiang,1 Jingwen Yang,1 Liming Wang,1 Ying Zou,1 Xiaoyu Lv,1 Xinguo Hou,1– 4 Li Chen1– 4
1Department of Endocrinology, Qilu Hospital of Shandong University, Jinan, 250012, People’s Republic of China; 2Institute of Endocrine and Metabolic Diseases of Shandong University, Jinan, 250012, People’s Republic of China; 3Key Laboratory of Endocrine and Metabolic Diseases, Shandong Province Medicine & Health, Jinan, 250012, People’s Republic of China; 4Jinan Clinical Research Center for Endocrine and Metabolic Disease, Jinan, 250012, People’s Republic of China
Correspondence: Li Chen, Department of Endocrinology, Qilu Hospital of Shandong University, Shandong, People’s Republic of China, Email [email protected]
Background: The depletion of β cell mass is widely recognized as a significant contributor to the progression of type 2 diabetes mellitus (T2DM). Exosomes derived from mesenchymal stem cells (MSC-EXOs) hold promise as cell-free therapies for treating T2DM. However, the precise effects and mechanisms through which MSC-EXO affects β cell function remain incompletely understood, and the limited ability of MSC-EXO to target β cells and the short blood circulation time hampers its therapeutic effectiveness.
Methods: The effects of MSC-EXO were investigated in T2DM mice induced by a high-fat diet combined with STZ. Additionally, the high glucose-stimulated INS-1 cell line was used to investigate the potential mechanism of MSC-EXO. Michael addition reaction-mediated chemical coupling was used to modify the surface of the exosome membrane with a β-cell-targeting aptamer and polyethylene glycol (PEG). The β-cell targeting and blood circulation time were evaluated, and whether this modification enhanced the islet-protective effect of MSC-EXO was further analyzed.
Results: We observed that the therapeutic effects of MSC-EXO on T2DM manifested through the reduction of random blood glucose levels, enhancement of glucose and insulin tolerance, and increased insulin secretion. These effects were achieved by augmenting β cell mass via inhibiting nuclear factor erythroid 2–related factor 2 (NRF2)-mediated ferroptosis. Mechanistically, MSC-EXOs play a role in the NRF2-mediated anti-ferroptosis mechanism by transporting active proteins that are abundant in the AKT and ERK pathways. Moreover, compared to MSC-EXOs, aptamer- and PEG-modified exosomes (Apt-EXOs) were more effective in islet protection through PEG-mediated cycle prolongation and aptamer-mediated β-cell targeting.
Conclusion: MSC-EXO suppresses NRF2-mediated ferroptosis by delivering bioactive proteins to regulate the AKT/ERK signaling pathway, thereby improving the function and quantity of β cells. Additionally, Apt-EXO may serve as a novel drug carrier for islet-targeted therapy.
Keywords: exosome, type 2 diabetes mellitus, β-cell-targeting, polyethylene glycol modification, ferroptosis
Graphical Abstract:

Introduction
Type 2 diabetes mellitus (T2DM) is a multifaceted metabolic condition marked by inadequate β cell mass and insulin resistance, which is on the rise in terms of occurrence and poses a significant risk to the well-being of the general population.1,2 Both the diminished quantity and impaired performance of β cells are responsible for the decline in β cell mass.3
Although apoptosis has been widely accepted as the main pathway leading to the reduction of β cell mass,4 the discovery of ferroptosis may challenge this view. Ferroptosis is defined as iron-dependent regulatory necrosis caused by membrane damage mediated by massive lipid peroxidation.5 During ferroptosis, cells often undergo various morphological alterations, including compromised plasma membrane integrity, moderate condensation of chromatin, and mitochondrial abnormalities.6–9 Notably, glutathione peroxidase 4 (GPX4) inhibition of lipid peroxidation is a significant defense mechanism against ferroptosis.10 Compared with other tissues, islets express lower peroxidase levels and are vulnerable to ferroptosis.11,12 The occurrence of ferroptosis in β cells impairs insulin secretion, whereas inhibition of ferroptosis improves islet function.13,14 Therefore, inhibition of β cell ferroptosis has become a novel therapeutic strategy.
The therapeutic use of mesenchymal stromal cells (MSCs) has great potential for mitigating diabetes. The low immunogenicity of the human umbilical cord renders it an ideal source of MSCs.15 Recent studies have indicated that mesenchymal stem cells primarily exert their therapeutic benefits via paracrine mechanisms. Exosomes are tiny extracellular vesicles that play a role in intercellular communication by carrying functional nucleic acids (mRNA and microRNAs), proteins, and lipids.16 Moreover, MSC-derived exosomes alleviate insulin resistance and enhance insulin secretion.17 Nonetheless, the full impact of MSC-EXO on β cell mass and ferroptosis remains undetermined. Notably, exosome therapy poses challenges. Biodistribution studies show that exosomes mainly accumulate in the liver and spleen, followed by the lungs, with few reaching the pancreas post-intravenous injection.18,19 Therefore, the targeting capability of exosomes must be improved for them to play a more effective therapeutic role. Aptamers obtained by SELEX are oligonucleotides composed of single-stranded DNA or RNA. These aptamers have a unique three-dimensional structure, exhibit specificity towards particular tissues or cells, and bind to target molecules.20,21 Exosomes loaded with drugs and modified with aptamers have been documented as a means of specifically treating cancer.22 In addition, the rapid clearance of exosomes in vivo also limits its application.23 Previous studies have demonstrated that PEGylation of cells, proteins, and nanoparticles reduces immunogenicity and prolongs circulation time.24,25
As shown in Scheme 1, we investigated the effects of MSC-EXO on β cell damage using a T2DM mouse model and INS-1 cells. Subsequently, we conjugated MSC-EXO with PEGylated β-cell-specific aptamers to form aptamer-PEG-exosome complexes (Apt-EXO) to investigate whether Apt-EXO could improve β-cell targeting and extend blood circulation time, and thereby further augment β cell quantity and function compared to MSC-EXO.
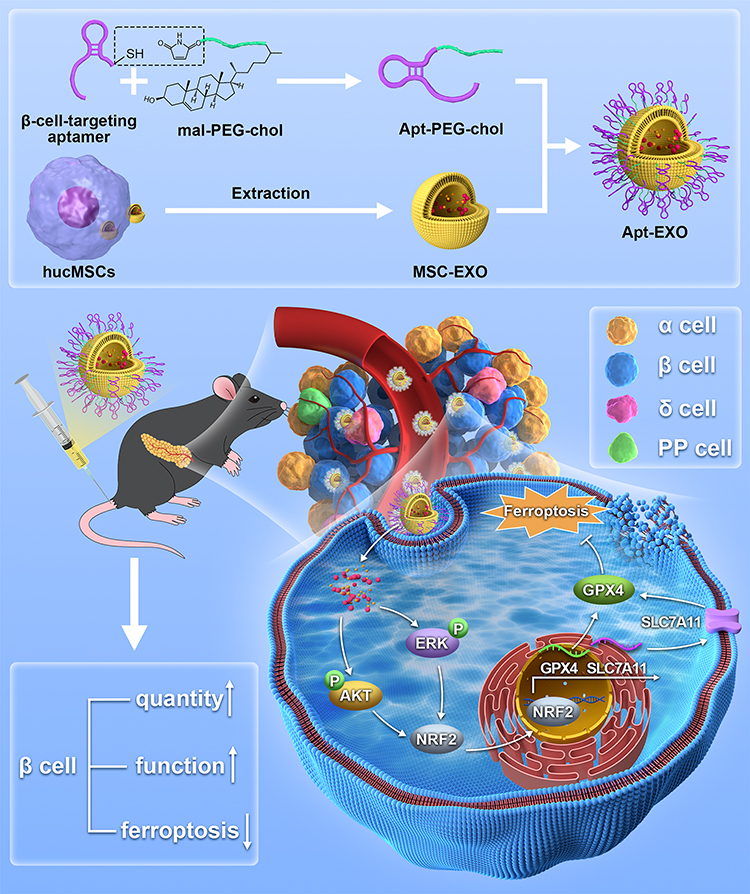 |
Scheme 1 Preparation process of Apt-EXO and mechanism of improving β cell function and quantity. |
Materials and Methods
Cell Culture and Treatment
The Ethics Committee of Qilu Hospital of Shandong University approved the acquisition of fresh umbilical cords from healthy newborns. This study complies with the Declaration of Helsinki. Consent was obtained from pregnant women after they were provided with the necessary information. Subsequently, the blood vessels were extracted from the umbilical cord, and Wharton’s jelly was broken down into tiny fragments. The fragments were subsequently cultivated in a medium that included 10% fetal bovine serum (FBS; Gibco), along with 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco). After the hucMSCs reached 80% confluency in the culture medium, flow cytometry analysis and induction of differentiation were conducted during the third to fifth passage of human umbilical cord mesenchymal stromal cells.
Nanjing Medical University, PR China, provided the INS-1 cell line. The use of the INS-1 cell line was approved by the Ethics Committee of Qilu Hospital of Shandong University. The cells were grown in RPMI 1640 solution (Gibco), with the addition of 15% FBS, 10 mM HEPES (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich), 2 mM L-glutamine (Gibco), and 50 μM β-mercaptoethanol (Sigma-Aldrich) at a temperature of 37°C and 5% CO2. The cells were seeded in a 6-well plate and grown in a medium containing 33.3 mM glucose (referred to as high glucose or HG) for 48 h; subsequently, they were exposed to hucMSC-derived exosomes (MSC-EXO) at a concentration of 50 μg/mL or a ferroptosis inhibitor (Fer-1, 10 μM, Selleck) for 24 h. The control group, RPMI-1640, consisted of 11.1 mM glucose. To explore the mechanism of action of MSC-EXOs, Lipofectamine 2000 Transfection Reagent (Invitrogen) was used to introduce small interfering RNA (siRNA) into INS-1 cells before MSC-EXO (50 μg/mL) treatment. Following this, INS-1 cells were subjected to MSC-EXO treatment along with either AKT inhibitor (LY294002, 10 μM, MCE) or ERK inhibitor (PD98059, 10 μM, MCE) for 24 h. GenePharma Co., Ltd. (Shanghai, China) synthesized the siRNA oligonucleotides listed in Table S1.
MIN-6 cells (Bena Culture Collection, China) were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco), which was enriched with 10% fetal bovine serum (FBS; Gibco) and 100 μg/mL L-glutamine (Gibco).
Isolation and Identification of Exosomes
Briefly, 48 h after refreshing the hucMSC culture medium with serum-free medium, we collected the conditioned medium. Subsequently, a series of centrifugations were performed to eliminate cells, apoptotic bodies, and debris. Specifically, the medium was first spun at 300 × g for 10 min, followed by 2000 × g for 20 min. To remove residual debris, further centrifugation was executed at 10,000 × g for 30 min. The resulting fluid was then filtered through a 0.22-micron filter. Finally, to isolate the exosomes, the remaining fluid was centrifuged at 120,000 × g for 70 min at 4°C.
All pellets containing exosomes were either dissolved in PBS or disrupted using an RNA lysis buffer for further examination. Transmission electron microscopy (TEM) and nanosight tracking analysis (NTA; ZetaVIEW S/N 21–674, PARTICLE METRIX, Germany) were used to observe the exosomes. Western blotting was performed to evaluate the protein expression of Calnexin, CD9, TSG101, and HSP70.
Animal Models and Treatments
Five-week-old male C57BL/6J mice were sourced from Jiangsu Huachuang Sino Pharmaceutical Co. Ltd. After a 1–2 week acclimation period, the mice underwent a 12-week high-fat diet (HFD) regimen with 60% fat content. In contrast, control mice received a regular chow diet. Following this, mice on the HFD were intraperitoneally administered 60 mg/kg STZ (Sigma-Aldrich, USA) for five consecutive days, commencing on Day 1. Mice were confirmed to have T2DM if their fasting glucose levels were ≥16.7 mmol/L. To investigate the impact of MSC-EXOs on islet function and underlying mechanisms, the mice were divided into three groups: control + PBS, T2DM + PBS, and T2DM + MSC-EXOs. To explore possible improvements in the islet-enhancing impact of exosomes using Apt-EXOs, the mice were divided into four groups: control +PBS, T2DM+ PBS, T2DM+EXO, and T2DM+ Apt-EXO. Twice a week for a duration of 6 weeks, a 200 μL PBS solution containing 200 μg of MSC-EXO (or Apt-EXO) was administered via the tail vein. Animal experimentation protocols were authorized by the Animal Ethics Committee of Shandong University (issue no. Dwll-2022-155) and all followed the guidelines formulated by the National Institutes of Health.
Metabolic Parameter Analysis
Weekly monitoring of body weight and random blood glucose levels was conducted from the initiation of the MSC-EXO or Apt-EXO therapy until two weeks after the conclusion of the intervention. The IPGTT (1.5 g/kg) and IPITT (0.75 U/kg) were conducted as previously mentioned.26 Serum insulin levels were measured by obtaining blood samples from the inner canthal vein before and 30 min after initiating IPGTT. The AccuChek® Performa was used to measure blood glucose levels at the tip of the tail.
Glucose-Stimulated Insulin Secretion (GSIS)
Following the required treatment, glucose-deprived INS-1 cells were incubated for 1 h at 37°C in KRBS-Ringer bicarbonate solution containing 2.5 mmol/L glucose, and the resulting supernatant was collected. Subsequently, the supernatant was further collected after another 1-hour incubation at 37°C in KRBS solution with 25 mmol/L glucose. Insulin release upon stimulation was quantified by measuring insulin levels in the collected supernatant using a kit (Blue Gene, Shanghai, China). Finally, insulin levels were normalized to the cellular protein concentrations.
Haematoxylin and Eosin (HE), Immunohistochemistry (IHC), and Immunofluorescence (IF) Staining
HE staining was performed following a standard protocol via Hematoxylin and Eosin Staining Kit (Beyotime, China). For IHC staining, the slides were dewaxed, and antigen retrieval was performed using citrate buffer. To deactivate natural peroxidases and avoid nonspecific attachment, a 3% solution of H2O2 was administered for 15 min, followed by incubation with goat serum for 30 min. The primary antibodies were subsequently left to incubate at a temperature of 4°C for the duration of the night. Secondary antibody and DAB staining were performed the following day, following the instructions of the IHC kit provided by Gene Tech Company (GK600505, China). For IF staining, after retrieving the antigen and blocking non-specific sites, antibodies were used for staining. A fluorophore-conjugated secondary antibody (ZSGB-Bio, China) was applied the following day at 37°C for 1 h. Following DAPI staining, fluorescence microscopy images were captured using a fluorescence microscope (BX53; Olympus, Japan). Table S2 contains a list of all antibodies.
Western Blotting
Proteins were extracted using a lysis buffer and subsequently separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After transferring the proteins onto PVDF membranes, the membranes were blocked and incubated overnight with primary antibodies. The next day, a horseradish peroxidase-conjugated secondary antibody (ZSGB-Bio) was applied. Immune complexes were detected using an enhanced chemiluminescence kit (Millipore, Billerica, MA, USA). Table S2 contains a list of all antibodies.
TEM
Mitochondrial morphology was assessed via TEM. After the designated treatment, the pancreas was incubated in 2.5% electron microscopy-grade glutaraldehyde. The cells were then fixed, dehydrated, embedded, sectioned, and stained. Finally, these samples were visualized using a transmission electron microscope.
CCK-8
Cell viability was assessed using a CCK-8 kit (Yeasen 40203ES76, China). After treating INS-1 cells exposed to HG with Fer-1 (10 μM, Selleck), Z-VAD (10 μM, Selleck), and Nec-1 (10 μM, Selleck), the medium was replaced with a CCK-8 working solution comprising 10% CCK-8 reagent. Afterward, the cells were placed in an incubator set at a temperature of 37°C for a duration of 2 h. The absorbance of each well was measured at 450 nm using a microplate reader.
Iron Measurement
An iron assay kit (Solarbio, BC5315, China) was used to detect the amount of total iron in the cells. A ferrous iron detection kit (BC5415; Solarbio, China) was used to measure the amount of ferrous iron in the pancreatic tissue. The experimental protocol was performed in strict accordance with the manufacturer’s guidelines.
Malondialdehyde (MDA) Measurement
The levels of MDA in the serum and pancreatic tissue were assessed using a Lipid Peroxidation MDA Assay Kit (A003-1-2, Nanjing Jiancheng Bioengineering Institute, China). A Cellular Lipid Peroxidation MDA Assay Kit (A003-4-1) from Nanjing Jiancheng Bioengineering Institute was used to analyze the levels of MDA in cells. The MDA measurement procedure adhered strictly to the manufacturer’s guidelines.
Chromatin Immunoprecipitation (ChIP)
The ChIP experiment was performed with the CST SimpleChIP® Plus Sonication Chromatin IP Kit (56383). In short, INS-1 cells were treated with 1% formaldehyde to create cross-links, fragmented into 150–900 bp pieces, and subsequently subjected to overnight incubation at 4°C with either an anti-NRF2 antibody (CST,12721) or a normal IgG antibody. Finally, cells were pulled down using magnetic beads. The ChIP assay was performed strictly according to the instructions provided by the manufacturer.
Finally, the enrichment of specific DNA sequences was evaluated by reverse transcription-quantitative polymerase chain reaction (RT-qPCR). Table S1 contains a list of all primer sequences.
Real-Time Quantitative Reverse Transcription PCR
Total RNA was extracted from INS-1 cells using an RNA Extraction Kit (AG21017; Accurate Biotechnology, China) and subjected to reverse transcription using an Evo M-MLV RT Kit (AG11705; Accurate Biotechnology, China). For RT-qPCR, a SYBR Green Premix Pro Taq HS qPCR Kit (AG11702; Accurate Biotechnology) was used. The 2−ΔΔCt method was utilized to calculate the alteration in mRNA expression. Table S1 contains a list of all primer sequences.
C11-BODIPY Staining
To evaluate cellular lipid peroxidation, a C11-BODIPY probe (D3861, Invitrogen, USA) was employed. Post-therapy, INS-1 cells were incubated with 5 μmol/L C11-BODIPY for 30 min, followed by a 10-minute Hoechst staining (C1027, Beyotime, China). Subsequently, confocal scanning microscopy was used to observe the stained cells.
Tunel
Death in INS-1 cells was evaluated through TUNEL assays employing TUNEL Assay Kits (G1501, Servicebio, China) in strict adherence to the guidelines. Fluorescence images were obtained using a fluorescence microscope (BX53; Olympus, Japan).
Conjugation of Aptamers to MSC-EXO
The 200 nM sulfhydryl and cy3-labeled aptamers (Sangon) were subjected to reduction using a TCEP solution on ice for 30 min. Subsequently, cells were incubated overnight with cholesterol-polyethylene glycol-maleimide (chol-PEG-mal) in HEPES buffer. Excess chol-PEG-mal was eliminated by filtration for 15 min at 8000 g, using a molecular weight cutoff of 5000. Afterward, 200 nM cy3-aptamer-PEG-chol was added to 1.0 mg/mL MSC-EXO in PBS at 4°C overnight. The unconjugated aptamers were washed thrice in PBS using a 100 kDa ultrafiltration tube. The fluorescence intensity of the cy3-aptamer exosome was detected using the microplate reader to confirm that the aptamer was successfully coupled to the exosome membrane. The aptamer sequences are shown in Table S1.
Cellular Internalization of Exosomes
MSC-EXOs and Apt-EXOs were labeled with DIO cell-labeling solution (Invitrogen, V22886, USA) for in vitro studies and with infrared fluorescent DIR Lipophilic dye (Invitrogen, D12731, USA) for in vivo studies. The uptake efficiency of the exosomes by MIN-6 cells was assessed using flow cytometry. Fluorescence imaging was employed to evaluate exosome uptake in the mouse pancreas. The staining protocol was meticulously followed according to the manufacturer’s guidelines.
Biodistribution Imaging
The 200 μg DIR-labeled exosomes and an equivalent amount of free exosomes were intravenously administered via the tail vein. Distribution was assessed through whole-body imaging at 2 h, 6 h, and 12 h, as well as fluorescence imaging of dissected organs, including the pancreas, liver, spleen, lung, kidney, and heart, after 12 h. Fluorescence signals were monitored by collecting blood samples from the inner canthus at 1 h, 3 h, and 6 h. A Tanon ABL-X5 in vivo fluorescence tomography imaging system was utilized for image capture.
Extraction of Primary Mouse Islets
The experimental methods were carried out according to standard or customized procedures. The detailed information was described in Supplementary material.
Molecular Docking of Protein to Protein
The experimental methods were carried out according to standard or customized procedures. The detailed information was described in Supplementary material.
Proteomic Data Analysis
The original proteomic data of the MSC-EXOs were downloaded from ProteomeXchange using the identifier PXD033899. The protein expression data from MSC-EXOs were processed and analyzed using DEP packages in R (version 4.2.2). Proteins meeting the criteria of P-value < 0.05 and |logFC| > 1 were deemed as standard. Subsequently, KEGG enrichment analysis was conducted using the R package clusterProfiler, with the significance threshold set at P < 0.05. A path bubble map was created using the ggplot2 package. The important differentially expressed genes were matched with the STRING database. A confidence threshold > 0.4 was applied to perform the protein–protein interaction (PPI) network analysis. The resulting network was visualized using the Cytoscape software (v3.9.1).
Statistical Analysis
The data is displayed as the mean ± SEM. GraphPad Prism 9 software was used to assess the disparities between groups using either a paired Student’s t-test or one-way ANOVA. Statistical significance was set at P < 0.05.
Results
Identification and Cellular Internalization of MSC-EXO
Oil Red O and Alizarin Red staining showed that the hucMSCs differentiated into adipocytes and osteoblasts, respectively (Figure S1A and B). HucMSCs expressed the stem cell markers CD105 and CD73 but did not express CD34 and HLA-DR (Figure S1C). The exosome cup-like membrane structure was detected using TEM (Figure 1A) and was measured to have a mode diameter of approximately 130 nm using a nanoparticle-tracking system (Figure 1B). Western blot analysis indicated that MSC-EXOs were positive for exosome markers, including CD9, TSG101, and HSP70. Conversely, calnexin, a marker of the endoplasmic reticulum membrane, was enriched in hucMSCs but not in MSC-EXOs (Figure 1C). As shown in Figure 1D, MSX-EXOs were successfully taken up by the INS-1 cells.
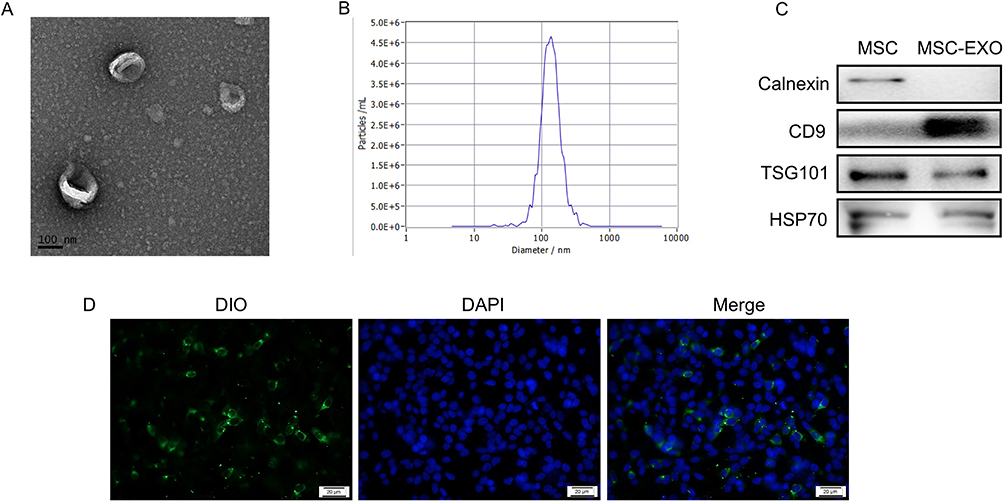 |
Figure 1 Identification and cellular internalization of MSC-EXO. (A) Transmission electron microscopy image of MSC-EXO. (B) Nanoparticle tracking analysis of MSC-EXO. (C) Western blot analysis of exosome markers CD9, TSG101, HSP70, and ER membrane marker Calnexin in MSC-EXO. (D) Representative picture of INS-1 cells stained with DiO-labeled exosomes (scale bar, 20μm). |
MSC-EXO Improves β Cell Function and Quantity
To examine whether MSC-EXOs enhanced islet dysfunction, a mouse model of T2DM was used (Figure 2A). As expected, random blood glucose levels were significantly reduced after the MSC-EXO intervention (Figure 2B). Moreover, MSC-EXOs alleviated weight loss in T2DM mice (Figure 2C). T2DM mice that received MSC-EXOs exhibited notable improvements in glucose tolerance (Figure 2D and E), insulin sensitivity (Figure 2F and G), and insulin secretion 30 min following IPGTT (Figure 2H). In T2DM mice, the size of islets and pancreatic β cells was significantly reduced compared to that of the normal control group. However, treatment with MSC-EXO helped mitigate the decrease in both the islet and β cell areas (Figure 2I–K and M). The ratio of insulin-positive cells was lower, and the ratio of glucagon-positive cells was higher in T2DM mice than in the control mice. Administration of MSC-EXOs reversed the loss of insulin-positive cells (Figure 2K and M). The data indicated that MSC-EXO improves pancreatic β cell function and quantity in T2DM.
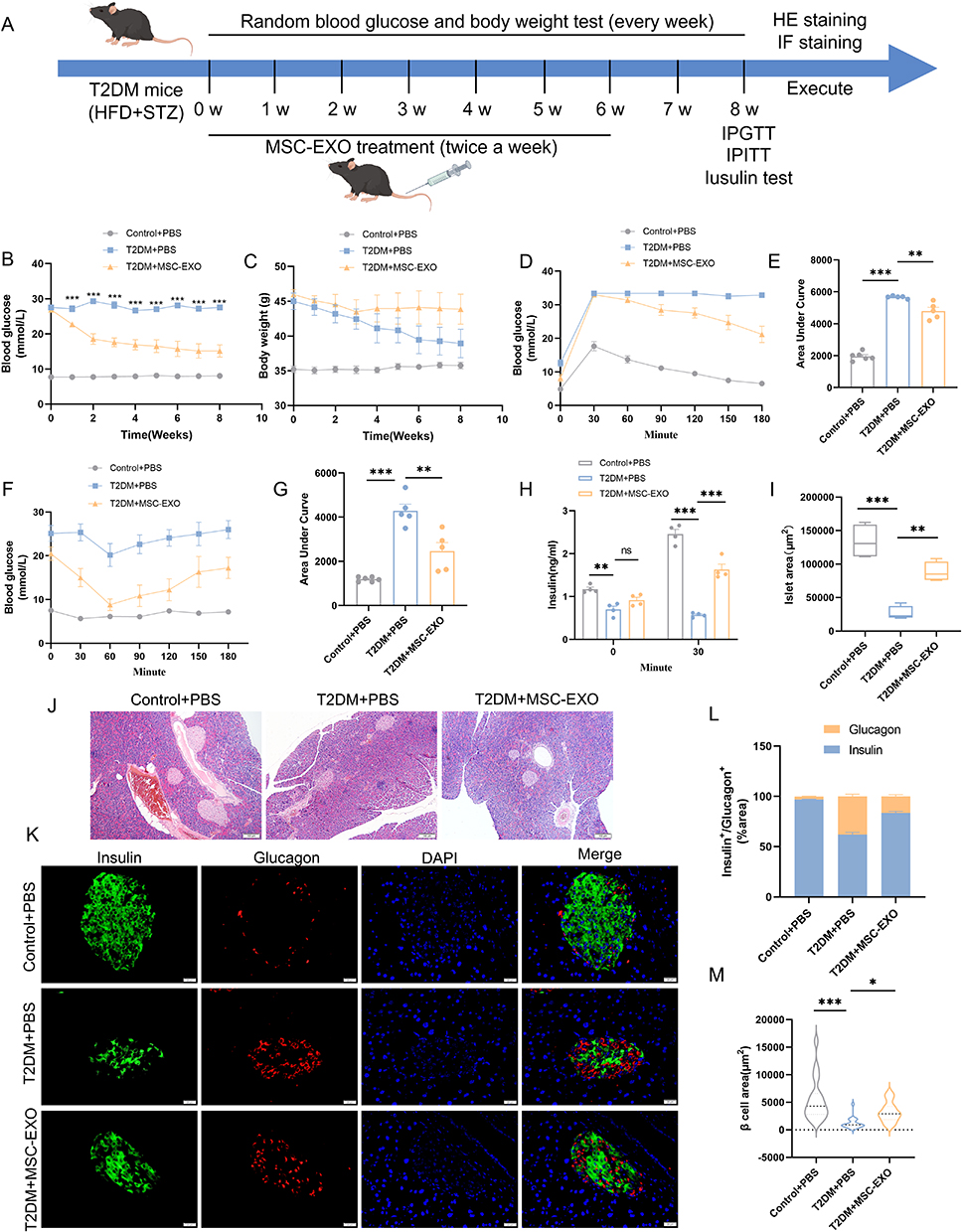 |
Figure 2 MSC-EXO improves β cell function and quantity in T2DM mice. (A) Study design for MSC-EXO to improve β cell function and quantity in mice. (B) Random blood glucose levels and (C) body weight in Control+PBS, T2DM+PBS, and T2DM+MSC-EXO treatment groups (n = 5–6 mice). (D) IPGTT and (E) Area Under Curve in T2DM mice following MSC-EXO injection (n = 5–6 mice). (F) IPITT and (G) Area Under Curve in T2DM mice post MSC-EXO injection (n = 5–6 mice). (H) Insulin secretion during fasting and 30 minutes of IPGTT (n = 4 mice). (I) Analysis of Langerhans islets area across all mouse groups (n = 4 mice). (J) H&E staining of pancreatic sections in each group (scale bar, 100 μm). (K) Insulin and glucagon staining in representative pancreatic sections (scale bar, 20 μm). (L) Proportion of insulin and glucagon in immune-positive areas (n = 3 mice and 20 islets). (M) Analysis of β-cell area in IF staining (n = 3 mice and 20 islets). Data are represented as the mean ± SEM. (*P < 0.05, **P < 0.01, ***P < 0.001). |
MSC-EXO Inhibits β Cell Ferroptosis
High glucose-induced Ferroptosis has the potential to result in β cell dysfunction.27 Hence, our investigation aimed to ascertain whether ferroptosis significantly impacts the function of β cells and whether MSC-EXO can mitigate the levels of ferroptosis in these cells. The effects of various inhibitors of programmed cell death were assessed using the CCK-8 assay in INS-1 cells. The inhibitors used were Z-VAD-FMK (apoptosis), Fer-1 (ferroptosis), and necrostatin-1 (Nec-1; necroptosis). Fer-1 demonstrated the most potent effect in preserving cell viability compared to the alternative inhibitors (Figure 3A). Furthermore, GSIS revealed that HG reduced insulin secretion in INS-1 cells. However, both Fer-1 and MSC-EXOs effectively mitigated this detrimental effect (Figure 3B). Subsequently, we assessed morphological and biochemical markers of ferroptosis. Analogous to Fer-1, MSC-EXO treatment significantly attenuated the HG-induced elevation of MDA, total iron and ferrous concentration (Figures 3C and D, S2A), and increased GSH content (Figure S2B). Significant lipid peroxidation was observed following HG stimulation, as indicated by C11-BODIPY staining. However, this effect was reversed upon treatment with Fer-1 or MSC-EXOs (Figure 3E and F). The TUNEL test verified that the administration of Fer-1 or MSC-EXO could effectively alleviate β cell death induced by HG (Figure 3E and G). The mRNA levels of PTGS2 and ACSL4, which were upregulated by HG, were rescued by either Fer-1 or MSC-EXOs treatment, as shown by RT-qPCR. (Figure 3H). MSC-EXO treatment significantly increased GPX4 and SLC7A11 protein expression, and similar results were observed for Fer-1 (Figure 3I).
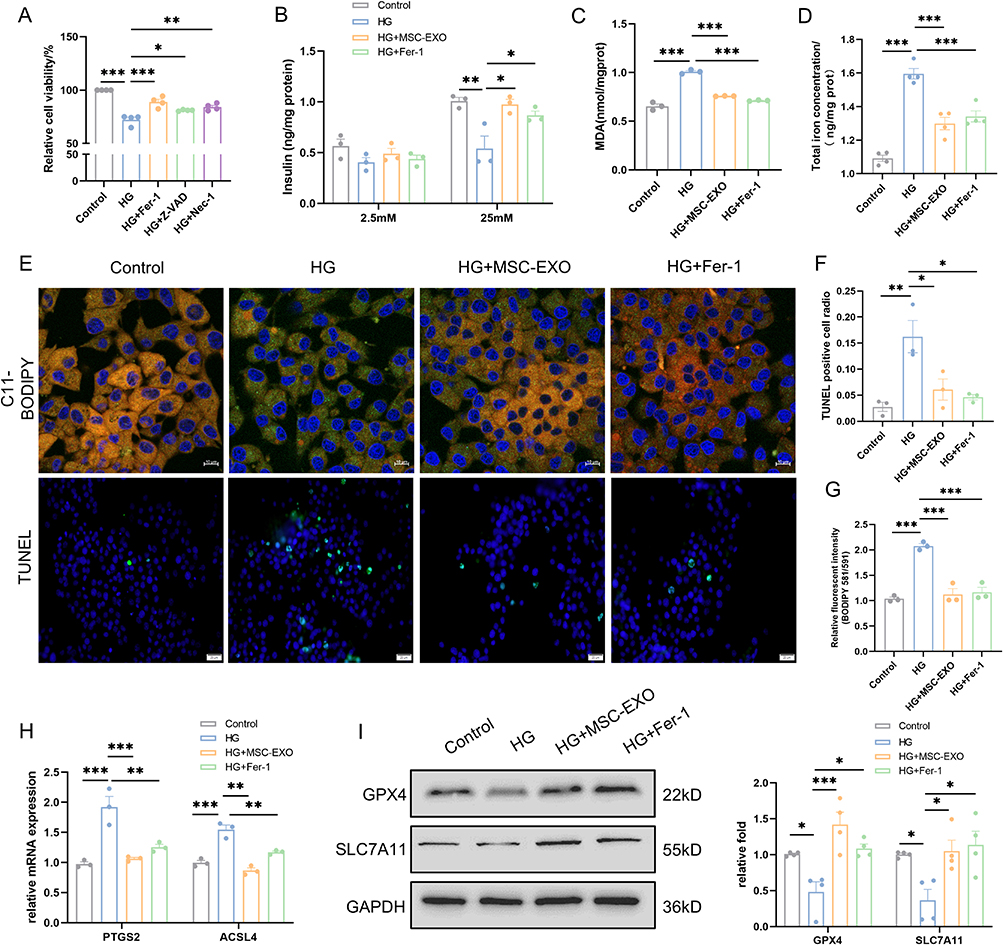 |
Figure 3 MSC-EXO inhibits β cell ferroptosis in INS-1 cells. (A) Cell viability of INS-1 cells was assessed using the CCK8 assay (n = 4). (B) GSIS test was conducted to evaluate insulin release in INS-1 cells (n = 3). (C) Quantitative measurement of MDA levels using an MDA assay kit in INS-1 cells (n = 3). (D) Quantitative determination of total iron concentrations in INS-1 cells (n = 4). (E) Representative C11-BODIPY staining (scale bar: 10 μm) and TUNEL assay images (scale bar: 20 μm) are presented. (F) Fluorescence intensity analysis of lipid peroxides (n = 3). (G) Analysis of TUNEL-positive cells (n = 3). (H) Detection of ferroptosis markers (PTGS2 and ACSL4) at mRNA levels by RT-qPCR (n = 3). (I) Detection of ferroptosis markers (GPX4 and SLC7A11) at protein levels by Western blot (n = 4). Data are represented as the mean ± SEM. (*P < 0.05, **P < 0.01, ***P < 0.001). |
Next, we examined the ferroptosis-related markers in T2DM mice. MDA levels were markedly increased in both the sera and pancreases of mice with diabetes but showed a remarkable decrease following intervention with MSC-EXOs (Figure 4A and B). Additionally, an identical pattern was noted in the identification of ferrous iron (Figure 4C). The TEM assay indicated a decrease or absence of mitochondrial spines in the T2DM group; however, these changes were reduced by MSC-EXO intervention (Figure 4D). Moreover, IHC staining revealed a notable increase in GPX4 levels and a decrease in ACSL4 levels in the pancreatic islet tissue of mice with T2DM after treatment with MSC-EXOs (Figure 4E). Western blotting results showed that MSC-EXO increased the expression of GPX4 and SLC7A11 in mouse primary islets (Figure S2C and D). The findings suggest that MSC-EXO suppresses ferroptosis in β cells both in vivo and in vitro.
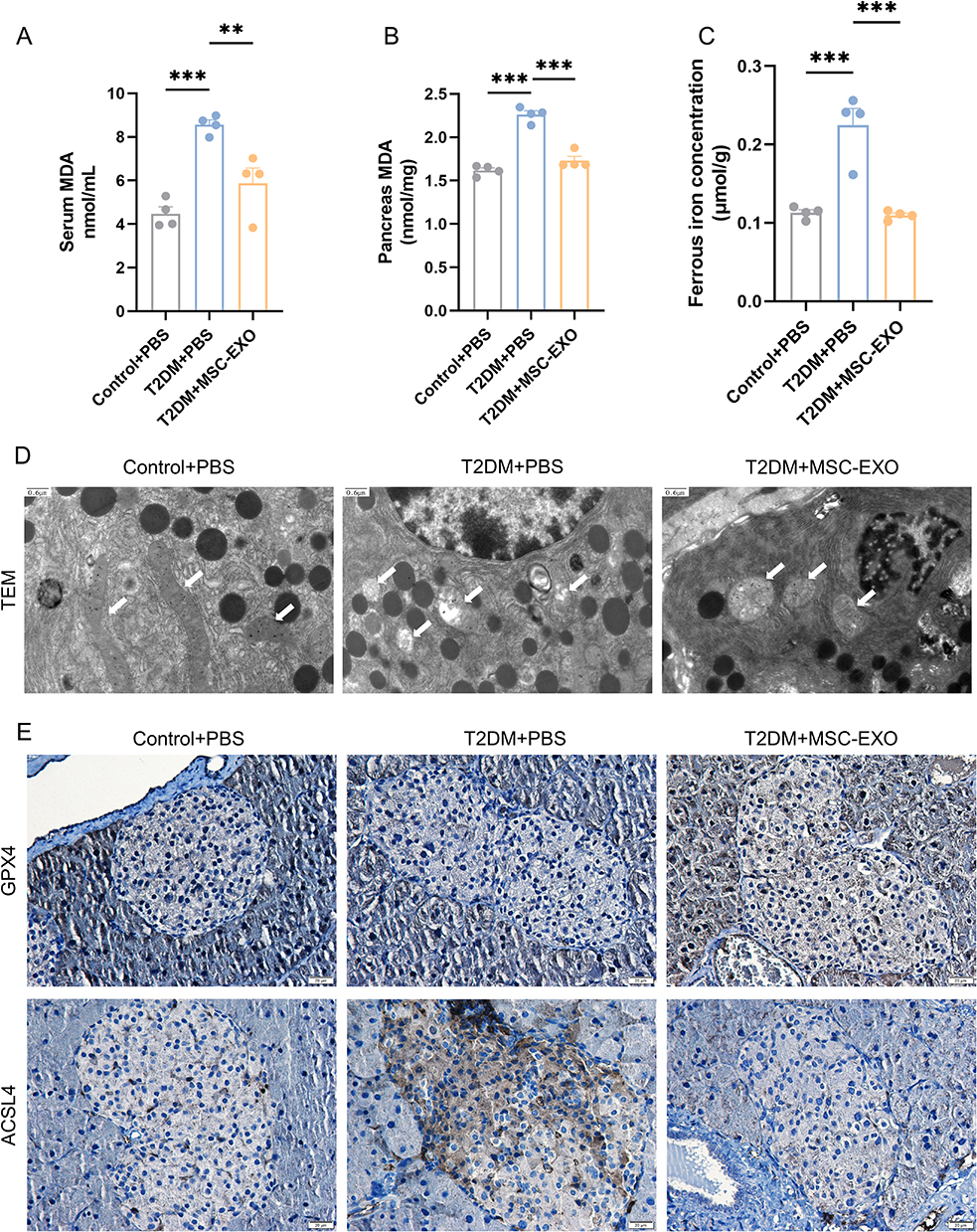 |
Figure 4 MSC-EXO inhibits β cell ferroptosis in T2DM mice. MDA levels in serum (A) and pancreas (B) (n = 4 mice). (C) Ferrous iron concentrations in the pancreas (n = 4 mice). (D) Representative TEM images (scale bar: 0.6 μm). (E) IHC staining of GPX4 and ACSL4 in pancreatic paraffin sections (scale bar: 20 μm). Data are represented as the mean ± SEM. (**P < 0.01, ***P < 0.001). |
NRF2 Was Essential for the Anti-Ferroptosis Effect of MSC-EXO
To identify the targets of MSC-EXOs in the inhibition of ferroptosis, NRF2, a key anti-ferroptosis molecule,28 was examined. The expression of NRF2, p-NRF2, and HO-1 was enhanced by MSC-EXOs treatment, and the mRNA expression of SLC7A11 and HO-1 increased, whereas KEAP1 expression remained unaffected (Figure 5A and B). Before its translocation to the nucleus, NRF2 is phosphorylated at Ser40 (which serves as a binding site for Keap1). Hence, the p-NRF2 antibody can be used to measure the ratio of NRF2 within the nucleus.29 Therefore, the NRF2-Ser40 (p-NRF2) antibody was used to quantify the proportion of nuclear NRF2, indicating that MSC-EXOs increased NRF2 expression, nuclear translocation, and transcriptional activity but did not exert a regulatory effect on NRF2 through the KEAP1 pathway. Moreover, we noted that the administration of MSC-EXO reversed the decline of phosphorylated NRF2 in the pancreatic β cells of mice with T2DM, as evidenced by immunofluorescence staining (Figure 5C). Subsequently, we introduced NRF2 siRNA into INS-1 cells. The highest knockdown efficiency was observed with the 1st siRNA sequence targeting NRF2 (Figure S1D). Therefore, this sequence was used in subsequent experiments. Western blotting results showed that NRF2 knockdown reversed NRF2 transport into the nucleus and reduced HO-1, GPX4, and SLC7A11 expression (Figure 5D). Meanwhile, suppressing NRF2 weakened the beneficial results of MSC-EXOs in reducing lipid peroxidation and mitigating cell death (Figure 5E–G). Finally, ChIP assays were performed using INS-1 cells. MSC-EXO treatment significantly promoted the enlistment of NRF2 to GPX4 and SLC7A11, indicating direct binding of NRF2 to this chromatin region (Figure 5H and I). These findings indicated that MSC-EXOs utilize NRF2 as a crucial focal point to elicit therapeutic benefits.
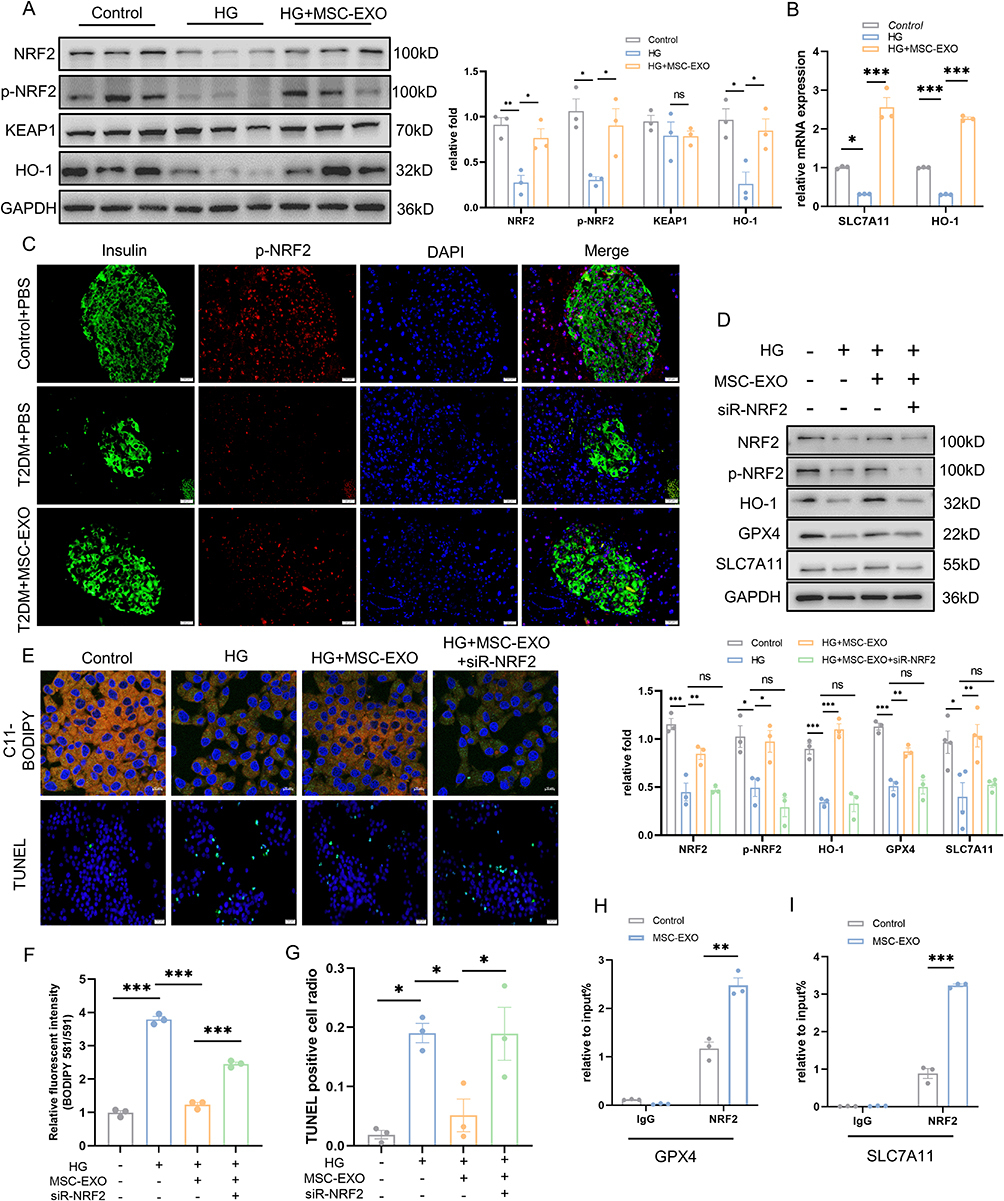 |
Figure 5 MSC-EXO exerts anti-ferroptosis effect through NRF2. (A) Western blot analysis of NRF2, p-NRF2, KEAP1, and HO-1 in INS-1 cells treated with HG and MSC-EXO (n = 3). (B) mRNA levels of SLC7A11 and HO-1 in INS-1 cells were assessed by RT-qPCR (n = 3). (C) Pancreatic section showing p-NRF2 (red) and insulin (green) IF staining (scale bar: 20 μm). (D) Western blot analysis of NRF2, p-NRF2, HO-1, GPX4, and SLC7A11 in INS-1 cells transfected with NRF2 siRNA and treated with MSC-EXO and HG (n = 3–4). (E) Representative C11-BODIPY staining images (scale bar: 10 μm) and TUNEL assay images (scale bar: 20 μm) in INS-1 cells. (F) Fluorescence intensity analysis of lipid peroxides (n = 3). (G) Quantification of TUNEL-positive cells (n = 3). ChIP assays using anti-NRF2 antibody or normal IgG detected GPX4 (H) and SLC7A11 (I) in INS-1 cells treated with MSC-EXO (n = 3). Data are presented as the mean ± SEM. (*P < 0.05, **P < 0.01, ***P < 0.001). |
MSC-EXO Exerts an Anti-Ferroptosis Effect by Delivering Active Proteins Abundant in AKT and ERK Pathways
In addition to KEAP1, NRF2 is regulated by various signaling kinases.29 To explore the mechanism of the islet-protective effect of MSC-EXOs by inhibiting ferroptosis, we analyzed the active proteins that may play a role through the proteomic data of MSC-EXOs. As shown in Figure 6A, KEGG pathway analysis demonstrated the enrichment of exosome proteins in the PI3K-AKT and MAPK signaling pathways. It has been previously established that NRF2 is regulated by these two pathways.29 The PPI network analysis of the MSC-EXO proteins in PI3K-AKT and MAPK signaling pathways identified the key proteins as ITGA5, ITGB3, HRAS, RAP1A, and RAP1B (Figure 6B and C). This finding was confirmed by Western blot analysis of MSC-EXOs (Figure 6D). Furthermore, the MAPK signaling pathway was significantly enriched in the ERK pathway (Figure 6C). To determine whether MSC-EXOs activated the AKT and ERK signaling pathways in vitro and in vivo, IHC and IF staining were employed. IHC staining results confirmed that MSC-EXOs caused an increase in the levels of AKT and ERK phosphorylation in mice with T2DM (Figure 6E). Additionally, we noted that MSC-EXO therapy led to elevated levels of p-AKT and p-ERK in INS-1 cells (Figure 6F), indicating the potential involvement of AKT and ERK signaling in the NRF2-mediated prevention of ferroptosis. As shown in Figure 6B and C, among the five major molecules we identified, only HRAS participated in both the ERK and AKT signaling pathways, so we assume that HRAS is the key molecule that plays a role. HRAS may interact with a variety of effectors, mainly PI3K and RAF, which are classical targets of AKT and ERK pathways.30 Subsequently, molecular docking techniques were used to evaluate the binding affinity of HRAS with PI3K and RAF. The results showed that HRAS had a high affinity for PI3K and RAF, with binding energies of −221.70 kcal/mol and −221.70 kcal/mol, respectively (Figure S3A and B).
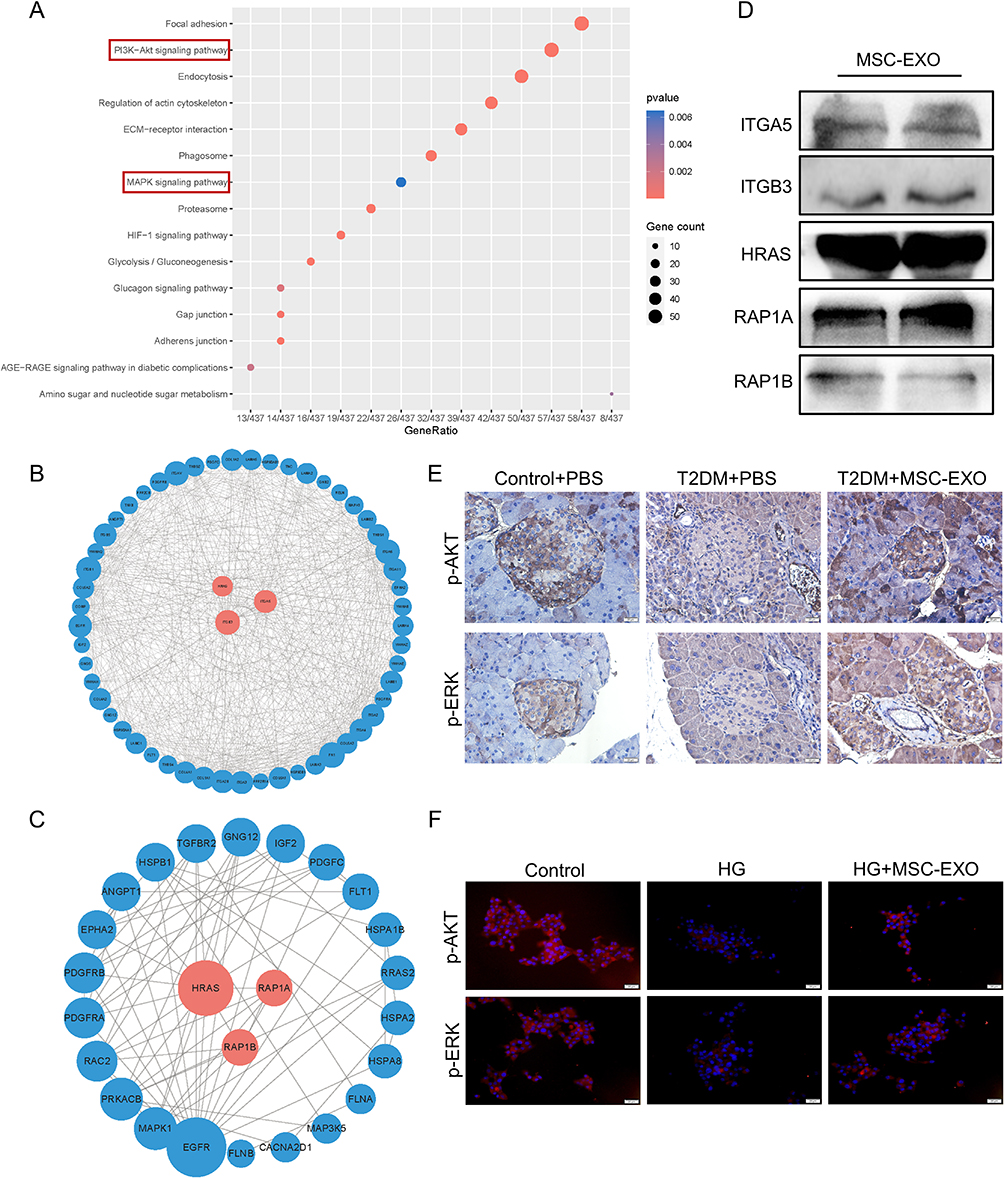 |
Figure 6 MSC-EXO activates AKT and ERK signaling pathways. (A) KEGG analysis of proteins in MSC-EXO. (B) PPI network of proteins involved in the AKT and (C) MAPK signaling pathways within MSC-EXO. (D) Western blot analysis showing levels of ITGA5, ITGB3, HRAS, RAP1A, and RAP1B in MSC-EXO. (E) IHC staining for p-AKT and p-ERK in mouse pancreatic paraffin sections (scale bar: 20 μm). (F) IF staining of p-AKT and p-ERK in INS-1 cells (scale bar: 20 μm). Data are represented as the mean ± SEM. |
To investigate if the AKT and ERK-mediated NRF2 activation is necessary for the anti-ferroptosis effect of MSC-EXO on β cells, we administered MSC-EXO to INS-1 cells along with LY294002 (AKT inhibitor) or PD98059 (ERK inhibitor) at the same time. As shown in Figure 7A, LY294002 and PD98059 successfully inhibited AKT and ERK phosphorylation in the MSC-EXOs. Suppressing AKT and ERK reversed the beneficial results of MSC-EXOs in enhancing NRF2, p-NRF2, GPX4, and SLC7A11 expression (Figure 7E). LY294002 and PD98059 eliminated the effects of MSC-EXOs treatment, including diminished lipid peroxidation and a decreased proportion of TUNEL-positive cells (Figure 7B and D). These findings indicate that MSC-EXOs control NRF2-mediated prevention of ferroptosis by transporting active proteins that are abundant in the AKT and ERK pathways.
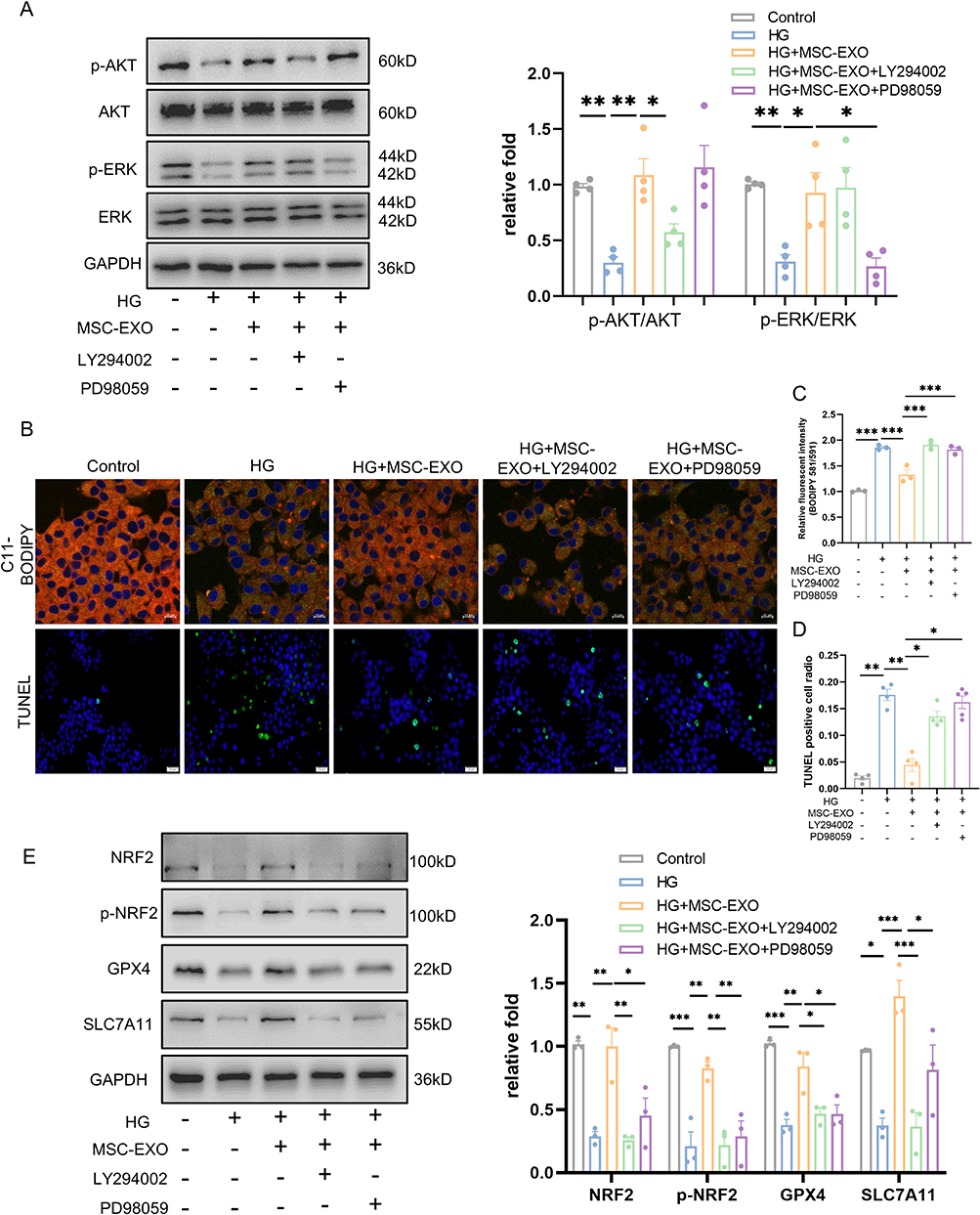 |
Figure 7 The inhibition of AKT and ERK impaired MSC-EXO anti-ferroptosis effects in INS-1 cells. (A) Western blot analysis of AKT, p-AKT, ERK, and p-ERK in INS-1 cells treated with LY294002, PD98059, MSC-EXO, and HG (n = 4). (B) Representative C11-BODIPY staining (scale bar: 10 μm) and TUNEL assay images (scale bar: 20 μm) in INS-1 cells. (C) Analysis of lipid peroxide fluorescence intensity (n = 3). (D) Analysis of TUNEL-positive cells (n = 4–5). (E) Western blot analysis of NRF2, p-NRF2, GPX4, and SLC7A11 in INS-1 cells (n = 3). Data are represented as the mean ± SEM. (*P < 0.05, **P < 0.01, ***P < 0.001). |
Construction and Identification of Apt-EXO
To further enhance the islet repair effects of exosomes, we propose that MSC-EXOs be modified with specific and recognizable ligands for targeted delivery. Previous studies have reported that an RNA aptamer can specifically identify human and mouse β cells in vitro and in vivo.31 The aptamer structure was determined based on predictions from the RNA structure website (Figure S4A). Using flow cytometry and IF staining analysis, we observed that the fluorescence signal of the cy3-labeled aptamer was significantly stronger than that of cy3-labeled Rd (random sequence) in MIN-6 cells (Figure S4B–D).Ex vivo tissue fluorescence imaging demo nstrated a significant accumulation of the aptamer in the pancreatic tissue (Figure S4E).
To enhance the delivery of MSC-EXO to β cells, the β-cell-specific aptamer was conjugated to MSC-EXO. As shown in Figure 8A, the aptamer’s 3’ terminal was altered with a thiol group capable of engaging in a Michael reaction with the maleimide group found in chol-PEG-mal. Additionally, the cholesterol moiety located at the opposite end can attach to the exosome membrane, conjugating the aptamer to the exosome and forming the Apt-EXO. To confirm whether chemical bonding occurred, the exosomes were incubated with the Cy3-labeled aptamer in the presence or absence of chol-PEG-mal, and the intensity of cy3 fluorescence emitted by the exosomes was measured. The results demonstrated that the fluorescence signal in the group treated with chol-PEG-mal was significantly elevated compared to that of the untreated group with chol-PEG-mal, suggesting successful coupling of the PEGylated aptamer to the exosome membrane (Figure 8B). Next, we utilized TEM, a nanoparticle tracking system, zeta potential, and Western blotting to analyze the structure, size, and protein makeup of Apt-EXOs. Apt-EXOs were observed using TEM and a nanoparticle tracking system, revealing their cup-shaped membrane structure and an approximate mode diameter of 130 nm (Figure 8C and D). Compared to the EXO, the zeta potential was slightly decreased by aptamer functionalization (Figure 8E). Moreover, Western blot analysis indicated that EXOs and Apt-EXOs were positive for exosome markers, including CD9, TSG101, and HSP70 (Figure 8F).
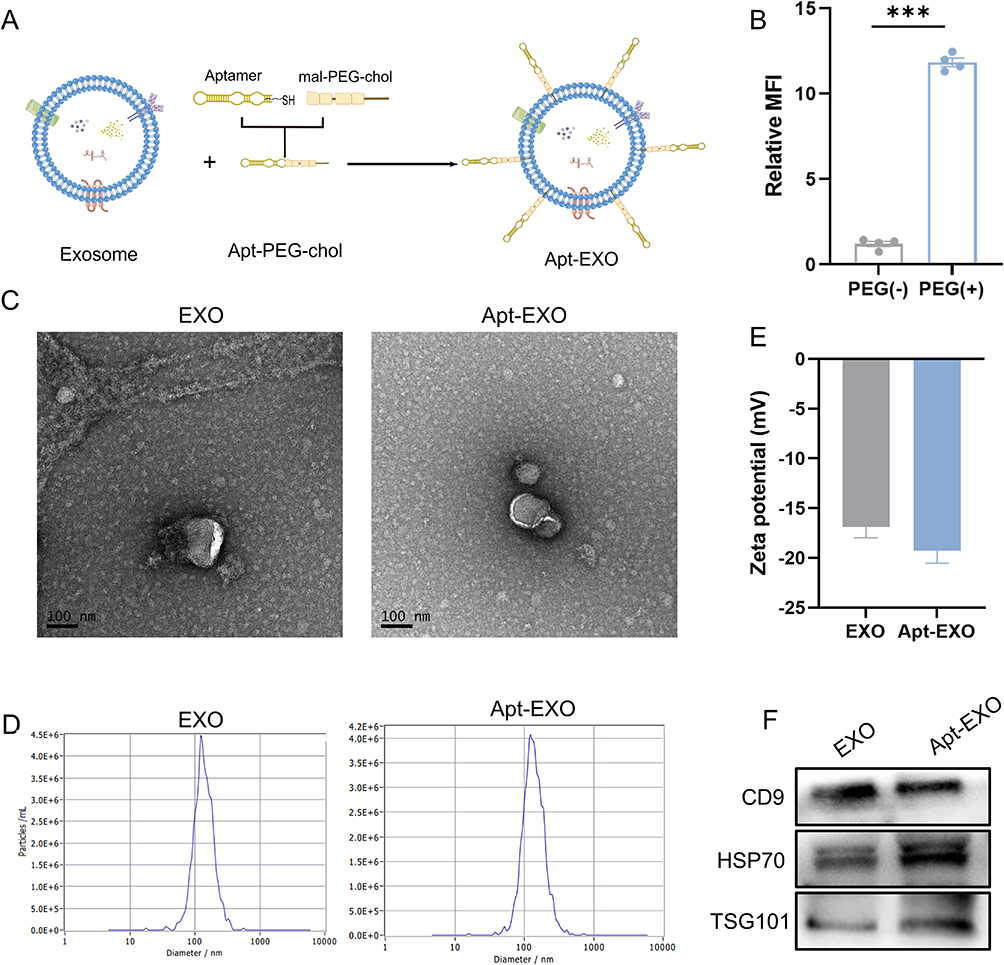 |
Figure 8 Identification of EXO and Apt-EXO. (A) Illustration depicting the conjugation process between the aptamer and MSC-EXO. (B) Fluorescence intensity assay assessing the reactivity between MSC-EXO and Cy3-labeled aptamer, either conjugated with chol-PEG-mal or not (n = 4). (C) Transmission electron microscopy image of EXO and Apt-EXO. (D) Nanoparticle tracking analysis of EXO and Apt-EXO. (E) Zeta potential analysis of EXO and Apt-EXO. (F) Western blot analysis of exosome markers CD9, TSG101, HSP70, and ER membrane marker Calnexin in EXO and Apt-EXO. Data are represented as the mean ± SEM. (***P < 0.001). |
Apt-EXO Has Enhanced β-Cell-Targeting and Prolonged Circulation
To further examine the localization of Apt-EXO in pancreatic β cells, the in vivo tissue distribution of Apt-EXO was confirmed, while the in vitro uptake efficiency of Apt-EXO by β cells was validated. Whole-body imaging of the mice was conducted at 2, 6, and 12 h following the infusion of DIR-labeled EXOs and Apt-EXOs through the caudal vein (Figure 9A). At 12h, organs were dissected and fluorescently imaged. The fluorescence intensity of the pancreas was notably greater in the Apt-EXO-treated group than in the EXO group (Figure 9B and C). Moreover, PEG modification has been reported to improve the blood circulation time of exosomes.23 Therefore, we collected blood samples at 1.3 and 6h after treatment with DIR-labeled Apt-EXO and EXO to detect the fluorescence signal in blood, and the results showed that the blood fluorescence signal in the Apt-EXO-treated group was significantly higher than that in the EXO-treated group(Figure 9D and E). For fluorescence imaging of other organs, as shown in Figure 9F, the liver fluorescence signal after Apt-EXO treatment was also stronger than that after EXO treatment, which may be due to the moderate uptake of aptamers in the liver (Figure S4E), and the blood fluorescence signal was also more abundant after Apt-EXO treatment. MIN-6 cells treated with the Apt-EXO exhibited increased uptake of exosomes compared to MIN-6 cells treated with EXO (Figure 9G–I). These results indicated that aptamer-functionalized exosomes have pancreatic β cell targeting ability and longer blood circulation time.
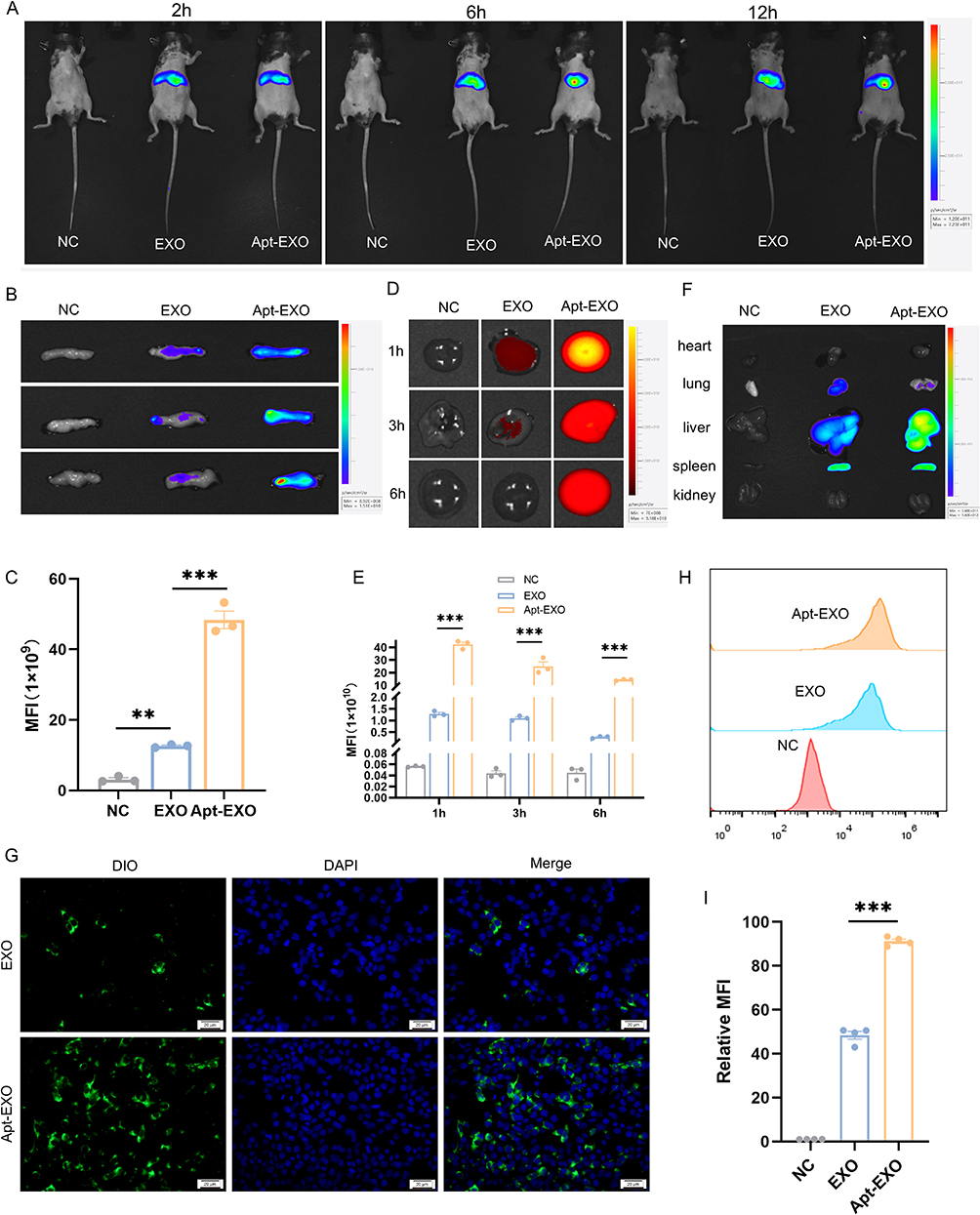 |
Figure 9 Apt-EXO displays enhanced β cell specificity and circulation time. (A) Whole-body imaging of the mice at 2, 6, and 12h after Tail vein infusion of EXO and Apt-EXO. (B) Ex vivo imaging of the pancreas 12h post-tail vein infusion of DiR-labeled EXO and Apt-EXO. (C) Quantitative analysis of pancreatic exosomes based on ex vivo fluorescence signals (n = 3). (D) Ex vivo imaging of blood at 1, 3, and 6h following tail vein infusion of DiR-labeled EXO and Apt-EXO. (E) Quantitative analysis of exosomes in the blood using ex vivo fluorescence signals (n = 3). (F)Ex vivo imaging of other major organs. (G) Representative images of DIO-labeled EXO and Apt-EXO internalization by MIN-6 cells. (H) Specificity of Apt-EXO to β cells analyzed via flow cytometry in MIN-6 cells. (I) Fluorescence intensity of DiO-labeled EXO and Apt-EXO cellular uptake in MIN-6 cells, as determined by flow cytometry (n = 3). Data are represented as the mean ± SEM. (**P < 0.01, ***P < 0.001). |
Apt-EXO Exhibited More Potent β Cell Function and Quantity Improvement Than Did EXO
To investigate whether Apt-EXOs have a stronger islet-protective effect than EXOs do, a T2DM mouse model was used(Figure 10A). The reduction in random glucose was greater with Apt-EXOs than with EXO, although the weight status was similar between the two groups (Figure 10B and C). T2DM mice treated with Apt-EXOs showed a more significant improvement in glucose tolerance (Figure 10D and E) and insulin secretion after fasting and 30 min in the IPGTT (Figure 10H) than did T2DM mice treated with standard EXOs. Furthermore, there was no notable disparity in insulin sensitivity between the Apt-EXO and EXO (Figure 10F and G). This finding aligns with the fact that Apt-EXO specifically targets pancreatic β cells, as insulin sensitivity indicates the level of insulin resistance in peripheral organs. The HE and IF staining analysis indicated that, compared to EXO treatment, treatment with Apt-EXO greatly improved the area of islets, the area of β cells, and the proportion of cells positive for insulin (Figure 10I–M). These results indicated that PEGylated aptamer modification enhanced the improvement of β cell function and quantity by MSC-EXOs.
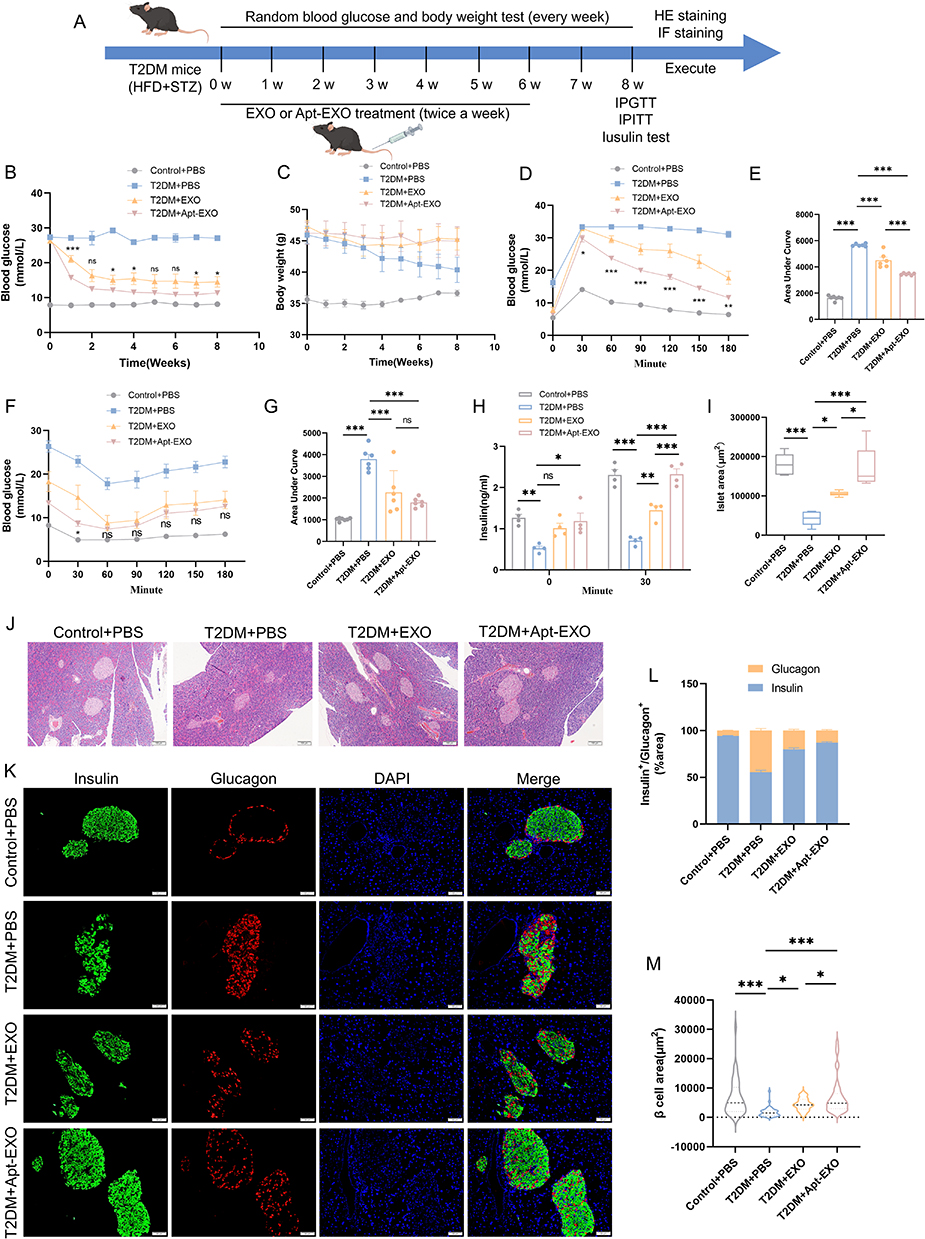 |
Figure 10 Apt-EXO is more effective than EXO in repairing the islet function. (A) Study design of PEGylated β-cell-targeting aptamer modification to enhance the islet repair effect of exosomes. Random blood glucose (B) and body weight (C) in control+PBS, T2DM+PBS, T2DM+EXO, and T2DM+Apt-EXO treated mice (n = 6–7 mice). IPGTT (D) and Area Under Curve (E) in T2DM mice following EXO or Apt-EXO injection (n = 6–7 mice). IPITT (F) and Area Under Curve (G) in T2DM mice after EXO and Apt-EXO treatment (n = 6–7 mice). Insulin secretion (H) during fasting and 30 min of IPGTT (n = 4 mice). Analysis of islet area (I) across all mouse groups (n = 5 mice). HE staining of pancreatic sections (J) in each group (scale bar, 100 μm). Representative pancreatic sections (K) stained for insulin and glucagon (scale bar, 50 μm). Proportion of insulin and glucagon in immune-positive areas (L) (n = 5 mice and 40 islets). Analysis of β-cell area (M) in IF staining (n = 5 mice and 40 islets). Data are represented as the mean ± SEM. (*P < 0.05, **P < 0.01, ***P < 0.001). |
Discussion
β cells are located in clusters called islets and help regulate blood glucose levels by producing insulin.32 The demise of β cells has been documented as a key factor in advancing T2DM.33 Although MSC-based therapies have shown promise for treating T2DM, their clinical application is hindered by the potential risk of tumorigenicity.34,35 Apart from being the main means by which MSCs repair damage, exosomes are easy to store and transport and have low immunogenicity.36 Therefore, exosome therapy has a promising clinical significance for managing diabetes. Previous studies have shown that systemic infusion of MSC-EXO can improve islet function and ameliorate hyperglycemia, but there are few studies on the mechanism of MSC-EXO inhibiting β cell death, and the effective contents of exosomes playing a therapeutic role have not been clarified. Sun et al reported that MSC-EXOs improve insulin resistance, and also promote insulin secretion and islet regeneration by inhibiting beta-cell apoptosis in high-fat diet combined with STZ-induced T2DM rats,17 while the specific mechanism of MSC-EXOs improving β cells has not been studied. Chen et al’s study suggested that MSC-EXOs alleviate hypoxia-induced ER stress, thereby protecting the β cell line βTC-6 from hypoxia-induced apoptosis.37 However, the active ingredient in MSC-EXOs that plays a therapeutic role has not been further studied, and corresponding animal studies are lacking. Our study provides evidence for the mechanism by which MSC-EXO improves islet function and identifies the effective molecule in MSC-EXO, that is, MSC-EXO activates the AKT and ERK signaling pathways through the secretion of active proteins such as HRAS, thereby exerting NRF2-mediated anti-ferroptosis effects.
To assess MSC-EXO’s impact on T2DM-related β cell damage, we treated T2DM mice with MSC-EXOs. This led to improved glucose and insulin tolerance, stable body weight, and reduced spontaneous blood glucose levels. Additionally, islet and β-cell areas increased, along with the proportion of insulin-producing cells, corroborating previous findings on MSCs’ beneficial effects on β-cell function.17,38–40
Ferroptosis, a novel form of controlled cellular death, is characterized by disrupted iron homeostasis and oxidative lipid degradation.5 The expression of antioxidant enzymes, like GPX4, is lacking in β cells, and GPX4 plays a crucial role in combating ferroptosis.11,12,14 Therefore, β cells are highly vulnerable to ferroptosis. The impact of β cell ferroptosis on insulin secretion has been verified, and the administration of the ferroptosis inhibitor Fer-1 can lower blood glucose levels, enhance insulin secretion, augment the islet area, and increase the proportion of insulin-positive cells.13,41,42 In T2DM mice and glucose-stimulated INS-1 cells, ferroptosis was detected. Notably, the ferroptosis inhibitor proved more effective in preserving INS-1 cell viability under high-glucose conditions than other cell-death inhibitors. Additionally, the administration of Fer-1 and MSC-EXOs enhanced insulin release from glucose-activated INS-1 cells. MSC-EXO effectively inhibited ferroptosis in β cells at both in vivo and in vitro levels. Importantly, NRF2 plays a critical role in the transcriptional regulation of anti-ferroptosis genes.28 Earlier research has shown that the complete absence of NRF2 in mice leads to decreased survival of islet cells, smaller islets, and increased susceptibility of β cells to arsenic damage.43–45 Our study showed that NRF2 knockdown reversed MSC-EXOs’ anti-ferroptotic effect, confirming that MSC-EXOs protect islet function by inhibiting NRF2-mediated ferroptosis.
The composition of MSC-EXOs has been extensively investigated because of their crucial role in intercellular communication. According to prior studies, MSC-EXOs predominantly comprise proteins, lipids, and nucleic acids.46 Identification of functional proteins within exosomes is vital for elucidating the mechanisms by which MSC-EXOs facilitate tissue regeneration.47 Intricately regulated by KEAP1 and a range of other signaling kinases, the activation of NRF2 is particularly significant.29 Our investigation disclosed that the active proteins transported by MSC-EXOs were principally enriched in the AKT and ERK pathways. These pathways modulate NRF2 activity through the phosphorylation of AKT and ERK, thereby inhibiting ferroptosis.
Although studies of MSC-EXO have yielded promising results, they are mostly preclinical studies. Therefore, more research is needed to optimize MSC-EXO-based therapies. For example, MSCs or MSC-EXO are specifically designed to maximize the therapeutic effects of exosomes.48 Lv et al reported that loading miR-21-5p mimics into exosomes via electroporation increased in vivo re-epithelialization, collagen remodeling, angiogenesis, and vascular maturation to accelerate diabetic wound healing.49 Wang et al showed that MSCs treated with inflammatory factors obtain MSC-EXO with high expression of PD-L1, which enhances the therapeutic effect of T1DM.50 Therefore, to promote the clinical application of MSC-EXO in T2DM, we further optimize the treatment strategy of MSC-EXO.
The effectiveness of MSC-EXO in improving islet function is limited due to the restricted quantity of exosomes that can reach the pancreas after being administered systemically.18 Previous research has successfully obtained specific nanoparticles targeting tumors through chemical modification using Tumor necrosis factor-related apoptosis-inducing ligands.51 The modification of PEG and C─C motif chemokine ligands in macrophage-derived exosomes is employed to enhance the accumulation of exosomes in joints, resulting in a significant reduction of joint inflammation.52 Compared to antibodies, aptamers have the advantages of small molecular weight, stable assembly, flexible chemical modification, and low immunogenicity.53 Additionally, aptamers are temperature insensitive; they can tolerate high temperatures up to 80 °C and maintain their 3D structure, whereas monoclonal antibodies denature at this temperature.54 Therefore, we modified MSC-EXO with islet β-cell-targeting aptamers and used PEG as an intermediary to prolong blood circulation. This enhanced the β cell repair efficacy of MSC-EXOs while limiting adverse effects.
This study has several limitations. First, the effects of MSC-EXO on islet β cells have not been further validated in human-derived islets or islet organoids. Secondly, a cycling-lengthening exosome with β cell targeting was constructed, but it was not equipped with drugs to further enhance its therapeutic effect. In future projects, we will build a targeted drug delivery system for islets based on MSC-EXO to bring exosome therapy closer to clinical application.
Conclusions
Collectively, our findings indicate that MSC-EXO effectively improves β cell function and quantity in T2DM mice through the delivery of bioactive proteins that are abundant in the AKT and ERK pathways, resulting in the phosphorylation of AKT and ERK and the subsequent inhibition of NRF2-mediated ferroptosis. Additionally, we introduce an innovative and safe method to improve the targeting abilities of MSC-EXO towards pancreatic β cells and circulation time, increasing its potential for therapy in cases of β cell function and quantity.
Acknowledgments
This work was supported by the National Nature Science Foundation [grant number 82070800], a Special fund for Taishan Industrial leading talent project, Major basic research project of Shandong Natural Science Foundation [grant number ZR2022ZD15]. We would like to thank Editage (www.editage.cn) for English language editing.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Poitout V, Robertson RP. Glucolipotoxicity: fuel Excess and β-cell dysfunction. Endocrine Rev. 2008;29(3):351–366. doi:10.1210/er.2007-0023
2. Porat S, Weinberg-Corem N, Tornovsky-Babaey S, et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 2011;13(4):440–449. doi:10.1016/j.cmet.2011.02.012
3. Prentki M, Matschinsky Franz M, Madiraju SRM. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18(2):162–185. doi:10.1016/j.cmet.2013.05.018
4. Wajchenberg BL. beta-cell failure in diabetes and preservation by clinical treatment. Endocrine Rev. 2007;28(2):187–218. doi:10.1210/10.1210/er.2006-0038
5. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
6. Berghe TV, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135–147. doi:10.1038/nrm3737
7. Dixon Scott J, Lemberg Kathryn M, Lamprecht Michael R, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
8. Yagoda N, von Rechenberg M, Zaganjor E, et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):865–869. doi:10.1038/nature05859
9. Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191. doi:10.1038/ncb3064
10. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–152. doi:10.1016/j.freeradbiomed.2018.09.014
11. Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20(3):463–466. doi:10.1016/0891-5849(96)02051-5
12. Koulajian K, Ivovic A, Ye K, et al. Overexpression of glutathione peroxidase 4 prevents β-cell dysfunction induced by prolonged elevation of lipids in vivo. Am J Physiol Endocrinol Metab. 2013;305(2):E254–E262. doi:10.1152/ajpendo.00481.2012
13. Bruni A, Pepper AR, Pawlick RL, et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. 2018;9(6):595. doi:10.1038/s41419-018-0506-0
14. Krümmel B, Plötz T, Jörns A, Lenzen S, Mehmeti I. The central role of glutathione peroxidase 4 in the regulation of ferroptosis and its implications for pro-inflammatory cytokine-mediated beta-cell death. Biochim Biophys Acta Mol Basis Dis. 2021;1867(6):166114. doi:10.1016/j.bbadis.2021.166114
15. Xie Q, Liu R, Jiang J, et al. What is the impact of human umbilical cord mesenchymal stem cell transplantation on clinical treatment? Stem Cell Res Ther. 2020;11(1):519. doi:10.1186/s13287-020-02011-z
16. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478). doi:10.1126/science.aau6977
17. Sun Y, Shi H, Yin S, et al. Human mesenchymal stem cell derived exosomes alleviate type 2 diabetes mellitus by reversing peripheral insulin resistance and relieving β-cell destruction. ACS Nano. 2018;12(8):7613–7628. doi:10.1021/acsnano.7b07643
18. Wiklander OPB, Nordin JZ, O’Loughlin A, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015;4:26316. doi:10.3402/jev.v4.26316
19. Lai CP, Mardini O, Ericsson M, et al. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano. 2014;8(1):483–494. doi:10.1021/nn404945r
20. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249(4968):505–510. doi:10.1126/science.2200121
21. Hermann T, Patel DJ. Adaptive recognition by nucleic acid aptamers. Science. 2000;287(5454):820–825. doi:10.1126/science.287.5454.820
22. Zou J, Shi M, Liu X, et al. Aptamer-functionalized exosomes: elucidating the cellular uptake mechanism and the potential for cancer-targeted chemotherapy. Anal Chem. 2019;91(3):2425–2430. doi:10.1021/acs.analchem.8b05204
23. Kooijmans SAA, Fliervoet LAL, van der Meel R, et al. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release. 2016;224:77–85. doi:10.1016/j.jconrel.2016.01.009
24. Giraldo JA, Molano RD, Rengifo HR, et al. The impact of cell surface PEGylation and short-course immunotherapy on islet graft survival in an allogeneic murine model. Acta Biomater. 2017;49:272–283. doi:10.1016/j.actbio.2016.11.060
25. Sun B, Chen Y, Yu H, et al. Photodynamic PEG-coated ROS-sensitive prodrug nanoassemblies for core-shell synergistic chemo-photodynamic therapy. Acta Biomater. 2019;92:219–228. doi:10.1016/j.actbio.2019.05.008
26. Hu H, Zhao R, He Q, et al. cGAS-STING mediates cytoplasmic mitochondrial-DNA-induced inflammatory signal transduction during accelerated senescence of pancreatic β-cells induced by metabolic stress. FASEB J. 2022;36(5):e22266. doi:10.1096/fj.202101988R
27. Ding Y, Wu Q. 1,25D/VDR inhibits pancreatic β cell ferroptosis by downregulating FOXO1 expression in diabetes mellitus. Cell Signal. 2023;105:110564. doi:10.1016/j.cellsig.2022.110564
28. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi:10.1016/j.redox.2019.101107
29. Baumel-Alterzon S, Katz LS, Brill G, Garcia-Ocaña A, Scott DK. Nrf2: the master and captain of beta cell fate. Trends Endocrinol Metab. 2021;32(1):7–19. doi:10.1016/j.tem.2020.11.002
30. Rosseland CM, Wierød L, Flinder LI, Oksvold MP, Skarpen E, Huitfeldt HS. Distinct functions of H-Ras and K-Ras in proliferation and survival of primary hepatocytes due to selective activation of ERK and PI3K. J Cell Physiol. 2008;215(3):818–826. doi:10.1002/jcp.21367
31. Van Simaeys D, De La Fuente A, Zilio S, et al. RNA aptamers specific for transmembrane p24 trafficking protein 6 and clusterin for the targeted delivery of imaging reagents and RNA therapeutics to human β cells. Nat Commun. 2022;13(1). doi:10.1038/s41467-022-29377-3
32. Prasad MK, Mohandas S, Ramkumar KM. Dysfunctions, molecular mechanisms, and therapeutic strategies of pancreatic β-cells in diabetes. Apoptosis. 2023;28(7–8):958–976. doi:10.1007/s10495-023-01854-0
33. Remedi MS, Emfinger C. Pancreatic β-cell identity in diabetes. Diabetes Obes Metab. 2016;18(Suppl 1):110–116. doi:10.1111/dom.12727
34. Si Y, Zhao Y, Hao H, et al. Infusion of mesenchymal stem cells ameliorates hyperglycemia in type 2 diabetic rats: identification of a novel role in improving insulin sensitivity. Diabetes. 2012;61(6):1616–1625. doi:10.2337/db11-1141
35. Zhai M, Zhu Y, Yang M, Mao C. Human mesenchymal stem cell derived exosomes enhance cell-free bone regeneration by altering their miRNAs profiles. Adv Sci. 2020;7(19):2001334. doi:10.1002/advs.202001334
36. Elahi FM, Farwell DG, Nolta JA, Anderson JD. Preclinical translation of exosomes derived from mesenchymal stem/stromal cells. Stem Cells. 2020;38(1):15–21. doi:10.1002/stem.3061
37. Chen J, Chen J, Cheng Y, et al. Mesenchymal stem cell-derived exosomes protect beta cells against hypoxia-induced apoptosis via miR-21 by alleviating ER stress and inhibiting p38 MAPK phosphorylation. Stem Cell Res Ther. 2020;11(1):97. doi:10.1186/s13287-020-01610-0
38. Khatri R, Mazurek S, Petry SF, Linn T. Mesenchymal stem cells promote pancreatic β-cell regeneration through downregulation of FoxO1 pathway. Stem Cell Res Ther. 2020;11(1):497. doi:10.1186/s13287-020-02007-9
39. Wang M, Song L, Strange C, Dong X, Wang H. Therapeutic effects of adipose stem cells from diabetic mice for the treatment of type 2 diabetes. Mol Ther. 2018;26(8):1921–1930. doi:10.1016/j.ymthe.2018.06.013
40. Yin Y, Hao H, Cheng Y, et al. Human umbilical cord-derived mesenchymal stem cells direct macrophage polarization to alleviate pancreatic islets dysfunction in type 2 diabetic mice. Cell Death Dis. 2018;9(7):760. doi:10.1038/s41419-018-0801-9
41. Markelic M, Stancic A, Saksida T, et al. Defining the ferroptotic phenotype of beta cells in type 1 diabetes and its inhibition as a potential antidiabetic strategy. Front Endocrinol. 2023;14:1227498. doi:10.3389/fendo.2023.1227498
42. Li D, Jiang C, Mei G, et al. Quercetin alleviates ferroptosis of pancreatic β cells in type 2 diabetes. Nutrients. 2020;12(10):2954. doi:10.3390/nu12102954
43. Li S, Vaziri ND, Masuda Y, et al. Pharmacological activation of Nrf2 pathway improves pancreatic islet isolation and transplantation. Cell Transplant. 2015;24(11):2273–2283. doi:10.3727/096368915X686210
44. Yang B, Fu J, Zheng H, et al. Deficiency in the nuclear factor E2-related factor 2 renders pancreatic β-cells vulnerable to arsenic-induced cell damage. Toxicol Appl Pharmacol. 2012;264(3):315–323. doi:10.1016/j.taap.2012.09.012
45. Uruno A, Furusawa Y, Yagishita Y, et al. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol. 2013;33(15):2996–3010. doi:10.1128/MCB.00225-13
46. Jin Q, Wu P, Zhou X, Qian H, Xu W. Extracellular vesicles: novel roles in neurological disorders. Stem Cells Int. 2021;2021:6640836. doi:10.1155/2021/6640836
47. Roefs MT, Sluijter JPG, Vader P. Extracellular vesicle-associated proteins in tissue repair. Trends Cell Biol. 2020;30(12):990–1013. doi:10.1016/j.tcb.2020.09.009
48. Liu J, Sun X, Zhang FL, et al. Clinical potential of extracellular vesicles in type 2 diabetes. Front Endocrinol. 2020;11:596811. doi:10.3389/fendo.2020.596811
49. Lv Q, Deng J, Chen Y, Wang Y, Liu B, Liu J. Engineered human adipose stem-cell-derived exosomes loaded with miR-21-5p to promote diabetic cutaneous wound healing. Mol Pharm. 2020;17(5):1723–1733. doi:10.1021/acs.molpharmaceut.0c00177
50. Wang L, Qi C, Cao H, et al. Engineered cytokine-primed extracellular vesicles with high PD-L1 expression ameliorate type 1 diabetes. Small. 2023;19(38):e2301019. doi:10.1002/smll.202301019
51. Feng X, Li F, Zhang L, et al. TRAIL-modified, doxorubicin-embedded periodic mesoporous organosilica nanoparticles for targeted drug delivery and efficient antitumor immunotherapy. Acta Biomater. 2022;143:392–405. doi:10.1016/j.actbio.2022.03.001
52. Wang Z, Zhang C, Meng J, et al. A targeted exosome therapeutic confers both CfDNA scavenging and macrophage polarization for ameliorating rheumatoid arthritis. Adv Mater. 2023;35(48):e2302503. doi:10.1002/adma.202302503
53. Narwade M, Shaikh A, Gajbhiye KR, Kesharwani P, Gajbhiye V. Advanced cancer targeting using aptamer functionalized nanocarriers for site-specific cargo delivery. Biomater Res. 2023;27(1):42. doi:10.1186/s40824-023-00365-y
54. Dhar P, Samarasinghe RM, Shigdar S. Antibodies, nanobodies, or aptamers-which is best for deciphering the proteomes of non-model species? Int J Mol Sci. 2020;21(7):2485. doi:10.3390/ijms21072485
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.