")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of Milvexian in Healthy Chinese Adults
Authors Luo Z, Wang J, Niu Z, Hu C , Chintala M, Luo X, Lee TI, Plotnikov AN, Zannikos P
Received 24 July 2024
Accepted for publication 24 January 2025
Published 1 March 2025 Volume 2025:19 Pages 1503—1514
DOI https://doi.org/10.2147/DDDT.S488414
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Zhu Luo,1 Jie Wang,2 Zhuolu Niu,3 Cuili Hu,4 Madhu Chintala,5 Xinchao Luo,3 Tsung-I Lee,3 Alexei N Plotnikov,5 Peter Zannikos5
1Clinical Trial Center, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 2Department of Hematology, West China Hospital, Sichuan University, Chengdu, People’s Republic of China; 3Johnson & Johnson, Shanghai, People’s Republic of China; 4Johnson & Johnson, Beijing, People’s Republic of China; 5Johnson & Johnson, Raritan, NJ, USA
Correspondence: Zhuolu Niu, Janssen Research & Development, 65 Gui Qing Road, Shanghai, 200233, People’s Republic of China, Email [email protected]
Background: Milvexian is a small molecule, selective factor XIa (FXIa) inhibitor being developed as an oral anticoagulant. This study assessed the pharmacokinetics, pharmacodynamics (activated partial thromboplastin time [aPTT]), and safety of milvexian in healthy Chinese subjects.
Methods: Part 1: Thirty subjects were randomly assigned 1:1:1 to receive milvexian 25 mg on Day 1 followed by 25 mg once daily (QD) on Days 5– 12; milvexian 25 mg twice daily at 12-hour intervals (BID) on Days 1– 8; or milvexian 100 mg BID on Days 1– 8. Part 2: Ten subjects received milvexian 200 mg on Day 1 followed by 200 mg BID on Days 5– 12. Plasma samples were collected for pharmacokinetics and aPTT assessments. Safety and tolerability were assessed.
Results: Milvexian was rapidly absorbed (median tmax of 3– 4 hours after a single dose and repeated administration). Mean maximum concentrations or area under the concentration-time curve values of milvexian in plasma after single doses or BID administration of 25 mg, 100 mg, or 200 mg increased in a dose-dependent manner. Steady state conditions were achieved within 6 days of repeated administration based on milvexian trough concentration values. Mean terminal half-life values (9– 10 hours) were independent of the dose. Milvexian reversibly prolonged aPTT in a manner that was directly related to milvexian dose and exposure. All milvexian regimens were safe and well tolerated, with only mild treatment-emergent adverse events and no clinically significant bleeding events. No new safety signals were identified.
Conclusion: The pharmacokinetic, pharmacodynamic, and safety profiles of milvexian demonstrate suitability for further clinical development in Chinese participants.
Keywords: anticoagulant, aPTT, FXIa inhibitor, milvexian, pharmacokinetics
Introduction
Anticoagulants are routinely used to reduce the risk of recurrent serious adverse atherothrombotic and thromboembolic events in patients with vascular and thromboembolic diseases.1–4 However, despite the proven effectiveness of these anticoagulant therapies in the prevention of thrombosis, they are associated with an increased, dose-dependent risk of major bleeding events.5,6 Therefore, it is crucial to develop new treatments for thrombotic disorders with improved benefit/risk profiles compared with current therapies.
Under normal physiologic conditions, hemostasis and thrombus formation are achieved through the coordinated activation of platelets and the coagulation cascade.7 Thrombin, which is generated upon vascular injury via the cleavage of prothrombin by factor Xa, plays a key role in thrombus formation, as it induces platelet aggregation, converts fibrinogen to fibrin, and activates factors XIII, V, VIII, and XI.8 The zymogen factor XI (FXI) is a component of the intrinsic pathway that is activated by thrombin to form FXIa, which in turn activates factors IX, X, and prothrombin to amplify thrombin generation to a level that is sufficient to achieve hemostasis.8–10
Epidemiologic studies have shown that individuals with FXI deficiency have a reduced risk of thrombus formation and cardiovascular and venous thromboembolism.11–13 Additionally, in preclinical models, inhibition of FXI/FXIa increased activated partial thromboplastin time (aPTT) without changing prothrombin time, reduced thrombus weight when the inhibition occurred during ongoing thrombus formation, and was effective in the treatment of existing thrombosis without increased bleeding.14–17 Current evidence suggesting that inhibition of FXIa may prevent thrombosis while preserving hemostasis, and yield a safer bleeding profile in patients at high risk of thrombotic or bleeding events, has made inhibition of FXIa an attractive therapeutic target.18
Milvexian (formerly referred to as BMS-986177/JNJ-70033093) is a small-molecule, FXIa inhibitor that is being developed as an oral anticoagulant for the prevention of thrombotic events in multiple patient populations.19 Milvexian selectively inhibits FXIa in the intrinsic pathway with high affinity and interrupts thrombin amplification without interfering with extrinsic pathway-initiated thrombin generation, which is critical for preserving hemostasis. In a phase 1 study of healthy, primarily White subjects, milvexian was safe and well tolerated at doses up to 500 mg daily or 200 mg twice daily for 14 days with no major bleeding, clinically significant bleeding events, or other concerning treatment-emergent adverse events (TEAEs).20 In a phase 1 study of healthy Japanese subjects, milvexian was well tolerated at regimens of up to 500 mg once daily for 14 days with no serious TEAEs; pharmacokinetic and pharmacodynamic profiles were similar to those observed in non-Asian subjects.21 Separate phase 1 studies have further demonstrated the safety of a single dose of 60 mg milvexian in subjects with mild to moderate hepatic impairment and moderate to severe renal impairment.22,23 Finally, two completed phase 2 studies evaluated the safety and efficacy of various milvexian regimens in patients undergoing total knee replacement surgery (AXIOMATIC-TKR) and patients with acute ischemic stroke or transient ischemic attack (AXIOMATIC-SSP).24,25 The Librexia program includes phase 3 studies that will evaluate the safety and efficacy of milvexian (i) for stroke prevention after an acute ischemic stroke or high-risk transient ischemic attack and (ii) in patients with atrial fibrillation, and (iii) in patients who have had a recent acute coronary syndrome at risk for thrombosis (ClinicalTrials.gov Identifiers: NCT05702034, NCT05757869, NCT05754957). These studies are enrolling patients in various countries worldwide, including China.
The primary objective of the current study was to assess the pharmacokinetics of single and multiple doses of milvexian in healthy Chinese subjects. Secondary objectives included assessment of pharmacodynamics (aPTT), safety, and tolerability of milvexian in this population.
Methods
Participants
Healthy Chinese males and females between 18 and 55 years of age were eligible to participate in the study if they had normal blood coagulation, serum biochemistry, and hematology. In addition, subjects were required to have a body mass index between 18 and 30 kg/m² (body weight of male subjects ≥50 kg and body weight of female subjects ≥45 kg), supine systolic blood pressure of 90 to 140 mmHg, and diastolic blood pressure of 60 to 90 mmHg. Potential subjects were excluded from the study if they had any significant acute or chronic medical conditions, evidence of bleeding disorders, history of head trauma or recurrent headaches, aneurysm, recent blood loss or blood donation, current hepatitis B, C, or human immunodeficiency virus infection, or abnormal laboratory values or electrocardiography results.
Study Design
This phase 1, randomized, open-label, single- and multiple-dose study (ClinicalTrials.gov identifier: NCT04569695) was conducted at one study center in China in accordance with the ethical principles of the Declaration of Helsinki, and was consistent with Good Clinical Practice and applicable regulatory requirements. The protocol and amendments were reviewed by an independent ethics committee at the study center, and all subjects provided written informed consent.
Part 1 of the study consisted of three cohorts (Cohorts A, B, and C) that each enrolled 10 subjects (Figure 1). Subjects were assigned 1:1:1 to the three cohorts in Part 1 based on a computer-generated randomization schedule prepared by the sponsor. Subjects in Cohort A received milvexian 25 mg on Day 1 followed by a 4-day washout period, and then milvexian 25 mg once daily on Days 5 to 12. Subjects in Cohort B received milvexian 25 mg twice daily (with doses in the morning and evening, 12 hours apart) on Days 1 to 8. Subjects in Cohort C received milvexian 100 mg twice daily on Days 1 to 8.
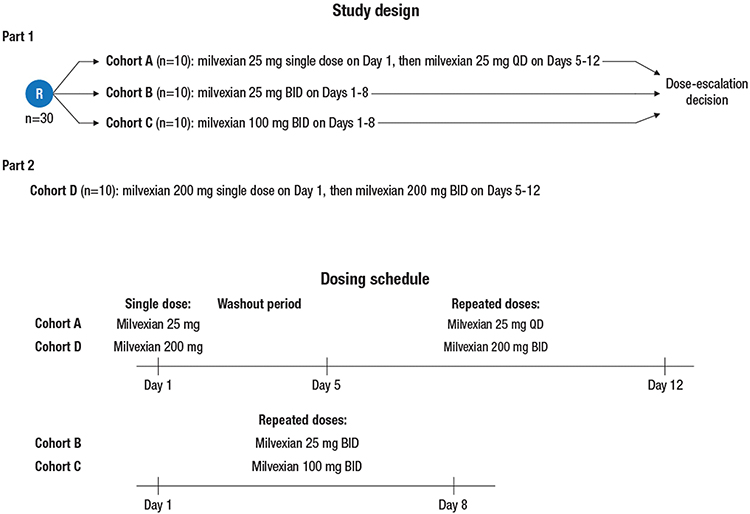 |
Figure 1 Study design and milvexian dosing schedule. Abbreviations: BID, twice daily (administration every 12 hours); QD, once daily (administration every 24 hours). |
In Part 2 of the study, 10 subjects in Cohort D were enrolled after the safety and tolerability data from Part 1 were deemed acceptable (Figure 1). These subjects received milvexian 200 mg on Day 1 followed by a 4-day washout period, and then milvexian 200 mg twice daily on Days 5 to 12.
All subjects fasted overnight (~10 hours) prior to administration of each morning dose of milvexian. All doses were taken with approximately 240 mL of water. On Days 1 and 12 for Cohorts A and D, Days 1 and 8 for Cohorts B and C, and Day 5 for Cohort D, subjects continued fasting for 4 hours postdose (lunch, dinner, and a snack were provided approximately 4, 8, and 12 hours, respectively, after the morning dose). On Days 5 to 11 for Cohort A, Days 2 to 7 for Cohorts B and C, and Days 6 to 11 for Cohort D, breakfast and lunch were provided at 1 hour and 4 hours after the morning dose, respectively. For the twice daily regimens, dinner was provided at 10 hours after the morning dose (ie, 2 hours prior to the evening dose) and a snack was provided 13 hours after the morning dose (ie, 1 hour after the evening dose). Milvexian was supplied as 25 mg and 100 mg capsules.
Assessments
Intensive serial blood sampling was performed for pharmacokinetic assessments in plasma. For Cohort A (25 mg single dose followed by 25 mg once daily), sampling occurred predose and for 72 hours after the morning dose on Day 1 and for 72 hours after the morning dose on Day 12. For Cohort B (25 mg twice daily) and Cohort C (100 mg twice daily), sampling occurred predose and for 12 hours after the morning doses on Day 1 and Day 8. For Cohort D (200 mg single dose followed by 200 mg twice daily), sampling occurred predose and for 72 hours after the morning dose on Day 1 and for 12 hours after the morning doses on Day 5 and Day 12. In addition, trough pharmacokinetic samples were collected for 2 consecutive days (Days 10 and 11 for Cohorts A and D and Days 6 and 7 for Cohorts B and C) for the assessment of steady state. Plasma samples were analyzed using a validated, specific, and sensitive liquid chromatography/tandem mass spectrometry (LC-MS/MS) method with an assay range from 1 to 1000 ng/mL (Labcorp Shanghai bioanalysis laboratory).
Pharmacokinetic parameters, calculated using noncompartmental methods (Phoenix™ WinNonlin® [Tripos L.P., version 6.4 or above]) and actual sampling times, included maximum plasma concentration (Cmax), time to maximum plasma concentration (tmax), trough (ie, predose) plasma concentration at the end of a dosing interval (Ctrough), AUC from time zero to the last measurable concentration (AUClast), AUC from time zero to infinite time (AUCinf), AUC during a dosing interval at steady state (AUCτ), and terminal elimination half-life (t1/2).
Plasma aPTT was assessed (Labcorp Shanghai) over the intervals described above for pharmacokinetic sampling (with the exception of aPTT samples that were not collected from subjects in Cohort D after the morning milvexian dose on Day 5). Observed values and the percentage change from baseline (prior to the first dose on Day 1 for all cohorts) in plasma aPTT were reported.
Safety and tolerability were assessed based on medical review of adverse event reports and the results of clinical laboratory tests, fecal occult blood assessments, vital sign measurements, electrocardiograms (ECGs), and physical examinations that were conducted prior to milvexian administration and at predetermined times postdose. TEAEs of special interest included bleeding events categorized as major bleeding (including fatal bleeding, critical organ bleeding, and bleeding leading to a transfusion), clinically relevant nonmajor bleeding (CRNM; hemorrhage necessitating medical intervention but not classified as major bleeding), and other bleeding.
Statistical Analyses
Sample size was determined based on milvexian Cmax and AUCinf of previous studies (data on file). Based on interindividual variability (percent coefficient of variation [%CV]) of 39.0% and 37.2% for Cmax and AUCinf after single dose administration, respectively, a sample size of 10 subjects with evaluable pharmacokinetic data per cohort was expected to provide point estimates for mean milvexian Cmax and AUCinf within 80% to 125% and 81% to 124%, respectively, of the true value with 90% confidence. Based on interindividual variability of 46.6% and 48.9% for Cmax and AUCτ after twice daily administration, respectively, a sample size of 10 subjects with evaluable pharmacokinetic data per cohort was expected to provide point estimates for mean milvexian Cmax and AUCτ within 76% to 131% and 75% to 133%, respectively, of the true value with 90% confidence.
Pharmacokinetic data were summarized using descriptive statistics that included all subjects who received ≥1 dose of study drug and had ≥1 measurable concentration in plasma. All subjects who received ≥1 dose of milvexian were included in the safety analysis. Safety and pharmacodynamic data were summarized using descriptive statistics.
Results
Participant Characteristics
In Part 1, 30 subjects were randomly assigned 1:1:1 to Cohorts A, B, or C, and in Part 2, 10 subjects were enrolled in Cohort D. All subjects completed the study. Demographics were balanced across all groups. Overall, 31 (77.5%) subjects were male and 9 (22.5%) were female, mean (standard deviation [SD]) age was 27.7 (6.8) years, and mean (SD) body mass index was 22.2 (2.2) kg/m2 (Table 1). All subjects were of Chinese ethnicity.
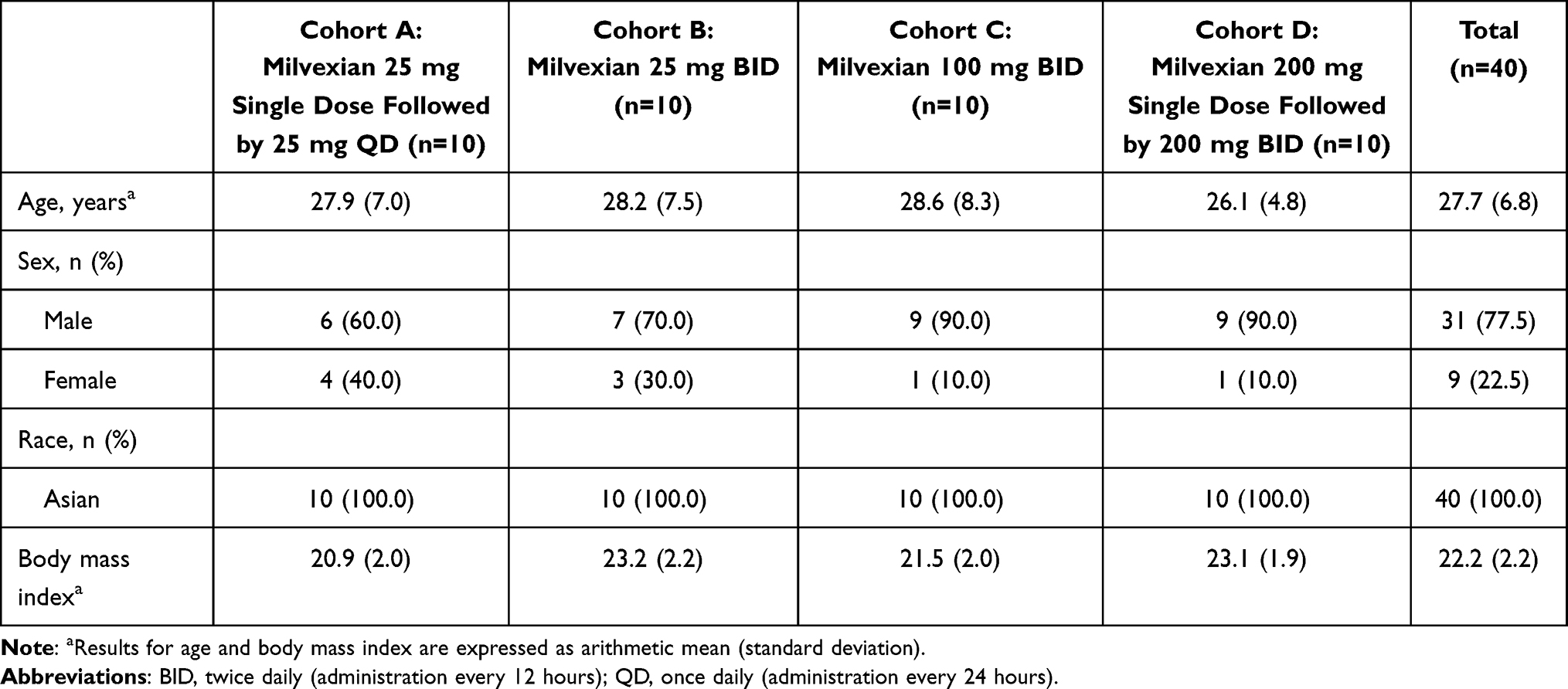 |
Table 1 Subject Demographics |
Pharmacokinetics
Plasma-concentration time profiles showed that milvexian was rapidly absorbed. Mean Cmax was reached at a median of approximately 3 to 4 hours after administration of a single dose and after repeated once daily (ie, every 24 hours) and twice daily (ie, every 12 hours) administration (Figure 2A, C and Table 2).
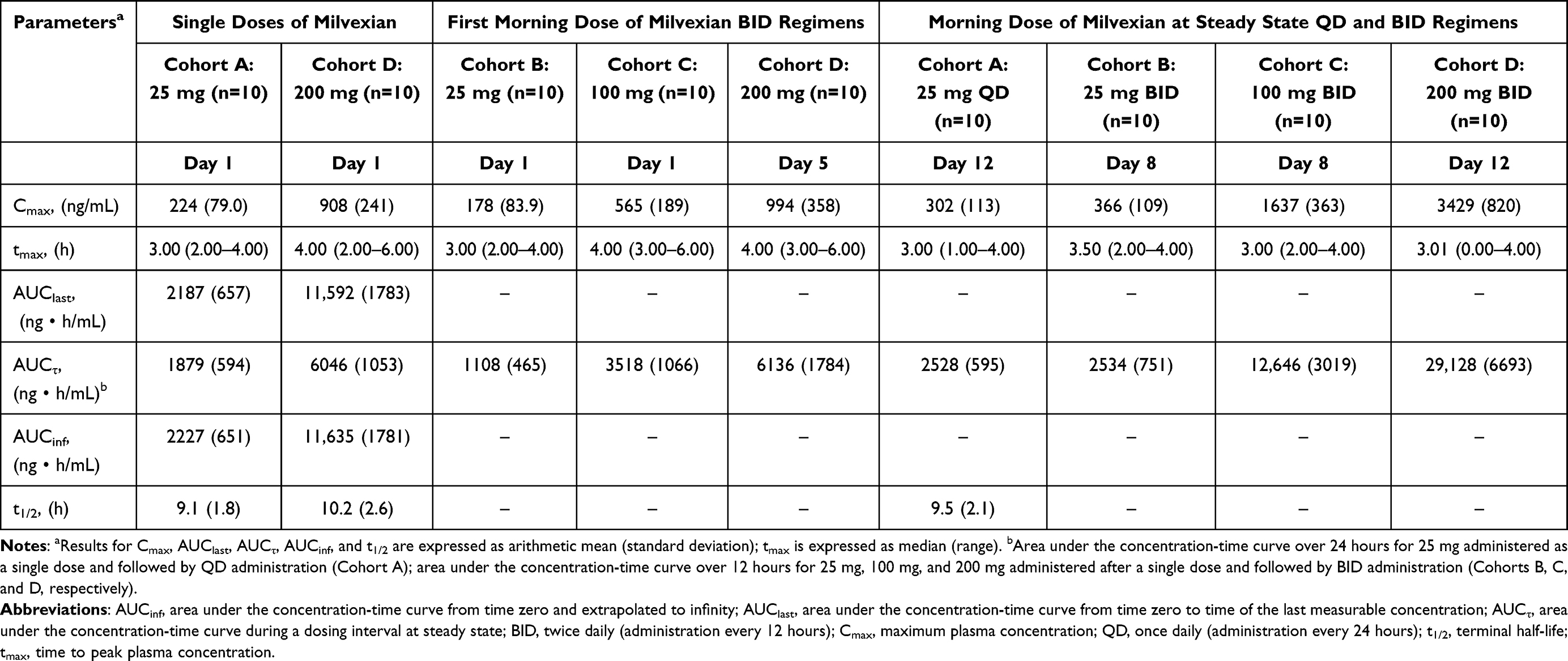 |
Table 2 Pharmacokinetic Parameters of Milvexian in Plasma After Single Dose Administration and After Once-Daily or Twice-Daily Administration |
 |
Figure 2 Plasma milvexian concentration versus time profiles (A and C) and trough concentrations (B) after single dose administration of milvexian (A) or repeated administration of milvexian for up to 8 consecutive days (B and C). Data are presented as arithmetic mean ± standard deviation. For (A), pharmacokinetic samples were collected predose and after the morning dose on Day 1 or Day 5. For Cohorts A and D, drug administration on Day 1 was followed by a 4-day washout. For (B), pharmacokinetic samples were collected prior to the morning dose on Days 10, 11, and 12 (Cohorts A and D) or Days 6, 7, and 8 (Cohorts B and C). For (C), pharmacokinetic samples were collected predose and after the morning dose on Day 12 (Cohorts A and D) or Day 8 (Cohorts B and C). Abbreviations: BID, twice daily (administration every 12 hours); QD, once daily (administration every 24 hours). |
Mean milvexian Cmax, AUClast, and AUCinf values increased in a dose-dependent manner after administration of single oral doses of milvexian 25 mg (Cohort A) and 200 mg (Cohort D). Mean t1/2 values were similar (9.1 and 10.2 hours, respectively; Table 2) for Cohorts A and D, and interindividual variability was low for milvexian Cmax and AUC values at doses of 25 mg and 200 mg.
After administration of milvexian 25 mg once daily (Cohort A), concentrations reached steady state within 6 days, with an approximate 1.3-fold accumulation in Cmax and AUCτ, relative to the initial dose (Figure 2B and Table 2). At steady state, milvexian concentrations declined with a mean t1/2 of 9.5 hours. Interindividual variability in milvexian pharmacokinetic parameters was low.
For the initial doses of the twice daily regimens (Cohorts B, C, and D), milvexian Cmax and AUCτ increased in a dose-dependent manner. Milvexian Ctrough values indicated that steady state was reached within 6 days of repeated administration (Figure 2B). At steady state, the increase in mean milvexian exposure was dose-dependent. For Cohorts B, C, and D, mean milvexian Cmax and AUCτ values were approximately 2.1- to 3.4-times and 2.3- to 4.7-times higher, respectively, at steady state after twice daily administration relative to the corresponding initial doses. Interindividual variability in milvexian pharmacokinetic parameters was low.
Pharmacodynamics
In general, higher doses and plasma concentrations of milvexian were associated with greater percent increases from baseline in aPTT (Figure 3). For a given dose, aPTT prolongation was greater after repeated administration relative to the initial dose. After administration of milvexian 25 mg on Day 1, a maximal mean increase of approximately 56% over baseline was observed at 4 hours (Cohort A). A maximal mean change versus baseline was observed at 3 hours postdose on Day 12 (~69%) after once daily administration of 25 mg (Cohort A). A maximal mean increase relative to baseline was observed at 3 hours postdose (~42%) after administration of milvexian 25 mg on Day 1 and at 4 hours postdose (~75%) in the morning on Day 8 after twice daily administration of 25 mg (Cohort B). After an initial 100 mg dose on Day 1, the aPTT mean increase from baseline peaked at 4 hours postdose (~104%) whereas it was at 4 hours (~164%) in the morning on Day 8 after twice daily administration of 100 mg (Cohort C). After a single 200 mg dose of milvexian, the maximal mean aPTT prolongation was observed at 6 hours (~113%) postdose, and on Day 12 after twice daily administration of 200 mg (Cohort D), the maximal aPTT prolongation was observed at 4 hours (~206%) postdose. aPTT values declined to approximately baseline levels by 72 hours after the last dose of milvexian was administered to subjects in all cohorts (data not shown).
 |
Figure 3 Percent change from baseline in aPTT versus time profiles after (A) single dose administration of milvexian or (B) repeated administration of milvexian for 8 consecutive days. Data are presented as arithmetic mean ± standard deviation. For (A), aPTT samples were collected predose and after the morning dose on Day 1. For Cohorts A and D, milvexian administration on Day 1 was followed by a 4-day washout. For (B), aPTT samples were collected predose on Day 1 and after the morning dose on Day 12 (Cohorts A and D) or Day 8 (Cohorts B and C). Abbreviations: aPTT, activated partial thromboplastin time; BID, twice daily (administration every 12 hours); QD, once daily (administration every 24 hours). |
Safety and Tolerability
Milvexian was generally well tolerated after single or multiple dose administration at doses of 25 to 200 mg. Overall, 21 (52.5%) of the 40 subjects had ≥1 TEAE, and in 10 (25.0%) subjects, these were considered to be related to milvexian (Table 3). Fecal occult blood positive (3 [7.5%]), mouth ulceration (3 [7.5%]), and epistaxis (3 [7.5%]) were the most common TEAEs (>5%) reported by preferred term. All TEAEs were considered mild in severity by the investigator. There were no serious adverse events, TEAEs leading to discontinuation of milvexian, or deaths. Ten (25.0%) of the 40 subjects had ≥1 bleeding TEAE, of which, 8 (20.0%) had bleeding events that were considered to be related to milvexian. Bleeding TEAEs included fecal occult blood positive (3 [7.5%]), epistaxis (3 [7.5%]), ecchymosis (2 [5.0%]), red blood cell positive urine (2 [5.0%]), and anal hemorrhage (1 [2.5%]). None of the subjects had major bleeding or CRNM bleeding events based on International Society of Thrombosis and Hemostasis (ISTH) assessment. Thirteen (32.5%) of 40 subjects had ≥1 nonbleeding TEAE, of which 2 were considered related to milvexian (data not shown). The most frequently reported nonbleeding TEAEs were blood uric acid increased (2 [5%]), pancreatic enzymes increased (2 [5%]), and mouth ulceration (3 [7.5%]). No notable changes in ECG measurements, vital signs, or physical examination results were observed or reported as adverse events during the study.
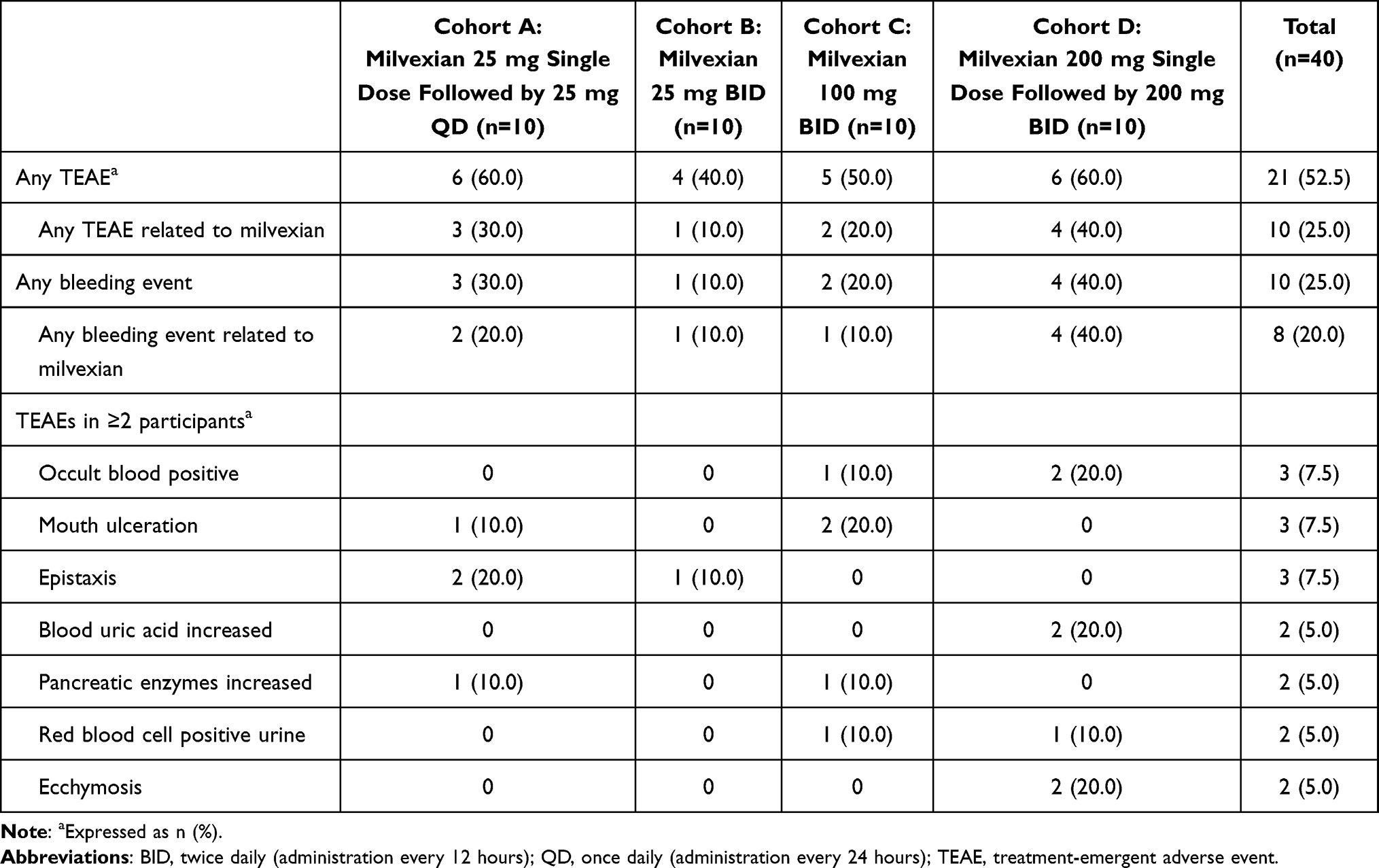 |
Table 3 Summary of Treatment-Emergent Adverse Events for Each Milvexian Regimen |
Discussion
This phase 1 study characterized the pharmacokinetic, pharmacodynamic, safety, and tolerability profiles of milvexian after single and multiple oral doses in healthy Chinese adult subjects. It was anticipated that the information obtained would support the inclusion of Chinese patients in clinical studies and the registration of milvexian in China.
The results of the present study demonstrated that milvexian is rapidly absorbed, with median tmax values of 3 to 4 hours after a single dose or repeated administration (ie, once or twice daily). Mean plasma Cmax and AUC values of milvexian dose-dependently increased with single doses of 25, 100, and 200 mg. A dose-dependent increase in mean milvexian exposure was also observed after milvexian was administered twice daily until steady state conditions were achieved. Steady state conditions were achieved within 6 days of repeated administration based on assessment of milvexian Ctrough values. Mean terminal t1/2 values across all doses were similar (~9 to 10 hours) when assessed after single doses of 25 mg and 200 mg and after once daily administration of 25 mg. In general, the interindividual variability in pharmacokinetic parameters was low.
The pharmacokinetic results from this study are generally in line with those observed in previous milvexian studies that enrolled healthy subjects who were not of Chinese ethnicity. For example, in studies that enrolled subjects who were predominately White, predominately White or Black/African American, or of Japanese ethnicity, milvexian was rapidly absorbed with median tmax values of approximately 3 to 4 hours following a single dose administered in the fasted state.20–23,26 Mean milvexian terminal t1/2 values were 8 to 14 hours for the non-Chinese subjects. These milvexian pharmacokinetic parameter values are consistent with corresponding values in the present study. The mean milvexian Cmax after a single dose of 100 mg that was administered to subjects who were predominately White or Black/African American was 599 ng/mL,26 similar to the mean Cmax of 565 ng/mL that was achieved after 100 mg was administered to subjects of Chinese ethnicity. Milvexian exposure after administration in the fasted state was similar after a single dose of 200 mg in non-Chinese subjects and Chinese subjects in the present study (mean Cmax: 1068 ng/mL versus 908/994 ng/mL; mean AUCinf: 13759 ng∙h/mL versus 11635 ng∙h/mL, respectively).20 Milvexian exposure was also similar after twice daily administration of 200 mg in non-Chinese subjects and Chinese subjects in the present study (mean Cmax: 3579 ng/mL versus 3429 ng/mL; mean AUCt: 30734 ng∙h/mL versus 29128 ng∙h/mL, respectively).20
Milvexian reversibly prolonged aPTT in a manner that was dependent on the dose administered and therefore the concentrations of milvexian achieved in plasma. The maximal mean percent change from baseline in aPTT after a single dose ranged from 56% or 42% at 25 mg (Cohorts A and B, respectively) to 113% at 200 mg (Cohort D). For a given initial dose, mean aPTT prolongation was noticeably greater after repeated administration, particularly for the twice daily regimens. The time that maximum prolongation of aPTT was achieved generally coincided with the tmax of milvexian in plasma. Subsequently, the extent to which aPTT was prolonged declined in a manner that was in accordance with the plasma milvexian concentration-time profile. The latter observations are consistent with the mechanism of reversible FXIa inhibition by milvexian.
It was previously demonstrated that the milvexian-induced prolongation in aPTT observed in healthy subjects who were not of Chinese ethnicity was also dose- and concentration-dependent. For example, in subjects who were predominately White or predominately White or Black/African-American, the maximal mean increase from baseline after single doses of milvexian 20 mg to 200 mg ranged from ~37% to 147%.20,22,23,26 The mean increase from baseline after a single dose of 100 mg was administered to subjects who were predominately White or Black/African American26 was ~126% at 4 hours postdose (data on file), similar to the mean of ~104% at 4 hours postdose after 100 mg was administered to subjects of Chinese ethnicity. After twice daily administration of 200 mg, the maximal mean increase in aPTT in predominately White subjects and Chinese subjects in the present study was ~240% (data on file) and ~206%, respectively. It was previously demonstrated that the prolongation of aPTT was reversible with a time course that was consistent with the plasma milvexian concentration-time profile, in agreement with the results of the present study.
Administration of single milvexian doses of 25 mg, 100 mg, or 200 mg, once daily administration of milvexian 25 mg, or twice daily administration of milvexian 25 mg, 100 mg, or 200 mg in the present study was safe and well tolerated, with no new or unanticipated safety signals compared with the phase 1 and phase 2 trials completed to date. The total number of subjects reporting adverse events was generally similar among cohorts. All TEAEs, including bleeding adverse events, were mild. There was no evidence to indicate any trend of relationship between dose levels and incidence and severity of adverse events, including bleeding events, whereas a dose-related increase in bleeding risk has been reported for other classes of oral anticoagulants.5,6 There were no serious TEAEs or study discontinuations due to TEAEs. The safety of milvexian will be more thoroughly evaluated in patients, including those of Chinese ethnicity, who are participating in ongoing global phase 3 studies for stroke prevention after an acute ischemic stroke or high-risk transient ischemic attack, in patients with atrial fibrillation, and in patients who have had a recent acute coronary syndrome at risk for thrombosis.
The current study, which included a relatively broad range of milvexian doses, provided evidence that the pharmacological properties of milvexian produced by a single dose and after once or twice daily administration were supportive of continued clinical development in subjects of Chinese ethnicity. A dose-dependent increase in milvexian exposure was observed. Furthermore, milvexian administration resulted in a reversible and dose-related prolongation of aPTT. A direct correlation between prolongation of aPTT and plasma milvexian concentration was evident. The study also showed that milvexian was generally safe and well tolerated in healthy Chinese subjects. The similarity of the current findings and those of previous studies suggests there are no clinically relevant effects attributable to Chinese ethnicity on the pharmacokinetic, pharmacodynamic, or safety properties of milvexian.
Limitations of this study include the open-label study design and absence of a placebo-controlled cohort to better interpret safety results. Although cohorts were small, a wide range of milvexian doses was evaluated. Larger studies are ongoing to investigate the safety and efficacy of treatment with milvexian in broader populations of patients who require thromboprophylaxis.
Conclusions
The results suggest that the pharmacokinetic and pharmacodynamic profiles of milvexian in subjects of Chinese ethnicity are similar to those observed in healthy subjects who were enrolled in previously completed phase 1 studies. In addition, milvexian was generally safe and well tolerated by subjects who were enrolled in the present study. Overall, the results are supportive of further clinical development of milvexian in Chinese subjects.
Abbreviations
aPTT, activated partial thromboplastin time; AUCinf, area under the concentration-time curve extrapolated to infinity; AUClast, area under the concentration-time curve from time 0 to last measurable concentration; AUCτ, area under the concentration-time curve during a dosing interval at steady state; BID, twice daily (administration every 12 hours); Cmax, maximum plasma concentration; CRNM, clinically relevant nonmajor; Ctrough, trough plasma concentration at the end of a dosing interval; CV, coefficient of variation; ECG, electrocardiogram; FXI, Factor XI; FXIa, Factor XIa; LC-MS/MS, liquid chromatography/tandem mass spectrometry; QD, daily (administration every 24 hours); SD, standard deviation; t1/2, terminal elimination half-life; TEAE, treatment-emergent adverse event; tmax, time to maximum plasma concentration.
Data Sharing Statement
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access [YODA] Project site at http://yoda.yale.edu.
Ethics Approval and Consent to Participate
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and was consistent with Good Clinical Practice and applicable regulatory requirements. The protocol and amendments were reviewed by the Independent Ethics Committee on Clinical Trials, West China Hospital of Sichuan University. Informed consent was obtained from all individual subjects included in the study.
Acknowledgments
This study was sponsored by Bristol Myers Squibb and Johnson & Johnson. Medical writing support was provided by Dana Tabor, PhD, of Lumanity Communications Inc., and was funded by Bristol Myers Squibb and Johnson & Johnson. The authors greatly appreciate the review of a draft version of the manuscript by Feng Xu (Johnson & Johnson).
Funding
This study was sponsored by Bristol Myers Squibb and Johnson & Johnson.
Disclosure
ZL and JW have no potential conflicts of interest for this work. ZN, CH, MC, XL, T-IL, ANP, and PZ are employees of and may hold stock in Johnson & Johnson.
References
1. Kapil N, Datta YH, Alakbarova N, et al. Antiplatelet and anticoagulant therapies for prevention of ischemic stroke. Clin Appl Thromb Hemost. 2017;23(4):301–318. doi:10.1177/1076029616660762
2. Gurbel PA, Fox KAA, Tantry US, Ten Cate H, Weitz JI. Combination antiplatelet and oral anticoagulant therapy in patients with coronary and peripheral artery disease. Circulation. 2019;139(18):2170–2185. doi:10.1161/CIRCULATIONAHA.118.033580
3. Eisen A, Giugliano RP, Braunwald E. Updates on acute coronary syndrome: a review. JAMA Cardiol. 2016;1(6):718–730. doi:10.1001/jamacardio.2016.2049
4. Chen A, Stecker E, Warden BA. Direct oral anticoagulant use: a practical guide to common clinical challenges. J Am Heart Assoc. 2020;9(13):e017559. doi:10.1161/JAHA.120.017559
5. Ezekowitz MD, Reilly PA, Nehmiz G, et al. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO Study). Am J Cardiol. 2007;100(9):1419–1426. doi:10.1016/j.amjcard.2007.06.034
6. Chen X, Huang W, Sun A, Wang L, Mo F, Guo W. Bleeding risks with novel oral anticoagulants especially rivaroxaban versus aspirin: a meta-analysis. Thromb J. 2021;19(1):69. doi:10.1186/s12959-021-00322-6
7. Cate HT, Hackeng TM, Garcia de Frutos P. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb Haemost. 2017;117(7):1265–1271. doi:10.1160/TH17-02-0079
8. Al-Amer OM. The role of thrombin in haemostasis. Blood Coagul Fibrinolysis. 2022;33(3):145–148. doi:10.1097/MBC.0000000000001130
9. Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. 2015;13(8):1383–1395. doi:10.1111/jth.13005
10. Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res. 2016;141(suppl 2):S8–S11. doi:10.1016/S0049-3848(16)30354-1
11. Preis M, Hirsch J, Kotler A, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129(9):1210–1215. doi:10.1182/blood-2016-09-742262
12. Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111(8):4113–4117. doi:10.1182/blood-2007-10-120139
13. Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105(2):269–273. doi:10.1160/TH10-05-0307
14. Wong PC, Crain EJ, Bozarth JM, et al. Milvexian, an orally bioavailable, small-molecule, reversible, direct inhibitor of factor XIa: in vitro studies and in vivo evaluation in experimental thrombosis in rabbits. J Thromb Haemost. 2022;20(2):399–408. doi:10.1111/jth.15588
15. Minnema MC, Friederich PW, Levi M, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101(1):10–14. doi:10.1172/JCI781
16. Heitmeier S, Visser M, Tersteegen A, et al. Pharmacological profile of asundexian, a novel, orally bioavailable inhibitor of factor XIa. J Thromb Haemost. 2022;20(6):1400–1411. doi:10.1111/jth.15700
17. Sakimoto S, Hagio T, Yonetomi Y, et al. ONO-8610539, an injectable small-molecule inhibitor of blood coagulation Factor XIa, improves cerebral ischemic injuries associated with photothrombotic occlusion of rabbit middle cerebral artery.
18. Hsu C, Hutt E, Bloomfield DM, Gailani D, Weitz JI. Factor XI inhibition to uncouple thrombosis from hemostasis: JACC review topic of the week. J Am Coll Cardiol. 2021;78(6):625–631. doi:10.1016/j.jacc.2021.06.010
19. Dilger AK, Pabbisetty KB, Corte JR, et al. Discovery of milvexian, a high-affinity, orally bioavailable inhibitor of factor XIa in clinical studies for antithrombotic therapy. J Med Chem. 2022;65(3):1770–1785. doi:10.1021/acs.jmedchem.1c00613
20. Perera V, Wang Z, Luettgen J, et al. First-in-human study of milvexian, an oral, direct, small molecule factor XIa inhibitor. Clin Transl Sci. 2022;15(2):330–342. doi:10.1111/cts.13148
21. Perera V, Wang Z, Lubin S, et al. Safety, pharmacokinetics, and pharmacodynamics of milvexian in healthy Japanese participants. Sci Rep. 2022;12(1):5165. doi:10.1038/s41598-022-08768-y
22. Perera V, Abelian G, Li D, et al. Single-dose pharmacokinetics of milvexian in participants with normal renal function and participants with moderate or severe renal impairment. Clin Pharmacokinet. 2022;61(10):1405–1416. doi:10.1007/s40262-022-01150-1
23. Perera V, Abelian G, Li D, et al. Single-dose pharmacokinetics of milvexian in participants with mild or moderate hepatic impairment compared with healthy participants. Clin Pharmacokinet. 2022;61(6):857–867. doi:10.1007/s40262-022-01110-9
24. Weitz JI, Strony J, Ageno W, et al. Milvexian for the prevention of venous thromboembolism. N Engl J Med. 2021;385(23):2161–2172. doi:10.1056/NEJMoa2113194
25. Sharma M, Molina CA, Toyoda K, et al. Safety and efficacy of factor XIa inhibition with milvexian for secondary stroke prevention (AXIOMATIC-SSP): a phase 2, international, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol. 2024;23(1):46–59. doi:10.1016/S1474-4422(23)00403-9
26. Perera V, Wang Z, Lubin S, et al. Effects of rifampin on the pharmacokinetics and pharmacodynamics of milvexian, a potent, selective, oral small molecule factor XIa inhibitor. Sci Rep. 2022;12(1):22239. doi:10.1038/s41598-022-25936-2
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.