")
Back to Journals » International Journal of Nanomedicine » Volume 20
Preparation and Characterization of Mitochondrial-Targeted Nitronyl Nitroxide Loaded PLGA Nanoparticles for Brain Injury Induced by Hypobaric Hypoxia in Mice
Authors Da Q , Xu M, Tian Y , Ma H, Wang H, Jing L
Received 19 November 2024
Accepted for publication 25 March 2025
Published 1 April 2025 Volume 2025:20 Pages 3999—4020
DOI https://doi.org/10.2147/IJN.S507315
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Dong Wang
Qingyue Da,1,2 Min Xu,3,4 Yiting Tian,2 Huiping Ma,2 Haibo Wang,3 Linlin Jing1,2
1Department of Pharmacy, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, 710061, People’s Republic of China; 2Department of Pharmacy, The 940th Hospital of Joint Logistics Support Force of PLA, Lanzhou, Gansu, 730050, People’s Republic of China; 3Department of Chemistry, School of Pharmacy, The Air Force Medical University, Xi’an, 710032, People’s Republic of China; 4The Third Stationed Outpatient Department, General Hospital of Central Theater Command, Wuhan, 430070, People’s Republic of China
Correspondence: Linlin Jing, Department of Pharmacy, The First Affiliated Hospital of Xi’an Jiaotong University, No. 277 Yanta West Road, Yanta District, Xi’an, Shaanxi, 710061, People’s Republic of China, Tel +86-029-85323537, Email [email protected] Haibo Wang, Department of Chemistry, School of Pharmacy, The Air Force Medical University, No. 169, Changle West Road, Xi’an, Shaanxi, 710032, People’s Republic of China, Tel +86-029-84774473, Email [email protected]
Background: Oxidative stress is considered an important mechanism of acute high-altitude brain injury. Imidazole nitronyl nitroxide radicals are a class of stable organic radical scavengers that contain single electrons in their molecules. Therefore, in order to search for compounds with low toxicity and better effect against high-altitude brain injury, the preparation methods of PLGA nanoparticles (TPP-C6-HPN@PLGA-NPs) loaded with a synthesized mitochondria targeting imidazole nitronyl nitroxide were emphasized and investigated. Furthermore, its protective effect on brain injury caused by low-pressure hypoxia (HH) in mice was evaluated.
Methods: Nanoparticles were prepared by emulsion solvent evaporation method, and the preparation method was optimized by Box Behnken design based on particle size, encapsulation efficiency (EE) and drug loading (DL). Physical characterization and release studies of the optimized NPs were conducted. The high altitude brain injury mice model was selected to evaluate the therapeutic effect of TPP-C6-HPN@PLGA-NPs in vivo. The histological and biochemical tests were conducted in serum and brain of mice exposed to HH condition.
Results: The nanoparticle size was 120.63 nm, the EE was 89.30%, the DL was 6.82%, the polydispersity index (PDI) was 0.172, and the zeta potential was − 22.67 mV under optimal preparation process. In addition, TPP-C6-HPN@PLGA-NPs owned good stabilities and sustained drug releases. TPP-C6-HPN@PLGA-NP exhibited lower toxicity than TPP-C6-HPN and was well uptaken by PC12 cells. Histological and biochemical analysis demonstrated that TPP-C6-HPN@PLGA-NPs significantly reduced HH induced pathological lesions, oxidative stress, energy dysfunction and inflammation response of brain tissue. Furthermore, nanoparticles did not show significant toxicity to major organs such as the liver and kidneys, as well as hematology in mice.
Conclusion: TPP-C6-HPN@PLGA-NPs exhibits good stability, low hemolysis rate, sustained release, low toxicity, and long residence time in brain tissue and can be used as a promising formulation for the proper treatment of HH-induced brain damage.
Keywords: nitronyl nitroxide, mitochondrial-targeted, PLGA nanoparticles, characterization, high altitude brain injury
Introduction
There is an increasing number of people, approximately 40 million or more, travelling to or visiting high-altitude (HA) areas each year.1–3 In high-altitude areas, the decrease in oxygen partial pressure caused by low air pressure leads to a reduction in the availability of oxygen absorbed by blood and tissues, which poses a significant challenge to people’s health.4 The brain is particularly susceptible to damage due to hypoxia, as an organ with high metabolic and oxygen consumption rates.5 Drugs such as acetazolamide, dexamethasone and edaravone have demonstrated efficacy in ameliorating high-altitude hypoxia-induced brain injury (HHBI).1,6 However, their widespread clinical adoption is significantly constrained by pronounced adverse effects. Therefore, there is currently a substantial lack of effective therapeutic approaches to HHBI.
The mechanism of acute high-altitude brain injury is relatively complex, and the reasons are not fully understood at present. Nevertheless, it is considered to involve many molecular pathways including oxidative stress (OS)7 and inflammation response.8 The state of low-pressure hypoxia can disrupt the balance between the oxidative and antioxidant systems in the brain, leading to an increase in reactive oxygen species (ROS) such as hydroxyl radicals (•OH), superoxide (O2•−), and hydrogen peroxide (H2O2), while endogenous enzyme antioxidants and non-enzyme antioxidants such as superoxide dismutase (SOD) and glutathione (GSH) decrease.9 Mitochondria are subcellular organelles in cells that contain DNA in addition to the nucleus. The mitochondrial respiratory chain is also the main site for ROS production. The low-pressure hypoxic environment leads to excessive production of ROS in mitochondria, causing mitochondrial damage.10
Therefore, the strategy of targeting drug delivery to mitochondria may provide a new therapeutic approach for combating HH-induced damage. So far, covalently linking drugs with lipophilic cations has been an effective method for delivering them specifically to mitochondria, for instance, linking a triphenylphosphonium (TPP) moiety to a central pharmacophore of interest.11 A very successful example is Mito Q, which is covalently bonded to coenzyme Q10 by triphenylphosphate cation (TPP+). Studies have shown that Mito Q can effectively block the production of ROS, prevent mitochondrial oxidative damage, and has a hundred of times stronger effect than untargeted coenzyme Q10.12 A significant challenge in utilizing TPP-conjugated antioxidants for therapeutic purposes lies in their potential mitochondrial toxicity.13 Consequently, during clinical evaluations of these compounds, precise dosage regulation is imperative to maintain MTA (mitochondria-targeted antioxidant) concentrations below levels that could compromise mitochondrial integrity and functionality. Conventional approaches to developing MTAs involved the conjugation of TPP with antioxidant moieties, including phenolic structures and flavonoid derivatives. However, these traditional agents typically exhibit a stoichiometric 1:1 interaction with ROS, necessitating administration of substantial quantities to achieve therapeutic efficacy.
We synthesized 4’-hydroxy-2-substituted phenyl nitro nitrogen oxide (HPN, Figure 1A) previously, which is an imidazole nitrogen oxygen free radical antioxidant. HPN has the ability to mimic SOD activity and react with OH radicals in cyclic and catalytic manners, blocking Fenton reaction and inhibiting ROS attacks on biomolecules and biofilms, thereby reducing cellular oxidative damage.14–16 We found that HPN was able to penetrate the blood–brain barrier and exerted excellent protective effects on HH-induced brain damage.17,18 However, similar to other nitroxyl radicals, HPN has a small molecular weight, high reactivity, and lacks targeting, resulting in a fast clearance rate and short plasma half-life.19 These factors limit its application in the prevention of HHBI.
 |
Figure 1 The chemistry structure of HPN (A) and TPP-C6-HPN (B). |
Nanotechnology has been widely applied in the development of modern medicine and pharmacy. Nanoparticles have significant advantages in achieving targeted drug delivery and sustained release, enhancing drug stability, improving pharmacokinetics, prolonging blood circulation time and reducing the toxic side effects of drugs.20 Due to the excellent biocompatibility, biodegradability, and unique physicochemical properties of poly(D, L-lactide-co-glycolide, PLGA) copolymers, NPs based on PLGA have been used for drug delivery.21,22 PLGA has been approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for medical use, and is one of the most widely used nanoparticle polymers.23
With the aim of improving protective efficiency of HPN against HH-induced brain damage, we designed and synthesized a new mitochondrial-targeting HPN derivative (TPP-C6-HPN, Figure 1B) using triphenylphosphine cations as carriers. In addition, we used PLGA as a nanocarrier to encapsulate the TPP-C6-HPN to form nanoparticles (TPP-C6-HPN@PLGA-NPs) for improving the stability and circulation time. Furthermore, we studied the protective effect of TPP-C6-HPN@PLGA-NPs on the HH-induced brain injury in vivo. Finally, we also monitored the toxic effects of TPP-C6-HPN@PLGA-NPs on mice.
Materials and Methods
Chemical Materials
The (4-hydroxyphenyl)-4,4,5,5-tetramethyl-4,5-dihydro imidazole radical (HPN) was prepared according to the previous synthetic method.17 (6-Bromohexyl) triphenylphosphonium bromide (CAS No. 83152–22-1) and Sodium cholate (CAS number: 361–09-1) were purchased from Aladdin Ltd. (Shanghai, China). Other chemical reagents, including potassium carbonate (K2CO3), potassium iodide (KI), acetonitrile (CH3CN), dichloromethane (CH2Cl2) and methanol (CH3OH), were purchased from commercial sources. PLGA (MW: 10000; 50:50) was purchased from Daigang Biomaterials Co., Ltd. (Jinan, China). Poloxamer 188 (P188; 5 mg/mL; CAS number: 9003–11-6) was purchased from Beijing Solarbio Science Technology Co., Ltd. (Beijing, China).
Synthesis of (4-(6-(Triphenylphosphonio)hexyl)oxy)phenyl)-4,4,5,5-Tetramethyl-4,5-Dihydro Imidazole Radical (TPP-C6-HPN)
Figure 2A shows the synthetic route of TPP-C6-HPN. Briefly, HPN (1.24 g, 5 mmol) and 6-bromohexylphosphine bromide (3.04 g, 6 mmol) was added to 50 mL CH3CN. After dissolution, K2CO3 (1.38 g, 10 mmol) and KI (0.017 g, 0.01 mmol) was also added and stirred well. The mixture was heated to 65°C, stirred, and reacted continuously for 12 hours. After the reaction was complete, CH3CN was removed under reduced pressure, and 20 mL of distilled water was added to the remaining mixture to dissolve K2CO3 and KI. The mixture was extracted with CH2Cl2 (20 mL) three times again. The organic phase was combined and dried over anhydrous sodium sulfate by overnight. The anhydrous sodium sulfate was filtered, and the solvent was removed to obtain a dark blue oily crude product. The crude product was purified by column chromatography using CH2Cl2:CH3OH=20:1 eluent, obtaining 2.87 g of dark blue solid with a yield of 85.3%. IR (KBr): ν 2860, 1603, 1490, 1380, 1080, 812 cm−1 (Figure S1). HRMS (ESI): m/z [M-Br]+ calcd for C38H43N2O5P+: 594.3006, Found: 594.3015 (Figure S2). EPR (CH3OH): five lines, g = 2.0064, |aN| = 7.71G (Figure S3).
 |
Figure 2 (A) The synthesis route for TPP-C6-HPN. (B) Flowchart of TPP-C6-HPN@PLGA-NPs Preparation. |
Determination of the TPP-C6-HPN Content
The content of TPP-C6-HPN was determined by HPLC chromatography. The chromatographic column was Symmetry® C18 (5 µm, 4.6 mm × 150 mm), the sample injection volume was 10 μL, and the detection wavelength was 275 nm. The mobile phase consisted of acetonitrile containing 0.1% trifluoroacetic acid and water containing 0.1% tetrafluoroacetic acid. Degree elution procedure: 0–12 minutes, 40–60% acetonitrile; 12–12.1 minutes, 6–40% acetonitrile, 12.1–15 minutes, 40% acetonitrile. The flow rate was set to 1.0 mL/min. The column temperature of the chromatography column was 25°C. Prepared solutions of TPP-C6-HPN reference, TPP-C6-HPN@PLGA-NPs, and PLGA-NPs were injected under the chromatographic conditions to assess the specificity of TPP-C6-HPN. The limit of detection (LOD) and limit of quantitation (LOQ) of TPP-C6-HPN were determined. TPP-C6-HPN solution was diluted with acetonitrile to prepare reference solutions of different concentrations (3.125, 6.25, 12.5, 25, 50, 100 μg/mL). And a linear regression equation was established between the concentration (X) and peak area (Y). Additionally, methodological validation was conducted using accuracy, precision and stability experiments to confirm the reliability of the established method.
Preparation of Nanoparticles (TPP-C6-HPN@PLGA-NPs)
Figure 2B showed the preparation method of TPP-C6-HPN@PLGA-NPs. TPP-C6-HPN was encapsulated in PLGA by an improved emulsion solvent evaporation method. In brief, the organic phase consists of a solution of PLGA and TPP-C6-HPN dissolved in CH2Cl2. A certain amount of sodium cholate (SC) and poloxamer 188 (P188) dissolve in water to form an aqueous phase. The organic phase is slowly fed into the aqueous phase one by one, and then the mixture is emulsified in the ice bath for 5 minutes with an ultrasonic homogenizer (Scientz IID, Xingzhi Biological, China), and the power is 200 W to obtain oil in water (O/W) lotion. Then, use a magnetic stirrer to stir the lotion for 5 h to remove the solvent. Use an ultrafiltration tube with a molecular weight of 10 kDa to carry out three cycles of centrifugation steps (13 000rpm at 4°C, 15min) for purification TPP-C6-HPN@PLGA-NPs. Finally, the purified TPP-C6-HPN@PLGA-NPs was redispersed in distilled water, which was stored at 4°C until further use.
Box-Behnken Experimental Design (BBD)
BBD was used to determine the most important influencing factors in the preparation process. In this work, three independent variables were chosen based on results of single-factor experiments (Table S1–S6) as follows: (X1) the ratio of PLGA to TPP-C6-HPN (w/w), (X2) the ratio of water volume to oil volume (v/v), and (X3) concentration of P188 (w/v, %). Three independent variables were established, namely the particle size of nanoparticles (nm) (Y1), the encapsulation efficiency of TPP-C6-HPN (EE) (Y2), and the drug loading capacity of TPP-C6-HPN (DL) (Y3). Based on the three independent variables, three different levels were set as test variables: low (−1), medium (0), and high (+1) levels (Table 1). Table 2 presents the matrices of 17 experimental formulations constructed. Variance analysis (ANOVA) was used to verify the polynomial equation generated after analysis, and the relationship between all variables and their influence on the response were predicted. Subsequently, the contour maps (2D) response surface maps (3D) were generated by the software to understand the relationships between variables and their interactions.
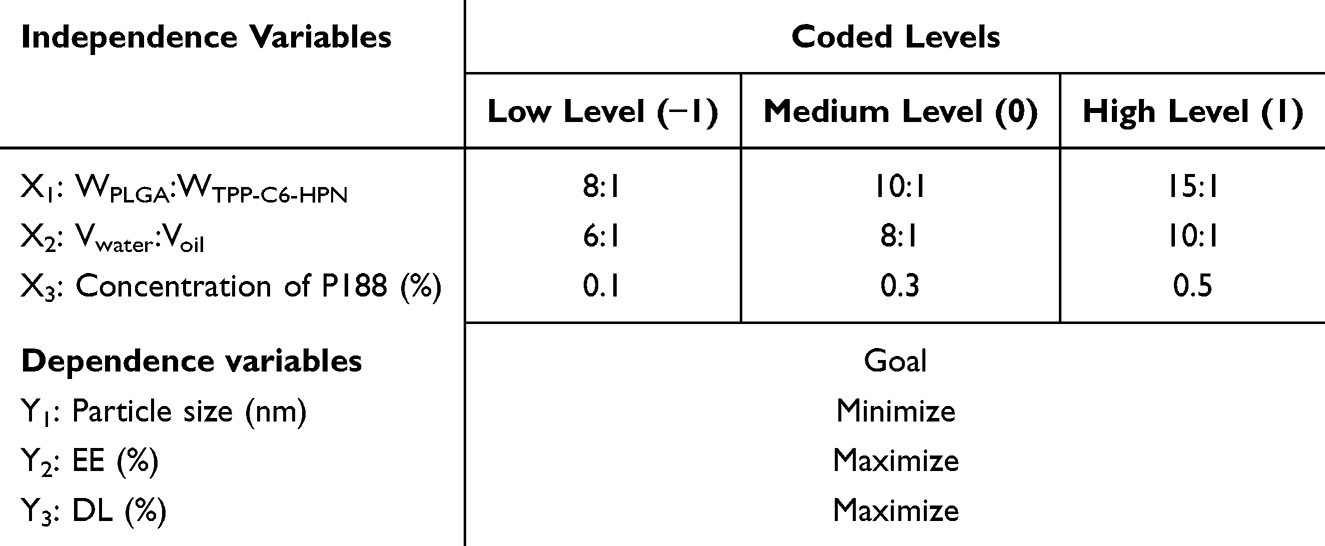 |
Table 1 Box-Behnken Design Used for Optimization of TPP-C6-HPN@PLGA-NPs |
 |
Table 2 BBD Experimental Design Results |
Nanoparticle Size, Zeta Potential and PDI Measurements
The particle size, polydispersity index (PDI) and zeta potential of the nanoparticles were measured by dynamic light scattering (DLS) using a nano Zetasizer (Zetasizer Pro, Malvern, UK) at room temperature. Measure each result three times.
Drug Encapsulation Efficiency (EE) and Drug Loading (DL) Measurements
0.2 mL of TPP-C6-HPN@PLGA-NPs was added in ultrafiltration tube, which was then centrifuged at 4°C, 13,000 rpm for 15 min. Then, the sediment was washed for three times. The solutions were collected and determine using HPLC to analyzed the content of TPP-C6-HPN. 0.2 mL of TPP-C6-HPN@PLGA-NPs was accurately measured and treated with 0.2 mL CH3CN to disrupt the nanoparticles. 0.2 mL of TPP-C6-HPN@PLGA-NPs was accurately measured and treated with 0.2 mL CH3CN to disrupt the nanoparticles. The mixture was sonicated for 30 minutes and then centrifuged at 12000 rpm for 10 minutes at 4°C. The supernatant was determined by HPLC to analyze the total content of drug dosage.


where Wtotal represents the total drug weight, Wfree represents the weight of free drug, WPLGA represents the weight of PLGA.
Morphological Observation by Scanning Electron Microscopy (SEM)
TPP-C6-HPN@PLGA-NPs (10 μL) was dropwised onto SiO2 pellets and dried at room temperature. Then, the particles were coated with gold sputtering and scanned using a scanning electron microscope (SEM, Quattro S; Thermo Fisher, Brno Chernovice, the Czech Republic).
Morphological Observation by Transmission Electron Microscopy (TEM)
The solution of TPP-C6-HPN@PLGA-NPs (10 μL) was dropped onto a copper grid, and the copper grid was dried at room temperature. The grid was negatively stained using 10 μL of 2% phosphotungstic acid. The sample was observed using a transmission electron microscope (TEM, JEM-1400, Japan Electron Optics Laboratory, Tokyo, Japan).
X-Ray Diffraction Analysis (XRD)
Appropriate quantities of TPP-C6-HPN, TPP-C6-HPN@PLGA-NPs and PLGA-NPs were individually freeze-dried to obtain freeze-dried powders. Freeze-dried powder was deposited on a glass slide and determined by an X-ray diffractometer (Bruker D8 Advance) with a scanning range of 10–90 and a diffraction scanning angle of 2θ, while observing the diffraction peaks of the samples.
Fourier Transform Infrared Spectroscopy Analysis (FTIR)
The Nicolet-iS5 infrared spectrometer was used to test the surface chemical structure of nanoparticles. At room temperature, TPP-C6-HPN, TPP-C6-HPN@PLGA-NPs, and PLGA-NPs were individually placed on the sample window. The infrared absorption peak of the sample was scanned within the range of 4000–400 cm−1.
Stability Assessment
The TPP-C6-HPN@PLGA NPs solution was stored at 4°C and its particle size and PDI were tested on days 0, 1, 3, 5, 7, 10, and 15, respectively. This investigation aimed to assess the storage stability of TPP-C6-HPN@PLGA-NPs at 4°C.
Hemolysis Assay
Red blood cells (RBC) were isolated from whole blood samples of 6-week-old healthy SD rats. Different concentrations of TPP-C6-HPN@PLGA-NPs solutions (6.25, 12.5, 25, 50, and 100 µg/mL) were mixed and incubated with equal volumes of diluted RBC suspension. Meanwhile, set up positive (+) and negative (-) control groups. The red blood cell suspension of the positive (+) control group was incubated with distilled water; and the red blood cell suspension of the negative (-) control group was incubated with physiological saline. After incubating each group of samples at 37°C for 1 hour, the supernatant was measured at 492 nm. The hemolysis percentage of red blood cells was calculated using the following formula:

In vitro Drug Release
1 mL of TPP-C6-HPN and 1 mL of TPP-C6-HPN@PLGA-NPs were loaded into separate dialysis bags (10 kDa) and immersed in 90 mL of pH 7.4 PBS solution at 37°C for continuous magnetic stirring. Samples (200 μL) were collected at 0, 0.08, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, 60, 72, 84, and 96 h, respectively. After each sampling, an equal volume of fresh PBS solution is added again. The content of TPP-C6-HPN was determined by HPLC method. The in vitro release curve was plotted as the cumulative drug release percentage versus dialysis time. The drug release behavior was analyzed by fitting the data with the drug release kinetics equation.
Cell Culture
PC12 cells (highly differentiated) were obtained from the Chinese Academy of Sciences (Shanghai, China) and cultured in the high sugar Dulbecco’s Modified Eagle medium (DMEM, Bios Biotechnology Hangzhou, China) containing 10% FBS and 1% penicillin/streptomycin at 37°C and 5% carbon dioxide.
CCK-8 Assay
Under normoxic conditions, PC12 cells were incubated with a certain concentration of TPP-C6-HPN@PLGA-NPs for 24 h. The culture medium was discarded, and 10 μL of CCK-8 reagent (Bioss Biotechnology, Hangzhou, China) was added to each well and incubated for another 2 h. Measure the absorbance at 450 nm using a microplate reader (Molecular Devices, Sunnyvale, USA). Cell viability is expressed as a percentage of normal cells.
Cell Uptake Studies
PC12 cells were cultured in 20 mm confocal culture dishes for 24 h. Then, Cy5.5@PLGA-NPs (prepared and characterized as described in Materials and Methods section 1.3 and Results section 2.4 of the supporting information) were added and then incubated for 4, 12 or 24 h at 37°C. The cells were washed three times with PBS to remove excess NPs that were not taken up by the cells. Then, 4% paraformaldehyde was added to fix the cells, washed three times with PBS, and stained the nuclei with DAPI reagent. The uptake of NPs by cells was observed using an Olympus DP80 fluorescence microscope. Green fluorescence of Cy5.5@PLGA-NPs was obtained from the green channel (excitation wavelength: 644 nm, emission wavelength: 655 nm). The blue fluorescence of nuclei was obtained from the blue channel (excitation wavelength: 364 nm, emission wavelength: 454 nm).
Animals
BALB/c male mice (weight 18–22 g, age 6–8 weeks, SPF grade) and SD rats (weight 180–220 g, age 6–8 weeks, positive protection grade) were provided by the Experimental Animal Center of the Air Force Medical University. Animals were kept in a constant environment with a temperature of 23 ± 2°C and a relative humidity of 60 ± 5%. The lighting conditions consist of a 12 h light/12 h dark cycle. Animals were allowed to freely access water and granular food. The pain experienced by animals and the number of animals used were minimized. The animal experiment has been approved by the Animal Care and Use Committee of 940th Hospital (certificate number 2019XYLL059), following the “Guidelines for the Care and Use of Laboratory Animals in China”.
Distribution in vivo of Cy5.5@PLGA-NPs
The Cy5.5-labeled NPs (10 mg/kg in 200 μL PBS) were administered to BALB/c mice intravenously (i.v). Then, at 0, 0.5, 1, 2, 4, 6, 12, 24, and 48 h, the brain tissues were harvested from the sacrificed mice. The Cy5.5 fluorescence imaging in these organs was observed and photographed using a Kodak in vivo imaging system (IVIS Spectrum; PerkinElmer Corp., Waltham, MA, USA).
Normobaric Hypoxia Test
The normobaric hypoxia test was conducted based on our earlier published method.17 Thirty male BALB/c mice were randomly allocated into five groups (n = 6 per group): hypoxia group, TPP-C6-HPN (5 mg/kg), and three TPP-C6-HPN@PLGA-NPs groups (1.25, 2.5 or 5 mg/kg of TPP-C6-HPN). The mice in the hypoxia group were injected with physiological saline (0.1 mL/20 g) via the tail vein (iv), while the mice in the other drug groups were intravenously injected with corresponding concentrations of drugs. After 30 min of administration, the mice were subjected to normobaric hypoxia test, and their survival time was recorded. Calculate the prolongation rate of drug on mouse survival:

Establishment of Acute Hypobaric Hypoxia Brain Injury (HHBI) Model
The HHBI model was established in accordance with our earlier published method.17 In brief, mice were randomly divided into a control group, HH group, HH+TPP-C6-HPN group, and high, medium, and low-dose TPP-C6-HPN@PLGA-NPs groups. The drug was administered to mice via tail vein injection. After 30 minutes of administration, the control group mice were fed under laboratory conditions of normal pressure and oxygen (altitude 1400 meters, temperature 23–25°C). Other groups of mice were fed in a simulated animal decompression chamber (DYC-3070, Fenglei, Guizhou, China) at an altitude of 8000 meters for 24 h (temperature 23°C to 25°C, humidity 40–50%, 18% oxygen, 0.035 MPa). After 24 h, the mice were euthanized by cervical dislocation, and their serum and brain tissue samples were collected and stored at −80°C for experimental testing.
HE Staining
The brain tissue was fixed with 4% paraformaldehyde for 24 h. After embedding and fixing in paraffin, the brain tissues were cut it into 5 μm thick sections. Slices were stained using the hematoxylin eosin staining method. Observe and capture images using an optical microscope.
Biochemical Analysis
The brain tissue homogenate (10% (w/v)) was centrifuged to obtain the supernatant. The levels of superoxide dismutase (SOD), glutathione (GSH), malondialdehyde (MDA), hydrogen peroxide (H2O2), adenosine triphosphate (ATP) and ATPases in the supernatant were tested using reagent kits (Nanjing Institute of Bioengineering, China) according to the manufacturer’s protocol.
Measurement of the Contents of Inflammatory Factors in Brain Tissue
The levels of interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) in mouse serum and brain tissue were evaluated using a commercial ELISA kit (Shanghai Jianglai Biotechnology Co., Ltd., China) according to the manufacturer’s protocol.
In vivo Toxicity Assay of NPs-TPP-NIT
BALB/c male mice were randomly divided into three groups: control group, TPP-C6-HPN group, and TPP-C6-HPNPLGA@NPs group, with 6 mice in each group. The mice were treated with PBS, TPP-C6-HPN (5 mg/kg) or TPP-C6-HPN@PLGA-NPs (5 mg/kg of TPP-C6-HPN) through iv twice a day, for 14 days consecutively. On the 14th day after administration, blood samples were collected from each group of mice to detect the levels of red blood cells (RBC), white blood cells (WBC), platelets (PLT), hemoglobin (HGB), aspartate aminotransferase (AST), alanine aminotransferase (ALT), albumin (ALB), and blood urea nitrogen (BUN). Simultaneously, the mice were dissected to obtain their main organs such as brain, heart, liver, lungs, spleen, kidney and subjected to HE staining histopathological examination.
Statistical Analysis
The data were statistically analyzed using GraphPad Prism 6 (GraphPad, California, USA) software. Each experiment should be repeated at least three times, and the results should be presented as mean ± standard deviation. Unpaired Student’s t-tests was used to compare the differences between the two groups; one way ANOVA was used for multiple comparisons, followed by a post-hoc Turkey analysis. P < 0.05 was regarded as statistically significant.
Results
Determination of TPP-C6-HPN Content in TPP-C6-HPN@PLGA-NPs
HPLC analysis was applied to access TPP-C6-HPN reference solution, TPP-C6-HPN@PLGA-NPs, and PLGA-NPs. TPP-C6-HPN reference solution and TPP-C6-HPN@PLGA-NPs, exhibited characteristic peaks of TPP-C6-HPN@PLGA-NPs, at the same retention time, whereas PLGA-NPs did not show any peak, as depicted in Figure S4. The results indicated that the introduction of PLGA into the nanoparticles did not interfere with the detection of TPP-C6-HPN. Table S7 showed the LOD and LOQ of TPP-C6-HPN, were 0.3 μg/mL and 1.0 μg/mL, respectively. Figure S5 showed that the regression equation exhibited a good linear relationship (R = 0.9999) within the testing range. Methodological investigations revealed that the accuracy, precision and stability experiments had RSD below 2% (Table S8–S9). These findings demonstrated the reliability of the TPP-C6-HPN content determination method established in this study.
Optimization of TPP-C6-HPN@PLGA-NPs by Response Surface Experiment of BBD
The function to predict the studied process variables affecting the size of nanoparticles (Y1), EE (Y2) and DL (Y3) was automatically generated by Design Expert software:
Y1=−432.9118+62.8047*X1+59.9346*X2+53.8515*X3+0.0448*X1X2+6.3953*X1X3-7.1188*X2X3-5.7533*X12-4.0092*X22-87.1688*X32 (R2 = 0.8772)
Y2=−79.5370+15.8086*X1+15.8511*X2+2.1415*X3-0.1930*X1X2-2.8801*X1X3+5.2312*X2X3-0.5207*X12-0.8546*X22-15.3312*X32 (R2 = 0.8696)
Y3=6.5620+0.5072*X1-0.6801*X2+4.0034*X3+0.0003*X1X2-0.3930*X1X3+0.5438*X2X3-0.0282*X12+0.0418*X22-5.4438*X32 (R2 = 0.9315)
The values of the determination coefficient R2 were 0.8772, 0.8696, and 0.9315, respectively. Meanwhile, the lack of fit was not significant. Using the equation fitted above, we can predict the best formulation of the TPP-C6-HPN@PLGA-NPs. According to the results of the statistical analysis (Tables 3 and S10-S12), it could be confirmed that Vwater:Voil ratio (X1) had significant effects on the particle size of the TPP-C6-HPN@PLGA-NPs, while WPLGA: WTPP-C6-HPN ratio (X2) had significant effects on the EE and DL of TPP-C6-HPN@PLGA-NPs. The concentration of P188 (X3) had no independent impact on the particle size, EE, and DL of TPP-C6-HPN@PLGA-NPs. Figure 3 showed the 2D and 3D response surfaces of the data. As shown in Figure 3A, the particle size of the nanoparticles appeared a trend of first increasing and then decreasing, as the ratio of Vwater: Voil (X1) decreases. As shown in Figure 3B and C, the EE of the nanoparticles gradually increased, while the DL gradually decreased, with an increase in the WPLGA: WTPP-C6-HPN ratio (X2). Based on the results of response surface optimization design, the final process for preparing nanoparticles was determined as follows: Vwater: Voil ratio of 10:1, WPLGA: WTPP-C6-HPN ratio of 10:1, P188 concentration of 0.3%. Three batches of nanoparticles were obtained using this preparation process, with an average particle size of 120.76 ± 3.82 nm, PDI of 0.302 ± 0.021 (Figure 4A), zeta potential of −27.93 ± 0.59 mV, EE of 89.30 ± 0.33%, and DL of 6.82 ± 0.26%. Moreover, the measured values were compared with the predicted values of the model, and the results showed that the values were very close.
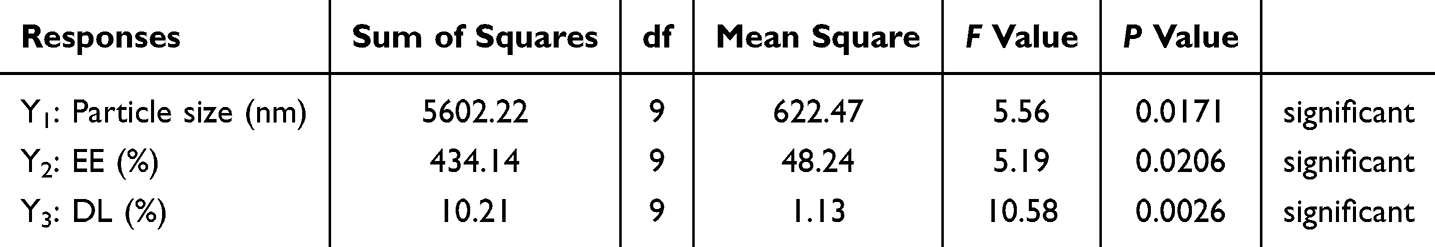 |
Table 3 The ANOVA Results for All Responses |
 |
Figure 3 2D contour plots and 3D response surface plot and for Particle size (A), EE (B) and DL (C). |
 |
Figure 4 Characterization of TPP-C6-HPN@PLGA-NPs. (A) particle size and zeta potential. (B) SEM image of TPP-C6-HPN@PLGA-NPs. (C) TEM image of TPP-C6-HPN@PLGA-NPs. Presents the XRD diffraction analysis (D) and FTIR spectra (E) of TPP-C6-HPN, PLGA-NPs, and TPP-C6-HPN@PLGA-NPs. (F) Changes in particle size and PDI of TPP-C6-HPN@PLGA-NPs over a period of 15 days. (G) Hemolysis rate analysis of TPP-C6-HPN@PLGA-NPs. (H) Drug release curve of TPP-C6-HPN and TPP-C6-HPN@PLGA-NPs at pH 7.4 in PBS. Date represented as mean ± SD, n = 3. |
Characterization of TPP-C6-HPN@PLGA-NPs
TPP-C6-HPN@PLGA-NPs prepared using the optimal fabrication process appeared as a pale blue milky solution without precipitation. The SEM and TEM images of TPP-C6-HPN @PLGA-NPs were shown in Figure 4B and C. The image showed that the nanoparticles were spherical and could remain dispersed in solution without aggregation. The results indicated that the distribution of NPs was uniform and their size distribution was narrow.
As indicated by XRD analysis (Figure 4D), the spectrum of TPP-C6-HPN@PLGA-NPs resembled the spectrum of PLGA-NPs. The characteristic peaks of TPP-C6-HPN almost disappeared within TPP-C6-HPN@PLGA-NPs. Similarly, FTIR spectroscopy results (Figure 4E) also indicated that TPP-C6-HPN@PLGA-NPs displayed similar peaks corresponding to PLGA, while the characteristic absorption peaks of TPP-C6-HPN were not distinct. The above results indicated that TPP-C6-HPN has been successfully encapsulated in the PLGA carrier.
As depicted in Figure 4F, the appearance of TPP-C6-HPN@PLGA-NPs remained unchanged within 15 days. The particle size and PDI of TPP-C6-HPN@PLGA-NPs did not show significant changes during this period. Therefore, TPP-C6-HPN@PLGA-NPs exhibited good stability over a 15-day period and could be stably stored during this time.
The hemolysis test was conducted to evaluate the effect of NPs on the red blood cell tension. In the present study, the hemolysis ratio of TPP-C6-HPN@PLGA-NPs was less than 3% (Figure 4G), meaning that TPP-C6-HPN@PLGA-NPs exhibited no indication of hemolysis and could be administered intravascularly within certain concentration limits.
In vitro Drug Release
The functional relationship between dialysis time and cumulative drug release percentage is used to reflect the release of drugs. As shown in Figure 4H, compared to TPP-C6-HPN@PLGA-NPs, the release rate of TPP-C6-HPN was faster, and it is almost completely released around 12 h. Within the first hour, only 4.14% of the TPP-C6-HPN@PLGA-NPs was released, demonstrating the absence of any burst release effect. Within 24 h, TPP-C6-HPN@PLGA-NPs exhibited a cumulative release of 39%. This observation suggested that the encapsulation of TPP-C6-HPN within PLGA could extend the drug’s action duration, achieving a sustained-release effect. The slow increase in cumulative release rate following 24h may be due to its instability under this system.
The drug release curves of TPP-C6-HPN and TPP-C6-HPN@PLGA-NPs under normal blood pH conditions were fitted using four different models: zero-order release, first-order release, Higuchi, and Ritger-Peppas models. As presented in Table S13 and S14, the first-order release model exhibited the highest fitting degree for the drug release kinetics of both TPP-C6-HPN (R2=0.9821) and TPP-C6-HPN@PLGA-NPs (R2=0.9858). The Ritger Peppas equation can reflect the mechanism of drug release. The results showed that the diffusion coefficient of TPP-C6-HPN@PLGA-NPs was in the range of 0.45–0.85, which indicated that the release mechanism was a combination of skeleton dissolution and Fick diffusion.
Cytotoxicity and Cellular Uptake Study
Prior to the cellular uptake study, we first evaluated the cytotoxicity of TPP-C6-HPN both as a free drug and when loaded into nanoparticles, at the same drug equivalent concentration of 1.5 × 10−12 to 1.5 × 10−7 mol/L using the CCK-8 assay in PC12 cells. As shown in Figure 5A, compared with control group, no significant changes in viability were observed within the selected concentration range of TPP-C6-HPN@PLGA-NPs, while a significant decrease in viability was observed in TPP-C6-HPN group when the concentration was higher than 1.5 × 10−9 mol/L, revealing that the TPP-C6-HPN@PLGA-NPs exhibited better biocompatibility than TPP-C6-HPN. Figure 5B showed confocal microscopic images of PC12 cells, respectively, after exposure of the cells to Cy5.5@PLGA-NPs. It was found that the cellular uptake of Cy5.5@PLGA-NPs by PC12 cells was observed following a 4 h incubation period. As the duration of time increased, Cy5.5@PLGA-NPs were accumulated within the cytoplasm of incubated PC12 cells, which demonstrated that NPs could rapidly cross cell membranes.
 |
Figure 5 The cytotoxicity of different concentrations of TPP-C6-HPN and TPP-C6-HPN@PLGA-NPs on PC12 cells (A). Data was presented as mean ± SD, n = 6. *P < 0.05 and **P < 0.01 vs the control group. (B) The cellular uptake of Cy5.5@PLGA-NPs in PC12 cells (Scale bar = 200 μm). (C) Biodistribution of Cy5.5@PLGA-NPs at 0, 0.5, 1, 2, 4, 6, 12, 24, 48 h administration in BALB/c mice. (D) Semi-quantitative results of the average fluorescence intensity. Data was presented as mean ± SD, n = 3. (E) Ex vivo fluorescent image of Cy5.5@PLGA-NPs in the brain at different time after administration. The color bar represents the average radiance efficiency (ph s−1 cm−2 s r−1)/(μW cm−2). (F) Semi-quantitative analysis of the fluorescent intensity. Data was presented as mean ± SD, n = 3. |
Distribution in vivo
Images were obtained, and fluorescence quantification was determined in mice and brain tissue at different time points after Cy5.5@PLGA-NPs administration to study the long-term biological distribution of Cy5.5@PLGA-NPs in mice (Figure 5C and D) and brain tissue (Figure 5E and F). The results indicate that Cy5.5@PLGA-NPs were rapidly distributed throughout in mice, within 1 h after administration. As Cy5.5@PLGA-NPs circulated, fluorescence signals in mouse brain gradually weakened, exhibiting time-dependent clearance. After 48 h of administration, weak fluorescence intensity can still be detected in mouse brain tissue, indicating that NPs have a longer retention time in brain tissue. This may be beneficial for enhancing the protective effect of drugs on the brain tissue of HH mice.
The Effect of TPP-C6-HPN@PLGA-NPs on the Survival Time of Normobaric Hypoxia Model Mice
As seen in Table 4, TPP-C6-HPN@PLGA-NPs administration dose-dependently extended the survival time of mice exposed to hypoxia environment. The survival rates of mice prolonged by low, medium, and high doses of TPP-C6-HPN@PLGA-NPs were 22.63%, 41.11%, and 82.64%, respectively, which showed statistical differences compared with the hypoxia group (P < 0.01 or P < 0.05).
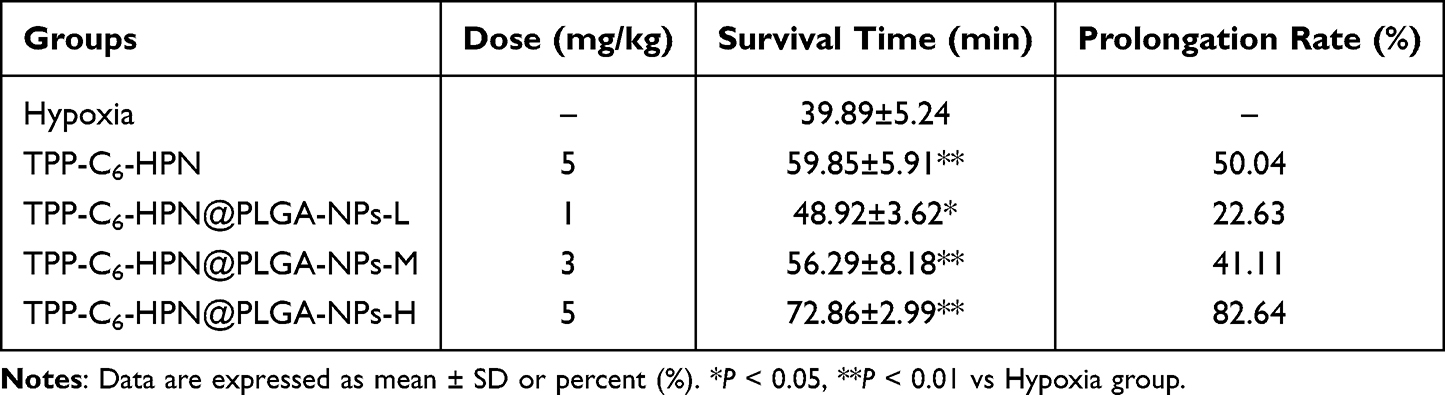 |
Table 4 Effect of TPP-C6-HPN@PLGA-NPs on the Survival Time of Mice in Normobaric Hypoxia Test ( |
TPP-C6-HPN@PLGA-NPs Mitigated Brain Injury of Mice Exposed to HH
As shown in Figure 6, the arrangement of brain tissue cells in the HH group of mice was disordered, the number of neuronal cells decreased, the nuclear cytoplasmic gap increased, and the nucleus condensed into irregular shapes, compared with the control group. After TPP-C6-HPN and TPP-C6-HPN@PLGA-NPs intervention, the pathological structure of brain tissue was improved to a certain extent; there is a tendency for the cell structure to return to normal, with an increase in the number of neurons and a decrease in nuclear staining.
 |
Figure 6 Effect of TPP-C6-HPN@PLGA-NPs on brain histopathological changes in mice exposed to HH by HE staining. Scale bar = 100 μm. |
TPP-C6-HPN@PLGA-NPs Mitigated HH Caused Oxidative Stress in Mice Brain
As shown in Figures 7A–D, the content of H2O2 and MDA in mouse brain tissue significantly increased under HH conditions; the SOD activity and GSH content were significantly decreased compared to the control group (P < 0.01). Compared with the HH group, the levels of H2O2 and MDA in the brain tissues of mice in the NPs-L, NPs-M, NPs-H, and TPP-C6-HPN groups were significantly reduced (P < 0.01), and the levels of GSH were significantly increased (P < 0.01). In addition, NPs-H could significantly increase the activity of SOD (P < 0.01). These data suggested that TPP-C6-HPN@PLGA-NPs supplication significantly alleviated HH caused oxidative stress.
 |
Figure 7 Effects of TPP-C6-HPN@PLGA-NPs on oxidative stress and energy metabolism in brain tissue of mice exposed to HH. The levels of H2O2 (A), MDA (B), SOD (C), GSH (D), ATP (E), Na+-K+-ATPase (F) and Ca2+-Mg2+-ATPase (G) in brain tissues. Data was presented as mean ± SD, n = 6. #P < 0.05 and ##P < 0.01 vs the Control group; *P < 0.05 and **P < 0.01 vs the HH group. |
TPP-C6-HPN@PLGA-NPs Attenuated HH Caused Energy Metabolism Disorder in Mice Brain
To assess the regulating effect of TPP-C6-HPN@PLGA-NPson energy metabolism, levels of ATP and ATPase in brain tissue were detected using commercial biochemical kits. As shown in Figure 7E–G, the activities of Na+-K+-ATPase and Ca2+-Mg2+-ATPase in mouse brain tissue were significantly reduced under HH conditions, and the content of ATP, suggesting that HH stimulation triggered energy metabolism dysfunction. After administration of drugs, the ATP content in the brain tissue of mice in the NPs-L, NPs-M, and NPs-H groups significantly increased (P < 0.01); The Na+-K+-ATPase activity of NPs-H groups also significantly increased (P < 0.01). The Ca2+-Mg2+-ATPase activity of NPs-L, NPs-M, NPs-H, and TPP-C6-HPN groups was significantly reduced (P < 0.01 or P < 0.05). These data suggested that TPP-C6-HPN@PLGA-NPs supplication attenuated the energy metabolism dysfunction caused by HH.
TPP-C6-HPN@PLGA-NPs Mitigated HH Caused Inflammatory Response in Mice Brain
The pro-inflammatory cytokines IL-6, TNF-α, and an anti-inflammatory factor IL-10 were measured using ELISA to evaluate the anti-inflammatory effect of TPP-C6-HPN@PLGA NPs. As shown in Figure 8, the levels of IL-6 and TNF-α in the serum and brain tissue of HH group mice significantly increased, while the level of IL-10 significantly decreased compared to the control group (P < 0.01). After drug intervention, compared with HH, the content of IL-6 in serum was significantly decreased in NPs-L, NPs-M, NPs-H, and TPP-C6-HPN groups (P < 0.01 or P < 0.05), while the content of IL-1β in brain was significantly decreased in NPs-M, NPs-H, and TPP-C6-HPN groups (P < 0.01). The levels of TNF-α in the serum and brain tissue of mice in the NPs-L, NPs-M, NPs-H, and TPP-C6-HPN groups were significantly reduced (P < 0.01 or P < 0.05). The serum IL-10 levels were significantly increased in the NPs-M and NPs-H groups. The IL-10 content in the TPP-C6-HPN group significantly increased. These data suggested that TPP-C6-HPN@PLGA-NPs supplication attenuated the inflammation response caused by HH.
 |
Figure 8 Effects of TPP-C6-HPN@PLGA-NPs on inflammatory response in serum and brain tissue of mice exposed to HH. The levels of IL-6 (A), TNF-α (B) and IL-10 (C) in serum. The levels of IL-6 (D), TNF-α (E) and IL-10 (F) in brain tissues. Data was presented as mean ± SD, n = 6. ##P < 0.01 vs the Control group; *P < 0.05 and **P < 0.01 vs the HH group. |
In vivo Toxicities of TPP-C6-HPN@PLGA-NPs
For the safety concerns, we further evaluated the long-term toxicity of TPP-C6-HPN@PLGA-NPs in mice (n = 6) after i.v. injection for up to 14 d. The effect of TPP-C6-HPN@PLGA-NPs (5 mg/kg) on the pathological changes in major organs on the 14th day was examined using an HE staining assay. As is described in Figure 9A–H, there were no significant differences compared to the control group in blood routine indexes, including the number of WBCs, RBCs and PLAs as well as the content of PLTs, and blood biochemistry parameters, such as the content of ALT, AST, ALB and BUN. Furthermore, no obvious damage was found in these major organs such as the heart, brain, liver, lung, kidney, and spleen in mice at the experimental time point (Figure 9I). In addition, no significant changes in body weight (Figure 9J) and no death were observed in mice of each group after various treatments during the observation. Overall, TPP-C6-HPN@PLGA-NPs did not show significant side effects.
 |
Figure 9 TPP-C6-HPN@PLGA-NPs shows good security in long-term use. The levels of WBC (A), RBC (B), HGB (C), PLT (D), ALT (E), AST (F), ALB (G) and BUN (H). Data was presented as mean ± SD, n = 6. (I) Represented HE images of the major organs of the mice after different treatments for 14 days. Scale bar = 50 μm, n = 3. (J) Body weight of mice. Data was presented as mean ± SD, n = 6. |
Discussion
HPN is a good free radical scavenger and exhibits significant protective effects on the brain damage induced by HH, leading to a surge in research focused on its potential applications. However, enhancing the stability and targeting of HPN remains a challenging issue that needs to be addressed. In this research, a mitochondria-targeted HPN (TPP-C6-HPN) was prepared to deliver HPN to mitochondria. Then, using PLGA as carrier, TPP-C6-HPN loaded nanoparticles were prepared by lotion solvent evaporation method to improve their stability. This novel drug delivery system exhibited excellent protective property against HH-induced brain injury and good safety.
Mitochondria are the main site of ROS production in cells.24 Hypoxia leads to mitochondrial damage, increased production of ROS, and irreversible cellular damage.25 Although HPN is effective in scavenging ROS in cells, it is unable to enter mitochondria. To address this issue, we designed and synthesized TPP-C6-HPN according the design concept for Mito-TEMPO, which is a mitochondrial targeted antioxidant. Although HPN can effectively eliminate ROS in cells, it cannot enter mitochondria. To address this issue, we designed and synthesized TPP-C6-HPN based on Mito-TEMPO’s design philosophy, which is a mitochondrial targeted antioxidant. It has been reported that the connection of TPP targeting groups can promote the accumulation of targeted molecules in mitochondria, enabling direct clearance of mitochondrial ROS.26
NPs offer a platform to encapsulate drugs within colloidal particles, thereby enhancing drug’s stability and extending the drug’s duration of action within the body. PLGA-NPs have been proved to be an effective approach for neuroprotective drug delivery.27 In this study, we employed PLGA as the carrier to construct the TPP-C6-HPN nanoparticle drug delivery system utilizing the emulsion solvent evaporation method for improve the stability and circulation time in the body of TPP-C6-HPN. In order to achieve an optimal formulation for the NPs, this study employed BBD to optimize the preparation process. BBD is the main response surface methodology (RSM) in the pharmaceutical field, which has been widely applied in bioengineering processes.28 In this article, BBD was used to optimize the preparation formula of NPs.29,30 Under this optimal condition, TPP-C6-HPN@PLGA-NPs were successfully produced with a small particle size (120.63 nm), PDI (0.302) and zeta potential (−27.93 mV), and a high EE (89.30%) and DL (6.82%). Subsequently, we conducted a series of characterizations on TPP-C6-HPN@PLGA-NPs. As seen from SEM and TEM, TPP-C6-HPN@PLGA-NPs were shown as a core-shell structure and had a smooth appearance. The results of XRD and FTIR confirmed that PLGA successfully encapsulated TPP-C6-HPN and successfully prepared TPP-C6-HPN@PLGA-NPs. We found that TPP-C6-HPN@PLGA-NPs also exhibited good stability. In addition, we also found that the concentration of TPP-C6-HPN@PLGA-NPs in the range of 6.25 to 100 µg/mL did not cause any significant hemolysis (less than 5%), which indicated that TPP-C6-HPN@PLGA-NPs was safe for intravenous injection.31 The release profile showed no initial burst phase but exhibited sustained release effect over time due to the nanoparticle degradation and relaxation. These characterization results collectively confirmed the successful preparation and good performance of TPP-C6-HPN@PLGA-NPs.
To further explore the potential of TPP-C6-HPN@PLGA-NPs in biomedical applications, we first employed an CCk-8 assay to study the toxicity of TPP-C6-HPN@PLGA-NPs on PC12 cells after 24 h of incubation. It is reported that when drugs are prepared into nanomedicines, their toxic side effects can be reduced.32 Similarly, in the current study, we found that TPP-C6-HPN@PLGA-NPs was safer for PC12 cells compared to TPP-C6-HPN. Treatment with TPP-C6-HPN@PLGA-NPs at the concentration tested did not result in a significant loss of PC12 cell viability, whereas the cell viability was significantly inhibited by TPP-C6-HPN at concentrations higher than 1.5 × 10−9 mol/L. In addition, incubation of PC12 cells with the Cy5.5@PLGA-NPs resulted in intracellular accumulation of the fluorophores over 24 h. Previous reports have shown that loading drugs with PLGA can improve the ability of drug delivery to the brain.33,34 Similarly, the small animal imaging results showed that the fluorescence could be quickly observed in the brain tissue following administration, and last for 24 hours, indicating that the nanoparticles could enter the brain tissue with long residence time. The above characteristics contributed to the protective effect of TPP-C6-HPN@PLGA-NPs on brain injury.
In the present study, we initially accessed the anti-hypoxia activity of TPP-C6-HPN@PLGA-NPs applying normobaric hypoxia test, and found that TPP-C6-HPN@PLGA-NPs could significantly extend the survival time of mice under hypoxia condition, exhibiting excellent anti-hypoxia capacity. Further, we examined the protective effect of TPP-C6-HPN@PLGA-NPs in a mice model of HH brain injury. Consistent with previous studies,35,36 our current experiment also observed that HH could induce pathological lesions of brain tissue. Whereas TPP-C6-HPN@PLGA-NPs treatment mitigated the brain histopathological harm, offering good protection against HH caused brain injury.
The role of oxidative stress in the brain damage induced by HH has been well documented.9,37 Maintaining cellular redox balance is crucial, and it relies on the equilibrium between ROS generation and their neutralization by the cell’s antioxidant defenses.38 However, during exposure to HH, an excessive amount of ROS is produced, resulting in an increase in lipid peroxidation products such as MDA and a decrease in the activity of endogenous antioxidant enzymes such as SOD and GSH-Px. This imbalance can lead to severe cellular damage.7 Similar changes were also observed in our study, whereas administration of TPP-C6-HPN@PLGA-NPs reduced the H2O2 and MDA production and raised the SOD activity and GSH content in the brains of mice exposed to HH. The results demonstrated that the inhibition of TPP-C6-HPN against HH-induced damage may be closely related to its effective free radical scavenging activity, which was consistent with our previous and other studies about the protective mechanism of NIT radicals.39–41 Mitochondria, which utilize oxygen to generate ATP through oxidative phosphorylation, are powerhouse of cells42 and the dominate oxygen-consuming organelles. Under hypoxic conditions, the aberrant increase of ROS triggers the disruption of mitochondrial structure and function, which has been confirmed to be associated with neuronal cell death.43 Studies have shown that mitochondrial dysfunction is one of the important causes of brain damage caused by high-altitude hypoxia. The damage process is often accompanied by mitochondrial oxidative phosphorylation and dysfunction of the tricarboxylic acid cycle, as well as an increase in mitochondrial free radical release.44–46 Consistent with previous research findings, in this study, we also observed energy metabolism disruption in the brain tissue of mice exposed to HH. TPP-C6-HPN@PLGA-NPs treatment could partly enhance the energy metabolism, evidenced by increasing the ATP content and the activities of Na+-K+-ATPase, Ca2+-Mg2+-ATPase. In addition, previous studies have also shown a close association between HH-induced brain damage and neuroinflammation. Uncontrolled neuro-inflammation can cause neuronal damage and induce neural cell death, which could ultimately lead to brain damage.8 In the present study, we also found that HH leads to a decrease in anti-inflammatory cytokines (IL-10) and an increase in pro-inflammatory cytokines (TNF-α and IL-6) in the serum as well as in brain tissue of mice. TPP-C6-HPN@PLGA-NPs treatment could prevent this abnormality.
The two important characteristics that drugs need to possess for the human body are safety and effectiveness. These above results indicated that TPP-C6-HPN@PLGA-NPs showed significant protection against HH-induced brain injury, proving the efficacy of TPP-C6-HPN@PLGA-NPs. PLGA NPs are rapidly recognized by the mononuclear phagocyte system (MPS), particularly Kupffer cells in the liver, following intravenous administration. This phenomenon is a well-documented challenge in nanoparticle-based drug delivery systems.47 For the safety concern, we conducted the potential toxicity study in vivo. No significant differences in body-weight of the mice were observed between control and TPP-C6-HPN@PLGA-NPs groups. The HE-stained pathological sections displayed that TPP-C6-HPN and TPP-C6-HPN@PLGA-NPs did not harm the histological structure of the major organs. Likewise, the hematological tests and serum biochemicals tests did not show any toxicity of TPP-C6-HPN@PLGA-NPs on the blood cells and the function of the liver (represented by ALT, AST and ALB) and kidney (represented by and BUN). These results suggested the safety of TPP-C6-HPN@PLGA-NPs, which would be a prerequisite for its practical use against HH-induced brain injury.
There are some limitations in this study. First, the stability and controlled release effect of TPP-C6-HPN@PLGA-NPs in body should be further substantiated by the pharmacokinetic experiments conducted in vivo. Second, besides directly scavenging free radical, whether there are other mechanisms of TPP-C6-HPN@PLGA-NPs against HH-induced brain injury remains to be elucidated. These issues will be solved in further study.
Conclusion
In the present study, the HPN derivative with mitochondrial targeting group TPP+ (TPP-C6-HPN) was successfully synthesized. In order to improve the stability of TPP-C6-HPN and prolong its circulation time in vivo, TPP-C6-HPN nanoparticles loaded with PLGA were developed, and the preparation process was optimized using BBD. Under optimal conditions, TPP-C6-HPN@PLGA-NPs exhibits good stability, low hemolysis rate, sustained release, low toxicity, and long residence time in brain tissue. Furthermore, TPP-C6-HPN@PLGA-NPs exhibited good safety and excellent protective effect against HH-induced brain damage. These results indicate that TPP-C6-HPN@PLGA-NPs have great potential as drug delivery systems and are promising formulations in the management of brain injury induced by HH.
Institutional Review Board Statement
The animal study protocol was approved by the Animal Care and Use Committee of 940th Hospital (2019XYLL059).
Data Sharing Statement
Data supporting the findings of this study are available from the corresponding author upon request.
Acknowledgments
We thank Professor Meng Li from the Air Force Medical University for assistance in technical support for in vivo imaging of small animals.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (82001994, 82241062, 81973236), the Institutional Foundation of The First Affiliated Hospital of Xi’an Jiaotong University (2022MS-11) and the Institutional Foundation of Air Force Medical University (2024JC054).
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. Ma YY, Li X, Yu ZY, et al. Oral antioxidant edaravone protects against cognitive deficits induced by chronic hypobaric hypoxia at high altitudes. Transl Psychiatry. 2024;14(1):415. doi:10.1038/s41398-024-03133-1
2. Chen X, Zhang J, Lin Y, et al. Mechanism, prevention and treatment of cognitive impairment caused by high altitude exposure. Front Physiol. 2023;14:1191058. doi:10.3389/fphys.2023.1191058
3. Burtscher M, Hefti U, Hefti JP. High-altitude illnesses: old stories and new insights into the pathophysiology, treatment and prevention. Sports Med Health Sci. 2021;3(2):59–69. doi:10.1016/j.smhs.2021.04.001
4. Luks AM, Swenson ER, Bärtsch P. Acute high-altitude sickness. Eur Respir Rev. 2017;26(143):160096. doi:10.1183/16000617.0096-2016
5. Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5(6):437–448. doi:10.1038/nrn1408
6. Subudhi AW, Dimmen AC, Julian CG, et al. Effects of Acetazolamide and dexamethasone on cerebral hemodynamics in hypoxia. J Appl Physiol. 2011;110(5):1219–1225. doi:10.1152/japplphysiol.01393.2010
7. Li X, Zhang J, Liu G, et al. High altitude hypoxia and oxidative stress: the new hope brought by free radical scavengers. Life Sci. 2024;336:122319. doi:10.1016/j.lfs.2023.122319
8. Li D, Zhang L, Huang X, et al. WIP1 phosphatase plays a critical neuroprotective role in brain injury induced by high-altitude hypoxic inflammation. Neurosci Bull. 2017;33(3):292–298. doi:10.1007/s12264-016-0095-9
9. Pena E, El Alam S, Siques P, Brito J. Oxidative stress and diseases associated with high-altitude exposure. Antioxidants. 2022;11(2):267. doi:10.3390/antiox11020267
10. Murray AJ, Horscroft JA. Mitochondrial function at extreme high altitude. J Physiol. 2016;594(5):1137–1149. doi:10.1113/jp270079
11. Zielonka J, Joseph J, Sikora A, et al. Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev. 2017;117(15):10043–10120. doi:10.1021/acs.chemrev.7b00042
12. Hu Q, Ren J, Li G, et al. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018;9(3):403. doi:10.1038/s41419-018-0436-x
13. Jiang Q, Yin J, Chen J, et al. Mitochondria-targeted antioxidants: a step towards disease treatment. Oxid Med Cell Longev. 2020;2020:8837893. doi:10.1155/2020/8837893
14. Wu Y, Bi L, Bi W, et al. Novel 2-substituted nitronyl nitroxides as free radical scavengers: synthesis, biological evaluation and structure-activity relationship. Bioorg Med Chem. 2006;14(16):5711–5720. doi:10.1016/j.bmc.2006.04.016
15. Luo H, Sun W, Shao J, et al. Protective effect of nitronyl nitroxide against hypoxia-induced damage in PC12 cells. Biochem Cell Biol. 2020;98(3):345–353. doi:10.1139/bcb-2019-0269
16. Qian J, Liang T, Xu Y, et al. Effect of the novel free radical scavenger 4’-hydroxyl-2-substituted phenylnitronyl nitroxide on oxidative stress, mitochondrial dysfunction and apoptosis induced by cerebral ischemia-reperfusion in rats. Neuroscience. 2024;540:1–11. doi:10.1016/j.neuroscience.2023.12.016
17. Fan PC, Ma HP, Jing LL, Li L, Jia ZP. The antioxidative effect of a novel free radical scavenger 4’-hydroxyl-2-substituted phenylnitronyl nitroxide in acute high-altitude hypoxia mice. Biol Pharm Bull. 2013;36(6):917–924. doi:10.1248/bpb.b12-00854
18. Jing L, Da Q, Zhang S, et al. Nitronyl nitroxide ameliorates hypobaric hypoxia-induced cognitive impairment in mice by suppressing the oxidative stress, inflammatory response and apoptosis. Neurochem Res. 2024;49(3):785–799. doi:10.1007/s11064-023-04080-x
19. Yoshitomi T, Nagasaki Y. Nitroxyl radical-containing nanoparticles for novel nanomedicine against oxidative stress injury. Nanomedicine. 2011;6(3):509–518. doi:10.2217/nnm.11.13
20. Beach MA, Nayanathara U, Gao Y, et al. Polymeric nanoparticles for drug delivery. Chem Rev. 2024;124(9):5505–5616. doi:10.1021/acs.chemrev.3c00705
21. El-Hammadi MM, Arias JL. Recent advances in the surface functionalization of PLGA-based nanomedicines. Nanomaterials. 2022;12(3):354. doi:10.3390/nano12030354
22. Danhier F, Ansorena E, Silva JM, et al. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. doi:10.1016/j.jconrel.2012.01.043
23. Guo X, Zuo X, Zhou Z, et al. PLGA-based micro/nanoparticles: an overview of their applications in respiratory diseases. Int J Mol Sci. 2023;24(5):4333. doi:10.3390/ijms24054333
24. Hernansanz-Agustín P, Enríquez JA. Generation of reactive oxygen species by mitochondria. Antioxidants. 2021;10(3): 415. doi:10.3390/antiox10030415
25. Chen R, Lai UH, Zhu L, et al. Reactive oxygen species formation in the brain at different oxygen levels: the role of hypoxia inducible factors. Front Cell Dev Biol. 2018;6:132. doi:10.3389/fcell.2018.00132
26. Shetty S, Kumar R, Bharati S. Mito-TEMPO, a mitochondria-targeted antioxidant, prevents N-nitrosodiethylamine-induced hepatocarcinogenesis in mice. Free Radic Biol Med. 2019;136:76–86. doi:10.1016/j.freeradbiomed.2019.03.037
27. Cunha A, Gaubert A, Latxague L, Dehay B. PLGA-based nanoparticles for neuroprotective drug delivery in neurodegenerative diseases. Pharmaceutics. 2021;13(7): 1042. doi:10.3390/pharmaceutics13071042
28. Bezerra MA, Santelli RE, Oliveira EP, Villar LS, Escaleira LA. Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta. 2008;76(5):965–977. doi:10.1016/j.talanta.2008.05.019
29. Abul Kalam M, Khan AA, Khan S, Almalik A, Alshamsan A. Optimizing indomethacin-loaded chitosan nanoparticle size, encapsulation, and release using Box-Behnken experimental design. Int J Biol Macromol. 2016;87:329–340. doi:10.1016/j.ijbiomac.2016.02.033
30. Kraisit P, Yonemochi E, Furuishi T, Mahadlek J, Limmatvapirat S. Chitosan film containing antifungal agent-loaded SLNs for the treatment of candidiasis using a Box-Behnken design. Carbohydr Polym. 2022;283:119178. doi:10.1016/j.carbpol.2022.119178
31. Zhang CX, Cheng Y, Liu DZ, et al. Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J Nanobiotechnol. 2019;17(1):18. doi:10.1186/s12951-019-0451-9
32. Wolfram J, Zhu M, Yang Y, et al. Safety of nanoparticles in medicine. Curr Drug Targets. 2015;16(14):1671–1681. doi:10.2174/1389450115666140804124808
33. Zhi K, Raji B, Nookala AR, et al. PLGA nanoparticle-based formulations to cross the blood-brain barrier for drug delivery: from R&D to cGMP. Pharmaceutics. 2021;13(4): 500. doi:10.3390/pharmaceutics13040500
34. Yavarpour-Bali H, Ghasemi-Kasman M, Pirzadeh M. Curcumin-loaded nanoparticles: a novel therapeutic strategy in treatment of central nervous system disorders. Int J Nanomed. 2019;14:4449–4460. doi:10.2147/ijn.S208332
35. Li N, Chen K, Bai J, et al. Tibetan medicine Duoxuekang ameliorates hypobaric hypoxia-induced brain injury in mice by restoration of cerebrovascular function. J Ethnopharmacol. 2021;270:113629. doi:10.1016/j.jep.2020.113629
36. Liu F, He H, Yang W, et al. Novel energy optimizer, meldonium, rapidly restores acute hypobaric hypoxia-induced brain injury by targeting phosphoglycerate kinase 1. Cell Commun Signal. 2024;22(1):383. doi:10.1186/s12964-024-01757-w
37. Gaur P, Prasad S, Kumar B, Sharma SK, Vats P. High-altitude hypoxia induced reactive oxygen species generation, signaling, and mitigation approaches. Int J Biometeorol. 2021;65(4):601–615. doi:10.1007/s00484-020-02037-1
38. Kozlov AV, Javadov S, Sommer N. Cellular ROS and antioxidants: physiological and pathological role. Antioxidants. 2024;13(5):602. doi:10.3390/antiox13050602
39. Jing L, Huang Y, Ma H, Wang H. A novel nitronyl nitroxide radical HPN-C(6) attenuates brain damage in an acute hypobaric hypoxia mouse model through inhibition of the oxidative stress. Neurosci Lett. 2022;782:136650. doi:10.1016/j.neulet.2022.136650
40. Wang H, Wang J, Yang Q, et al. Synthesis of a novel nitronyl nitroxide radical and determination of its protective effects against infrasound-induced injury. Neurochem Res. 2015;40(7):1526–1536. doi:10.1007/s11064-015-1602-5
41. Luo G, Sun L, Li H, et al. The potent radioprotective agents: novel nitronyl nitroxide radical spin-labeled resveratrol derivatives. Fitoterapia. 2021;155:105053. doi:10.1016/j.fitote.2021.105053
42. Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020;37:101674. doi:10.1016/j.redox.2020.101674
43. Burtscher J, Mallet RT, Pialoux V, Millet GP, Burtscher M. Adaptive responses to hypoxia and/or hyperoxia in humans. Antioxid Redox Signal. 2022;37(13–15):887–912. doi:10.1089/ars.2021.0280
44. Hou Y, Fan F, Xie N, et al. Rhodiola crenulata alleviates hypobaric hypoxia-induced brain injury by maintaining BBB integrity and balancing energy metabolism dysfunction. Phytomedicine. 2024;128:155529. doi:10.1016/j.phymed.2024.155529
45. Murray AJ. Energy metabolism and the high-altitude environment. Exp Physiol. 2016;101(1):23–27. doi:10.1113/ep085317
46. Wang X, Tang Y, Xie N, et al. Salidroside, a phenyl ethanol glycoside from Rhodiola crenulata, orchestrates hypoxic mitochondrial dynamics homeostasis by stimulating Sirt1/p53/Drp1 signaling. J Ethnopharmacol. 2022;293:115278. doi:10.1016/j.jep.2022.115278
47. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348. doi:10.1016/j.jconrel.2016.01.020
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.