")
Back to Journals » Drug Design, Development and Therapy » Volume 19
Population Pharmacokinetics and Pharmacodynamics with Enterohepatic Recirculation of Co-Medication of Rosuvastatin and Ezetimibe
Received 6 March 2025
Accepted for publication 28 May 2025
Published 4 June 2025 Volume 2025:19 Pages 4777—4787
DOI https://doi.org/10.2147/DDDT.S522863
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Hyungmi An,1 Dongseong Shin2
1Department of Statistics and Data Science, Korea National Open University, Seoul, Korea; 2Department of Clinical Pharmacology and Therapeutics, Gil Medical Center, Gachon University College of Medicine, Incheon, Korea
Correspondence: Dongseong Shin, Department of Clinical Pharmacology and Therapeutics, Gachon University College of Medicine, Clinical Trials Center, Gachon University Gil Medical Center, 21, Namdong-daero 774 beon-gil, Namdong-gu, Incheon, 21565, Korea, Tel +82-32-460-9459, Fax +82-32-460-9443, Email [email protected]
Objective: Combination therapy with rosuvastatin and ezetimibe is generally administered to patients with high cardiovascular risk. The objective of this study was to develop a population pharmacokinetic/pharmacodynamic (PK/PD) model of the interaction between rosuvastatin and ezetimibe that incorporates enterohepatic recirculation (EHC).
Methods: Concentration–time data were obtained from a two-part, open-label, multiple-dose crossover, drug interaction study. In total, 50 healthy male subjects received both monotherapy and co-therapy (Part A: rosuvastatin and co-therapy; Part B: ezetimibe and co-therapy). Rosuvastatin (20 mg) or ezetimibe (10 mg) were administered once daily for 7 days as monotherapy or co-therapy. Plasma concentrations were measured for PK analysis until 72 h post-dose at steady state. The changes in low-density lipoprotein cholesterol (LDL-C) levels from baseline to steady state at 24 h after the last administration were measured. A population PK/PD model incorporating EHC was developed using Monolix 2024R1. Covariate effects were explored, and the final model was evaluated through goodness-of-fit diagnostics and visual predictive checks. Model-based simulations were conducted to compare the LDL-C lowering effects of monotherapy and co-therapy.
Results: A population PK/PD model was established using a two-compartment model for rosuvastatin and a four-compartment model for ezetimibe incorporating EHC via intermittent gallbladder emptying. No significant PK interaction was observed. An indirect response PD model reflected the independent LDL-C lowering effects of both drugs. Simulations showed LDL-C reductions of − 51.0% (rosuvastatin), − 25.3% (ezetimibe), and − 60.7% (co-therapy), supporting the additive efficacy of co-therapy. EHC increased the exposure of total ezetimibe with limited LDL-C lowering effects.
Conclusion: The overall PK interaction between rosuvastatin and total ezetimibe was not significant. The developed PK/PD model incorporating EHC successfully described the independent LDL-C lowering effects. These findings support the additive benefit of co-therapy of rosuvastatin and ezetimibe and may guide future research toward personalized lipid-lowering strategies.
Keywords: pharmacokinetics, pharmacodynamics, modeling, rosuvastatin, ezetimibe
Introduction
Statin monotherapy is the primary pharmaceutical option for dyslipidemias.1 Moreover, patients with high risk of coronary artery disease or high low-density lipoprotein cholesterol (LDL-C) levels require combination drug therapy including ezetimibe.1 Rosuvastatin is a moderate- to high-intensity 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor.2 Rosuvastatin is metabolized by cytochrome P450 2C9 and 2C19 and excreted by the organic anion transporter protein 1B1.2,3 Ezetimibe inhibits the intestinal absorption of dietary and biliary cholesterol by interacting with Niemann–Pick C1-like protein 1 (NPC1L1) at the brush border.1 After oral administration, ezetimibe is biotransformed into an active metabolite, ezetimibe-glucuronide, by the UDP glycosyltransferases 1A1, 1A3, and 2B15 in the intestinal mucosa and liver.3,4 Ezetimibe co-therapy can lead to an additional 20% reduction in LDL-C levels compared with statin monotherapy.1,3
Ezetimibe shows multiple peaks in plasma concentration–time profiles, suggesting enterohepatic circulation (EHC).5 EHC involves the transport of bile acids and xenobiotics from the liver to the bile, followed by biliary excretion and intestinal reabsorption.6,7 Drug pharmacokinetics (PK) and pharmacodynamics (PD) are affected by biliary excretion and reabsorption during the EHC process, resulting in a prolonged elimination half-life and an increased area under the plasma concentration–time curve (AUC) with multiple peaks after specific time intervals.7 EHC can be affected by several factors, including drug physicochemical properties, co-medication, use of bile acid sequestrants, genetic variations, species, sex, and diseased conditions.7 However, the evaluation of EHC remains challenging owing to the lack of suitable models, the complexity of physiological processes, and ethical constraints.7
In spite of the clinical relevance, few PK/PD modeling studies have mechanistically incorporated EHC to evaluate drug–drug interactions in combination therapies—particularly for rosuvastatin and ezetimibe. Existing models have showed the difficulty in representing the physiological dynamics of biliary recycling and are limited in simulating the additive or synergistic lipid-lowering effects of co-administered agents.8 Moreover, most previous models have focused on single-agent profiles or employed simplified EHC assumptions, limiting their ability to accurately capture the interaction dynamics and delayed reabsorption processes relevant to combination therapy.5,9
In a previous study, the co-administration of rosuvastatin with ezetimibe revealed bioequivalent PK interactions between rosuvastatin and total ezetimibe, with the exception of free ezetimibe, which showed additive effects in the reduction of LDL-C.3 Based on these results, we developed a population PK/PD model of rosuvastatin and ezetimibe at steady state to mechanistically incorporate EHC via intermittent gallbladder emptying and account for the independent lipid-lowering actions of rosuvastatin and ezetimibe.8
To our knowledge, in this study, a physiologically relevant EHC model was novelly embedded within a PK/PD framework for rosuvastatin–ezetimibe combination therapy, enabling the simulation of LDL-C responses with monotherapy and co-therapy. In light of the growing interest in individualized therapy, such mechanistic models may support future covariate-based approaches to optimizing lipid-lowering strategies.
Methods
Study Population and Design
The concentration–time data used in this analysis were obtained from a two-part clinical trial involving an open-label, multiple-dose, two-treatment, two-period, two-sequence crossover study in each part.3 The study was performed in accordance with the recommendations of the Korean Good Clinical Practice and the Declaration of Helsinki (https://clinicaltrials.gov/ registry number: NCT02289430). The institutional review board of Gachon University Gil Medical Center approved the study protocol and ensured informed consent (GCIRB2014-324). The drug interaction study consisted of two parts: part A involved the evaluation of the PKs of rosuvastatin, and part B the evaluation of the PKs of ezetimibe. In total, 56 healthy male participants aged 19–45 years of age and within ± 20% of their ideal body weight (28 participants in each part) were eligible if they did not have clinically significant medical histories, physical examination findings, 12-lead electrocardiogram (ECG) readings or clinical laboratory testing results. Subjects who showed creatinine clearance under 80 mL/min by the Cockcroft-Gault equation were also excluded from this study. Enrolled subjects were randomly assigned to one of the two sequences. In part A, rosuvastatin 20 mg or a combination of rosuvastatin 20 mg and ezetimibe 10 mg was administered once daily for 7 days. In part B, ezetimibe 10 mg or a combination of rosuvastatin 20 mg and ezetimibe 10 mg was administered once daily for 7 days. A standard meal was provided at 4, 10, and 24 h after the final dose during the hospitalization period. After a 14-day washout period, the subject received the other treatment and the same procedures were followed.
To evaluate the drug interactions in the steady state, rosuvastatin and ezetimibe were co-administered once daily for 7 days. Blood samples for PK evaluation in the steady state were obtained at 16 time points: prior to dosing (0 h) and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 12, 24, 48, and 72 h after dosing on day 7 considering the longer mean half-life of total ezetimibe with approximately 20 h. The LDL-C level for the therapeutic effect was measured before dosing (day 1) and 24 h after the final dose in the perspective of clinical usefulness with once daily administration.
Population PK/PD Analyses
The population PK/PD model was developed using a nonlinear mixed effects modeling approach with Monolix 2024R1 software (Lixoft, Orsay, France). The stochastic approximation expectation maximization algorithm in Monolix was used throughout the model-building process. Concentrations below the lower limit of quantification (LLOQ) were observed in 8.38% of rosuvastatin samples and 1.25% of ezetimibe samples, all of which occurred after 48 h after the final dosing in each period. Those data were regarded as missing and excluded from the analysis, where observations below the LLOQ are ignored in the likelihood.
Population PK Model for Rosuvastatin
Different structural and residual error models were explored during the model-building process, including one- and two-compartment models with additive, proportional, or combined residual error models. The random effects accounting for variability in rosuvastatin included parameters for interindividual variability and residual unexplained variability. All parameters were assumed to follow a log-normal distribution, and an exponential error model was used for interindividual variability.
Population PK Model for Ezetimibe
The concentration–time profiles of ezetimibe showed enterohepatic reabsorption with multiple peaks.3 Different structural models, including one-, two-, and three-compartment models, were explored during the model-building steps using the EHC process. A two-compartment model with an additional gallbladder (GB) component provided the best fit to the data. GB emptying times were standardized based on meal times as defined in the protocol (4, 10, and 24 h after the last drug administration on day 7) and the appearance of a secondary peak in the obtained concentration–time profiles. Interindividual variability and residual unexplained variability were described using an exponential model. A combined proportional and additive residual error model was used to describe random errors in the plasma concentrations. All PK parameters were assumed to be log-normal, and correlations between the random effects of the parameters were tested in the model.
PD Model for LDL-C
The final PK models for rosuvastatin and ezetimibe were combined with an indirect effects model to describe the LDL-C data. Rosuvastatin exerts lipid-lowering effects by inhibiting HMG-CoA reductase, whereas ezetimibe inhibits cholesterol uptake by binding to NPC1L1. Considering the decrease in plasma levels of LDL-C following these independent lipid-lowering mechanisms of rosuvastatin and ezetimibe, the variation in LDL-C levels over time is described as follows:10

where  and
and  denote the LDL-C production and elimination rates, respectively.
denote the LDL-C production and elimination rates, respectively.  and
and  are the PK model-predicted rosuvastatin and ezetimibe plasma concentrations at time t, respectively, and
are the PK model-predicted rosuvastatin and ezetimibe plasma concentrations at time t, respectively, and  and
and  are the respective rosuvastatin and ezetimibe concentrations that lead to 50% maximum inhibition of LDL-C production.
are the respective rosuvastatin and ezetimibe concentrations that lead to 50% maximum inhibition of LDL-C production.
The model assumes  (full inhibitory effect), which was fixed rather than estimated, owing to the lack of observable saturation and to reduce model complexity under sparse PD sampling. This approach allowed for robust parameter estimation while maintaining mechanistic interpretability of the combined effects of the two drugs.
(full inhibitory effect), which was fixed rather than estimated, owing to the lack of observable saturation and to reduce model complexity under sparse PD sampling. This approach allowed for robust parameter estimation while maintaining mechanistic interpretability of the combined effects of the two drugs.
Covariate Analysis and Model Evaluation
Along with the base structural model selections, the effects of potential covariates were graphically explored and tested in both PK and PD models. These covariates included baseline characteristics, such as age and weight, and laboratory measurements, such as serum creatinine, albumin, alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, and gamma-glutamyl transferase. Continuous variables were centered on mean values. Covariates were initially tested using univariate analysis and then using a combination of stepwise forward addition and backward elimination. The final model selection was based on goodness-of-fit plots and precision of the parameter estimates, as well as numerical criteria, such as objective function value (−2 log-likelihood) change, Akaike information criteria, and Bayesian information criteria. The visual predictive check (VPC) involved overlaying the observed data with the median, 5th, and 95th percentile curves of the prediction using 1,000 simulated replicates.
Model Based Simulation
The LDL-C level changes from the baseline to the steady state 24 h after the last administration of rosuvastatin (20 mg), ezetimibe (10 mg), or a combination of these two drugs were simulated and compared based on the final PK-PD model.
Results
Study Population and Baseline Characteristics
A total of 50 subjects (25 each in Parts A and B) were finally included in the population PK/PD analysis, providing 25, 25, and 50 concentration–time profiles for rosuvastatin, ezetimibe, and a combination of the two drugs, respectively. Demographic and biochemical parameters are presented in Table 1. The mean plasma concentration–time profile plots of rosuvastatin and ezetimibe are displayed in Figure 1. Generally, the plasma concentration–time curves of ezetimibe exhibited two absorption peaks typically at 1–2 h and around 5 h post-dose, although the second peak was relatively less pronounced in the current study population.3
 |
Table 1 Demographics and Baseline Characteristics of Study Population |
 |
Figure 1 Mean and individual plasma concentration–time profiles of rosuvastatin and total ezetimibe at steady state after 7-day treatment administration. (a) Rosuvastatin profiles (b) Total ezetimibe profiles. Rosuvastatin (20 mg) and ezetimibe (10mg) monotherapy are presented in black and cotherapy of rosuvastatin and ezetimibe is presented in grey. |
PK Analysis for Rosuvastatin
A two-compartment model with first-order absorption was used to describe the PK profile of rosuvastatin (Figure 2). The estimated PK parameters for the absorption rate ( ), clearance (
), clearance ( ), central volume of distribution (
), central volume of distribution (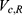 ), peripheral volume of distribution (
), peripheral volume of distribution ( ), and inter-compartmental clearance (
), and inter-compartmental clearance (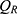 ), along with their interindividual variability and percent relative standard error values (RSE%), are summarized in Table 2. Between-subject variability for the estimated rosuvastatin PK parameters was moderate, whereas the model parameters were precisely estimated with a relatively low RSE%. A substantial correlation of random effects between
), along with their interindividual variability and percent relative standard error values (RSE%), are summarized in Table 2. Between-subject variability for the estimated rosuvastatin PK parameters was moderate, whereas the model parameters were precisely estimated with a relatively low RSE%. A substantial correlation of random effects between  and
and 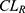 was observed and was included in the final model, significantly improving the goodness-of-fit criteria. The residual variability was described using a combined error model consisting of the additive component
was observed and was included in the final model, significantly improving the goodness-of-fit criteria. The residual variability was described using a combined error model consisting of the additive component  and the multiplicative coefficient
and the multiplicative coefficient  :
:



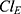

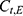
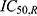
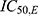
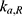

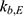
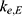
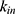
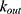
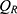
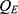

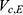

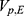
 |
Table 2 Parameter Estimates of the Final Pharmacokinetic/Pharmacodynamic Model |
where  and
and  represent the
represent the 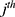 observed and model-predicted concentrations of rosuvastatin in the
observed and model-predicted concentrations of rosuvastatin in the 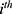 individual, respectively. The residual error term
individual, respectively. The residual error term 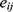 is assumed to be standard and normally distributed with a mean of 0 and variance of 1. No covariate effect was significant for the estimated PK parameters, possibly because the volunteers were all young, healthy males. Supplementary Figure 1 presents goodness-of-fit (GOF) plots for the final PK model of rosuvastatin, including observed vs predicted values (both individual and population), residuals vs predictions, residuals vs time, and normalized prediction distribution errors (NPDE) distributions. The residuals are symmetrically and randomly distributed across time and predicted values, indicating the absence of systematic bias and supporting the adequacy of model performance.
is assumed to be standard and normally distributed with a mean of 0 and variance of 1. No covariate effect was significant for the estimated PK parameters, possibly because the volunteers were all young, healthy males. Supplementary Figure 1 presents goodness-of-fit (GOF) plots for the final PK model of rosuvastatin, including observed vs predicted values (both individual and population), residuals vs predictions, residuals vs time, and normalized prediction distribution errors (NPDE) distributions. The residuals are symmetrically and randomly distributed across time and predicted values, indicating the absence of systematic bias and supporting the adequacy of model performance.
PK Analysis for Ezetimibe
A population PK model was developed to simulate the EHC of ezetimibe by incorporating an intermittent GB emptying process (Figure 2). The structure of this model included four compartments: central, peripheral, gastrointestinal (GI) track, and GB.8 The central compartment was reversibly connected to the peripheral compartment. A hypothetical GB compartment was introduced to link the central and GI tract compartments and operate the EHC loop. Biliary secretion was modeled using a first-order process with a switch on/off function in order to mimic intermittent GB emptying after a meal. Assuming that all EHC processes after a meal were identical, the duration of bile release in each EHC cycle was set to 0.75 h.8 The bile release rate constant 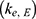 was fixed as the inverse of duration, 1.33 h - 1 = (0.75 h) - 1. The set of ordinary differential equations for the final PK model of ezetimibe was defined as follows:
was fixed as the inverse of duration, 1.33 h - 1 = (0.75 h) - 1. The set of ordinary differential equations for the final PK model of ezetimibe was defined as follows:







where  represents the amount of ezetimibe in a specific compartment: (1) GI represents the gastrointestinal tract compartment, (2) c represents the central compartment, (3) p represents the peripheral compartment, and (4) GB represents the gallbladder compartment. The term
represents the amount of ezetimibe in a specific compartment: (1) GI represents the gastrointestinal tract compartment, (2) c represents the central compartment, (3) p represents the peripheral compartment, and (4) GB represents the gallbladder compartment. The term 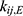 denotes the transfer rate constant between compartments i and j, which includes the absorption
denotes the transfer rate constant between compartments i and j, which includes the absorption 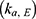 , elimination
, elimination  , and bile transfer
, and bile transfer 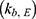 rate constants. The term GBE is an indicator function with a value 0 or 1, where GBE = 1 when GB emptying occurs within a bile release duration of 0.75 h after a meal; otherwise, GBE = 0. Three intermittent bile release periods (at 4, 10, and 24 h post-dose) were considered according to mealtime. The duration of bile release was assumed to be 0.75 h, which approximates the mean duration of GB emptying in healthy subjects. The residual variability was described using an additive error model with zero proportional error (b = 0) in Equation 2.
rate constants. The term GBE is an indicator function with a value 0 or 1, where GBE = 1 when GB emptying occurs within a bile release duration of 0.75 h after a meal; otherwise, GBE = 0. Three intermittent bile release periods (at 4, 10, and 24 h post-dose) were considered according to mealtime. The duration of bile release was assumed to be 0.75 h, which approximates the mean duration of GB emptying in healthy subjects. The residual variability was described using an additive error model with zero proportional error (b = 0) in Equation 2.
PK parameters showed no significant covariates. The final model estimates, including the correlations of the random effects between 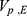 –
–  ,
, 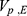 –
–  , and
, and  –
– 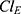 , are summarized in Table 2. Supplementary Figure 2 presents the GOF plots for total ezetimibe, which show strong agreement between the predicted and observed concentrations. The residuals versus predictions and versus time plots exhibit random scatter without apparent trends, and both PWRES and IWRES, and NPDEs are centered around zero, suggesting robust model performance.
, are summarized in Table 2. Supplementary Figure 2 presents the GOF plots for total ezetimibe, which show strong agreement between the predicted and observed concentrations. The residuals versus predictions and versus time plots exhibit random scatter without apparent trends, and both PWRES and IWRES, and NPDEs are centered around zero, suggesting robust model performance.
PK/PD Analysis for LDL-C
An indirect model with two independent drug mechanisms was used to describe changes in LDL-C levels (Figure 2). PK/PD modeling was performed sequentially, incorporating the results of the two PK models. The individual PK parameters of rosuvastatin and ezetimibe were set to their estimated values and used as constants in the PK/PD model to fit the PD data. The residual error for LDL-C levels was best described by the combined error model in Equation (2).
Parameter estimates from the final full PK/PD model are presented in Table 2. All parameters were estimated with good precision (relative standard error <35%). Rosuvastatin and ezetimibe independently inhibited the production rate  of LDL-C without any drug–drug interactions. Due to the homogeneity of the study population, no significant covariates were identified, and the random effects showed no notable correlations. The goodness-of-fit plots in Supplementary Figure 3 demonstrate linear agreement without bias, and both the WRES and NPDEs are centered around zero with homogenous spread, indicating a good fit. No time-dependent or prediction-dependent error patterns were observed, supporting the adequacy of the PD model. The VPC plots in Figure 3 show that most of the observed concentrations were within the 5th and 95th percentiles of the simulated concentrations, suggesting that the final model was not unspecified.
of LDL-C without any drug–drug interactions. Due to the homogeneity of the study population, no significant covariates were identified, and the random effects showed no notable correlations. The goodness-of-fit plots in Supplementary Figure 3 demonstrate linear agreement without bias, and both the WRES and NPDEs are centered around zero with homogenous spread, indicating a good fit. No time-dependent or prediction-dependent error patterns were observed, supporting the adequacy of the PD model. The VPC plots in Figure 3 show that most of the observed concentrations were within the 5th and 95th percentiles of the simulated concentrations, suggesting that the final model was not unspecified.
 |
Figure 3 Visual predictive check plots for the final pharmacokinetic-pharmacodynamic model for (a) Rosuvastatin, (b) Total ezetimibe, (c) Low-density lipoprotein cholesterol (LDL-C) under rosuvastatin monotherapy, (d) LDL-C under ezetimibe monotherapy, and (e) LDL-C under co-therapy with rosuvastatin and ezetimibe. Solid lines represent the 5th, 50th, and 95th percentiles of the observed data. Blue areas represent 95% confidence intervals for 5th and 95th percentiles of simulated data. Red areas represent 95% confidence intervals for median of simulated data. The time of drug administration at steady state (Day 7) was set to 0 h. |
Simulation Based on the PK/PD Model
Simulations were performed to compare the LDL-C-lowering effects of rosuvastatin or ezetimibe monotherapy and combined therapy based on the final PK/PD model. The mean percentage reduction in LDL-C levels measured 24 h after dosing from baseline (pre-dose) was −51.0 ± 15.0% for rosuvastatin monotherapy, −25.3 ± 16.2% for ezetimibe monotherapy, and −60.7 ± 13.9% for combination therapy (Figure 4). All three cases showed statistically significant reductions; however, combination therapy resulted in a greater reduction than monotherapy (Figure 4).
 |
Figure 4 Distribution of low-density lipoprotein cholesterol (LDL-C) levels at baseline (yellow) and 24 hours after administration (blue) of rosuvastatin (20 mg), ezetimibe (10 mg), and their combinations at steady state, simulated in 1,000 individuals using the final pharmacokinetic/pharmacodynamic model. (a) % change of LDL-C (b) LDL-C level. |
Discussion
This study represents the first population PK/PD modeling analysis of the drug–drug interaction between rosuvastatin and ezetimibe at steady state, incorporating a mechanistic representation of EHC. Compared to previous studies where PK or PD endpoints were evaluated separately, our approach enables the simultaneous modeling of exposure–response relationships for both drugs using minimal model complexity. The integration of a physiologically meaningful EHC component and the indirect response model for LDL-C allows for the evaluation of additive pharmacodynamic effects under clinically relevant dosing regimens. This modeling strategy may provide a quantitative foundation for future investigations into individualized lipid-lowering therapy and can aid clinicians in evaluating the LDL-C response to combination therapy involving drugs undergoing enterohepatic recirculation.
After oral intake, foreign compounds entering the GI tract are absorbed into the portal vein by enterocytes and delivered to hepatocytes.6 They are then secreted into the bile and reabsorbed across the intestinal lumen during EHC.6 EHC involves biliary excretion, intestinal reabsorption with or without hepatic conjugation, intestinal deconjugation, and systemic circulation.6,7 In general, compounds with molecular weight greater than 400 g/mol, polarity, and biotransformation, including conjugation, can undergo EHC.6,7 Co-medication including bile-sequestrating agents, age, sex, genetic variations, disease conditions, and diurnal variations may affect biliary excretion during the EHC process, which may prolong the pharmacological effect.7 The clinical significance of EHC, including its pharmacological and toxicological effects, is related to the extent of biliary excretion and efficiency of reabsorption.7 In this study of a recommended combination therapy, a population PK/PD model was developed for rosuvastatin and ezetimibe with respect to drug-drug interactions via EHC.3 After a meal, fatty acids and amino acids from the stomach stimulate the I-cells in the duodenum and jejunum to release cholecystokinin, which contract the GB and secrete bile into the biliary tree.11 As a result, multiple peaks in the concentration–time plots related to GB emptying are observed around mealtime.8 Since multiple peaks in concentration–time profiles of ezetimibe were more remarkable in the previous study, the effect of EHC on the PKs of ezetimibe was explored.3
Ezetimibe is absorbed and metabolized in the intestinal wall, and ezetimibe and ezetimibe phenolic glucuronide are transported through the portal vein.8 After further glucuronidation of ezetimibe in the liver and biliary secretion into the intestinal lumen, ezetimibe-glucuronide is hydrolyzed to the parent drug and reabsorbed into systemic circulation.4,12,13 Total ezetimibe is the sum of unchanged ezetimibe and ezetimibe-glucuronide. Total ezetimibe is considered the primary endpoint in PK/PD modeling, because ezetimibe-glucuronide which accounts for 80–90% of the total ezetimibe in plasma, has a potent therapeutic effect similar to that of its parent compound.4 This model consisted of four compartments including the GB for EHC, considering two pathways from GB; the one is to central compartment, including blood and liver and the other is to GI compartment.8 Therein, the GI compartment-linked model provided a better description than the central compartment-linked model based on the goodness-of-fit plots. Considering the ezetimibe metabolism, physiological process, and pattern of the secondary EHC peak, drug transport with bile release to the intestinal lumen could contribute to this model.14
Although ezetimibe exhibits distinct secondary peaks owing to enterohepatic recirculation, the second peak at approximately 5 h was modest and not clearly observed in the present study. This may be attributed to low reabsorption dynamics in healthy subjects and the relatively smoothened average profile. In addition, the limited sensitivity of plasma sampling to detect subtle postprandial fluctuations could have contributed to this visual subtlety. Nevertheless, the structural PK model incorporating GB emptying adequately captured the EHC effect, even if the secondary peak was less visually prominent in the mean profile (Figure 1).
The overall PK parameters of rosuvastatin and ezetimibe between monotherapy and co-therapy were consistent in this study, and the effect of comedication on the PK parameters including 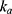 ,
, 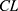 and
and  was limited. Although a slightly increased AUC of total ezetimibe was observed in the NCA analysis, the current PK model showed that the PK interaction between rosuvastatin and ezetimibe at the steady state, including EHC, is not significant.3
was limited. Although a slightly increased AUC of total ezetimibe was observed in the NCA analysis, the current PK model showed that the PK interaction between rosuvastatin and ezetimibe at the steady state, including EHC, is not significant.3
Applying the Bliss independence method would be appropriate given the independent therapeutic mechanisms of rosuvastatin and ezetimibe in lowering LDL-C.3,10 Multicompartmental models for EHC could have issues, including increased complexity, parameter identifiability, and estimation difficulties, so minimal model complexity with assumed variables was required.8,15 The basic model of indirect responses assumes that the value of Imax is 1.16 Hill coefficient, which represents the steepness of the concentration–response curve, is simply fixed at 1.9 Although the mechanisms of cholesterol synthesis inhibition and LDL receptor upregulation have been reported, statins have decreased the LDL-C synthesis rate constant  in previous PD models.17,18 Considering the pharmacological effect of ezetimibe as an inhibitor of intestinal cholesterol uptake, its concentration was incorporated within
in previous PD models.17,18 Considering the pharmacological effect of ezetimibe as an inhibitor of intestinal cholesterol uptake, its concentration was incorporated within 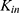 . The final PD model adequately described the LDL-C changes and supported the additive effects of rosuvastatin and ezetimibe without drug interactions.3,18,19
. The final PD model adequately described the LDL-C changes and supported the additive effects of rosuvastatin and ezetimibe without drug interactions.3,18,19
The recycled amount of ezetimibe was calculated using several methods to evaluate the effect of EHC. With 17% of the absorbed dose in the previous study, the AUC was estimated to increase by approximately 86% compared to the value in the absence of EHC.5 When the drug fraction excreted into bile was indirectly explored using the ratio between 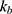 and
and 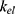 (elimination rate), approximately 30% of the absorbed amount was recirculated.8 In the current model, the effect of EHC on ezetimibe exposure was explored based on the AUC ratio.13 The AUC ratio in the absence of EHC was estimated assuming that reabsorption was incorporated into the elimination process. In the present study, the AUC ratio was estimated to be 0.94, which significantly decreased in the absence of EHC. However, this difference did not significantly affect LDL-C level changes. The results of this modeling estimated that exposure to total ezetimibe was less affected by EHC compared to previous studies.5,8,13 This phenomenon was possibly caused by the concentration–time profiles, where the second and third peaks were not prominently observed (Figure 1). In addition, total ezetimibe quantitatively insensitive to EHC and infrequent sampling during reabsorption in healthy subjects may have contributed to these results, which could be a limitation of this study.
(elimination rate), approximately 30% of the absorbed amount was recirculated.8 In the current model, the effect of EHC on ezetimibe exposure was explored based on the AUC ratio.13 The AUC ratio in the absence of EHC was estimated assuming that reabsorption was incorporated into the elimination process. In the present study, the AUC ratio was estimated to be 0.94, which significantly decreased in the absence of EHC. However, this difference did not significantly affect LDL-C level changes. The results of this modeling estimated that exposure to total ezetimibe was less affected by EHC compared to previous studies.5,8,13 This phenomenon was possibly caused by the concentration–time profiles, where the second and third peaks were not prominently observed (Figure 1). In addition, total ezetimibe quantitatively insensitive to EHC and infrequent sampling during reabsorption in healthy subjects may have contributed to these results, which could be a limitation of this study.
The PK/PD profiles of the study drug, particularly rosuvastatin, possibly show variability according to ethnic variability including genetic polymorphism, co-medication, age, and gender.20 These potential covariates were explored and found to be insignificant in our model. The absence of significant covariate effects may partly be due to the homogeneity of the study population. In particular, the dataset was derived from a clinical trial in healthy volunteers consisting exclusively of young adult males, thereby limiting the extrapolation of our findings to other populations such as females, older adults, or individuals with underlying disease. This demographic restriction may have affected the sensitivity of the model to detect covariate influences, as well as the observable impact of EHC on ezetimibe exposure.
Future studies, including individuals with comorbidities or genetic polymorphisms, could provide further insights into inter-individual variability in drug response. A broader demographic representation will also be essential to evaluating whether the current PK/PD relationships remain consistent across patient subgroups and to support individualized lipid-lowering strategies.
Conclusion
At the steady state, rosuvastatin and total ezetimibe showed no significant interaction in modeling analysis. The EHC PK process was well described by a four-compartment model with intermittent GB emptying and incorporated in the LDL-C synthesis process in a PD interaction model. This PK/PD model of co-medication led to a reasonable LDL-C-lowering effect, with adequate assumptions and minimal complexity. The PD model supported the independent lipid-lowering mechanisms of rosuvastatin and ezetimibe. The EHC process increased the exposure to total ezetimibe with a limited effect on LDL profiles.
The lack of significant covariate effects, possibly due to the homogeneous study population, may highlight the importance of considering EHC processes when evaluating drug interactions. Researchers should in future studies investigate the influence of covariates including various demographic factors, comorbidities or genetic polymorphisms on drug response, and explore the utility of mechanistic or physiologically based PK/PD models in guiding personalized treatment strategies.
Data Sharing Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.
Author Contributions
All authors contributed significantly to the study, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; participated in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal for submission; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Gachon University research fund of 2023 (GCU- 202310260001) and 2024 (GCU-202410190001). The investigation was conducted at the Clinical Trials Center, Gachon University Gil Medical Center.
Disclosure
The authors have no conflicts of interest to disclose.
References
1. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111–188. doi:10.1093/eurheartj/ehz455
2. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Circulation. 2019;139(25):e1082–e1143. doi:10.1161/CIR.0000000000000625
3. Kim CH, An H, Kim SH, Shin D. Pharmacokinetic and pharmacodynamic interaction between ezetimibe and rosuvastatin in healthy male subjects. Drug Des Devel Ther. 2017;11:3461–3469. doi:10.2147/DDDT.S146863
4. Kosoglou T, Statkevich P, Johnson-Levonas AO, Paolini JF, Bergman AJ, Alton KB. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clin Pharmacokinet. 2005;44(5):467–494. doi:10.2165/00003088-200544050-00002
5. Ezzet F, Krishna G, Wexler DB, Statkevich P, Kosoglou T, Batra VK. A population pharmacokinetic model that describes multiple peaks due to enterohepatic recirculation of ezetimibe. Clin Ther. 2001;23(6):871–885. doi:10.1016/s0149-2918(01)80075-8
6. Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. 2002;41(10):751–790. doi:10.2165/00003088-200241100-00005
7. Malik MY, Jaiswal S, Sharma A, Shukla M, Lal J. Role of enterohepatic recirculation in drug disposition: cooperation and complications. Drug Metab Rev. 2016;48(2):281–327. doi:10.3109/03602532.2016.1157600
8. Soulele K, Karalis V. On the population pharmacokinetics and the enterohepatic recirculation of total ezetimibe. Xenobiotica. 2019;49(4):446–456. doi:10.1080/00498254.2018.1463117
9. Yang J, Li LJ, Wang K, et al. Race differences: modeling the pharmacodynamics of rosuvastatin in Western and Asian hypercholesterolemia patients. Acta Pharmacol Sin. 2011;32(1):116–125. doi:10.1038/aps.2010.169
10. Niu J, Straubinger RM, Mager DE. Pharmacodynamic Drug-Drug Interactions. Clin Pharmacol Ther. 2019;105(6):1395–1406. doi:10.1002/cpt.1434
11. Jones MW, Small K, Kashyap S, Deppen JG. Physiology, Gallbladder. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
12. van Heek M, Davis H. Pharmacology of ezetimibe. Eur Heart J Suppl. 2002;4(J):J5–J8. doi:10.1016/S1520-765x(02)90076-3
13. Chen W, Ruan Z, Chen J, et al. Population pharmacokinetics and enterohepatic recirculation of hyzetimibe and its main metabolite in Chinese healthy subjects. Br J Clin Pharmacol. 2022;88(7):3153–3161. doi:10.1111/bcp.15187
14. Tong X, Zhou D, Savage A, et al. Population pharmacokinetic modeling with enterohepatic circulation for AZD3241 in healthy subjects and patients with multiple system atrophy. J Clin Pharmacol. 2018;58(11):1452–1460. doi:10.1002/jcph.1134
15. Soulele K, Karalis V. Development of a joint population pharmacokinetic model of ezetimibe and its conjugated metabolite. Eur J Pharm Sci. 2019;128:18–26. doi:10.1016/j.ejps.2018.11.018
16. Deschamps C, Dubruc C, Mentre F, Rosenzweig P. Pharmacokinetic and pharmacodynamic modeling of mizolastine in healthy volunteers with an indirect response model. Clin Pharmacol Ther. 2000;68(6):647–657. doi:10.1067/mcp.2000.112341
17. Jadhav SB, Amore BM, Bockbrader H, et al. Population pharmacokinetic and pharmacokinetic-pharmacodynamic modeling of bempedoic acid and low-density lipoprotein cholesterol in healthy subjects and patients with dyslipidemia. J Pharmacokinet Pharmacodyn. 2023;50(5):351–364. doi:10.1007/s10928-023-09864-w
18. Kakara M, Nomura H, Fukae M, et al. Population pharmacodynamic analysis of LDL-cholesterol lowering effects by statins and co-medications based on electronic medical records. Br J Clin Pharmacol. 2014;78(4):824–835. doi:10.1111/bcp.12405
19. Olsson AG, Pears J, McKellar J, Mizan J, Raza A. Effect of rosuvastatin on low-density lipoprotein cholesterol in patients with hypercholesterolemia. Am J Cardiol. 2001;88(5):504–508. doi:10.1016/s0002-9149(01)01727-1
20. Karlson BW, Palmer MK, Nicholls SJ, Barter PJ, Lundman P. Effects of age, gender and statin dose on lipid levels: results from the VOYAGER meta-analysis database. Atherosclerosis. 2017;265:54–59. doi:10.1016/j.atherosclerosis.2017.08.014
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Pharmacokinetic-Pharmacodynamic Comparison of Recombinant and Plasma-Derived von Willebrand Factor in Patients with von Willebrand Disease Type 3
Bauer A, Friberg-Hietala S, Smania G, Wolfsegger M
Journal of Blood Medicine 2023, 14:399-411
Published Date: 13 June 2023
Olanzapine Pharmacokinetics: A Clinical Review of Current Insights and Remaining Questions
Kolli P, Kelley G, Rosales M, Faden J, Serdenes R
Pharmacogenomics and Personalized Medicine 2023, 16:1097-1108
Published Date: 21 December 2023
A Phase 1a/1b Study of Fostroxacitabine Bralpamide (Fostrox) Monotherapy in Hepatocellular Carcinoma and Solid Tumor Liver Metastases
Plummer R, Greystoke A, Naylor G, Sarker D, Anam ANMK, Prenen H, Teuwen LA, Van Cutsem E, Dekervel J, Haugk B, Ness T, Bhoi S, Jensen M, Morris T, Baumann P, Sjögren N, Tunblad K, Wallberg H, Öberg F, Evans TRJ
Journal of Hepatocellular Carcinoma 2024, 11:2033-2047
Published Date: 24 October 2024
A Pharmacokinetic/Pharmacodynamic Study of Esomeprazole Comparing a Dual Delayed-Release Formulation (YYD601) to a Conventional Formulation Following Multiple Administrations in Healthy Adult Subjects
Lee HW, Kang WY, Park JS, Lee JH, Park JJ, Gwon MR, Yoon YR, Seong SJ
Drug Design, Development and Therapy 2025, 19:97-110
Published Date: 8 January 2025
Pharmacokinetics, Pharmacodynamic, Safety and Tolerability of Fazamorexant, a Novel Dual Orexin Receptor Antagonist: Report of the First-in-Human Study
Ni J, Jin L, Zhao D, Zhang W, Li B, Huang X, Hao X
Drug Design, Development and Therapy 2025, 19:5271-5282
Published Date: 19 June 2025